DOI:
10.1039/D4MH01047G
(Communication)
Mater. Horiz., 2025,
12, 227-237
Research on advanced photoresponsive azobenzene hydrogels with push–pull electronic effects: a breakthrough in photoswitchable adhesive technologies†
Received
9th August 2024
, Accepted 18th October 2024
First published on 25th October 2024
Abstract
Smart materials that adapt to various stimuli, such as light, hold immense potential across many fields. Photoresponsive molecules like azobenzenes, which undergo E–Z photoisomerization when exposed to light, are particularly valuable for applications in smart coatings, light-controlled adhesives, and photoresists in semiconductors and integrated circuits. Despite advances in using azobenzene moieties for stimuli-responsive adhesives, the role of push–pull electronic effects in regulating reversible adhesion remains largely unexplored. In this study, we investigate for the first time photo-controlled hydrogel adhesives of azobenzene with different push–pull electronic groups. We synthesized the monomers 4-methoxyazobenzene acrylate (ABOMe), azobenzene acrylate (ABH), and 4-nitroazobenzene acrylate (ABNO2), and examined their effects on reversible adhesion properties. By incorporating these azobenzene monomers into acrylamide, dialdehyde-functionalized poly(ethylene glycol), and [3-(methacryloylamino)propyl]-trimethylammonium chloride, we prepared ABOMe, ABH, and ABNO2 ionic hydrogels. Our research findings demonstrate that only the ABOMe ionic hydrogel exhibits reversible adhesion. This is due to its distinct transition state mechanism compared to ABH and ABNO2, which enables efficient E–Z photoisomerization and drives its reversible adhesion properties. Notably, the ABOMe ionic hydrogel reveals an outstanding skin adhesion strength of 360.7 ± 10.1 kPa, surpassing values reported in current literature. This exceptional adhesion is attributed to Schiff base reactions, monopole–quadrupole interactions, π–π interactions, and hydrogen bonding with skin amino acids. Additionally, the ABOMe hydrogel exhibits excellent reversible self-healing capabilities, significantly enhancing its potential for injectable medical applications. This research underscores the importance of integrating multifunctional properties into a single system, opening new possibilities for innovative and durable adhesive materials.
New concepts
This study pioneers the integration of push–pull electronic effects with azobenzene-based ionic hydrogels, marking a breakthrough in photo-controlled, reversible adhesion. Unlike previous work that explored azobenzene for stimuli-responsive adhesives, our approach uniquely regulates adhesion by modifying the electronic structure of azobenzene monomers. Among the synthesized ionic hydrogels-ABOMe, ABH, and ABNO2 – only the ABOMe ionic hydrogel demonstrates efficient, reversible adhesion due to its distinct rotation-type E–Z photoisomerization. With exceptional skin adhesion strength (360.7 ± 10.1 kPa), underwater adhesive properties, and self-healing capabilities, ABOMe ionic hydrogels set a new benchmark for smart adhesives in biomedical and wearable applications. This innovative system leverages Schiff base reactions, cation–π interactions, π–π stacking, and hydrogen bonding, presenting a new framework for multifunctional materials. Our findings highlight how electronic modifications can significantly enhance the design of stimuli-responsive materials for dynamic environments, paving the way for next-generation adhesive technologies.
|
1. Introduction
Smart materials, also called advanced materials or intelligent materials, can alter or modify their physical and/or chemical properties in response to internal or external stimuli, such as changes in light, temperature, pH, or magnetism.1–5 This adaptability allows them to be widely applied in fields like supramolecular chemistry, biochemistry, and molecular machines.6–10 Among these stimuli, light is especially attractive due to its non-invasive and low-pollution nature, as well as its ability to be applied externally with high spatial and temporal resolution, enabling selective, fast, and high-yielding photochemical processes, such as isomerization.11 Numerous photoresponsive or photochromic molecules, including azobenzenes, stilbenes, spiropyrans, and diarylethenes, are recognized to undergo conformational changes or bond breaking/formation under specific wavelengths of light.12–18 This makes them promising candidates for engineering applications such as smart coatings for optical devices, light-controlled adhesives, photothermal sensors, robotic fingers, and photoresists in semiconductors and IC applications.19–26
Azobenzene is a photoswitchable molecule that undergoes E–Z photoisomerization under light irradiation, allowing it to switch its glass transition temperature or melting point. It is well-known for its photochemical solid–liquid transition properties. Recently, research has focused on utilizing the solid–liquid transition characteristics of azobenzene to develop stimuli-responsive smart switchable adhesives.27–31 For instance, Kong and Wu investigated azobenzene-based polymers as light-controlled adhesives with photoinduced reversible solid–liquid transitions. Their materials demonstrated reversible adhesion to various substrates with different wettability, chemical composition, and surface roughness.27 Akiyama's research team synthesized side chain-type azobenzene polymers and tested the solid–liquid transitions of different alkyl chain azobenzene polymers. They found that polymers with longer alkyl chains exhibited greater adhesive strength in photosetting samples compared to thermosetting samples, achieving light-reversible or heat-reversible adhesion.28 Yu and Wang designed and synthesized dendritic azobenzene polymers, which served as adhesives with strong adhesive strength under visible light and reduced adhesion under UV irradiation. Their tests on the adhesion properties of varying amounts of dendritic polymers showed that adhesive strength increased with the number of dendritic polymers.29 Despite these advances, the use of the push–pull electronic effects of azobenzene to regulate reversible adhesion remains unexplored.
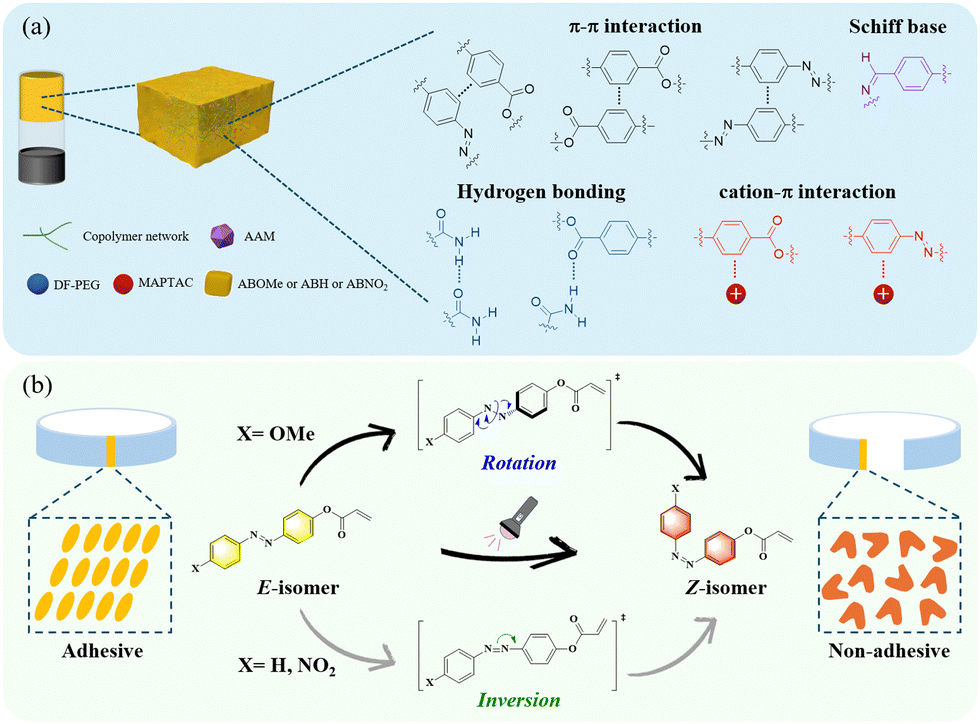
In this work, we investigated for the first time the photo-controlled hydrogel adhesives of azobenzene with different push–pull electronic groups. We systematically explored the effects of substituents on the reversible adhesion properties to substrates and the underlying mechanisms by synthesizing the monomers 4-methoxyazobenzene acrylate (ABOMe), azobenzene acrylate (ABH), and 4-nitroazobenzene acrylate (ABNO2). On the other hand, cation–π interactions, also known as monopole–quadrupole interactions, are a type of electrostatic attraction between positively charged cations and negatively charged electron-rich π systems. These interactions are widely used in nature to regulate and stabilize the structure and function of various biomolecules.32–35 Utilizing cation–π interactions, recent studies by Gu and Yan have shown that strong and reversible non-covalent cation–π interactions exist between amine and aromatic components in adjacent polymer chains, imparting excellent self-healing properties and tissue adhesiveness to hydrogels.36 To broaden the potential applications of our hydrogels, we also incorporated the cationic monomer [3-(methacryloylamino)propyl]-trimethylammonium chloride (MAPTAC).37 We anticipated that MAPTAC would form cation–π interactions with the aromatic moiety, enhancing the mechanical properties of the hydrogels and providing excellent self-healing capabilities. Based on the above discussion, we successfully prepared ABOMe, ABH, and ABNO2 ionic hydrogels and found that only the ABOMe ionic hydrogel exhibited reversible adhesion properties (Fig. 1). This reversibility is due to the E–Z photoisomerization of the azobenzene moiety. Upon further investigation of the photoisomerization mechanism of these azobenzene derivatives, our research revealed that ABOMe undergoes a different photoisomerization process compared to ABH and ABNO2. The transition state mechanism of ABOMe is rotation-type, which enables rapid E–Z photoisomerization and thus confers reversible adhesion properties. Notably, the ABOMe ionic hydrogel demonstrates an outstanding skin adhesion strength of 360.7 ± 10.1 kPa, surpassing the values reported in the current literature. This strong adhesion is likely due to Schiff base reactions, monopole–quadrupole (cation–π) interactions, π–π interactions, and hydrogen bonding between the ionic hydrogels and the amino acids on the skin surface. Furthermore, the ABOMe ionic hydrogel exhibits excellent reversible self-healing capabilities, ensuring the longevity and durability of the hydrogel, which brings additional advantages for practical applications. The ABOMe ionic hydrogel's unique combination of high skin adhesion strength, rapidly reversible adhesion, and excellent reversible self-healing capabilities makes it a promising candidate for applications requiring dynamic and durable adhesive materials.
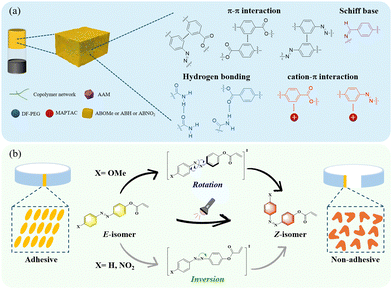 |
| | Fig. 1 (a) The proposed possible intermolecular interactions of azobenzene-based ionic hydrogels (AAM is acrylamide, DF-PEG is dialdehyde-functionalized poly(ethylene glycol), and MAPTAC is [3-(methacryloylamino)propyl]-trimethylammonium chloride; different substituents of the azobenzene monomer are ABOMe, ABH, and ABNO2, and the copolymer network is formed by the copolymerization of AAM, MAPTAC, and the azobenzene monomer.). (b) The driving forces generating reversible adhesion and their photoisomerization mechanism process. | |
2. Results and discussion
2.1. Hydrogel preparation and mechanical property analysis
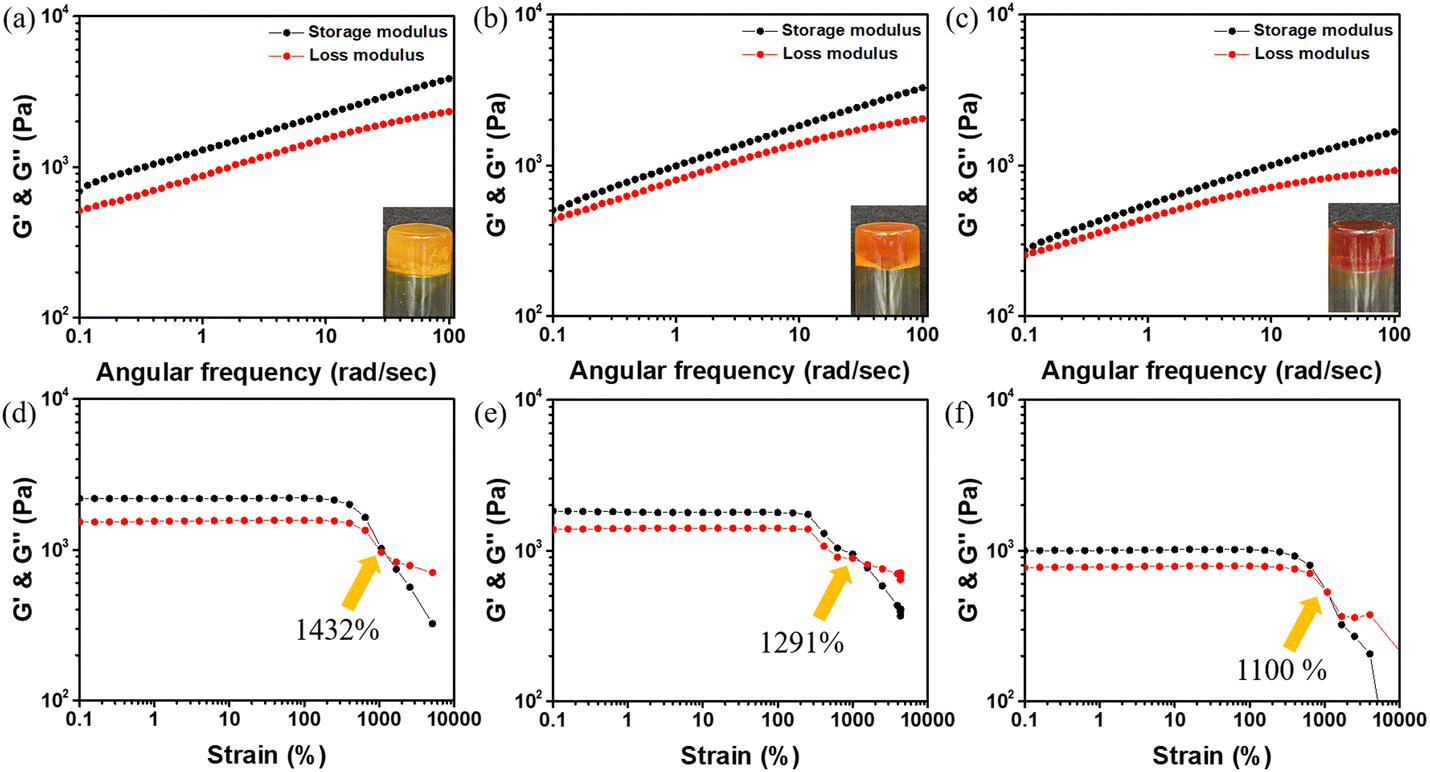
The synthesis methods for the monomers ABOMe, ABH, and ABNO2 are shown in Scheme S1 (ESI†). First, p-anisidine undergoes a diazotization reaction by treatment with sodium nitrite in the presence of an acid, followed by the addition of phenol to obtain (E)-4-((4-methoxyphenyl)diazenyl)phenol. Finally, esterification using acryloyl chloride produces the ABOMe monomer as a yellow solid. By replacing p-anisidine with aniline or p-nitroaniline, the monomers ABH (an orange solid) and ABNO2 (a red solid) can be synthesized, respectively. After obtaining azobenzene monomers with different substituents, we proceeded with the preparation of hydrogels. Here, we use acrylamide (AAM) and dialdehyde-functionalized poly(ethylene glycol) (DF-PEG) as the base gel, as this combination has been proven to exhibit dynamic covalent (Schiff base reaction) and noncovalent (hydrogen bonding) interactions.38 Based on AAM and DF-PEG, we synthesized ABOMe, ABH, and ABNO2 hydrogels by incorporating the respective monomers. To further enhance the multifunctionality of these hydrogels, we integrated MAPTAC into the azobenzene-based hydrogels, creating novel ABOMe, ABH, and ABNO2 ionic hydrogels. In total, we prepared six azobenzene-based hydrogels, with detailed formulations provided in Table S1 (ESI†). Fig. 2 and Fig. S1 (ESI†) show the optical images of the six hydrogels and their corresponding rheological properties. It was observed that the ABOMe hydrogel and ABOMe ionic hydrogel appeared as yellow opaque hydrogels, the ABH hydrogel and ABH ionic hydrogel appeared as orange opaque hydrogels, and the ABNO2 hydrogel and ABNO2 ionic hydrogel appeared as red opaque hydrogels. To evaluate the effect of different azobenzene substituents and the addition of MAPTAC on the mechanical properties of the hydrogels, rheological tests were conducted.39,40 As shown in Fig. 2 and Fig. S1 (ESI†), the G′ (storage modulus) values of the ABOMe hydrogel, ABH hydrogel, ABNO2 hydrogel, ABOMe ionic hydrogel, ABH ionic hydrogel, and ABNO2 ionic hydrogel were 1285 Pa, 1067 Pa, 709 Pa, 1909 Pa, 1540 Pa, and 830 Pa, respectively. All six hydrogels exhibited G′ values greater than G′′ (loss modulus), indicating their elastic behavior.41 From the G′ values, it was detected that the hydrogel with the methoxy group on azobenzene exhibited the highest G′, followed by the unsubstituted azobenzene, while the nitro-substituted azobenzene had the lowest G′. This is likely due to the hydrogen bonding provided by the methoxy group, which enhances the strength of the hydrogel.42,43 Additionally, the ionic hydrogels showed higher G′ values than their corresponding non-ionic hydrogels, and the G′/G′′ ratio was also higher, indicating that the incorporation of MAPTAC provides additional ionic electrostatic interactions and cation–π interactions to stabilize the hydrogel system.44–46 The strain amplitude sweep data revealed that the G′ and G′′ curves intersected at strains of 680%, 646%, 411%, 1432%, 1291%, and 1100% for the ABOMe hydrogel, ABH hydrogel, ABNO2 hydrogel, ABOMe ionic hydrogel, ABH ionic hydrogel, and ABNO2 ionic hydrogel, respectively (Fig. 2 and Fig. S1, ESI†). This indicates that the critical transition point between the solid and fluid state occurs in the order of the ABOMe ionic hydrogel, ABH ionic hydrogel, ABNO2 ionic hydrogel, ABOMe hydrogel, AB hydrogel, and ABNO2 hydrogel. These findings suggest that the ionic hydrogels, particularly those with the methoxy group, exhibit greater resistance to deformation and maintain their solid-like behavior over a broader range of strains. The incorporation of the ionic component MAPTAC enhances the structural integrity and stability of the hydrogels, likely due to the synergistic effects of cation–π interactions and electrostatic interactions.44–46 This makes the ionic hydrogels more robust and versatile for potential applications where mechanical resilience and flexibility are required. Therefore, we proceeded to investigate the adhesive performance of ABOMe, ABH, and ABNO2 ionic hydrogels and evaluate their versatility and practicality in real-world scenarios. Moreover, we conducted cytotoxicity tests on these ionic gels (see details in the ESI†). Fig. S2 (ESI†) shows that the hydrogels maintained cell viability above 85% over five days, indicating excellent biocompatibility and potential for expansion into biomedical applications.
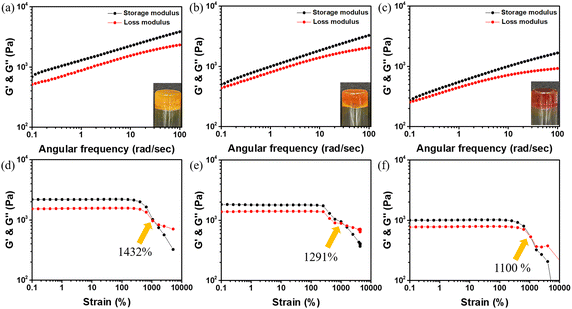 |
| | Fig. 2 Frequency dependence (ω = 0.1–100 rad s−1) of rheology measurement of (a) ABOMe ionic hyderogel, (b) ABH ionic hydrogel, and (c) ABNO2 ionic hydrogel. Inset: optical images of the corresponding hydrogels. Strain dependence of G′ (black) and G′′ (red) of the (d) ABOMe ionic hydrogel, (e) ABH ionic hydrogel, and (f) ABNO2 ionic hydrogel. | |
2.2. Effect of azobenzene substituent groups on reversible adhesive properties
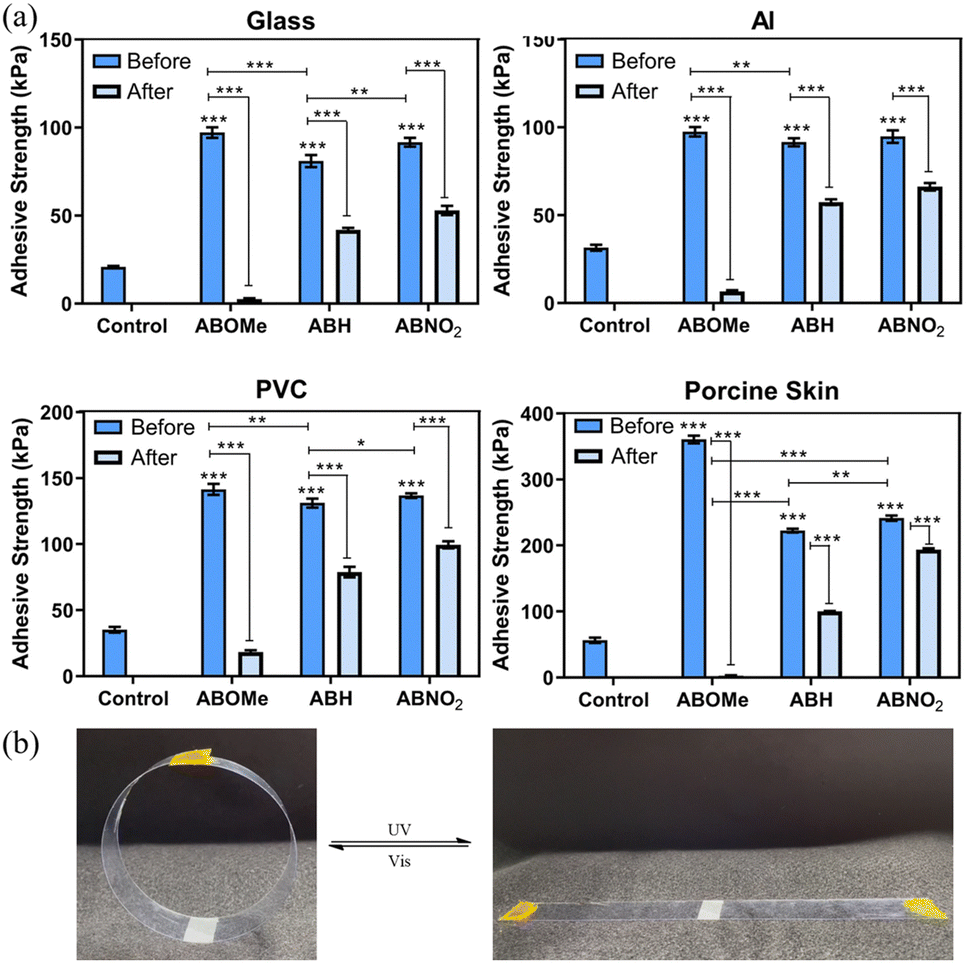
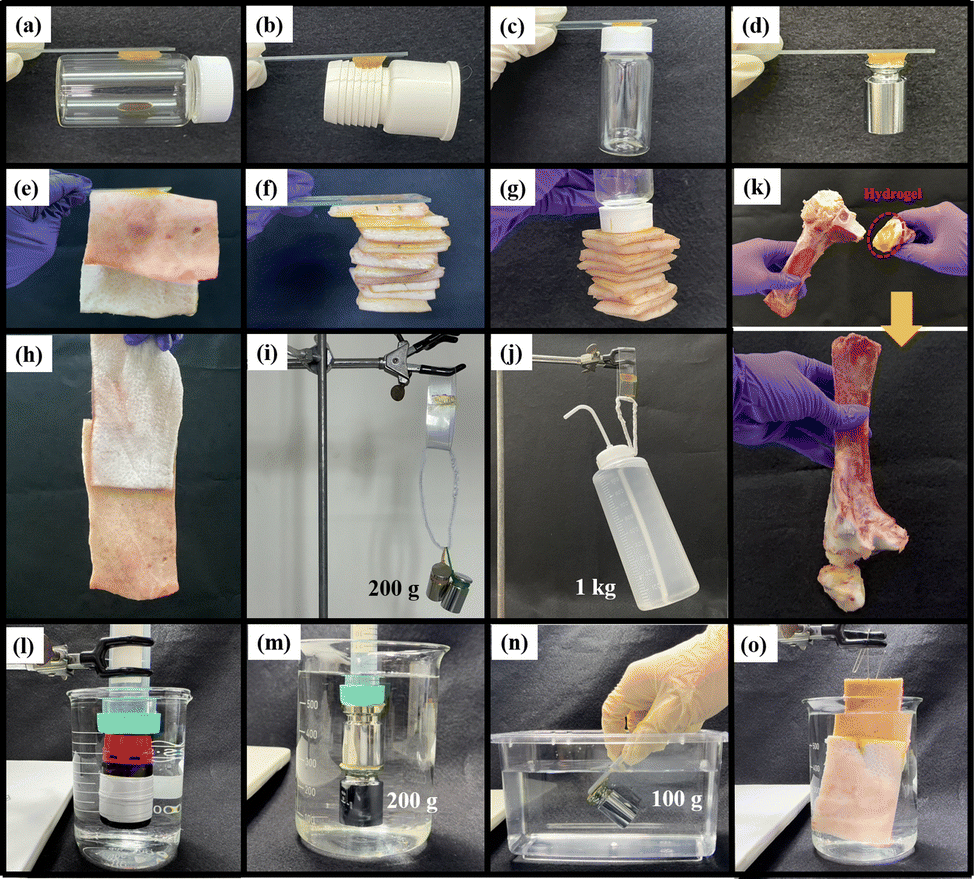
To objectively demonstrate the adhesion properties of the hydrogels, we used lap shear strength testing to measure the adhesive capabilities of ABOMe, ABH, and ABNO2 ionic hydrogels. We used pairs of glass slides, aluminum (Al) sheets, polyvinyl chloride (PVC) sheets, and porcine skin, fixing a 2 cm × 2 cm area of hydrogel between each pair to investigate the degree of adhesion. For comparison, we used ionic hydrogels without azobenzene derivatives as the control. As shown in Fig. 3a and Fig. S3, Table S2 (ESI†), ABOMe ionic hydrogels exhibited the highest adhesion across all substrates, followed by ABNO2, with ABH displaying slightly less adhesion than ABNO2, and the control showing minimal adhesion. Among the different materials, the adhesion to skin was the highest, with the ABOMe gel showing an adhesion strength of 360.7 ± 10.1 kPa. Compared to the adhesion strength of hydrogel materials on porcine skin reported in the literature (Table S3, ESI†), the ABOMe ionic hydrogel demonstrates exceptional adhesion to porcine skin. This strong adhesion may be due to the presence of various amino acids on the skin surface, such as phenylalanine, histidine, and tyrosine, which have the potential to form cation–π interactions with MAPTAC and/or π–π stacking interactions with the aromatic components of azobenzene and DF-PEG.47–50 Additionally, the amino groups of amino acids can form C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N covalent interactions with DF-PEG, while the hydrophilic amino acid side chains may establish hydrogen bonds with the methoxy group.38,51 These intermolecular interactions are likely the driving force behind the enhanced adhesion of the materials to the substrate surface. Given the superior adhesion properties of ABOMe ionic hydrogels, we conducted a series of practical tests to evaluate their performance (Fig. 4). We adhered one end of the hydrogel to glass and attached the other end to various materials, including glass, rubber, plastic, stainless steel, and porcine skin. In another set of tests, we adhered one end to plastic and the other to porcine skin, or both ends to porcine skin. The results were impressive: even when lifting and shaking these samples, the ABOMe ionic hydrogels maintained a strong bond across all these substrates, showcasing their excellent adhesive capabilities. Furthermore, we tested the load-bearing capacity of the ABOMe ionic hydrogels by adhering them to a PVC ring. Remarkably, the hydrogels could support a 200 g weight and even a 1 kg water bottle, highlighting their robust adhesive strength. In addition, the potential applications of ABOMe ionic hydrogels extend to the biomedical field, as they could effectively function as bone adhesives (Fig. 4k). Significantly, the images in Fig. 4l–o demonstrate that the prepared ABOMe ionic hydrogel adhesive also exhibits strong underwater adhesion on a range of substrates. The adhesion of the ABOMe ionic hydrogel in water is likely driven by two factors: topologically enhanced cation–π interactions that provide strong cohesive ability and the aromatic components of azobenzene and DF-PEG, along with the cationic groups in the hydrogel, which create multiple interactions with the substrates, resulting in tight interfacial adhesion.52,53 On the other hand, some studies suggest that azopolymers with E–Z photoisomerization properties can serve as switchable adhesives.27–30 The ability to manipulate adhesion using light is particularly significant for applications requiring reversible bonding and debonding, such as in temporary manufacturing processes, biomedical devices, and smart adhesives. Consequently, we also tested whether photoresponsive adhesion is applicable to ABOMe, ABH, and ABNO2 ionic hydrogels on different substrates (Fig. 3a). The results revealed that the adhesion of ABOMe, ABH, and ABNO2 ionic hydrogels was higher before UV light exposure compared to their corresponding gels after UV light exposure. Surprisingly, only the ABOMe ionic hydrogel became almost non-adhesive upon UV light irradiation (Video S1, ESI†), probably due to the most efficient E–Z photoisomerization of ABOMe. When exposed to visible light, the ABOMe ionic hydrogel regained its adhesion properties. We then utilized PVC sheets to validate the lap shear strength measurements, thoroughly examining the adhesive properties of the hydrogels under various conditions. As shown in Fig. 3b and Video S2 (ESI†), the (E)-ABOMe ionic hydrogel demonstrated excellent adhesion, allowing the PVC sheet ends to stick together, forming a PVC ring. Upon exposure to UV light, the ABOMe ionic hydrogel switched to the Z-isomer, losing its adhesive properties and causing the PVC sheet to unfold. Further exposure to visible light allowed the PVC sheet to adhere together again, reforming the PVC ring. Additionally, the ABOMe ionic hydrogel can undergo reversible photoisomerization multiple times through continuous UV and visible light exposure, with no significant performance degradation (Fig. S4, ESI†). This reversible adhesion demonstrates the potential of using ABOMe ionic hydrogels in applications requiring switchable adhesion properties. Their light-responsive nature provides a versatile, non-invasive method to manage material interactions, enhancing the functionality and applicability of azobenzene-based ionic hydrogels across various fields. As a comparison, the control sample lacking azobenzene did not exhibit sufficient adhesion to bond the PVC sheet ends together (Video S3, ESI†). This underscores the crucial role of azobenzene in serving as an effective substrate adhesive.
N covalent interactions with DF-PEG, while the hydrophilic amino acid side chains may establish hydrogen bonds with the methoxy group.38,51 These intermolecular interactions are likely the driving force behind the enhanced adhesion of the materials to the substrate surface. Given the superior adhesion properties of ABOMe ionic hydrogels, we conducted a series of practical tests to evaluate their performance (Fig. 4). We adhered one end of the hydrogel to glass and attached the other end to various materials, including glass, rubber, plastic, stainless steel, and porcine skin. In another set of tests, we adhered one end to plastic and the other to porcine skin, or both ends to porcine skin. The results were impressive: even when lifting and shaking these samples, the ABOMe ionic hydrogels maintained a strong bond across all these substrates, showcasing their excellent adhesive capabilities. Furthermore, we tested the load-bearing capacity of the ABOMe ionic hydrogels by adhering them to a PVC ring. Remarkably, the hydrogels could support a 200 g weight and even a 1 kg water bottle, highlighting their robust adhesive strength. In addition, the potential applications of ABOMe ionic hydrogels extend to the biomedical field, as they could effectively function as bone adhesives (Fig. 4k). Significantly, the images in Fig. 4l–o demonstrate that the prepared ABOMe ionic hydrogel adhesive also exhibits strong underwater adhesion on a range of substrates. The adhesion of the ABOMe ionic hydrogel in water is likely driven by two factors: topologically enhanced cation–π interactions that provide strong cohesive ability and the aromatic components of azobenzene and DF-PEG, along with the cationic groups in the hydrogel, which create multiple interactions with the substrates, resulting in tight interfacial adhesion.52,53 On the other hand, some studies suggest that azopolymers with E–Z photoisomerization properties can serve as switchable adhesives.27–30 The ability to manipulate adhesion using light is particularly significant for applications requiring reversible bonding and debonding, such as in temporary manufacturing processes, biomedical devices, and smart adhesives. Consequently, we also tested whether photoresponsive adhesion is applicable to ABOMe, ABH, and ABNO2 ionic hydrogels on different substrates (Fig. 3a). The results revealed that the adhesion of ABOMe, ABH, and ABNO2 ionic hydrogels was higher before UV light exposure compared to their corresponding gels after UV light exposure. Surprisingly, only the ABOMe ionic hydrogel became almost non-adhesive upon UV light irradiation (Video S1, ESI†), probably due to the most efficient E–Z photoisomerization of ABOMe. When exposed to visible light, the ABOMe ionic hydrogel regained its adhesion properties. We then utilized PVC sheets to validate the lap shear strength measurements, thoroughly examining the adhesive properties of the hydrogels under various conditions. As shown in Fig. 3b and Video S2 (ESI†), the (E)-ABOMe ionic hydrogel demonstrated excellent adhesion, allowing the PVC sheet ends to stick together, forming a PVC ring. Upon exposure to UV light, the ABOMe ionic hydrogel switched to the Z-isomer, losing its adhesive properties and causing the PVC sheet to unfold. Further exposure to visible light allowed the PVC sheet to adhere together again, reforming the PVC ring. Additionally, the ABOMe ionic hydrogel can undergo reversible photoisomerization multiple times through continuous UV and visible light exposure, with no significant performance degradation (Fig. S4, ESI†). This reversible adhesion demonstrates the potential of using ABOMe ionic hydrogels in applications requiring switchable adhesion properties. Their light-responsive nature provides a versatile, non-invasive method to manage material interactions, enhancing the functionality and applicability of azobenzene-based ionic hydrogels across various fields. As a comparison, the control sample lacking azobenzene did not exhibit sufficient adhesion to bond the PVC sheet ends together (Video S3, ESI†). This underscores the crucial role of azobenzene in serving as an effective substrate adhesive.
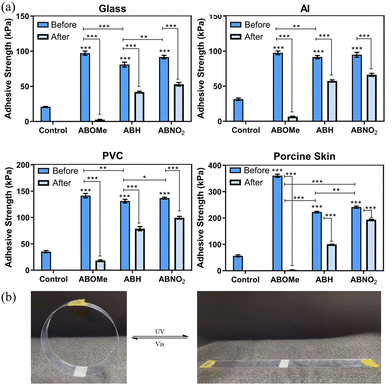 |
| | Fig. 3 (a) Adhesive strength of control, ABOMe, ABH, and ABNO2 ionic hydrogels on various substrates, including glass, Al, PVC, and porcine skin. Statistical significance is indicated by *, **, or ***, representing p < 0.05, p < 0.01, and p < 0.001, respectively. (Dark blue: before UV light irradiation; light blue: after UV light irradiation.) (b) Photoswitchable adhesion of ABOMe ionic hydrogels on PVC (UV light irradiation for 35 seconds; visible light irradiation for 95 seconds). | |
 |
| | Fig. 4 The adhesive properties of the ABOMe ionic hydrogel were evaluated on various substrates: (a) glass/glass, (b) glass/rubber, (c) glass/plastic, (d) glass/stainless steel, (e) and (f) glass/porcine skin, (g) plastic/porcine skin, (h) porcine skin/porcine skin (porcine skin dimensions: 14 cm × 16 cm). (i) and (j) Photographs of the ABOMe ionic hydrogel firmly adhered to PVC, and (k) ABOMe ionic hydrogels are capable of gluing bone together. Underwater adhesive behavior of the ABOMe ionic hydrogel adhesive to various substrates: (l) plastic/plastic, 50 mL bottle filled with water; (m) plastic/stainless steel, stainless steel/stainless steel; (n) glass/stainless steel, inclined at a 45-degree angle in water; and (o) porcine skin/porcine skin. | |
2.3. Impact of azobenzene substituent groups on photoswitchable properties
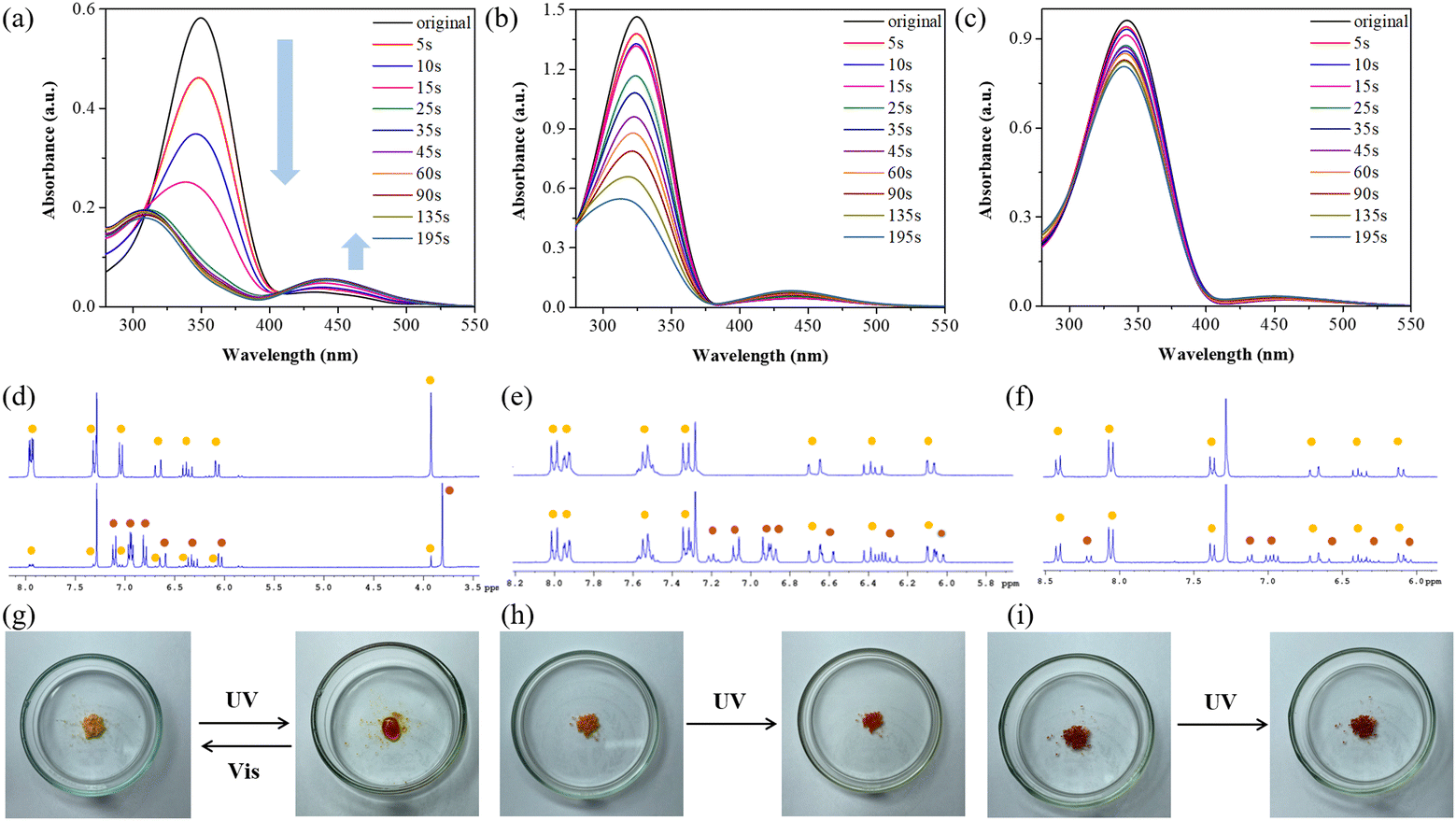
Based on the results of the adhesion experiments discussed earlier, it is evident that azobenzene acts as the primary adhesive source. The difference in adhesion before and after UV light exposure is significant and is strongly influenced by the substituents on azobenzene. To investigate the impact of substituents on the adhesion mechanism of azobenzene-based molecules, we analyzed their UV-vis spectra under UV light irradiation in CHCl3, as illustrated in Fig. 5a–c. Remarkably, ABOMe was the only one to exhibit distinct E–Z isomerization. The absorption spectrum of (E)-ABOMe shows a strong π → π* absorption band at 350 nm and a weaker n → π* absorption band at 425 nm.54,55 Upon conversion to the Z-isomer via UV irradiation, the π → π* transition blue-shifts significantly to 309 nm, while the n → π* transition red-shifts to 441 nm. To determine the time required for the Z-form to reach maximum saturation, the sample was exposed to UV light in 5 or 10-second intervals, with UV-vis spectra recorded after each interval until the conversion was complete. After approximately 35 seconds, the Z-state was observed to be nearly saturated (Fig. 5a). The E–Z isomerization of ABH showed a moderate effect, suggesting a less prominent transition compared to ABOMe. In contrast, the isomerization of ABNO2 was minimal, indicating a minor structural change under the same conditions (Fig. 5b and c). We further used proton nuclear magnetic resonance (1H NMR) spectroscopy to identify the molecular structural changes of these azobenzene derivatives with different electron-donating and electron-withdrawing groups under UV light irradiation. To ensure sufficient UV light exposure time, we chose to irradiate the materials for 195 seconds before performing NMR analysis. This duration was selected based on preliminary tests to ensure that E–Z isomerization had reached a significant extent, allowing for clear differentiation in the NMR spectra (Fig. 5a). As shown in Fig. 5d–f, the integration of the corresponding E–Z isomer proton signals allows us to calculate the conversion ratios. Specifically, (E)-ABOMe converted to the Z-form at a remarkable ratio of 93% under UV light, suggesting that the methoxy substituent on the azobenzene significantly facilitates the isomerization process. Alternatively, (E)-ABH, which lacks the methoxy group, showed a lower conversion ratio of 42%, indicating less efficient isomerization. The electron-withdrawing nitro group in (E)-ABNO2 resulted in the lowest conversion ratio of 20%, demonstrating that the presence of such a group hinders the isomerization process. The NMR analysis results are consistent with the UV-vis spectral experiments, confirming the reliability of our observations. These findings highlight the significant impact of substituents on the photoisomerization behavior of azobenzene derivatives. The high conversion efficiency of ABOMe could be advantageous in applications requiring rapid and efficient switching, while the lower efficiency in ABH and ABNO2 might be useful in scenarios where a more gradual or controlled isomerization is desired. We also irradiated the materials with visible light and used NMR to confirm their molecular structures, demonstrating that ABOMe exhibits reversible E–Z photoisomerization properties. Moreover, considering that azobenzene derivatives are known for their solid-to-liquid transition capabilities, we examined the light-stimulated responses of ABOMe, ABH, and ABNO2 in their solid states.27 Interestingly, only ABOMe transformed from a yellow powder into a viscous liquid after 10 minutes of UV light irradiation. When exposed to visible light, the liquid reverted to a solid within 30 minutes, highlighting its photoinduced reversible solid-to-liquid transition due to reversible E–Z photoisomerization. However, ABH and ABNO2 did not exhibit any solid-to-liquid transition under similar conditions (Fig. 5g–i). These experimental results, including UV-vis and NMR spectra, solid-to-liquid transitions, and corresponding changes in material adhesion before and after UV light exposure, consistently support that the adhesion mechanism is due to the E–Z photoisomerization of azobenzene-based molecules. This capability likely arises because E-isomers, when in their solid state, lack sufficient chain mobility for separation, while Z-isomers, in their liquid state, exhibit necessary chain mobility for reversible bonding and debonding processes.27,56
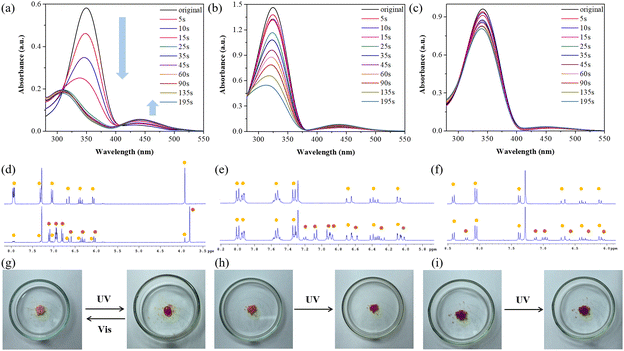 |
| | Fig. 5 Photoisomerization studies of ABOMe, ABH, and ABNO2. (a)–(c) UV-vis spectral changes of (a) ABOMe, (b) ABH, and (c) ABNO2 under UV light irradiation at 500 μM in CHCl3. (d)–(f) Partial 1H NMR spectra of (d) ABOMe, (e) ABH, and (f) ABNO2 after UV light irradiation in CDCl3. Upper: NMR for the E-isomer (yellow mark); lower: NMR for the E-isomer (yellow mark) and Z-isomer (brown mark). (g)–(i) Solid-to-liquid transition experiments of (g) ABOMe, (h) ABH, and (i) ABNO2 under UV and visible light irradiation. | |
2.4. Theoretical calculations
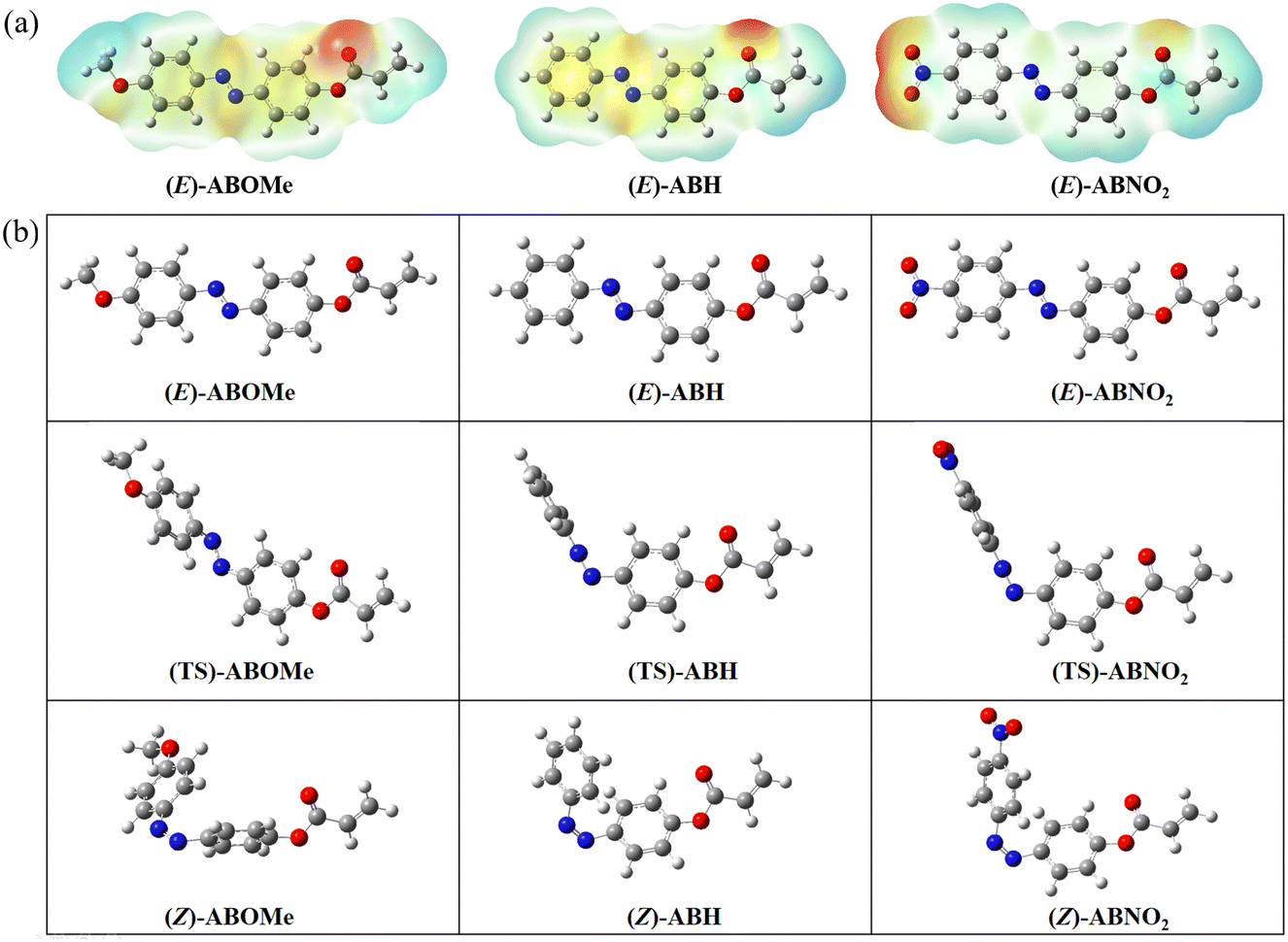
To gain deeper insight into the photophysical and photoisomerization differences among ABOMe, ABH, and ABNO2 at the molecular level, we utilized theoretical calculations. The E- and Z-isomers of these molecules, along with their corresponding transition states (TSs), were optimized using density functional theory (DFT) at the B3LYP/6-31G(d) level.57,58 Fig. S5 and S6 (ESI†) illustrate the energy diagrams of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for the E- and Z-forms of ABOMe, ABH, and ABNO2, respectively. A comparison of the HOMO and LUMO energies reveals that ABOMe has a higher HOMO compared to the other molecules, while ABNO2 has a lower LUMO, which can be attributed to the electron-donating group on ABOMe and the electron-withdrawing group on ABNO2. The calculated band gaps for the E-isomers of ABOMe, ABH, and ABNO2 are 3.62, 3.79, and 3.50 eV, respectively. For the Z-isomers of ABOMe, ABH, and ABNO2, the band gaps are 3.73, 3.82, and 3.58 eV, respectively. These findings are consistent with the experimentally observed photophysical properties (Fig. 5), further validating our theoretical calculations. The electrostatic potential maps (EPMs) of the E-isomers of ABOMe, ABH, and ABNO2 are shown in Fig. 6a. EPMs are highly useful molecular three-dimensional representations that allow us to visualize the charge distribution and related properties of molecules, which can be used to determine how molecules interact with each other.59 From the EPMs, we observe that ABNO2 has less electron cloud overlap in the NN region. Consequently, when ABNO2 is excited, the transition from the NN double bond to the N–N single bond is relatively minimal, making molecular rotation less likely.60 Conversely, ABOMe exhibits more electron cloud overlap in the NN region, facilitating the formation of a N–N single bond upon excitation, which in turn makes molecular rotation easier. This single bond rotation is crucial for the E–Z isomerization process, explaining why ABOMe readily undergoes this transformation. Experimentally, we observed that ABOMe has a Z-isomer conversion rate of 93%, ABH has 42%, and ABNO2 has 20%. This charge distribution elucidates why ABOMe has the highest Z-isomer conversion rate, followed by ABH, with ABNO2 having the lowest conversion rate. We further analyzed the E–Z isomerization mechanism of ABOMe, ABH, and ABNO2. It is known that the isomerization of azobenzene mainly occurs via two mechanisms: rotation and inversion.61 The rotation mechanism involves the transformation of the NN double bond into an N–N single bond, allowing free rotation and changing the C5–N7–N8–C9 dihedral angle, with the C5–N7–N8 angle being approximately 120°. For the inversion mechanism, the C5–N7N8–C9 dihedral angle is fixed, and the C5–N7–N8 angle increases through the transition state of the double bond, resulting in a C5–N7–N8 angle of 180°.62 As shown in Fig. 6b and Table S4 (ESI†), ABOMe exhibits a longer N–N bond length (1.29 Å) in the transition state, indicating more single-bond characteristics of the N–N bond. This suggests that its transition state mechanism is rotation-type, with a C5–N7–N8 angle of 123.99°. On the other hand, the bond lengths of ABH and ABNO2 are around 1.23 Å, with bond angles close to 180°, indicating that their mechanism is inversion-type. These findings provide a molecular-level explanation for the observed photophysical behaviors and isomerization efficiencies of these compounds. The rotational mechanism observed in ABOMe is consistent with its higher Z-isomer conversion rate, as the free rotation around the N–N single bond facilitates E–Z transition. The inversion mechanism in ABH and ABNO2, characterized by the fixed dihedral angle and a significant increase in the C5–N7–N8 angle during transition, presents a more energy-intensive pathway, resulting in lower isomerization efficiencies. Understanding these mechanisms provides valuable insights into designing azobenzene-based materials with tailored photophysical properties. By manipulating the substituents on the azobenzene core, we can control the isomerization pathway and efficiency, optimizing these materials for specific applications such as light-responsive adhesives, molecular switches, and photo-controlled drug delivery systems. The clear distinction between rotation and inversion mechanisms also aids in predicting the behavior of other azobenzene derivatives, guiding future synthetic strategies to achieve desired functional outcomes.
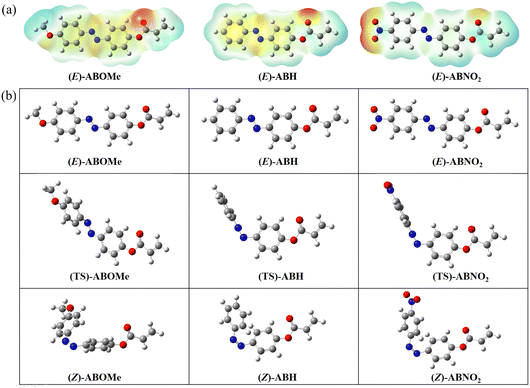 |
| | Fig. 6 (a) 3D representation of the electrostatic potentials around the molecules of E-isomers of ABOMe, ABH, and ABNO2, calculated using DFT at the B3LYP/6-31G(d) level. The red surface indicates a region of negative electrostatic potential, while the blue surface indicates a region of positive electrostatic potential. (b) Optimized structures of the E- and Z-isomers, as well as the transition states (TS) of ABOMe, ABH, and ABNO2, determined using DFT at the B3LYP/6-31G(d) level. Carbon, hydrogen, nitrogen, and oxygen atoms are depicted in gray, white, blue, and red, respectively. | |
2.5. Study of self-healing capabilities
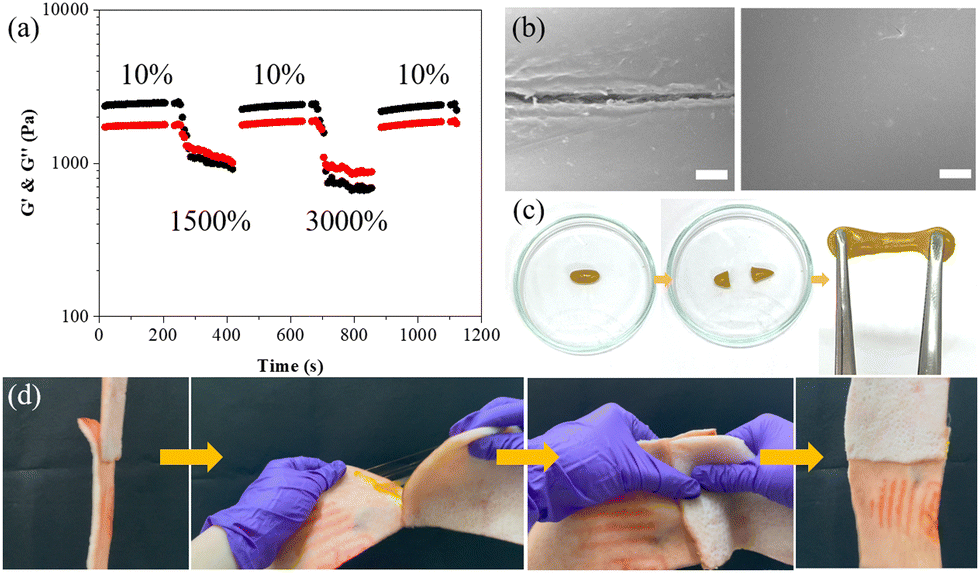
After investigating the fundamental physical properties, adhesive performance, and photoresponsive characteristics of the ABOMe ionic hydrogel, we proceeded to examine its self-healing capability to expand its potential applications. We evaluated the self-healing ability of the hydrogel through alternating strain measurements (Fig. 7a).63 As depicted in Fig. 2d, the G′ and G′′ values of the ABOMe ionic hydrogel remain nearly constant up to a strain of approximately 400%. Beyond this point, both G′ and G′′ drop sharply and become equal at around 1432% strain, indicating that the hydrogel network breaks down into a sol state once the critical strain is exceeded. Consequently, we performed continuous cyclic strain tests (10% strain → 1500% strain → 10% strain → 3000% strain → 10% strain) to assess the recoverability of the hydrogel (Fig. 7a). The results show that when the strain exceeds the critical threshold of 1432% (i.e., 1500% or 3000%), the hydrogel transitions to a sol state (G′ < G′′). Upon returning the strain to 10%, the gel reassembles, and the G′ and G′′ values quickly return to their initial levels (G′ > G′′). Throughout the cyclic tests, the rapid sol–gel transition behavior of the hydrogel network is fully reversible and reproducible, demonstrating that the ABOMe ionic hydrogel exhibits excellent self-healing properties. In addition to rheological testing, we conducted both microscopic and macroscopic experiments to verify the self-healing performance of the ABOMe ionic hydrogel. Fig. 7b shows the scanning electron microscopy (SEM) images of the ABOMe ionic hydrogel. We introduced a scratch on the sample, and after 10 minutes, the crack was sealed and healed, indicating its high self-repair capability. This rapid healing process is crucial for applications where maintaining the structural integrity is essential. To further test the self-healing properties, we performed a macroscopic experiment where we cut the hydrogel into two approximately equal pieces and placed the cut surfaces together to observe their self-healing ability. After 10 minutes, we pulled the hydrogel from both ends, and the cut surface did not separate, as shown in Fig. 7c. Furthermore, we were able to stretch the healed hydrogel from 3 cm to 22 cm without any breakage (Fig. S7, ESI†). This demonstrated that the hydrogel can autonomously heal and restore its original mechanical properties after being damaged. Additionally, we explored the potential of the ABOMe ionic hydrogel as an adhesive for biological tissues (Fig. 7d). We used the hydrogel as an adhesive for porcine skin, repeatedly bonding, tearing, bonding, and shaking the porcine skin pieces to simulate real-life conditions. The experiments showed that the ABOMe ionic hydrogel effectively bonded the two pieces of porcine skin, maintaining a strong adhesive bond throughout the process. We repeated these actions 10 times (Video S4, ESI†), and each time, the hydrogel successfully re-bonded the porcine skin without losing its adhesive strength. This repetitive testing highlights the hydrogel's ability to maintain its self-healing properties over multiple cycles, ensuring long-term reliability and durability. The combination of microscopic and macroscopic experiments provides comprehensive evidence of the exceptional self-healing abilities of the ABOMe ionic hydrogel. These properties make it a promising candidate for various applications, including wound healing, tissue engineering, and other injectable biomedical uses where reliable and durable adhesion is required. The hydrogel's ability to repeatedly heal itself and maintain its adhesive properties over multiple cycles ensures that it can withstand the demands of practical use, offering a versatile solution for a wide range of applications.
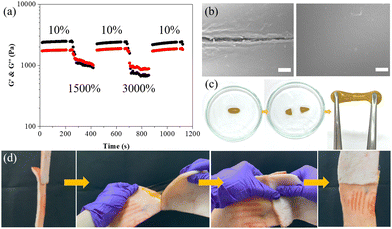 |
| | Fig. 7 (a) The cyclic G′ (black) and G′′ (red) values of the ABOMe ionic hydrogel under alternate-step strain sweep. (b) SEM images of self-healing tests on the ABOMe ionic hydrogel showing the cut sample (left) and the healed cut (right). Scale bar: 10 μm. (c) Photographs of self-healing tests on damaged and healed ABOMe ionic hydrogels. (d) Photographs of self-healing tests of the ABOMe ionic hydrogel as a skin adhesive. | |
3. Conclusions
In summary, we synthesized the monomers ABOMe, ABH, and ABNO2 to investigate their effects on reversible adhesion properties. By incorporating these azobenzene monomers into AAM, DF-PEG, and MAPTAC, we prepared ABOMe, ABH, and ABNO2 ionic hydrogels. Our findings reveal that only the ABOMe ionic hydrogel exhibits reversible adhesion, driven by the E–Z photoisomerization of the azobenzene moiety. Further investigation showed that ABOMe undergoes a different photoisomerization process compared to ABH and ABNO2, characterized by a rotation-type transition state mechanism, enabling rapid E–Z photoisomerization and thus conferring reversible adhesion properties. In addition, a series of experiments confirmed that the ABOMe ionic hydrogel demonstrates excellent adhesion to a variety of materials and maintains strong adhesive properties even under underwater conditions. The observed skin adhesion strength of 360.7 ± 10.1 kPa is a testament to the hydrogel's exceptional performance, which is largely due to the interplay of Schiff base reactions, cation–π interactions, π–π stacking, and hydrogen bonding with skin amino acids. These interactions enhance the adhesion and bonding capacity of the hydrogel, making it highly effective for applications requiring strong, yet reversible adhesion. Additionally, the excellent reversible self-healing capabilities of the ABOMe ionic hydrogel are crucial for its practical use in dynamic and durable adhesive applications. The ability to self-heal and maintain functionality over multiple cycles of use further extends the potential applications of ABOMe hydrogels in fields such as biomedical adhesives, wearable electronics, and smart coatings. This research highlights the importance of optimizing both photoresponsive mechanisms and material interactions to develop advanced adhesive technologies that meet the demands of diverse applications. The innovative approach of integrating push–pull electronic effects with photoresponsive azobenzene moieties opens new avenues for designing multifunctional materials with tailored properties for specific uses.
4. Experimental section
4.1. Materials and methods
p-Anisidine, aniline, p-nitroaniline, sodium nitrite, phenol, and acryloyl chloride were purchased from Alfa Aesar. [3-(Methacryloylamino)propyl]-trimethylammonium chloride solution (50 wt% in H2O, MAPTAC), 2,2′-azobis(2-methyl-propionitrile) (AIBN), 4-formylbenzoic acid, 4-(dimethylamino)-pyridine (DMAP), N,N′-dicyclohexylcarbodiimide (DCC), and ethanol were obtained from Sigma-Aldrich. Acrylamide and poly(ethylene glycol) (PEG) with an average MW of 2000 were sourced from Acros Organics. Deionized water was used in all experiments. The detailed synthesis methods for ABOMe, ABH, and ABNO2 monomers are provided in the ESI.† DF-PEG was synthesized following the literature protocol.38
4.2. Preparation of ABOMe, ABH, and ABNO2 hydrogels
ABOMe (0.02 g) was dissolved in ethanol (1.00 mL) and sonicated until fully dissolved, and then AIBN (0.008 g), acrylamide (0.9 g), and DF-PEG (0.04 g) were added. Deionized water (1.00 mL) was added and sonicated at room temperature until the mixture was homogeneous. The mixture was placed in an oven set to 70 °C for 6 hours. Afterward, the gel was removed and placed in deionized water, changing the water every hour until the ethanol was completely replaced with deionized water, resulting in ABOMe as a yellow hydrogel. Following a similar procedure, replacing ABOMe with ABH produces an orange ABH hydrogel, and replacing ABOMe with ABNO2 yields a red ABNO2 hydrogel.
4.3. Preparation of ABOMe, ABH, and ABNO2 ionic hydrogels
In a similar manner, ABOMe (0.02 g) was dissolved in ethanol (1.00 mL), and then AIBN (0.008 g), acrylamide (0.9 g), DF-PEG (0.04 g), and MAPTAC (0.08 g) were added. Next, deionized water was added and sonicated at room temperature until the mixture is homogeneous. The mixture was placed in a 70 °C oven for 6 hours, and then the gel was transferred to deionized water; the water was changed hourly until the ethanol was replaced, yielding a yellow ABOMe ionic hydrogel. Using ABH instead of ABOMe produces an orange ABH ionic hydrogel, while replacing ABOMe with ABNO2 results in a red ABNO2 ionic hydrogel.
4.4. Characterization
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded using a Bruker AVANCE II-400 MHz spectrometer with DMSO-d6 and CDCl3 as solvents to elucidate the molecular structures and confirm the E–Z photoisomerization efficiencies of the azobenzene derivatives. UV-vis absorption spectra were recorded with a JASCO V-750 spectrometer to investigate the photoswitching behavior of the azobenzene moieties. Rheological assessments were conducted using a TA rheometer (DHR-1) with a parallel plate setup (20 mm diameter and 1 mm gap) to characterize the viscoelastic properties of the hydrogels. The storage modulus (G′) and loss modulus (G′′) were assessed as a function of angular frequency (ω) across the range of 0.1 to 100 rad s−1, under a strain (γ) of 1% at 25 °C. The reported values of G′ and G′′ represent the averages of the measurements taken within the 0.1 to 100 rad s−1 range. The strain amplitude sweep test (γ = 0.1–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000%, ω = 10 rad s−1) at 25 °C was performed to evaluate the mechanical responses of the hydrogels under varying strain conditions. Amplitude oscillatory strains were switched from small strain (γ = 10%) to subsequent large strains (γ = 1500% and 3000%) with 200 s intervals for each strain. This testing protocol allowed for the examination of the recovery and durability of the hydrogels under cyclic loading conditions. The lap-shear test was performed on a tensile tester (Gotech AI-3000-U) at a rate of 10 mm min−1 to measure the adhesive strength of the hydrogels. Scanning electron microscopy (SEM) using a JEOL JSM-7600F model was utilized to examine the morphologies of the hydrogels before and after the scratch test. Test samples were prepared via freeze-vacuum drying to preserve their microstructures.
000%, ω = 10 rad s−1) at 25 °C was performed to evaluate the mechanical responses of the hydrogels under varying strain conditions. Amplitude oscillatory strains were switched from small strain (γ = 10%) to subsequent large strains (γ = 1500% and 3000%) with 200 s intervals for each strain. This testing protocol allowed for the examination of the recovery and durability of the hydrogels under cyclic loading conditions. The lap-shear test was performed on a tensile tester (Gotech AI-3000-U) at a rate of 10 mm min−1 to measure the adhesive strength of the hydrogels. Scanning electron microscopy (SEM) using a JEOL JSM-7600F model was utilized to examine the morphologies of the hydrogels before and after the scratch test. Test samples were prepared via freeze-vacuum drying to preserve their microstructures.
4.5. Statistical analysis
Statistical analysis of cell viability over different days, as well as adhesion values before and after UV light irradiation, was conducted using a parametric one-way ANOVA with Tukey's multiple comparisons post-test.64 All conditions were compared, and the data are presented as mean ± standard deviation (n = 3). Statistical significance was indicated by *, **, or ***, representing p < 0.05, p < 0.01, and p < 0.001, respectively.
4.6. Computational methodology
Geometry optimization of the azobenzene derivatives was conducted using density functional theory (DFT) with the B3LYP method, which is Becke's three-parameter hybrid functional combined with the Lee, Yang, and Parr correlation functional that includes both local and non-local correlation terms, at the 6-31G(d) level.57,58 The geometries of the transition state and Z-isomer were constructed by constraining the C–NN–C dihedral angle to 90° and 0°, respectively, while optimizing all other structural parameters. The highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) analysis was performed to elucidate the electronic properties and charge transfer characteristics of the molecules. Moreover, theoretical electrostatic potential maps were generated to analyze the charge distribution and understand the intra- and intermolecular interactions.
Author contributions
Y.-Y. W. and M.-Y. Y.: conceptualization, investigation, and methodology. Y.-Y. W. and P.-W. C.: data curation and formal analysis. Y.-H. C.: data curation. M.-Y. Y.: funding acquisition, resources, supervision, and writing – review and editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The research presented in this work was supported by grants from the National Science and Technology Council of the Republic of China, Taiwan (NSTC 113-2113-M-033-004-).
References
- J. Hwang, D. Lim, G. Lee, Y. E. Kim, J. Park, M.-J. Baek, H.-S. Kim, K. Park, K. H. Ku and D. W. Lee, Mater. Horiz., 2023, 10, 2013–2023 RSC.
- Z. Yang, L. Chen, D. J. McClements, C. Qiu, C. Li, Z. Zhang, M. Miao, Y. Tian, K. Zhu and Z. Jin, Food Hydrocolloids, 2022, 124, 107218 CrossRef CAS.
- M. Neumann, G. di Marco, D. Iudin, M. Viola, C. F. van Nostrum, B. G. P. van Ravensteijn and T. Vermonden, J. Biol. Macromol., 2023, 56, 8377–8392 CrossRef CAS PubMed.
- A. Roy, K. Manna and S. Pal, Mater. Chem. Front., 2022, 6, 2338–2385 RSC.
- Q. Shi, H. Liu, D. Tang, Y. Li, X. Li and F. Xu, NPG Asia Mater., 2019, 11, 64 CrossRef CAS.
- Q. Zhang, D.-H. Qu, H. Tian and B. L. Feringa, Matter, 2020, 3, 355–370 CrossRef.
- T. Kakuta, T.-a Yamagishi and T. Ogoshi, Acc. Chem. Res., 2018, 51, 1656–1666 CrossRef CAS.
- M. Bril, S. Fredrich and N. A. Kurniawan, Smart Mater. Med., 2022, 3, 257–273 CrossRef.
- N. Du, F. Ye, J. Sun and K. Liu, ChemBioChem, 2022, 23, e202100416 CrossRef CAS PubMed.
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, Adv. Mater., 2020, 32, 1906036 CrossRef CAS PubMed.
- R. Liu, X. Zhang, F. Xia and Y. Dai, J. Catal., 2022, 409, 33–40 CrossRef CAS.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef PubMed.
- G. Cabré, A. Garrido-Charles, M. Moreno, M. Bosch, M. Porta-de-la-Riva, M. Krieg, M. Gascón-Moya, N. Camarero, R. Gelabert, J. M. Lluch, F. Busqué, J. Hernando, P. Gorostiza and R. Alibés, Nat. Commun., 2019, 10, 907 CrossRef PubMed.
- H.-B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef CAS.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- J. Keyvan Rad, Z. Balzade and A. R. Mahdavian, J. Photochem. Photobiol., C, 2022, 51, 100487 CrossRef CAS.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef PubMed.
- S. Bakker, E. de Korver, M. Fransen, E. Kamer, G. A. Metselaar, A. C. C. Esteves and A. P. H. J. Schenning, ACS Appl. Opt. Mater., 2023, 1, 403–411 CrossRef CAS.
- W. Shen and G. Li, Laser Photonics Rev., 2023, 17, 2200207 CrossRef CAS.
- A. M. Asadirad, S. Boutault, Z. Erno and N. R. Branda, J. Am. Chem. Soc., 2014, 136, 3024–3027 CrossRef CAS.
- J. Jiang, S. Xu, H. Ma, C. Li and Z. Huang, Mater. Today Bio, 2023, 20, 100657 CrossRef PubMed.
- Q. Yang, H. Shahsavan, Z. Deng, H. Guo, H. Zhang, H. Liu, C. Zhang, A. Priimagi, X. Zhang and H. Zeng, Adv. Funct. Mater., 2022, 32, 2206939 CrossRef CAS.
- S. Hu, L. Tong, J. Wang, X. Yi and J. Liu, Anal. Chem., 2019, 91, 15418–15424 CrossRef CAS PubMed.
- S.-L. Xiang, P.-F. Luo, Y.-Y. Ren, L.-Y. Peng, H. Yin, J.-D. Huang, P. Hong, Q. Yu, R. Tian, C. Li, J. Liu and M.-Q. Zhu, ACS Appl. Polym. Mater., 2024, 6, 2496–2503 CrossRef CAS.
- Z. Sekkat and S. Kawata, Laser Photonics Rev., 2014, 8, 1–26 CrossRef CAS.
- Y. Zhou, M. Chen, Q. Ban, Z. Zhang, S. Shuang, K. Koynov, H.-J. Butt, J. Kong and S. Wu, ACS Macro Lett., 2019, 8, 968–972 CrossRef CAS PubMed.
- H. Akiyama, T. Fukata, A. Yamashita, M. Yoshida and H. Kihara, J. Adhes., 2017, 93, 823–830 CrossRef CAS.
- X. Xu, P. Zhang, B. Wu, Y. Xing, K. Shi, W. Fang, H. Yu and G. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 50135–50142 CrossRef CAS.
- Z. Wu, C. Ji, X. Zhao, Y. Han, K. Müllen, K. Pan and M. Yin, J. Am. Chem. Soc., 2019, 141, 7385–7390 CrossRef CAS.
- Y.-F. Chen, M.-R. Huang, Y.-S. Hsu, M.-H. Chang, T.-Y. Lo, B. Gautam, H.-H. Hsu and J.-T. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 29153–29161 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
- E. V. Pletneva, A. T. Laederach, D. B. Fulton and N. M. Kostić, J. Am. Chem. Soc., 2001, 123, 6232–6245 CrossRef CAS PubMed.
- L. Xiang, J. Zhang, W. Wang, L. Gong, L. Zhang, B. Yan and H. Zeng, Acta Biomater., 2020, 117, 294–301 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- C. He, S. Bi, R. Liu, H. Zhao, C. Chen, X. Zhao, J. Gu and B. Yan, ACS Appl. Mater. Interfaces, 2024, 16, 35887–35897 CrossRef CAS PubMed.
-
Y. Azim, in Smart Polymer Nanocomposites, ed. S. A. Bhawani, A. Khan and M. Jawaid, Woodhead Publishing, 2021, pp. 117–143 Search PubMed.
- H.-J. Wang, Y.-Z. Chu, C.-K. Chen, Y.-S. Liao and M.-Y. Yeh, RSC Adv., 2021, 11, 6620–6627 RSC.
- H. A. Barnes, Rheol. Rev., 2003, 1–36 Search PubMed.
- S. O. Ilyin, V. G. Kulichikhin and A. Y. Malkin, Rheol. Acta, 2016, 55, 223–233 CrossRef CAS.
- A. B. Kousaalya, B. I. Biddappa, S. Rai and S. Pilla, Biocomposites, 2015, 161–200 CAS.
- M. Palusiak and S. J. Grabowski, J. Mol. Struct., 2002, 642, 97–104 CrossRef CAS.
- R. Wu and B. Brutschy, Chem. Phys. Lett., 2004, 390, 272–278 CrossRef CAS.
- G. Bovone, O. Y. Dudaryeva, B. Marco-Dufort and M. W. Tibbitt, ACS Biomater. Sci. Eng., 2021, 7, 4048–4076 CrossRef CAS PubMed.
- R. J. Aguado, A. Mazega, Q. Tarrés and M. Delgado-Aguilar, Ind. Crops Prod., 2023, 201, 116898 CrossRef CAS.
- J. Zhang, H. Lei, M. Qin, W. Wang and Y. Cao, Supramol. Chem., 2022, 1, 100005 CrossRef.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- D. A. Dougherty, J. Nutr., 2007, 137, 1504S–1508S CrossRef CAS PubMed.
- S.-M. Liao, Q.-S. Du, J.-Z. Meng, Z.-W. Pang and R.-B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- Y. A. Amador-Sánchez, P. López-Mendoza, M. V. Mijangos and L. D. Miranda, Chem. – Eur. J., 2022, 2022, e202200080 Search PubMed.
- M. A. Gebbie, W. Wei, A. M. Schrader, T. R. Cristiani, H. A. Dobbs, M. Idso, B. F. Chmelka, J. H. Waite and J. N. Israelachvili, Nat. Chem., 2017, 9, 473–479 CrossRef CAS PubMed.
- H. Pang, P. Yu, A. Huang, X. Li, Z. Xu and S. Yin, Macromol. Mater. Eng., 2022, 307, 2200531 CrossRef CAS.
- L. H. Urner, B. N. S. Thota, O. Nachtigall, S. Warnke, G. von Helden, R. Haag and K. Pagel, Chem. Commun., 2015, 51, 8801–8804 RSC.
- C. Fedele, T.-P. Ruoko, K. Kuntze, M. Virkki and A. Priimagi, Photochem. Photobiol. Sci., 2022, 21, 1719–1734 CrossRef CAS PubMed.
- C. Heinzmann, S. Coulibaly, A. Roulin, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2014, 6, 4713–4719 CrossRef CAS PubMed.
- J. Chen, X. Chen, L. Cao, H. Deng, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2022, 61, e202200343 CrossRef CAS PubMed.
- H.-T. Feng, X. Zheng, X. Gu, M. Chen, J. W. Y. Lam, X. Huang and B. Z. Tang, Chem. Mater., 2018, 30, 1285–1290 CrossRef CAS.
- H.-Y. Chang, L.-C. Fu, H.-S. Huang, Y.-C. Yeh and M.-Y. Yeh, Dyes Pigm., 2023, 219, 111579 CrossRef CAS.
- T.-T. Yin, Z.-X. Zhao and H.-X. Zhang, RSC Adv., 2016, 6, 79879–79889 RSC.
- M. Zhu and H. Zhou, Org. Biomol. Chem., 2018, 16, 8434–8445 RSC.
- J. Dokić, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef.
- L. Zhou, C. Dai, L. Fan, Y. Jiang, C. Liu, Z. Zhou, P. Guan, Y. Tian, J. Xing, X. Li, Y. Luo, P. Yu, C. Ning and G. Tan, Adv. Funct. Mater., 2021, 31, 2007457 CrossRef CAS.
- M. F. Martiz, R. M. Ray, A. J. Bick, M. Tomasicchio, J. G. Woodland, Y. Govender, C. Avenant and J. P. Hapgood, PLoS One, 2018, 13, e0196043 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*