
Quantitative photoacoustic spectral transformations in theranostic solid lipid nanoparticles labelled with increasing concentrations of a photoacoustic NIR BODIPY†
Clément
Linger
 ab,
Giulia
Maccini
a,
Gilles
Clavier
c,
Rachel
Méallet
d,
Nicolas
Tsapis‡
*a and
Jérôme
Gateau‡
*b
ab,
Giulia
Maccini
a,
Gilles
Clavier
c,
Rachel
Méallet
d,
Nicolas
Tsapis‡
*a and
Jérôme
Gateau‡
*b
aUniversité Paris-Saclay, CNRS, Institut Galien Paris-Saclay, 91400 Orsay, France. E-mail: nicolas.tsapis@universite-paris-saclay.fr
bSorbonne Université, CNRS, Inserm, Laboratoire d'Imagerie Biomédicale, 75006 Paris, France. E-mail: jerome.gateau@sorbonne-universite.fr
cUniversité Paris-Saclay, ENS Paris-Saclay, CNRS, PPSM, 91190 Gif-sur-Yvette, France
dUniversité Paris-Saclay, CNRS, Institut des Sciences Moléculaires d'Orsay, 91405 Orsay, France
First published on 5th November 2024
Abstract
Solid lipid nanoparticles (SLNs) have shown great capabilities for drug delivery and are therefore attractive theranostic candidates when labelled with an imaging contrast agent. This work aims to create the first SLNs labelled for photoacoustic (PA) imaging by encapsulating a specially designed and near-infrared absorbing BODIPY dye (BY-aniline-Palm) into SLNs of dexamethasone palmitate. A one-pot formulation protocol enabled us to replace the prodrug with the BY-aniline-Palm label in various proportions up to 100%. Increasing the dye content resulted in complex but gradual transformations of the SLNs in terms of optical absorption and PA spectra, and the formation of aggregates at high concentration. A comprehensive and quantitative PA spectrometric study revealed a photoacoustic generation efficiency (PGE) that was spectrally varying and notably greater than 1. A joint spectral decomposition of the absorption and PA spectra into the sum of three Gaussian functions displayed a per-band evolution of the PGE when the concentration of BY-aniline-Palm varied and showed an interplay between the bands with a constant spectrum area. Finally, a novel quantitative PA spectroscopic approach, involving measurements at three different ambient temperatures, demonstrated that the remarkable PGE values arose from a significant thermo-elastic expansion of the SLNs during PA signal generation independently of the absorption band. This study highlights that labeled SLNs are promising agents for PA imaging and also unveils complex transformations that can occur in such nanosystems with a dye prone to aggregation.
1. Introduction
Solid lipid nanoparticles (SLNs) are increasingly studied for therapeutic applications because of their ability to deliver mostly hydrophobic drugs.1 SLNs consist of a matrix of biocompatible solid lipids stabilized by surfactants. They are highly stable thanks to their solid structure, show poor toxicity and are easily scalable at low cost.1 Lorscheider et al. recently developed a simple one-pot formulation protocol for dexamethasone palmitate (DXP) SLNs.2,3 DXP is a prodrug of dexamethasone, an anti-inflammatory drug, released by enzymatic cleavage once these SLNs are administered in vivo. The anti-inflammatory activity of DXP SLNs was demonstrated in vivo in a murine model of rheumatoid arthritis and the nanoparticles exhibited very promising therapeutic effects. SLNs can become theranostic agents with the co-encapsulation of imaging contrast agents, and have already been used for MRI image-guided therapy.4,5 Investigations for optical imaging are just emerging, with at least one study6 in which hydrophobic methine dyes were encapsulated in SLNs for fluorescence imaging.Fluorescence imaging and photoacoustic imaging (PAI) are the main optical bioimaging modalities for applications at centimetre depths,7 when operating in the near infrared (NIR), also called the optical window of biological tissues (typically between 650 and 1300 nm). The high sensitivity of these modalities to molecular contrast is a real asset to monitor the accumulation of drug-loaded nanoparticles. PAI is based on the transformation of absorbed optical energy into ultrasound, through photothermal conversion and thermo-elastic expansion.8 This ultrasound generation and subsequent detection yields images with submillimetre spatial resolution, which is particularly suitable for theranostic applications in focal lesions. Photoacoustic imaging at different successive excitation wavelengths (called multispectral PAI) enables the spectral separation of absorbers.9 At each wavelength, the amplitude of the emitted ultrasound signal is proportional to the absorbed energy converted into heat, which calls, in terms of optical properties of theranostic SLNs, for a high photothermal conversion efficiency (PTCE) and a large absorption cross section in the NIR range. As for the absorption spectrum, NIR dyes are attractive PAI molecular contrast agents because of the presence of absorption bands providing a spectral signature different from that of haemoglobin,9 and thereby facilitating spectral unmixing from the main endogenous absorber.
Among the variety of NIR dyes developed for PAI,10 BODIPYs hold a promising position. Indeed, the BODIPY scaffold is a versatile electron-withdrawing platform, and the addition of electron-donating groups enables the fine tuning of the maximum absorption wavelength in the visible and NIR ranges and favours fluorescence quenching. The high molar extinction coefficients of BODIPY dyes, their strong photostability, and low de-excitation through triplet state transitions also favour the development of molecular photoacoustic agents.10 Several studies reported the use of BODIPY in nanoparticle assemblies,11 with possibly high molecular loading to enhance the per-particle absorption, and thus PAI detectability. Unlike widespread heptamethine cyanine dyes,9 such as indocyanine green (ICG) or cyanine-7 (Cy7), BODIPY derivatives are not amphiphilic but highly hydrophobic, which proves to be advantageous for their encapsulation into hydrophobic particles. In this context, Bodin et al.12 developed a BODIPY-aniline (BY-aniline) presenting a high molar extinction coefficient at 753 nm (∼8 × 105 M−1 cm−1) and a superior photostability compared to Cy7. BY-aniline was grafted with a poly(lactic acid) (PLA) chain by ring-opening polymerization and formulated into polymer nanoparticles. The nanoparticles did not show cytotoxicity and, once injected intravenously into mice, provided a detectable contrast agent for multispectral PAI.
To label SLNs with a BODIPY dye for PAI-based theranostic applications, we investigate here the BY-aniline loading in DXP SLNs. For this purpose, BY-aniline was grafted to a palmitate chain and loaded into DXP SLNs at different BY-aniline-palmitate/DXP ratios. Then, formulated SLNs were thoroughly characterized in terms of optical and photoacoustic (PA) properties. Quantitative PA characterization was performed with a recently developed and novel calibrated photoacoustic spectrometer13 based on a conventional PA imaging system. The great strength of this PA spectrometer lies in the calibration of the system, which yields quantitative spectral measurements directly comparable to the absorption spectrum, unlike prevalent PA spectra in arbitrary units.14 The global and per-band quantitative comparison between PA and absorption spectra of BY-aniline loaded PLA particles recently revealed the appearance of non-linearity in the PA spectrum. This non-linearity was caused by a reduction in the photoacoustic efficiency for one absorption band when the laser fluence increased and the dye concentration decreased. The phenomenon was attributed to ground state depopulation of the fluorescent BY-aniline and was reduced by the ‘aggregation caused quenching’ effect.15 These novel results in the domain of dye loaded nanoparticles for PAI have encouraged the present study on new particles labelled with BY-aniline, especially with a much higher dye loading, which would strongly increase the per-particle photoacoustic brightness15 and could cause further aggregation of the dye.
In this paper, we present the first SLNs tailored for PAI using a NIR BODIPY. The versatility of the formulation enabled us to create SLNs with an extended range of BY-aniline concentrations, reaching very attractive absorption and PA molar coefficients for PAI detectability. Strong spectral modifications were also revealed with increasing dye concentration, up to the appearance of a new spectral band at high concentration. Moreover, quantitative PA coefficients unveiled a photoacoustic efficiency higher than that expected and spectrally varying. These surprising PA features were further characterized with additional PA measurements at different temperatures and a band decomposition of the spectra to investigate their origin.
2. Results
2.1. Formulation and colloidal suspension characterisation
The formulation of solid lipid nanoparticles (SLNs) was adapted from Lorscheider et al.2,3 and relied on an emulsion–evaporation protocol. The PA BODIPY-aniline dye (BY-aniline) synthesized by Bodin et al.12 was esterified on its alcohol function, using a palmitate chain, to yield BY-aniline-Palm. Chemical grafting prevents dye leakage from the SLNs and limits burst release.Dye loading in the SLNs was investigated over a wide range by varying the molar ratio of BY-aniline-Palm and DXP, up to completely replacing the prodrug with the dye. A first batch (Batch #1) containing nine molar percentages of BY-aniline-Palm compared to DXP was formulated: SLN-0%, SLN-1%, SLN-2%, SLN-6%, SLN-12%, SLN-25%, SLN-50%, SLN-75% and SLN-100%. All nanoparticles were prepared on the same day. Fig. 1(c) shows the nine suspensions, which appear darker as the BY-aniline-Palm percentage increases.
 | ||
Fig. 1 (a) SLN hydrodynamic diameter measured by dynamic light scattering and (b) SLN polydispersity index (PdI) as a function of the molar ratio of BY-aniline-Palm versus DXP. Mean values and error bars (standard deviation) of triplicate measurements. Markers are blue circles for Batch #1 and orange diamonds for Batch #2. (c) Picture of the BY-aniline-Palm concentration ladder created by the 9 SLN formulations of Batch #1 (percentages referring to the molar proportion of BY-aniline-Palm versus DXP). For the sake of visibility, each sample is a 50-fold dilution of the formulation result. (d) SLN TEM images with negative staining (uranyl acetate 2% w/w) obtained with a JEOL JEM-1400 microscope at an acceleration voltage of 120 kV. Magnifications for SLN-0%, SLN-1%, SLN-25% and SLN-100% are 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 12000, 25000 and 15000, respectively. White scale bars in the top right corners represent 200 nm. 000, 12000, 25000 and 15000, respectively. White scale bars in the top right corners represent 200 nm. | ||
Each formulation was characterized by zeta potential measurements, dynamic light scattering (DLS) and transmission electron microscopy (TEM). The characteristics of SLN-0% (without BY-aniline-Palm) match those reported by Lorscheider et al.,2 with a hydrodynamic diameter of 160 nm, a polydispersity index (PdI) of 0.18 and a zeta potential of −58 mV. For all SLNs, the zeta potential was found to be negative, with values slightly increasing with the BY-aniline-Palm percentage from −58 mV for SLN-0% to −45 mV for SLN-100% (Fig. S7†), suggesting a colloidal stability through electrostatic and steric repulsions of all suspensions. The hydrodynamic diameter determined by DLS (Fig. 1(a)) is stable, ∼170 nm, for BY-aniline-Palm percentages up to 50% and then decreases down to 125 nm for SLN-100%. The PdI is below 0.2 for BY-aniline-Palm percentages up to 25% and then increases to reach 0.39 for SLN-100% (Fig. 1(b)). The stability over time was assessed through DLS measurements: all the formulations are stable for at least one month when stored at 4 °C and for at least 4 days at 37 °C. The TEM images in Fig. 1(d) show quite monodisperse spherical particles for SLN-0% and SLN-1% (small dots in the background are likely to be DSPE-PEG2000 micelles). However, the appearance and size of individual SLNs become increasingly heterogeneous as the BY-aniline-Palm percentage increases. The global shape of the particles becomes less spherical and the surface becomes more facetted for BY-aniline-Palm contents above 25%, as shown in Fig. 1(d) and Fig. S8.† The increase in polydispersity is confirmed by TEM with smaller egg-shaped particles for SLN-100%.
To assess the formulation reproducibility and perform additional characterization studies, a second batch (Batch #2) was formulated 8 months later for five molar percentages: SLN-0%, SLN-2%, SLN-25%, SLN-50% and SLN-100%. While not exactly identical to Batch #1, SLNs of Batch #2 have globally similar characteristics to those of Batch #1, and follow the same trends. Indeed, zeta potential (Fig. S7†), hydrodynamic diameter (Fig. 1(a)) and PdI (Fig. 1(b)) results showed identical variations to those of Batch #1 as a function of BY-aniline-Palm ratio.
Therefore, the developed PA-tailored SLN formulation is reproducible in terms of nanoparticle size, dispersity and surface charge.
2.2. Colloidal suspension comparison with BY-aniline-PLA particles
In a previous study by our consortium,12 BY-aniline was grafted to polylactide (PLA) and BY-aniline-PLA was formulated into polymeric nanoparticles (NPs) with PLA–PEG5000. As the BY-aniline-PLA concentration increased, the molar absorption coefficient of the nanoparticles increased linearly up to a BY-aniline-PLA to PLA-PEG ratio of 50%. The number of BY-aniline-PLA molecules per particle was then estimated from the ratio between the nominal BY-aniline-PLA concentration and the number density (or concentration) of nanoparticles evaluated by nanoparticle tracking analysis (NTA). For a BY-aniline-PLA to PLA-PEG ratio of 50%, around 3.1 × 103 BY-aniline-PLA molecules per particle were found, for a number density of 2.8 × 1016 particles per L. We reproduced this result by formulating BY-aniline-PLA-47% NPs with the same initial material concentrations. We found a number of BY-aniline-PLA molecules per particle of around 7.5 × 103 for approximately half the number density (Table 1). The difference can be explained by the slightly larger diameter of our particle (∼145 nm compared to ∼120 nm in ref. 12). The size and number density variations might arise from different formulation solvents. Indeed, Bodin et al.12 used dichloromethane while we used chloroform to unify the formulation process with SLN formulation.| NP | BY-aniline (mM) | #part. ×1016 L−1 | #BY-aniline/part. | ε particle (M−1 cm−1) |
|---|---|---|---|---|
| a ε particle: absorption cross section per mole of particles (in M−1 cm−1) evaluated at the maximum absorption wavelength. | ||||
| BY-aniline-PLA-47% | 0.16 | 1.3 ± 0.1 | 7.5 × 103 | 4.6 × 108 |
| SLN-2% | 0.16 | 1.8 ± 0.1 | 5.4 × 103 | 2.2 × 108 |
| SLN-25% | 1.7 | 0.96 ± 0.03 | 1.1 × 105 | 3.6 × 109 |
| SLN-50% | 3.0 | 1.0 ± 0.1 | 1.8 × 105 | 7.7 × 109 |
| SLN-100% | 4.9 | 0.92 ± 0.03 | 3.2 × 105 | 1.7 × 1010 |
Table 1 summarises the main parameters determined by NTA. We first notice that the number density of particles is stable and close to 1016 particles per L in each SLN formulation. It means that the number density is not influenced by the dye proportion. The estimated number of BY-aniline-Palm molecules per particle in SLN formulations is also given in Table 1 and ranges from 5.4 × 103 BY-aniline-Palm molecules per particle for SLN-2% to 3.2 × 105 BY-aniline-Palm molecules per particle for SLN-100%. Thus, labelling SLNs instead of PLA NPs allows us to multiply the maximum number of BY-aniline dye per particle by 50 to 100. Indeed, the long chain length of PLA (15 × 103 g mol−1) compared to BY-aniline (790 g mol−1) limited the actual dye loading to 2.9% (w/w). BY-aniline-Palm has a much lower molecular weight (1029 g mol−1) than BY-aniline-PLA and a much greater dye loading can be achieved. One can notice that SLN-2% and PLA-47% correspond to the same quantity of BY-aniline in the formulation and yield a similar number of BY-aniline per particle.
Finally, SLNs are intrinsically theranostic particles, as the prodrug DXP takes part in the formulation and is present in the core of the particle. For comparison, drug encapsulation during the formulation was not implemented for BY-aniline-PLA particles.
2.3. Characterization of the optical properties of SLNs
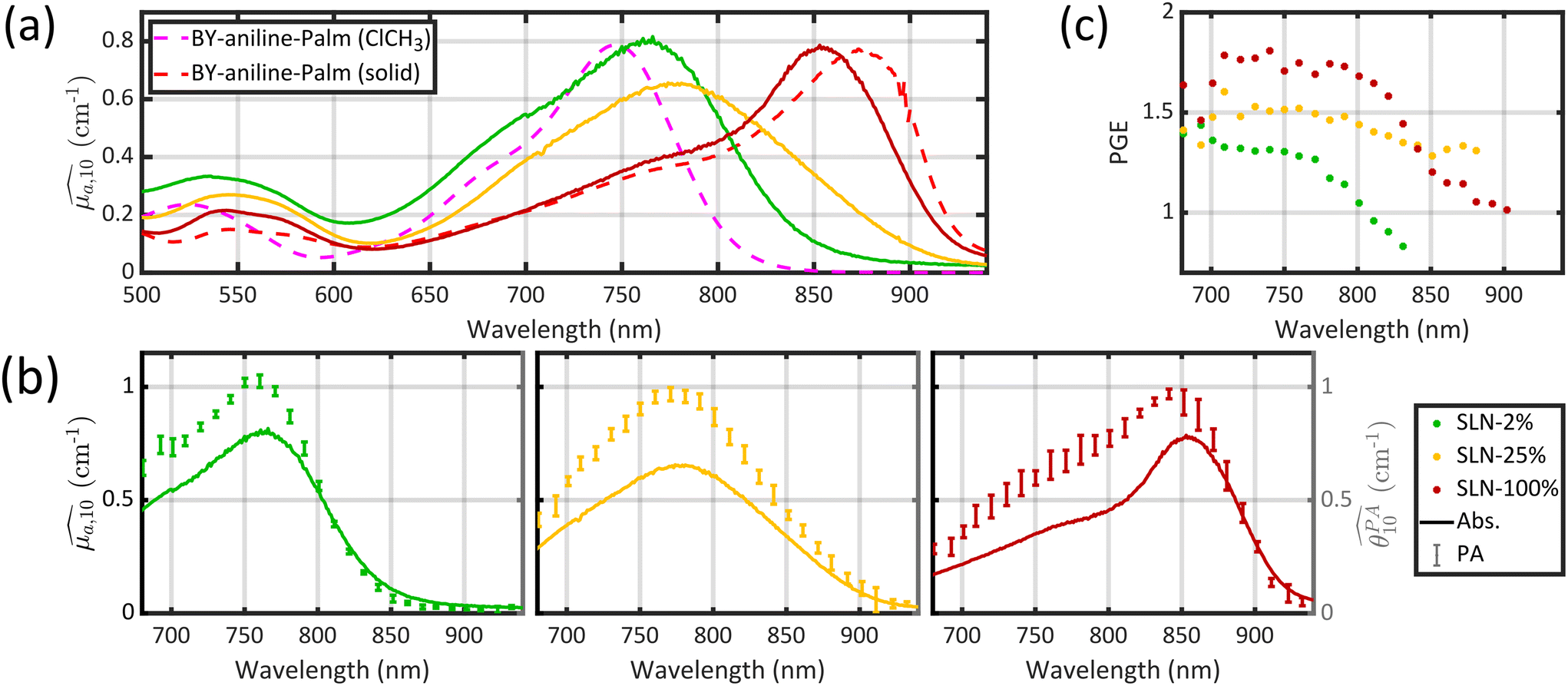
Optical absorption properties of SLN formulations were measured by absorbance spectrophotometry in the visible and NIR range at ambient temperature. First, the BY-aniline-Palm spectrum was measured in solution (Fig. 2(a)). Chloroform was used as a solvent as BY-aniline-Palm was not hydrosoluble. No spectral differences between BY-aniline and BY-aniline-PLA in the same solvent were observed. In the range of 650–980 nm, the spectrum comprises a band around a maximum at 745 nm and a vibronic shoulder around 690 nm. Then, the absorption spectra of the SLN formulations were acquired with a spectrophotometer equipped with an integrating sphere and compared to the spectra in solution. SLN-0% at the same dilution factor was used as a reference for the spectrometer (blank) to limit the influence of scattering on the absorbance spectra. To avoid potential bias due to the increasing absorption of the SLN suspensions with the BY-aniline-Palm concentration, all batches were diluted in Milli-Q water to obtain a constant BY-aniline-Palm concentration (1.73 × 10–2 mM), as detailed in Table S1.† Decadic absorption coefficients,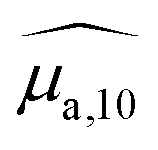 , were obtained by dividing the absorbance by the length of the cuvette. The hat over the coefficient symbols indicates that the quantities are measured at the defined BY-aniline-Palm concentration. In Fig. 2(a),
, were obtained by dividing the absorbance by the length of the cuvette. The hat over the coefficient symbols indicates that the quantities are measured at the defined BY-aniline-Palm concentration. In Fig. 2(a), 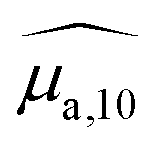 spectra reveal a progressive modification of the absorption spectrum with increasing percentage of BY-aniline-Palm in the SLNs. For SLN-1% and SLN-2%, the spectra are close to that of BY-aniline-Palm in solution (absorption band around 753 nm). As the BY-aniline-Palm percentage increases, the absorption band shifts gradually towards longer wavelengths and becomes more symmetrical, as shown in Fig. S13,† until a new absorption band centred around 860 nm appears.
spectra reveal a progressive modification of the absorption spectrum with increasing percentage of BY-aniline-Palm in the SLNs. For SLN-1% and SLN-2%, the spectra are close to that of BY-aniline-Palm in solution (absorption band around 753 nm). As the BY-aniline-Palm percentage increases, the absorption band shifts gradually towards longer wavelengths and becomes more symmetrical, as shown in Fig. S13,† until a new absorption band centred around 860 nm appears.
 | ||
| Fig. 2 (a) Absorption spectra of SLN-2% (green), SLN-25% (yellow) and SLN-100% (dark red) compared to BY-aniline-Palm spectra in chloroform (magenta, dashed line) and in the solid state (red dashed line). For the sake of readability, the absorption spectra in chloroform and in the solid state were scaled to have a comparable maximum value, around 0.8 cm−1. (b) Absorption (solid line) and PA (dots with error bars) spectra of SLN-2%, SLN-25% and SLN-100%. (c) Ratio between the PA coefficient and the absorption coefficient for each PA wavelength: photoacoustic generation efficiency (PGE). To limit artefacts related to the ratio at low absorbances, the ratio was calculated only for absorbance and PA coefficients greater than 0.15 cm−1. The colour code for SLN is applied in all subfigures. | ||
The absorption cross section per mole of particles, expressed in M−1 cm−1 and denoted as εparticle, is defined as the decadic absorption coefficient divided by the molar concentration of particles, and is equivalent to the molar absorption coefficient for molecular dyes. Besides the bathochromic shift of the maximum absorption wavelength with increasing BY-aniline concentration, εparticle evaluated at this shifting wavelength increases linearly with the BY-aniline concentration: starting from around 2 × 108 M−1 cm−1 for SLN-2% and reaching 2 × 1010 M−1 cm−1 for SLN-100% (Table 1 and Fig. S10†). As a comparison, εparticle of BY-aniline-PLA-47% is measured to be equal to 5 × 108 M−1 cm−1, similarly to that reported by Bodin et al.12 Thus, εparticle values are consistent with the number of BY-aniline per particle. Thereby, εparticle of SLNs can be up to 40 times higher than that for BY-aniline-PLA-47% and up to 100 times higher than that of the BY-aniline-PLA NPs of Bodin et al.,12 which have been successfully detected by multispectral PAI in vivo. εparticle of SLN-100% even falls within the same order of magnitude as that of gold nanoparticles such as gold nanorods, nanocubes and nanoshells commonly detected in biomedical multispectral PAI.9
The appearance of the 100 nm redshifted band (corresponding to a 1650 cm−1 shift) is accompanied by several pieces of evidence pointing to BY-aniline-Palm aggregation within J-aggregates. BODIPY dyes are known to form J-type aggregates when present in a condensed form and a classical method to assess the formation of J-type aggregates is to increase gradually the water percentage in an organic dye solution.16–18 The absorbance spectrum gradually evolves with the formation of a new band, very similar to the one we observed, and an isosbestic point appears. However, this method can be applied to quite amphiphilic dyes (and is applicable to indocyanine green (ICG) for instance), which is not the case for BY-aniline. No new band was observed by Bodin et al.15 in the BY-aniline spectrum in water/THF solution even at 70% water15 and precipitation was observed for higher percentages. We therefore decided to analyse the solid directly as a reference for the condensed form of BY-aniline-Palm. First, when BY-aniline-Palm is isolated and after evaporation of the organic purification solvent, the remaining solid forms flakes, suggesting the supramolecular self-organisation of BY-aniline-Palm molecules. This supramolecular structure is also suggested in SLNs by the facetted particles in the TEM images (Fig. 1(d)) and a cryogenic electron-microscopy (cryo-EM) image of SLN-100% further reveals a highly organized structure (Fig. S9†). To follow this path, the solid-state absorption spectrum of a dried thin film of BY-aniline-Palm was measured. The spectrum is presented in Fig. 2(a) and displays a different shape than the spectrum in solution: the two bands observed in solution (vibronic shoulder and absorption band) form a large and poorly defined band from 650 to 800 nm, and a new band is observed around 875 nm. Spectral decomposition as a Gaussian function (see section 4.9) allows us to identify the three bands. The position and width of the new band in the solid-state spectrum are close to those observed for the 860 nm band in SLN-50% to SLN-100%. This similarity suggests that the BY-aniline-Palm molecules form the same aggregate type in SLN-50% to SLN-100% and in the solid form, with only more partial aggregation in the SLNs. This aggregate type has its own spectrum with a bathochromic shift, which seems to correspond, at least partially, to J-type aggregation, even if this 860 nm band is not as thin as those usually observed for isolated J-aggregates.19 Indeed, a bathochromic shift of the absorption maximum by around a hundred nanometres through the appearance of a symmetrical and thin band is among the main characteristic of J-aggregates.19 No fluorescence enhancement was observed, with either a small or large Stokes shift, as could be the case with J-aggregates.19 In contrast, as shown in Fig. S12,† all nanoparticles were found to be poorly fluorescent, and this fluorescence intensity decreases even further as the BY-aniline concentration increases, along with a small bathochromic effect. This decrease in fluorescence follows the aggregation-caused quenching effect proposed by Bodin et al.15
Likewise, when aggregation is perfect, an isosbestic point is generally observed between the monomer absorption band and the aggregation band. Here, it appears that the monomer band corresponding to the main S0–S1 transition widens before transitioning to the aggregate band. This is particularly obvious in the spectrum of SLN-25% in Fig. 2(a), which is much broader than that of SLN-2% and the monomer spectrum. This could correspond to partial aggregation or random aggregation of BY-aniline-Palm as the highly organized structure visible for SLN-100% on the cryo-EM image is not observed for SLN-25% but the particles have a rough surface compared to SLN-2% (Fig. S9†). Modification of the electronic levels by poorly organized aggregation changes the absorption wavelengths and distorts the spectrum. To clarify these phenomena and simplify the study of these spectral modifications, a spectral decomposition as a sum of Gaussian functions is presented in section 2.6.
However, even though the obtained aggregation state is likely imperfectly, incompletely organized, and somewhat random, it nonetheless remains stable over time for at least one month when stored in a parafilm-sealed vial at 4 °C.
The solid spectrum of BY-aniline-PLA (Fig. S11†) was found to be similar to the spectrum in DCM and did not display a 100 nm redshifted band, which indicated that the grafted chain influenced the aggregation capabilities of BY-aniline.
2.4. Characterization of the photoacoustic properties at 25 °C
Photoacoustic (PA) decadic coefficients, correspond to the optical absorption decadic coefficients, but restricted to the portion of the absorption that is converted into a PA signal.13 They were determined using the home-made PA spectrometer described by Lucas et al.13 at a temperature of 25 °C. This spectrometer is based on a conventional photoacoustic imaging system and
correspond to the optical absorption decadic coefficients, but restricted to the portion of the absorption that is converted into a PA signal.13 They were determined using the home-made PA spectrometer described by Lucas et al.13 at a temperature of 25 °C. This spectrometer is based on a conventional photoacoustic imaging system and  is derived from amplitudes measured on images of tubes containing the nanoparticle suspension (Fig. S6(b)†). At a given wavelength, the amplitude of the image varies with the sample absorption. The solutions prepared for the absorption measurements (section 2.3) were used for a direct comparison between the absorption and quantitative photoacoustic spectra, and because these solutions allow us to operate in the same sensitivity range of the instruments for all SLNs.
is derived from amplitudes measured on images of tubes containing the nanoparticle suspension (Fig. S6(b)†). At a given wavelength, the amplitude of the image varies with the sample absorption. The solutions prepared for the absorption measurements (section 2.3) were used for a direct comparison between the absorption and quantitative photoacoustic spectra, and because these solutions allow us to operate in the same sensitivity range of the instruments for all SLNs.
First, the photostability of BY-aniline-Palm in particles was verified. For this purpose, we used the fact that 15 sweeps of the optical wavelengths were performed consecutively for the PA spectrum measurement and we assessed  for each sweep independently.
for each sweep independently.  was found to be constant over the 15 sweeps at the maximum absorption wavelength (Fig. S23†), which indicated the photostability of the dye. Next, each PA coefficient was computed from an average over the 15 sweeps, as explained in section 4.8. Statistical analysis in the following were then performed on iterations of the measurement using the median and the median absolute deviation, MAD (cf. section 4.8.1).
was found to be constant over the 15 sweeps at the maximum absorption wavelength (Fig. S23†), which indicated the photostability of the dye. Next, each PA coefficient was computed from an average over the 15 sweeps, as explained in section 4.8. Statistical analysis in the following were then performed on iterations of the measurement using the median and the median absolute deviation, MAD (cf. section 4.8.1).
Three typical spectra, corresponding to SLN-2%, SLN-50% and SLN-100%, are displayed in Fig. 2(b), together with their respective absorption spectra. The PA and absorption spectra of SLNs exhibit similar primary trends such as the redshift of the maximum absorption wavelength and the appearance of a new band around 860 nm for SLN-100%. However, the most striking result is that PA and absorption spectra are not superimposed for the SLNs, contrary to what is observed with a solution of nigrosine (Fig. S6(a)†) or a suspension of small (2 nm) gold nanoparticles (Fig. S22† and ref. 20). The PA spectra have significantly higher values than the absorption spectra, especially below 800 nm. PA spectra at 25 °C and absorption spectra for all SLNs are displayed in Figs. S13 and S14† for Batch #1 and Batch #2, respectively.
To analyse the difference between the absorption and PA spectra, one should recall that the measured PA coefficient  is related to the decadic absorption coefficient
is related to the decadic absorption coefficient 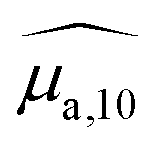 by the following formula:
by the following formula:
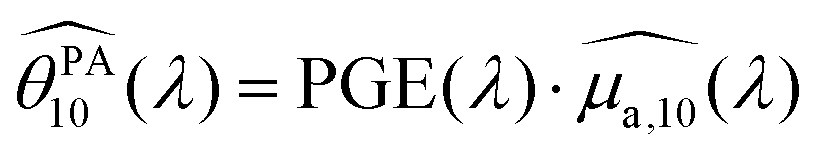 | (1) |
In the next section, we discuss the reasons that can explain the PGE above 1 for the SLNs.
2.5. Effect of the SLN matrix on the photoacoustic generation efficiency
To understand the observed PGE values above 1, we need to go into further detail on PGE. The measured PGE is the product of the photothermal conversion efficiency (PTCE) and the effective Grüneisen coefficient of the solution relative to water.13,22 The PTCE represents the conversion efficiency of absorbed energy into heat. Thereby, PTCE ≤ 1 respects the law of conservation of energy. The Grüneisen coefficient describes the conversion of heat energy into acoustic pressure. The evaluation relative to water here is linked to the calibration process. Thereby, PGE can be expressed as:
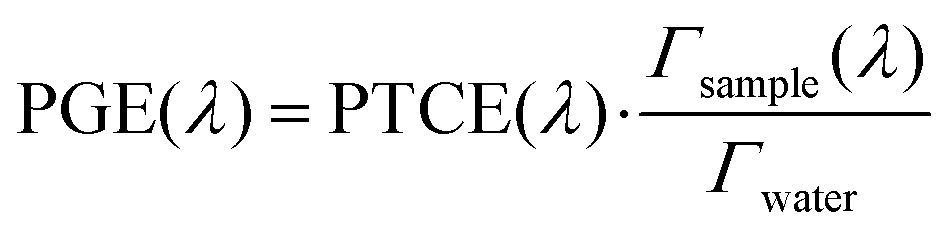 | (2) |
Having PGE > 1 implies that Γsample > Γwater. It is worth noting that PGE(760 nm) increases with the BY-aniline percentage while the dilution of the SLN suspensions in water increases by a factor 31 between the suspensions of SLN-2% and SLN-100%. Therefore, unformulated reagents in the continuous phase (water) are unlikely to be responsible for PGE > 1. However, the Grüneisen coefficient of water is around 0.12 at 25 °C while it varies between 0.5 and 1.1 for lipids.24 In the case of thermal and stress confinements at the scale of a single SLN particle during laser pulse excitation, photoacoustic generation would mainly occur inside the nanoparticle and the ratio of Γsample/Γwater would be on the order of 4 to 10. However, with a laser pulse duration of τp ∼ 7 ns and a spherical particle radius of R ∼ 50 nm, the stress confinement condition is not verified at the spatial scale of the particle and the thermal diffusivity in the surrounding medium cannot be neglected during the laser pulse. To limit the particle radius to the optically absorbing core, R was taken to be smaller than the hydrodynamic radius and on the order of the size observed in the TEM images (Fig. 1(d)). The thermal diffusivities in solid palmitic acid,25 in PLA26 and in water are on the order of χ ∼ 10−7 m2 s−1, which implies that the heat diffusion length in these materials is about 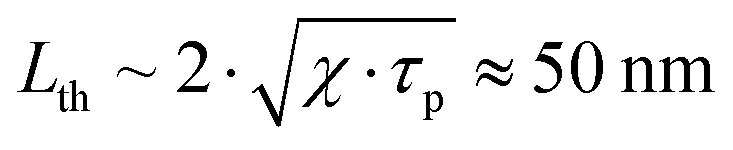 . Lth cannot be considered small compared to the size of the particle, which implies significant thermal diffusion in the surrounding water during laser excitation. But Lth is also not large compared to R so the particle experiences partial thermal confinement during the laser pulse. The interparticle distance for SLN-2% can be estimated to be on the order of 800 nm (3 μm for SLN-100%) and is much larger than Lth + R; therefore, the particles can be considered to be isolated and synergistic effects between particles can be excluded. A 50 nm thick heated layer of water around the particle implies a volume about 7 times larger for this layer than for the particle itself and suggests a significant contribution of this water layer to the photoacoustic signal and therefore to Γsample. One can then understand why the measured PGE is not larger than 2.
. Lth cannot be considered small compared to the size of the particle, which implies significant thermal diffusion in the surrounding water during laser excitation. But Lth is also not large compared to R so the particle experiences partial thermal confinement during the laser pulse. The interparticle distance for SLN-2% can be estimated to be on the order of 800 nm (3 μm for SLN-100%) and is much larger than Lth + R; therefore, the particles can be considered to be isolated and synergistic effects between particles can be excluded. A 50 nm thick heated layer of water around the particle implies a volume about 7 times larger for this layer than for the particle itself and suggests a significant contribution of this water layer to the photoacoustic signal and therefore to Γsample. One can then understand why the measured PGE is not larger than 2.
Assuming perfect thermal contact between a single spherical particle and its surrounding water, the characteristic time for heat exchange27 is τE ∼ ρNP·CNPp·R2/(3·kwater), where ρNP and CNPp are the density and specific heat capacity of the particle, respectively, and kwater is the thermal conductivity of water. Interestingly, this formula depends on the parameters of both the nanoparticle and the surrounding medium. For SLN and PLA particles, τE ∼ 2 ns when taking the parameters for solid palmitic acid25 and PLA.26,28τE < τp confirms the partial confinement of heat inside the particle. It is worth noting that τE depends quadratically on the radius of the particle. For much smaller particles, such as the gold nanoparticles used here as a reference (R ∼ 1 nm), one can have τE ≪ τp, meaning that the particle is mostly a heat source for the surrounding water. For these gold particles, the PGE was found to equal 1.20
Nanoparticles with a PGE larger than 1 were also observed by Aoki et al.29 but the PGE was not evaluated quantitatively. In their study, polymer nanoparticles (PNPs) containing an organic dye were exposed to laser pulses with τp ∼ 1 ns. Similarly to our study, the thermal diffusivity of the polymer materials was on the order of 10−7 m2 s−1, but because of the shorter pulse duration, the heat diffusion length was Lth ≈ 20 nm. They demonstrated that the photoacoustic signal arose almost entirely from water for small particles (R ∼ 20 nm, τE ∼ 0.3τp) and a significant contribution of the particle to the photoacoustic signal could be observed for larger particles (R ∼ 60 nm, τE ∼ 3τp) and increased with the particle radius. Polymers with Grüneisen coefficients ranging from 0.7 to 1.4 at 20 °C were used for PNP synthesis and the photoacoustic signal generated by the solutions increased with the Grüneisen coefficient for larger particles while it remained constant for smaller ones. The PGE above 1 was attributed to partial thermal confinement inside the larger PNPs and to the large Grüneisen coefficient relative to that of water of the matrix polymers.
Here, we compare PLA particles loaded with BY-aniline-PLA and SLN loaded with BY-aniline-Palm, which have similar hydrodynamic sizes and thermal properties. To compare particles with a similar dye loading and concentration (Table 1), we compare SLN-2% and BY-aniline-PLA-47%. The fluorescence can be considered negligible for the two particles, leading to PTCE ≈ 1. The particles are therefore expected to experience similar partial thermal confinement inside the particle and to generate similar heating of the surrounding water layer. At 25 °C, we determined GPAE = 1.05 for BY-aniline-PLA-47% and GPAE = 1.2 for SLN-2% (Table S4†). The Grüneisen coefficient of PLA was not found in the literature. However, the large Grüneisen coefficient of lipids relative to water can explain PGE > 1.
Theoretical considerations and comparisons with other experimental studies strongly suggest that the solid lipid matrix of the particle is responsible for PGE > 1. Numerical simulation of photoacoustic generation for the particle would require the determination of several thermodynamic and acoustic parameters of the different materials composing the particle and the implementation of a complex model;30 these are beyond the scope of our experimental study. However, following the approach of Aoki et al.29 and Simandoux et al.,31 we further demonstrate that PGE is linked to the matrix of the particle by investigating the PA emission of the particles at different water temperatures. The Grüneisen coefficient of water is temperature-dependent, and the Grüneisen coefficient of the lipid core is not expected to vary in the same proportion24 in the range 15 °C to 35 °C. Therefore, the effective Grüneisen coefficient should manifest a dependence on temperature only if the lipid core is involved in photoacoustic signal generation.
First, at the laser fluence and ultrasound frequencies involved in this study, we consider that thermal nonlinear contributions to the photoacoustic signal are negligible and cannot explain PGE > 1. Indeed, the PA coefficient was not enhanced at higher fluence for SLN-2% (Fig. S24†). Thereby, the Grüneisen coefficient of the water layer surrounding the particle is expected to be determined by the temperature of the thermostatic water tank in which the sample containers are immersed. The temperature of the water tank was set successively at three different temperatures, 15 °C, 25 °C and 35 °C, and quantitative PA spectra were acquired. The Grüneisen coefficient of water increases from 0.07 to 0.18 over this temperature range.24
All previous measurements with the photoacoustic spectrometer were performed at a fixed temperature.12,13,15,20 To obtain, for the first time, quantitative PA spectra of the same sample but at different temperatures, we determined that the calibration process needed to be repeated for each temperature. Thereby, computed PGE values are relative to water at the same temperature. The absorbance spectra of the three samples were found to be independent of temperature (Figs. S21 and S22†). Validation of our experimental method was performed using a suspension of small GNPs. Indeed, the photoacoustic generation of these GNPs arises from the surrounding water32 and, as a consequence, the calibrated PA spectrum is expected to match the absorption spectrum for all temperatures. The superimposition is verified visually (Fig. 3) and by a GPAE on the order of 1 for the three temperatures (Table S5†).
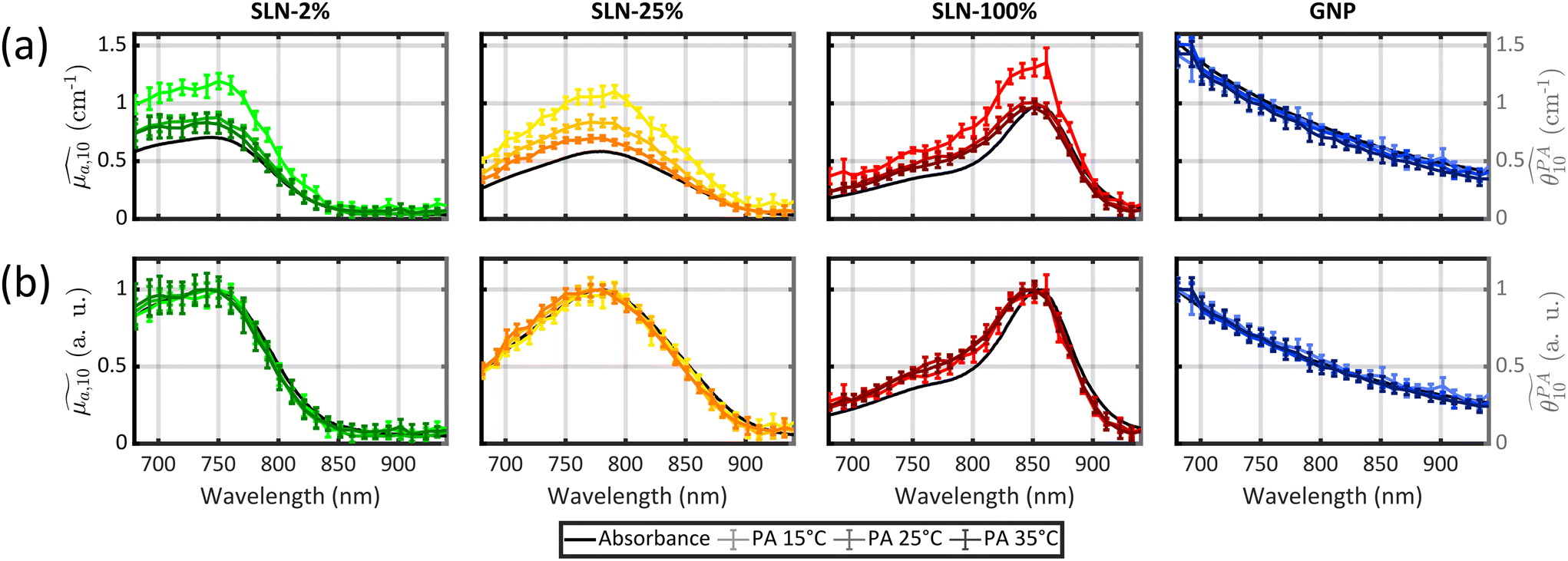 | ||
| Fig. 3 (a) Absorption spectra (black) and PA spectra at three temperatures (15 °C, 25 °C and 35 °C) of SLN-2% (shades of green), SLN-25% (shades of yellow), SLN-100% (shades of red) of Batch #2, gold nanoparticles (shades of blue). (b) Absorption spectra (black) and PA spectra at three temperatures (15 °C, 25 °C and 35 °C) normalized by the spectrum maximum. The colour code is the same as in (a). | ||
Following this validation, the quantitative PA spectra of SLN-2% and PLA-47% can be compared. The first result common to the two particles is that the spectra at the three temperatures are not superimposed although their spectral shape is conserved. Normalisation to the maximum value shows that the spectra differ by multiplicative factors. These factors are given by the GPAE (Table S5†). The GPAE increases with decreasing temperature, that is to say with decreasing Γwater. While Γwater decreases, one can hypothesise that the relative weight of the heated water layer to Γsample would decrease and thereby the relative weight of the particle would increase. However, the PA spectra at 25 °C and 35 °C are almost superimposed, indicating that Γsample/Γwater is nearly constant between the two temperatures while Γwater increases from 0.12 to 0.18. In contrast, the PA coefficients were found to be larger at 15 °C than at 25 °C with GPAE(15 °C)/GPAE(25 °C) = 1.38 for SLN-2% and GPAE(15 °C)/GPAE(25 °C) = 1.18 for PLA-47%, while Γwater(15 °C) = 0.06 and Γwater(25 °C) = 0.12. Aoki et al.29 demonstrated the contribution of the particle to the photoacoustic signal by further decreasing the water temperature down to 4 °C where Γwater(4 °C) = 0. However, the calibration of our photoacoustic spectrometer would not have been possible under these conditions, which is why we limited our investigations to 15 °C for this first exploration of the spectrometer performance at different temperatures. The larger difference in the PA coefficient values when the temperature was set below an ambient temperature of 25 °C than when it was set above it may indicate a saturation process or a limit. This result is observed for all the other SLNs of Batch #2 (Fig. 3). Measurements at more temperatures, especially below 25 °C, are beyond the scope of this study but will be investigated in future studies to better understand the variations of Γsample/Γwater with water temperature.
The significant increase of the PA coefficient at 15 °C for the SLN particle is an additional argument that the particle matrix is involved in photoacoustic generation. The higher GPAE values for SLN-2% than for BY-aniline-PLA-47% provide further evidence that the Grüneisen coefficient of the solid lipid core is larger than the polymer one.
2.6. Per-band analysis of the absorption and PA spectra and PGEs
While PGE values above 1 could be explained by the lipid matrix of the SLN particles in section 2.5, the wavelength-dependency of PGE, especially for SLN-50% to 100%, requires further investigation. Band-dependent PGEs have been reported for fluorescent dyes in solution. The poor dye solubility in water leads to a mixture of different species (monomers and aggregates) that exhibit different absorption bands with various photoacoustic properties.22In this section, we perform a per-band analysis of the absorption and PA spectra, and subsequently of the PGE. The decomposition of the spectra in three absorption bands and the derived band photoacoustic efficiencies (BPAEs) are first presented. Then, each band is attributed to an energy transition or species. Finally, further investigations of the band-dependent PGE are revealed.
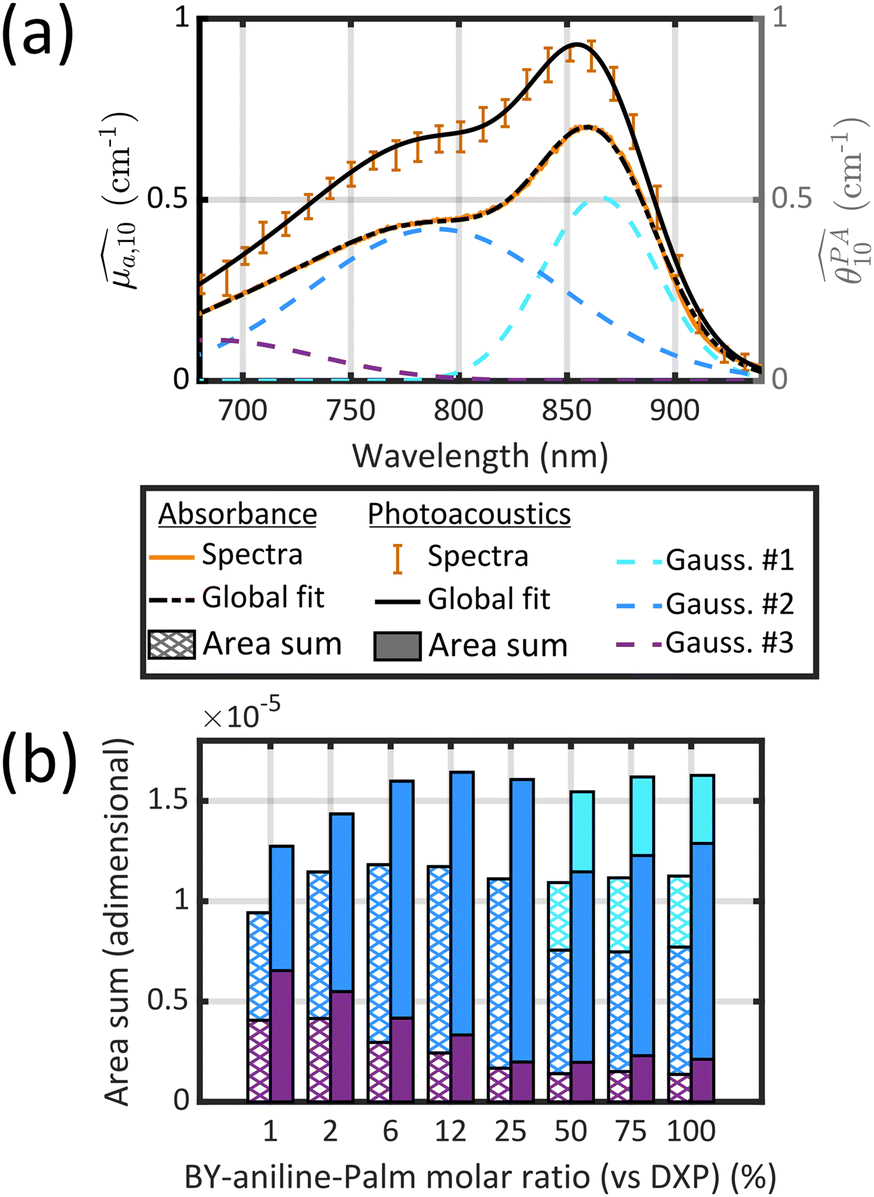
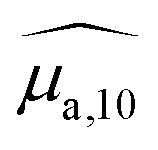 spectra, in the range 640–980 nm, and
spectra, in the range 640–980 nm, and  spectra (at 25 °C), in the range 680–930 nm, as the sum of three Gaussian functions. A simpler Gaussian decomposition of both absorption and photoacoustic spectra, with only two Gaussian functions, was performed previously on BY-aniline-PLA particles15 and showed the feasibility of such decomposition for BY-aniline and its interest for band analysis. This decomposition was reproduced here for BY-aniline-PLA-47% (Fig. S20 and Table S3†). Within a complete study including various concentrations, PA spectra at different laser fluences as well as ns-transient absorption measurements, the decomposition in two bands mainly enabled a ground state depopulation phenomenon in one of the two bands to be highlighted.
spectra (at 25 °C), in the range 680–930 nm, as the sum of three Gaussian functions. A simpler Gaussian decomposition of both absorption and photoacoustic spectra, with only two Gaussian functions, was performed previously on BY-aniline-PLA particles15 and showed the feasibility of such decomposition for BY-aniline and its interest for band analysis. This decomposition was reproduced here for BY-aniline-PLA-47% (Fig. S20 and Table S3†). Within a complete study including various concentrations, PA spectra at different laser fluences as well as ns-transient absorption measurements, the decomposition in two bands mainly enabled a ground state depopulation phenomenon in one of the two bands to be highlighted.
The three Gaussian functions are ordered by decreasing centre wavelength and named Gaussian #1 to Gaussian #3. Pure BY-aniline-Palm spectra in chloroform and in the solid state were first decomposed, as illustrated in section 4.9. The width and centre wavelength of Gaussian #1 evaluated from the Gaussian decomposition of the solid-state spectrum and the centre wavelength of Gaussian #3 evaluated from the solution spectrum were used as references for the Gaussian decomposition of the SLN suspension spectra. Moreover, the centre wavelength and the width of Gaussian #2 and the width of Gaussian #3 were jointly optimized for the absorption and PA spectra of SLN suspensions. Amplitudes were optimized independently. Fig. 4 illustrates that the Gaussian decomposition enables a satisfactory fitting of both the absorption and the PA spectra. The quality of the decomposition was verified for all the formulations, as shown in Figs. S16 and S17† for Batch #1 and in Fig. S19† for Batch #2. Detailed results of the model fitting parameters are given in Table S2† for Batch #1 and in Table S3† for Batch #2.
 | ||
| Fig. 4 (a) Decomposition of the absorption spectrum for SLN-75% in the sum of three Gaussian functions. From left to right, the Gaussian functions are centred at 685 nm (purple), 789 nm (blue) and 865 nm (cyan). The sum is given by the black dotted line. For the sake of readability, only the PA spectrum (with error bars) and its global fit (black continuous line) are shown. For details see Figs. S16 and S17.† (b) Contribution of each Gaussian function (area) in the total spectrum area for each SLN. Absorption spectra are on the left-hand side as hatched bars while PA spectra are on the right-hand side as coloured bars. The colour code is the same as in (a) and we are considering Batch #1 (for Batch #2, see ESI†). | ||
It is remarkable that despite the spectral deformation between the PA and absorption spectra, they could both be decomposed with the same three Gaussian functions by mainly varying the weighting factors. For the rest of this study, we only use these 3 bands, which are sufficient to describe the spectrum instead of the 30 wavelengths of the PA spectrum. To consider both the amplitude and the width of each band, we compute the area of each Gaussian. Those areas allow us to compare the weight of each band in one formulation, and also to evaluate the weight of each band in relation to the BY-aniline percentage. The computed areas are displayed in a vertical stacked bar chart in Fig. 4(b). This figure focuses on Batch #1, but an equivalent figure is given in the ESI† for Batch #2 (Fig. S18†). The first striking observation is that besides the spectral transformation, the total area (sum of the areas for the three Gaussian functions) is constant for the 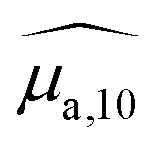 spectra, and constant and 1.4 times higher for the
spectra, and constant and 1.4 times higher for the  spectra for BY-aniline-Palm percentages above 6%. The constant total areas imply an interplay and a balance between absorption bands. For
spectra for BY-aniline-Palm percentages above 6%. The constant total areas imply an interplay and a balance between absorption bands. For 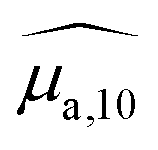 , the purple band (Gaussian #3) represents around 50% of the total area for SLN-1% and gradually diminishes to reach a plateau around 15% from SLN-25%. The blue band (Gaussian #2) first increases with the BY-aniline-Palm percentage, reaching 85% of the total area for SLN-25%. From SLN-25%, it gradually decreases, while the cyan band (Gaussian #1) appears. To compare absorbance and PA spectra, we use here the band photoacoustic efficiency (BPAE) defined by Bodin et al.15 as the ratio of the areas of each band between the photoacoustic spectrum and the absorption spectrum (cf. section 4.9). It corresponds to a weighted mean PGE of the band. The results are presented in Tables S2 and S3.† For the BPAE of Gaussian #3, no real trend seems to emerge with values of about 1.3. The BPAE of Gaussian #1 is remarkably stable with a value around 1. On the other hand, for Gaussian #2, the BPAE increases with the BY-aniline-Palm ratio, between 1.2 for SLN-1% and 1.7 for SLN-100%.
, the purple band (Gaussian #3) represents around 50% of the total area for SLN-1% and gradually diminishes to reach a plateau around 15% from SLN-25%. The blue band (Gaussian #2) first increases with the BY-aniline-Palm percentage, reaching 85% of the total area for SLN-25%. From SLN-25%, it gradually decreases, while the cyan band (Gaussian #1) appears. To compare absorbance and PA spectra, we use here the band photoacoustic efficiency (BPAE) defined by Bodin et al.15 as the ratio of the areas of each band between the photoacoustic spectrum and the absorption spectrum (cf. section 4.9). It corresponds to a weighted mean PGE of the band. The results are presented in Tables S2 and S3.† For the BPAE of Gaussian #3, no real trend seems to emerge with values of about 1.3. The BPAE of Gaussian #1 is remarkably stable with a value around 1. On the other hand, for Gaussian #2, the BPAE increases with the BY-aniline-Palm ratio, between 1.2 for SLN-1% and 1.7 for SLN-100%.
Despite some differences in the distribution of areas between the bands, very similar trends and values are observed in Fig. S18† for Batch #2.
The Gaussian functions of Fig. 4 can be interpreted in terms of different transitions. Gaussian #3 (purple) models the shoulder (a priori S0–S1 vibronic state transition37), which is mostly visible at BY-aniline-Palm percentages below 6%, as illustrated in Fig. S16.† Gaussian #2 (blue) is centred around 760–800 nm and could correspond to the S0–S1 0–0 electronic excitation,37 which is the main absorbing band in chloroform. These two bands were also identified in polymer particles with BY-aniline-PLA studied by Bodin et al.15 and in BY-aniline-PLA-47% (Fig. S20†). The enlargement and the bathochromic shift of Gaussian #2 in SLNs, when the BY-aniline-Palm percentage increases, could be explained by the gradual environment change for BY-aniline. Indeed, solvatochromism experiments by Bodin et al.15 showed that the absorption spectrum of BY-aniline changed with its environment polarity. Strong bathochromic and hypsochromic shifts of the band maximum were observed depending on the polarity of the solvent: from 728 nm in cyclohexane to 772 nm in DMSO. As the BY-aniline-Palm percentage increases in SLNs, the environment polarity of BY-aniline is necessarily modified, as aromatic rings become more numerous and interact with each other. For low concentrations of BY-aniline-Palm, each BY-aniline-Palm molecule is expected to mainly interact with aliphatic chains (typically from DSPE or palmitate from DXP), whereas interactions with aromatic rings should increase with the increasing number of BY-aniline-Palm per particle even when structured aggregates are not formed. Random aggregation could also cause band enlargement. Gaussian #1 (cyan) appears only for BY-aniline-Palm percentages above 25% and models a new band with a 100 nm (or 1650 cm−1) bathochromic shift with regard to SLN-1%, likely corresponding to the partial J-type aggregation of BY-aniline.19
The attribution of the absorption band gives indications that compared to the low BY-aniline concentration in PLA particles, for which the weight of the vibronic shoulder and S0–S1 0–0 electronic transition remain stable while the dye concentration increases, the high concentration in the SLN particles strongly impacts the weight of the vibronic shoulder and S0–S1 0–0 electronic transition. This impact suggests strong intermolecular interactions and coupling between BY-aniline, up to the formation of J-aggregates.
Surprisingly, the band associated with J-aggregates has a lower BPAE than the other two bands. Moreover, the BPAE is around 1 at 25 °C and this value is also observed for GNPs for instance. In previous studies,15,22 the reported BPAE involved fluorescent dyes and the band-dependency of the PGE was attributed to a band-dependent PTCE. Additionally, the BPAE was below 1 because of the quantum yield and the energy ‘stored’ in excited states with lifetimes on the same order of magnitude as or larger than the laser pulse duration could further lower the PTCE.
Since the BPAE is larger than 1 and because a very weak fluorescence was recorded (Fig. S12†), even for a low BY-aniline concentration, we investigate independently the possibility of a band-dependent effective Grüneisen coefficient and a band-dependent PTCE.
To test this hypothesis, we measured PA spectra at three different temperatures, 15 °C, 25 °C and 35 °C, for the SLNs of Batch #2 (Fig. 3 and Fig. S21† for SLN-50%). The temperature variation allows us to modify the relative contribution of water. Moreover, the absorption spectra were recorded at three temperature (Fig. S21†), with no significant changes. DLS measurements at three temperatures were also performed, as object size can influence thermoelastic expansion.29 A small decrease in size was observed at 15 °C (about 5%) but should not significantly change the thermal behaviour.
The results for SLN-50% and SLN-100% at 15 °C showed that the BPAE for the J-aggregate band could reach 1.5 and 1.2, respectively, which indicated that the BPAE for this band was not limited to 1 and that the particle matrix contributed to the photoacoustic signal for this band. More globally, one can clearly observe that the PA coefficients increase with decreasing temperature for all the particles (Fig. 3(a)). However, for each formulation, PA spectra at different temperatures are superimposed when normalized to the maximum value (Fig. 3(b)). This implies that the PA spectral shape is independent of temperature. Thereby, for each SLN, the 2 or 3 different BPAEs vary as the GPAE. The relative variation of the BPAEs at 15 °C and 35 °C compared to 25 °C is therefore not influenced by the BPAE value at 25 °C. As a consequence, the relative contributions of the particles and their surrounding water layers do not depend on the absorption band; this invalidates the hypothesis of a band-dependent effective Grüneisen coefficient. Thereby, the effective Grüneisen coefficient is constant over all the spectra. We have demonstrated that PGE is larger than 1 due to a thermoelastic expansion of the SLN matrix, but the per-band variability of PGE should be attributed to light-to-heat energy conversion.
The relative change of the GPAE with temperature was found to be dependent on the BY-aniline-Palm concentration for Batch #2 (Table S5†). However, the small number of water temperatures, batches and BY-aniline-Palm concentrations do not allow us to speculate on trends.
The stability of the PA signal over the sweeps in Fig. S23† indicates that the photochemical pathway is unlikely. For BY-aniline-PLA, Bodin et al.15 showed that long lifetime excited states leading to ground state depopulation were responsible for a decrease of the PA signal especially at a BY-aniline-PLA percentage below 50%. As SLN-2% has the same number of BY-aniline per particle as BY-aniline-PLA-50%, ground state depopulation is expected to be limited for the SLN investigated here. A ground state depopulation would depend on the deposited optical energy and would result in the PTCE decreasing with the laser fluence. In addition to the measurements performed with a laser fluence of ∼3.5 mJ cm−2, we measured the calibrated PA spectrum with a laser fluence of 2.5 mJ cm−2 (Fig. S24†). For SLN-25% and SLN-100%, the difference between the two spectra is not significant, and the PGE is not lower for higher fluence, especially for the J-aggregate band, which has a lower BPAE.
Additionally, we investigate the presence of triplet states and long lifetime non-radiative pathways in SLNs using transient absorbance spectroscopy with nanosecond pulses. In this spectroscopic technique,38 a nanosecond pump laser is used to achieve an excited state of the molecule, the spectrum of which is measured by a probe light beam, similarly to a spectrophotometer. The wavelength of the pump laser was set to 680 nm, which was the maximum wavelength available for the apparatus we used. Such a spectroscopic system is homebuilt and somewhat unique. The spectrum obtained without the pump laser is subtracted from the one obtained with it to identify absorption variation between fundamental and excited states. For the wavelength of this subtracted spectrum, a positive signature means that the excited state absorbs more than the ground state, and a negative signature indicates the opposite (referred to as bleaching). The spectrum can be measured at different times after excitation to determine the decay lifetime and the de-excitation rates of the excited species. With a pump at 680 nm, we expect to probe the excited states linked to the S0–S1 0–0 electronic transition band. The S0–S1 vibronic state transition band cannot directly yield long-lived states.
Before focusing on SLNs, the transient absorption spectrum of BY-aniline-Palm in DCM was measured (Fig. S25†) and showed that the excited state returned to the ground state in around 5 ns. This result was similar to the results of Bodin et al.15 on BY-aniline, but we found a shorter excited state lifetime for the grafted dye (∼5 ns compared to a few tens of nanoseconds). Regarding SLN-2%, we observe a disappearance of the positive and negative signatures in a few nanoseconds after laser excitation and lower transient absorption values compared to BY-aniline-Palm in DCM, as shown in Fig. S25.† Given the very short de-excitation times, the signal was disrupted by the presence of fluorescence. Therefore, a measurement of the spectrum without a source beam was performed to correct the signal for fluorescence. For SLN-25% and SLN-100%, the subtraction of the excited/non-excited spectra has an even smaller value and the difference can be considered negligible even at 0 ns delay between pump and probe. Therefore, no energy storage should hinder PA generation for SLNs with a BY-aniline-Palm ratio higher than 25%. Furthermore, bubbling with inert gas (argon) for 10 min did not change the transient spectroscopic signal, indicating the absence of triplet state species for the excited band.
A decrease in the excited state lifetime with increasing concentration of BY-aniline-PLA was already observed by Bodin et al.15 for PLA particles and was associated with a decrease in the fluorescence quantum yield and an increase of the PGE. For SLNs, the BPAE of Gaussian #2 increases from SLN-2% to SLN-25%. However, no saturation of this BPAE was observed for higher concentrations for Batch #1. Therefore, the reduced lifetime of the excited state and the weaker fluorescence signal may only partially explain the BPAE increase with increasing BY-aniline-Palm concentration for this band.
Unfortunately, the upper wavelength of the pump laser at 680 nm did not enable the direct excitation of the aggregation band, and no indirect excitation was observed. Therefore, we could not evaluate the existence of longer-lived excited species in this band, nor explain the lower BPAE for Gaussian #1. Extension of the wavelength range of the nanosecond-transient absorption spectrometer requires significant investment and could not be performed for this study.
3. Discussion
The goal of this work was to create new theranostic agents for photoacoustic imaging and to characterize their spectral properties and efficiency as imaging agents. Using a BY-aniline compound grafted on a palmitate chain, we formulated the first solid lipid nanoparticles tailored for PAI, employing a straightforward and one-step formulation process. The exquisite and versatile dye loading capability enabled us to increase the per-particle absorption with small modifications of the particle size. Interestingly, we succeeded in formulating SLNs with up to 100% BY-aniline-Palm. In the absence of the prodrug, SLN-100% can be considered as a pure contrast agent. The interchangeability of the dye and the prodrug is certainly due to identical palmitate grafting. From the application perspective, for percentages above 50% SLNs can be considered as dominantly contrast agents while below 50% they are dominantly drug carriers. No toxicity tests or animal injections have been performed so far. However, DXP-loaded SLNs2 and PLA nanoparticles12 containing BY-aniline were not toxic to cells up to a particle concentration of about 0.45 mg mL−1. Furthermore, PLA particles were visible in photoacoustic imaging12 and the accumulation of SLN in arthritic paws of mice was confirmed with fluorescent contrast agents.2 We can be confident that these SLNs will be detectable and usable in multispectral photoacoustic imaging (PAI) in vivo. The determined absorption cross section per mole of particle place the high-loading SLNs in the same range as gold particles commonly used for biomedical preclinical imaging studies.9The quantitative characteristic of the PA spectrometer allowed us to observe that the photoacoustic efficiency was higher for the SLN suspensions than for an aqueous solution of molecular compound with the same absorption. The novel approach of acquiring calibrated PA spectra at three different temperatures enabled us to demonstrate that the enhancement of the PGE was indeed linked to a partial expansion of the lipid matrix coupled with the expansion of the surrounding water layer. This demonstration highlights that, in addition to the absorbing molecule, the material of the nanoparticle may play an important role in the design of organic particles for PAI, especially for a material with a Grüneisen coefficient much higher than that of water.
The paired optical and PA spectra revealed spectral transformations with increasing BY-aniline-Palm concentration, and a band-dependency of the photoacoustic efficiency. The fact that the PA and absorption spectral shapes are not always superimposed, even after correction for scattering, shows the importance of an upstream characterization of the nanoparticle. Indeed, considering the accurate PA spectrum of a contrast agent may prove crucial for its discrimination from endogenous absorbers and for the quantitative evaluation of its distribution in multispectral PAI. Concerns regarding the upstream PA characterisation of contrast agents rather than relying only an optical absorbance spectrum have already been shown for molecules,22 and we highlight here that it is all the more essential for nanoparticular agents. With the three-temperature approach, we were able to show that the spectrum distortion was necessarily linked to per-band variations in the energy conversion between light and heat generation and not to spectral variations in the thermoelastic expansion. Despite our attempts to understand band-dependent photothermal conversion, we were not able to determine the associated photophysical phenomenon. PA spectra at different light fluences and transient absorption spectra could not reveal a long lifetime excited state that would result in ground state depopulation. However, reporting our findings and investigations is of strong interest since similar behaviour may occur in different nanoconstructs for PAI.
Regarding the spectral transformation, we demonstrated that both absorption and PA spectra could be interpreted as a balance between two different bands at low concentrations of BY-aniline-Palm and three different bands at high concentrations of BY-aniline-Palm. The absorption band centred at 860 nm (corresponding to Gaussian #1) was attributed to the generation of J-type-like aggregates. This attribution is supported by the highly organized structures observed on a cryo-EM image of SLN-100% (Fig. S9†). The aggregation phenomenon, particularly J-type aggregates, is sought after by various research teams working on the development of PAI contrast agents. Indeed, J-aggregation can increase the molar absorption of a compound with a great compactness and often improves the photostability.19,39 Moreover, the redshifted maximum absorption wavelength allows us to use higher excitation wavelengths for a better penetration into biological tissues and the usually observed thin absorption band facilitates separation from endogenous absorbers with spectral unmixing algorithms. Enhanced fluorescence is not desired, as a competitive deexcitation pathway, but is not systematic with J-aggregates, especially if aggregation is imperfect. Indeed, random molecule organization in poorly organized aggregates can even lead to a reduction in fluorescence through the effect of aggregation caused quenching.15 For instance, here, no fluorescence was recorded at high concentrations of BY-aniline-Palm. The formulation principle with dye organized aggregates (such as J-type) generally relies on pre-aggregation by progressive evaporation of the organic phase followed by inclusion of the aggregates in nanovectors. For example, organized aggregates can be inserted into a lipid film subsequently formulated into liposomes40 or they can be encapsulated after the liposome formulation.41 Pre-formed organized aggregates can also be included by a double emulsion process.42,43 Another organized aggregation method relies on the maturation of dye-loaded particles either by waiting for a long period of time (about 60 days for PAtrace44) or by favouring molecular interactions (reduced pore spaces, addition of metal ions, etc.).45 All these processes enable organized aggregation of quality but require a very long preparation time and/or numerous steps. Moreover, it is difficult to control the quantity of formed organized aggregates and to halt the aggregation process. With the SLN formulation presented here, self-organized aggregation occurs in situ for sufficient BY-aniline-Palm concentrations, and the nanoparticles can be used in less than 4 hours. We evaluated the potential maturation of the J-aggregates after long-term conservation (3 months). We observed the appearance of large crystallized objects in the suspension but optical spectra were similar to those of fresh suspensions with no enhancement of the optical absorption around 860 nm compared to 760 nm. With BY-aniline-Palm and our one-pot rapid SLN formulation, the appearance of a perfect J-aggregate was not shown. The J-aggregation did cause a redshift of the maximum absorption wavelength beyond 800 nm, which was interesting from a detection perspective in biological tissue,9 but no superior enhancement of the absorption or PA cross section per mole of particles was observed. Moreover, the absence of a perfect control of organized aggregation level, as opposed to long maturation processes44 and as seen in batch-to-batch comparison, requires an absorption and PA spectral characterization of each batch. However, the stability over time of the absorption and photoacoustic spectra make it possible to use a batch for at least a month when stored at 4 °C.
Despite the simplicity of the nanoparticle formulation proposed here and the excellent dye loading capabilities of the SLNs, the use of BY-aniline revealed complex photophysical phenomena in addition to the surprising PA properties such as a PGE above 1. This complexity was found to be linked to the aggregation properties. The solid-state absorption spectrum indicates similar J-type aggregation in the BY-aniline-Palm solid. The supramolecular arrangement in the solid results in flakes. Moreover, the TEM images of SLNs showed facetted surfaces and less spherical particles as the BY-aniline-Palm proportion increased. The spatial arrangement of the structured aggregates in the SLNs and the nanostructuration of the particles could be of interest to better understand the particular properties of BY-aniline-Palm SLNs. Only a few cryo-EM images have been obtained so far but they show a variation of the nanostructure with different BY-aniline-Palm proportions. The current study demonstrated the strong involvement of the lipid matrix in the PGE value. Indeed, a comparison of SLN-2% and BY-aniline-PLA-47%, which contained the same concentration of BY-aniline but different matrices, showed an enhancement of PGE in the case of the lipids. However, the contribution of aggregation to the observed PA properties at high BY-aniline concentrations remains an open question. To investigate this issue, it would be useful to synthesize a similar molecular agent with non-aggregative properties to reach the same BY-aniline high concentrations in SLNs, but without generating aggregates. A comparative study of the nanostructural, optical, and PA properties between the two SLN types would likely isolate the structural effect of the matrix on PGE from the aggregation effect. Such a dye is currently under study, and experiments in this regard are being conducted by our consortium.
4. Materials and methods
4.1. Materials
Dexamethasone palmitate (DXP) was purchased from Interchim (France). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol))-2000 (ammonium salt)] (DSPE-PEG2000) was obtained from Avanti Polar Lipids, Inc. (USA). Palmitoyl chloride was purchased from Sigma-Aldrich. Milli-Q water was purified using a RIOS system from Merck-Millipore (France). All experiments except BODIPY-aniline-palmitate synthesis were performed in amber vials due to the photosensitivity of dexamethasone palmitate.464.2. Synthesis of BODIPY-aniline-palmitate
1H NMR (400 MHz, CDCl3): δ 7.63 (d, 2H, J = 14.5 Hz, HC = CH), 7.55 (d, 4H, J = 8.7 Hz, HAr), 7.26 (d, 2H, J = 14.2 Hz, ![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) C = CH), 6.74 (d, 4H, J = 8.2 Hz, HAr), 4.22 (t, 2H, J = 6.4 Hz, C2–O), 3.25 (t, 2H, J = 6.4 Hz, C2–S), 3.04 (s, 9H, NC3), 2.65 (q, 4H, J = 6.4 Hz, C2–CH3), 2.32 (t, 2H, J = 7.6 Hz, C = OC2), 1.64 (m, 2H, C2), 1.55 (s, 6H, C3), 1.23–1.37 (m, 24H, C2), 1.19 (t, 6H, J = 7.5 Hz, C3–CH2), 0.88 (t, 3H, J = 6.9 Hz, C3) ppm.
C = CH), 6.74 (d, 4H, J = 8.2 Hz, HAr), 4.22 (t, 2H, J = 6.4 Hz, C2–O), 3.25 (t, 2H, J = 6.4 Hz, C2–S), 3.04 (s, 9H, NC3), 2.65 (q, 4H, J = 6.4 Hz, C2–CH3), 2.32 (t, 2H, J = 7.6 Hz, C = OC2), 1.64 (m, 2H, C2), 1.55 (s, 6H, C3), 1.23–1.37 (m, 24H, C2), 1.19 (t, 6H, J = 7.5 Hz, C3–CH2), 0.88 (t, 3H, J = 6.9 Hz, C3) ppm.
13C NMR (100 MHz, CDCl3): δ 173.5, 151.8, 151.1, 137.3, 129.2, 125.9, 115.8, 112.3, 62.0, 40.4, 34.2, 33.2, 32.1, 29.8, 29.7, 29.6, 29.5, 29.4, 29.3, 25.0, 22.8, 18.7, 14.3, 14.2, 10.7 ppm.
19F NMR (376 MHz, CDCl3): δ −132.3 (m, 2F), −138.6 (m, 2F), −139.3 (q, 2F, JF–B = 30.7 Hz) ppm.
11B NMR (128 MHz, CDCl3): δ 0.1 (t, JB–F = 34.3 Hz) ppm.
HRMS (ESI): m/z: calculated for [C59H75BF6N4O2S] 1028.5608, found 1028.5592 [M+].
4.3. Nanoparticle formulation
Nanoparticles were formulated in two batches by an emulsion–evaporation process,2 as illustrated in Fig. 5, from BY-aniline-Palm (1029 g mol−1), DXP (631 g mol−1) and DSPE-PEG2000 (2806 g mol−1). 5 mL of Milli-Q water was prechilled at 4 °C and stock solutions of DXP and BY-aniline-Palm were prepared in chloroform.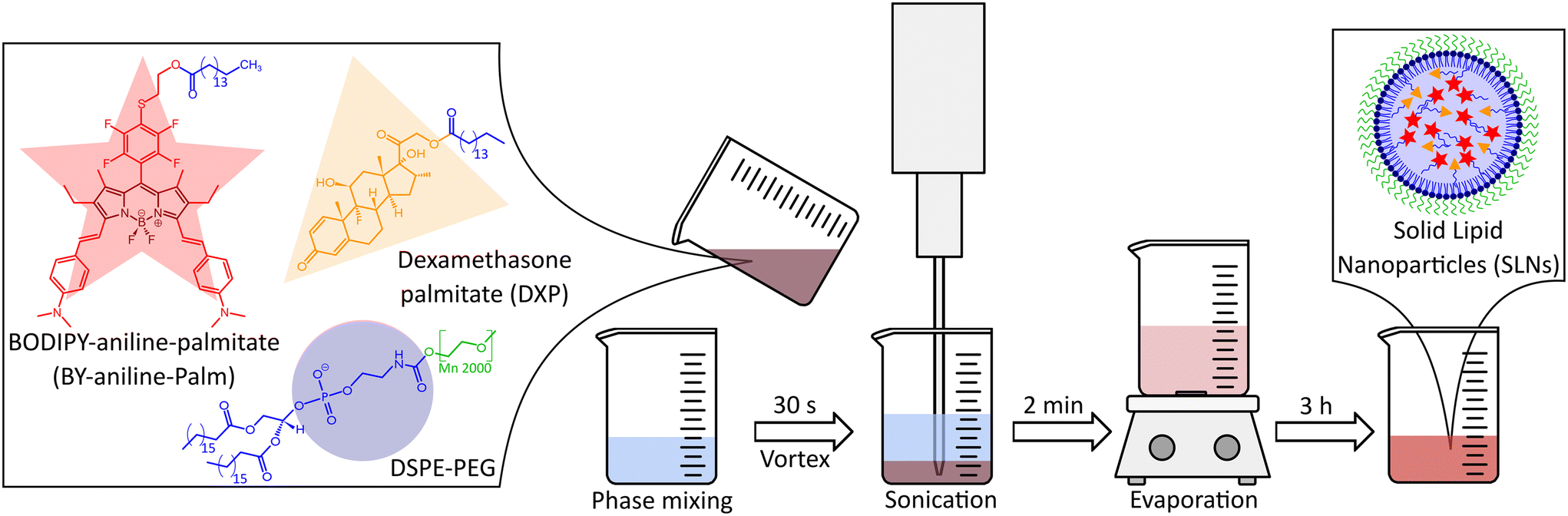 | ||
| Fig. 5 Scheme of the solid lipid nanoparticle (SLN) formulation process. All lipids are dissolved in chloroform (shown here with their schematic representations) and poured into water after vortexing for 30 s, evaporation for 2 min and evaporation for 3 h, the SLNs (supposed structure shown in schematic representation: red star for BODIPY-aniline, yellow triangle for dexamethasone, blue circle for the phosphatidylcholine head of DSPE-PEG, blue line for the lipidic chain and green line for the PEG chain) are available. | ||
The first batch, named Batch #1, was designed to obtain nine target final concentrations of BY-aniline-Palm with the following BY-aniline-Palm/DXP/DSPE-PEG2000 mass ratios: 0.00/25.00/12.50; 0.41/24.59/12.50; 0.81/24.19/12.50; 2.36/22.64/12.50; 4.55/20.45/12.50; 8.81/16.19/12.50; 15.50/9.50/12.50; 20.76/4.24/12.50; and 25.00/0.00/12.50 in mg per 5 mL. This corresponds to a constant mass of DSPE-PEG2000 (12.50 mg per 5 mL) for a molar ratio of BY-aniline-Palm/DXP of 0/100; 1/99; 2/98; 6/94; 12/88; 25/75; 50/50; 75/25 and 100/0. DXP solution was prepared at 20.0 mg mL−1 (2.53.10−2 mmol mL−1) and the BY-aniline-Palm one at 15.2 mg mL−1 (1.48.10−2 mmol mL−1).
The second batch, named Batch #2, was designed to obtain five target final concentrations of BY-aniline-Palm (among the nine concentrations of Batch #1) with the following BY-aniline-Palm/DXP/DSPE-PEG2000 mass ratios: 0.00/25.00/12.50; 0.81/24.19/12.50; 8.81/16.19/12.50; 15.50/9.50/12.50; and 25.00/0.00/12.50 in mg per 5 mL. This corresponds to a constant mass of DSPE-PEG2000 (12.50 mg per 5 mL) for a molar ratio of BY-aniline-Palm/DXP of 0/100; 2/98; 25/75; 50/50 and 100/0.
For each formulation, an appropriate volume of each solution was taken through precision Hamilton syringes and the mixture was completed with chloroform to reach 2 mL. 12.5 mg of DSPE-PEG2000 were added, and the organic phase was poured into the 5 mL water phase. The mixture was then pre-emulsified by vortexing for 30 s before being placed under ultrasonication in an ice bath for 2 min at an amplitude of 40% with a Digital Sonifier 450 instrument (Branson, USA) for Batch #1 and a Sonifier SFX150 instrument for Batch #2 (Branson, USA). The organic-phase evaporation was notably performed under ambient conditions in a chemical hood for 3 h with 300 rpm stirring to ensure homogeneous soft evaporation conditions. After full evaporation of the solvent, the suspension volume was completed to 5 mL with Milli-Q water in a volumetric flask, and nanoparticles were stored at 4 °C protected from light.
The SLN-2% formulation corresponds to a BY-aniline-Palm molar concentration of 1.56 × 10–1 mM, which is comparable to the BY-aniline-PLA-50% formulation (BY-aniline-PLA molar concentration of 1.67 × 10–1 mM) as a reference.12 We reproduced the formulation protocol of ref. 12 to get the BY-aniline-PLA-47% formulation (corresponding to a BY-ani-PLA molar concentration of 1.56 × 10–1 mM). Briefly, 11.75 mg of BY-ani-PLA and 13.25 mg of DSPE-PEG5000 were put in 2 mL of chloroform and poured into 5 mL of a cold solution of sodium cholate (15 g L−1). After 30 s of vortexing and ultrasonication in an ice bath for 1 min at an amplitude of 30% with a Sonifier SFX150 instrument (Branson, USA), the organic phase was evaporated under a chemical hood for 3 h with 300 rpm stirring. Sodium cholate was removed by ultracentrifugation for 1 h at 4 °C and 40000 rpm (Optima LE-80K ultracentrifuge, Beckman Coulter, USA). The obtained solid was resuspended in Milli-Q water to a final polymer concentration of 5 mg mL−1.
4.4. Size and ζ potential measurements
Size and ζ potential were determined with a Zetasizer Nano-ZS from Malvern Instrument (UK), based on quasi-elastic light scattering. The measurements were performed with a 633 nm laser in triplicate at a backscattered angle of 173° at 25 °C but also, when mentioned for the size measurements, at 15 °C and 35 °C. Hydrodynamic diameter (dH) and polydispersity index (PdI) were recorded on 1/20 diluted samples in Milli-Q water, and ζ potential measurements on 1/10 diluted samples in 1 mM NaCl.4.5. Nanoparticle tracking analysis (NTA)
Nanoparticle tracking analysis (NTA) using a NanoSight LM10 instrument (NanoSight, Amesbury, UK) was carried out to assess the number of nanoparticles per mL after a 10000-fold or 20000-fold dilution in water. The signal was collected using a sCMOS camera and a 405 nm monochromatic laser. For all the experiments, the following parameters were defined in the acquisition software: a controlled temperature of 25.0 °C, a camera level of 13 and a detection threshold of 5. Measurements were done in quintuplicate; mean values and standard deviations are indicated. By dividing the BY-aniline-PLA or -Palm concentration in the formulation by the number of particles, we obtain the number of BY-aniline per particle.
4.6. Transmission electron microscopy (TEM)
Transmission electron microscopy was performed at I2BC (CNRS, Gif-sur-Yvette, France). A volume of 5 μL of the nanoparticle suspension with 3.5 mg mL−1 lipid was deposited for 1 min on glow-discharged 400 mesh formvar-coated copper grids. Negative staining was performed by the addition of a drop of uranyl acetate at 2% w/w for 15 s repeated twice. Excess solution was removed, and grids were left to dry before observation. The observations were carried out on a JEOL JEM-1400 microscope (Japan) at an acceleration voltage of 120 kV. Images were acquired using an Orius camera (Gatan Inc., USA).4.7. Photophysical measurements
Scattering was suppressed by the use of the integration sphere and by the use of unloaded particles as the spectral blank (Fig. S5†). Spectra were measured at a constant concentration of BY-aniline-Palm (1.73.10−2 mM) corresponding to an optical density around 1. The dilutions in Milli-Q water required are given in Table S1† and the spectra are then corrected from the real BY-aniline-Palm concentration to the targeted one.
For the second batch, supplementary absorption spectra were recorded using a V-650 spectrophotometer (Jasco, Japan) with a Peltier system to precisely control the acquisition temperature. Spectra were recorded at 15 °C, 25 °C and 35 °C consistently with photoacoustic spectra measurements.
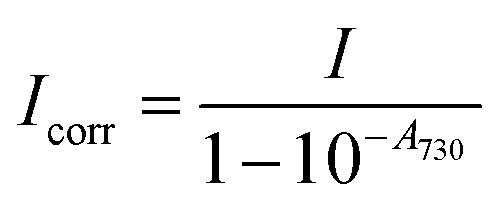 .
.
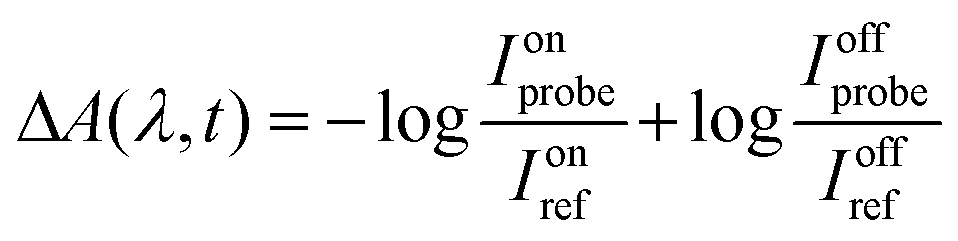 with Ionprobe and Ionref as the probe and reference signals when the pump laser is on, and Ioffprobe and Ioffref as the probe and reference signals when the pump is off. Spectra were obtained in 10 mm path length quartz cuvettes with a volume of 1 mL. For all samples, absorbance spectra were checked before and after the measurements to ensure that the samples had not been degraded by the pump laser (which was never the case).
with Ionprobe and Ionref as the probe and reference signals when the pump laser is on, and Ioffprobe and Ioffref as the probe and reference signals when the pump is off. Spectra were obtained in 10 mm path length quartz cuvettes with a volume of 1 mL. For all samples, absorbance spectra were checked before and after the measurements to ensure that the samples had not been degraded by the pump laser (which was never the case).
4.8. Photoacoustic spectra measurements
The samples were injected twice into the four tubes for a total of 8 measurements per sample. The median value and the median absolute deviation with a scaling factor 1.4826 were computed over the 8 measurements to obtain the photoacoustic coefficient and its deviation. In this paper, we call MAD the median absolute deviation with a scaling factor of 1.4826.
Gold nanoparticles were also used as an external reference compound. Au@DTDTPA 8 nm particles, synthesized at Université de Franche-Comté (CNRS, Institut UTINAM, Besançon, France),20 were selected because of their PGE being equal to 1 over the entire wavelength range according to Lucas et al.20
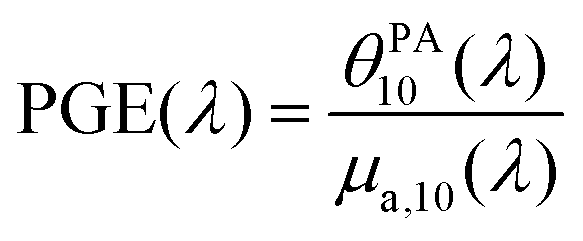 | (3) |
We also use the global photoacoustic efficiency (GPAE), as defined by Bodin et al.15 GPAE is the ratio between the integral of the PA spectrum and the integral of the corresponding absorption spectrum on the interval [λ1 = 680 nm, λ2 = 920 nm]. It can be seen as the weighted mean PGE over the whole spectrum.
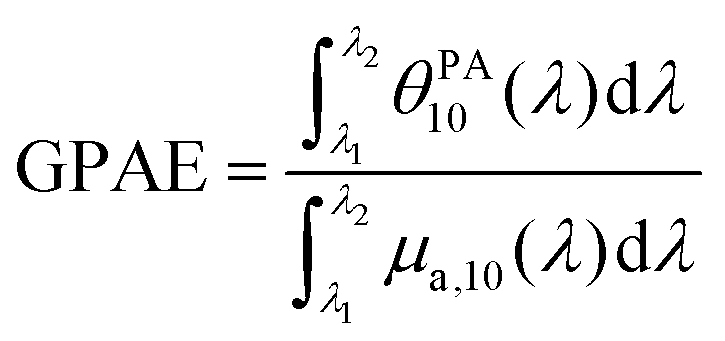 | (4) |
The integrals were calculated on the experimental data with the trapezoidal rule.
4.9. Spectra decomposition
We implemented a decomposition of the absorption spectra and the PA spectra into the sum of three Gaussian functions. We used a fitting algorithm based on a nonlinear least-squares solver (trust-region-reflective method) to determine the amplitude ai (cm−1), the central wavelength λi (nm) and the Gaussian root mean square (RMS) width ωi (nm) of each Gaussian function of the sum: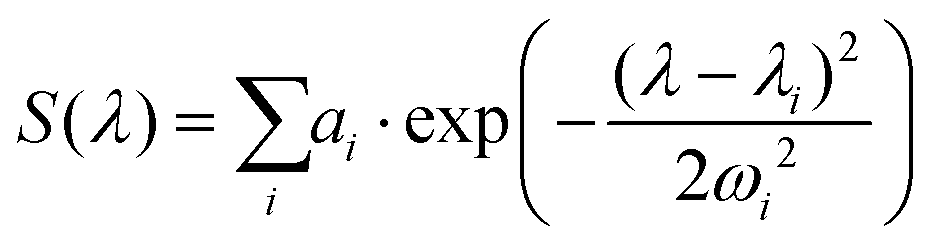 with i ∈ ⟦1,3⟧. ωi is proportional to the full width at half maximum (FWHM) of the Gaussian function
with i ∈ ⟦1,3⟧. ωi is proportional to the full width at half maximum (FWHM) of the Gaussian function  .
.
Absorption spectra between 640 nm and 980 nm were decomposed into the sum of 3 Gaussian functions. The Gaussian functions were numbered by decreasing central wavelength: λ1 ≈ 865 nm, λ2 ≈ 780 nm and λ3 = 685 nm. An illustration of this decomposition is given in Fig. 6 for the absorption spectra of BY-aniline-Palm in solution (Gaussian #2 and #3) and in the solid state (Gaussian #1 to #3). PA spectra between 680 nm and 930 nm were modelled for the SLN suspensions by the sum of 3 Gaussian curves with the same central wavelengths λ1, λ2 and λ3. For each SLN batch, the parameter determination was achieved by simultaneous and joint optimization of the curve fitting for both the absorption spectrum and the PA spectrum.
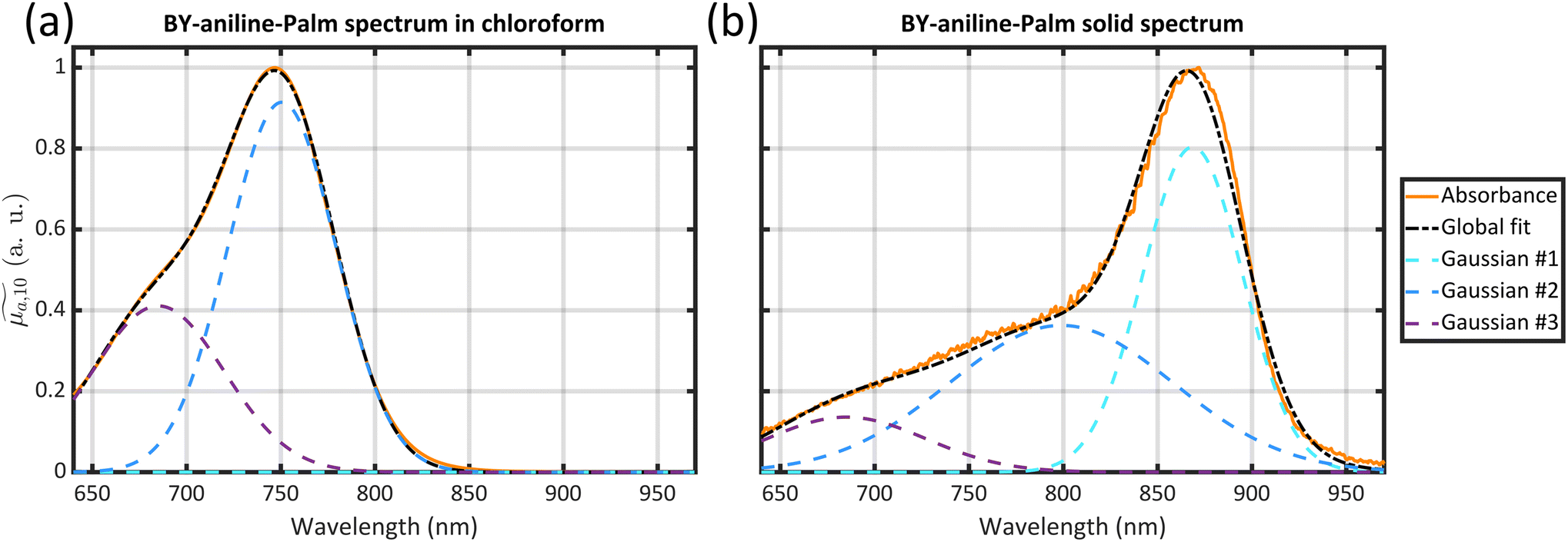 | ||
| Fig. 6 Decomposition, as the sum of three Gaussian functions, of the absorption spectrum of BODIPY-aniline-Palm (a) in chloroform and (b) in the solid state after drop casting. From left to right, the Gaussian functions are centred at 685 nm (purple), 750–799 nm (blue) and 860–869 nm (cyan). | ||
In the following, parameters for the PA spectrum are identified with a prime symbol, while the parameters for the absorption spectrum have no prime symbol. To help algorithm convergence, central wavelengths for Gaussian functions #1 and #2 were set to be equal with a ± 5 nm tolerance between the absorption spectrum and the PA spectrum, especially because the absorption spectrum has a 0.5 nm step while the PA spectrum has a 10 nm step. Therefore, a first constraint was 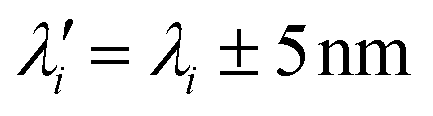 for i < 3. The RMS widths were set to be equal:
for i < 3. The RMS widths were set to be equal:  . Amplitudes
. Amplitudes  and ai were not coupled. Additional constrains were applied for the different bands but remained the same for all SLN batches. For Gaussian function #3, the central wavelength was set to
and ai were not coupled. Additional constrains were applied for the different bands but remained the same for all SLN batches. For Gaussian function #3, the central wavelength was set to 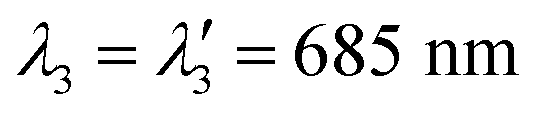 , because of the vicinity between the central wavelength and the lower wavelength limit of the PA spectrum. For Gaussian function #2, the central wavelength and the RMS width were bounded by the results of solid-state spectrum decomposition, i.e. λ2 < 799 nm and ω2 < 59.5 nm. Finally, for Gaussian function #1, we determined that the amplitude a1 had non-significant values for BY-aniline-Palm percentages below 50% and it was set to a1 = 0. The RMS width was set to equal the RMS width found in the solid-state spectrum decomposition, i.e. ω1 = 26.5 nm.
, because of the vicinity between the central wavelength and the lower wavelength limit of the PA spectrum. For Gaussian function #2, the central wavelength and the RMS width were bounded by the results of solid-state spectrum decomposition, i.e. λ2 < 799 nm and ω2 < 59.5 nm. Finally, for Gaussian function #1, we determined that the amplitude a1 had non-significant values for BY-aniline-Palm percentages below 50% and it was set to a1 = 0. The RMS width was set to equal the RMS width found in the solid-state spectrum decomposition, i.e. ω1 = 26.5 nm.
The areas of the Gaussian functions were calculated with the analytical expression of the adimensional integral:
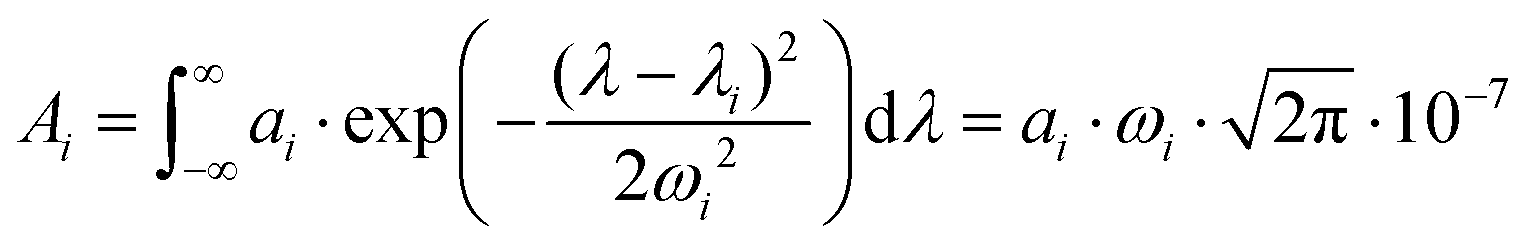 | (5) |
We then use the band photoacoustic efficiency (BPAE) defined in ref. 15 as the ratio of the PA band area to the corresponding absorption band area. In our case, as the RMS widths of the Gaussian functions were set to be equal for the PA and absorption decompositions, the BPAE can be computed by a ratio of amplitudes (Table S3†).
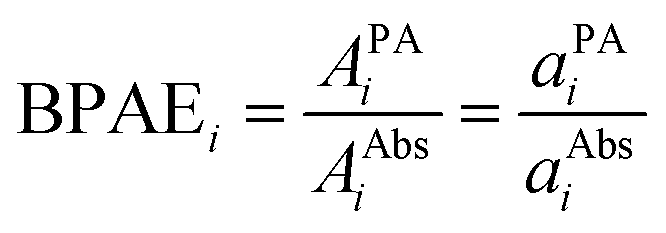 | (6) |
5. Conclusions
We have successfully labelled SLNs with a BODIPY-aniline for PAI, using a simple and direct one-pot formulation protocol. Our particle design enabled us to study the absorption and PA spectral transformations as a function of the percentage of dyes incorporated into the SLN. Highly absorbing SLNs with a remarkable absorption cross section per mole of particles were obtained. Despite significant spectral modifications, modelling the spectrum as the sum of three Gaussian functions allowed us to simplify the study to the interplay of three spectral bands, which were attributed to different photophysical phenomena (vibration, absorption, and aggregation). The interplay was quantitatively demonstrated. A precise measurement of the PA generation efficiency over the entire spectrum, per band, and per wavelength highlighted the enhancement of the PA signal caused by the lipid core (PGE above 1), in comparison with previously described polymer particles, and raised questions about the involvement of different energy conversions (light-to-heat and thermoelastic expansion) in the per-band variations of the efficiency. Additional PA experiments were conducted by varying the ambient temperature and the excitation fluence to clarify these points. This study clearly opens the way to careful consideration of the optical and photothermal phenomena at stake in optically labelled nanoparticles for photoacoustic imaging, in particular when building materials, such as lipids, possess a Grüneisen coefficient larger than that of water.Author contributions
J. G., N. T., C. L., R. M. and G. C. are responsible for the conceptualization of the project and experiments. C. L., N. T. and J. G. developed the SLN formulation. C. L. and G. M. performed the SLN formulations. C. L. and G. M. performed the photoacoustic and spectrophotometric measurements. C. L., J. G., N. T. and R. M. analysed and evaluated the data. G. C. developed, prepared and characterized the BY-aniline. G. C. and C. L. synthesized and characterized BY-aniline-Palm. All authors discussed and contributed to the preparation of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received financial support from the French National Research Agency under the program ANR-21-CE09-0024-01 and from the CNRS through the MITI interdisciplinary programs. It has also benefited from the Imagerie–Gif core facility supported by L'Agence Nationale de la Recherche (ANR-11-EQPX-0029/Morphoscope, ANR-10-INBS-04/FranceBioImaging; ANR-11-IDEX-0003-02/Saclay Plant Sciences), with the help of C. Gillet and C. Boulogne. The authors warmly thank G. Laurent, R. Bazzi and S. Roux from the Institut UTIMAM Université de Franche-Comté/CNRS for the gold nanoparticles. The authors sincerely thank M.-H. Ha-Thi and M. Dang from the ISMO Université Paris-Saclay/CNRS for the transient absorption experiments. The authors warmly thank F. Gobeaux from LIONS-NIMBE UMR 3685 CEA/CNRS for the cryo-EM images. The authors acknowledge B. Prost, SAMM core facility from IPSIT, for providing HRMS analysis. This work was supported in part by France Life Imaging under grant no. ANR-11-INBS-0006. RM thanks the Région Ile-de-France and DIM NanoK for financial support as well as CHARMMMAT LabEx (11-LABX-0039).References
- J. Akbari, M. Saeedi, F. Ahmadi, S. M. H. Hashemi, A. Babaei, S. Yaddollahi, S. S. Rostamkalaei, K. Asare-Addo and A. Nokhodchi, Pharm. Dev. Technol., 2022, 27, 525–544 CrossRef CAS PubMed
.
- M. Lorscheider, N. Tsapis, R. Simón-Vázquez, N. Guiblin, N. Ghermani, F. Reynaud, R. Canioni, S. Abreu, P. Chaminade and E. Fattal, Mol. Pharm., 2019, 16, 2999–3010 CrossRef CAS
- M. Lorscheider, N. Tsapis, M. ur-Rehman, F. Gaudin, I. Stolfa, S. Abreu, S. Mura, P. Chaminade, M. Espeli and E. Fattal, J. Controlled Release, 2019, 296, 179–189 CrossRef CAS PubMed
- K. Oumzil, M. A. Ramin, C. Lorenzato, A. Hémadou, J. Laroche, M. J. Jacobin-Valat, S. Mornet, C.-E. Roy, T. Kauss, K. Gaudin, G. Clofent-Sanchez and P. Barthélémy, Bioconjugate Chem., 2016, 27, 569–575 CrossRef CAS PubMed
- E. Andreozzi, P. Wang, A. Valenzuela, C. Tu, F. Gorin, M. Dhenain and A. Louie, Bioconjugate Chem., 2013, 24, 1455–1467 CrossRef CAS
- G. Chinigò, A. Gonzalez-Paredes, A. Gilardino, N. Barbero, C. Barolo, P. Gasco, A. Fiorio Pla and S. Visentin, Spectrochim. Acta, Part A, 2022, 271, 120909 CrossRef PubMed
- V. Ntziachristos, Nat. Methods, 2010, 7, 603–614 CrossRef CAS PubMed
- P. C. Beard, Interface Focus, 2011, 1, 602–631 CrossRef PubMed
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed
- R. E. Borg and J. Rochford, Photochem. Photobiol., 2018, 94, 1175–1209 CrossRef CAS PubMed
- P. Ramezani, S. C. De Smedt and F. Sauvage, Bioeng. Transl. Med., 2024, e10652 CrossRef CAS
- J.-B. Bodin, J. Gateau, J. Coïs, T. Lucas, F. Lefebvre, L. Moine, M. Noiray, C. Cailleau, S. Denis, G. Clavier, N. Tsapis and R. Méallet-Renault, ACS Appl. Mater. Interfaces, 2022, 40501–40512 CrossRef CAS PubMed
- T. Lucas, M. Sarkar, Y. Atlas, C. Linger, G. Renault, F. Gazeau and J. Gateau, Sensors, 2022, 22, 6543 CrossRef CAS PubMed
- Q. Fu, R. Zhu, J. Song, H. Yang and X. Chen, Adv. Mater., 2019, 31, 1805875 CrossRef PubMed
- J.-B. Bodin, C. Linger, J. Gateau, T. Beguin, T. Lucas, L. Moine, D. Chapron, M.-H. Ha-Thi, A. Fatima, G. Clavier, N. Tsapis and R. Méallet, J. Phys. Chem. C, 2023, 127, 18971–18985 CrossRef CAS
- W. Hu, X. Miao, H. Tao, A. Baev, C. Ren, Q. Fan, T. He, W. Huang and P. N. Prasad, ACS Nano, 2019, 13, 12006–12014 CrossRef CAS PubMed
- S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751–755 RSC
- K. Li, X. Duan, Z. Jiang, D. Ding, Y. Chen, G.-Q. Zhang and Z. Liu, Nat. Commun., 2021, 12, 2376 CrossRef CAS PubMed
- J. L. Bricks, Y. L. Slominskii, I. D. Panas and A. P. Demchenko, Methods Appl. Fluoresc., 2017, 6, 012001 CrossRef PubMed
- T. Lucas, C. Linger, T. Naillon, M. Hashemkhani, L. Abiven, B. Viana, C. Chanéac, G. Laurent, R. Bazzi, S. Roux, S. Becharef, G. Avveduto, F. Gazeau and J. Gateau, Nanoscale, 2023, 15, 17085–17096 RSC
- T. Stahl, T. Allen and P. Beard, Characterization of the thermalisation efficiency and photostability of photoacoustic contrast agents, Proc. SPIE 8943, Photons Plus Ultrasound: Imaging and Sensing, 2014, 89435H, DOI:10.1117/12.2039694.
- J. P. Fuenzalida Werner, Y. Huang, K. Mishra, R. Janowski, P. Vetschera, C. Heichler, A. Chmyrov, C. Neufert, D. Niessing, V. Ntziachristos and A. C. Stiel, Anal. Chem., 2020, 92, 10717–10724 CrossRef CAS PubMed
- T. Stahl, R. Bofinger, I. Lam, K. J. Fallon, P. Johnson, O. Ogunlade, V. Vassileva, R. B. Pedley, P. C. Beard, H. C. Hailes, H. Bronstein and A. B. Tabor, Bioconjugate Chem., 2017, 28, 1734–1740 CrossRef CAS PubMed
- S. Liang, B. Lashkari, S. S. S. Choi, V. Ntziachristos and A. Mandelis, Photoacoustics, 2018, 11, 56–64 CrossRef PubMed
- M. Duquesne, C. Mailhé, S. Doppiu, J.-L. Dauvergne, S. Santos-Moreno, A. Godin, G. Fleury, F. Rouault and E. Palomo Del Barrio, Materials, 2021, 14, 4707 CrossRef CAS
- O. Zmeskal, L. Marackova, T. Lapcikova, P. Mencik and R. Prikryl, AIP Conf. Proc., 2020, 2305, 020022 CrossRef CAS
- V. K. Pustovalov, Nanotechnol. Precis. Eng., 2024, 7, 015001 CrossRef CAS
- M. Pyda, R. C. Bopp and B. Wunderlich, J. Chem. Thermodyn., 2004, 36, 731–742 CrossRef CAS
- H. Aoki, M. Nojiri, R. Mukai and S. Ito, Nanoscale, 2014, 7, 337–343 RSC
- G. A. Pang, F. Poisson, J. Laufer, C. Haisch and E. Bossy, J. Phys. Chem. C, 2020, 124, 1088–1098 CrossRef CAS
- O. Simandoux, A. Prost, J. Gateau and E. Bossy, Photoacoustics, 2015, 3, 20–25 CrossRef PubMed
- A. Prost, F. Poisson and E. Bossy, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 115450 CrossRef
- A. Kumari, K. Kumari and S. Gupta, Sci. Rep., 2019, 9, 20334 CrossRef CAS PubMed
- S. Jaiswal, S. B. Dutta, D. Nayak and S. Gupta, ACS Omega, 2021, 6, 34842–34849 CrossRef CAS PubMed
- G. Marbán, T. T. Vu and T. Valdés-Solís, Appl. Catal., A, 2011, 402, 218–223 CrossRef
- D. K. Das, K. Makhal, S. N. Bandyopadhyay and D. Goswami, Sci. Rep., 2014, 4, 6097 CrossRef CAS
- K. Zlatić, H. B. E. Ayouchia, H. Anane, B. Mihaljević, N. Basarić and T. Rohand, J. Photochem. Photobiol., A, 2020, 388, 112206 CrossRef
- A. Fatima, J. Rabah, E. Allard, H. Fensterbank, K. Wright, G. Burdzinski, G. Clavier, M. Sliwa, T. Pino, R. Méallet-Renault, K. Steenkeste and M.-H. Ha-Thi, Photochem. Photobiol. Sci., 2022, 21, 1573–1584 CrossRef CAS PubMed
- C. Gómez-Gaete, N. Tsapis, L. Silva, C. Bourgaux, M. Besnard, A. Bochot and E. Fattal, Eur. J. Pharm. Biopharm., 2008, 70, 116–126 CrossRef PubMed
- C. C. L. Cheung, G. Ma, K. Karatasos, J. Seitsonen, J. Ruokolainen, C.-R. Koffi, H. A. F. M. Hassan and W. T. Al-Jamal, Nanotheranostics, 2020, 4, 91–106 CrossRef PubMed
- D. Miranda, H. Huang, H. Kang, Y. Zhan, D. Wang, Y. Zhou, J. Geng, H. I. Kilian, W. Stiles, A. Razi, J. Ortega, J. Xia, H. S. Choi and J. F. Lovell, Theranostics, 2019, 9, 381–390 CrossRef CAS PubMed
- B. Changalvaie, S. Han, E. Moaseri, F. Scaletti, L. Truong, R. Caplan, A. Cao, R. Bouchard, T. M. Truskett, K. V. Sokolov and K. P. Johnston, ACS Appl. Mater. Interfaces, 2019, 11, 46437–46450 CrossRef CAS PubMed
- M. R. Kawelah, S. Han, C. Atila Dincer, J. Jeon, J. Brisola, A. F. Hussain, A. S. Jeevarathinam, R. Bouchard, A. E. Marras, T. M. Truskett, K. V. Sokolov and K. P. Johnston, ACS Appl. Mater. Interfaces, 2024, 16, 5598–5612 CrossRef CAS PubMed
- C. A. Wood, S. Han, C. S. Kim, Y. Wen, D. R. T. Sampaio, J. T. Harris, K. A. Homan, J. L. Swain, S. Y. Emelianov, A. K. Sood, J. R. Cook, K. V. Sokolov and R. R. Bouchard, Nat. Commun., 2021, 12, 5410 CrossRef CAS PubMed
- W. Xu, J. Leskinen, T. Sahlström, E. Happonen, T. Tarvainen and V.-P. Lehto, Photoacoustics, 2023, 33, 100552 CrossRef PubMed
- R. Canioni, F. Reynaud, T. Leite-Nascimento, C. Gueutin, N. Guiblin, N.-E. Ghermani, C. Jayat, P. Daull, J.-S. Garrigue, E. Fattal and N. Tsapis, Int. J. Pharm., 2021, 600, 120509 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02880e |
| ‡ Equal leading contribution. |
| This journal is © The Royal Society of Chemistry 2025 |