
Synergistic dual-layer passivation boosts efficiency and stability in perovskite solar cells using naphthol sulfonate†
Hao
Liu‡
a,
Ning
Jiang‡
a,
Jintao
Wang
*b,
Shuming
Chen
c,
Jian
Zhang
*a and
Yu
Duan
 *a
*a
aCollege of Electronic Science and Engineering, Jilin University, Changchun 130012, China. E-mail: zhangjian@jlu.edu.cn; duanyu@jlu.edu.cn
bCollege of Information Engineering, Yantai Institute of Technology, Yantai 264005, China. E-mail: wangjintao@yitsd.edu.cn
cCollege of Physics, Changchun University of Science and Technology, Changchun 130012, China
First published on 23rd October 2024
Abstract
The performance and stability of perovskite solar cells (PSCs) are critically influenced by the interfacial properties between the perovskite absorption layer and the electron transport layer (ETL). This study introduces a novel interfacial engineering approach using dipotassium 7-hydroxynaphthalene-1,3-disulfonate (K-NDS) as a multifunctional passivator to enhance both the SnO2 ETL and the perovskite absorber layer. The sulfonic acid groups (–SO3−) in K-NDS effectively fill oxygen vacancies on the SnO2 surface, while the hydroxyl groups (–OH) passivate dangling bonds, improving the crystallinity of the perovskite film. Additionally, the diffusion of K+ from the SnO2 ETL into the perovskite layer optimizes energy level alignment, thereby enhancing charge carrier extraction and transport. This bifacial passivation strategy has significantly improved both the power conversion efficiency (PCE) and long-term stability of PSCs. The modified devices achieved a champion PCE of 23.00% and an open-circuit voltage (VOC) of 1.172 V. Furthermore, these devices maintained 75% of their initial PCE even after 1000 hours of storage under indoor environmental conditions. This work demonstrates the effectiveness of synergistic interfacial passivation in advancing the performance and durability of PSCs.
New conceptsIn this study, we introduce a novel dual-sided interfacial engineering approach using dipotassium 7-hydroxynaphthalene-1,3-disulfonate (K-NDS) as a multifunctional passivator to enhance perovskite solar cells (PSCs). Unlike traditional methods that focus on either the electron transport layer (ETL) or the perovskite layer separately, our strategy simultaneously targets both layers. This is achieved by filling oxygen vacancies in SnO2 and passivating defects in the perovskite, which collectively improve crystallinity and energy level alignment through K+ diffusion. This innovative approach not only boosts the power conversion efficiency (PCE) but also significantly enhances long-term stability. Our work provides new insights into the role of multifunctional materials in optimizing PSC performance, highlighting the potential for materials with versatile active groups to set new benchmarks for efficiency and stability in photovoltaic materials science. This concept differentiates itself by offering a synergistic solution that addresses multiple challenges concurrently, paving the way for more integrated and effective strategies in solar cell technology. |
Introduction
Organic–inorganic metal halide perovskite solar cells (PSCs) exhibit excellent optoelectronic properties,1,2 including tunable band gap,3 low exciton binding energy4 and long carrier diffusion length.5 Significant progress has been achieved in enhancing their stability and scalability.6–9 The power conversion efficiency (PCE) has impressively increased from the initial 3.9%10 to the current record of 26.7%,11 approaching the highest efficiency of monocrystalline silicon solar cells. However, the commercialization of perovskite photovoltaic technology still faces considerable challenges, with one of the most critical obstacles being the presence of defects and recombination centres at the device interface.12 Research indicates that the defect densities at the interface can be significantly higher than within the perovskite film.13 These interfacial defects and recombination centres serve as traps, capturing charge carriers and leading to non-radiative recombination, which ultimately limits the enhancement of the PSC device efficiency.12,14Within n–i–p type PSCs, SnO2 has become a popular choice for the electron transport layer (ETL), owing to its favourable characteristics, including great visible light transmittance, excellent carrier mobility, and tunable electronic properties. However, SnO2 films tend to surface defects and dangling bonds, especially at the energy level disparity and interface junction with the perovskite layers. These imperfections can adversely affect charge transport efficiency, potentially limiting the overall performance of the PSCs.15–17
To optimize the interface between the SnO2 ETL and the perovskite absorber layer, various materials can be employed. One approach involves introducing organic molecules having specific functional groups, such as carboxylic acid (–COO−)18 and sulfonic acid (–SO3−),19 which can be either incorporated into the SnO2 material or deposited on its surface. These organic molecules interact with the dangling bonds on the SnO2 surface, leading to improved SnO2 properties and promoting the crystallization of the perovskite layer. Additionally, ionic salts, including alkaline halide salts (e.g., KCl20 and KI21) and ammonium halide salts (e.g., NH4F (ref. 22) and MDACl2 (ref. 23)), can be integrated into the SnO2 ETL. These ionic salts effectively passivate surface defects on SnO2 through the action of hydrogen bonding and electrostatic interactions. Moreover, the anions and cations in these salts can diffuse into the perovskite absorber layer, where they interact with uncoordinated Pb2+, thereby passivating organic vacancy defects, reducing non-radiative recombination at the interface, and ultimately enhancing the device's performance and stability.24
Current defect passivation and interface modification methods often focus on a single functional layer, which limits their ability to achieve optimal passivation. Therefore, a synchronized and coordinated passivation of the functional layers and their interfaces is crucial for reducing defect density within films and at the interfaces, while simultaneously improving interlayer contact to enhance the performance of PSCs.
In this work, we introduced a dipotassium 7-hydroxynaphthalene-1,3-disulfonate (K-NDS) as the modification layer between the SnO2 and the perovskite. The –SO3− in K-NDS can coordinate with SnO2, while the hydroxyl groups (–OH) coordinate with uncoordinated Pb and I ions in the perovskite. Furthermore, K+ can diffuse at the interface between the ETL and perovskite layers, leading to a dual-sided interface passivation effect that enhances the performance of the PSC devices. Thus, the device modified with K-NDS achieved a champion PCE of 23.00%, compared to 21.81% for the pristine PSC device. Furthermore, this bilateral interface passivation strategy also enhanced the long-term stability of the PSC device, with unencapsulated devices maintaining 75% of their original PCE even after 1000 hours of storage under indoor environmental conditions.
Results and discussion
K-NDS (Fig. S1, ESI†) was dissolved in deionized water at different concentrations and subsequently spin-coated on the surface of the SnO2 ETL, followed by annealing to form the SnO2/K-NDS (Fig. S2, ESI†). The interaction mechanism between K-NDS and SnO2 was investigated by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR) spectroscopy. Fig. 1a displays the XPS full spectra of SnO2 and SnO2/K-NDS. Compared to the SnO2 full XPS spectrum, the SnO2/K-NDS exhibits additional S 2p characteristic peaks and stronger K 2p peaks (the high-resolution spectra are shown in Fig. S3, ESI†). This preliminary analysis confirms that the K-NDS compound was successfully incorporated on the SnO2 surface. The weak K 2p peaks observed in the SnO2 spectrum are due to potassium hydroxide (KOH), which was used as a stabilizer for the commercial Alfa-SnO2.25 Compared to the pristine SnO2, the Sn 3d3/2 and Sn 3d5/2 peaks for the SnO2/K-NDS are shifted to high binding energy by approximately 0.20 eV (Fig. 1b), indicating the coordination of –SO3− with SnO2. Specifically, the negative charge of the –SO3− passivates the uncoordinated Sn atoms on the SnO2 surface, leading to an accumulation of extra electrons around the Sn atoms.26,27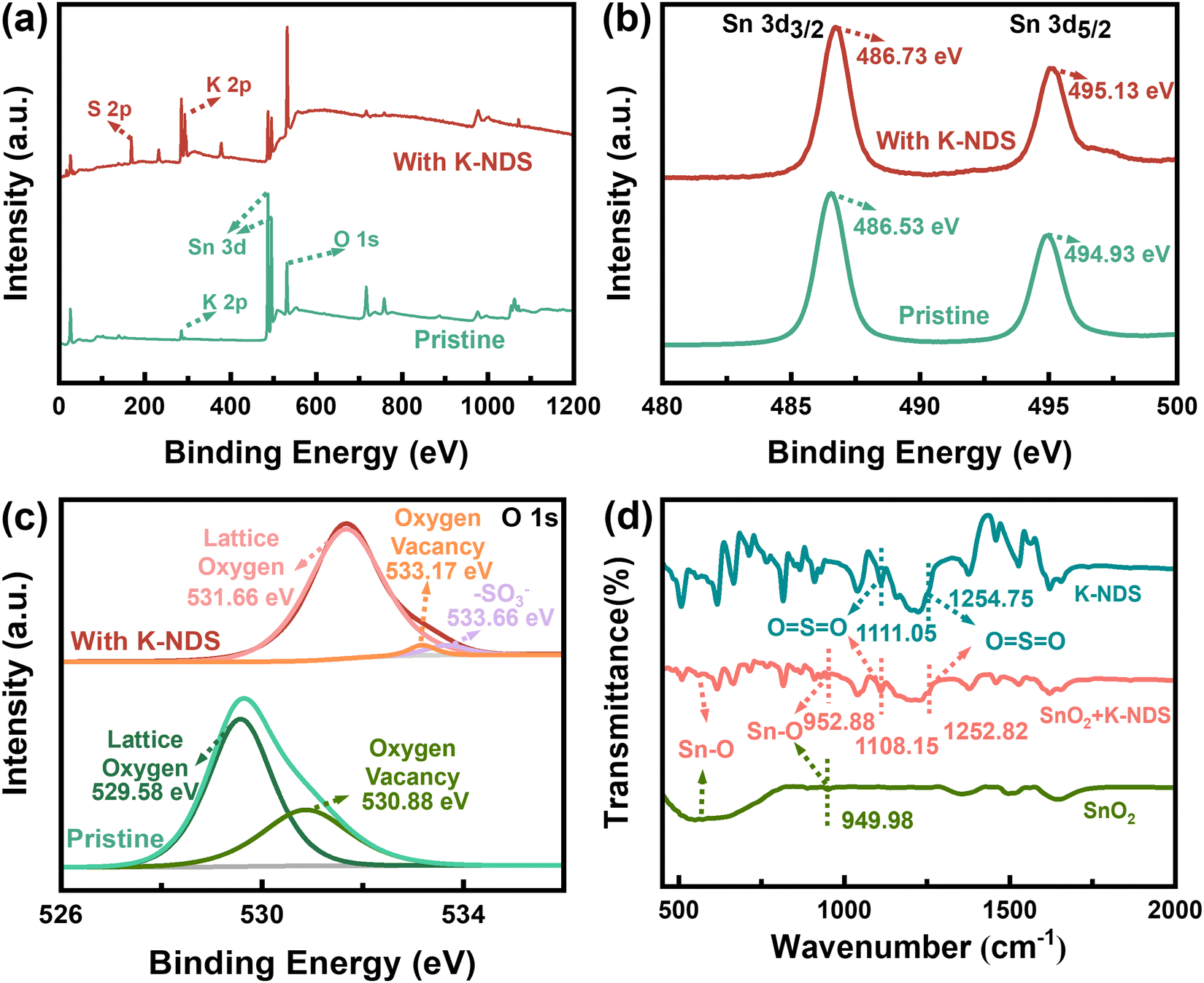 | ||
| Fig. 1 (a) XPS full spectra of SnO2 and SnO2/K-NDS. XPS high-resolution spectra of SnO2 and SnO2/K-NDS for (b) Sn 3d, and (c) O 1s. (d) FTIR spectra of SnO2, K-NDS, and SnO2/K-NDS. | ||
The O 1s XPS peaks for both the SnO2 and the SnO2/K-NDS are shown in Fig. 1c. Compared to the O 1s peak of the SnO2, the O 1s peak of the SnO2/K-NDS sample has moved to a high binding energy by about 2.05 eV. This notable shift may be attributed to the coordination between the –SO3− and the SnO2 film, which enhances the electron cloud density around the SnO2 oxygen atoms.26,27 Furthermore, the O 1s peak of the pristine SnO2 could be deconvoluted into two distinct components. The peak at 529.58 eV corresponded to the Sn–O bond, while the peak at 530.88 eV was associated with oxygen vacancies (OV) or surface adsorbed hydroxyls (OOH).28 After the K-NDS modification, these peaks shifted to a high binding energy of 531.66 eV and 533.17 eV. The calculation result of the area ratio of the O 1s peaks is shown in Table S1 (ESI†). We found that the pristine SnO2 ratio (OV + OOH)/Oall of 33.99% was significantly reduced to 2.40% for SnO2/K-NDS, indicating a drastic decrease in surface defects, which is expected to minimize trap-assisted recombination. This reduction in defect density suggests that K-NDS effectively passivates oxygen vacancies, which is further supported by the reduced non-radiative recombination observed in subsequent photoluminescence (PL) and time resolved photoluminescence (TRPL) measurements.29
The FTIR spectra of SnO2, K-NDS, and SnO2/K-NDS are shown in Fig. 1d. The FTIR spectrum of the pristine SnO2 showed Sn–O absorption band at 500 cm−1, along with Sn–O asymmetric stretching vibration at 949.98 cm−1.30 The FTIR spectrum of the K-NDS shows characteristic peaks at 1111.05 cm−1 and 1254.75 cm−1, which can be assigned to the stretching vibrations of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO. Following the K-NDS deposited on the SnO2 surface, the FTIR spectrum of the SnO2/K-NDS exhibits a notable change. In comparison with the unmodified SnO2, the SnO2/K-NDS demonstrates that the Sn–O absorption band is observed near 500 cm−1. However, the Sn–O asymmetric stretching vibration has shifted to a high wavenumber of 952.88 cm−1. Furthermore, the stretching vibration OSO has shifted to 1108.15 cm−1 and 1252.82 cm−1. The shifts of characteristic peaks confirm the effect of coordination between the K-NDS and SnO2. These can be attributed to the –SO3− from Sn–O bands with the uncoordinated Sn on the SnO2 surface.26,27,31,32
SO. Following the K-NDS deposited on the SnO2 surface, the FTIR spectrum of the SnO2/K-NDS exhibits a notable change. In comparison with the unmodified SnO2, the SnO2/K-NDS demonstrates that the Sn–O absorption band is observed near 500 cm−1. However, the Sn–O asymmetric stretching vibration has shifted to a high wavenumber of 952.88 cm−1. Furthermore, the stretching vibration OSO has shifted to 1108.15 cm−1 and 1252.82 cm−1. The shifts of characteristic peaks confirm the effect of coordination between the K-NDS and SnO2. These can be attributed to the –SO3− from Sn–O bands with the uncoordinated Sn on the SnO2 surface.26,27,31,32
The properties of visible light transmission of the SnO2 and SnO2/K-NDS films were analyzed using ultraviolet-visible (UV-vis) spectroscopy, as shown in Fig. 2a. It can be observed that the K-NDS modification layer has no impact on the light absorption of the perovskite layer deposited subsequently. The X-ray diffraction (XRD) patterns of the SnO2 and SnO2/K-NDS films are presented in Fig. S4 (ESI†). The diffraction peaks of the two films are nearly identical, indicating that K-NDS modification does not affect the crystal structure of SnO2. Ultraviolet photoelectron spectroscopy (UPS) was conducted on the SnO2 and SnO2/K-NDS films to investigate their energy level characteristics. As shown in Fig. 2b and c, the secondary electron cut-off energies (Ecut-off) for SnO2 and SnO2/K-NDS are 16.93 eV and 17.07 eV, while the valence band edges (EVBE) are 4.08 eV and 3.87 eV. These values were then calculated using the following formula:
EF = Ecut![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) off − 21.22 off − 21.22 | (1) |
| EF − EVBM = EVBE | (2) |
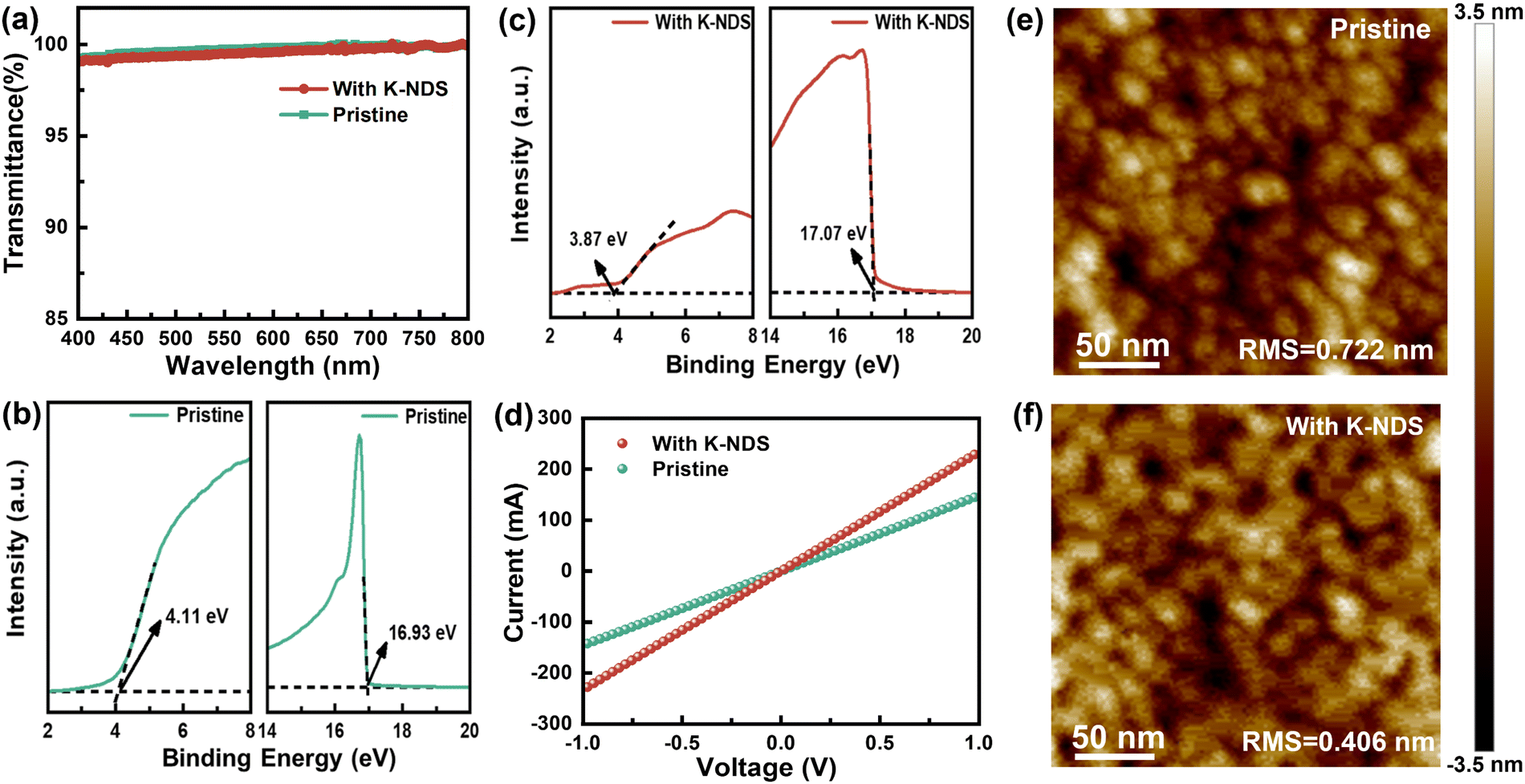 | ||
| Fig. 2 (a) Transmission spectroscopy of SnO2 and SnO2/K-NDS films. (b) UPS measurements of the SnO2 film and (c) SnO2/K-NDS film. (d) I–V curves of SnO2 and SnO2/K-NDS films. (e) AFM measurements of SnO2 film and (f) SnO2/K-NDS film. | ||
The Fermi levels (EF) of the SnO2 and SnO2/K-NDS are −4.29 eV and −4.15 eV, while the valence band maximums (VBM) are −8.37 eV and −8.02 eV. By combining the Tauc plots of SnO2 and SnO2/K-NDS (shown in Fig. S5, ESI†), the conduction band minimums (CBM) of SnO2 and SnO2/K-NDS were measured as −4.21 eV and −3.87 eV.33 These results are summarized in Table S2 (ESI†). The energy level alignment diagram derived from these findings is shown in Fig. S6 (ESI†). With the K-NDS modification, both the VBM and CBM of SnO2 shift to higher energy levels. Moreover, the transmission of electrons at the interface has been optimized. These observations can be attributed to the fact that the K+ in K-NDS optimizes the energy level alignment between the SnO2 ETL and the perovskite layer.34
To investigate the impact of the K-NDS modified layer on the conductivity of the SnO2 ETL, we fabricated ITO/SnO2/without or with K-NDS/Ag single-electronic devices, the current–voltage (I–V) curves are presented in Fig. 2d. Compared to the SnO2 device, the I–V curve of the SnO2/K-NDS device exhibits a steeper profile, indicating a remarkable improvement in the conductivity of the SnO2 film after the K-NDS modification.35 As shown in the atomic force microscope (AFM) images in Fig. 2e and f, the root-mean-square (RMS) roughness of the SnO2 film was 0.722 nm. In contrast, the SnO2/K-NDS film had a roughness of 0.406 nm. These results demonstrate that the K-NDS modification can enhance the flatness of the electron transport layer (ETL).32 Compared to ITO (78.3°) and ITO/SnO2 (46.3°), ITO/SnO2/K-NDS exhibits a smaller water contact angle (19.9°), as shown in Fig. S7 (ESI†). This reduction in contact angle lowers the Gibbs free energy during nucleation, facilitating subsequent film growth.36
To obtain a deeper understanding of the interaction between K-NDS and the perovskite films, XPS analysis was conducted on the exposed perovskite bottom interface without the SnO2 layer. Fig. 3a shows the full XPS spectra of the perovskite and K-NDS/perovskite films. Compared to the perovskite sample, a weak K 2p characteristic peak is observed in the K-NDS/perovskite (the high-resolution spectrum in Fig. S8, ESI†). The observation of a weak K 2p peak in the K-NDS/perovskite spectrum suggests that during the annealing process, K+ can diffuse from the SnO2 ETL into the perovskite layer. This diffusion likely passivates the grain boundaries, thereby reducing defect sites and enhancing the overall quality of the perovskite film. Compared to the perovskite film, the spectrum of the K-NDS/perovskite shows that the Pb 4f and I 3d characteristic peaks exhibit a negative shift of about 0.13 eV and 0.12 eV, as shown in Fig. 3b and c. The observed shift indicates a stronger bonding environment, which can be attributed to the interaction between the hydroxyl groups (–OH) from K-NDS and the uncoordinated Pb and I in the perovskite. This interaction leads to the effective passivation of dangling bonds on the perovskite surface, thereby reducing the defect density within the perovskite film.35,37,38Fig. 3d shows the FTIR spectra of PbI2, K-NDS, and K-NDS + PbI2. Compared to the FTIR spectrum of PbI2, the Pb–I peak in the K-NDS + PbI2 shifts from 1308.76 cm−1 to 1306.76 cm−1. Additionally, the –OH peak shifts from 3532.14 cm−1 to 3525.49 cm−1 with K-NDS modification, which indicates that the –OH passivates the uncoordinated Pb and I on the perovskite surface, consistent with the XPS analysis. To further elucidate the interaction mechanism between the –OH of K-NDS and PbI2, we conducted 1H NMR measurements on K-NDS and K-NDS + PbI2 samples. The –OH proton signal in K-NDS + PbI2 showed a downfield shift of Δδ = 0.02 ppm compared to that in K-NDS (Fig. S9, ESI†). This downfield shift indicates hydrogen bond formation between the –OH and PbI2, enhancing their interaction and improving perovskite defect passivation.35,38,39 The schematic diagram in Fig. 3e illustrates the K-NDS interface modification mechanism. The –SO3− passivates the SnO2 and fills the oxygen vacancies in the SnO2. Meanwhile, the –OH forms hydrogen bonds with the uncoordinated Pb and I on the perovskite surface, effectively passivating the interfacial defects. Furthermore, the free K+ may reside at the SnO2/perovskite interface, thereby simultaneously passivating both layers.20,26,32,35,38
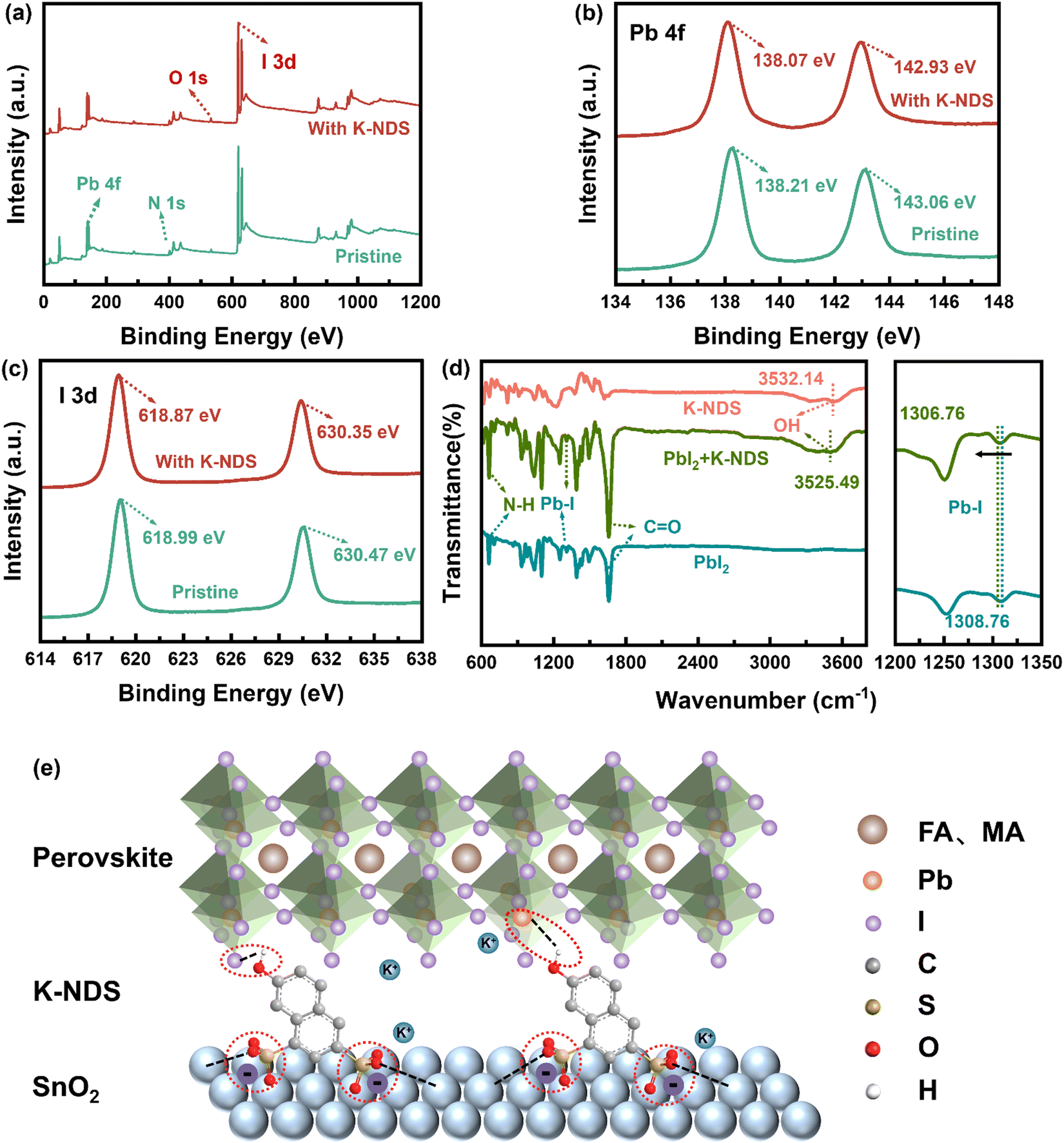 | ||
| Fig. 3 (a) XPS full spectra of the perovskite and K-NDS/perovskite. XPS high resolution spectra of the perovskite and K-NDS/perovskite for (b) Pb 4f, and (c) I 3d. (d) FTIR spectra of the perovskite, K-NDS, and K-NDS/perovskite. (e) The schematic diagram of K-NDS passivation of the SnO2 ETL and perovskite layer. | ||
The modification layer below the perovskite film can significantly impact the crystal growth and overall quality of the perovskite layer, which is crucial for achieving high performance PSC devices.40 Shown in Fig. 4a are the UV-vis absorption spectra of the perovskite and K-NDS/perovskite films, which indicates that the visible light absorption of the perovskite film is not influenced by the K-NDS modification layer. The XRD patterns of the perovskite films deposited on the SnO2 and SnO2/K-NDS substrates were then acquired to investigate the crystallization behavior of the perovskite. As shown in Fig. 4b, compared to the perovskite film deposited on the SnO2 substrate, the perovskite film deposited on the SnO2/K-NDS substrate exhibits a reduced PbI2 diffraction peak intensity, while the (100) peaks are enhanced, without any shift in the overall peak positions. These results indicate that the K-NDS modified layer promotes the crystallization of the perovskite film. Furthermore, as shown in Fig. S10 (ESI†), the full width at half maximum (FWHM) of the (100) peak is lower for the perovskite film deposited on the SnO2/K-NDS substrate, compared to the film deposited on the pristine SnO2 substrate. The narrower FWHM values indicate improved crystallinity of the K-NDS modified perovskite film, which can further enhance the efficiency and stability of the PSCs.19,28
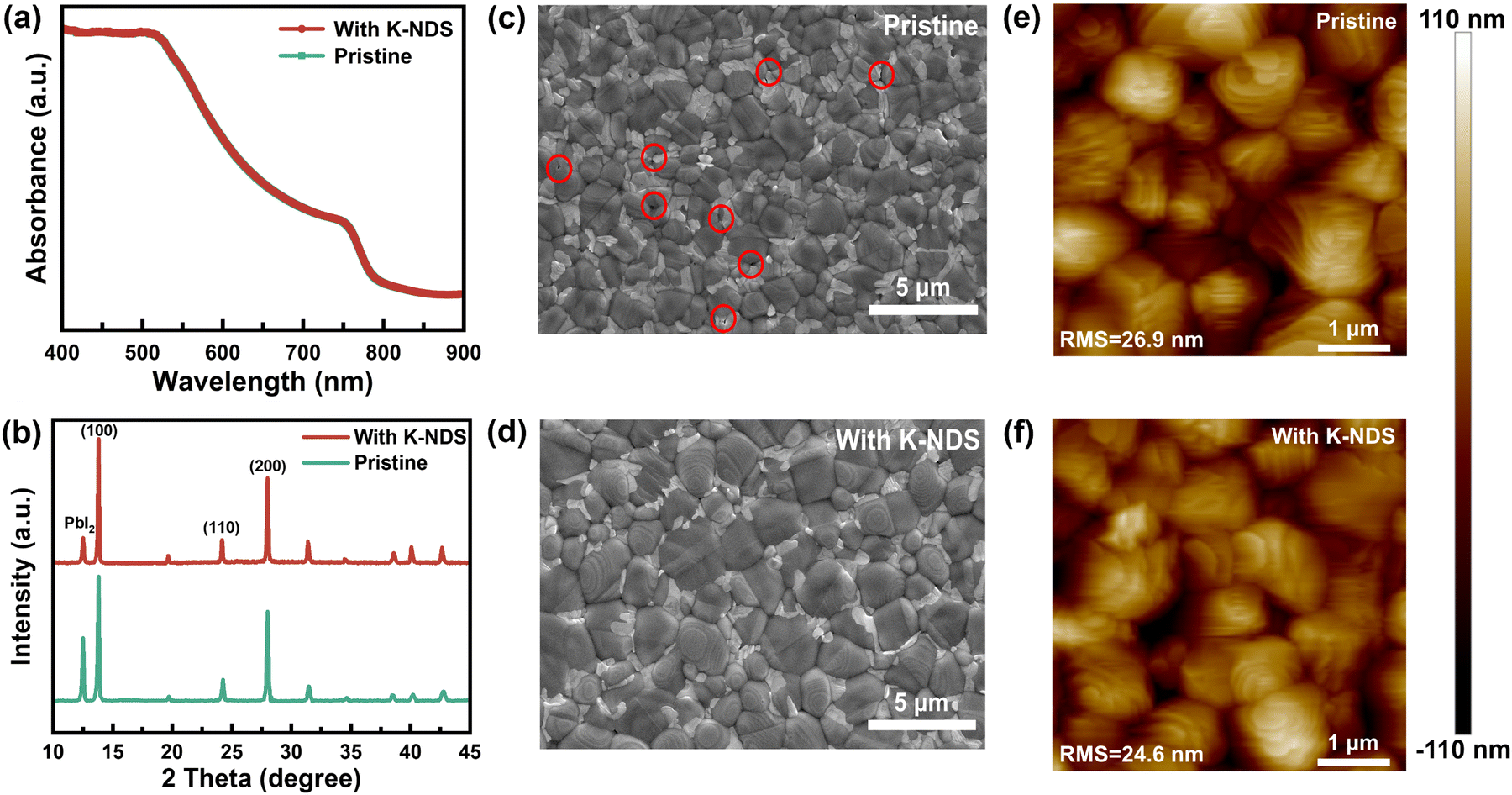 | ||
| Fig. 4 (a) Absorbance spectroscopy and (b) XRD patterns of the perovskite films deposited on SnO2 and SnO2/K-NDS substrates. SEM images of perovskite films deposited on (c) SnO2 and (d) SnO2/K-NDS. AFM images of perovskite films deposited on (e) SnO2 and (f) SnO2/K-NDS. | ||
The impact of the K-NDS modified layer on the perovskite film quality was then investigated using scanning electron microscopy (SEM). As shown in Fig. 4c and d, the SnO2 base perovskite film exhibits some pinholes, while the SnO2/K-NDS based perovskite film shows more uniform overall film morphology. The statistical histogram of the grain size of the perovskite films (Fig. S11, ESI†) reveals that the K-NDS modification can enlarge the grain size of the perovskite and reduce the grain boundaries of the perovskite films.41 The cross-sectional SEM images are shown in Fig. S12 (ESI†), demonstrating that the SnO2/K-NDS-based perovskite exhibits flatter and more uniform morphology, compared to the SnO2-based perovskite. These results confirm that the K-NDS modification enables the formation of higher-quality perovskite films, consistent with the surface SEM results.33,42
The AFM images of the perovskite films deposited on the SnO2 and SnO2/K-NDS substrates are presented in Fig. 4e and f. The RMS roughness of the perovskite film on the SnO2/K-NDS decreased from the initial 26.9 nm to 24.6 nm, indicating an enhancement in the surface morphology of the K-NDS modified perovskite films. The 3D-AFM images shown in Fig. S13 (ESI†) further corroborate the smaller roughness of the perovskite film on the SnO2/K-NDS substrate. These results can be attributed to the formation of hydrogen bonds between the –OH and the uncoordinated Pb and I. As shown in Fig. S14 (ESI†), the hydrogen bonds at the interface retard the perovskite crystallization and effectively passivate defects on the perovskite surface.35,38 Furthermore, during the annealing process after the deposition of the perovskite film, K+ diffuses from the SnO2 layer to the perovskite side and passivates the grain boundaries on the perovskite surface, thus enhancing the overall quality of film formation.20,41
The enhanced interfacial contact and quality of both the SnO2 and perovskite films can have an impact on the extraction and transport of carriers at the interface. We performed PL and TRPL measurements on devices with two configurations: SnO2/perovskite and SnO2/K-NDS/perovskite. In both configurations, the light was incident from the perovskite side. As shown in the PL spectrum in Fig. 5a, compared to the perovskite based on SnO2, the perovskite based on SnO2/K-NDS exhibits pronounced PL quenching, indicating that the SnO2/K-NDS ETL is more efficient in extracting carriers. Subsequently, the charge transport and recombination dynamics within the perovskite films were further analyzed using TRPL spectroscopy, and the results were fitted using an exponential decay function:
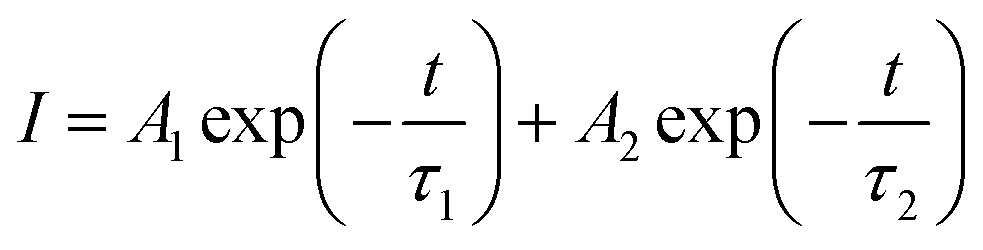 | (3) |
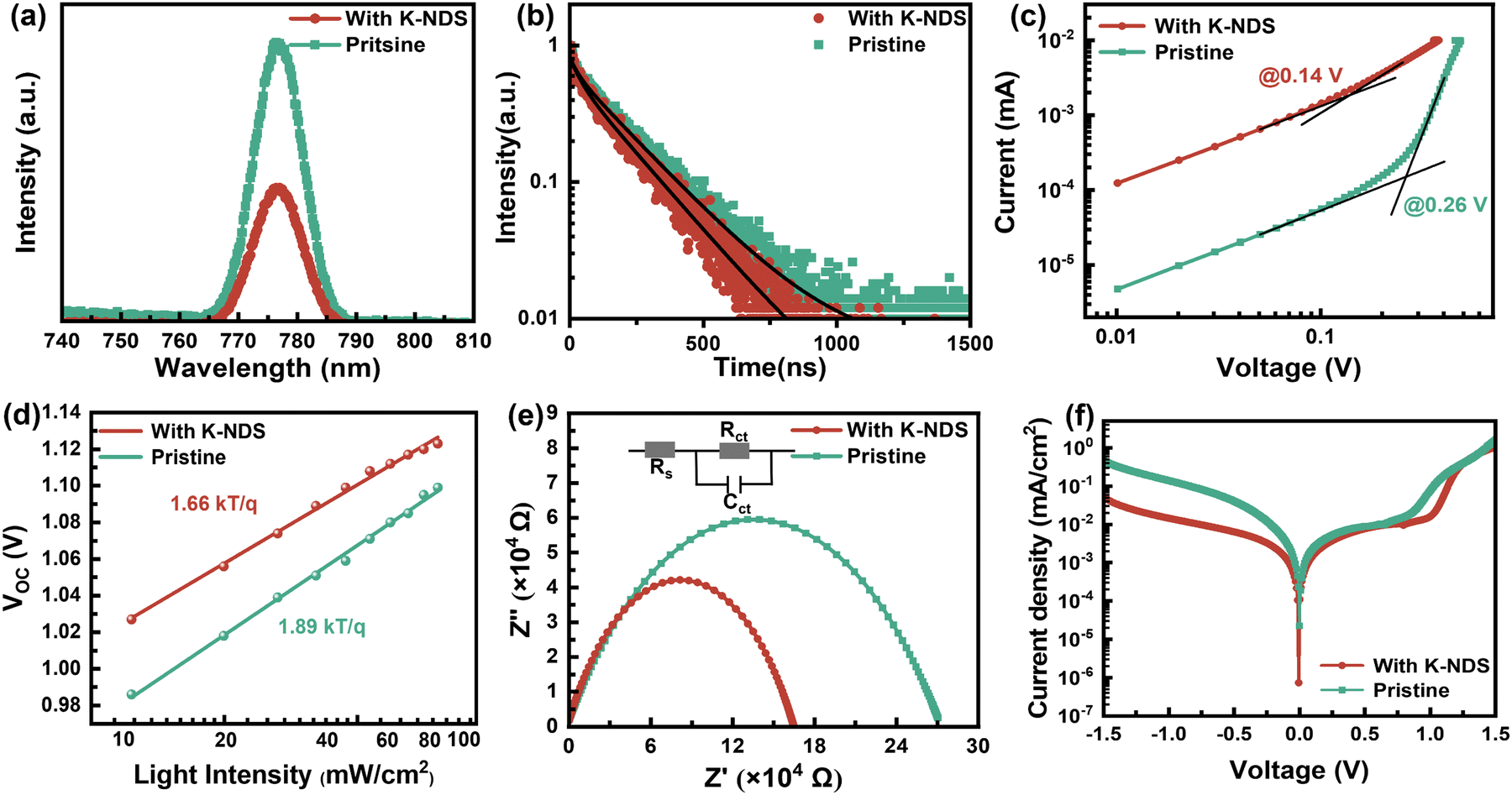 | ||
| Fig. 5 (a) PL and (b) TRPL of perovskite films deposited on the SnO2 and SnO2/K-NDS substrates. (c) Dark J–V curves, (d) VOCversus light intensity plots, (e) Nyquist plots, and (f) dark current characteristics of SnO2 and SnO2/K-NDS based PSCs device. | ||
The carrier lifetimes obtained from the exponential decay function fitting are shown in Fig. 5b and Table S3 (ESI†). Specifically, for the SnO2/K-NDS/perovskite film, the average carrier lifetime (τave) decreases from 203.07 ns for the pristine SnO2/perovskite film to 181.27 ns. This shorter carrier lifetime is consistent with the steady state PL results, proving that the K-NDS modification effectively enhances electron extraction and transport at the interface.43
The space charge-limited current (SCLC) technique was utilized to calculate the trap state density in the electron-only devices of ITO/SnO2/without or with K-KDS/perovskite/PCBM/Ag. As shown in Fig. 5c, the linear response area observed under low bias voltage corresponds to the ohmic contact regime of the device. Nevertheless, as the applied bias voltage goes up, the current increases rapidly and linearity vanishes, suggesting that the traps in the perovskite film are being filled. This voltage point is defined as the trap-filling voltage (VTFL),28 which can then be utilized to calculate the electron trap density in the perovskite film according to the following equation:
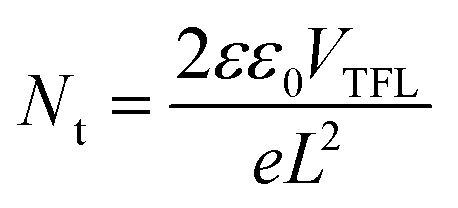 | (4) |
In this equation, e is the elementary charge, Nt denotes the trap state density in the corresponding perovskite film, L represents the thickness of the perovskite layer, and ε and ε0 represent the relative dielectric constant and vacuum dielectric constant of the perovskite. The calculation results in Table S4 (ESI†) showed that for the K-NDS modified device, the trap state density in the perovskite film dropped from 3.063 × 1015 cm−3 to 1.636 × 1015 cm−3. This reduction can be attributed to the improved electron transport layer, enhanced perovskite film quality, and elevated interfacial contact.43,44
As shown in Fig. 5d, the open-circuit voltage (VOC) of perovskite films deposited on different substrates is dependent on light intensity. The deviation of the slope from the ideal value of kT/q indicates the presence of trap-assisted recombination centers within the solar cell, leading to non-radiative recombination of charge carriers.41 These can be calculated by using the following formula:
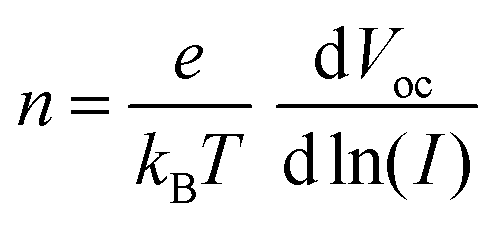 | (5) |
In this formula, n represents the ideality factor, k is the Boltzmann constant, T is the absolute temperature, and e is the elementary charge. The slope of the SnO2/K-NDS based PSC device is 1.66kT/q, which is lower than the 1.89kT/q of the SnO2-based PSC device. These results indicate that the SnO2/K-NDS-based PSC device has less non-radiative recombination caused by defects. Furthermore, changes in the charge transport within the perovskite devices were investigated using electrochemical impedance spectroscopy (EIS). As shown in Fig. 5e, the corresponding Nyquist plots indicated that the semicircle in the high-frequency region represents the charge transfer resistance (Rct).45 Compared to the original PSC device, the Rct of the SnO2/K-NDS-based PSC device is significantly reduced, indicating that the K-NDS modification enables more efficient extraction and transport of charge carriers, consistent with the TRPL result.
Finally, dark current measurements were carried out on the PSCs devices fabricated on different substrates. As shown in Fig. 5f, the dark current density of the SnO2/K-NDS-based PSC device was effectively reduced, indicating a significant suppression of the leakage current within the device.46 This confirms that the incorporation of K-NDS passivates defects at the interface, thereby reducing the trapping of photogenerated carriers by defect states and promoting the extraction and transport of carriers at the interface.
To further validate the advantages of the K-NDS modification and to explore the optimal K-NDS concentration for high efficiency PSCs, we set up a K-NDS solution concentration gradient of 0, 2, 4, and 6 mg ml−1. Twenty groups of PSC devices were fabricated by spin-coating the K-NDS solution onto the SnO2 substrate. The performance characteristics of the PSC devices fabricated with different K-NDS concentrations are summarized in Fig. S15 and Table S5 (ESI†). The champion device, achieved with a K-NDS concentration of 4 mg ml−1, exhibited a VOC of 1.172 V, JSC of 25.44 mA cm−2, FF of 77.15%, and PCE of 23.00%. In comparison, the original device showed a VOC of 1.150 V, JSC of 25.20 mA cm−2, FF of 75.27%, and PCE of 21.81%, as shown in Fig. 6a.
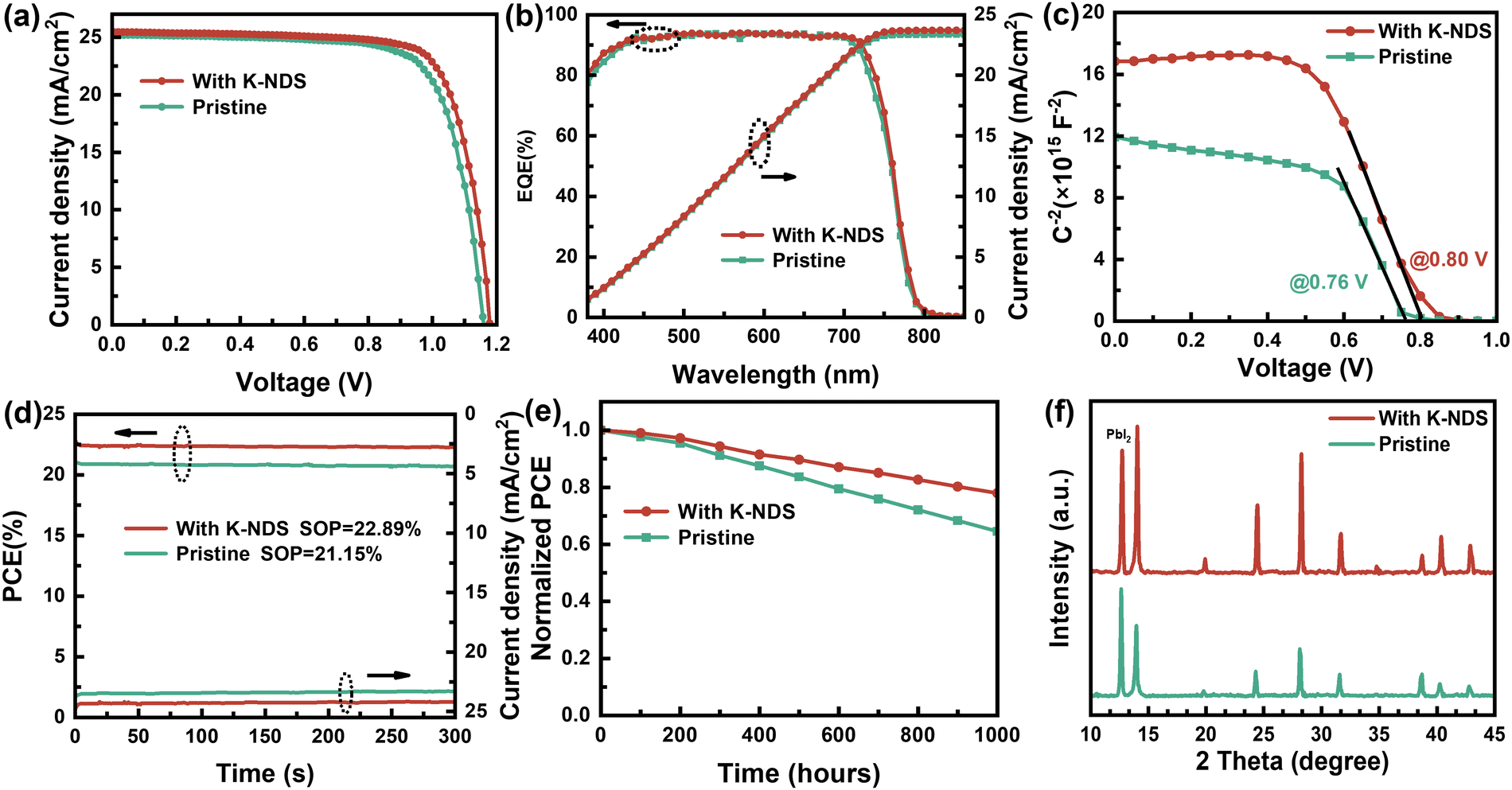 | ||
| Fig. 6 (a) J–V curves of champion PSCs devices with or without the K-NDS passivation layer. (b) EQE and current density spectra, (c) Mott–Schottky curves, (d) steady output of PSC devices deposited on SnO2 and SnO2/K-NDS ETL. (e) Long-term stability of without encapsulation PSC devices via storage under indoor conditions. (f) The XRD patterns of perovskite films deposited on SnO2 and SnO2/K-NDS via storage under indoor environment. | ||
The external quantum efficiency (EQE) spectra of the corresponding devices are shown in Fig. 6b. The integrated currents calculated from the EQE spectra are 23.39 mA cm−2 for the SnO2-based PSC device and 23.73 mA cm−2 for the SnO2/K-NDS-based PSC device, which are consistent with the J–V measurements. Furthermore, capacitance–voltage (C–V) measurements were used to investigate the changes in the built-in potential (Vbi) of the perovskite devices. The corresponding Mott–Schottky plots are presented in Fig. 6c. Compared to the original device, the Vbi of the K-NDS modified device increased from 0.76 V to 0.80 V. The increased Vbi can enhance the extraction and transport of photogenerated carriers at the interface. The improvement in the PSC device performance, as reflected by the increase in VOC and FF, can be attributed to the introduction of the K-NDS interface modification layer, which improves the film quality and optimizes the interlayer contact and energy level alignment within the device.
To assess device stability, we measured the steady output (SOP) under maximum power point (MPP) conditions. The SnO2/K-NDS based PSC device achieved a steady efficiency of 22.89% at MPP (@1 V), while the SnO2 based PSC device reached 21.15% at MPP (@0.92 V), as shown in Fig. 6d. Neither device exhibited significant performance degradation during the 300 s tracking test. This demonstrates that the working stability of the device can be improved after K-NDS modification. Additionally, the device J–V curves obtained during both reverse scan (RS) and forward scan (FS) are shown in Fig. S16 and in Table S6 (ESI†). The hysteresis observed in the SnO2/K-NDS based PSC device is reduced, indicating that the trap density is decreased, and the electron extraction efficiency is improved after the K-NDS modification.
To investigate the influence of the K-NDS bottom interface modification on the stability of PSC devices, unencapsulated SnO2-based and SnO2/K-ND- based PSCs devices were conducted to long-term efficiency tests. As shown in Fig. 6e, the SnO2/K-NDS-based PSC device could maintain 75% of the initial PCE after 1000 hours of storage at RH = 45 ± 5% and 20 ± 5 °C (indoor conditions). In contrast, the original PSC device could only retain less than 60% of the initial PCE under the same conditions. Additionally, XRD patterns were obtained on the perovskite films that had been stored in an indoor environment for 30 days. As shown in Fig. 6f, the perovskite film fabricated on the SnO2/K-NDS structure exhibited slow decomposition and maintained its original shape, while the crystal structure of the SnO2-based perovskite film was severely degraded. Furthermore, water contact angle measurements were carried out in the SnO2/perovskite and SnO2/K-NDS/perovskite films, as shown in Fig. S17 (ESI†). The results indicate that the K-NDS modified perovskite film performs better hydrophobically, which can be attributed to the K-NDS modification improving the quality of the perovskite film and making it more compact to prevent moisture ingress. These results prove that the K-NDS modification effectively improves the film quality and reduces the defect state density at the interface, thereby enhancing the stability of the PSC devices.
Conclusions
In summary, we present a novel synergistic passivation approach utilizing naphthol sulfonate potassium salt to enhance the performance and stability of the PSCs. Our comprehensive experimental investigations reveal that K-NDS modification significantly improves the structural and electronic properties of the SnO2 ETL. This modification not only optimizes the energy level alignment and interfacial contact between SnO2 and the perovskite absorber, but also markedly increases the conductivity of the SnO2 layer. The incorporation of K-NDS at the SnO2/perovskite interface effectively passivates interfacial defects through the synergistic action of the –SO3−, –OH, and K+ functional groups, leading to reduced trap state density and enhanced crystallinity of the perovskite films. These improvements facilitate more efficient carrier extraction and transport, culminating in an increase in both VOC and fill factor. As a result, PSCs incorporating the K-NDS-modified SnO2 ETL achieved a champion PCE of 23.00%. Moreover, unencapsulated devices demonstrated exceptional long-term stability, retaining 75% of their initial PCE after 1000 hours of storage under indoor conditions. This work establishes a robust and scalable strategy for interfacial engineering in PSCs, offering a promising pathway toward the development of high-efficiency, stable perovskite-based photovoltaics.Author contributions
Hao Liu and Ning Jiang contributed equally to this work. Hao Liu: conceptualization, data curation, investigation, writing – original draft, writing – review & editing. Ning Jiang: conceptualization, formal analysis, software, writing – review & editing. Jintao Wang: methodology, resources, visualization, writing – original draft. Shuming Chen: data curation, validation, supervision. Jian Zhang: funding acquisition, project administration, supervision, writing – original draft. Yu Duan: resources, funding acquisition, visualization, writing – review & editing.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Natural Science Foundation project (Grant No. 62374070) and the Project of Science and Technology Development Plan of Jilin Province (Grant No. 20230201040GX, 20240302068GX), Changchun Science and Technology Development Plan Project (Grant No. 23JQ02).References
- Z.-W. Gao, Y. Wang and W. C. H. Choy, Adv. Energy Mater., 2022, 12, 2104030 CrossRef CAS.
- J. Wang, G. Jin, Q. Zhen, C. He and Y. Duan, Adv. Mater. Interfaces, 2021, 8, 2002078 CrossRef CAS.
- M. R. Filip, G. E. Eperon, H. J. Snaith and F. Giustino, Nat. Commun., 2014, 5, 5757 CrossRef CAS PubMed.
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T.-W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582–587 Search PubMed.
- Z. Guo, J. S. Manser, Y. Wan, P. V. Kamat and L. Huang, Nat. Commun., 2015, 6, 7471 Search PubMed.
- C. Yu, B. Zhang, C. Chen, J. Wang, J. Zhang, P. Chen, C. Li and Y. Duan, Nano Res., 2022, 15, 4431–4438 CrossRef CAS.
- J. Wang, Z. Wang, S. Chen, N. Jiang, L. Yuan, J. Zhang and Y. Duan, Adv. Mater. Interfaces, 2023, 10, 2202266 CrossRef CAS.
- S. Chen, J. Wang, C. Yu, N. Jiang, Z. Wang, Y. Zhou, C. He, K. Fang, B. Liu, J. Zhang, Y. Li, C. Li, P. Chen and Y. Duan, Sol. RRL, 2022, 6, 2200405 CrossRef CAS.
- K. Fang, J. Wang, H. Liu, S. Chen, N. Jiang, Z. Wang, C. Li, J. Zhang and Y. Duan, Sol. RRL, 2023, 7, 2300766 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- National Renewable Energy Laboratory, “Best Research-Cell Efficiency Chart”, 2024, https://www.nrel.gov/pv/cell-efficiency.html.
- C. M. Wolff, P. Caprioglio, M. Stolterfoht and D. Neher, Adv. Mater., 2019, 31, 1902762 CrossRef CAS PubMed.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2020, 5, 44–60 CrossRef CAS.
- L. Xiong, Y. Guo, J. Wen, H. Liu, G. Yang, P. Qin and G. Fang, Adv. Funct. Mater., 2018, 28, 1802757 CrossRef.
- Y. Bai, X. Meng and S. Yang, Adv. Energy Mater., 2018, 8, 1701883 CrossRef.
- S. Y. Park and K. Zhu, Adv. Mater., 2022, 34, 2110438 CrossRef CAS PubMed.
- S. You, H. Zeng, Z. Ku, X. Wang, Z. Wang, Y. Rong, Y. Zhao, X. Zheng, L. Luo, L. Li, S. Zhang, M. Li, X. Gao and X. Li, Adv. Mater., 2020, 32, 2003990 CrossRef CAS PubMed.
- K. Choi, J. Lee, H. I. Kim, C. W. Park, G.-W. Kim, H. Choi, S. Park, S. A. Park and T. Park, Energy Environ. Sci., 2018, 11, 3238–3247 RSC.
- P. Zhu, S. Gu, X. Luo, Y. Gao, S. Li, J. Zhu and H. Tan, Adv. Energy Mater., 2020, 10, 1903083 CrossRef CAS.
- D.-Y. Son, S.-G. Kim, J.-Y. Seo, S.-H. Lee, H. Shin, D. Lee and N.-G. Park, J. Am. Chem. Soc., 2018, 140, 1358–1364 CrossRef CAS PubMed.
- E. H. Jung, B. Chen, K. Bertens, M. Vafaie, S. Teale, A. Proppe, Y. Hou, T. Zhu, C. Zheng and E. H. Sargent, ACS Energy Lett., 2020, 5, 2796–2801 CrossRef CAS.
- K. Huang, L. Chang, Y. Hou, W. Ji, T. Tran-Phú, A. D. Bui, A. O. Mayon, W. Wang, O. L. C. Lem, D. Nguyen, G. D. Tabi, L. Duan, Y. Liu, H. Shen, J. Yang, T. P. White, K. R. Catchpole, K. J. Weber and T. Duong, Adv. Energy Mater., 2024, 2304073 CrossRef CAS.
- X. Shang, X. Ma, F. Meng, J. Ma, L. Yang, M. Li, D. Gao and C. Chen, J. Colloid Interface Sci., 2023, 630, 155–163 CrossRef CAS PubMed.
- T. Bu, J. Li, F. Zheng, W. Chen, X. Wen, Z. Ku, Y. Peng, J. Zhong, Y.-B. Cheng and F. Huang, Nat. Commun., 2018, 9, 4609 CrossRef PubMed.
- Z. Xiong, X. Chen, B. Zhang, G. O. Odunmbaku, Z. Ou, B. Guo, K. Yang, Z. Kan, S. Lu, S. Chen, N. A. N. Ouedraogo, Y. Cho, C. Yang, J. Chen and K. Sun, Adv. Mater., 2022, 34, 2106118 CrossRef CAS PubMed.
- J. Yan, Z. Lin, Q. Cai, X. Wen and C. Mu, ACS Appl. Energy Mater., 2020, 3, 3504–3511 CrossRef CAS.
- Y. Dong, W. Shen, W. Dong, C. Bai, J. Zhao, Y. Zhou, F. Huang, Y. Cheng and J. Zhong, Adv. Energy Mater., 2022, 12, 2200417 CrossRef CAS.
- J. H. Lee, S. Lee, T. Kim, H. Ahn, G. Y. Jang, K. H. Kim, Y. J. Cho, K. Zhang, J.-S. Park and J. H. Park, Joule, 2023, 7, 380–397 CrossRef CAS.
- H.-Q. Zhan, C. Deng, Q. Liu, X.-H. Li, Z.-P. Xie and C.-A. Wang, Chin. J. Inorg. Chem., 2020, 36, 1605–1612 CAS.
- J. Chang, E. Feng, X. Feng, H. Li, Y. Ding, C. Long, S. Lu, H. Zhu, W. Deng, J. Shi, Y. Yang, S. Xiao, Y. Yuan and J. Yang, Nano Res., 2024, 17, 8068–8076 CrossRef CAS.
- Y. Li, D. Yao, Z. Tang, B. Jiang, C. Li, Y. Gao, N. Tian, J. Wang, G. Zheng and F. Long, ACS Appl. Mater. Interfaces, 2024, 16, 9388–9399 CrossRef CAS PubMed.
- X. Wang, H. Huang, M. Wang, Z. Lan, P. Cui, S. Du, Y. Yang, L. Yan, Q. Zhang, S. Qu and M. Li, Adv. Mater., 2024, 2310710 CrossRef CAS PubMed.
- R. Zhao, Z. Deng, Z. Zhang, J. Zhang, T. Guo, Y. Xing, X. Liu, L. Huang, Z. Hu and Y. Zhu, ACS Appl. Mater. Interfaces, 2022, 14, 36711–36720 CrossRef CAS PubMed.
- P. Hu, W. Zhou, J. Chen, X. Xie, J. Zhu, Y. Zheng, Y. Li, J. Li and M. Wei, Chem. Eng. J., 2024, 480, 148249 CrossRef CAS.
- D. Yang, R. Yang, K. Wang, C. Wu, X. Zhu, J. Feng, X. Ren, G. Fang, S. Priya and S. (Frank) Liu, Nat. Commun., 2018, 9, 3239 CrossRef PubMed.
- L. Lin, T. W. Jones, T. C.-J. Yang, X. Li, C. Wu, Z. Xiao, H. Li, J. Li, J. Qian, L. Lin, J. Q. Shi, S. D. Stranks, G. J. Wilson and X. Wang, Matter, 2024, 7, 38–58 CrossRef CAS.
- Q. Yang, X. Liu, S. Yu, Z. Feng, L. Liang, W. Qin, Y. Wang, X. Hu, S. Chen, Z. Feng, G. Hou, K. Wu, X. Guo and C. Li, Energy Environ. Sci., 2021, 14, 6536–6545 RSC.
- X. Li, C. Chen, M. Cai, X. Hua, F. Xie, X. Liu, J. Hua, Y. Long, H. Tian and L. Han, Adv. Energy Mater., 2018, 8, 1800715 CrossRef.
- J. Guo, G. Meng, X. Zhang, H. Huang, J. Shi, B. Wang, X. Hu, J. Yuan and W. Ma, Adv. Mater., 2023, 35, 2302839 CrossRef CAS PubMed.
- Q. Xiao, Y. Zhao, Z. Huang, Y. Liu, P. Chen, S. Wang, S. Zhang, Y. Zhang and Y. Song, Adv. Funct. Mater., 2024, 2314472 CrossRef CAS.
- H. Yang, R. Li, S. Gong, H. Wang, S. M. H. Qaid, Q. Zhou, W. Cai, X. Chen, J. Chen and Z. Zang, Nano Lett., 2023, 23, 8610–8619 CrossRef CAS PubMed.
- M. Wu, Y. Duan, L. Yang, P. You, Z. Li, J. Wang, H. Zhou, S. Yang, D. Xu, H. Zou and Z. Liu, Adv. Funct. Mater., 2023, 33, 2300128 CrossRef CAS.
- Y. Cao, J. Feng, M. Wang, N. Yan, J. Lou, X. Feng, F. Xiao, Y. Liu, D. Qi, Y. Yuan, X. Zhu and S. (Frank) Liu, Adv. Energy Mater., 2023, 13, 2302103 CrossRef CAS.
- W. E. I. Sha, H. Zhang, Z. S. Wang, H. L. Zhu, X. Ren, F. Lin, A. K.-Y. Jen and W. C. H. Choy, Adv. Energy Mater., 2018, 8, 1701586 CrossRef.
- Y. Wang, B. Zhou, M. Han, J. Zhao, R. Wang, J. Zhang, H. Ren, G. Hou, Y. Ding, Y. Zhao and X. Zhang, Nano Energy, 2023, 118, 108981 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01311e |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2025 |