
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Measurement of large ribosomal subunit size in cytoplasm and nucleus of living human cells†
Aneta
Magiera
 ,
Karolina
Kucharska
,
Tomasz
Kalwarczyk
,
Patrycja
Haniewicz
,
Karina
Kwapiszewska
* and
Robert
Hołyst
*
,
Karolina
Kucharska
,
Tomasz
Kalwarczyk
,
Patrycja
Haniewicz
,
Karina
Kwapiszewska
* and
Robert
Hołyst
*
Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Poland. E-mail: kkwapiszewska@ichf.edu.pl; rholyst@ichf.edu.pl
First published on 11th December 2024
Abstract
Ribosomes are the most essential macromolecules in cells, as they serve as production lines for every single protein. Here, we address the demand to study ribosomes in living human cells by applying time-resolved microscopy. We show that oxazole yellow iodide (YO-PRO-1 dye) intercalates tRNA and rRNA with a determined equilibrium constant of 3.01 ± 1.43 × 105 M−1. Fluorescence correlation spectroscopy (FCS) is used to measure both the rotational (∼14 ms−1) and translational (∼4 μm2 s−1) diffusion coefficients of the 60S ribosomes directly within living human cells. Furthermore, we apply the empirical length-scale dependent viscosity model to calculate the hydrodynamic radius of 60S ribosomes, equal to ∼15 nm, for the first time determined inside living cells. The FCS in YO-PRO-1 stained cells is used to assess ribosome abundance changes, exemplified in rapamycin-treated HeLa cells, highlighting its potential for dynamic ribosome characterization within the cellular environment.
New conceptsWe present a method for staining ribosomes directly inside living human cells, creating the capability for monodisperse nanoprobing of complex biological fluids. We demonstrate that a DNA intercalator, YO-PRO-1 dye, specifically binds to tRNA and the large ribosomal subunit inside human cells. Based on free diffusion in combination with a nanoviscosity model, we use this selective labeling to measure the size of large ribosomal subunits for the first time directly inside cells. Contrary to literature reports, we prove that YO-PRO-1 freely penetrates living cells. Our approach brings new insight into nanoscience, creating a tool to study the behavior of nanoparticles in complex biological systems at the nanoscale by investigating naturally occurring, abundant ribosomes as a model for nanostructures. Until now, the intracellular interactions of YO-PRO-1, commonly used so far as a marker for apoptotic cells, were unrecognized, and thus, the existence of a specific dye on tRNA and rRNA was not known. |
Introduction
Ribosomes are complex machinery essential for life. Different biochemical processes occurring inside the human body require different proteins synthesized during translation on ribosomes. The total intracellular protein concentration ranges to 250 g L−1, while the nucleic acid amount varies between 20 and 50 g L−1.1–3 In the last few years, scientists have been widely studying ribosomes' importance in cancer development,4 gene regulation,5 or even age-related diseases.6 Along with investigating the advanced role of ribosomes, there are still gaps in the knowledge of their exact structure or size.7,8Like proteins or nucleic acids, ribosomes freely diffuse inside a cell due to Brownian motions. By studying the movement of molecules within a cell, we can get insight into its structure, size (the bigger the size, the smaller the diffusion is), and even the interactions it undergoes.9,10 However, to obtain such information, we must have a method allowing us to measure individual molecules directly inside the cytoplasm/cell nucleus. Moreover, the movement of an object depends on the system's viscosity. In a medium with higher viscosity, a molecule will move more slowly. In our previous studies, we proved that the cytoplasm/nucleoplasm viscosity depends on a length scale and provided the length-scale dependent nanoviscosity (LSDV) model.11,12 The LSDV model describes viscosity at different length scales (1–100 nm) inside cells, providing the possibility of predicting diffusion coefficients of biomolecules based on their sizes. This model has become a robust tool for supporting FCS (fluorescence correlation spectroscopy) measurements at a single-cell and single-molecule level.9,10,13 Analyzing diffusion coefficients in cells, we can precisely identify the interactions a molecule introduced into a cell undergoes, determining what it is bound to and at what concentration. Based on the cytoplasm/nucleoplasm nanoviscosity curve, we know the tested molecule's diffusion coefficient inside the cell. Thus, if this coefficient is higher than predicted, the molecule is bound to an intracellular component. Conversely, if the movement of the molecule is faster than expected – we are probably observing a reduction of the size, indicating degradation of the molecule.
In this paper, using FCS coupled with the nanoviscosity model, we examined the interactions of the YO-PRO-1 dye in vivo.‡ According to the literature, this positive-charged dye is a molecule that intercalates two adjacent DNA base pairs, which induces fluorescence.14,15 The YO-PRO-1 structure consists of benzoxazole and quinoline rings connected by a cyanine-based linker (Fig. 1).16 In 2014, Sho Fujisawa and others suggested that YO-PRO-1 is a very sensitive marker of early apoptosis.17 The mechanism allowing the dye to enter early apoptotic cells is likely based on the P2X7 receptors’ activation due to the secretion of ATP and UTP molecules outside the cell.18 This process opens up membrane channels that allow the transport of cations (including YO-PRO-1) and larger molecules.
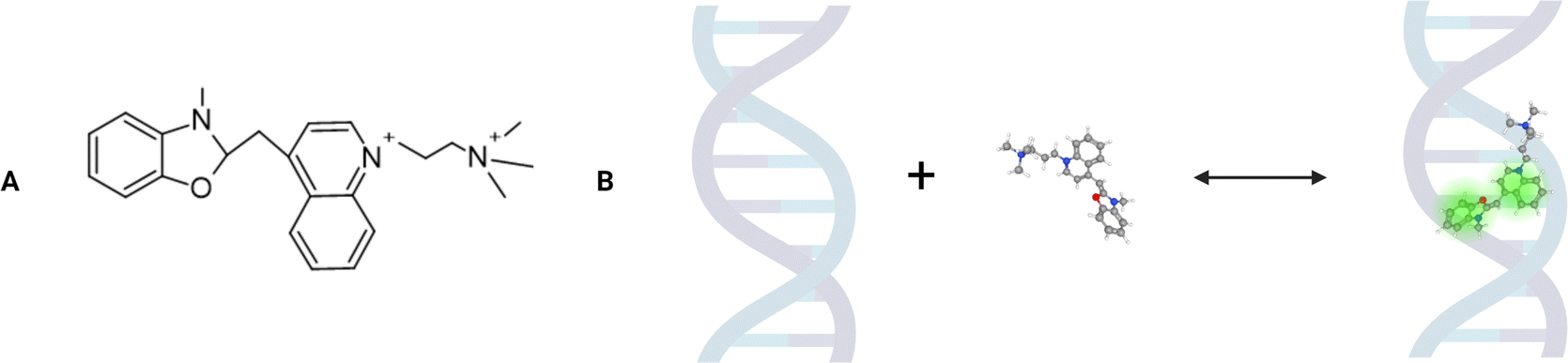 | ||
| Fig. 1 The scheme of the YO-PRO-1 structure and its intercalation reaction into double stranded nucleic acids. (A) Structure of the YO-PRO-1 dye. The image was created with the Chemsketch program. (B) The reaction between YO-PRO-1 (a cyanine-based linker connects benzoxazole and quinoline rings) and a double-stranded DNA fragment. The image was created with the BioRender program and PubChem Compound Summary for CID 6439500, YO-PRO-1. | ||
Contrary to the literature data,19 we demonstrate that the dye also penetrates living cells and forms fluorescent complexes in the cytoplasm and nucleoli. We performed FCS measurements directly inside these two cellular compartments, fitting the data with the model describing free, three-dimensional diffusion of two components, one exhibiting detectable rotations. Following ex vivo controls, we proved that YO-PRO-1 formed complexes with tRNA and ribosomes inside living cells. After identifying YO-PRO-1 targets, we determined the hydrodynamic radius of the large ribosomal subunit based on simultaneous translational and rotational diffusion measurements. It was the first experimental proof of the model presented by Makuch and others20 predicting nanoviscosity for rotational and translational mobility at different length scales in complex liquids. Moreover, we performed measurements on the 40S, 60S, and 80S ribosomes isolated from HeLa cells, proving the reaction between YO-PRO-1 dye and the rRNA. In addition, we calculated that the 60S subunit represented 52% and 75% of the mobile components present in the cytoplasm and cell nucleus, respectively. We also observed the decrease of large ribosomal subunit contribution into the acquired signal in rapamycin-induced inhibition of ribosomes' biosynthesis conditions. We also determined the equilibrium constants between YO-PRO-1 and tRNA, which equals 3.01 ± 1.43 × 105 M−1. Contrary to the literature reports, we defined the actual number of dye molecules interacting with the probe instead of analyzing the fluorescent intensity. Additionally, we investigated the use of YO-PRO-1 as a marker of early apoptosis. Based on the experiments in which we induced the intrinsic and extrinsic apoptosis pathway, we conclude that YO-PRO-1 should not be considered a universal marker of early apoptotic cells. Our research has shown that YO-PRO-1 can be a selective dye for ribosomes and tRNA, making it unique from those commonly used.
Experimental
Reagents
YO-PRO-1 dye was purchased from Invitrogen (United States) and was diluted in phosphate-buffered saline, PBS (pH = 7.4, a phosphate buffer concentration of 0.01 mM, 0.0027 M potassium chloride, and 0.137 M sodium chloride). In ex vivo experiments (outside the cell, with biologically derived components), the buffer was supplemented with 0.002% Tween20 surfactant to avoid absorption of the compounds on the sample well chamber.21We used propidium iodide (Sigma-Aldrich), DAPI (Thermo Fisher Scientific), formaldehyde (Sigma-Aldrich), and Triton X-100 (Thermo Fisher Scientific) in the colocalization experiment. In apoptosis-related experiments, we utilized TRAIL (Sigma-Aldrich) to induce the extrinsic pathway and staurosporine (Selleck Chemicals) for the intrinsic pathway.22 RNA control 250 (Thermo Fisher Scientific, 2000 nt RNA transcript; the sequence is proprietary), hairpins (IBA GmbH; TTT AAT TAA TCG TTG TTA AAA AAA TTT TAA CCC CTT AAA ATT TTT TTA ACA ACG ATT AAT TAA A sequence), single-stranded DNA (IBA GmbH; TAG CAC ATC CGT A sequence), Human Serum Albumin (Sigma-Aldrich), FBS (Gibco), the total pool of RNA (Thermo Fisher Scientific, catalog number: 4307281) and tRNA (Thermo Fisher Scientific, CAS number: 9014-25-9) was used to determine YO-PRO-1 target.
For drug experiments, we used rapamycin (Sigma-Aldrich) and doxorubicin hydrochloride (Sigma-Aldrich).
Cell culture
HeLa cells were purchased from the American Type Culture Collection (ATCC, Manassas, USA) and cultured as a monolayer in standard conditions (37 °C, 5% CO2). The cells were growing in the complete growth medium, based on Dulbecco's modified Eagle's medium (DMEM) with 1 g L−1 of glucose (Institute of Immunology and Experimental Technology, Wrocław, Poland), supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v L-glutamine (Sigma-Aldrich), and 1% v/v solution of streptomycin (10 mg mL−1) (Merck) and penicillin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1) (Sigma-Aldrich). Using regular passages, the cell culture was maintained in a logarithmic growth phase. The passages were conducted utilizing 0.25% Trypsin-EDTA solution (Sigma-Aldrich) to detach cells from the surface. The trypsinization process was controlled by light microscopy.
000 U mL−1) (Sigma-Aldrich). Using regular passages, the cell culture was maintained in a logarithmic growth phase. The passages were conducted utilizing 0.25% Trypsin-EDTA solution (Sigma-Aldrich) to detach cells from the surface. The trypsinization process was controlled by light microscopy.
Cell lysate
Cells detached from the culture surface after passage were centrifuged. The supernatant was removed, and 1 mL of a non-denaturing lysis buffer consisting of 10 mM imidazole, 0.5 M sodium chloride, 1% Triton X-100 (Sigma-Aldrich), 0.2 mM sodium ortho-vanadate, 0.2 mM phenylmethylsulfonyl fluoride was added to the cell pellet. We prepared an analogous buffer enriched with 10 mg mL−1 RNAase (Sigma-Aldrich) to obtain a cell lysate with degraded RNA. In both cases, we incubated the cells with the buffer for 15 minutes.FCS setup
Experiments were performed on an inverted confocal microscope (Nikon EZ-145 C1) integrated with time-correlated single-photon counting (TCSPC) data acquisition system (PicoHarp 300, PicoQuant). All measurements were carried out using Nikon PlanApo 60× water immersion objective (NA = 1.2). The laser diode emitting picosecond pulses with a wavelength of 485 ± 3 nm was used for excitation. The fluorescence signal was recorded by a detector based on a Single Photon Avalanche Diode (SPAD) (PerkinElmer Optoelectronics). The fluorescence was detected through a 488 long-pass filter (Chroma) positioned in the optical path in front of the detector.Data acquisition was performed using Symphotime 64 software (PicoQuant). For temperature control, we used the Okolab Cage Incubation System, which stabilizes the temperature with an accuracy of 0.5 °C.
Confocal setup
We used a Nikon A1 confocal microscope operated by NIS Elements software (Nikon) to visualize the internalization of YO-PRO-1 into cell cultures. YO-PRO-1 dye emits radiation at a wavelength λmax = 491 nm. We exited the tested probe using a 485 nm laser.FCS measurements in a buffer
We used the laser power of 30 μW (measured before the objective by power meter PM400, Thorlabs). At such low laser power, the photobleaching of the fluorophore and the contribution of the triplet state in the acquired fluorescence signal were reduced. At the beginning of every experiment, equipment was calibrated using 5 nM rhodamine 110 (Sigma-Aldrich) dissolved in water, and parameters of focal volume were established. For all measurements, we used the 8 well-chambered cover glass plates (Cellvis). FCS measurements were performed 10 μm above the glass cover at 25 ± 0.5 °C for YO-PRO-1 targets’ determination and 36 ± 0.5 °C for brightness experiments. Acquired data were analyzed by using self-written Python scripts.FCS measurements directly in living cells (in vivo)
We performed intracellular FCS measurements at 36.0 ± 0.5 °C. Before each experiment, we determined the size and shape of the confocal volume by doing the calibration. As a calibration solution, we used rhodamine 110 dissolved in 2.5% glucose in PBS to keep a similar refractive index as the cell interior.23 The laser power was equal to 10 μW. We started cellular measurements by positioning a confocal volume in the cytoplasm or nucleus of the selected cell (approximately 2 μm above the glass surface). Cells were selected based on spindle-shaped morphology. Then, SymphoTime software captured the fluorescence fluctuation record of the YO-PRO-1 dye present in the focal volume. This record was mathematically processed to obtain an autocorrelation curve. Next, autocorrelation curves were fitted with normal diffusion with a rotations model using QuickFit 3.0 software.Cell death observations
Extrinsic apoptosis was induced using human recombinant TRAIL protein (TNF-Related Apoptosis Inducing Ligand), applied at a final concentration of 9 μg mL−1 in a cell culture medium. Intrinsic apoptosis was induced using Staurosporine antibiotic, applied at a final concentration of 2 μM in a cell culture medium. Both reagents were used in concentrations 1000× higher than IC50. HeLa cells were seeded to 8 well-chambered cover glass plates (Cellvis) 24 hours before the experiment. Then, the cell culture medium was replaced with 500 nM YO-PRO-1 in PBS containing magnesium and calcium ions for 30 minutes. Next, TRAIL or staurosporine was added to the medium, and immediate observation started. Cells were imaged using a time-stack mode. Two data channels were acquired: a transmitted light detector (TD) for quasi-brightfield images of cellular morphology and a green fluorescence channel (λex = 485 nm) for YO-PRO-1 fluorescence.Confocal imaging
HeLa cells were seeded to 8 well-chambered cover glass plates (Cellvis) 24 hours before the experiment. After this period, cells were washed with PBS and then fixed with 4% formaldehyde in PBS for 10 minutes. Fixed cells were washed with PBS and then subjected to permeabilization using 0.2% Triton X-100 in PBS for 5 minutes. Permeabilization buffer was washed out with fresh PBS, and samples were ready to be subjected to dyes. YO-PRO-1 was used in the final concentration of 200 nM. Propidium Iodide (PI) was used in a final 100 μg mL−1 concentration. DAPI was used in a final concentration of 300 nM. Depending on the dyes used, two out of three channels were acquired: blue fluorescence channel (λex = 405 nm) for DAPI, green fluorescence channel (λex = 485 nm) for YO-PRO-1, and orange channel (λex = 561 nm) for PI. Images were further processed using ImageJ,24 including JACoP plugin25 and Imaris (Oxford Instruments) software.Treating cells with doxorubicin and rapamycin
We investigated whether the treatment of cells with doxorubicin or rapamycin may translate into changes in LSU radius as well as the abundance of tRNA and LSU in the cell nucleus and cytoplasm. For that, we incubated HeLa cells with 100 nM of doxorubicin or 1 μM of rapamycin for 6 hours. After this time, we removed the tested drug from cells and added 40 nM of YO-PRO-1 for 30 minutes. Then, we performed FCS measurements inside living cells (in vivo) according to the description above.Ribosome purification from HeLa cells
HeLa cells from the ATCC were grown under standard conditions (according to the description in cell culture – Experimental section). Once confluent, they were serum-starved for 6 hours to get a synchronized cell population.Cells were harvested by centrifugation. Pellets were resuspended in lysis buffer containing 15 mM Tris–HCl pH 7.5, 300 mM NaCl, 6 mM MgCl2, with freshly added 0.5% w/v Nonidet P-40 and RNasin® Plus RNase Inhibitor (Promega). Resuspension was incubated 20 minutes on ice. All subsequent steps were performed at 4 °C. Cell debris was removed by centrifugation at 6000g for 1 hour. The supernatant was centrifuged 5.5 hours in T-865 Rotor at 45000 rpm (Ultracentrifuge Sorvall WX Ultra, Thermo Scientific).
Next, the crude ribosome pellet was resuspended in buffer A containing 20 mM Tris–HCl pH 7.5, 50 mM KCl, 4 mM MgCl2, 2 mM DTT, 250 mM sucrose to which a puromycin (Carl Roth) was added to a final concentration of 1 mM. It was incubated for 10 minutes at 0 °C and then for 10 minutes at 37 °C. To separate the ribosomes into subunits, dissociation was used by adding KCl to a final concentration of 500 mM (this step was omitted to obtain whole ribosomes). The solution was then centrifuged in a T-865 rotor at 45000 rpm in an Ultracentrifuge Sorvall WX Ultra (Thermo Scientific) for 4.5 hours, yielding a ribosome precipitate, which was purified of initiating factors and other cellular factors.
The ribosome pellet was resuspended in a small volume of buffer A in case of purification of whole units or in buffer A supplemented with 500 mM KCl in case the aim was to get pure subunits. Suspension was gently loaded onto the prepared early 10–30% sucrose gradient and separated by centrifugation in a buffer A (+500 mM KCl) in a Surespin 630 rotor (Thermo Scientific) at 22000 rpm for 18 hours. Gradients were fractionated in 1-mL fractions, which were analyzed by DeNovix DS-11 Spectrophotometer.
Fractions containing 80S or 40S and 60S folded separately and ultracentrifuged at 100000g in a Thermo Scientific Fiberlite F50L-24 × 1.5 rotor for 2 hours. The pure ribosomal precipitates were resuspended for further analysis in buffer B containing 20 mM Tris–HCl pH 7.5, 2 mM DTT, 2 mM MgCl2, 100 mM KCl, 250 mM sucrose.
Brightness analysis method
The brightness analysis method (BAM) is a quantitative technique established in our group to obtain equilibrium constants of biochemical reactions.9,26–28 The method is based on the change of intrinsic molecular brightness of the reactants in the reaction upon complex formation. Molecular Brightness (MB) is defined as the number of photons emitted by a single fluorophore in a second. In all BAM experiments, the dye was added 2 hours before the measurements at a concentration of 40 nM.We studied the reactions between tRNA and YO-PRO-1 and DNA and YO-PRO-1 (ESI,† S2), performing experiments at 36 °C in a PBS buffer. The reaction formula can be written as:
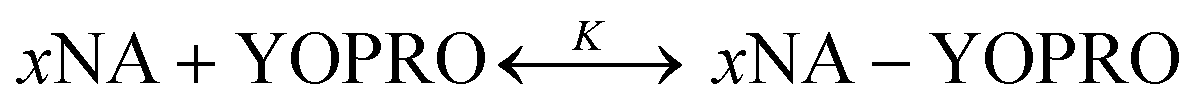 | (1) |
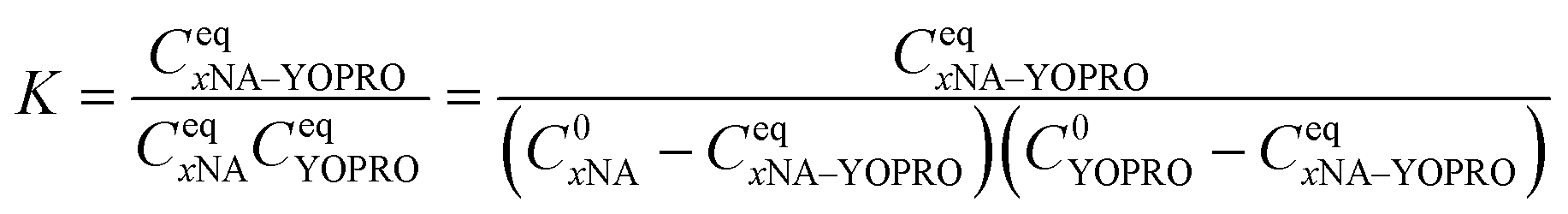 | (2) |
The BAM allows for determining the equilibrium constant. For this purpose, we performed experiments preparing a titration curve, keeping a 40 nM concentration of YO-PRO-1 constant and varying concentrations of biomolecules in ranges 1–100 nM and 1–200 nM for DNA and tRNA, respectively. Every sample was measured in 6 separate 20 s measurements, during which the fluorescence signal was recorded as a fluorescence intensity I. In every experiment, we measured intensity I0 for a free dye eqn (3):
| I0 = V0·NA·CYOPRO·α | (3) |
We recorded fluorescence intensity I1 for all the samples with the dye and biomolecules eqn (4):
| I1 = V0·NA·(CeqYOPRO·α + CeqxNA–YOPRO·γ) | (4) |
:1 – one biomolecule per one complex with the dye.
We solved the quadratic equation derived from eqn (2) to determine the concentration of the complex in an equilibrium state eqn (5):
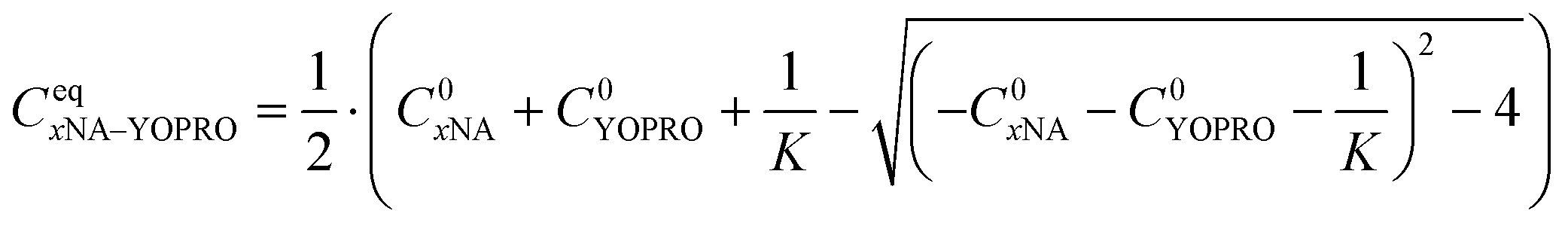 | (5) |
We rewritten eqn (4), incorporating eqn (2):
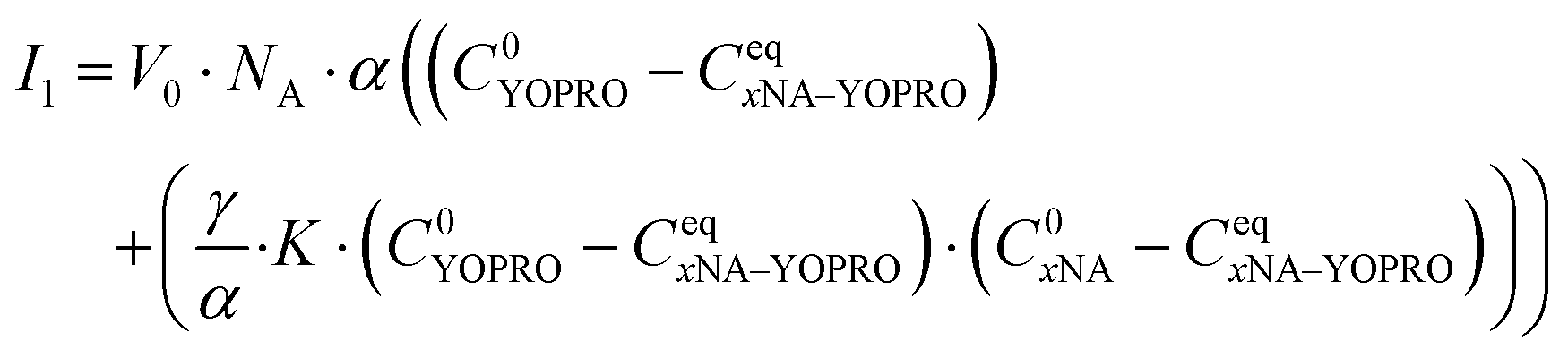 | (6) |
We fitted eqn (6) for every experiment in a self-written Python script.
Results & discussion
Research on YO-PRO-1 targets
According to widespread literature, YO-PRO-1 intercalates between DNA double strands, emitting fluorescence. After incubating HeLa cells with the dye, we observed that positively charged YO-PRO-1 freely penetrates the cells. Our in vivo studies showed that YO-PRO-1 stains structures in the nucleus (nucleoli), and at the same time, a large amount of scattered signal came from the cytoplasm (Fig. 2(A)). Thus, we started testing in a buffer to understand what can be a potential target for YO-PRO-1. As an experimental approach, we expected an increase in the fluorescence intensity in the presence of interaction.28 We investigated different biological molecules, including nucleic acid types (mRNA, DNA hairpins, and ssDNA), proteins, and cell lysate. As a control, we performed an experiment without tested dye for all probes (Table 1).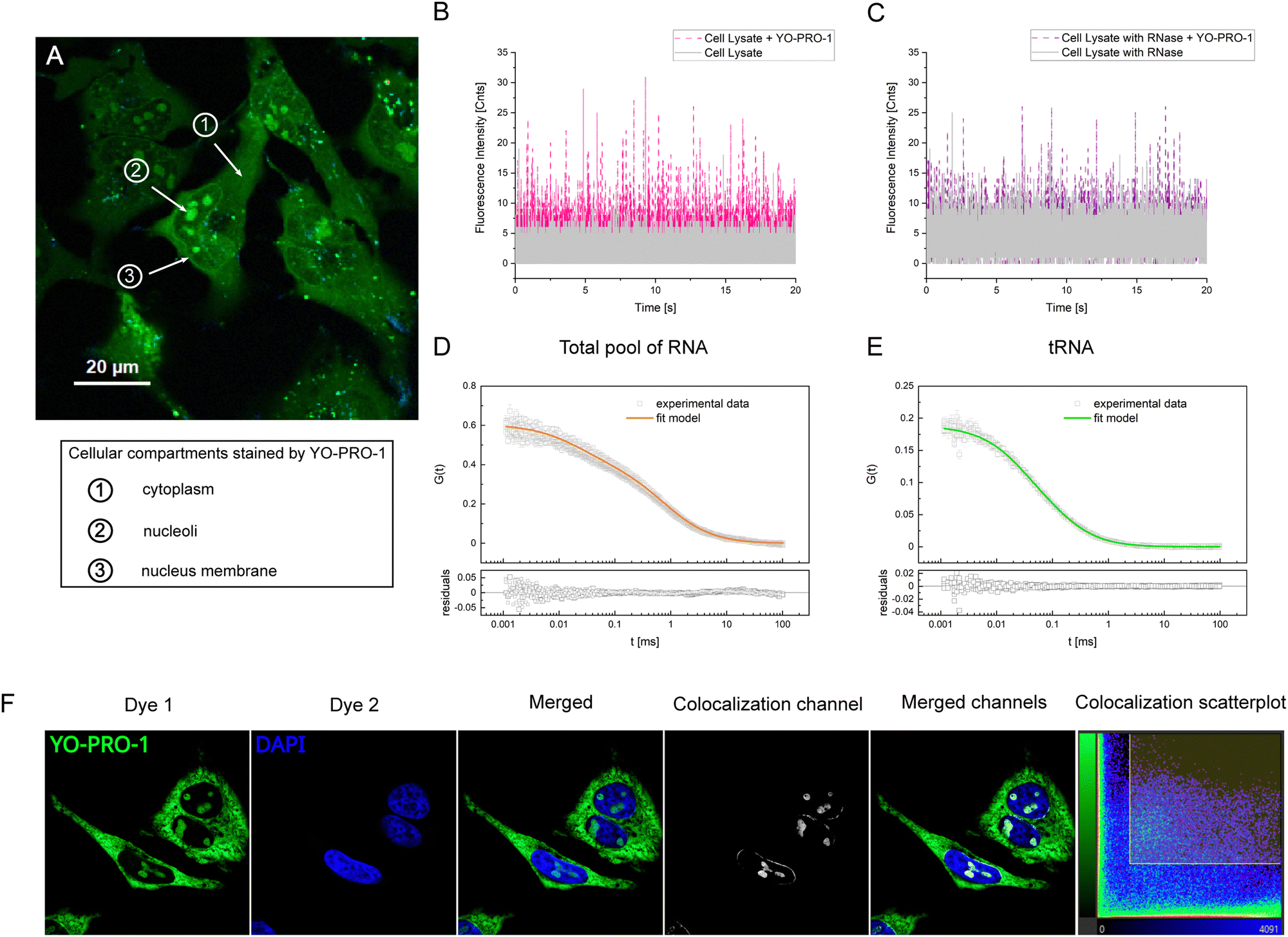 | ||
| Fig. 2 (A) The exemplary confocal image of HeLa cells stained with YO-PRO-1 in vivo. We can distinguish three separate cellular compartments within the cells: cytoplasm, which is dispersal labeled with the dye, nucleoli, and nucleus membrane. (B) Fluorescence intensity dependence on time (time trace graph) for HeLa cells lysate (in vitro experiments). The dye reacts with the components of the lysate as the signal coming from the complex with YO-PRO-1 is visibly higher than cell lysate alone. (C) Fluorescence intensity dependence on time for the YO-PRO-1 dye and HeLa cells lysate treated with RNAse enzyme (in vitro experiments). As in the case of cell lysate without the enzyme, we can see that the recorded fluorescence intensity is bigger for the sample with the dye compared with cell lysate + RNase alone. (D) FCS curve for the reaction between YO-PRO-1 dye and the total pool of RNA in vitro. Two separate fluorescent specimens contribute to the autocorrelation function: one small and one large. Based on the hydrodynamic radius values, we assumed it was tRNA and ribosomes, respectively. (E) FCS autocorrelation function acquired for the reaction between YO-PRO-1 dye and tRNA in vitro. The fitted value of tRNA's hydrodynamic radius equals 2 nm. (F) Fixed and permeabilized HeLa cells stained with two nucleic acid intercalators in vivo. A colocalization channel was created based on thresholds marked at colocalization scatterplots. Scale bars correspond to 10 μm. | ||
| Type of probe | Tested probe | [1] Count rate without dye [cnts per s] | [2] Count rate with dye [cnts per s]a | Ratio [2]/[1] | Binding |
|---|---|---|---|---|---|
| a Unbound YO-PRO-1 dye count rate = 623 cnts per s. | |||||
| Nucleic acid | mRNA | 552 | 657 | 1.19 | No |
| Nucleic acid | Hairpin (DNA) | 410 | 671 | 1.64 | No |
| Nucleic acid | ssDNA | 545 | 644 | 1.18 | No |
| Nucleic acid | dsDNA | 616 | 29117 |
47.27 | |
| Nucleic acid | The total pool of RNA | 2124 | 40348 |
19.00 | Yes |
| Nucleic acid | rRNA | 855 | 19050 |
22.28 | Yes |
| Nucleic acid | Large ribosomal subunit | 824 | 2133 | 2.59 | Yes |
| Nucleic acid | Small ribosomal subunit | 797 | 1585 | 1.99 | Yes |
| Nucleic acid | tRNA | 970 | 63271 |
94.43 | Yes |
| Protein | HSA | 6066 | 6869 | 1.13 | No |
| Protein | FBS | 1301 | 1319 | 1.01 | No |
| Cell interior content | Lysate | 1503 | 2747 | 1.63 | Yes |
| Cell interior content | Lysate after RNAse treatment | 3128 | 3860 | 1.23 | Yes |
We did not notice any significant increase in the detected count rate nor the presence of autocorrelation function for tested nucleic acid molecules: mRNA, hairpin, and single-stranded DNA (ssDNA). For more details, please see ESI,† S1. It proves that there were no fluorescent particles in the focal volume, so YO-PRO-1 did not intercalate between these molecules. YO-PRO-1 intercalates in double-stranded DNA and double-stranded RNA, inserting its flat aromatic part between base pairs of double-stranded fragments. mRNA and ssDNA are types of single-stranded nucleic acids, which explains the lack of interaction with the tested dye. In the case of the hairpin having double-stranded regions, we assume the probable explanation for the lack of reaction is the short half-life of the double-stranded hairpin's regions. We also neglected YO-PRO-1-protein binding in the example of HSA, one of the most abundant proteins in the human body – the addition of the dye did not increase the count rate significantly, meaning there was no reaction between protein and YO-PRO-1. We also excluded the interactions between YO-PRO-1, the cell culture medium components, and fetal bovine serum (FBS) (please see S1, ESI†). No reaction in any investigated cases was observed.
As our previous ex vivo tests did not point to any potential target for YO-PRO-1, we decided to perform an experiment with HeLa cells lysate. Apart from DNA, cell lysates obtained using a non-denaturing buffer (please see Experimental section) include a pool of RNAs: rRNA, tRNA, mRNA, and also mitochondrial RNA. Suspecting that the dye reacts with some type of RNA, as tRNA and rRNA have double-stranded regions in their structures, we added RNAse enzyme to one of the lysate samples. In this way, we could check whether there will be any visible impact of RNAse on the results. We collected the fluorescence intensity graphs for the samples without (Fig. 2(B)) and with the addition of the enzyme (Fig. 2(C)). As expected, the reaction between cell lysate components and YO-PRO-1 occurred. We observed an increase in the fluorescence count rate. We have attempted to conduct FCS measurements, but the autocorrelation curves we observed were significantly stretched due to the presence of many fluorescent species, and they could not be fitted with available models. In the sample with the RNAse enzyme, we noticed a lower increase in the count rate than in the sample without the enzyme (Table 1). The explanation for that might be that the RNAze enzyme cut RNA molecules into shorter fragments, which increased the number of fluorescent species in the focal volume. These results suggest that YO-PRO-1 reacts with some type of RNA.
In the next step, we focused directly on RNA. We measured the total pool of RNA (including mainly tRNA, rRNA, and mRNA) with YO-PRO-1. The presence of the FCS autocorrelation function (Fig. 2(D)) proved the dye's affinity to RNA molecules. We fitted the data, obtaining two separate YO-PRO-1 targets: tRNA and ribosomes (rRNA). Due to the presence of fluorescent species with various sizes in a sample, we got averaged hydrodynamic radiuses (21 ± 2 nm for ribosomes and 2 nm for tRNA). Then, we performed an experiment with tRNA only, confirming the reaction between tRNA and YO-PRO-1. The obtained hydrodynamic radius from fitting the FCS curve (Fig. 2(E)) was equal to 2 nm. The interaction between the tested dye and ribosomes was confirmed by isolating 80S, 60S, and 40S ribosomes from living HeLa cells (see Experimental section and S8, ESI†). The lack of the reaction between YO-PRO-1 and mRNA was excluded previously. The count rate values are collected in Table 1.
The equilibrium constant of YO-PRO-1 with tRNA
YO-PRO-1 dye is an intercalator – it inserts the planar aromatic moiety between two adjacent base pairs of double-stranded DNA and RNA, as we proved above. Most quantitative studies applying YO-PRO-1 are based on the comparison of intensity analysis, omitting precise determination of the actual number of dye molecules interacting with the tested probe. Here, we investigated YO-PRO-1 interactions with tRNA using the brightness analysis method (BAM).9,26–28We performed a series of fluorescence titration experiments for tRNA and YO-PRO-1 reactions in a nanomolar range of concentrations. In every experiment, we determined molecular brightnesses of the free dye α and formed complex γ. The ratio of averaged γ to averaged α equals 195. For comparison, for DNA, this ratio was 64 (please see ESI,† S2). Such a difference in γ value between the YO-PRO-1-tRNA complex and YO-PRO-1-DNA may be associated with the rotations. Rotations increase the probability of excitation and fluorescence emission events due to a higher chance for the dye to be oriented parallel to the excitation beam vector. Thus, the higher number of photons is coming from a single molecule in a given time unit (precise calculations for YO-PRO-1 stained tRNA are presented in Table S6, ESI†).
The calculated equilibrium constant was defined for the concentration of binding sites, applying the size of a binding site as 5 base pairs for a single YO-PRO-1 molecule.29 Considering the number of base pairs involved in a tRNA double-strand sequence (see S6, ESI†), the binding sites for the tested dye on one tRNA molecule equals ∼4. The exemplary curve fitted with the brightness model is shown in Fig. 3. Averaged equilibrium constant of the reaction between tRNA and YO-PRO-1 equals 3.01 ± 1.43 × 105 M−1. The equilibrium constant obtained for the complex formation between DNA and YO-PRO-1 equals 7.48 ± 1.95 × 105 M−1 (for further data, see S2, ESI†). Despite differences in structures, charges, and sugars in the backbone, equilibrium constants are in the same order. Thus, we assume the equilibrium constant for the reaction between YO-PRO-1 and LSU is in the same range.
 | ||
| Fig. 3 Equilibrium constant determination of YO-PRO-1-tRNA complex formation (in vitro experiments). The dotted line corresponds to the fit of eqn (6). CYO-PRO-1 is a fitted value of initial dye concentration, K stands for the equilibrium constant, α is the molecular brightness for a free dye, and γ – the molecular brightness of the formed complex. | ||
Subcellular localization of YO-PRO-1
YO-PRO-1 up-taken to untreated, viable cells was accumulated in three main locations: (1) nucleoli, (2) nuclear membrane, and (3) dispersed in the cytosol (Fig. 2(A)). This observation is counter-intuitive, as YO-PRO-1 is widely described as nucleic acid stain, that intercalates double-helix structures,17,30 which suggests accumulation in the cell nucleus, where double-stranded DNA is mostly located. We confronted this observation with another nucleic acid intercalator – DAPI. Fixed and permeabilized HeLa cell samples were exposed to these dyes, and the result is presented in Fig. 2(F). It was observed that only DAPI is selective to the DNA localized in the cell nucleus, whereas YO-PRO-1 stains both nuclear and cytoplasmic structures. In the nucleus, YO-PRO-1 localizes mostly in nucleoli, while DAPI is spread through the whole nucleus area. Moreover, the nuclear envelope could have been distinguished in the YO-PRO-1 micrograph. Qualitative colocalization analysis revealed almost no colocalization of YO-PRO-1 and DAPI-stained cellular regions. These observations were confirmed by quantitative assessment of Pearson's correlation coefficient (p, please see S3, ESI†). DAPI seems selective to DNA, as it cannot be observed in the cytoplasm, while YO-PRO-1 stains both DNA and RNA. Another question is why YO-PRO-1 does not colocalize with DAPI in the regions where DNA is localized (p < 0, see Table S1, ESI†), while YO-PRO-1 binding to DNA in a buffer was confirmed (see section Research on YO-PRO-1 targets). The most probable explanation is that DNA fragments in the buffer have a different structure than chromatin, where some regions are in the form of nucleosomes while others are processed by proteins, causing decreased double-strand accessibility. Moreover, assuming that protein-encoding DNA (2% of the whole genome) is an active part of DNA, accessible for intercalators to bind, we obtained a number of potential binding sites 2.5 × 107 binding sites per cell. In comparison, the number of available binding sites for YO-PRO-1 dye on LSU and tRNA is, respectively, ∼10 times and 3.5 times higher. That indicates that dye might be already bound to RNA structures before reaching nuclear DNA.FCS revealed rotations of ribosomal large subunits (LSU) in both nucleus and cytoplasm
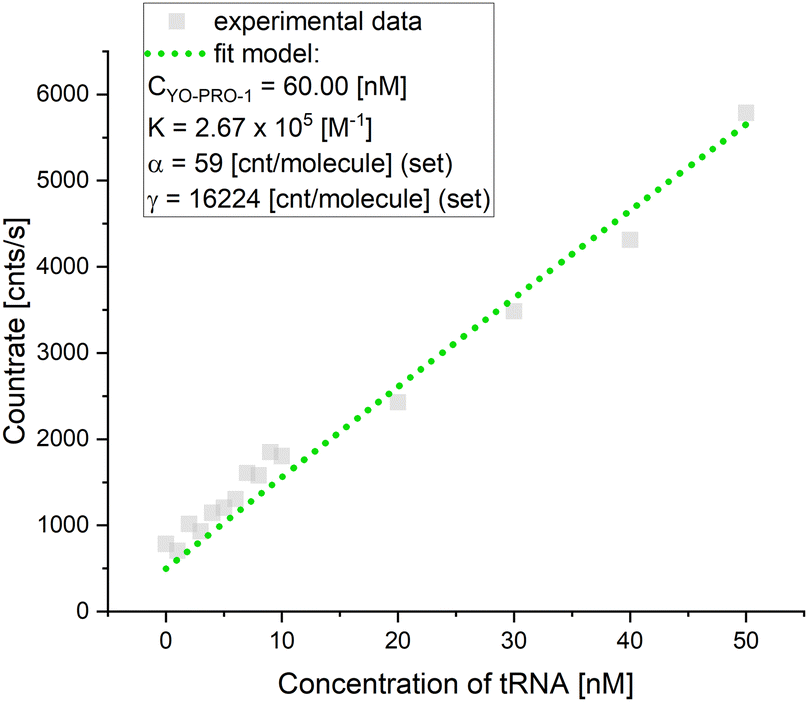
The affinity of YO-PRO-1 to certain intracellular structures was analyzed by FCS, which is used to measure diffusion coefficients of fluorescent molecules. Importantly, the diffusion, and thus FCS results, directly depends on the size of the tested molecule. This is particularly visible in the rotational diffusion.31 Diffusion coefficients measured in the cell interior can be further analyzed using a length-scale dependent viscosity model32,33 to calculate the sizes of the diffusing objects. Consequently, size-based identification of intracellular species is possible. In our previous work, we developed detailed length-scale viscosity curves for HeLa cell cytoplasm,10,12 and nucleus11 (see S4, ESI† for details) as well as the cytoplasm of MDA-MB-231 cells.9 We use this model to identify and measure concentrations of intracellular species.9,13First, FCS curves were acquired in the cytoplasm and nuclei of YO-PRO-1 stained living HeLa cells. Detection spots were chosen randomly, except for areas of the highest brightness (Fig. 4(A)). FCS curves acquired in the nucleus and cytoplasm had similar shapes, with distinct two parts (Fig. 4(A)). Part of the autocorrelation curve at 0.1–10 ms is expected for translational diffusion of intracellular species.10 On the other hand, a clear FCS signal present in times shorter than 10 μs suggested detectable rotations of the registered objects.34,35 The experimental data was fitted with the 2-component free diffusion model with rotational diffusion term (eqn (S12), ESI†). Example fit is presented in Fig. 4(B). Number of fitting parameters was reduced to (1) radius of the rotating object, and (2) relative concentrations (amplitudes) of the registered species. The rest of the model parameters were fixed, with respect to the following assumptions: (a) one of the components were tRNAs, of the radii rp = 2 nm, as measured in the buffer (see section Research on YO-PRO-1 targets), rotations of tRNAs were too fast to be observed with the used equipment (characteristic time for rotations ∼7 ns); (b) amplitudes of the rotational term were fixed according to the theoretical model;35 and (c) nanoviscosity sensed by the diffusing objects followed length-scale dependent viscosity model for cytoplasm and nucleus of HeLa cells. Previously, it was theoretically proved that molecules in complex liquids sense different nanoviscosities for their rotational and translational diffusion.20 The values of the nanoviscosities: ηrot (for rotational diffusion) and ηtrans (for translational diffusion) depend on rp and can be calculated using eqn (S11) (ESI†) (see S5, ESI† for more details). Although the authors of the original paper provided theoretical evidence for ηtrans(rp) ∼ ηrot(rp), no experimental proof has been published so far. Here, we show the applicability of the formula in living cells: rotational and translational parts of the FCS curves fitted well, both in the nucleus and the cytoplasm (see S5, ESI†).
 | ||
| Fig. 4 (A) Example measurement spots and autocorrelation curves acquired in HeLa cytoplasm and nucleus (in vivo). (B) 2-component free diffusion model with rotations fitted to experimental data from in vivo experiments. Parts of the curve corresponding to rotational and translational diffusion are marked with dotted rectangles. (C) Schematic view of types of rRNAs and tRNAs present in cells and stained with YO-PRO-1. Insert graph presents relative brightness and mobility of the species, justifying their impact on the FCS autocorrelation curve – presence in the ‘FCS detection window’ (see S6, ESI†). Due to the binding between mRNA (∼100 nm in diameter itself9) and a ribosome, a transcripting ribosome is non-detectable in timescales for FCS as well as nucleolus (from 500 nm in diameter36). | ||
Data fitting resulted in the conclusion that two components are detectable by FCS in YO-PRO-1 stained cells. The first component was initially identified as tRNA, the most abundant RNA molecule in the cell (including the nucleus37),38 possessing conserved double-strand domains. The size of tRNA was set to 2 nm based on in vitro experiments. The second component, the bigger one, was measured to have a hydrodynamic radius of 14.6 ± 1.7 nm in the cytoplasm (14.8 ± 1.0 nm in the nucleus). This component diffused and rotated freely in both examined compartments. We stated that the second diffusing component was the ribosomal large subunit (LSU), which is the biggest, freely diffusing structure containing rRNA and present both in the nucleus and cytosol39 (80S ribosomes are not freely diffusing particles, as they are assembled on mRNA molecules, and form translation complexes40 – for more details see S6, ESI†). To confirm our assumption, we performed ex vivo experiments based on isolating LSU and SSU from HeLa living cells (see Experimental section). Sizes of isolated ribosome subunits in a buffer and living human cells were in perfect agreement (see S8, ESI†). In addition to this, we analyzed crystallography structures of pre-60S ribosomal subunits,39 in terms of their expected hydrodynamic radii. We also calculated the expected hydrodynamic radii of the LSU using formulas provided by Kalwarczyk and others.41 The calculated values of rLSU varied between 10 and 17 nm (see S5, ESI†). In the literature, we only found measurements of rLSU from the species of Artemia, and it was equal to 13.3 ± 0.3 nm, measured in a buffer.42 Following knowledge on the evolution of ribosomes,43 we can expect human LSU to be bigger than nonvertebrate one. Thus, after the comparison with the radius fitted from the ex vivo experiment (15.0 ± 2.7 nm), we state that the objects of rp = 14.6 (14.8) nm were free large ribosomal subunits (or their precursors in the nucleus). The effect of the crowded cell interior on the diffusion measurements was taken into account through the effective viscosity parameters (for more details, please see ESI,† S5). The selectivity of YO-PRO-1 binding to nucleic acids also excluded the possible influence of other cell components on the obtained FCS signal. Visible in confocal imaging, the YO-PRO-1-stained nuclear membrane is the result of dotting the outer layer of the membrane with ribosomes.44
FCS curves acquired in YO-PRO-1 stained MDA-MB-231 cells showed the same shape as in HeLa cells. For fitting the autocorrelation curves from the nuclei, we fixed the value of hydrodynamic radii of tRNA and LSU. We achieved perfect agreement as compared with the nuclei of the HeLa cells (see Fig. S7, ESI†). In the case of the cytoplasm, fitting resulted in slightly larger radii of LSU (17.1 ± 1.9 nm). The possible explanation can be a relatively large uncertainty of the MDA-MB-231 viscosity curve for the probes larger than 10 nm9 – the theoretical model based on which we determine the size of objects diffusing into the cytoplasm. Thus, the obtained slight difference in LSU size is within the error.
The remaining question was why other nucleic acid-based molecules are not detectable with FCS. However, chromatin chains should be considered immobile in timescales of the FCS experiment (seconds). According to RNAs, three main types must be taken into account (due to their abundance and existence of double-stranded domains): mRNA, tRNA, and rRNA. Results of our buffer experiments confirmed YO-PRO-1 binding to tRNA, while excluded binding to mRNA molecules (see section Research on YO-PRO-1 targets). Thus, the remaining rRNAs and tRNA were discussed in terms of their relative brightness and mobility in S6 (ESI†). In brief, the molecule contributes to fluorescence fluctuations (base of FCS) if they are mobile and exhibit enough brightness (Fig. 4(C)).45 Number of molecules detectable by FCS in our experiments was ∼500 times lower than expected abundance of rRNA and tRNA, thus we assumed single-molecule staining of each particle (S6, ESI†). However, possible brightness of each molecule can also rely on its rotation frequency, and this value is three orders of magnitude higher for tRNA than ribosomal subunits (see Table S5, ESI†). Additional factor to be considered is the probability of binding of a single YO-PRO-1 molecule to the certain RNA type, calculated as an abundance of accessible base pairs. Assuming 1:1:10 ratio of numbers of molecules of LSU:SSU:tRNA, we can expect this probability to be 0.45:0.14:0.41, respectively (see S6, ESI†). Bringing these estimations together, we displayed RNA molecules in Fig. 4(C), with respect to their contribution to FCS curves.
The measured FCS amplitudes in HeLa cells (proportional to the abundance of stained molecules) of LSU:tRNA were 0.59:0.41 in the cytoplasm and 0.58:0.42 in the nucleus. Lack of the signal resulting from SSU is most probably attributed to higher than 1:1 ratio of freely diffusing LSU:SSU (see simulations in Fig. S9, ESI†). This observation is consistent with a known mechanism of translation initiation,46 where small ribosomal subunits are the first to be recruited to the mRNA molecules, and spend considerably longer time in a bound state than large ribosomal subunits.
The experiments were performed on the population of cells at different phases of the cell cycle. To address the influence of cell-cycle phase impact on LSU:tRNA ratio, we synchronized HeLa cells with Aphidicolin (see SI13, ESI†).47 We observed small, but significant changes in the LSU:tRNA ratio, both in cytoplasm and the nucleus. However, there was a more significant difference between unsynchronized and synchronized cells, suggesting that cell cycle arrest influences the measured parameters (S13, ESI†).
Increased contribution of tRNA in rapamycin-treated cells
We investigated the abundance of stained molecules in conditions of ribosome biosynthesis inhibition. To inhibit ribosome biosynthesis, we used rapamycin.48 We performed FCS measurements inside HeLa cells after drug treatment, both in cytoplasm and nucleus, and determined the abundance of stained molecules. The abundance ratio in the control cells (stained with YO-PRO-1) was 1:0.6 (LSU:tRNA). After the treatment, we observed increased contribution of tRNA, both in cytoplasm and nucleus, resulting in the abundance ratio 1:1 (Fig. 5(B)). We also performed a similar experiment, using doxorubicin as an inhibition-causing reagent,49 however, we did not observe any changes (for more information see S9, ESI†).
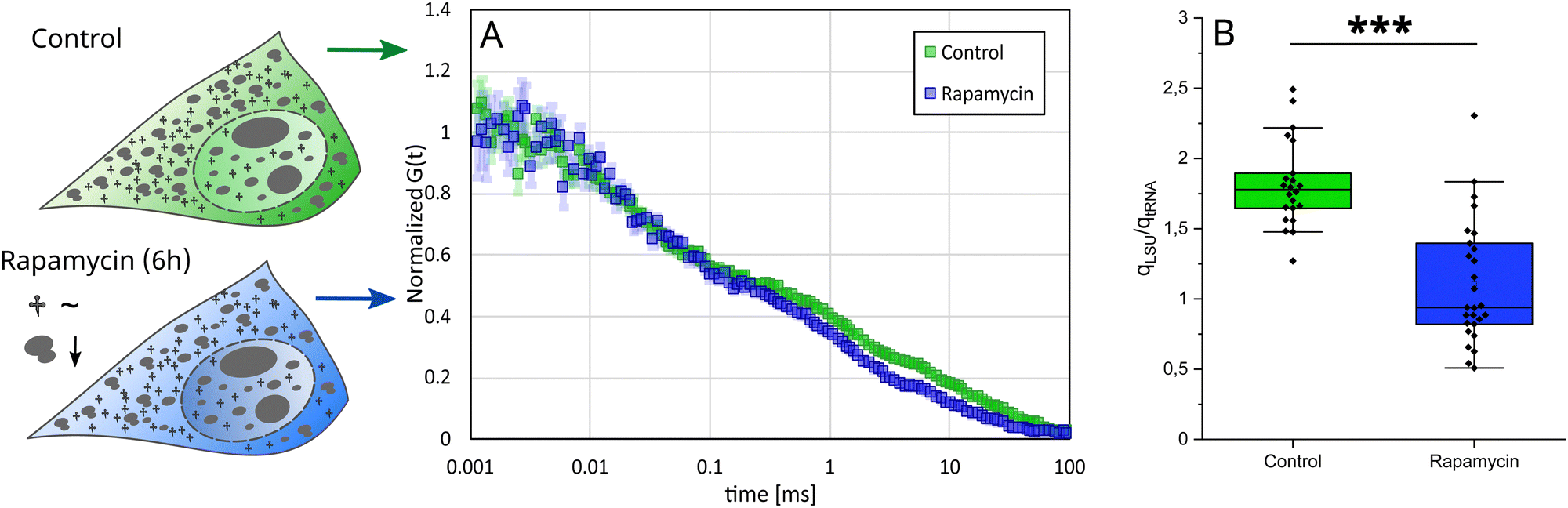 | ||
| Fig. 5 (A) HeLa cells were exposed to rapamycin for 6 h and then stained with YO-PRO-1 (in vivo experiments). FCS curves obtained in the cytoplasm of those cells differed in the translational diffusion part compared to control cells. (B) Ratios of qLSU (fraction of 60S ribosomes) and qtRNA (fraction of tRNA) were calculated for control and rapamycin-treated cells. The difference between these two populations of cells was significant (p < 0.0005). | ||
The obtained difference between rapamycin and doxorubicin lies in the mechanism of their influence on ribosomes. Rapamycin is an inhibitor of the Target of Rapamycin (TOR) pathway. TOR pathways are identified to regulate ribosome production, adjusting ribosomal content based on nutrient availability and environmental conditions.50 Therefore, we can expect a quick response in the number of ribosomes upon rapamycin treatment. Conversely, doxorubicin disrupts ribosome biosynthesis through a combination of transcriptional repression and structural degradation of ribosomal proteins. Doxorubicin blocks ribosomal RNA transcription and simultaneously inhibits the formation of the 80S ribosome complex. In our study, we observe only 60S ribosomes, not assembled into the 80S ribosomes. Therefore, the reduction in ribosome number that occurred in doxorubicin-treated cells was most probably compensated by the increased fraction of free 60S ribosomes. This can be visible in Fig. S15 (ESI†), where slight disturbances in tRNA and rRNA fractions are visible.
Extrinsic apoptosis does not increase YO-PRO-1 uptake
After incubation HeLa cells with YO-PRO-1, we observed that the dye cellular uptake is not the same for all cells. Uneven staining of native cell populations raised a question on the influence of apoptosis on cell membrane permeability to YO-PRO-1. Two scenarios were analyzed: intrinsic and extrinsic apoptosis. Intrinsic apoptosis was induced by staurosporine,51,52 and extrinsic apoptosis was induced by TRAIL peptide.53 The experiment was designed to capture the process of apoptosis in real time by time-lapse microscopy. Cell populations were exposed to 0.5 μM YO-PRO-1 for 30 minutes, and after that, a drug – apoptosis-inducing agent was added to the culture media to the final concentration of 3 orders of magnitude higher than IC50. Image recording started immediately after adding the drug.Cells exposed to staurosporine (intrinsic apoptosis pathway) show an increase in cell membrane permeability during the first 20 minutes of the experiment (Fig. 6). Cells, initially unevenly stained with YO-PRO-1, after exposure to 2 μM staurosporine gradually increased uptake and reached saturation within the first hour. Simultaneous observation of a transmitted light channel did not show any morphological changes in the cells. The experiment was continued for another 2 hours, and no further changes in either fluorescence or morphology were observed. This result is consistent with literature data, claiming YO-PRO-1 as a marker of early apoptotic cells.54,55 Staurosporine, which is supposed to induce apoptosis, simultaneously triggered increased YO-PRO-1 uptake. These two processes, however, are largely separated in time.
 | ||
| Fig. 6 Time-lapse microscopy of HeLa cells exposed to apoptosis-inducing agents: staurosporine (intrinsic) and TRAIL (extrinsic). Transmitted light images represent cell morphology, while fluorescence represents YO-PRO-1 bound to intracellular nucleic acids (λex = 485 nm). Staurosporine induces uniform uptake of YO-PRO-1 into cells. On the other hand, cells treated with TRAIL do not change their fluorescence, and cells stained with the dye from the very beginning (green asterisks) remain viable, while cells with a lower amount of YO-PRO-1 undergo apoptotic blebbing (red asterisk). Scale bars correspond to 20 μm. | ||
Different cellular behavior was observed for the extrinsic apoptosis pathway. Cells exposed to 9 μg mL−1 TRAIL did not show higher permeability to YO-PRO-1 within the first hour after exposure (Fig. 6). On the other hand, this time was sufficient for the first morphological symptoms of apoptosis to be visible – cells shrinked and blebbed after 40 minutes (marked with the red asterisk in Fig. 6). What is significant – apoptotic cell did not show increased YO-PRO-1 uptake. On the contrary, cells with spontaneous higher amount of uptake YO-PRO-1 did not undergo visible apoptosis (marked with green asterisks in Fig. 6).
Our observations raise a question about the usability of a YO-PRO-1 dye as an ‘early apoptotic marker’.54,55 It can be helpful in applications where the possible apoptosis pathway is well described, and a linkage between increased membrane permeability and subsequent apoptosis is certain. However, our TRAIL experiment showed that (1) YO-PRO-1 positive cells are not always on their way to apoptosis, and (2) ongoing apoptosis is not always linked with increased YO-PRO-1 uptake. Thus, we state that YO-PRO-1 should not be claimed as a universal early apoptotic cell marker.
Conclusions
In this study, we used YO-PRO-1 dye to specifically stain ribosomal RNA and transfer RNA inside living human cells at the level of both single-molecule and single-cell. YO-PRO-1 is an intercalator that can insert itself between two adjacent DNA or RNA base pairs. The equilibrium constant of YO-PRO-1 for DNA and tRNA is 7.48 ± 1.95 × 105 M−1 and 3.01 ± 1.43 × 105 M−1, respectively. Previous literature suggests that YO-PRO-1 is impermeable to cells with intact cell membranes.19 In Sho Fujisawa and others' experiments, very few HeLa cells with functional mitochondria were stained with YO-PRO-1.17 However, we show that it passively penetrated living HeLa and MDA-MB-231 cells, staining the cytoplasm, nucleoli, and nuclear membrane. To determine the targets of YO-PRO-1, we conducted ex vivo measurements, considering nucleic acids, ribosome subunits isolated from HeLa cells, proteins, serum, and cell lysate as potential YO-PRO-1 targets. The results of the FCS and BAM method revealed that YO-PRO-1 intercalation was detected in tRNA- and rRNA-consisting samples (see Table 1 and S8, ESI†).Narrowing targets for YO-PRO-1 ex vivo, we conducted quantitative FCS measurements inside YO-PRO-1 stained HeLa and MDA-MB-231 cells. We measured the diffusion coefficients of the components present inside cells. Upon in-depth analysis, we discovered that tRNA molecules and the 60S large ribosomal subunit (LSU) formed complexes with the tested dye both in the cytoplasm and nucleus. Through the FCS technique, we were able to measure the size of the LSU: its hydrodynamic radius in HeLa was equal to 14.6 ± 1.7 nm in the cytoplasm (14.8 ± 1.0 nm in the nucleus), while measured values in MDA-MB-231 were 17.1 ± 1.9 nm in the cytoplasm (14.8 nm in the nucleus). The size of LSU obtained inside living human cells was in agreement with the ex vivo results. The hydrodynamic radius of LSU isolated from HeLa cells was 15.0 ± 2.7 nm (see S8, ESI†). This research is the first to accurately determine the size of 60S ribosomes in human cells. Furthermore, we were able to define the abundance ratio between tRNA and freely diffusing large ribosomal subunits, which was 0.59:0.41 in the HeLa cytoplasm and 0.58:0.42 in the HeLa nucleus. Moreover, we have shown that this ratio is altered in rapamycin-treated cells.
In summary, through the FCS technique, we were able to observe the rotational diffusion of ribosomes. Importantly, we were also able to measure the size of LSU directly in living mammalian cells with unaltered metabolism, which was beyond the limits of alternative techniques so far. To validate our findings, we conducted a buffer experiment and found perfect agreement in the size of LSU ex vivo and in vivo. Recent literature reports9,56,57 confirm the growing interest in methods that investigate the molecules' movement inside cells, for example, to understand the interactions occurring in cells and learn more about them.
We state that YO-PRO-1-based staining of ribosomes can provide new opportunities for biological studies.
Author contributions
Aneta Karpińska: conceptualization, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Karolina Kucharska: conceptualization, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Tomasz Kalwarczyk: formal analysis, methodology, software, writing – review & editing; Patrycja Haniewicz: investigation, methodology, writing – review & editing; Karina Kwapiszewska: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review & editing; Robert Hołyst: conceptualization, methodology, project administration, supervision, validation, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI† and will be available at RepOD.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland, within the grant OPUS UMO-2019/33/B/ST4/00557. We acknowledge the Centre of New Technologies, University of Warsaw, Poland for providing access to an ultracentrifuge.Notes and references
- B. J. Zeskind, C. D. Jordan, W. Timp, L. Trapani, G. Waller, V. Horodincu, D. J. Ehrlich and P. Matsudaira, Nat. Methods, 2007, 4, 567–569 CrossRef PubMed.
- F. X. Theillet, A. Binolfi, T. Frembgen-Kesner, K. Hingorani, M. Sarkar, C. Kyne, C. Li, P. B. Crowley, L. Gierasch, G. J. Pielak, A. H. Elcock, A. Gershenson and P. Selenko, Chem. Rev., 2014, 114, 6661–6714 CrossRef PubMed.
- I. Conlon and M. Raff, J. Biol., 2003, 2, 1–10 CrossRef PubMed.
- J. Pelletier, G. Thomas and S. Volarevic, Nature, 2018, 18, 51–63 CAS.
- Z. Han, Q. Zhang, Y. Zhu, J. Chen and W. Li, Stem Cells Int., 2020, 2020, 1–12 Search PubMed.
- J. Yin, S. Ibrahim, F. Petersen and X. Yu, Front. Aging Neurosci., 2021, 13, 1–12 Search PubMed.
- G. Yusupova and M. Yusupov, Philos. Trans. R. Soc., B, 2017, 372, 20160184 CrossRef PubMed.
- D. N. Wilson and J. H. Doudna Cate, Cold Spring Harb. Perspect. Biol., 2012, 4, a011536 Search PubMed.
- A. Karpińska, M. Pilz, J. Buczkowska, P. J. Żuk, K. Kucharska, G. Magiera, K. Kwapiszewska and R. Hołyst, Analyst, 2021, 146, 7131–7143 RSC.
- K. Kwapiszewska, T. Kalwarczyk, B. Michalska, K. Szczepański, J. Szymański, P. Patalas-Krawczyk, T. Andryszewski, M. Iwan, J. Duszyński and R. Hołyst, Sci. Rep., 2019, 9, 1–9 CrossRef.
- G. Bubak, K. Kwapiszewska, T. Kalwarczyk, K. Bielec, T. Andryszewski, M. Iwan, S. Bubak and R. Hołyst, J. Phys. Chem. Lett., 2021, 12, 294–301 CrossRef PubMed.
- K. Kwapiszewska, K. Szczepański, T. Kalwarczyk, B. Michalska, P. Patalas-Krawczyk, T. Szymański, J. Andryszewski, M. Iwan, J. Duszyński and R. Hołyst, J. Phys. Chem. Lett., 2020, 11, 6914–6920 CrossRef PubMed.
- N. Kleczewska, P. J. Sikorski, Z. Warminska, L. Markiewicz, R. Kasprzyk, N. Baran, K. Kwapiszewska, A. Karpinska, J. Michalski, R. Holyst, J. Kowalska and J. Jemielity, Chem. Sci., 2021, 12, 10242–10251 RSC.
- D. Marie, D. Vaulot and F. Partensky, Appl. Environ. Microbiol., 1996, 62, 1649–1655 CrossRef PubMed.
- T. L. Netzel, K. Nafisi, M. Zhao, J. R. Lenhard and I. Johnson, J. Phys. Chem., 1995, 99, 17936–17947 CrossRef.
- C. Jin, D. Cerutti and R. I. Cukier, J. Phys. Chem. B, 2017, 121, 10242–10248 CrossRef PubMed.
- S. Fujisawa, Y. Romin, A. Barlas, L. M. Petrovic, M. Turkekul, N. Fan, K. Xu, A. R. Garcia, S. Monette, D. S. Klimstra, J. P. Erinjeri, S. B. Solomon, K. Manova-Todorova and C. T. Sofocleous, Cytotechnology, 2014, 66, 259–273 CrossRef PubMed.
- C. Virginio, A. MacKenzie, R. A. North and A. Surprenant, J. Physiol., 1999, 519, 335–346 CrossRef PubMed.
- T. Idziorek, J. Estaquier, F. De Bels and J. C. Ameisen, J. Immunol. Methods, 1995, 185, 249–258 CrossRef PubMed.
- K. Makuch, R. Hołyst, T. Kalwarczyk, P. Garstecki and J. F. Brady, Soft Matter, 2020, 16, 114–124 RSC.
- K. Bielec, K. Sozanski, M. Seynen, Z. Dziekan, P. R. Ten Wolde and R. Holyst, Phys. Chem. Chem. Phys., 2019, 21, 10798–10807 RSC.
- N. J. Ernest, C. W. Habela and H. Sontheimer, J. Cell Sci., 2008, 121, 290–297 CrossRef PubMed.
- T. Kalwarczyk, K. Kwapiszewska, K. Szczepanski, K. Sozanski, J. Szymanski, B. Michalska, P. Patalas-Krawczyk, J. Duszynski and R. Holyst, J. Phys. Chem. B, 2017, 121, 9831–9837 CrossRef PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef PubMed.
- S. Bolte and F. P. Cordelières, J. Microsc., 2006, 224, 213–232 CrossRef.
- K. Bielec, G. Bubak, T. Kalwarczyk and R. Holyst, J. Phys. Chem. B, 2020, 124, 1941–1948 CrossRef.
- Y. Zhou, K. Bielec, P. Pasitsuparoad and R. Hołyst, Analyst, 2020, 145, 6600–6606 RSC.
- K. Kucharska, M. Pilz, K. Bielec, T. Kalwarczyk, P. Kuźma and R. Hołyst, Molecules, 2021, 26, 1–8 Search PubMed.
- A. Larsson, C. Carlsson, M. Jonsson and B. Albinssont, J. Am. Chem. Soc., 1994, 116, 8459–8465 CrossRef CAS.
- C. Flors, Photochem. Photobiol. Sci., 2010, 9, 643–648 CrossRef CAS.
- J. Michalski, T. Kalwarczyk, K. Kwapiszewska, J. Enderlein, A. Poniewierski, A. Karpińska, K. Kucharska and R. Hołyst, Soft Matter, 2024, 20, 5810–5821 RSC.
- T. Kalwarczyk, N. Ziębacz, A. Bielejewska, E. Zaboklicka, K. Koynov, J. Szymański, A. Wilk, A. Patkowski, J. Gapiński, H.-J. Butt and R. Hołyst, Nano Lett., 2011, 11, 2157–2163 CrossRef CAS.
- M. Tabaka, T. Kalwarczyk, J. Szymanski, S. Hou and R. Holyst, Front. Phys., 2014, 2, 1–14 Search PubMed.
- M. Oura, J. Yamamoto, H. Ishikawa, S. Mikuni, R. Fukushima and M. Kinjo, Sci. Rep., 2016, 6, 1–7 CrossRef.
- S. R. Aragón and R. Pecora, Biopolymers, 1975, 14, 119–137 CrossRef.
- D. A. Jackson, in Meyers: Encyclopedia of Molecular Cell Biology and Molecular Medicine: Epigenetic Regulation and Epigenomics, ed. R. A. Meyers, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2011 Search PubMed.
- K. Chatterjeea, R. T. Nostramo, Y. Wan and A. K. Hopper, Biochim. Biophys. Acta, 2018, 1861, 373–386 CrossRef.
- S. Kirchner and Z. Ignatova, Nat. Rev. Genet., 2015, 16, 98–112 CrossRef.
- X. Liang, M. Q. Zuo, Y. Zhang, N. Li, C. Ma, M. Q. Dong and N. Gao, Nat. Commun., 2020, 11, 1–14 CrossRef.
- H. Yoshikawa, M. Larance, D. J. Harney, R. Sundaramoorthy, T. Ly, T. Owen-Hughes and A. I. Lamond, eLife, 2018, 7, 1–26 CrossRef PubMed.
- T. Kalwarczyk, M. Tabaka and R. Holyst, Bioinformatics, 2012, 28, 2971–2978 CrossRef.
- P. Nieuwenhuysen and J. Clauwaert, J. Biol. Chem., 1981, 256, 9626–9632 CrossRef.
- J. C. Bowman, A. S. Petrov, M. Frenkel-Pinter, P. I. Penev and L. D. Williams, Chem. Rev., 2020, 120, 4848–4878 CrossRef.
- G. Cooper, The Cell: a Molecular Approach, Sinauer Associates, United States, 2000 Search PubMed.
- J. R. Lakowicz, Principles od Fluorescence Spectroscopy, Springer Science + Business Media, LLC, 3rd edn, 2006, pp. 797–840 Search PubMed.
- R. J. Jackson, C. U. T. Hellen and T. V. Pestova, Nat. Rev. Mol. Cell Biol., 2010, 10, 113–127 CrossRef.
- K. Szczepański, K. Kwapiszewska and R. Hołyst, Sci. Rep., 2019, 9, 1–8 CrossRef.
- D. G. Pestov and N. Shcherbik, Mol. Cell. Biol., 2012, 32, 2135–2144 CrossRef.
- K. Burger, B. Mühl, T. Harasim, M. Rohrmoser, A. Malamoussi, M. Orban, M. Kellner, A. Gruber-Eber, E. Kremmer, M. Hölzel and D. Eick, J. Biol. Chem., 2010, 285, 12416–12425 CrossRef PubMed.
- M. Ren, S. Qiu, P. Venglat, D. Xiang, L. Feng, G. Selvaraj and R. Datla, Plant Physiol., 2011, 155, 1367–1382 CrossRef.
- S. L. Spencer and P. K. Sorger, Cell, 2011, 144, 926–939 CrossRef PubMed.
- Z. Wang, H. Jiang, S. Chen, F. Du and X. Wang, Cell, 2012, 148, 228–243 CrossRef.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. Alnemri, E. Baehrecke, M. Blagosklonny, W. El-Deiry, P. Golstein, D. Green, M. Hengartner, R. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nuñez, M. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky and G. Melino, Cell Death Differ., 2009, 16, 3–11 CrossRef.
- J. M. Reinhart, W. Rose, D. J. Panyard, M. A. Newton, T. K. Liebenstein, J. Yee and L. A. Trepanier, Pharmacol. Res. Perspect., 2018, 6, 1–11 Search PubMed.
- M. Kubczak, A. Szustka, J. Z. Błoński, T. Gucký, M. Misiewicz, V. Krystof, P. Robak and M. Rogalińska, Mol. Med. Rep., 2019, 49, 3593–3603 Search PubMed.
- D. T. Mcswiggen, H. Liu, R. Tan, S. A. Puig, L. B. Akella, R. Berman, M. Bretan, H. Chen, X. Darzacq, K. Ford, R. Godbey, E. Gonzalez, A. Hanuka, A. Heckert, J. J. Ho, S. L. Johnson, R. Kelso, A. Klammer, R. Krishnamurthy, J. Li, K. Lin, B. Margolin, P. McNamara, L. Meyer, S. E. Pierce, A. Sule, C. Stashko, Y. Tang, D. J. Anderson and H. P. Beck, eLife, 2024, 12, RP9318 Search PubMed.
- A. Karpinska, G. Magiera, K. Kwapiszewska and R. Hołyst, J. Phys. Chem. Lett., 2023, 14, 1272–1278 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00214h |
| ‡ For the purpose of this paper we use “in vivo” in the meaning “in single living cell”. |
| This journal is © The Royal Society of Chemistry 2025 |