
Flexible, self-healing and portable supramolecular visualization smart sensors for monitoring and quantifying structural damage†
Dezhi
Jiao
 a,
Sihan
Gu
a,
Li
Cheng
b,
Shuoqi
Li
a and
Chengbao
Liu
*a
a,
Sihan
Gu
a,
Li
Cheng
b,
Shuoqi
Li
a and
Chengbao
Liu
*a
aCollege of Materials Science and Engineering, Shandong University of Science and Technology, Qingdao 266590, China. E-mail: chengbaocl@163.com
bLaboratory of Advanced Rubber Material, Ministry of Education (Type B), Qingdao University of Science and Technology, Qingdao 266042, China
First published on 11th October 2024
Abstract
Visually monitoring micro-crack initiation and corrosion failure evolution is crucial for early diagnosis of structural health and ensuring safe operation of infrastructures. However, existing damage detecting approaches are subject to the limited-detection of heterogeneous structures, intolerance of harsh environments, and challenge of quantitative analysis, impeding applications in structural health monitoring (SHM). Herein, we present a stretchable, semi-quantitative, instrument-free, supramolecular SHM sensor by integrating a polyurea elastomer with sensitive corrosion-probes, enabling localized corrosion monitoring and quantification of failure dynamics. Initially, a correlation between visual monitoring signals and structural health status is proposed, and sensor-based image processing software that accurately quantifies structural failure indicators (crack scale, corrosion reactivity and deterioration status) is proposed. Moreover, this sensor can be fabricated as multiple derivatives: a coating or patch covered on metallic substrates and an ionic-responsive test strip, ensuring real-time detection of the initiation of pitting, degradation events of metallic components and convenient monitoring of ion concentrations in corrosive media. Furthermore, the inherent geometric plasticity and dynamic hydrogen-bonded network validates the reliability for heterogeneous components and stability under extreme environments of sensors. This portable, smart SHM strategy established the channel-transformation model from corrosion dynamics to visual signals, exhibiting prospects for structural monitoring in offshore energy-harvesting equipment.
New conceptsImplanting self-monitoring capability into protective materials is of great significance to detect the occurrence of stress, damage and interfacial corrosion before critical events and catastrophic failure. Structural health monitoring (SHM) materials can provide pivotal damage related signals and are expected to assess the service conditions and ensure the operational safety of equipment. However, most SHM materials demonstrate an inability to monitor heterogeneous structures, are unstable in harsh environments, and their visual and quantitative evaluations are challenging, impeding their practical applications. To overcome this conundrum, we propose an in situ and real time self-monitoring strategy based on a rationally designed ionochromism sensor to achieve flexible, intrinsic self-healing and on-site damage visualization properties. The core concept is to incorporate a special ion sensitive corrosion probe into substantial urea groups engineered supramolecularly. The SHM mechanism is based on a corrosion-induced chromophoric reaction initiated by surface damage exposed to a marine environment. Benefiting from the unique structure of designed polyurea, the resultant SHM sensors demonstrate superior adaptability to heterogeneous components and excellent stability in harsh environments. In brief, this research offers a novel strategy to visually and quantitatively monitor the failure and structural deterioration states of structural materials. |
Introduction
Advanced materials are the basis and guarantee for achieving high performing, reliable and versatile industrial equipment. Metallic engineering equipment, including a marine drilling platform, oil/gas transportation pipelines and offshore wind turbines, are important prerequisites for the efficient exploitation and large-scale utilization of marine resources. However, corrosion and wear are inevitable for metallic materials especially in harsh marine environments,1–4 which are liable to initiate small-scale damage during long-term service, such as micropores or cracks. With the accumulation and development of this microdamage, the generation of macroscopic defects ultimately leads to performance deterioration, severe interfacial corrosion and ultimately catastrophic failure of marine infrastructures.5–7 Therefore, the demand for timely and accurate monitoring of structural microdamage and quantifying the degree of failure is therefore increasing, with the necessary goal for ensuring the safe and stable operation of marine equipment.Structural health monitoring (SHM) refers to the strategy and process that utilize integrated sensors to inspect and estimate the variations in the physical, chemical and electrical features of components, allowing the detection of damages and assessing the health condition of engineering structures.8–11 Currently, several external sensors have been developed based on a magnetic field,12 acoustic waves,13 electrical flux14 and infrared rays.15 These damage monitoring methodologies are limited by their intricate structures and prohibitive cost, and in particular their difficulty in heterogeneous marine components’ examination and inability to capture damage information over a large area. Polymer-based SHM materials have been employed due to their flexibility, convenient implementation and the fact they are independent of the geometrical shape and size of components.16,17 Piezoresistive composites are the most common kind of SHM material, which are constructed by incorporating conductive ingredients into a polymer matrix. Based on the response in conductivity, the occurrence of damage and geometric change of components can be detected.18 While piezoresistive composites are capable of reflecting the average damage events of the entire material, the corrosion pits or failure sites are still difficult to precisely locate. However, for marine metallic structures, a corrosion reaction often occurs at a localized position, which is one of the ubiquitous and severe failure modes. Consequently, sensing the corrosion initiation, determining the size and location of the damage, and predicting the corrosion evolution trend should be persued.
Self-warning or self-monitoring polymers, as a smart material, are sensitive to changes of compositional fluctuation in environment and physical/chemical characteristics in a material itself.19–22 Usually, the progression of metal corrosion is accompanied by metal ion enrichment and acid/alkaline fluctuation at localized regions.23–25 Based on the specific recognition between functional molecular probes and localized corrosive media, corrosion self-warning coatings have been designed, which exhibit conspicuous colorimetric/fluorescent visual signals to indicate the corroded behavior. By incorporating functional molecular probes,26,27 including pH indicators,28,29 ionic responders30 and AIEgens,31 into polymeric coatings, the interfacial corrosion sites and oxidation–reduction processes can be visually detected and a warning given. For marine installations, steel structural materials are the main components, which are confronted with severe local corrosion failure. Phenanthroline, a double-toothed heterocyclic compound ligand, can easily form red complexes with trace ferrous ions and strong chelating ability. To monitor the steel local corrosion initiation and study the propagation behavior, several phenanthroline-based self-warning anticorrosion coatings were constructed by chemical grafting or nanocarrier embedding phenanthroline molecules into a coating matrix in our previous works. Also, the responsive mechanisms and correlation to localized corrosion have also been demonstrated. However, the reported self-warning coatings mainly focus on qualitative corrosion judgment, which is unable to provide quantitative information and internal connections between visual signal and the failure degree. Additionally, the fabricated corrosion self-warning coatings have to be applied to the metal equipment in advance, which is unsuitable for corrosion detection in existing devices already in use. Furthermore, the corrosion of marine engineering equipment often occurs in harsh environments (high salinity, high humidity and temperature difference), posing a challenge to the sensitivity stability and environmental adaptability of corrosion sensors.
In this work, an innovative supramolecular visualization smart sensor was developed for monitoring and evaluating the localized corrosion reactions occurring in marine steel structures (Fig. 1). The smart sensor (defined as AP-SHM) was constructed through introducing functional nanocarriers, dendritic mesoporous silica loaded with 1,10-phenanthrolin-5-amine (NH2-Phen), into the well-designed polyurea (PU) resin. Combined with image acquisition devices (mobile phone lenses, cameras, etc.), the developed sensor presents a significant advancement in the field of corrosion detection, particularly in its accurate quantitative identification of corrosion failure extension. Contrary to prior works, the sensing system not only acts as a coating but also serves as a flexible detecting patch on metallic substrates, which significantly allows for the maximum utilization of the sensors and effectively recognizes corrosive byproducts under atmosphere or aqueous conditions. The multiple dynamic hydrogen bonds between PU molecules enabled the supramolecular sensor with a self-healing capability. Importantly, the optical feedback of the sensors can be readily recognized with the naked eye, and the detailed failure information, such as corrosion rate, distribution and deterioration degree, can be quantified after input into self-developed software. The pioneering damage monitoring strategy and the great advancements made with the supramolecular smart sensor highlight the design and development of next-generation SHM materials.
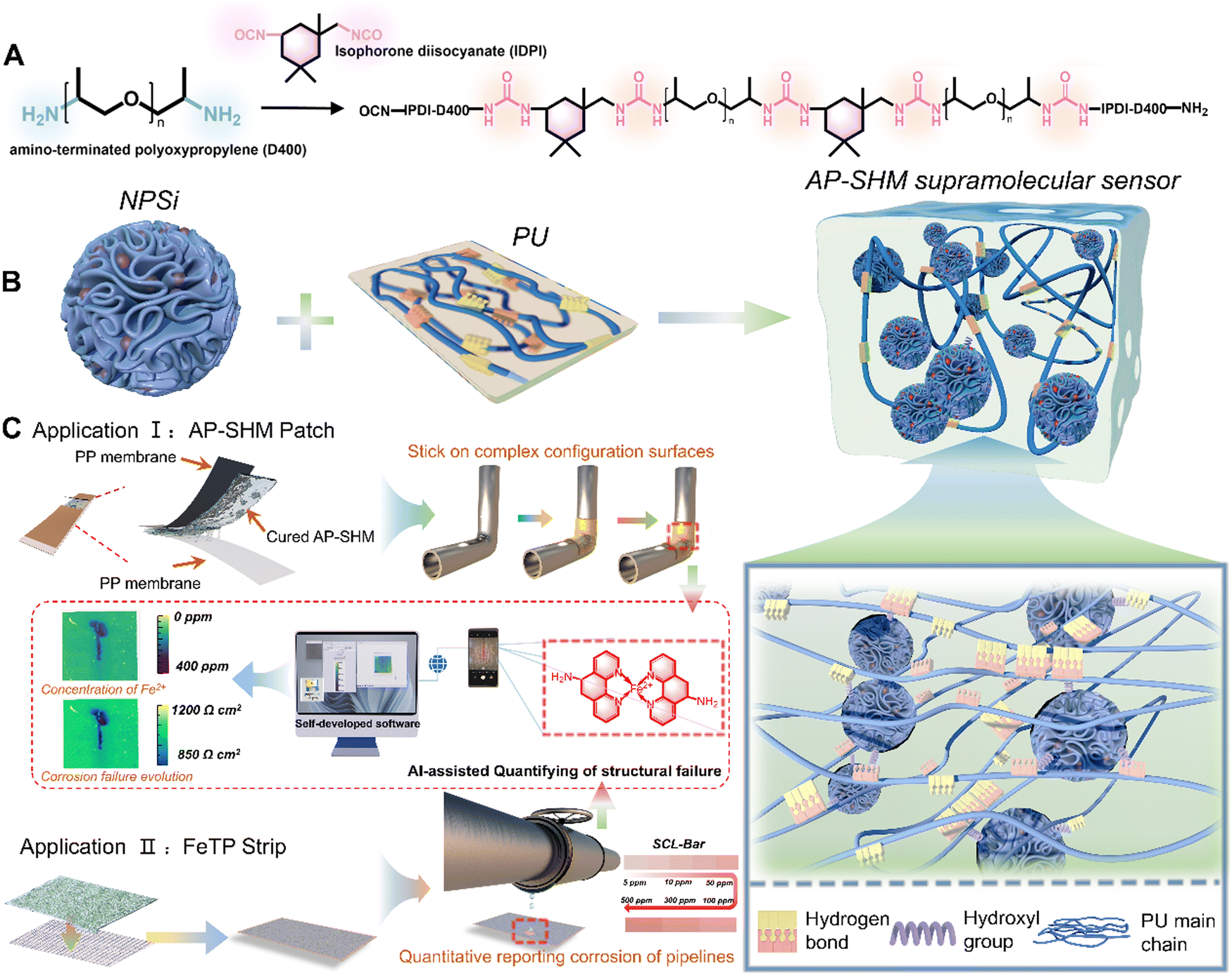 | ||
| Fig. 1 (a) Synthetic route of the PU; (b) schematic illustration of the fabrication of AP-SHM sensors and a cross-linking network of NPSi and PU resin molecules; (c) the application of an AP-SHM flexible patch and FeTP strip, and the semi-quantitative transformation process of structural failure information. | ||
Results and discussion
Synthesis and applications of AP-SHM sensors
For the early diagnosis of structural health and visual monitoring of corrosion failure in metallic engineering equipment in marine atmospheres, a stretchable, supramolecular SHM sensor is developed, assembled by the corrosion probes NPSi and PU elastomer. Within, the embedded NPSi with its abundant hydroxyl groups of the dendritic mesoporous silica surface, achieve a non-covalent interaction with the PU molecular chains. Meanwhile, the incorporation of NPSi greatly enhanced the mechanical performance of the supramolecular elastomers. The multiple dynamic hydrogen bonds within the elastomer itself confer the sensor with excellent self-healing properties, enabling it to withstand harsh environments with high-salinity and high temperature fluctuations. Moreover, the smart supramolecular AP-SHM sensor can be fabricated as a coating or patch to cover metallic substrates for the full-field monitoring of equipment throughout service life or real-time diagnosis of localized structural health, respectively (Fig. 1(c)). Notably, with the introduction of NPSi, the supramolecular sensor is endowed with an ionic-recognition property. By capturing the color evolution of Fe2+–NH2-Phen chelate during the corrosion process, a visual transformation model from corrosion dynamics to chromaticity signal is established. Meanwhile, the inherent stretchability of the supramolecular sensor allows it to adhere to the complex configuration surfaces.Nevertheless, for metallic equipment in corrosive media, quantitative and indirect structural inspection with an ionic responsiveness test strip addresses the limitations of the AP-SHM sensor patch. Initially, by attaching the supramolecular AP-SHM sensor on the test paper, an ionic responsiveness test strip (FeTP Strip) is successfully prepared. By determining the content of free Fe element in a corrosive medium system, then comparing it with a standard colorimetric bar (SCL-Bar), portable and indirect monitoring of metallic structural health in a marine environment can be achieved. Furthermore, image acquisition platforms (mobile phone lens or camera) can be utilized to record the sensor responsive results (the image of post-responded AP-SHM coating, AP-SHM patch and FeTP Strip), which can be input into self-design software. Then the structural failure information, including structural-damage scale, deterioration-status mapping and local corrosion dynamics, can be semi-quantitatively and intuitively assessed in real-time.
Synthesis mechanism and characterization of NPSi
To ensure the real-time and sensitive corrosion monitoring of the AP-SHM sensor, the content and distribution status of the corrosion probes NPSi in the supramolecular sensor were investigated. Firstly, Material Studio software was employed with molecular dynamics simulations to analyze the adsorption behavior between NH2-Phen chromophore molecules and SiO2 (100).32,33 As shown in Fig. 2(a), the chromophore molecules are tightly absorbed on the surface of SiO2 (100) in a parallel orientation, exhibiting the attachment of NH2-Phen to dendritic mesoporous silica (DMS). Furthermore, to analyze the forms of intermolecular interactions in such spontaneous adsorption, the radial distribution function (RDF) was used to describe the appearance probability of other molecules around the Si atoms. In general, a peak of small bond length is indicated at 1–3.5 Å, which is related to the chemisorption (covalent bond and Coulomb force). Whereas the peaks outside 3.5 Å are attributed to the hydrogen bond and van der Waals forces (physical interactions).34 As shown in Fig. S1 (ESI†), the function g(r) is examined for the surface Si atoms with the main heteroatoms of chromophore molecules (N1 and N2). The bond lengths of Si–N1 (2.86 Å) and Si–N2 (3.12 Å) exhibited the highest peaks. The coulombic interactions are the main non-bonding interactions between the molecules and Si atoms.35 The bonding mechanisms of NH2-Phen to DMS were confirmed. The surface of DMS has many active centers with great ability to donate and accept electron, and the chromophore is adsorbed into the dendritic mesopores by electrostatic forces. Subsequently, transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were employed to observe the surface morphology of the DMS and NPSi. As can be seen, DMS (Fig. 2(b) and Fig. S3a, S4a, ESI†) presents a dendritic mesopore spherical structure, while a smooth surface was observed for NPSi, as shown in Fig. 2(c) and Fig. S3b, S4b (ESI†). From the corresponding energy-dispersive X-ray spectroscopy (EDS) images, the distribution of the C and N elements on the internal of the dendritic mesopore and the surface of DMS was revealed. The mapping results of C and N elements of NPSi proved the successful attachment of the NH2-phen on MSN.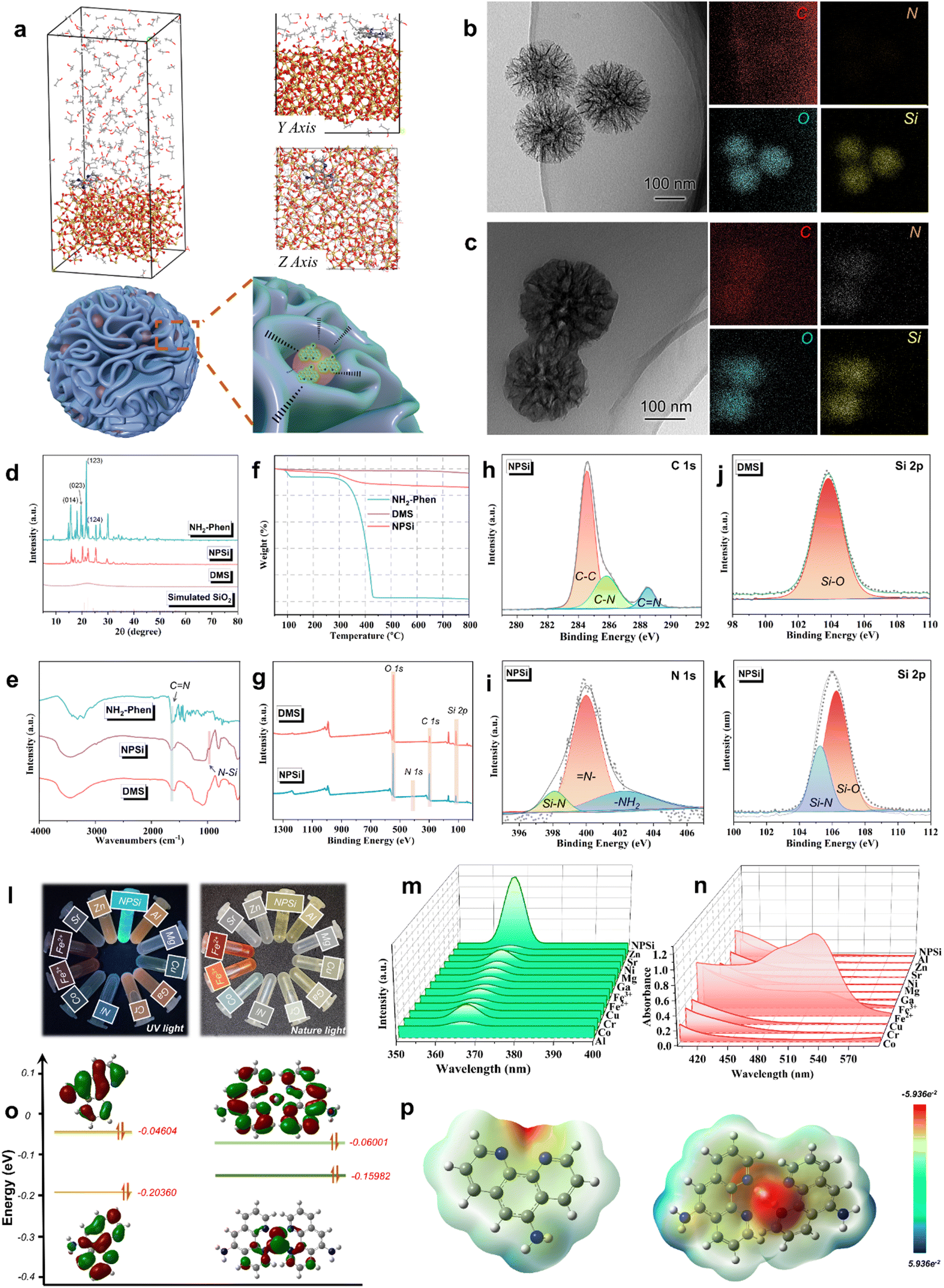 | ||
| Fig. 2 (a) Equilibrium adsorption configuration of NH2-Phen on SiO2 (110) substrate (inset: on-top view and main view); schematic illustration of the interaction mechanism between NPSi intelligent corrosion probes and DMS; TEM images and corresponding element distribution mappings of (b) DMS and (c) NPSi; (d) XRD patterns, (e) FTIR spectra and (f) TGA curves of NH-Phen, DMS and NPSi; (g) XPS full spectra of DMS and NPSi; fine spectra (NPSi) of (h) C 1s and (i) N 1s; fine spectra (Si 2p) of (j) DMS and (k) NPSi. (l) optical photograph of the addition of the NH2-Phen solution (1 × 10−3 mol·L−1) with the equal concentration cation (Al3+, Mg2+, Cu2+, Cr3+, Ni2+, Co2+, Fe3+, Fe2+, Sr2+, Zn2+) in UV mode and vis mode; the corresponding (m) fluorescence excitation spectrum and (n) UV-visible spectrum. (o) The HOMO and LUMO electron distribution and (p) electrostatic potential maps of NH2-Phen and Fe2+–NH2-Phen chelates. | ||
The crystalline properties of the NPSi were analyzed by wide-angle X-ray scattering (Fig. 2(d)). The pattern of DMS exhibits an obvious non-crystal diffraction peak of SiO2 in the 2θ range of 20–30°, which is consistent with the standard card data (Si24.00O48.00) (COD database code: 96-900-5270), indicating the successful preparation of DMS.36 Accordingly, the typical diffraction peaks of NH2-Phen were detected in the XRD curve of NPSi at 15–30°, indicating that NH2-Phen chromophore was introduced into the DMS.37,38 Additionally, to identify the changes of chemical bonds before and after encapsulation of NH2-Phen by DMS, Fourier-transform infrared (FT-IR) spectroscopy was used. As shown in Fig. 2(e), NPSi exhibits an absorption peak at 1637 cm−1, which is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of the phenyl ring in NH2-Phen. Importantly, the spectrum of NPSi displays a typical peak at 873 cm−1, which belongs to the symmetric stretching vibration of the Si–N bond.39,40 In conclusion, due to the formation of Si–N, the NH2-Phen molecule successfully attached onto DMS.
N stretching vibration of the phenyl ring in NH2-Phen. Importantly, the spectrum of NPSi displays a typical peak at 873 cm−1, which belongs to the symmetric stretching vibration of the Si–N bond.39,40 In conclusion, due to the formation of Si–N, the NH2-Phen molecule successfully attached onto DMS.
To estimate the loading capacity of DMS for NH2-Phen, thermogravimetric analysis (TGA) was conducted in the temperature range of 50–800 °C under a nitrogen atmosphere (20 mL min−1). As shown in Fig. 2(f), NH2-Phen was rapidly decomposed in the temperature range of 300–450 °C. Accordingly, an apparent weight loss of NPSi around 300–450 °C was corresponded to the thermal decomposition of NH2-Phen, revealing the successful loading of the NH2-Phen on DMS. Based on the above TGA results, the loading amount of NH2-Phen in DMS was approximately calculated to be 9.2 wt%. X-ray photoelectron spectroscopy (XPS) was used to further characterize the elemental composition of DMS and NPSi. In the full spectrum of NPSi and DMS (Fig. 2(g)), an apparent difference in O and Si content was observed, resulting from the attachment of NH2-Phen. As presented in Fig. 2(h), for NPSi, three peaks with binding energies of about 283.8, 286.5 eV and 288.6 eV were observed in the C 1s spectrum of NPSi, corresponding to the C–C, C–C and CN bonds.41 The N 1s spectrum can be fitted into three peaks (Fig. 2(i)) assigned to the Si–N (398.1 eV), –NH2 (402.6 eV), and N– (399.4 eV) orbits.42 Meanwhile, compared with the spectrum of Si 2p (Fig. 2(j) and (k)), a notable Si–N (105.1 eV) peak was observed for NPSi.43 In brief, these findings were further confirmed to the chemisorption of the NH2-Phen on the DMS.
The color variation in the NPSi ionic complex
To explore the selectivity of NPSi for different cations (Al3+, Mg2+, Cu2+, Cr3+, Ni2+, Co2+, Fe3+, Fe2+, Sr2+, Zn2+), a fluorescence spectrophotometer and UV-vis spectroscopy were employed. As shown in Fig. 2(m) and (n), the solution of NH2-Phen shows a blue-green color in UV mode and its fluorescence excitation characteristic peak is located at 370 nm. Then, as presented in the optical photographs of UV mode (Fig. 2(l)), the addition of ions resulted in an immediate fluorescence quenching phenomenon. Compared with other cations (Al3+, Mg2+, Cu2+, Cr3+, Ni2+, Co2+, Fe3+, Sr2+, Zn2+), Fe2+ exhibited the most pronounced chromaticity change in the Nature mode and significant fluorescence quenching in the UV mode. Meanwhile, the strongest absorption peak manifested in the UV absorption spectra was due to the formation of the Fe2+–NH2-Phen red complex. Thus, the specific ionic responsiveness of NPSi was confirmed, and it can serve as an ideal steel corrosion probe.Furthermore, to verify the complex mechanism of NH2-Phen with the target-ion, the NH2-Phen molecule and Fe2+–NH2-Phen clathrate were optimized by Gaussian 05. The electron cloud profiles of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), and electrostatic potential maps (ESP) are also presented in Fig. 2(o) and (p). In general, the HOMO is the orbit of electron donor due to its outermost orbitfilled with electrons, LUMO is the orbit as an electron acceptor that still has available space for electrons acceptance in its innermost orbit.44 Therefore, the EHOMO, ELUMO and the energy gap between the HOMO and LUMO (ΔELUMO–HOMO) are the important parameters for predicting chemical reactivity and molecular stability. As calculated, the ΔELUMO–HOMO of NH2-Phen (0.16) was higher than the complex (0.09), indicating that the chelate was more stable than the NH2-Phen ligand. Meanwhile, the HOMO orbital of the chelate was mainly focused on the two pyridine rings and CN–NC fragment of the ligand molecule, while the LUMO orbital was localized over the Fe2+ cation, which demonstrated the presence of π electron charge transfer. Furthermore, the NH2-Phen ligand molecule and Fe2+–NH2-Phen chelate and their protonated ESP maps are presented in Fig. 2(p). In the ESP maps, the regions of red color and blue color correspond to the high and low electron density representing a nucleophilic nature and electrophilic nature, respectively.45 All of the red regions were distributed in the electronegative groups, having heteroatoms (N atoms), which provided active centers for electrophiles (Fe atom).33 The chemical stability of the chelate and the reactive sites of the NH2-Phen ligand for Fe atom attack were further determined in the ESP maps.
Characterization of PU elastomer and AP-SHM supramolecular sensors
To confer the intermolecular interactions, infrared spectroscopy was carried out. In the full FT-IR (Fig. S6, ESI†), the significant peaks at 3335 cm−1 and 1535 cm−1 belong to the N–H stretching and scissoring vibrations of the urea groups.46 Meanwhile, the chemical structure characteristics of the polymer are presented in the 1H NMR spectra (Fig. S7, ESI†). The characteristic peak in the range of 5.51–5.96 ppm corresponds to the urea group of the elastomer.47 In brief, the FT-IR and 1H NMR results firmly demonstrated the successful preparation of the PU elastomers.To analyze the thermal property of PU, differential scanning calorimetry (DSC) was employed. As shown in Fig. S8 (ESI†), the glass transition temperature (Tg) is 24.3 °C, indicating the sufficient mobility of molecular chains at room temperature, which promotes chain migration and rearrangement and contributes to the self-healing process of the elastomers. Additionally, as shown in Fig. S9 (ESI†), the AP-SHM sensor exhibited a yellow-green color compared to the optical photograph of PU, which is attributed to the introduction of the corrosion probe loaded chromophores. This chromatic supramolecular elastomers with high transparency exhibited promising application potential in the SHM field.
As a damage sensing film for heterogeneous components, the mechanical tensile properties were investigated using a universal stretching machine. As shown in Fig. 3(a), the PU and AP-SHM exhibit different fracture behavior when subjected to tensile forces in opposite directions at both ends. In the stress–strain curves of PU, the mechanical tensile strength and elongation at break are 24.34 MPa and 431.2%, respectively, and the toughness is 40.2 MJ m−3 (Fig. S10, ESI†). With the introduction of NPSi, the fracture strength and elongation of AP-SHM reached 34.26 MPa and 397.75%, resulting in a toughness of 93.2 MJ m−3, indicating a more stable structure.48–50 The effect of the dynamic hydrogen bonds of AP-SHM on the self-healing ability was further determined. As shown in the optical photograph (Fig. 3(b)), two AP-SHM samples, one of which was dyed red, were subjected to contact at mild temperature (60 °C) for 3 h. The incision was completely healed together, and meanwhile a color transition zone was exhibited in the optical micrograph. The mechanical properties of AP-SHM are basically restored after a healing process, and its ultimate tensile strength, elongation, and toughness are 30.73 MPa, 383.33%, and 83.4 MJ m−3, respectively. The self-healing efficiency is close to 85%, revealing an outstanding self-healing ability. Meanwhile, the self-healing mechanism of the AP-SHM material is illustrated in Fig. 3(c). The original sample was ruptured, then the segments were brough into contact. At the fractured surface, the abundant dynamic H-bonds located on the polymer chain reconstruct an interface, allowing the supramolecular sensor to repair itself.
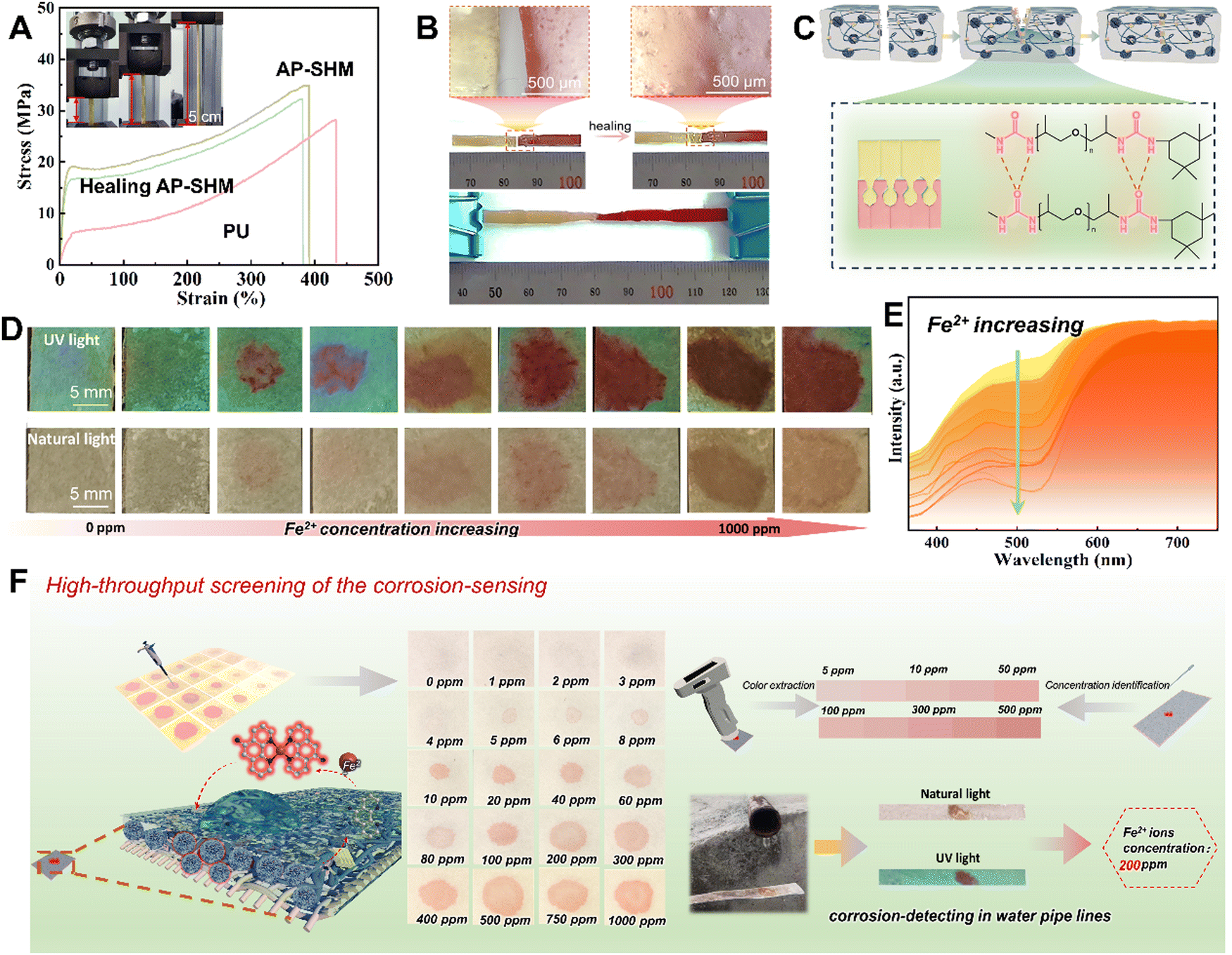 | ||
| Fig. 3 (a) Stress–strain curves of the PU, AP-SHM and the healing AP-SHM; (b) optical photos of the repaired AP-SHM stretching process and the optical microscope photographs at the interface fracture; (c) a schematic diagram of the self-healing mechanism of the AP-SHM material; (d) optical images of different concentrations of Fe tested on FeTP; (e) spectra of UV-vis results; (f) schematic diagram of FeTP quantitative analysis of solution Fe2+ content. | ||
To analyze the interfacial compatibility of the NPSi corrosion probes with the elastomer matrix, the dispersion of NPSi in elastomer was studied by observing the cross-section of the composites. The supramolecular sensor was firstly immersed in liquid nitrogen and then promptly fractured to achieve the cross-section surfaces. As shown in Fig. S11 (ESI†), the surface of pure PU was comparatively glazed, whereas after incorporation of NPSi it presented a rough morphology. Due to the abundance of hydroxyl groups on the surface of DMS, realizing an excellent non-covalent interaction with groups of the resin molecular chain (Fig. S12, ESI†). Thus, the corrosion probes can resist external forces extended during the fracture process, resulting in a rougher cross-section morphology. Briefly, excellent interfacial bonding is exhibited between NPSi and a PU molecular chain.
Determination of structural failure in corrosive media
For rapidly determining the states of corrosion failure evolution of metallic materials in corrosive marine media, a portable sensing device with easy-to-operate, real-time and visual Fe ion detection is prepared to monitor variations in ion concentrations. By introducing a supramolecular sensor on tester paper, a Fe2+ quantitative variation recognition strip was developed. Specifically, benefiting from the color changes of the NH2-Phen–Fe2+ complex with FeCl2 concentration, AP-SHM based test paper strips with quantitative monitoring capability can be achieved, which were defined as FeTP. With the cationic solution contacts with the FeTP, the NH2-Phen is immediately reacted to form a red complex within 2 min. This variation of chromaticity was fixed in the AP-SHM, facilitating the color visual analysis and quantitative assessment of the Fe content in corrosive media.To determine the chromaticity-response range of FeTP for the detection of Fe2+, a high-throughput assay of FeTP was performed. As shown in Fig. 3(d), a gradient solution of Fe2+, ranging from 0 ppm to 1000 ppm was prepared. Then, 5 μL of this solution was pipetted onto the 1 × 1 cm FeTP surface. The colorimeter was then used to capture the color of the complex on the FeTP strip surface, as depicted in Fig. 3(e). Firstly, a blank FeTP strip was selected as a specimen, and the color of other tested FeTP was recorded. Obviously, for these tested strips, the color of the red complex deepens with increasing Fe2+ concentration. Meanwhile, in the corresponding UV reflectance curve, the gap between the high points (600–700 nm) and low points (450–550 nm) exhibited a gradually increasing trend, which means that the color saturation of the tested strips is gradually enhanced. This color change is more intuitively displayed in the color coordinate (Fig. S13, ESI†). The chromaticity information of the standard concentration-colorimetric bar (SCL-bar) is derived from the UV reflectance spectroscopy, which is attributed to the human color vision of real scenes in accordance with the results of spectrophotometric colorimetry. The establishment of the SCL-bar realized the quantitative determination of Fe2+ in unknown solutions.
Furthermore, as exhibited in Fig. S14 (ESI†), there was a different chromaticity change of FeTP to various metal ions, with the most apparent color change for Fe2+ from white to red in natural light, demonstrating high sensitivity to specific ionic-responsiveness. These color changes were also recorded by the colorimeter in Fig. S15 (ESI†). Under uniform concentration, the highest gap is shown in the reflectance curve of Fe2+ ion. Meanwhile, its high points mean the highest saturation compared with other ions, and the lowest low point also predicts that its color has the greatest brightness. Moreover, the solution-response detection procedure for the complete FeTP strip is indicated in Fig. 3(f). The FeTP was placed at the outlet of a ferrous sewer drain and a clear color reaction was demonstrated within 60 s, revealing the concentration Fe2+ in the corrosive medium was around 200 ppm. In summary, it is found that the FeTP (with 1 wt% NPSi) have distinguished visual recognition to Fe2+ at 5–500 ppm, and exhibited high sensitivity, easy implementation, rapid response time, meager detection limits and naked eye detection ability. The practical results of detecting corrosion in pipelines also demonstrated the sensitivity and portability of the ionic response strips for corrosion-related failure monitoring.
The dual-responsiveness of AP-SHM coating
Owing to the incorporation of the NPSi corrosion probes, the AP-SHM sensors exhibited a dual-responsive function, ensuring rapid localization of film damage and interfacial corrosion. To validate the effectiveness of the film damage sensing, the AP-SHM sensor was coated on a Q235 steel surface. A scratch was introduced on the coated samples using customized tester blades, which were immersed in 3.5 wt% NaCl solution. Under 365 nm UV light irradiation, the AP-SHM emitted blue-green fluorescence, attributed to the incorporation of the photoluminescent of the NPSi. For shallow surface scratches (Fig. S16, ESI†), a noticeable fluorescence color difference was observed between the damaged and intact regions with 365 nm excitation, which was attributed to the damage-induced fluorescence enhancement (DIE) effect. As illustrated in Fig. S17 (ESI†), the roughness of the film increases at the damaged area after the film is subjected to mechanical forces and scratches. The internal reflection of the film is inhibited and the refraction of the scratch surface is immediately enhanced.51 Whereas, for deeper scratches, the steel substrate was directly exposed to the corrosive medium. After 5 mins, an obvious fluorescence quenching phenomenon was observed in the scratched area. Thus, the corrosion points of the substrate can be quickly and accurately identified under UV light. Due to the appearance of an electrochemical corrosion reaction occurring at the metal/film interface, the chromophore molecules complexed with the generated Fe2+ ions, leading to fluorescence quenching of the chromophore, which is defined as the ionic-recognition induced quenching effect (RIQ).52Furthermore, the corrosion monitoring process was revealed by an optical microscope. Before and after the corrosion initiation, a thin layer of corrosion products was generated and caused a color change, as exhibited in Fig. S20 (ESI†). Then, after removal of the coating, severe steel corrosion was observed around and within the scratches, which was ascribed to the permeation of NaCl solution. As intuitively illustrated in Fig. S18 (ESI†), the damage warning mechanism is based on the complexation between NH2-Phen and Fe2+ generated from the steel corrosion, triggering an RIQ effect. Once the NPSi was exposed to the electrolyte, the release of NH2-Phen from the NPSi was stimulated. This chroma transformation during corrosion was attributed to the formation of the red NH2-Phen–Fe2+ chelate. In contrast, the pure PU coated samples showed no noticeable visual changes after the application of shallow damage, as shown in Fig. S19a (ESI†). Meanwhile, for deep scratches, no corrosion response was exhibited before and after corrosion immersion. Therefore, the surface damage and interfacial corrosion are effectively demonstrated and warned by incorporating the synthesized NPSi, and the AP-SHM sensor exhibits a dual-responsive sensing capability.
Establishment and optimization of a channel-transformation model
Although the corrosion failure state of a metallic infrastructure in corrosive marine media can be indirectly determined using FeTP strips, ionic-responsiveness strips for offshore equipment are relatively limited in other marine corrosive zones (marine atmospheric corrosion zone, splash zone, tidal range zone, etc.). Accordingly, a multifunctional, semi-quantitative, algorithm-based AP-SHM patch is proposed for a quantitative assessment strategy of a component surface. The AP-SHM sensor is pre-cured onto a polypropylene (PP) membrane with an inherent adhesive interlayer, assembling a “binder” structure for rapid attachment of the supramolecular sensor onto the component surface. The multiple structural failure indicators, including crack scale, corrosion reactivity and deterioration status can be accurately quantified using sensor-based image processing software. Meanwhile, this smart patch can be rapidly geometrically cut and covered onto the surface of the metallic equipment. Due to the inherent excellent stretchability of the supramolecule patch, it can be adapted to monitor damage events for heterogeneous components.To evaluate the Fe2+ quantitative colorimetric sensing efficiency of the AP-SHM sensor, the correlation between chromaticity signals and Fe2+ concentration (cFe) is established. As demonstrated in Fig. 4(a) and (b), the fluorescence emission intensity at 370 nm decreased significantly as the Fe2+ concentration gradually increased in the range of 0–1000 ppm, which was confirmed by optical photos. In contrast to the characteristic peak (520 nm), the UV-vis curve showed an enhanced trend with the increase of the Fe2+ concentration. Then, the NH2-Phen–Fe2+ complex chromaticity information (RGB, HSV) was obtained from UV-vis spectroscopy, utilizing a grey-level co-occurrence matrix for the color extraction and defined as GV (Fig. 4(d)). Fitting the cFe with GV (Yc-G) using Levenberg–Marquardt (LM) algorithms (R2 = 0.998) (eqn (1)).53–55
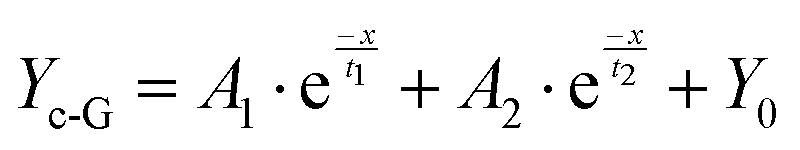 | (1) |
 | ||
| Fig. 4 (a) Optical images, (b) luminescence emission spectra and (c) spectra of UV-vis results of NPSi with the addition of different concentrations of Fe2+; (d) spectra of concentration versus grey-level value fitting results; (e) corrosion release process for carbon steel (exposed area = 1 cm2), and the corresponding (f) Nyquist and (g) Bode plots; histogram of Zf=0.01 corresponding to the release process of Q235; (i) spectra of concentration versus Zf=0.01 fitting results; (j) schematic diagram for digital analysis of corrosion in AP-SHM sensors. | ||
The GV–cFe function is successfully established and the detailed parameters are described in Table S3 (ESI†).
Meanwhile, for predicting the service life of metallic equipment, the corrosion rate of the steel components is considered as an important standard to evaluate its failure evolution stages. The polished Q235 steel electrode (with an exposed area of 1 cm2) was immersed in 3.5 wt% NaCl solution, and the corrosive medium was sampled at various intervals. The flame atomic absorption spectrophotometer (FAAS) was employed to determine the Fe content in the corrosive media at the corresponding immersion times (Fig. S22, ESI†). Therefore, the cFe–time relation curve was obtained (Fig. 4(e)), which can be divided into three phases: (I) slow release, (II) rapid release, and (III) gradual release. In Stage I, the iron and carbon of Q235 steel with a compact surface formed a primary cell in the 3.5 wt% NaCl solution, leading to electrochemical corrosion. In this reaction, iron acts as the cathode and carbon serves as the anode. By losing electrons, Fe is oxidized to generate Fe2+, which increases the Fe content in the corrosive medium. As the reaction continues, ferric ions continue to be generated and enter into the electrolyte with loose corrosion products forming on the iron surface, as shown in stage II. As the immersion time continued to increase, a gradual release was observed in stage III, which was attributed to a thick layer of corrosion products covering the iron surface.
To estimate the above corrosion rate of Q235 steel electrode immersed during these time periods, the electrochemical workstation (the frequency range of 105 to 10−2 Hz with a sinusoidal perturbation amplitude of 20 mV) was employed for the electrochemical impedance spectroscopy (EIS) test (Fig. S23, ESI†). To investigate the electrochemical corrosion process, the collected EIS data were fitted with the corresponding equivalent circuit (EC) models depicted in Tables S1 and S2 (ESI†). In the EC models, the electrochemical elements contained solution resistance (Rs), corrosion product film resistance (Rf), charge transfer resistance (Rct), surface film capacitance (Qf) and double-layer capacitance (Qdl). Meanwhile, to investigate the double layer property at the electrode–solution interface, constant phase elements (Qf and Qdl) were used, where n is the degree to which the element deviates from pure capacitance. When n = 1, the constant phase element is equivalent to capacitance; while n = 0, it is the same as resistance.56,57 Typically, the size of the reactance arc can provide a criterion for local corrosion of the matrix.58 As can be seen in Fig. 4(f), electrochemical corrosion just occurred at the initiation of the immersion, and the single capacitive arc was decreasing with increasing time, exhibiting the enhancement of local corrosion. As the corrosion product film accumulated on the alloy surface, two capacitive arcs were observed, indicating the deepening of localized corrosion.
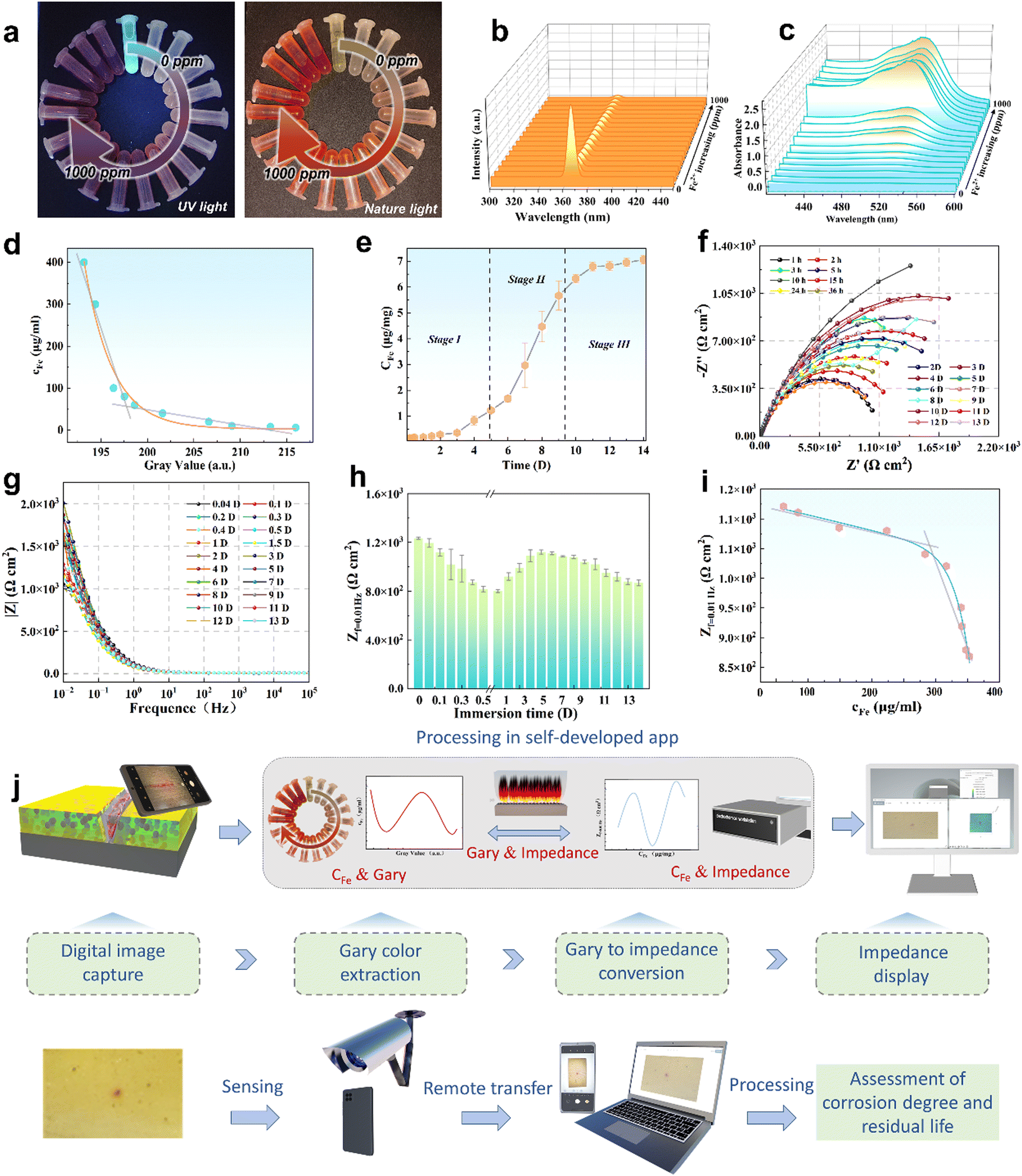
Furthermore, to evaluate the corrosion rate of Q235, the low frequency impedance value (Zf=0.01![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Hz) was considered to represent the corrosion failure evolution,59 as illustrated in Fig. 4(g). As presented in Fig. 4(h), the Zf=0.01Hz showed a large fluctuation in the beginning, while exhibiting a gradual downward trend (after 4 days of immersion), belonging to the stabilization of the corrosion product film. Therefore, it is valuable to select the Zf=0.01Hz in this period, and establish the correlation between cFe and Zf=0.01Hz (YZ-c) (R2 = 0.987) (eqn (2)) (LM algorithm). Thus, the relationship between GV and Zf=0.01Hz was obtained in terms of the GV–cFe function.
Hz) was considered to represent the corrosion failure evolution,59 as illustrated in Fig. 4(g). As presented in Fig. 4(h), the Zf=0.01Hz showed a large fluctuation in the beginning, while exhibiting a gradual downward trend (after 4 days of immersion), belonging to the stabilization of the corrosion product film. Therefore, it is valuable to select the Zf=0.01Hz in this period, and establish the correlation between cFe and Zf=0.01Hz (YZ-c) (R2 = 0.987) (eqn (2)) (LM algorithm). Thus, the relationship between GV and Zf=0.01Hz was obtained in terms of the GV–cFe function.
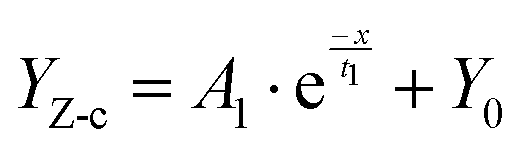 | (2) |
Based on these specific intrinsic links (GV–cF and GV–Zf=0.01Hz), a self-developed sensor-based image processing software was designed to semi-quantitatively assess the corrosion failure states of Q235 steel. By inputting an ionochromic sensor image, the Fe2+ concentration mapping, micro-crack scale and localized-corrosion evaluation reactivity can be quantified. A low-cost, easy-operation and real-time corrosion sensing method is realized to on-site monitor the corrosion.
The digital analysis procedures of the AP-SHM smart supramolecular structural health monitoring sensor are schematically illustrated in Fig. 4(j). When the electrochemical reaction occurs at the interface between the AP-SHM sensor and substrate, the apparent chromogenic reaction follows, which is visible to the naked eye and black in UV mode for the quick location of pitting (Fig. S24, ESI†). Subsequently, the ionochromic sensor image is captured and transferred back to the software (Fig. S25, ESI†). The image is firstly primary processed (select an appropriate chromatic threshold), then the desired module for structural failure analysis is selected. Finally, multiple structural failure indicators can be quantified by comparison with the corresponding standard colorimetric axes (Fig. S26, ESI†). In brief, the AP-SHM patch can achieve a fast responsive, real-time, intuitive, visual, algorithm-based, semi-quantitative corrosion analysis for the assessment of corrosion degree and residual life.
Structural damage monitoring and quantifying of AP-SHM sensors in real applications
To assess the structural failure responsiveness in the practical application of this AP-SHM sensor, artificial damage was introduced to the Q235 coated AP-SHM. The three-dimensional morphological analysis was carried out to determine the depth of the scratches (Fig. 5(a)). Subsequently, the damaged area was immersed in 3.5 wt% NaCl solution, and red complex appeared within 3 min, indicating the initiation of corrosion. Under 365 nm UV excitation, black precipitate was exhibited around the scratch, corresponding to the Fe–Phen complex. By inputting the ionochromic sensor image into image processing software, a clear color difference occurred from the intact areas in the concentration mapping, ensuring quick determination of the crack scale. Besides, the degree of corrosion increased with increasing immersion time, and there were no discernible color changes under direct observation, but this corrosion deepening evolution was captured though the image prolongation software. The corrosion concentration mapping monitored an exact concentration of Fe ion. Initially, a low concentration (0–5 ppm) of Fe2+ is present in corrosive media, but with further prolonging the immersion time, the concentration of Fe2+ in electrolyte was up to 200 ppm. Meanwhile, the corresponding impedance ranged from 1200 Ω cm2 to 900 Ω cm2, revealing the deepening of the corrosion and the severe localized failure of components.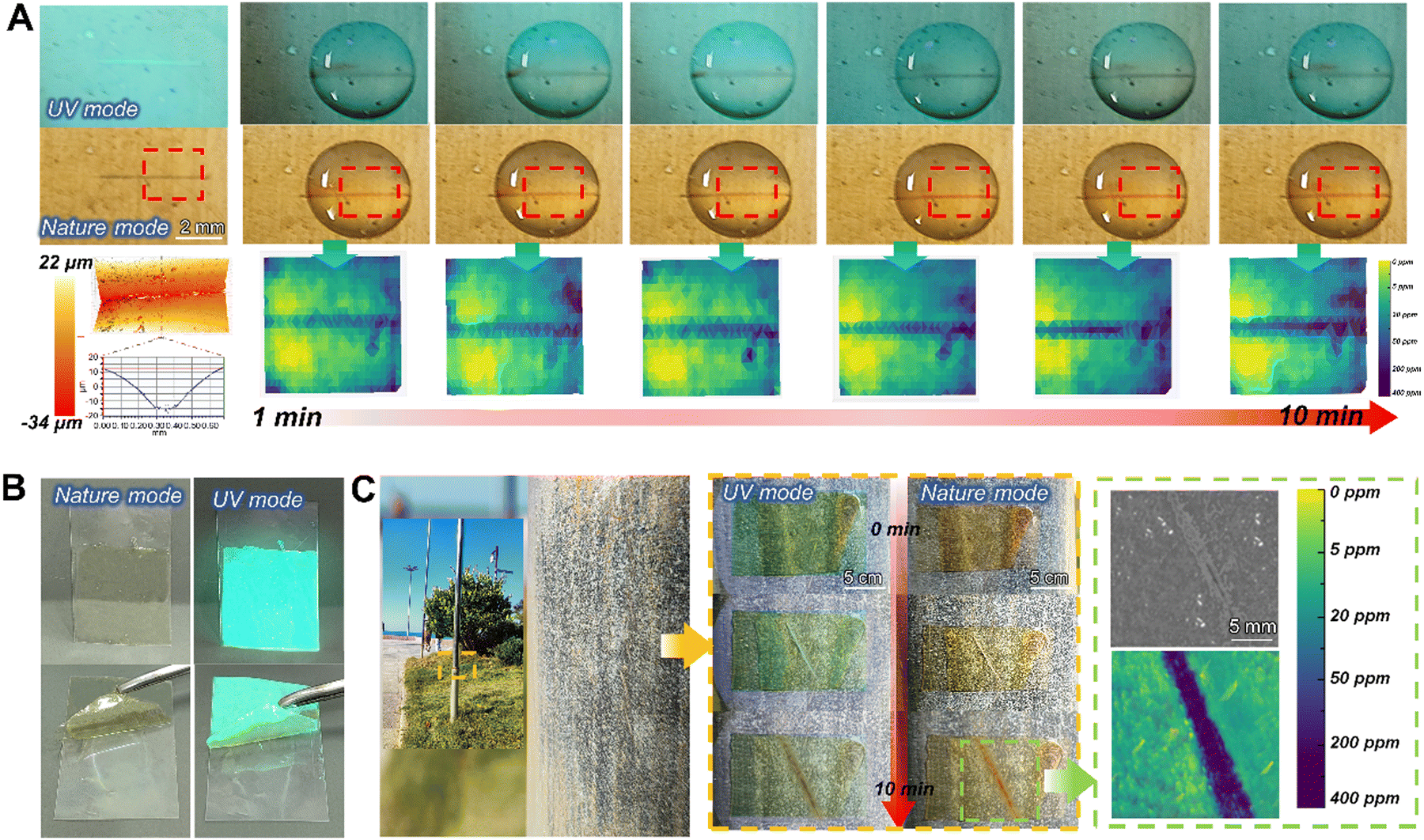 | ||
| Fig. 5 (a) Photographs in UV and nature modes, and concentration mapping of scratched AP-SHM on steel substrates with 10 min corrosion response during their immersion in a 3.5% wt solution; (b) optical photograph of the AP-SHM patch; (c) photograph of the seaside exposure test of AP-SHM and the final concentration mapping. | ||
To further validate the corrosion-sense of the AP-SHM performance in real environments, the AP-SHM patch was covered on a coastal steel column at a distance of 10 m from the coastline (Location Qingdao, China, temperature 25 °C, humidity 74%) (Fig. 5(c)). Then a scratch was introduced on the surface of the film and directly exposed to the marine atmospheric environment. The optical photo was acquired by a mobile phone lens. The areas of damage were recognized in the UV mode, and after 3 min red complexes appeared in the damaged area. Due to the high-temperature, high-humidity marine salt-spray atmosphere, the steel column triggered an entire ionic-response color change while the scratch induced a significant color deepening, indicating a severe corrosion failure of columns. When processed using software, the crack scale, concentration of Fe2+ (approximately 300 ppm) and low frequency impedance (820 Ω cm2) can be accurately quantified, demonstrating the ionic response result of the AP-SHM patch in actual service environments.
To demonstrate the adaptability to harsh marine environments, the AP-SHM sensor was exposed in high salinity, high humidity and temperature difference to explore the failure visualization monitoring behavior. The salt spray test was carried out in a chamber with continuous spray 5 wt% NaCl solution at 35 ± 2 °C, and the optical images of coated samples during a 168 h test are presented in Fig. 6(a) and (b). With the extension of exposure time, films still exhibited integration, and the corrosion products were not observed in UV mode or visible mode. When artificial damage was introduced to the AP-SHM surface, the fluorescence enhancement of the damaged area immediately appeared under UV irradiation. Once exposed to corrosive media, the corrosion-induced chromaticity reaction occurred within 10 min, then input it into the software and corrosion concentration mapping was obtained. Meanwhile, the sample was placed at 60 °C (for 3 h), due to the intrinsic dynamic hydrogen bonding of the AP-SHM, which demonstrated almost restoration within 180 min under optical microscope in situ observation (the specific healing process is shown in Fig. S27, ESI†).
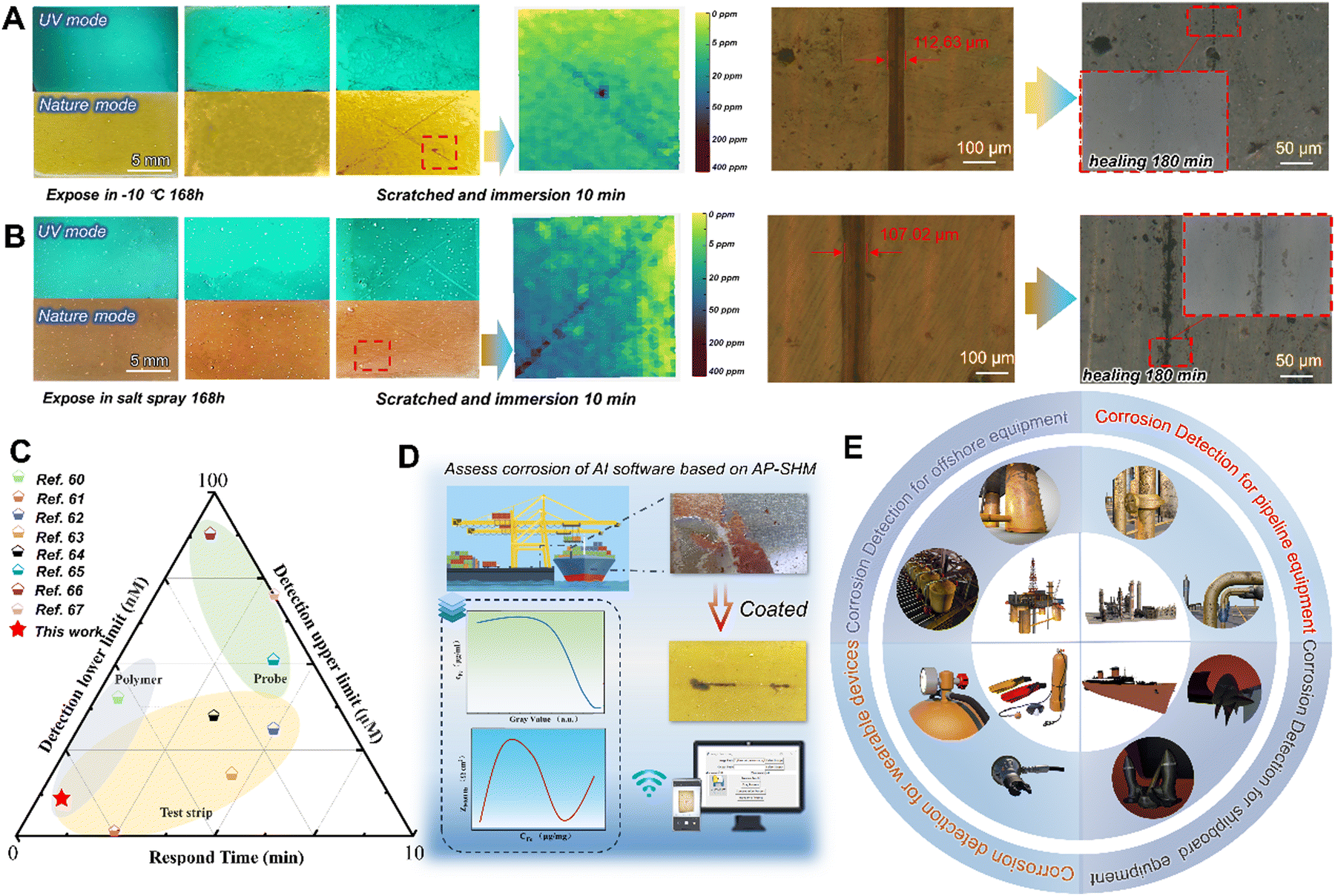 | ||
| Fig. 6 (a) The optical photograph and optical microscopic images of the coated AP-SHM samples after exposure in 5% NaCl (35 ± 2 °C) for 168 h; (b) the optical photograph and optical microscopic images of the coated AP-SHM samples after exposure to −10 °C for 168 h; (c) comparison of the detection limit and response time of AP-SHM with those of other reported smart sensors; (d) schematic diagram of the quantitative method analyses of corrosion digitally according to AP-SHM; (e) a schematic diagram of the AP-SHM structural health monitoring materials in real life and their manufacturing. | ||
The movement of the molecular chains of elastomer resins is affected by the temperature, which determines the self-healing efficiency of the supramolecular sensor. Therefore, to confirm the sensitive corrosion responsiveness and self-healing performance of the AP-SHM sensors at low temperature, the supramolecular sensor was exposed to −10 °C for a week. After the introduction of scratches, the hydrogen bonding of the sensor made the fracture reconstruct once placed under 60 °C for 3 h. Meanwhile, the ionochromic sensor can still be used for the assessment of deterioration degree and corrosion dynamics. Moreover, for other visual sensors based on spectral change detection (Fig. 6(c)), AP-SHM demonstrates high accuracy and readability over a wide range of monitoring limits, while maintaining the excellent response time and chromogenic stability. The multi-application design (patch and strip) and construction of multiple quantitative relationships (GV–cF and GV–Zf=0.01Hz) makes it more flexible and promising than other single types of sensors (polymer,60 test paper61–64 or probe65–67).
Furthermore, to verify the influence of environments (temperature and humidity) and state of the AP-SHM on failure diagnosis, the supramolecular sensor was subjected to ionochromic diagnosis with different degrees of tensile deformation. Specifically, 5 μL of Fe2+ ion solution was added dropwise to the surface of sensors with different degrees of deformation (0%, 50%, 100%, 150%), as shown in Fig. S28 (ESI†). The ionochromic sensor image is then captured and transferred back to the software, and read out (about 60 ppm), which indicates the tensile stability of the sensor. Importantly, to verify the accuracy of the sensor responsiveness for Fe2+, O is introduced as the ratio of the known concentration (cknow) to the test concentration (ctest) and using the following equation:
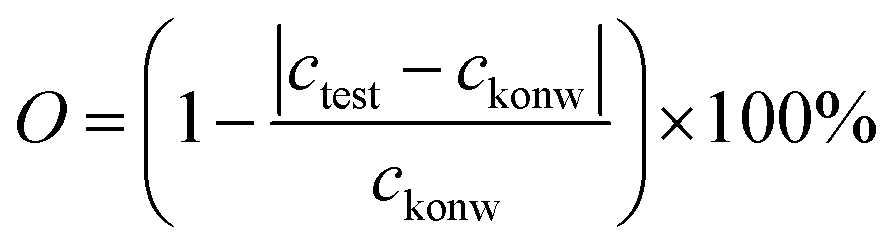 | (3) |
Eventually, the accuracy of the stretching of the AP-SHM sensor responsiveness for Fe2+ with testing multiple sensors is approximately 91%.
Subsequently, to evaluate the color change of the sensor with the environment conditions, the AP-SHM sensor (about 100 μm) was fully cured on a PP film, then peeled and cut. To investigate the effect of humidity changes on the ionochromism of the AP-SHM sensor, the sensor was placed in 5 humidity conditions that simulate marine atmospheric corrosion and titrated with equal concentrations of Fe2+ ions at room temperature. As shown in Fig. S29 (ESI†), the color change response of the sensor is little affected by humidity (Ohumidity ≈ 95%), except under water. The dispersed Fe2+ ions result in a wide range of color responses, affecting sensor failure diagnosis. The Fe responsive strips are designed for the ionic response of corrosive media in underwater environments, which are prepared by applying the sensor to the strip surface. To assess the effect of temperature on the responsiveness of AP-SHM, the same concentration of Fe2+ ions was titrated on the sensor of 5 temperature environments. As shown in Fig. S30 (ESI†), the temperature does not affect the ionochromism of the AP-SHM sensor (Otemperature is about 96%). In conclusion, the effectiveness of monitoring the structural health status of objects is slightly influenced by the state of the sensor (degree of stretching) and the environmental conditions (varying temperatures and humidity levels).
The temperature and humidity have little effect on the responsiveness stability of the sensor. Meanwhile, various temperature and humidity scenarios have occurred over the course of a year of actual marine atmospheric exposure and ultimately did not affect the structural failure diagnosis of the AP-SHM sensing layer. Thus, the long-term stability of the sensor under stressed tensile conditions is focused on. As shown in Fig. S31 (ESI†), the sensor undergoes continuous stretching after one week with different degrees of deformation. Due to the stretchable properties of the supramolecular itself, the sensor undergoes a certain degree of recovery at the end of stretching. Then the titration is performed with Fe2+ ions, the ionochromism of the sensor is still responsive and the titrated ion concentration was read out (60 ppm), demonstrating the long-term stability of the supramolecular sensor for structural health monitoring.
Conclusions
In summary, we have developed a universal, self-healing, and intelligent SHM sensor to quantify the health status of metallic materials. Benefiting from the incorporation of NPSi, the AP-SHM acquires inherent photoluminescence and ionic responsiveness. Meanwhile, the intrinsic self-healing properties of PU elastomers ensure the responsiveness of the supramolecular sensor in extreme environments. The supramolecular AP-SHM sensor has excellent geometric plasticity, which can be fabricated as a coating or a patch covered on metallic substrates for the full-field monitoring throughout service life. Correlations were established between visual monitoring signals and structural failure status based on the supramolecular sensor, providing the quantification of multiple structural failure indicators. In aquatic corrosive medium, the integrated AP-SHM strips exhibit a wide range (5–500 ppm), and immediate responsiveness (within 2 min) for the indirect assessment of structural failure. Through quantification of the concentration of Fe2+ (from 0–200 ppm) and corresponding impedance (from 1200 Ω cm2 to 900 Ω cm2) within 10 mins in a marine salt-spray atmosphere, the function of the AP-SHM patch in dynamically tracing structural failure evolution is demonstrated. The utilization of this method of universal, rapid, non-specialized machine analysis for quantified corrosion evaluation avoids the subjective judgement of human experts relying on Standards (GB, ISO) (Fig. 6(d) and (e)). Therefore, this work constructs potential, algorithm-based, integrated quantitative intelligent structural health monitoring materials for assessment of the residual life of heterogeneous engineering equipment. Furthermore, it offers a rapid and quantifiable SHM strategy for future perspectives of offshore energy-harvesting equipment.Experimental section
Materials
Triethanolamine (AR, 99%), sodium salicylate (≥99.5%), cetyltrimethylammonium bromide (CTAB) (99%), 1,10-phenanthrolin-5-amine (NH2-Phen) (97%), isophorone diisocyanate (IPDI, 99.9%), poly(propylene glycol) bis(2-aminopropyl ether) (D400, average Mn ∼ 400), dopamine hydrochloride (HCl DA, 98%), and N,N-dimethylformamide (DMF, 99.8%) were obtained from Aladdin Industrial Co. Tetraethyl orthosilicate (TEOS) (98%) was supplied by Shanghai Macklin Biochemical Co. Triethylamine (TEA, ≥99.0%) was bought from Chengdu Kelong Chemical Reagent Factory. Mild steel (Q235) was purchased from Shengxin Technology Co. and selected as the metal substrate for anticorrosion evaluations.Synthesis of NPSi
Triethanolamine (0.9 mmol) and sodium salicylate (2.1 mmol) were first added to deionized water (50 mL) and stirred at 80 °C for 30 min, and then cetyltrimethylammonium bromide (CTAB) (2 mmol) was mixed with the above solution. The resulting solution was stirred vigorously for 1 h to dissolve the solid completely. Then 8 mL of tetraethyl orthosilicate (TEOS) was added slowly and stirred for 2 hours. The final solution was centrifuged (6000 rpm) for 8 min, washed several times with ethanol and dried at 60 °C for 24 hours. Template (DMS) was obtained and subsequently placed in a muffle furnace at 600 °C for 6 h. Dendritic mesoporous silica spheres (DMS) were obtained. The DMS were then homogeneously dispersed in ethanol solution, and then poured into the ethanol solution of NH2-Phen, co-mixed for 30 min, centrifuged (8000 rpm) for 9 min, washed with ethanol several times and dried at 60 °C for 24 h. The final yellowish-green powder NH2-Phen was obtained as a result. The final yellow-green powder NH2-Phen@SiO2 (NPSi) was obtained.Synthesis of PU
The preparation procedure for PU can be described as follows. Firstly, D400 (16.00 g, 0.04 mol) was added into a three-necked flask and completely dispersed in 20 mL of DMF. Then, a mixture of IPDI (8.88 g, 0.04 mol) and 10 mL of DMF was slowly added dropwise into the flask at room temperature with stirring (200 rpm) and the reaction lasted for 2 h. And finally, the PU was obtained.Fabrication of highly sensitive test paper and flexible film for steel corrosion detection
The NPSi (0.05 g) was homogeneously dispersed in 5 g of polyurethane (PU) followed by ultrasonic shaking for 5 min, and the mixture was coated onto a polypropylene (PP) membrane with an inherent adhesive interlayer, assembling a “binder” structure. A flexible AP-SHM patch was obtained by placing it in an oven at 60 °C and pre-cured for 40 min. Then the patch was coated on the test strip, and the fast response Fe2+ standard test strips (FeTP) were developed by placing it in an oven at 40 °C and cured thoroughly for 60 min.Characterization of NPSi
Scanning electron microscopy (SEM, Nova NanoSEM 450, FEI, USA) and transmission electron microscopy (TEM, Tecnai F20, USA) were used to characterize the morphology and structure of the synthesized DMS and NPSi. Besides, the element compositions of DMS and NPSi were investigated by energy-dispersive X-ray spectroscopy (EDS, matched with FE-SEM). In addition, the functional groups and chemical composition of the DMS and NPSi were evaluated by Fourier-transform infrared (FT-IR, Nicolet 380, Thermo Electron Corporation, USA) spectroscopy and X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA), respectively. To analyze the crystal structures of NH2-Phen, DMS and NPSi, X-ray diffraction (XRD, Rigaku D/MAX 2500 PC, Rigaku Corporation, Japan) was carried out on a powder diffractometer. The loading capacity of the DMS for NH2-Phen molecules was determined using a thermogravimetric analyzer (TGA, TA Instruments Q500, USA), which was conducted at 50–700 °C under a nitrogen atmosphere (20 mL min−1). A UV-vis spectrometer (UV-vis, Agilent CARY 60, USA) and a fluorescence spectrophotometer (Hitachi F-4600, Japan) were used to explore the complexation interaction between NPSi and Fe2+ and the selectivity of NPSi for different cations (Al3+, Mg2+, Cu2+, Cr3+, Ni2+, Co2+, Fe3+, Fe2+, Sr2+, and Zn2+).Characterization of PU and AP-SHM sensors
1H NMR spectra (Bruker Avance NEO, 400 MHz, Germany) were obtained to determine the chemical structure of PU, using deuterated DMSO-d6 as a solvent. The glass transition temperature of PU was determined using a differential scanning calorimeter (DSC) (METTLER TOLEDO, Switzerland), which was conducted on a TGA/DSC I under a nitrogen atmosphere (20 mL min−1) and at a heating rate of 10 °C min−1 within the range of −60 to 100 °C. The Fourier transform infrared spectra (FT-IR) (Thermo Scientific Nicolet S10 FTIR spectrometer equipped with a diamond ATR crystal) were further used to determine the structure of PU. Moreover, the typical tensile–stress curves of PU, AP-SHM and healing AP-SHM were measured on an electronic universal testing machine (GT-AI7000S) equipped with a 2000 N load cell. In the strain–stress tests, the deformation rate is set to 20 mm min−1. For the elastomer self-healing ability characterization, the elastomers with rectangular shape were cut and the cut surface was put in contact to heal at 60 °C for 3 h. The healed specimens then underwent tensile–stress tests. The healing efficiency (η) is usually defined as the ratio of the tensile strength of the healed (σheal) and original samples (σori) and the equation is as follows: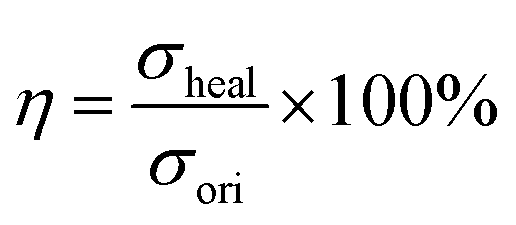 | (4) |
The tensile splines were obtained by using a custom cutting knife with a standard dumbbell shape. A metallographic microscope (OM, Axio Lab A1, ZEISS, Germany) was employed to observe the results of the AP-SHM cutting repair process and corrosion extent of steel surfaces.
To investigate the damage responsiveness of the AP-SHM, an artificial scratch was made on the steel coated with AP-SHM using a home-made machine. The excitation wavelength of the ultraviolet lamp in the fluorescence spectrophotometers was set at 365 nm to record the fluorescence emission intensity of the coatings before and after corrosion. Meanwhile, the metallographic microscope was employed to observe the corrosion products generated on the steel surface. The specific chromatic transformation of coatings was evaluated using a spectrophotometer (TS7700, China) during the immersion process. Meanwhile, the spectrophotometer was used to record the chromaticity change of the FeTP response to Fe2+ and the selectivity for different cations.
The color change of the sensor with environment changes was evaluated. Firstly, to verify the effect of the degree of stretching of the AP-SHM on failure diagnosis, the supramolecular sensor was subjected to ionochromic diagnosis with different degrees of tensile deformation (0%, 50%, 100%, 150%). Meanwhile, to investigate the effect of humidity changes on the ionochromism of the AP-SHM sensor, the sensor was placed in 5 humidity conditions (20%, 40%, 60%, 80% and Underwater) that simulate marine atmospheric corrosion and titrated with equal concentrations of Fe2+ ions at room temperature. Subsequently, the same concentration of Fe2+ ions was titrated on the sensor with 5 temperature environments (−18 °C, 0 °C, 27 °C, 42 °C and 60 °C). The temperature of the sensors was measured and collected by using a thermal imager (UTi260B).
The evaluation of corrosion behavior of the Q235 substrate in 3.5 wt% NaCl solution was investigated using electrochemical impedance spectroscopy (EIS, CHI-660E, China). The counter electrode and reference electrode were a platinum sheet and a saturated calomel electrode (SCE), respectively. Q235 steel with an examined area of 1 cm2 corresponds to the working electrode. An EIS test was performed on an electrochemical workstation in the frequency range of 105 to 10−2 Hz with a sinusoidal perturbation amplitude of 20 mV. The analyte quantification analysis of electrolyte was undertaken using a flame atomic absorption spectrometry (FAAS) (TAS-986F, China) model with an air–acetylene burner.
Molecular dynamics simulations
As shown in Fig. 2(a), in the following system, the periodic boundary conditions and COMPASSII force fields were employed for high accuracy. Firstly, a simulation box of 26.8 Å × 26.8 Å × 62.9 Å dimension was established, containing 5 layers of SiO2 (100), 1 NH2-Phen molecule, 200 CH3CH2OH and a 20 Å vacuum layer. Afterward, the models were first placed under NPT for 100 ps, which can ensure the simulation system was close to the real environment. Furthermore, the models were put in NVT for 2000 ps to reach the equilibrium simulation state.Quantum chemistry calculations
The relevant parameters were established as follows. The working type was OPT + Frep. The calculation method was Ground and DFT, the Basis Set was 6-311+G*d for C H O and Lanl2 for Fe. The corresponding parameters of NH2-Phen and Fe2+–NH2-Phen under liquid phase (solvent = water) conditions were calculated.Author contributions
Dezhi Jiao: investigation, methodology, writing original draft. Chengbao Liu: supervision, conceptualization, funding acquisition, writing – review & editing. All authors discussed the results and have given their approval to the final version of the manuscript.Data availability
The data that support the findings of this study are available within the paper and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully appreciate the financial support from the National Natural Science Foundation of China (52201077, 52401096), the Natural Science Foundation of Shandong Province (ZR2022QE191, ZR2024QE462), and project 24-4-4-zrjj-54-jch supported by Qingdao Natural Science Foundation and the State Key Laboratory of Marine Coatings Funded Project (02030124902).References
- Y. Yang, Y. Li, D. Chen and G. Shen, Mater. Horiz., 2024, 11, 1934–1943 RSC.
- G. Wang, G. Wang, L. Fei, L. Zhao and H. Zhang, Nano-Micro Lett., 2024, 16, 150 CrossRef CAS.
- A. B. Tesler, S. Kolle, L. H. Prado, I. Thievessen, D. Böhringer, M. Backholm, B. Karunakaran, H. A. Nurmi, M. Latikka, L. Fischer, S. Stafslien, Z. M. Cenev, J. V. I. Timonen, M. Bruns, A. Mazare, U. Lohbauer, S. Virtanen, B. Fabry, P. Schmuki, R. H. A. Ras, J. Aizenberg and W. H. Goldmann, Nat. Mater., 2023, 22, 1548–1555 CrossRef CAS.
- M. Li, H. Sun, X. Tan, H. Zhang and J. Liu, Mater. Today, 2024, 74, 46–57 CrossRef.
- J. Zhao, D. Wang, F. Zhang, J. Pan, P. Claesson, R. Larsson and Y. Shi, Nano-Micro Lett., 2022, 14, 160 CrossRef CAS.
- T. Yimyai, D. Crespy and M. Rohwerder, Adv. Mater., 2023, 35, 2300101 CrossRef CAS.
- H. Wang, B. Liu, D. Chen, Z. Wang, H. Wang, S. Bao, P. Zhang, J. Yang and W. Liu, Mater. Horiz., 2024, 11, 2628–2642 RSC.
- S. Ramesh, S. Khan, Y. Park, E. Ford, S. Menegatti and J. Genzer, Mater. Today, 2022, 54, 90–109 CrossRef CAS.
- L. Luo, Z. Wu, Q. Ding, H. Wang, Y. Luo, J. Yu, H. Guo, K. Tao, S. Zhang, F. Huo and J. Wu, ACS Nano, 2024, 18, 15754–15768 CrossRef CAS PubMed.
- L. Jia, J. Xiao, Y. Tan, K. Zhang, Y. Liu and X. Wang, Small, 2024, 20, 2309231 CrossRef CAS PubMed.
- M. Inês Silva, E. Malitckii, T. G. Santos and P. Vilaça, Prog. Mater. Sci., 2023, 138, 101155 CrossRef.
- M. D. Crall, S. G. Laney and M. W. Keller, Adv. Funct. Mater., 2019, 29, 1806634 CrossRef.
- H. Mei, M. F. Haider, R. James and V. Giurgiutiu, Composites, Part B, 2020, 189, 107906 CrossRef.
- O. Rifaie-Graham, E. A. Apebende, L. K. Bast and N. Bruns, Adv. Mater., 2018, 30, 1705483 CrossRef.
- W. Wang, D. Yao, H. Wang, Q. Ding, Y. Luo, H. Ding, J. Yu, H. Zhang, K. Tao, S. Zhang, F. Huo and J. Wu, Adv. Funct. Mater., 2024, 34, 2316339 CrossRef CAS.
- H. He, Y. Qin, Z. Zhu, Q. Jiang, S. Ouyang, Y. Wan, X. Qu, J. Xu and Z. Yu, Nano-Micro Lett., 2023, 15, 226 CrossRef CAS.
- X. Rong, Q. Ding, L. Chen, S. Yang, J. Lou, Z. Liu, X. Li, Y. Jiang, X. Wang and W. Han, Mater. Horiz., 2024, 11, 2420–2427 RSC.
- B. Zhong, X. Qin, H. Xu, L. Liu, L. Li, Z. Li, L. Cao, Z. Lou, J. A. Jackman, N.-J. Cho and L. Wang, Nat. Commun., 2024, 15, 624 CrossRef CAS PubMed.
- R. Huang, Y. He, J. Wang, J. Zou, H. Wang, H. Sun, Y. Xiao, D. Zheng, J. Ma, T. Yu and W. Huang, Nat. Commun., 2024, 15, 1596 CrossRef CAS PubMed.
- C. Lv, Z. Zhou, Y. Li, S. Lu and Y. Bai, Chem. Eng. J., 2023, 477, 147059 CrossRef CAS.
- X. Chen, Y. Hui, J. Zhang, Y. Wang, J. Zhang, X. Wang, S. Cheng, K. Wen, Z. Li, C. Yi and J. Shao, Composites, Part B, 2023, 259, 110713 CrossRef CAS.
- V. Balasubramanian, O. Niksan, M. C. Jain, K. Golovin and M. H. Zarifi, Nat. Commun., 2023, 14, 4916 CrossRef CAS PubMed.
- L. Ma, R. Wu, A. Patil, J. Yi, D. Liu, X. Fan, F. Sheng, Y. Zhang, S. Liu, S. Shen, J. Wang and Z. L. Wang, Adv. Funct. Mater., 2021, 31, 2102963 CrossRef CAS.
- Y. Hou, X. Dong, D. Li, D. Shi, W. Tang and Z. L. Wang, Adv. Funct. Mater., 2023, 33, 2305719 CrossRef CAS.
- Z. Huang, W. Shi, S. Wu, Y. Wang, S. Yang and H. Chen, Sci. Adv., 2024, 10, eado8516 CrossRef PubMed.
- M. Babazadeh-Mamaqani, D. Razzaghi, H. Roghani-Mamaqani, A. Babaie, M. Rezaei, R. Hoogenboom and M. Salami-Kalajahi, Prog. Mater. Sci., 2024, 146, 101312 CrossRef CAS.
- A. Kumar, K. Preeti, S. P. Singh, S. Lee, A. Kaushik and S. K. Sharma, Mater. Today, 2023, 69, 262–286 CrossRef CAS.
- H. Nawaz, S. Chen, X. Zhang, X. Li, T. You, J. Zhang and F. Xu, ACS Nano, 2023, 17, 3996–4008 CrossRef CAS.
- X. Ma, Y. Wang, T. Zhao, Y. Li, L.-C. Su, Z. Wang, G. Huang, B. D. Sumer and J. Gao, J. Am. Chem. Soc., 2014, 136, 11085–11092 CrossRef CAS PubMed.
- Y. Liu, C. Wang, Z. Liu, X. Qu, Y. Gai, J. Xue, S. Chao, J. Huang, Y. Wu, Y. Li, D. Luo and Z. Li, Nat. Commun., 2024, 15, 663 CrossRef CAS PubMed.
- Y. Zheng, J. Jiang, M. Jin, D. Miura, F. X. Lu, K. Kubota, T. Nakajima, S. Maeda, H. Ito and J. P. Gong, J. Am. Chem. Soc., 2023, 145, 7376–7389 CrossRef CAS PubMed.
- B. Tan, S. Zhang, H. Liu, Y. Guo, Y. Qiang, W. Li, L. Guo, C. Xu and S. Chen, J. Colloid Interface Sci., 2019, 538, 519–529 CrossRef CAS PubMed.
- Y. Qiang, L. Guo, H. Li and X. Lan, Chem. Eng. J., 2021, 406, 126863 CrossRef CAS.
- A.-M. Li, Z. Wang, T. P. Pollard, W. Zhang, S. Tan, T. Li, C. Jayawardana, S.-C. Liou, J. Rao, B. L. Lucht, E. Hu, X.-Q. Yang, O. Borodin and C. Wang, Nat. Commun., 2024, 15, 1206 CrossRef CAS PubMed.
- S.-G. Kang, W. Jeong, J. Paeng, H. Kim, E. Lee, G.-S. Park, S. Han, H. Nam Han and I.-S. Choi, Mater. Today, 2023, 66, 62–71 CrossRef CAS.
- D. Yan, X. Liu, Z. Chen, Y. Wang, M. Zhang, T. Zhang and J. Wang, Chem. Eng. J., 2023, 451, 138995 CrossRef CAS.
- L. Cheng, C. Liu, H. Zhao and L. Wang, J. Mater. Chem. A, 2021, 9, 22509–22521 RSC.
- W. Zhao, P. Guo, J. Su, Z. Fang, N. Jia, C. Liu, L. Ye, Q. Ye, J. Chang and H. Wang, Adv. Funct. Mater., 2022, 32, 2200534 CrossRef CAS.
- T. Liu, D. Zhang, L. Ma, Y. Huang, X. Hao, H. Terryn, A. Mol and X. Li, Corros. Sci., 2022, 200, 110254 CrossRef CAS.
- T. Liu, D. Zhang, R. Zhang, J. Wang, L. Ma, P. Keil, A. Mol and X. Li, Chem. Eng. J., 2023, 454, 140335 CrossRef CAS.
- S. Guo, X. Wang, Z. Gao, G. Wang and M. Nie, Ultrason. Sonochem., 2018, 48, 19–29 CrossRef CAS PubMed.
- M. N. Masood, E. T. Carlen and A. van den Berg, Appl. Surf. Sci., 2015, 337, 105–110 CrossRef CAS.
- J. Liao, W. Cui, J. Li, J. Sheng, H. Wang, X. A. Dong, P. Chen, G. Jiang, Z. Wang and F. Dong, Chem. Eng. J., 2020, 379, 122282 CrossRef CAS.
- G. Chilkoor, K. Jawaharraj, B. Vemuri, A. Kutana, M. Tripathi, D. Kota, T. Arif, T. Filleter, A. B. Dalton, B. I. Yakobson, M. Meyyappan, M. M. Rahman, P. M. Ajayan and V. Gadhamshetty, ACS Nano, 2020, 14, 14809–14819 CrossRef CAS PubMed.
- X. Wang, X. Li, H. Fan and L. Ma, Nano-Micro Lett., 2022, 14, 205 CrossRef CAS PubMed.
- X. Wang, J. Xu, Y. Zhang, T. Wang, Q. Wang, S. Li, Z. Yang and X. Zhang, Nat. Commun., 2023, 14, 4712 CrossRef CAS PubMed.
- A. Docker, I. Marques, H. Kuhn, Z. Zhang, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 14778–14789 CrossRef CAS PubMed.
- X. Zhu, W. Zheng, H. Zhao and L. Wang, J. Mater. Chem. A, 2021, 9, 20737–20747 RSC.
- X. Zhu, W. Zhang, G. Lu, H. Zhao and L. Wang, ACS Nano, 2022, 16, 16724–16735 CrossRef CAS PubMed.
- J.-H. Gao, B. Wan, M.-S. Zheng, L. Luo, H. Zhang, Q.-L. Zhao, G. Chen and J.-W. Zha, Mater. Horiz., 2024, 11, 1305–1314 RSC.
- C. Liu, L. Cheng, L.-Y. Cui, P. Hou, B. Qian and R.-C. Zeng, J. Mater. Sci. Technol., 2023, 152, 169–180 CrossRef CAS.
- L. Cheng, C. Liu, H. Zhao and L. Wang, Chem. Eng. J., 2023, 467, 143463 CrossRef CAS.
- Y.-L. Kou, J. Tong, C. Meng, Q. Yuan, J. Wang and S.-Y. Yu, ACS Appl. Mater. Interfaces, 2023, 15, 40828–40838 CrossRef CAS PubMed.
- M. Theerasilp and D. Crespy, Nano Lett., 2021, 21, 3604–3610 CrossRef CAS.
- L. Yan, B. Zhang, Z. Zong, W. Zhou, S. Shuang and L. Shi, J. Colloid Interface Sci., 2023, 651, 59–67 CrossRef CAS.
- S. Chen and D. Zhang, Corros. Sci., 2019, 148, 71–82 CrossRef CAS.
- C. Liu, P. Hou, B. Qian and X. Hu, J. Ind. Eng. Chem., 2023, 118, 109–118 CrossRef CAS.
- Y. Zhang, J. Tian, J. Zhong and X. Shi, ACS Nano, 2018, 12, 10189–10200 CrossRef CAS.
- T. Zhao, Z. Jia, J. Liu, Y. Zhang, G. Wu and P. Yin, Nano-Micro Lett., 2023, 16, 6 CrossRef.
- Y. Huang, S. Chen, W. Huang, X. Zhuang, J. Zeng, M. Rong and L. Niu, Food Chem., 2024, 432, 137292 CrossRef CAS PubMed.
- R. Jiang, D. Lin, Q. Zhang, L. Li and L. Yang, Sens. Actuators, B, 2022, 350, 130902 CrossRef CAS.
- G. Kumar, K. Paul and V. Luxami, Sens. Actuators, B, 2018, 263, 585–593 CrossRef CAS.
- S. Lai, Y. Jin, L. Shi, R. Zhou and Y. Li, ACS Sens., 2023, 8, 3812–3823 CrossRef CAS.
- J. Liu, Y. Zhan, B. Qiu, Z. Lin and L. Guo, ACS Sens., 2023, 8, 884–892 CrossRef CAS.
- X.-Q. Ma, Y. Wang, T.-B. Wei, L.-H. Qi, X.-M. Jiang, J.-D. Ding, W.-B. Zhu, H. Yao, Y.-M. Zhang and Q. Lin, Dyes Pigm., 2019, 164, 279–286 CrossRef CAS.
- Y. Qiu, L. Xia, R. Shi, L. Yuan, Y. Zhang, A. Chen, K. Zhou, H. Wu, K. Zhang, Z. Xia and Q. Fu, Sens. Actuators, B, 2024, 401, 134958 CrossRef CAS.
- Y. Wu, G. Hou, H. Cui, Y. Zhang, L. Chen, Y. An, H. Zhou and J. Chen, Composites, Part B, 2023, 266, 111021 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01233j |
| This journal is © The Royal Society of Chemistry 2025 |