
A bioactive multifunctional dressing with simultaneous visible monitoring of pH values and H2O2 concentrations for promoting diabetic wound healing†
Jimin
Huang
ab,
Jinzhou
Huang
ab,
XinXin
Zhang
ab,
Qinyi
Xie
c,
Yi
Zheng
ab,
Chaoqin
Shu
a,
Zhe
Shi
a,
Xiao
Wang
ab,
Jiajie
Chen
 ab,
Bing
Ma
a,
Chengtie
Wu
ab and
Yufang
Zhu
*ab
ab,
Bing
Ma
a,
Chengtie
Wu
ab and
Yufang
Zhu
*ab
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, P. R. China. E-mail: zjf2412@163.com
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, 19A Yuquan Road, Beijing 100049, P. R. China
cSchool of Pediatrics, Chongqing Medical University, 136 Zhongshan 2nd Road, Chongqing 400014, P. R. China
First published on 18th October 2024
Abstract
Wound healing in diabetes is a complex physiological process with risks of ulceration and amputation. Real-time monitoring and treatment of diabetic wounds is an effective prevention of further deterioration. Herein, a multifunctional dressing is developed by encapsulating europium-containing bioactive glass (EuBG) and MoO3−x in a biocompatible sodium alginate (SA) dressing (MoO3−x–EuBG–SA), aiming to simultaneously monitor the wound pH value and hydrogen peroxide (H2O2) concentration, establish an anti-inflammatory microenvironment, and promote wound healing. The pH-responsive fluorescence quenching effect of EuBG contributes to visible monitoring of pH values, and MoO3−x enables high responsiveness and accurate colorimetric indication of H2O2 variability to understand the dynamic wound status. Then, these fluorescent and colorimetric changes can be captured using a smartphone and transformed into signals for wound pH values and H2O2 concentrations, enabling rapid and convenient real-time assessment of wound dynamics. Furthermore, the MoO3−x–EuBG–SA dressing can reduce inflammation and promote the proliferation, differentiation, and migration of fibroblasts and endothelial cells in vitro with the help of the released bioactive Si, Ca, and Mo ions. Notably, the MoO3−x–EuBG–SA dressing exhibits excellent abilities to monitor a wound microenvironment and immune regulation and promote diabetic wound healing in vivo. Overall, the multifunctional dressing has great potential for managing diabetic wounds and promoting wound healing.
New conceptsThe integration of monitoring and treatment is an important strategy for diabetic wound management. Here, we report a multifunctional wound dressing by encapsulating europium-containing bioactive glass (EuBG) and MoO3−x nanosheets in a biocompatible sodium alginate dressing (MoO3−x–EuBG–SA) for diabetic wound healing and monitoring. We have confirmed that the MoO3−x–EuBG–SA dressing could monitor a wound microenvironment due to the pH-responsive fluorescence quenching effect of EuBG particles and the H2O2-dependent color fading effect of MoO3−x nanosheets. Additionally, the MoO3−x–EuBG–SA dressing had a noteworthy function of accelerating diabetic wound healing through promoting vascular regeneration, collagen deposition and reducing inflammation. Hence, MoO3−x–EuBG–SA can be used as an ideal dressing to monitor diabetic wound status and accelerate wound healing. |
1. Introduction
Delayed or inadequate treatment of diabetic wounds can lead to disability or even death in diabetic patients.1 Wound healing can typically be divided into four phases: hemostasis, inflammation, proliferation, and remodeling.2,3 However, wounds in diabetic patients often fail to heal due to bacterial infections or stagnation in the pathological inflammatory phase.4,5 The dysregulation of immune cells causes inflammation, which hinders wound healing and leads to susceptibility to infection.6 The activation of ROS production exacerbates the inflammatory response and contributes to the difficulty in diabetic wound healing.7,8 During the various stages of the healing process of diabetes, wound exudates undergo specific changes in pH, hydrogen peroxide (H2O2), blood glucose, oxygen levels, etc.9 These wound biomarkers can indicate the healing status of the wound, which is crucial for effective wound management.10 The development of a multifunctional material that integrates diabetic wound repair and monitoring can alleviate patient pain and reduce pressure on the healthcare system.11pH is a fundamental index of the physiological microenvironment of diabetic wounds, and can significantly affect many physiological processes including inflammatory response, collagen formation, and angiogenesis.12,13 There is increasing evidence that the pH of a wound microenvironment is closely linked to wound infection and healing status.14,15 During the healing process, the pH of the exudates in diabetic wounds undergoes dynamic changes.16 Generally, the pH of normal skin tissue is usually between 4 and 6.17 However, upon damage or infection, the pH of the diabetic wound microenvironment usually approaches the physiological pH of 7.4 due to microvascular leakage and wound tissue hypoxia.18 Consequently, the pH value of wound skin tissue significantly increases to 7.5–8.9, which can cause inflammation and lead to delays in wound healing.19 Therefore, monitoring wound pH can provide early warning of the risk of infection and indicate the different stages of healing. This undoubtedly contributes to effective wound management. Various methods have been developed for monitoring pH values in wounds, such as fluorescence detection, electrochemical monitoring, and surface plasmon resonance.20–22 However, most of the materials used for pH monitoring only serve the purpose of monitoring and do not actively promote effective wound healing.
Bioactive glass (BG) is biocompatible and can promote wound healing by releasing bioactive ions to stimulate vascularization and significantly increase the activity of cells.23,24 In addition, BG can be doped with metal ions, such as copper (Cu), zinc (Zn), cerium (Ce), strontium (Sr), and manganese (Mn), to acquire antimicrobial, nerve regeneration, and antioxidant properties.25–29 Lanthanide-based fluorescent probes have attracted much attention due to abundant narrow-band emission peaks, long fluorescence lifetimes, and superior photostability.30,31 Europium-based fluorescent materials, including nanoparticles and organic complexes, have been found to exhibit pH-responsive properties and have been investigated for monitoring the change of pH values.32,33 Both promoting tissue regeneration and monitoring microenvironment are required for diabetic wound healing. Here, europium-containing bioactive glasses (EuBGs) with the abilities to monitor pH changes and promote diabetic wound healing were prepared for the first time. The incorporation of Eu ions endowed the bioactive glass with the function of monitoring pH, achieving the integration of monitoring the wound microenvironment and promoting wound healing.
In addition to susceptibility to bacterial infections, the inflammation and oxidative stress in the wound microenvironment are significant factors that contribute to the challenges in healing diabetic wounds.34,35 Immune cells and factors are activated upon skin tissue injury, initiating an inflammatory process.36 This is followed by the production of reactive oxygen species (ROS), which prevents bacterial infection and promotes wound repair.37–39 However, as a principal member of ROS, H2O2 can accumulate in large amounts because the immune system becomes dysregulated during diabetic wound healing.40,41 Excessive H2O2 can lead to an exaggerated inflammatory response in macrophages by initiating the NF-kB transcription factor pathway, and thereby inhibit diabetic wound recovery.42,43 It has been demonstrated that inflammation and oxidative stress have a reciprocal relationship.44 Inflammation leads to an increase in H2O2 production, which in turn exacerbates inflammation.8 Therefore, the elimination of inflammation and continuous monitoring the concentration of H2O2 are meaningful for wound management.
To date, various materials have been developed for monitoring the dynamic changes of the H2O2 concentration in wound microenvironment, such as quantum dots (QDs), metal–organic framework (MOF)-based biosensors, and near-infrared dye-sensitized upconversion nanoparticles (UCNPs).45–47 However, these sensors typically do not possess the functions that integrate the H2O2 monitoring with reduction of inflammation. MoO3−x nanomaterials have been reported to be used in colorimetric monitoring of H2O2 due to the oxidation of the oxygen vacancies by H2O2.48 Interestingly, Mo ions can polarize macrophages into the M2 phenotype to establish an anti-inflammatory microenvironment of wound.49 Therefore, MoO3−x nanomaterials may have excellent capacity to monitor the concentration of H2O2 and reduce inflammation for diabetic wound healing.
Herein, we reported a multifunctional wound dressing by encapsulating EuBG particles and MoO3−x nanosheets in a biocompatible sodium alginate dressing (MoO3−x–EuBG–SA) for diabetic wound healing and monitoring the pH value and H2O2 concentration of wounds. As shown in Scheme 1, the fluorescence of EuBG is continuously quenched as the pH of the microenvironment decreases, which facilitates monitoring of the pH variations based on the change of fluorescence intensity. Meanwhile, EuBG possesses the function of releasing bioactive ions to promote wound healing. Furthermore, the color of MoO3−x nanomaterials can fade as the concentration of H2O2 increases. Also, the Mo ions could polarize macrophages into the M2 phenotype to secrete anti-inflammatory cytokines. In detail, the bioactivity of the MoO3−x–EuBG–SA dressing for wound healing was investigated via evaluating cell proliferation, cell migration, angiogenesis-related gene expression and macrophage phenotype differentiation in vitro. The pH value and H2O2 concentration monitoring and the therapeutic effect on diabetic wound were also studied in vivo. Overall, the MoO3−x–EuBG–SA dressing could monitor the diabetic wound healing process and promote wound healing.
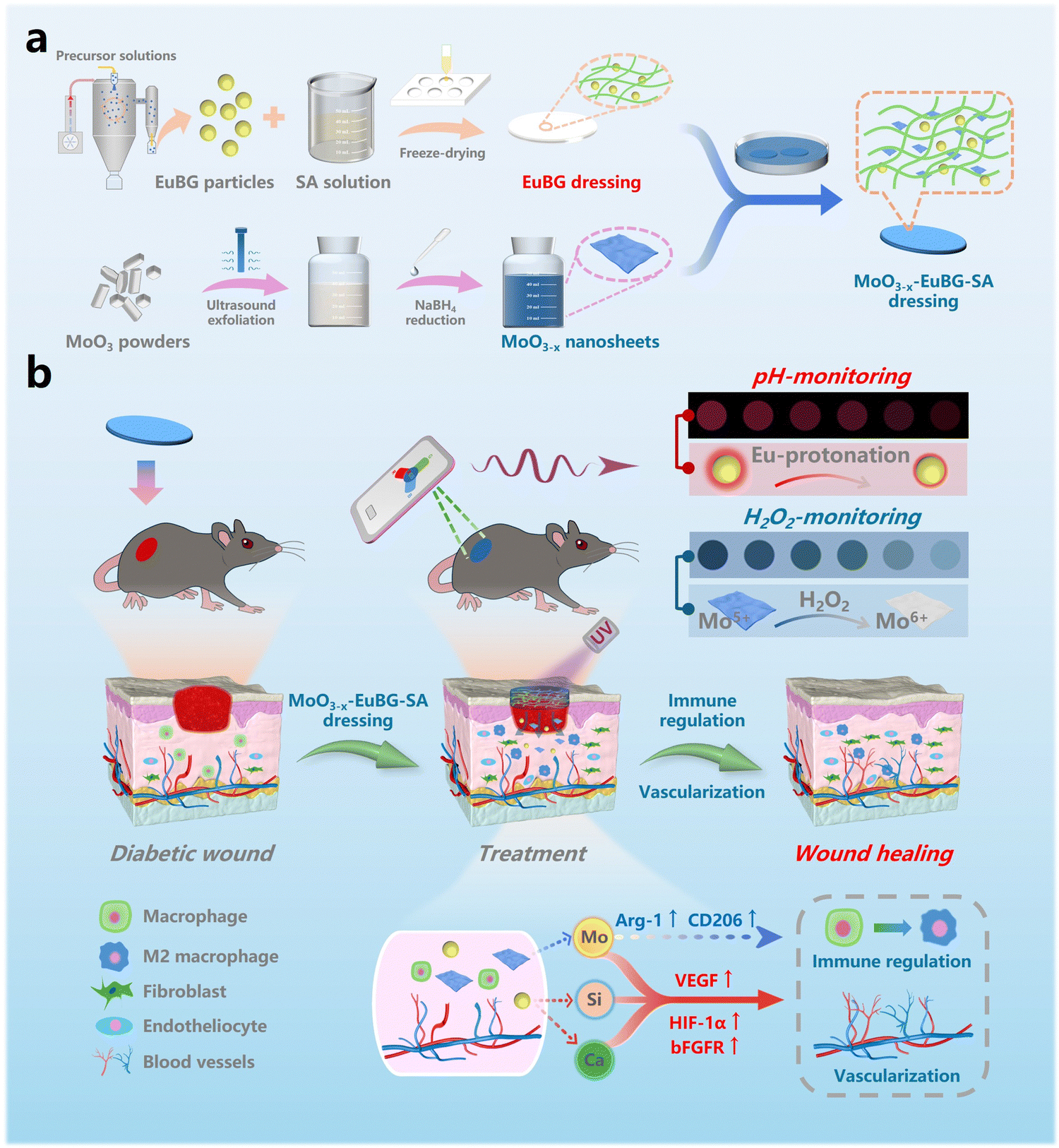 | ||
| Scheme 1 (a) Schematic diagram of materials preparation. (b) Schematic illustration of the MoO3−x–EuBG–SA dressing for simultaneous visible pH and H2O2 monitoring and accelerating diabetic wound healing. | ||
2. Results and discussion
2.1. Synthesis and characterization of EuBG particles and EuBG–SA dressings
EuBG particles containing different contents of Eu were successfully synthesized using a spray drying method. The EuBG particles exhibited a microsphere morphology with a relatively uniform distribution of elements, and the concentration of the Eu element in the particles increased with the addition of Eu during the preparation process, as indicated by both the SEM-EDS images and the ICP test results (Fig. 1a and Fig. S1, S2, ESI†). The statistical results of the particle size demonstrated that the microspheres were below 6 μm (Fig. S3, ESI†). The XRD patterns showed that the Eu addition did not alter the amorphous state of the BG particles, as all microspheres only displayed a broadened diffraction peak within 2θ = 20°–35° (Fig. 1b). Therefore, the above results indicated the successful synthesis of Eu incorporated bioactive glass.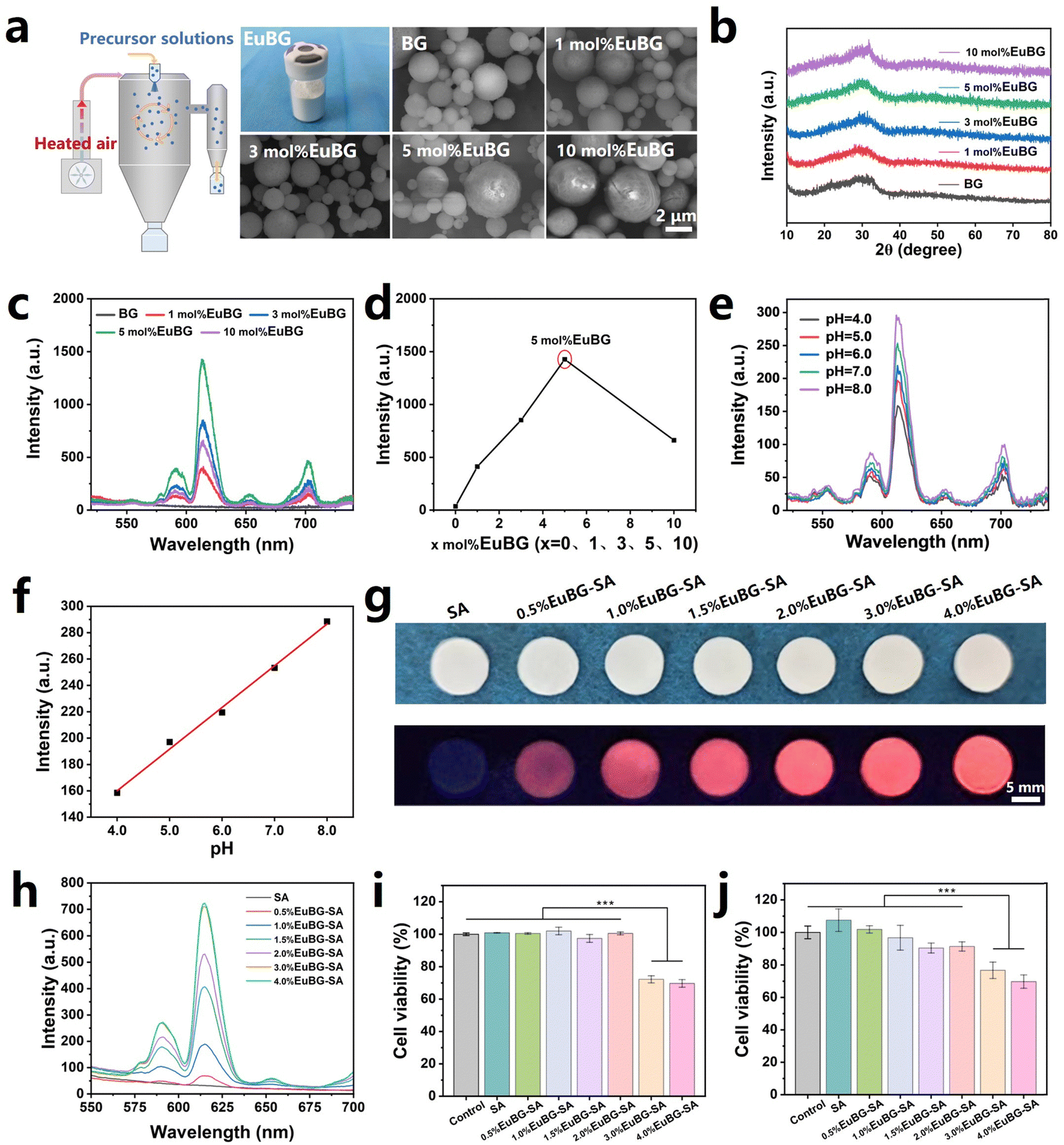 | ||
| Fig. 1 (a) Schematic diagram of bioactive glass particles prepared by the spray drying method and representative SEM images. (b) XRD patterns of EuBG particles. (c) Fluorescence spectrum of EuBG particles and (d) the emission intensities at 614 nm. (e) Fluorescence spectrum of 5 mol% EuBG particles treated with different pHs of PBS and (f) the emission intensities at 614 nm. (g) The photograph of EuBG–SA with different contents of 5 mol% EuBG particles under 254 nm and (h) corresponding fluorescence spectra. Cytotoxicity of (i) HDFs and (j) HUVECs cultured with EuBG–SA dressings (n = 5). All data are expressed as means ± standard deviation, one-way ANOVA with Bonferroni multiple comparison corrections, ***p < 0.001. | ||
It has been reported that the pH value of a wound microenvironment can serve as a forewarning of infection risks and an indicator of different healing phases.50 This is due to the pH of a wound microenvironment varies depending on the stage of healing.51 Therefore, monitoring the pH of wound undoubtedly assists effective wound management especially for diabetic wound. Additionally, repairing diabetic wounds is challenging due to vascular rupture and decreased collagen production.52 To overcome these hurdles, we incorporated Eu into BG to monitor pH and promote wound healing. The fluorescence intensity of BG with different Eu contents was measured. As shown in Fig. 1c and d, the emission intensities of EuBG particles increase with increasing Eu content, reaching a maximum at 5 mol% and then declining due to the concentration quenching effect.53 Then, 5 mol% EuBG particles were selected to measure the performance of pH monitoring. The performance of 5 mol% EuBG particles in monitoring pH was measured by recording the emission spectra of 5 mol% EuBG particles treated with different pH levels of PBS (Fig. 1e). It could be seen that the emission spectra of the 5 mol% EuBG particles showed a gradual decrease as the pH value decreased. The intensities at 614 nm for 5 mol% EuBG particles treated with different pH values of PBS are also shown in Fig. 1f. The fitting results indicated that the fluorescence intensity of 5 mol% EuBG particles exhibited a good linear correlation with pH in the range of 4–8 with a correlation coefficient of R2 = 0.99, suggesting that 5 mol% EuBG particles had a wide linear fluorescence response for pH sensing. Therefore, 5 mol% EuBG particles were selected as the inorganic component of the EuBG–SA dressings.
The lyophilized hydrogel has a porous structure and good flexibility, allowing efficient absorption of wound exudates, making it an excellent vehicle for collection of wound metabolites.54,55 In this study, we creatively prepared EuBG–SA dressings with different mass percentages of 5 mol% EuBG particles in SA by freeze drying to verify dual roles in pH monitoring and promoting wound healing. The optical and fluorescence photographs of EuBG–SA indicated that the dressings prepared by freeze-drying possessed a flat surface, which is conducive to monitoring (Fig. 1g). The EuBG–SA dressings exhibited strong red fluorescence under 254 nm UV irradiation, and the fluorescence intensities of dressings increased with increasing content of 5 mol% EuBG particles (Fig. 1h). The biocompatibility of the EuBG–SA dressings was then evaluated (Fig. 1i and j). It could be known from the cell viabilities of human dermal fibroblasts (HDFs) and human umbilical vein endothelial cells (HUVECs) that the EuBG–SA dressings possessed good biocompatibility when the content of 5 mol% EuBG was within 2%. The above results showed that 2%EuBG–SA had great potential for monitoring the pH of diabetic wound and possessed excellent biocompatibility. Thus, 2%EuBG–SA was selected as the substrate for the subsequent preparation of MoO3−x–EuBG–SA dressings.
2.2. Synthesis and characterization of MoO3−x and MoO3−x–EuBG–SA dressings
MoO3−x nanosheets were successfully synthesized by an improved ultrasound exfoliation-reduction method.56,57 After ultrasound exfoliation-reduction treatment, bulk MoO3 is transformed into MoO3−x nanosheets and the physical phase changes from crystalline to amorphous (Fig. 2a and Fig. S4, ESI†). MoO3−x nanosheets exhibit a thickness of less than 50 nm, which was analyzed by atomic force microscopy (AFM) (Fig. 2b and c). It has been reported that oxygen vacancies were formed when Mo6+ was partially reduced to Mo5+ and MoO3−x nanosheets have adequate surface free carriers to support LSPR absorption in the visible region.48 As illustrated in Fig. 2d, MoO3−x nanosheets showed strong absorption in the near-infrared region from 700 to 900 nm. Then, X-ray photoelectron spectroscopy (XPS) was employed to characterize Mo5+ and Mo6+ in the MoO3−x nanosheets. As displayed in Fig. 2e, the ratio between Mo5+ and Mo6+ was estimated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.67 from fitting the curves. More importantly, MoO3−x nanosheets can be used for monitoring the H2O2 concentration. The color of aqueous dispersions of MoO3−x nanosheets got lightened with the increase of the H2O2 concentration and became eventually colorless, which was attributed to the oxidation of Mo5+ in the nanosheets into Mo6+ by H2O2 (Fig. 2f and Fig. S5, ESI†). And the linear fitting results between the absorption at 750 nm of MoO3−x nanosheets and the H2O2 concentration demonstrated that MoO3−x nanosheets exhibited the ability to monitor the concentration of H2O2 (Fig. 2g).
:2.67 from fitting the curves. More importantly, MoO3−x nanosheets can be used for monitoring the H2O2 concentration. The color of aqueous dispersions of MoO3−x nanosheets got lightened with the increase of the H2O2 concentration and became eventually colorless, which was attributed to the oxidation of Mo5+ in the nanosheets into Mo6+ by H2O2 (Fig. 2f and Fig. S5, ESI†). And the linear fitting results between the absorption at 750 nm of MoO3−x nanosheets and the H2O2 concentration demonstrated that MoO3−x nanosheets exhibited the ability to monitor the concentration of H2O2 (Fig. 2g).
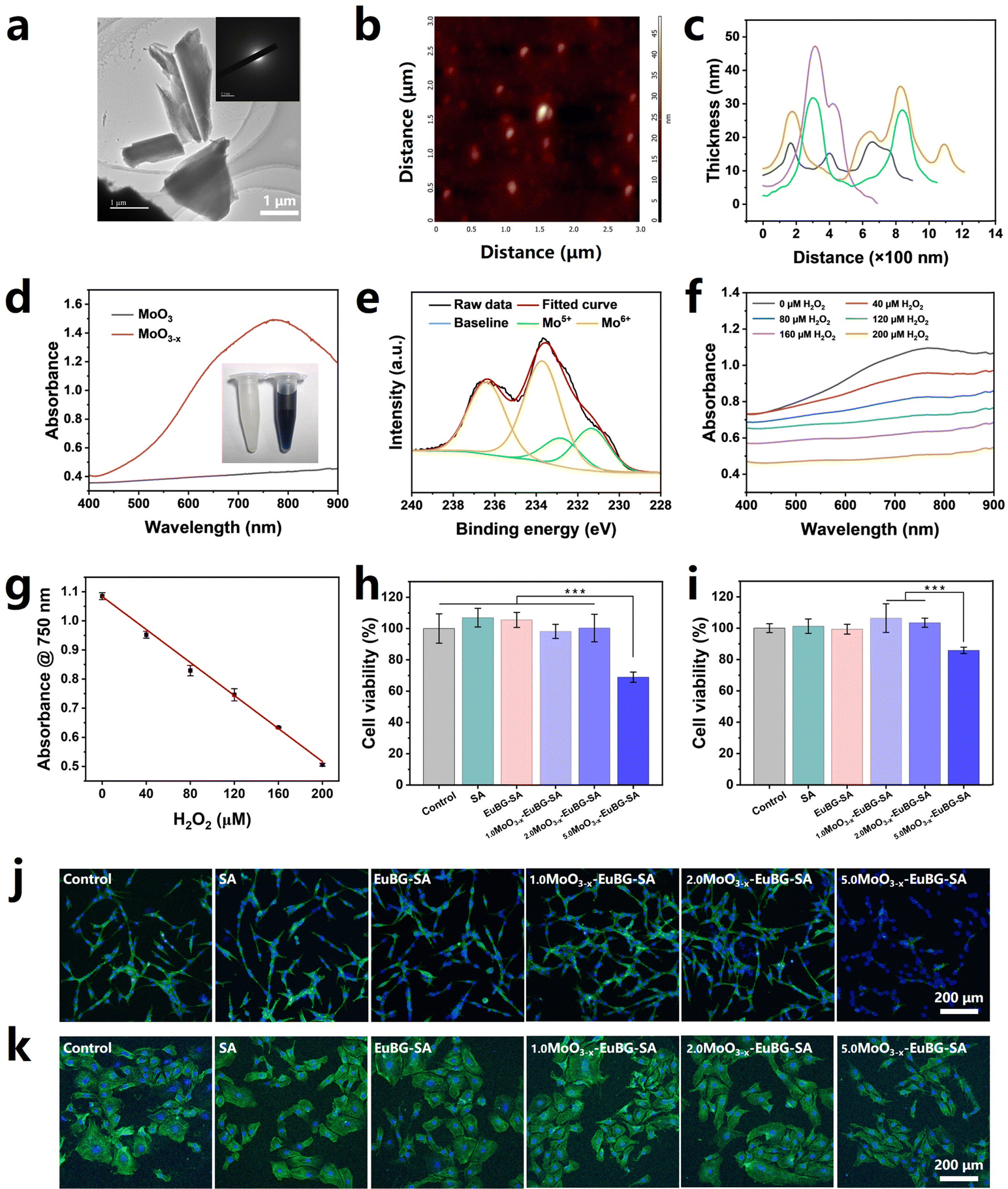 | ||
| Fig. 2 Synthesis and characterization of MoO3−x nanosheets and MoO3−x–EuBG–SA dressings. (a) High-resolution TEM image, (b) atomic force microscopy image, (c) thickness distribution, (d) ultraviolet-visible absorption spectrum and optical photographs, and (e) XPS spectrum of MoO3−x nanosheets. (f) The ultraviolet-visible absorption spectrum of MoO3−x nanosheets treated with various concentrations of H2O2 and (g) the absorbance at 750 nm. Cell viabilities of (h) HDFs and (i) HUVECs cultured with MoO3−x–EuBG–SA dressings (n = 5). Confocal laser scanning microscopy (blue: cell nuclei; green: cytoskeleton) images of (j) HDFs and (k) HUVECs. All data are expressed as means ± standard deviation, one-way ANOVA with Bonferroni multiple comparison corrections, ***p < 0.001. | ||
MoO3−x–EuBG–SA dressings were successfully fabricated by immersing 2%EuBG–SA dressings in different concentrations of MoO3−x nanosheets. The biocompatibility of a material is a crucial requirement for its use in biomedical applications. Thus, HDFs and HUVECs were chosen to evaluate the cytotoxicity of MoO3−x–EuBG–SA through a classical CCK-8 assay, live/dead staining, and nucleus/skeleton staining. As shown in Fig. 2h, i and Fig. S6 (ESI†), the cell viability and live/dead staining results both reveal that MoO3−x–EuBG–SA dressings had good biocompatibility when the MoO3−x concentration was within 2 mg mL−1. The nucleus/skeleton staining shown in Fig. 2j and k also exhibited that the HDFs and HUVECs treated with 2.0MoO3−x–EuBG–SA dressings had a very good state of skeleton spreading. The above results suggested that 2.0MoO3−x–EuBG–SA may be used as a multifunctional dressing to promote diabetic wound healing and monitor the wound state. Thus, the MoO3−x–EuBG–SA dressing treated with 2 mg mL−1 MoO3−x solution was chosen to conduct the following studies.
Stability and bioactivity are crucial factors to consider when evaluating wound dressings. And as is known that bioactive ions can promote the regeneration of blood vessels and collagen, which is crucial for diabetic wound repair.58 Thus, the in vitro degradation performance of composite dressings was detected by observing the morphological changes and measuring the releases of Si, Ca, and Mo ions. According to the EDS images of Mo and Si elements, we could know that MoO3−x nanosheets and EuBG particles were uniformly dispersed in the grid structure of the MoO3−x–EuBG–SA dressing (Fig. S7 and S8a, ESI†). And the SEM and EDS images before and after immersing in Tris–HCl (pH = 7.4) for 5 days showed that the MoO3−x–EuBG–SA dressing maintained the basic surface morphology, indicating that the MoO3−x–EuBG–SA dressing had good stability (Fig. S8b, ESI†). The results of ion releasing are shown in Fig. 3b–d, and it could be found that the MoO3−x–EuBG–SA dressing exhibited fast release of Si, Ca, and Mo ions. Additionally, there was no obvious difference among SA, EuBG–SA, and MoO3−x–EuBG–SA in the swelling ratio and all the dressings were hydrophilic, indicating good exudate absorption (Fig. S9 and S10, ESI†). From the results described above, the MoO3−x–EuBG–SA dressing possessed stability, good exudate absorption and could release bioactive ions.
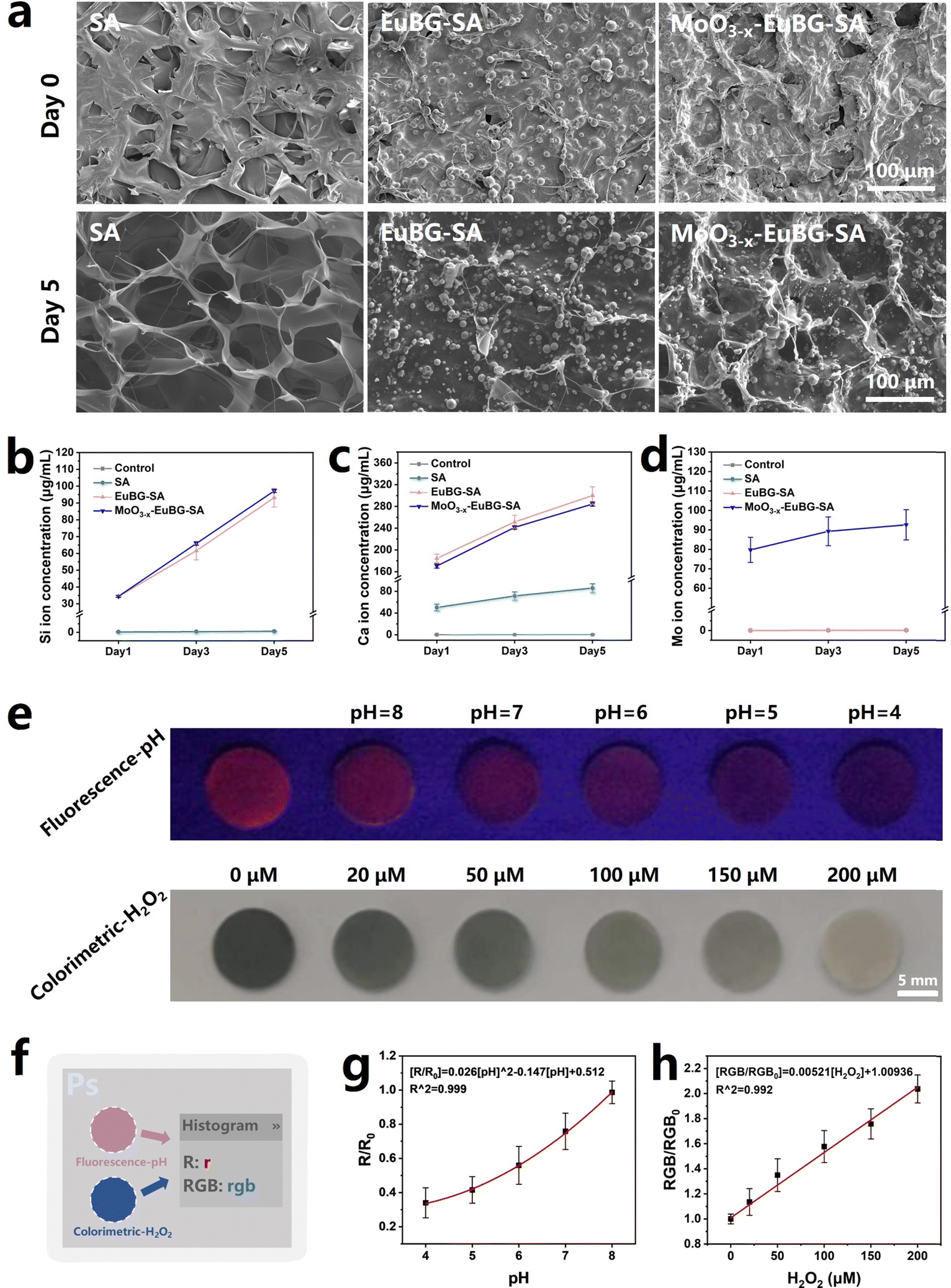 | ||
| Fig. 3 (a) SEM image of the surface of SA, EuBG–SA, and MoO3−x–EuBG–SA dressings at day 0 and after immersing in Tris–HCl for 5 days. (b) Si ion, (c) Ca ion, and (d) Mo ion releasing of SA, EuBG–SA, and MoO3−x–EuBG–SA dressings. (e) Visible color changes of the MoO3−x–EuBG–SA dressing upon ultraviolet irradiation at 254 nm with different pHs of PBS and optical photographs of the MoO3−x–EuBG–SA dressing treated with various concentrations of H2O2. (f) The process of analyzing the R value and RGB average value of the MoO3−x–EuBG–SA dressing via Photoshop software. (g) Stern–Volmer plot of pH sensing over the range of pH = 4–8. (h) Stern–Volmer plot of H2O2 sensing over the range of 0–200 μmol L−1 H2O2. All data are expressed as means ± standard deviation, one-way ANOVA with Bonferroni multiple comparison corrections. | ||
Then, the function of pH value and H2O2 concentration monitoring of MoO3−x–EuBG–SA dressings was evaluated. To investigate the correlation between the pH values and fluorescence intensity, the MoO3−x–EuBG–SA dressings were immersed in PBS solutions with varied pH levels. Fluorescence photographs were taken under 254 nm UV light irradiation and then the RGB data were analyzed using Photoshop software (Fig. 3e and f). The R data of fluorescence photographs can represent the intensity of red fluorescence. Hence, the correlation between the changes in fluorescence and pH values was quantitatively reflected by the change of R values of the fluorescence photographs (Fig. 3g). There is a good functional relationship between the pH values and fluorescence intensity, and the regression equation for pH was R0/R = 0.026[pH]2 − 0.147[pH] + 0.512. As a result, the obtained functional equation can be utilized to assess the pH values of the wound microenvironment. Similarly, to determine the correlation between H2O2 and color depth, the MoO3−x–EuBG–SA dressings were immersed in different concentrations of H2O2. Also, the optical photographs were taken and then handled with software Photoshop to readout the RGB data. The color depth of an optical photograph can be assessed by examining the average RGB value. There is a good linear relationship between the H2O2 levels and the color depth, and the linear regression equation for H2O2 was RGB0/RGB = 0.00521[H2O2 (μmol L−1)] + 1.00936. Therefore, these results suggest that the MoO3−x–EuBG–SA dressings possess the function of pH and H2O2 monitoring.
2.3. Bioactivity assay in vitro
To evaluate the effectiveness of various wound dressings, multiple types of assessments are necessary. In wound healing, intrinsic factors such as the proliferation, differentiation, and migration of cells play a crucial role.59 Additionally, it is important to modulate the immune system to prevent persistent inflammation and promote timely wound healing.60 Bioactive ions have shown the effects of accelerating vascularization and promoting tissue regeneration.61 Moreover, MoO3−x–EuBG–SA dressings could continuously release bioactive Si, Ca, and Mo ions. Thus, the effect of MoO3−x–EuBG–SA dressings on the proliferation of HDFs and HUVECs in vitro was firstly investigated (Fig. 4a and b). The HDFs and HUVECs cultured with EuBG–SA and MoO3−x–EuBG–SA groups exhibited a more significant proliferation effect than those cultured with the Control and SA groups according to the results of CCK-8 assay. The ability of MoO3−x–EuBG–SA dressings to regulate gene expression in HUVECs was also investigated. As shown in Fig. 4c, both EuBG–SA and MoO3−x–EuBG–SA groups could up-regulate the angiogenesis-related gene expression of VEGF, HIF-1α, and bFGFR in HUVECs. It is noteworthy that the gene expression of VEGF and HIF-1α was up-regulated to a higher degree in the MoO3−x–EuBG–SA group compared to the EuBG–SA group. The results suggested that not only Si and Ca ions but also Mo ions could promote angiogenesis-related gene expression. Subsequently, we explored the effect of the MoO3−x–EuBG–SA dressing on the migration ability of HDFs and HUVECs through cell scratch experiments. A plastic tip (200 μL) was used to scratch the cell layers of HDFs and HUVECs, the cells were then cultured in a non-serum cell culture medium, and the migration of the cells was recorded at 0 h and 24 h (Fig. 4d and f). From the photographs, it was found that both EuBG–SA and MoO3−x–EuBG–SA groups had observable cell migration. The cell migration area was calculated as shown in Fig. 4e and g, EuBG–SA and MoO3−x–EuBG–SA groups showed a higher migration ratio, compared with the Control and SA groups. Therefore, the MoO3−x–EuBG–SA dressing possessed excellent bioactivities and could promote the proliferation, differentiation, and migration of HDFs and HUVECs in vitro.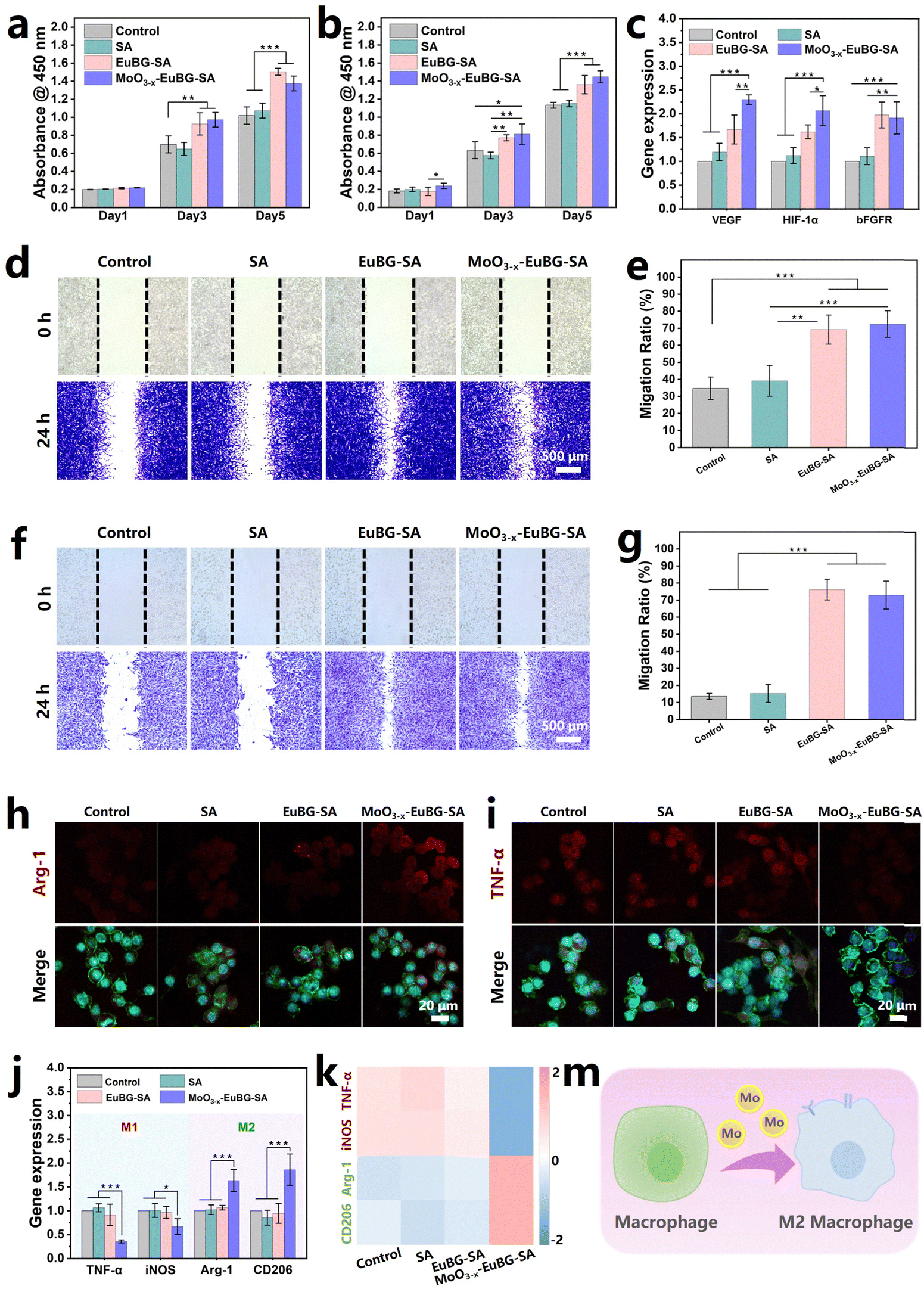 | ||
| Fig. 4 In vitro bioactivity evaluations. Cell proliferation of (a) HDFs and (b) HUVECs (n = 5). (c) Angiogenesis-related gene expression of VEGF, HIF-1α, and bFGFR in HUVECs (n = 4). Images of the migration state and the corresponding migration ratio of (d) and (e) HDFs and (f) and (g) HUVECs (n = 4). The representative (h) Arg-1 and (i) TNF-α fluorescence images of the RAW 264.7 cells. (j) Inflammation-related gene expression of TNF-α, iNOS, Arg-1, and CD206 in RAW 264.7 cells (n = 4). (k) Heatmap analysis of differentially expressed genes between samples. The blue represents downregulated mRNA, while pink indicates upregulated mRNA. (m) Schematic diagram of the Mo element regulates macrophage phenotypes. All data are expressed as means ± standard deviation, one-way ANOVA with Bonferroni multiple comparison corrections, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Molybdenum (Mo), an essential trace element found in nearly all organisms, has been demonstrated to be effective in regulating macrophage phenotypes.62 RAW 264.7 cells were used to investigate the expression of inflammatory phenotypes in immune cells mediated by the MoO3−x–EuBG–SA dressing. The cytotoxicity assay revealed that the MoO3−x–EuBG–SA dressing had good biocompatibility on RAW 264.7 cells (Fig. S11, ESI†). Then the expression of proteins related to inflammatory regulation in RAW 264.7 cells, such as tumor necrosis factor-α (TNF-α) and arginase-1 (Arg-1), was investigated. Immunofluorescence staining images showed that the MoO3−x–EuBG–SA group exhibited stronger expression of M2 macrophage marker Arg-1 and weaker expression of M1 macrophage marker TNF-α, compared with the Control, SA, and EuBG–SA groups (Fig. 4h, i and Fig. S12, ESI†). It is worth noting that the MoO3−x–EuBG–SA group exhibited a significant modulation of anti-inflammatory phenotypes in comparison to the EuBG–SA group, which is mainly attributable to the presence of Mo ions.
In particular, the MoO3−x–EuBG–SA group had the strongest Arg-1 expression and the weakest TNF-α expression, and the results of gene analysis were consistent with the immunofluorescence staining results (Fig. 4j). As shown in Fig. 4k, the heatmap exhibited that M1 genes (TNF-α and iNOS) were down-regulated and M2 genes (Arg-1 and CD206) were up-regulated in the MoO3−x–EuBG–SA group. Collectively, the results indicated that MoO3−x–EuBG–SA dressing had the ability to stimulate the production of the M2 macrophage marker and anti-inflammatory cytokines by releasing Mo ions (Fig. 4m). Therefore, the MoO3−x–EuBG–SA dressing was found to be significantly effective in stimulating macrophages towards the anti-inflammatory M2 phenotype, which was beneficial to prevent persistent inflammation and promote diabetic wound healing.
2.4. Monitoring performance and wound healing in vivo
pH is a crucial wound index as it significantly affects various physiological processes, such as the collagen formation and angiogenesis.63 Intact skin is naturally acidic, and the pH value of the wound surface increases owing to the the microvascular leakage upon injury, which facilitates bacterial infections and causes inflammation.64 Therefore, real-time pH monitoring of the diabetic wound microenvironment can clarify the status of the wound healing and provide early warning of the risk of infection.65 In addition, the level of the H2O2 concentration directly affects the oxidative stress in the diabetic wound, serving as a crucial indicator for the assessment of wound healing.66 Therefore, monitoring the pH values and H2O2 concentration in the wound microenvironment is beneficial to wound management. In this work, to evaluate the real wound-monitoring and wound-healing effects of the MoO3−x–EuBG–SA dressing, in vivo experiments were performed via a diabetic wound model. According to the process illustrated in Fig. 5a, the diabetic mice model was established by STZ injection firstly, and then the full-thickness wounds (Φ = 1 cm) were created on the back of mice. The pH and H2O2 monitoring performance of wounds was evaluated by applying the MoO3−x–EuBG–SA dressings onto the wounds, and then optical and fluorescence photographs were taken at certain intervals of time. The real wound-healing effects were investigated by applying different dressings (SA, EuBG–SA, and MoO3−x–EuBG–SA) to the wounds respectively, and the untreated group was used as the Control group.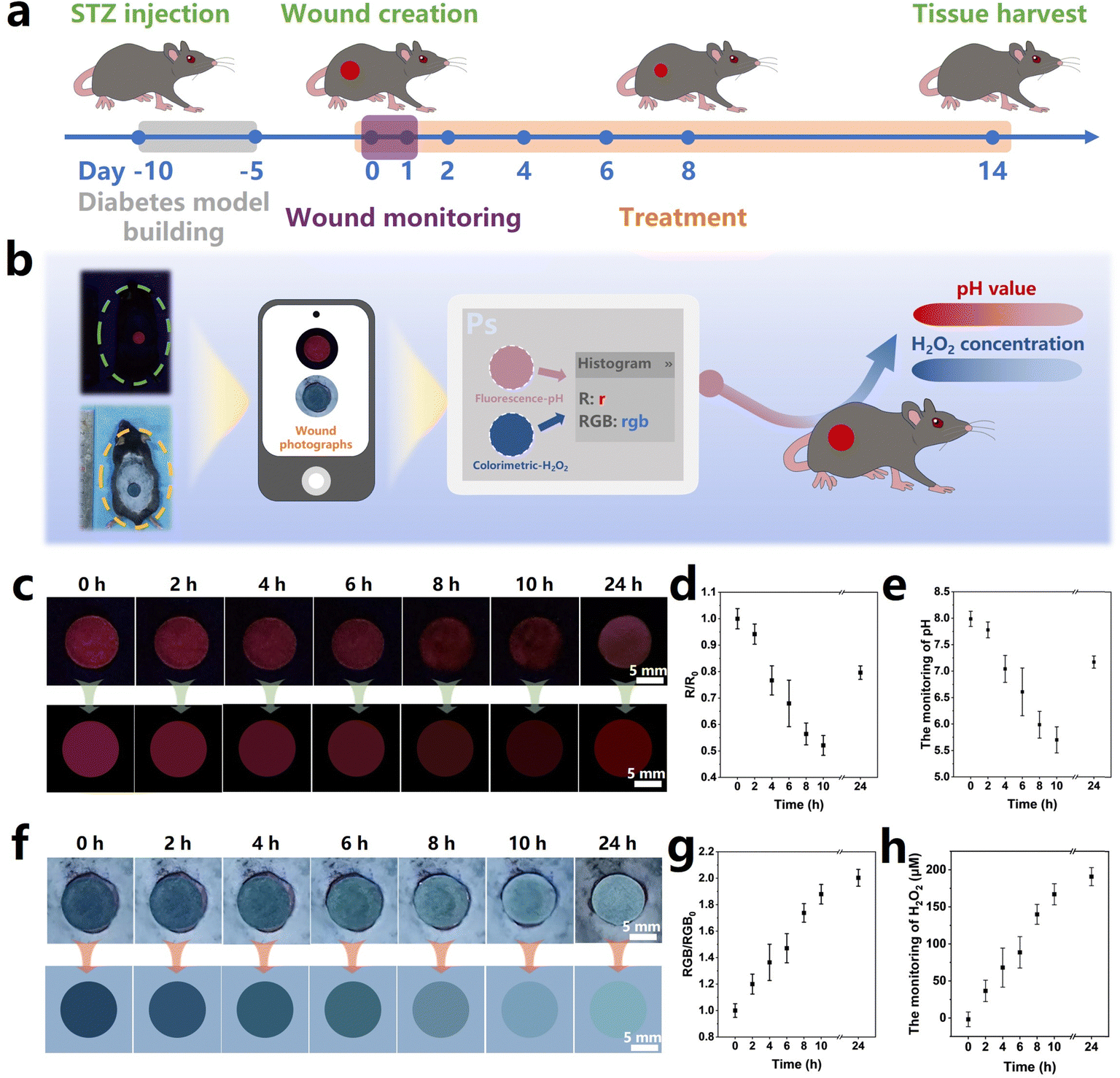 | ||
| Fig. 5 (a) In vivo monitoring and therapeutic protocol of the MoO3−x–EuBG–SA dressing on the diabetic C57BL/6 mice. (b) The schematic diagram of monitoring the pH value and H2O2 concentration in vivo. (c) Visible fluorescence changes of the MoO3−x–EuBG–SA dressing on the mice wound upon ultraviolet irradiation at 254 nm (upper) and the corresponding R values obtained from the images extracted by Photoshop software (down). (d) The corresponding R values within 24 h after the wounds treated with MoO3−x–EuBG–SA dressings (n = 4). (e) The monitoring of the pH value obtained by calculating the relationship between R0/R and pH value (n = 4). (f) Visible color changes of the MoO3−x–EuBG–SA dressing on the mice wound (upper) and the corresponding RGB values obtained from the images extracted by Photoshop software (down). (g) The corresponding RGB values within 24 h after the wounds treated with MoO3−x–EuBG–SA dressings (n = 4). (h) The monitoring of the H2O2 concentration obtained by calculating the relationship between RGB0/RGB and the H2O2 concentration (n = 4). All data are expressed as means ± standard deviation. | ||
The process of monitoring performance is shown in Fig. 5b. Briefly, a portable smartphone was used to collect visible photographs, which were then analyzed using Photoshop software to read the RGB data. After that, the pH values and H2O2 concentration at the wound sites could be calculated by substituting the RGB values into the corresponding equation. As shown in Fig. 5c, the red fluorescence of the MoO3−x–EuBG–SA dressing on the wound of diabetic mice gradually decreased within 10 h, and then the red fluorescence of the MoO3−x–EuBG–SA dressing appeared slightly brighter at 24 h compared to that at 10 h. The RGB data of the fluorescence photographs were extracted with software Photoshop, and the trend of R values was consistent with the red fluorescence (Fig. 5d). Additionally, pH values could be calculated according to the corresponding relationship between the R0/R values and pH values (R0/R = 0.026[pH]2 − 0.147[pH] + 0.512) (Fig. 5e). With increasing exudates after skin trauma, the pH monitoring results revealed that the original pH value of the wound tissue in mice was 5.7, which corresponds to an acidic inflammation stage. After 24 h, there was a significant increase in pH of the mice wound surface to 7.2. Meanwhile, the H2O2 concentration of wound in diabetic mice was also measured by the colorimetric method. The color of the MoO3−x–EuBG–SA dressing faded gradually within 24 h after wound creation (Fig. 5f). The average RGB of the MoO3−x–EuBG–SA dressing was also readout and then the H2O2 concentration was calculated according to the corresponding relationship: RGB0/RGB = 0.00521[H2O2(μmol L−1)] + 1.00936 (Fig. 5g and h). From the results, we could observe that the H2O2 concentration of wound gradually increased and reached 190.7 μmol L−1 at 24 h because of inflammation and oxidative stress. Furthermore, the pH value and H2O2 concentration in mice wound were also measured using a micro pH probe (AAT Bioquest, USA) and a H2O2 content detection kit (Solarbio, China), and the results had a good agreement with the monitoring results of the MoO3−x–EuBG–SA dressing (Fig. S13, ESI†). The above results indicated that the MoO3−x–EuBG–SA dressing could monitor pH values and H2O2 concentrations of wounds by fluorescence and colorimetry, respectively.
To evaluate the effect of the MoO3−x–EuBG–SA dressing in promoting wound healing, the wounds were observed at a determined time for 14 days, and the closure rate of the wound area, the collagen deposition, the neovascularization, and the expression of inflammation-related proteins were systematically analyzed. Representative pictures of the wounds at scheduled time intervals are shown in Fig. 6a and the closure rate of the wound area was also measured (Fig. 6b). The wounds treated with MoO3−x–EuBG–SA and EuBG–SA were almost completely healed after 14 days, and the healing rates were 99.6% and 97.5%. The results indicated that MoO3−x–EuBG–SA and EuBG–SA dressings could release bioactive ions to promote the proliferation and migration of tissue cells. Notably, the MoO3−x–EuBG–SA group not only possessed the most positive effect on diabetic wound healing, but also the wound closure rate had reached 85.4% compared with the Control group (35.4%) on day 8, indicating that the MoO3−x–EuBG–SA dressing could accelerate diabetic wound healing especially in the early stage of wound healing. It was since the MoO3−x–EuBG–SA dressing could reduce inflammation by modulating the immune system through the release of Mo. And the superimposed diagram simulated the changes in the size and morphology of wounds at six different time points (Fig. 6c). This vividly presented the satisfactory wound healing ability of the MoO3−x–EuBG–SA dressings. Additionally, there were no significant differences in blood glucose or body weight among the four groups during the treatment period, suggesting that the blood glucose levels and body weight did not influence the assessment of wound healing in each group (Fig. S14, ESI†).
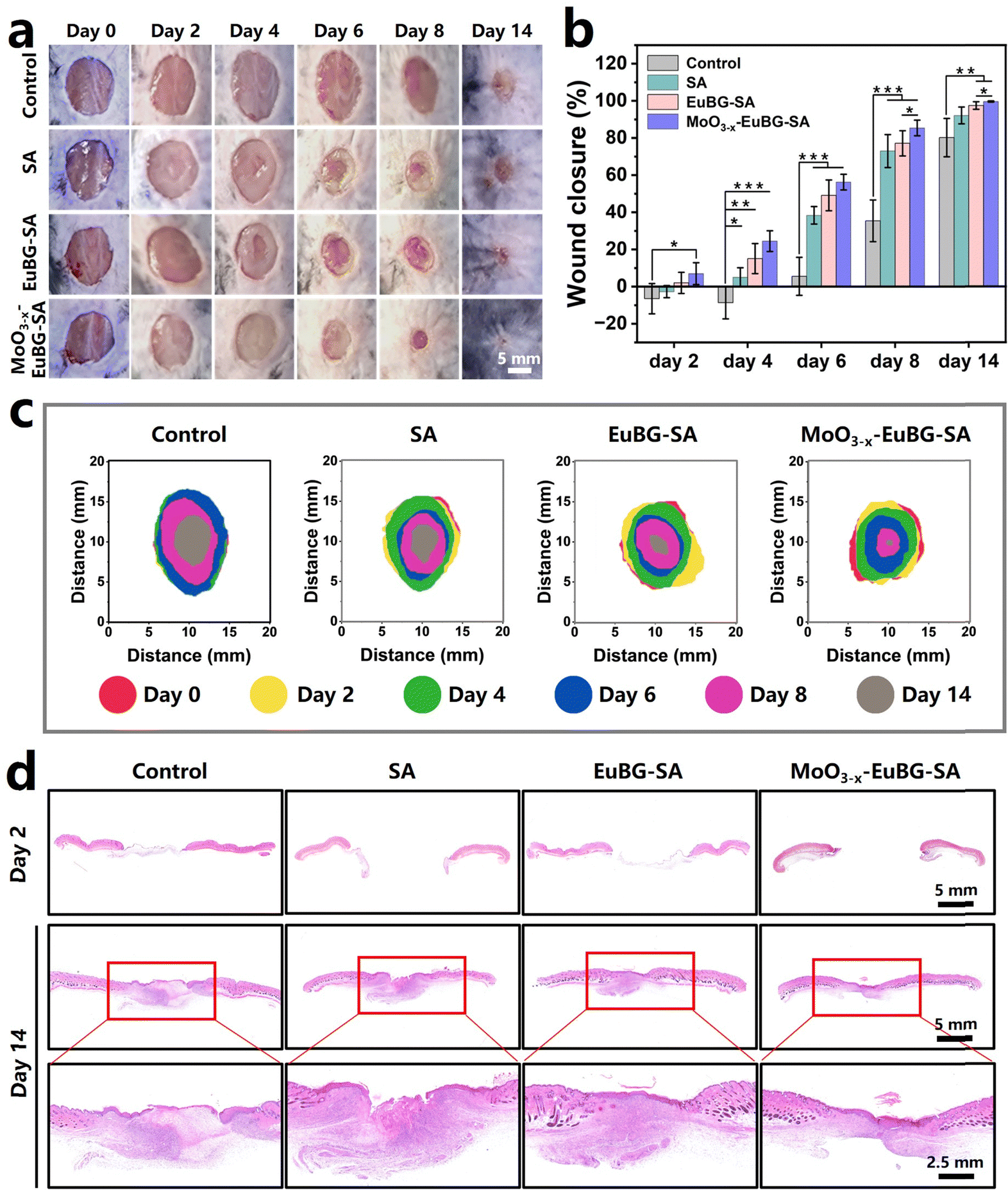 | ||
| Fig. 6 Diabetic wound healing ability in vivo. (a) Photographs of the wound healing process in diabetic mice treated with different dressings from day 0 to day 14. (b) Statistics of the wound closure rate in different groups from day 0 to day 14 (n = 5). (c) Schematic diagram of changes in the wound morphology. (d) H&E staining images of diabetic wounds on day 2 and day 14 after injury. All data are expressed as means ± standard deviation, one-way ANOVA with Bonferroni multiple comparison corrections, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
In addition, the histological analysis of wound tissue samples on day 2 and day 14 was performed. The hematoxylin and eosin (H&E) and Masson trichrome staining methods were used to observe the epidermal regeneration and collagen deposition in the wound tissue (Fig. 6d and 7a). The results showed that the MoO3−x–EuBG–SA group exhibited intact epidermal layers, visible dermal tissue, and more collagen fibers. Although the EuBG–SA group also presented almost complete coverage of the epithelium, the healing rate was slower than that of the MoO3−x–EuBG–SA group. Besides, the Control and SA groups presented incomplete epidermal layers, fewer collagen fibers, and a loose mesh arrangement. The H&E and Masson staining results indicated that the MoO3−x–EuBG–SA dressing could effectively promote wound healing. It is well known that the angiogenesis is necessary for skin wound healing. Therefore, the neovascularization of the four groups was assessed by CD31 immunofluorescence staining. As shown in Fig. 7b, the CD31 immunofluorescence staining showed that the MoO3−x–EuBG–SA group exhibited the most neovascularization at day 14, followed by EuBG–SA, SA, and Control, respectively. In addition, the statistical analysis of the number of blood vessels in the dermis of the four groups showed that the MoO3−x–EuBG–SA dressing could significantly promote angiogenesis during the wound healing process (Fig. 7d). The releasing of Mo ions could establish an anti-inflammatory microenvironment, thus the immunofluorescence staining method was used to evaluate the expression of the inflammatory factor in skin tissue.29 Judging from the immunofluorescence staining photographs and fluorescence intensity statistics of Arg-1and TNF-α on day 6 (Fig. 7e and f), the MoO3−x–EuBG–SA dressing exhibited higher expression of Arg-1 and lower expression of TNF-α. In accordance with the in vitro results, the MoO3−x–EuBG–SA dressing could induce M2 macrophage polarization and establish an anti-inflammatory microenvironment around wound. In conclusion, the above results suggested that the MoO3−x–EuBG–SA dressing had a noteworthy effect on epidermal regeneration, collagen deposition, vascular repairment, and anti-inflammation.
 | ||
| Fig. 7 (a) Masson's trichrome staining and (b) immunofluorescence staining of CD31 (red) was performed on tissue slices after 2 days and 14 days treatment of four groups. (c) Statistics of the collagen fiber content (% area, n = 5). (d) The number of newly formed blood vessels measured from the immunohistochemical images (n = 5). (e) Representative immunofluorescence images and (f) statistical results of the corresponding fluorescence intensity of Arg-1 and TNF-α in sections of skin tissue obtained from all groups on day 6 (n = 4). All data are expressed as means ± standard deviation, one-way ANOVA with Bonferroni multiple comparison corrections, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
3. Conclusion
In summary, a multifunctional SA dressing containing EuBG particles and MoO3−x nanosheets (MoO3−x–EuBG–SA dressing) was successfully prepared, with the functions of monitoring pH and H2O2, inducing immunomodulation, and promoting diabetic wound healing. Benefiting from the pH-responsive fluorescence quenching effect of EuBG particles and the H2O2-dependent color fading effect of MoO3−x nanosheets, the MoO3−x–EuBG–SA dressing could simultaneously monitor the pH values and H2O2 concentrations in wounds. Additionally, the MoO3−x–EuBG–SA dressing had a noteworthy function of promoting the proliferation, differentiation, and migration of HDFs and HUVECs in vitro by releasing bioactive ions. Furthermore, the MoO3−x–EuBG–SA dressing had the ability to release Mo, which created an adaptable anti-inflammatory immune microenvironment by inducing a switch from the M1 to M2 phenotype. Most importantly, in vivo evaluations confirmed that the MoO3−x–EuBG–SA dressing possessed the function of simultaneous visible monitoring of pH and H2O2 and accelerating diabetic wound healing through promoting vascular regeneration, collagen deposition and reducing inflammation. Hence, the MoO3−x–EuBG–SA dressing could be applied as an ideal dressing to accelerate diabetic wound healing and assist wound management through timely wound monitoring.4. Experimental section
4.1. Materials
Tetraethyl orthosilicate ((C2H5O)4Si, TEOS) and absolute ethanol were purchased from Lingfeng Chemical Reagent Co., Ltd, Shanghai, China. Triethyl phosphate (C6H15O4P, TEP), nitric acid (HNO3) and calcium chloride (CaCl2) were purchased from Sinopharm Chemical Reagent Co., Ltd, Shanghai, China. Calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), europium nitrate (Eu(NO3)3·6H2O), molybdenum trioxide (MoO3), sodium borohydride (NaBH4) and sodium alginate (SA, low viscosity, 30–90 mPa·s) were purchased from Aladdin Biochemical Technology Co., Ltd, Shanghai, China. All commercial reagents and solvents were used as received without further purification.4.2. Synthesis of materials
:1. After that, 0.15 mL of the above solutions with different contents of 5 mol% EuBG particles were dropped into the mold and kept in −20 °C refrigerator to freeze overnight and then freeze-dried for 5 hours. Finally, the dressings were cross-linked with 4% CaCl2 solution and freeze-dried again. According to the mass and total volume ratio of 5 mol% EuBG particles and SA solution (0%, 0.5%, 1.0%, 1.5%, 2.0%, 3.0%, and 4.0%), the obtained EuBG–SA dressings were denoted as yEuBG–SA (y = 0%, 0.5%, 1.0%, 1.5%, 2.0%, 3.0%, and 4.0%).
4.3. Characterization
The morphology of the EuBG particles, SA, EuBG–SA, and MoO3−x–EuBG–SA dressings was observed using a scanning electron microscope (SEM) (Hitachi SU 8220, Japan). The morphological and high-resolution images of MoO3−x nanosheets were obtained using a transmission electron microscope (TEM) (JEM-2100F, Japan). The crystal phases of EuBG and MoO3−x were characterized using an X-ray diffraction (XRD) instrument (D8 ADVANCE, Germany). The thickness of MoO3−x nanosheets was measured using an atomic force microscope (nanoIR2-FS, Germany) and the valence distribution of Mo ions was analyzed by X-ray photoelectron spectroscopy (ESCAlab250, England). UV-vis absorption spectra were recorded on a UV-vis spectrophotometer (RUNQEE, China). The fluorescence spectrum of EuBG was determined using an RF6000 fluorescence spectrophotometer (SHIMADZU CORPORATION, Japan). The absolute Si, Ca, and Mo ion concentrations were measured using an inductively coupled plasma optical emission spectrometer (ICP-OES) (Varian 715-ES, China). The contact angles and contact angle images were recorded by using a contact angle meter (JC2000C1, Powereach, China).4.4. Monitoring performance of materials
Fluorescence monitoring of pH by MoO3−x–EuBG–SA dressings and the fluorescence variation of MoO3−x–EuBG–SA dressings involved collecting visible pictures via a portable smartphone and then analyzing the RGB data using Photoshop software. The fluorescence variations of MoO3−x–EuBG–SA dressings against different pH of PBS was monitored objectively and reflected quantitatively by the change of R0/R values in the obtained fluorescence photographs. Briefly, the MoO3−x–EuBG–SA dressings were firstly prepared, and then varying pH levels (4.0, 5.0, 6.0, 7.0, and 8.0) of 50 μL of PBS were added to the dressings. The visible pictures were collected using a smartphone at predetermined time intervals under uniform shooting conditions.
The colorimetric monitoring of H2O2 by MoO3−x–EuBG–SA dressings was achieved by capturing visible images using a portable smartphone. The RGB data were then analyzed using Photoshop software. The monitored color variation of MoO3−x–EuBG–SA dressings against different concentrations of H2O2 was quantitatively reflected by the change in the RGB0/RGB values in the obtained photographs. Briefly, the MoO3−x–EuBG–SA dressings were firstly prepared, and then different concentrations (0, 40, 80, 120, 160, and 200 μmol L−1) of 50 μL of H2O2were added to the dressings. The visible pictures were collected using a smartphone at predetermined time intervals under uniform shooting conditions.
4.5. The cytotoxicity assay of EuBG–SA and MoO3−x–EuBG–SA dressings
CCK-8 assay was used to investigate the effect of yEuBG–SA and zMoO3−x–EuBG–SA dressings on the cytotoxicity of HDFs, HUVECs, and RAW264.7 cells (y = 0%, 0.5%, 1.0%, 1.5%, 2.0%, 3.0%, and 4.0%; z = 0, 1.0, 2.0, and 5.0). HDFs were cultured in DMEM medium (HyClone, high glucose) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). HUVECs were cultured in endothelial cell culture medium (ECM, Sciencell, USA) supplemented with 5% v/v FBS, 1% v/v P/S and 1% v/v endothelial cell growth factor (ECGS). RAW 264.7 cells were cultured in DMEM medium containing 10% FBS and 1% P/S. HDFs and HUVECs were seeded in 48-well plates and cultured for 24 h, then incubated with the yEuBG–SA dressings, respectively. And cells cultured without dressing were used as the Control group. After 24 h incubation, the medium with 10% CCK-8 was added and incubated for 1.5 h. Then the OD value at 450 nm was measured using a microplate reader (Epoch, BioTek, Winooski, VT, USA). The cytotoxicity assessment procedure of MoO3−x–EuBG–SA dressings on HDFs, HUVECs, and RAW264.7 cells was the same as above. The cell viability was calculated using the formula: cell viability = (ODsample − ODcck-8)/(ODBlank − ODcck-8) × 100%.4.6. Bioactivity assay in vitro
The bioactivities of different types of dressings (SA, EuBG–SA, and MoO3−x–EuBG–SA) were evaluated by cell proliferation, cell migration and cell expression, and cells cultured without dressings were used the Control group.
For the inflammation-related cytokine expression, TNF-α and Arg-1 were detected by immunofluorescent protein staining. Briefly, RAW 264.7 cells were fixed with 4% paraformaldehyde solution for 30 min, then incubated in 0.1% Triton-X100 for 5 min to permeabilize the cell membrane and soaked in 5% bovine serum albumin (BSA) solution for 1 h. Next, cells were respectively incubated with a primary antibody at 4 °C overnight. Subsequently, the samples were incubated with a fluorescent antibody at 37 °C for 1 h. Then the cytoskeleton was stained with FITC, and the nucleus was stained with DAPI. After washing with PBS, samples were then observed with a fluorescence microscope (DMi8 S, Leica, Germany).
4.7. Wound monitoring and healing assay
All in vivo experiments were conducted in accordance with the protocols approved by the Institutional Animal Care and Utilization Committee of Nanjing First Hospital, Nanjing Medical University (Nanjing, China) (permission number DWSY-22030156). And all procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals and the Regulation of Animal Protection Committee. Male C57BL/6 mice (8 weeks old, 20–25 g) were used to establish a diabetic model through intraperitoneal injection of streptozotocin (STZ) at a dose of 50 mg kg−1 for five consecutive days, as previously reported.67 Briefly, the back hair was removed and sterilized after the mouse was anesthetized. Subsequently, a rounded full-thickness cutaneous wound area (1.0 cm × 1.0 cm) was created on the back of each mouse, and then divided into four groups randomly: Control (no treatment), SA, SA–EuBG, and MoO3−x–EuBG–SA.4.8. Animal experiments
All animal experiments were conducted in accordance with the protocols approved by the Institutional Animal Care and Utilization Committee of Nanjing First Hospital, Nanjing Medical University (Nanjing, China) (permission number DWSY-22030156). And all procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals and the Regulation of Animal Protection Committee.4.9. Statistical analysis
All data were expressed as means ± standard deviation and analyzed using Origin 2024 software (Origin Lab, USA). Statistically significant differences between groups were analyzed by one-way ANOVA with Bonferroni multiple comparison corrections. The p value under 0.05 is considered statistically significant (*p < 0.05, **p < 0.01, and ***p < 0.001).Author contributions
Conceptualization: Jimin Huang and Yufang Zhu. Methodology: Jimin Huang and Yufang Zhu. Investigation: Jimin Huang and Yufang Zhu. Validation: Jimin Huang. Animal experiments: Jimin Huang, Jinzhou Huang, Xinxin Zhang, and Yi Zheng. Data collection and processing: Jimin Huang, Qinyi Xie, Chaoqin Shu, Zhe Shi, Xiao Wang, and Jiajie Chen. Visualization: Jimin Huang. Writing – original draft: Jimin Huang. Writing – review and editing: Yufang Zhu. Funding acquisition: Bing Ma and Chengtie Wu. Supervision: Yufang Zhu.Data availability
All data needed to evaluate the conclusions in the paper are presented in the manuscript and the supporting information. Additional data related to this paper may be provided on request.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
We greatly acknowledge the support by the National Natural Science Foundation of China (No. 32225028), the Science and Technology Commission of Shanghai Municipality (No. 22S31902800), and the Space Application System of China Manned Space Program (KJZ-YY-NCL05).References
- S. Gao, Y. Rao, X. Wang, Q. Zhang, Z. Zhang, Y. Wang, J. Guo and F. Yan, Adv. Mater., 2024, 15, 36–48 Search PubMed.
- J. Ma, Y. Fang, H. Yu, J. Yi, Y. Ma, P. Lei, Q. Yang, L. Jin, W. Wu, H. Li and D. Sun, Adv. Funct. Mater., 2023, 34, 2308387 CrossRef.
- H. Z. Wang and L. M. Zhang, Chem. Eng. J., 2024, 484, 149493 CrossRef CAS.
- M. Deng, M. Zhang, R. Huang, H. Li, W. Lv, X. Lin, R. Huang and Y. Wang, Biomaterials, 2022, 289, 121790 CrossRef CAS.
- S. Chen, Y. Zhu, Q. Xu, Q. Jiang, D. Chen, T. Chen, X. Xu, Z. Jin and Q. He, Nat. Commun., 2022, 13, 5684 CrossRef CAS PubMed.
- X. Qi, E. Cai, Y. Xiang, C. Zhang, X. Ge, J. Wang, Y. Lan, H. Xu, R. Hu and J. Shen, Adv. Mater., 2023, 35, 2306632 CrossRef CAS.
- D. Furman, J. Campisi, E. Verdin, P. Carrera-Bastos, S. Targ, C. Franceschi, L. Ferrucci, D. W. Gilroy, A. Fasano, G. W. Miller, A. H. Miller, A. Mantovani, C. M. Weyand, N. Barzilai, J. J. Goronzy, T. A. Rando, R. B. Effros, A. Lucia, N. Kleinstreuer and G. M. Slavich, Nat. Med., 2019, 25, 1822–1832 CrossRef CAS.
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Antioxid. Redox Signaling, 2014, 20, 1126–1167 CrossRef CAS.
- T. Xiang, Q. Guo, L. Jia, T. Yin, W. Huang, X. Zhang and S. Zhou, Adv. Healthcare Mater., 2023, 13, 202301885 Search PubMed.
- Y. Liang, J. He and B. Guo, ACS Nano, 2021, 15, 12687–12722 CrossRef CAS.
- D. Q. Gao, Y. D. Zhang, D. T. Bowers, W. J. Liu and M. L. Ma, APL Bioeng., 2021, 5, 031503 CrossRef CAS.
- M. M. Fu, Y. T. Zhao, Y. Wang, Y. Li, M. Wu, Q. Liu, Z. G. Hou, Z. H. Lu, K. K. Wu and J. S. Guo, Small, 2023, 19, 202205489 Search PubMed.
- Y. N. Zhu, J. M. Zhang, J. Y. Song, J. Yang, Z. Du, W. Q. Zhao, H. S. Guo, C. Y. Wen, Q. S. Li, X. J. Sui and L. Zhang, Adv. Funct. Mater., 2020, 30, 1905493 CrossRef CAS.
- Z. S. Hou, T. J. Wang, L. Wang, J. J. Wang, Y. Zhang, Q. Zhou, Z. H. Zhang, P. Li and W. Huang, Mater. Horiz., 2024, 11, 1997–2009 RSC.
- D. A. Jankowska, M. B. Bannwarth, C. Schulenburg, G. Faccio, K. Maniura-Weber, R. M. Rossi, L. Scherer, M. Richter and L. F. Boesel, Biosens. Bioelectron., 2017, 87, 312–319 CrossRef CAS PubMed.
- N. Tang, R. J. Zhang, Y. B. Zheng, J. Wang, M. Khatib, X. Jiang, C. Zhou, R. Omar, W. Saliba, W. W. Wu, M. M. Yuan, D. X. Cui and H. Haick, Adv. Mater., 2022, 34, 2106842 CrossRef CAS.
- T. T. Cui, J. F. Yu, C. F. Wang, S. Chen, Q. Li, K. Guo, R. K. Qing, G. F. Wang and J. A. Ren, Adv. Sci., 2022, 9, 2201254 CrossRef CAS PubMed.
- K. K. Zheng, Y. Tong, S. H. Zhang, R. Y. He, L. Xiao, Z. Iqbal, Y. H. Zhang, J. Gao, L. Zhang, L. B. Jiang and Y. L. Li, Adv. Funct. Mater., 2021, 31, 2102599 CrossRef CAS.
- X. Zhao, Y. P. Liang, Y. Huang, J. H. He, Y. Han and B. L. Guo, Adv. Funct. Mater., 2020, 30, 1910748 CrossRef CAS.
- S. X. Zhang, L. R. Wang, T. L. Xu and X. J. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 9110–9119 CrossRef CAS.
- S. Kalasin, P. Sangnuang and W. Surareungchai, Anal. Chem., 2022, 94, 6842–6852 CrossRef CAS PubMed.
- P. Salvo, V. Dini, A. Kirchhain, A. Janowska, T. Oranges, A. Chiricozzi, T. Lomonaco, F. Di Francesco and M. Romanelli, Biosensors, 2017, 17, 2952 Search PubMed.
- C. Fan, Q. Xu, R. Q. Hao, C. Wang, Y. M. Que, Y. X. Chen, C. Yang and J. Chang, Biomaterials, 2022, 287, 121652 CrossRef CAS PubMed.
- X. Y. Lu, Z. R. Liu, Q. R. Jia, Q. S. Wang, Q. Zhang, X. R. Li, J. Y. Yu and B. Ding, ACS Nano, 2023, 17, 11507–11520 CrossRef CAS PubMed.
- G. Y. Jian, D. Z. Li, Q. W. Ying, X. Chen, Q. M. Zhai, S. Wang, L. Mei, R. D. Cannon, P. Ji, W. Z. Liu, H. N. Wang and T. Chen, Adv. Healthcare Mater., 2023, 12, 202300469 Search PubMed.
- S. C. Huo, S. Liu, Q. Q. Liu, E. Xie, L. C. Miao, X. Y. Meng, Z. H. Xu, C. Zhou, X. S. Liu and G. H. Xu, Adv. Sci., 2024, 11, 202302674 Search PubMed.
- H. Chang, P. F. Tian, L. Z. Hao, C. W. Hu, B. Liu, F. Z. Meng, X. Yi, X. H. Pan, X. H. Hu, H. Wang, X. Y. Zhai, X. Cui, J. P. Y. Cheung, X. Y. Liu, H. B. Pan, S. Q. Bian and X. L. Zhao, Chem. Eng. J., 2024, 481, 148768 CrossRef CAS.
- M. Chen, D. D. Winston, M. Wang, W. Niu, W. Cheng, Y. Guo, Y. D. Wang, M. Luo, C. X. Xie, T. T. Leng, X. Y. Qu and B. Lei, Mater. Today, 2022, 53, 27–40 CrossRef CAS.
- J. M. Huang, Y. Zheng, H. C. Niu, J. Z. Huang, X. X. Zhang, J. J. Chen, B. Ma, C. T. Wu, Y. Cao and Y. F. Zhu, Adv. Healthcare Mater., 2024, 13, 202302328 Search PubMed.
- T. Jia and G. Y. Chen, Coord. Chem. Rev., 2022, 471, 214724 CrossRef CAS.
- X. Z. Luo, C. L. Zhang, Z. H. Yu, S. H. Wen and Y. Z. Xian, TrAC, Trends Anal. Chem., 2023, 169, 117368 CrossRef CAS.
- H. Jeong, B. I. Lee and S. H. Byeon, ACS Appl. Mater. Interfaces, 2016, 8, 10946–10953 CrossRef CAS.
- T. Wang, C. Liao, Z. K. Jiang, J. Wang, Y. Y. Ma, H. T. Lin, Y. M. Zhang, H. M. Lv, X. A. Zhang, Y. M. Hu, Y. D. Yang and G. J. Zhou, Talanta, 2024, 266, 125041 CrossRef CAS PubMed.
- Y. X. Cai, K. L. Chen, C. S. Liu and X. Qu, Bioact. Mater., 2023, 28, 243–254 Search PubMed.
- L. L. Deng, C. Z. Du, P. Y. Song, T. Y. Chen, S. L. Rui, D. G. Armstrong and W. Q. Deng, Oxid. Med. Cell. Longevity, 2021, 8852759 CrossRef CAS PubMed.
- Z. Wang, F. Qi, H. Luo, G. C. Xu and D. L. Wang, Front. Immunol., 2022, 13, 789274 CrossRef CAS.
- Y. Sun, Y. F. Lu, J. Saredy, X. W. Wang, C. Drummer, Y. Shao, F. Saaoud, K. M. Xu, M. Liu, W. Y. Yang, X. H. Jiang, H. Wang and X. F. Yang, Redox Biol., 2020, 37, 101696 CrossRef CAS.
- L. Y. Zhou, Y. Deng, Y. J. Ren, H. L. Poon, W. Y. Chu, H. Wang and Y. K. Chan, Chem. Eng. J., 2024, 482, 148978 CrossRef CAS.
- S. Bekeschus, T. von Woedtke, S. Emmert and A. Schmidt, Redox Biol., 2021, 46, 102116 CrossRef CAS.
- Y. X. Cui, W. Duan, Y. Jin, F. J. Wo, F. N. Xi and J. M. Wu, Acta Biomater., 2021, 131, 544–554 CrossRef CAS PubMed.
- Y. Xiong, L. Chen, P. Liu, T. Yu, C. C. Lin, C. C. Yan, Y. Q. Hu, W. Zhou, Y. Sun, A. C. Panayi, F. Q. Cao, H. Xue, L. C. Hu, Z. Lin, X. D. Xie, X. F. Xiao, Q. Feng, B. B. Mi and G. H. Liu, Small, 2022, 18, 2104229 CrossRef CAS PubMed.
- R. R. Manoharan, A. Prasad, P. Pospisil and J. Kzhyshkowska, Front. Immunol., 2024, 15, 1359600 CrossRef CAS.
- P. Tang, J. M. Gu, Z. A. Xie, Y. Gu, Z. W. Jie, K. M. Huang, J. Y. Wang, S. W. Fan, X. S. Jiang and Z. J. Hu, Free Radical Biol. Med., 2018, 120, 368–379 CrossRef CAS PubMed.
- R. B. Zhou, A. S. Yazdi, P. Menu and J. Tschopp, Nature, 2011, 469, 221–225 CrossRef CAS.
- J. L. Zhou, R. X. Zhao, Y. Q. Du, S. K. Liu, W. T. Li, S. L. Gai, F. He, L. L. Feng and P. P. Yang, Adv. Funct. Mater., 2022, 32, 2112083 CrossRef CAS.
- Y. Yan, M. Ni, F. Wang, Y. Yu, X. Gong, Y. Huang, W. Tao, C. Li and F. Wang, ACS Nano, 2022, 16, 15175–15187 CrossRef CAS PubMed.
- X. D. Wang, J. Fu, C. Jiang, X. H. Liao, Y. J. Chen, T. Jia, G. Y. Chen and X. Feng, Adv. Mater., 2023, 35, 2210948 CrossRef CAS.
- G. X. Duan, L. Wen, X. W. Sun, Z. X. Wei, R. X. Duan, J. F. Zeng, J. B. Cui, C. Y. Liu, Z. P. Yu, X. F. Xie and M. Y. Gao, Small, 2022, 18, 2107137 CrossRef CAS.
- J. F. Wu, J. G. Ma, H. Zhuang, H. S. Ma and C. T. Wu, Nano Today, 2023, 51, 101917 CrossRef CAS.
- C. L. Li, J. Y. Chen, G. Y. Deng, P. L. Wang, H. Zhang, K. Zhu, Q. T. Hu, S. Q. Feng, Q. G. Wang and H. D. Lin, Composites, Part B, 2023, 266, 110984 CrossRef CAS.
- Y. E. Zong, B. G. Zong, R. Y. Zha, Y. Zhang, X. H. Li, Y. Y. Wang, H. F. Fang, W. L. Wong and C. Y. Li, Adv. Healthcare Mater., 2023, 12, 202300431 Search PubMed.
- P. Y. Wang, L. L. Peng, J. Y. Lin, Y. Li, Q. Luo, S. H. Jiang, H. N. Tian, Y. Zhang, X. L. Liu and J. F. Liu, Chem. Eng. J., 2021, 415, 128901 CrossRef CAS.
- S. J. Wu, X. F. Fu, D. H. Zhang, Y. F. Sun, X. Lu, F. L. Lin, L. Y. Meng, X. L. Chen and C. Z. Lu, Adv. Mater., 2024, 36, 2401724 CrossRef CAS PubMed.
- Y. Zheng, W. P. Ma, Z. B. Yang, H. J. Zhang, J. G. Ma, T. Li, H. C. Niu, Y. L. Zhou, Q. Q. Yao, J. Chang, Y. F. Zhu and C. T. Wu, Chem. Eng. J., 2022, 430, 132912 CrossRef CAS.
- W. S. Gong, L. L. Liu, L. P. Luo and L. J. Ji, J. Biomed. Mater. Res., Part B, 2023, 111, 173–183 CrossRef CAS PubMed.
- A. K. Sharma, S. Pandey, K. H. Sharma, Y. Nerthigan, M. S. Khan, D. R. Hang and H. F. Wu, Anal. Chim. Acta, 2018, 1015, 58–65 CrossRef CAS PubMed.
- H. L. Yu, F. Q. Dong and B. W. Li, Sens. Actuators, B, 2020, 320, 128368 CrossRef CAS.
- Y. Li, Y. Miao, L. N. Yang, Y. T. Zhao, K. K. Wu, Z. H. Lu, Z. Q. Hu and J. S. Guo, Adv. Sci., 2022, 9, 2202684 CrossRef CAS PubMed.
- W. Ma, H. Ma, P. Qiu, H. Zhang, Z. Yang, B. Ma, J. Chang, X. Shi and C. Wu, Biomaterials, 2021, 279, 121225 CrossRef CAS PubMed.
- Z. W. B. Zhang, W. B. Li, D. Chang, Z. Q. Wei, E. D. Wang, J. Yu, Y. Z. Xu, Y. M. Que, Y. X. Chen, C. Fan, B. Ma, Y. L. Zhou, Z. G. Huan, C. Yang, F. Guo and J. Chang, Bioact. Mater., 2023, 24, 81–95 CAS.
- L. L. Ma, Y. L. Zhou, Z. W. B. Zhang, Y. Q. Liu, D. Zhai, H. Zhuang, Q. Li, J. D. Yuye, C. T. Wu and J. Chang, Sci. Adv., 2020, 6, eabb1311 CrossRef CAS PubMed.
- X. T. He, X. Li, M. Zhang, B. M. Tian, L. J. Sun, C. S. Bi, D. K. Deng, H. Zhou, H. L. Qu, C. T. Wu and F. M. Chen, Biomaterials, 2022, 283, 121439 CrossRef CAS PubMed.
- Y. G. Chen, C. X. Li, Y. Zhang, Y. D. Qi, X. H. Liu, J. Feng and X. Z. Zhang, Mater. Horiz., 2022, 9, 2824–2834 RSC.
- Y. Zhu, J. Zhang, J. Song, J. Yang, Z. Du, W. Zhao, H. Guo, C. Wen, Q. Li, X. Sui and L. Zhang, Adv. Funct. Mater., 2019, 30, 1905493 CrossRef.
- H. X. Xie, Z. Wang, R. Z. Wang, Q. Q. Chen, A. X. Yu and A. Lu, Adv. Funct. Mater., 2024, 2401209 CrossRef CAS.
- A. H. Wang, G. S. Fan, H. L. Qi, H. Y. Li, C. C. Pang, Z. K. Zhu, S. C. Ji, H. Liang, B. P. Jiang and X. C. Shen, Biomaterials, 2022, 289, 121798 CrossRef CAS PubMed.
- L. Sheng, Z. Zhang, Y. Zhang, E. Wang, B. Ma, Q. Xu, L. Ma, M. Zhang, G. Pei and J. Chang, Biomaterials, 2021, 264, 120414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01142b |
| This journal is © The Royal Society of Chemistry 2025 |