
Controlled bioorthogonal catalyst self-assembly and activity using rationally designed polymer scaffolds†
Rui
Huang
a,
Cristina-Maria
Hirschbiegel
 a,
David C.
Luther
a,
Cheng-Hsuan
Li
a,
Ahmed
Nabawy
a,
Jungmi
Park
a,
Alexander
Ribbe
b,
Yisheng
Xu
c and
Vincent M.
Rotello
*a
a,
David C.
Luther
a,
Cheng-Hsuan
Li
a,
Ahmed
Nabawy
a,
Jungmi
Park
a,
Alexander
Ribbe
b,
Yisheng
Xu
c and
Vincent M.
Rotello
*a
aDepartment of Chemistry, University of Massachusetts Amherst, 710 North Pleasant Street, Amherst, Massachusetts 01003, USA. E-mail: rotello@umass.edu
bDepartment of Polymer Science and Engineering, University of Massachusetts Amherst, 120 Governors Dr, Amherst, MA 01003, USA
cState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai, 200237, P. R. China
First published on 13th November 2024
Abstract
Polymer-based nanocatalysts have been extensively utilized in advanced drug delivery due to their versatility and high reactivity. Incorporating bioorthogonal transition metal catalysts (TMCs) into polymers generates bioorthogonal nanocatalysts capable of producing therapeutic agents in situ, minimizing off-target effects. The supramolecular interactions within the polymer matrix, including hydrophobic interactions and aromatic stacking, play a crucial role in catalytic properties. Our study focuses on co-engineering the host polymer structure and the catalyst encapsulation process to achieve precise control over the supramolecular interactions within the nanoenvironment. By carefully engineering these interactions, we successfully generate thermo-responsive nanocatalysts with a resolution of 6 °C. These nanocatalysts demonstrate thermal activation of the catalytic deprotection of a pro-antibiotic, with concomitant external control of bacterial biofilm eradication.
Introduction
Nanocatalysts are versatile tools with diverse applications in chemical synthesis,1,2 pharmaceuticals,3–5 pollution remediation,6,7 and the food industry.8,9 Polymer-based nanocatalysts combine the ease of functionalization of polymers with the high reactivity of nanocatalysis.10–13 The encapsulation of bioorthogonal transition metal catalysts (TMCs) into polymers generates nanocatalysts capable of catalyzing abiotic reactions without interfering with endogenous processes.14–17 These bioorthogonal nanocatalysts enable the generation of therapeutic agents in situ, minimizing off-target effects.18–21The nanoenvironment within the polymer matrix significantly impacts the catalytic properties of nanocatalysts.22–24 Supramolecular interactions govern the spatial arrangement and mobility of the embedded catalyst molecules.25–29 Precise control of these supramolecular interactions allows the regulation of substrate accessibility to the catalytic sites in response to different physicochemical conditions, opening up the possibility of creating stimuli-responsive nanocatalysts.30–32 We report here the synergistic control of supramolecular interactions within a polymer nanoenvironment, leading to the generation of thermo-responsive nanocatalysts. Nanocatalysts were fabricated by encapsulating a bioorthogonal TMC, iron(III) tetraphenyl porphyrin (FeTPP), into the hydrophobic pocket of a poly(oxanorbornene)imide-based random co-polymer (PONI) (Fig. 1). By carefully engineering the architecture of the host polymer with hydrophilic functionalities, we modulated the degree of hydrophobic chain packing, confirmed by nuclear magnetic resonance (NMR). Furthermore, by strategically engineering the catalyst encapsulation process, we regulated the aromatic stacking between the catalyst molecules, confirmed by UV-Vis spectroscopy. The combined effect of these factors synergistically influenced the degree of aggregation in FeTPP self-assemblies, creating a thermally regulated ‘turn-on’ catalytic system with a resolution of 6 °C. Notably, we tailored the activation temperature of these nanocatalysts within the physiological range (25–37 °C), ensuring their compatibility with biologically relevant systems. The nanocatalysts demonstrated thermal activation of a pro-antibiotic in E. coli bacterial biofilms, allowing for precise externally controlled eradication of bacterial biofilm.
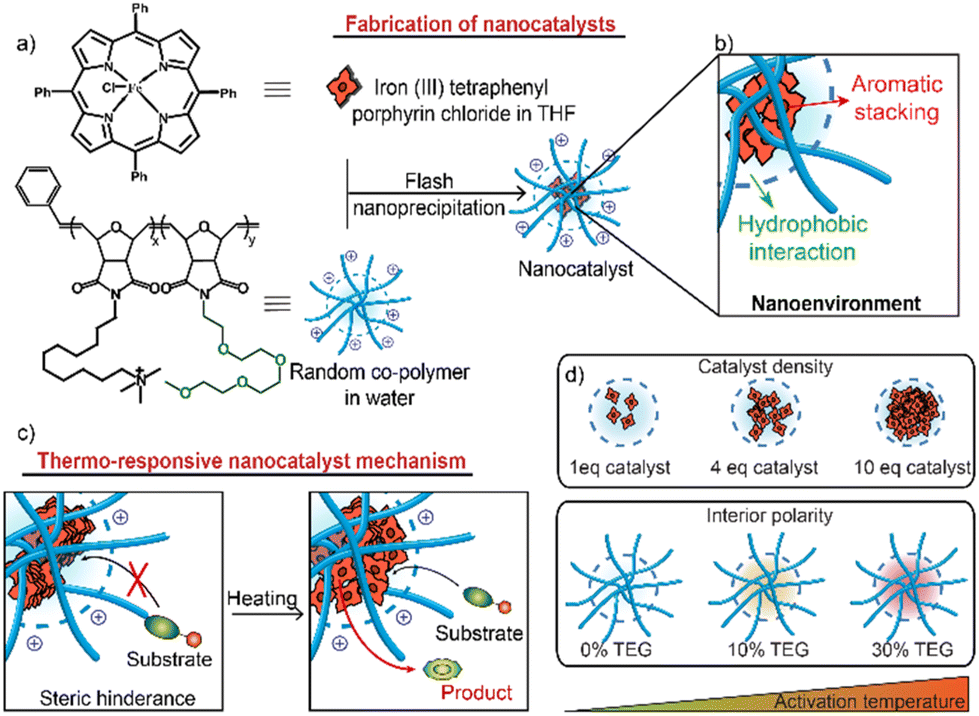 | ||
| Fig. 1 (a) Structural compositions of random co-polymers and FeTPP and illustration of the nanocatalyst fabrication process. (b) Supramolecular interactions within the nanoenvironment of nanocatalysts. (c) Demonstration of thermo-responsive mechanism: FeTPP self-assembles within the polymer hydrophobic pocket, thus blocking the substrate access. An increase in temperature facilitates the dissociation of FeTPP assemblies, restoring catalysis. (d) Depiction of two key parameters influencing the activation temperature of thermo-responsive nanocatalysts. | ||
Results and discussion
Design and synthesis of thermo-responsive nanocatalysts
FeTPP is a bioorthogonal catalyst capable of reducing aryl azides to their respective amines.10,33,34 FeTPP reduces the azide moieties in solution, as demonstrated by Meggers and co-workers.33 However, while FeTPP is catalytically active in organic solvents, it is insoluble in water (Fig. S10 and S11†) and therefore benefits from encapsulation into nanomaterials. When encapsulated into polymers, FeTPP molecules self-assemble through aromatic stacking interactions between the aromatic rings, blocking the access of the substrate to their metal center and rendering them inactive. However, when subjected to heat, the aromatic stacking interactions can be disrupted, leading to the activation of FeTPP and the initiation of catalytic reactions.35,36The hosting random co-polymer for FeTPP was designed with monomers featuring two crucial components: (i) a C11-bridged trimethylammonium (C11-TMA) chain to foster the self-assembly of the polymer, stabilizing and solubilizing FeTPP molecules and (ii) a hydrophilic tetraethylene glycol monomethyl ether chain (TEG-OMe) to regulate the polarity within the polymer interior (Fig. 1a).
The supramolecular interactions within the nanoenvironment of nanocatalysts cooperatively dictate their catalytic behavior (Fig. 1b). Hydrophobic interactions drive the formation of a hydrophobic pocket within the polymer matrix to encapsulate FeTPP molecules. The aromatic stacking between FeTPPs governs their self-assembly within this hydrophobic pocket. Incorporating TEG-OMe groups enhances the overall polarity within the polymer interior, resulting in tighter packing of hydrophobic chains to minimize their exposure to this polar nanoenvironment. Upon heating, the nanoenvironment induces a concurrent disaggregation of FeTPP assemblies and an alteration of the hydrophobic pocket, facilitating the access of the substrate to catalyst active centers.28 This temperature-dependent accessibility enables the generation of thermally regulated ‘turn-on’ nanocatalysts.
Higher proportions of TEG-OMe and increased catalyst loading were expected to lead to a higher activation temperature of thermo-responsive nanocatalysts due to the tighter hydrophobic chain packing and higher catalyst density within the nanoenvironment (Fig. 1d). We generated a random co-polymer library where we parametrically varied the proportion of TEG-OMe (0%, 10%, and 30%) within the host polymer structures to dictate the interior polarity in regulating the compactness of the hydrophobic pocket. This library was expanded by adjusting the polymer-to-catalyst feed ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:4, and 1:10) to explore the impact of the catalyst loading on its thermally induced disaggregation (Fig. 2a).
:1, 1:4, and 1:10) to explore the impact of the catalyst loading on its thermally induced disaggregation (Fig. 2a).
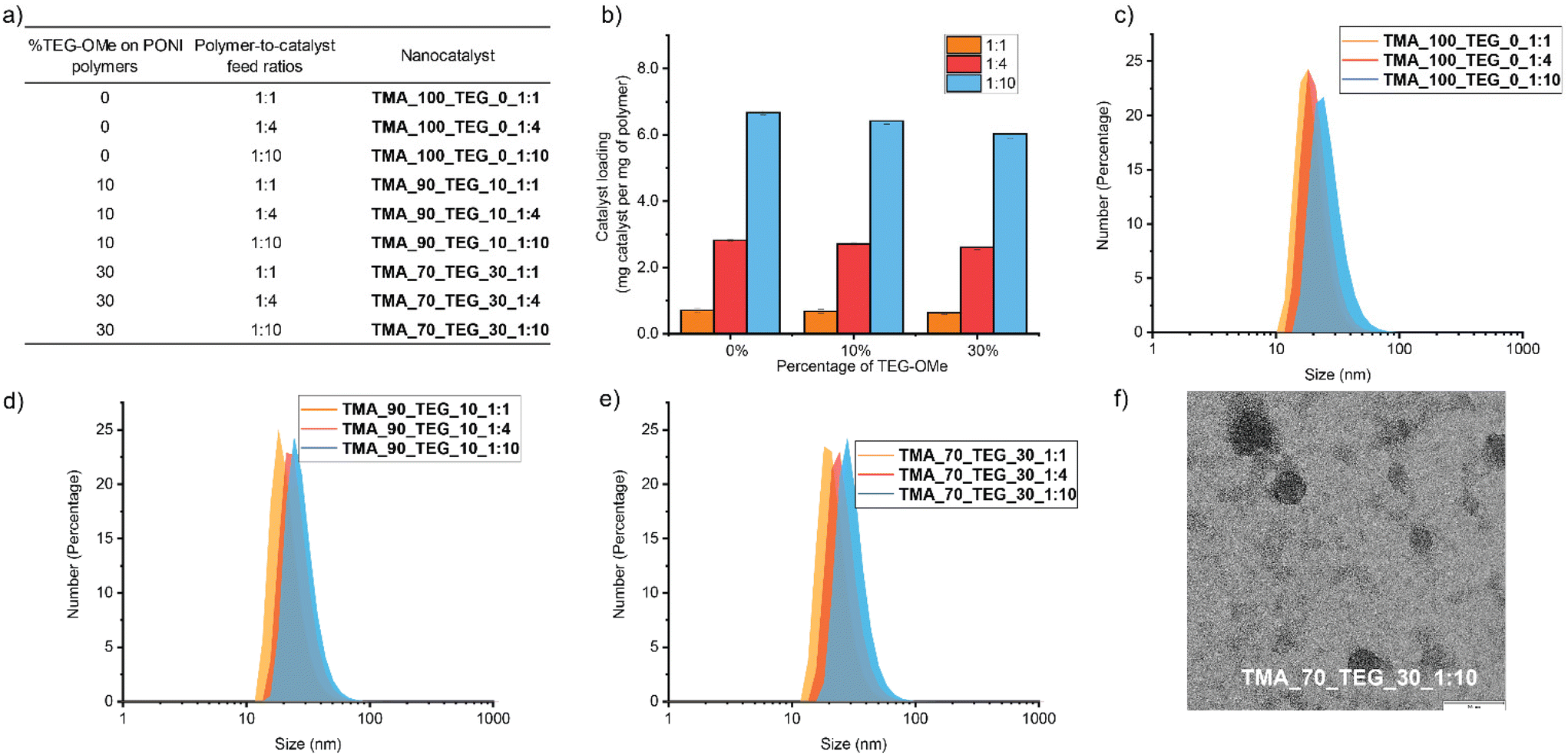 | ||
| Fig. 2 (a) Overview of the nomenclature system for polymer samples: TMA and TEG monomers with composition percentage, followed by polymer-to-catalyst feed ratio. (b) Catalyst loading of nanocatalysts with different TEG-OMe proportions in polymer structures and polymer-to-catalyst feed ratios. Each experiment was performed in triplicate. (c)–(e) Dynamic light scattering (DLS) measurements of nanocatalysts. (f) Transmission electron microscopy (TEM) image of TMA_70_TEG_30_1:10 at low and high magnifications. The scale bar is 90 nm. | ||
Nanocatalysts were prepared by microfluidically mixing 0.6 mL polymer aqueous solutions (1 mg mL−1) with 0.6 mL FeTPP in THF (1, 4, and 10 mg mL−1) through flash nanoprecipitation (FNP). FNP is a simple and efficient process for stabilizing insoluble compounds into nanosized polymer-based delivery vehicles.37–39 The high flow rate of two impingent streams enables rapid mixing and creates supersaturation conditions, generating monodispersed nanocatalysts with high loading capacity.40,41 Compared to traditional mixing methods, FNP exhibited enhanced encapsulation efficiency due to less precipitation of the catalyst during the mixing process (Fig. S4†), offering precise control over the catalyst density within the nanoenvironment. The nanocatalyst products were centrifuged with a filter (10 kDa) five times to remove any unbound catalyst. The amounts of FeTPP encapsulated in each polymer were quantified using UV-Vis spectroscopy (Fig. S5† and Fig. 2b).
Dynamic light scattering (Fig. 2c–e) measurements on the nanocatalysts showed the formation of nanocatalysts with no aggregation observed after the encapsulation of FeTPP. Minor particle size increase was observed when the polymer-to-catalyst feed ratio was increased across all three polymers (0%, 10%, and 30% TEG-OMe). This observation aligns with the FNP principle that a higher solute concentration prompts a higher degree of supersaturation, accelerating solute particle growth and resulting in larger particle sizes.39,42,43 The spherical morphology of the nanocatalyst was verified using transmission electron microscopy (TEM) imaging (Fig. 2f and Fig. S7†) and energy dispersive X-ray spectrometry (EDS) (Fig. S8 and 9†) to confirm the presence of encapsulated FeTPP.
Thermally activated catalytic bioorthogonal pro-dye deprotection
The catalytic activity of nanocatalysts was assessed by activating a rhodamine-based pro-dye (Fig. 3a). The pro-dye, initially non-fluorescent due to the masking of bis-azide groups, was rendered fluorescent upon the FeTPP-mediated catalytic reduction. The inherent fluorogenic property of the activated rhodamine provided a convenient and direct method for evaluating the catalytic kinetics.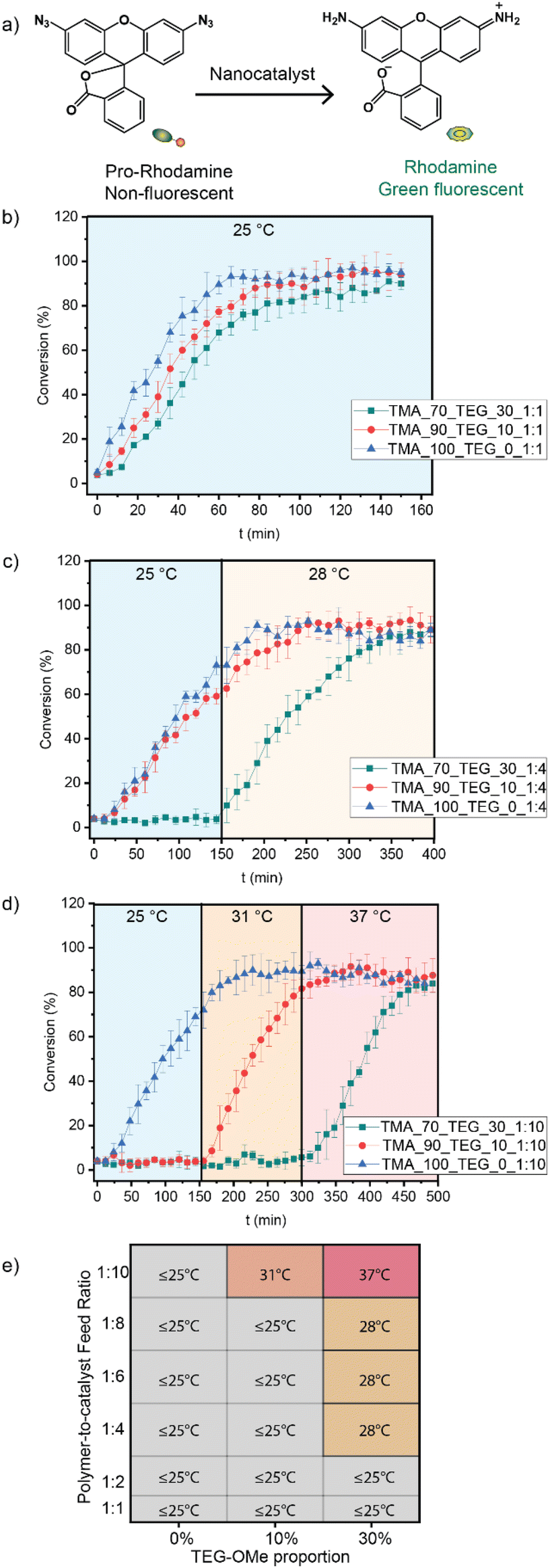 | ||
| Fig. 3 (a) Demonstration of the pro-rhodamine activation process, mediated by FeTPP through the reduction of aryl azide. The thermo-responsive behavior of different nanocatalysts (200 nM) was studied by measuring pro-rhodamine activation (10 μM) at different temperatures. Polymer-to-catalyst feed ratios are (b) 1:1, (c) 1:4, and (d) 1:10. We used 1 mM GSH as a reducing agent. Each experiment was performed in triplicate. (e) Heat map illustrating the nanocatalyst library. The activation temperature of each thermo-responsive nanocatalyst is labeled. The color provides a visual representation of the activation temperatures (increase in red indicates higher activation temperature). | ||
The activation of pro-rhodamine (Ex. 488 nm and Em. 521 nm) was performed by the different nanocatalyst variants at different temperatures (Fig. 3b–d). When the polymer-to-catalyst feed ratio is kept constant at 1:1, all three variants of the nanocatalysts (0%, 10%, and 30% TEG-OMe) activated pro-rhodamine at 25 °C without thermo-responsive control (Fig. 3b). However, by increasing the polymer-to-catalyst feed ratio to 1:4, a thermo-responsive nanocatalyst was obtained using the polymer containing 30% TEG-OMe (TMA_70_TEG_30_1:4), with an activation temperature of 28 °C (Fig. 3c). Higher activation temperatures were achieved by further increasing the catalyst loading. Increasing the polymer-to-catalyst feed ratio to 1:10 provided thermo-responsive nanocatalysts in polymers containing 10% and 30% TEG-OMe (TMA_90_TEG_10_1:10 and TMA_70_TEG_30_1:10), with the activation temperature at 31 °C and 37 °C, respectively (Fig. 3d). We further expanded the nanocatalyst library by systematically screening more polymer-to-catalyst ratios and generated a comprehensive heat map (Fig. 3e).
These results demonstrate that the proportion of TEG-OMe in the polymer structures and the polymer-to-catalyst feed ratio significantly impact the aggregation of the catalyst molecules within the polymer scaffold, which consequently modulation the activation temperature of the thermo-responsive nanocatalysts. Depending on the polymer structure and feed ratio, disaggregation of the catalyst is achieved at different temperatures, demonstrating the ability of polymer structure to dictate thermal properties of the nanocatalysts Next, we performed a series of experiments to study the effect of these parameters on supramolecular interactions within the nanoenvironment. We focused on TMA_70_TEG_30_1:10, as 37 °C is considered an optimal temperature for bacterial and cell-culture studies.
UV-Vis spectroscopy was used to investigate the supramolecular interactions between the encapsulated FeTPP assemblies. FeTPP typically exhibits a characteristic absorption band (Soret band), usually observed at around ∼410 nm.44,45 The dissociation of FeTPP assemblies into smaller structures is indicated by an increase in intensity and a blue shift of the Soret band.46,47 No significant changes in the Soret bands of the FeTPP assemblies were observed until the temperature reached the activation threshold of the nanocatalyst (Fig. 4a). This observation suggests a correlation between the activation of catalytic activity and the dissociation of FeTPP.
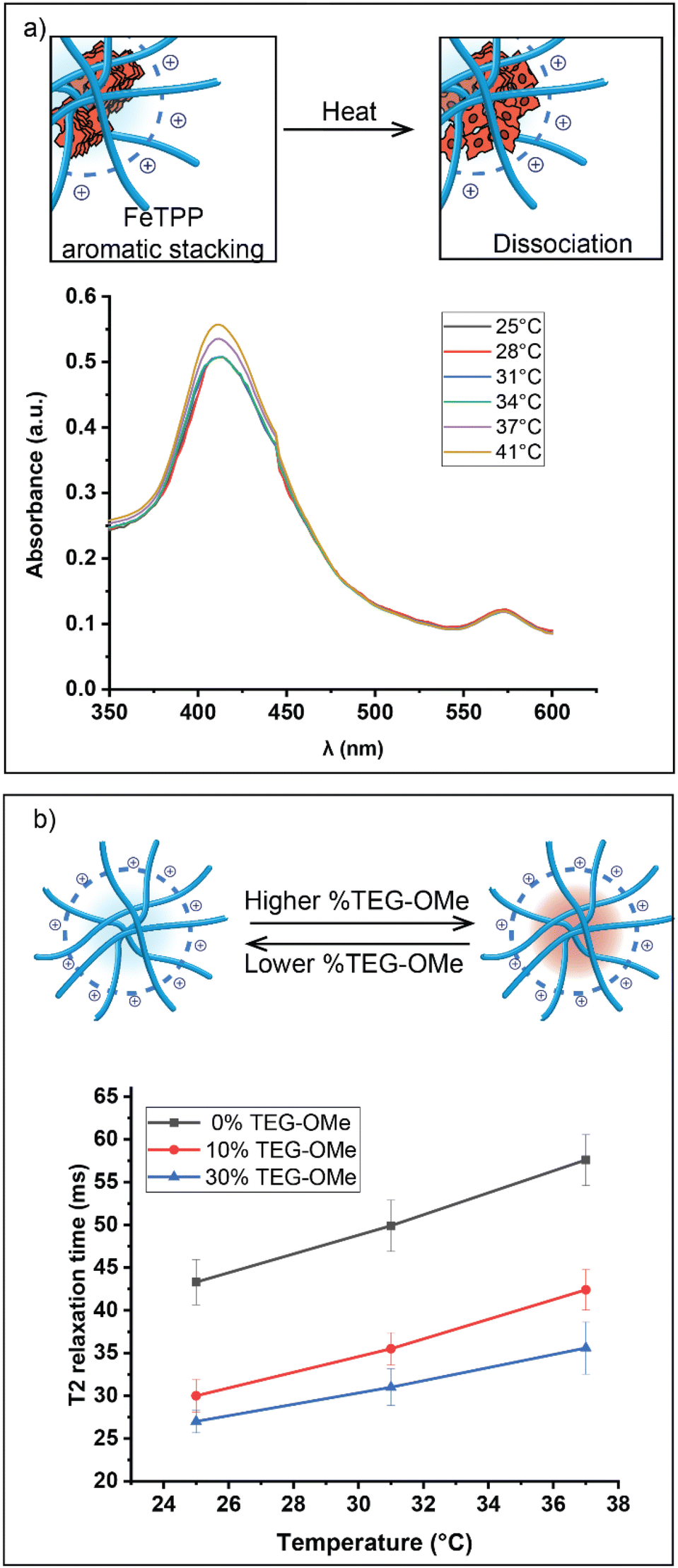 | ||
| Fig. 4 (a) Schematic representation of the dissociation of FeTPP assemblies upon increasing temperature. This dynamic process in TMA_70_TEG_30_1:10 was monitored using UV-Vis spectroscopy. The intensity of the Soret band showed changes only when the nanocatalyst reached the thermal threshold. (b) Schematic illustration of hydrophobic pocket conformations regulated by TEG-OMe proportions on the polymer structures. This conformational difference was studied by measuring the T2 relaxation time in D2O using the CPMG pulse sequence. | ||
DLS measurements of TMA_70_TEG_30_1:10 showed minimal changes in particle sizes across the investigated temperature range, indicating that the restoration of catalytic activity was not induced by particle aggregation (Fig. S12†). Additionally, catalyst loadings of TMA_70_TEG_30_1:10 measured by UV-Vis spectroscopy exhibited negligible changes after incubation at 37 °C for 4 hours and 24 hours, confirming the absence of catalyst leakage into the solution.
These results demonstrated that the activation temperature of nanocatalysts is primarily governed by the dissociation processes of FeTPP assemblies within the nanoenvironment. Adjusting the polymer-to-catalyst feed ratio can achieve precise control over FeTPP dissociation. Increasing the catalyst loading fosters stronger aromatic stacking interactions owing to higher catalyst density and tighter packing within the hydrophobic pocket due to larger assembly size. Consequently, higher activation temperatures are required to overcome these enhanced intermolecular interactions to expand the hydrophobic pocket and disaggregate FeTPP assemblies.
We next used T2 relaxation analysis to validate the impact of TEG-OMe on the degree of hydrophobic chain packing.48,49T2 relaxation, measured by nuclear magnetic resonance (NMR) spectroscopy, quantifies the loss of phase coherence among nuclear spins in the transverse plane.50 The T2 relaxation time is inversely correlated with the rate of molecular motion, providing a sensitive and robust approach to monitoring the packing degree of hydrophobic chains within the nanoenvironment.51
Our investigation primarily concentrated on the broad NMR resonance visible at 0.5–2 ppm in 1H NMR spectroscopy. This resonance correlates with the non-substituted segment of the C11-bridged alkyl chain, which is crucial in forming the hydrophobic pocket. Polymers with a higher proportion of TEG-OMe ligands displayed a shorter T2 relaxation time, indicating a more compact hydrophobic pocket (Fig. 4b). This effect can be attributed to the inherently strong polarity of ether bonds present in TEG-OMe groups. Introducing these polar groups amplifies the overall polarity of the nanoenvironment, leading to a significant solvophobic effect on the C11-chain. As a result, the hydrophobic chains tightly pack together to minimize their exposure to the polar surroundings. Moreover, the increase in T2 relaxation time at elevated temperatures confirmed that the hydrophobic pocket tends to expand when heated.
Harnessing thermo-responsive nanocatalysts for pro-drug activation
Having characterized our thermo-responsive nanocatalysts in solution, we explored their potential application in eradicating bacterial biofilms. We selected ciprofloxacin (Cip), a potent broad-spectrum antibiotic, as our antimicrobial agent (Fig. 5a). Cip is a broad-spectrum antibiotic quinolone that inhibits bacterial DNA topoisomerase II and IV.52 However, quinolones have been associated with adverse effects on the central nervous system, such as headaches, dizziness, and agitation due to the binding of quinolones to GABA receptors in the brain.53 Therefore, antibacterial quinolones such as Cip benefit from the local activation in bacterial biofilms.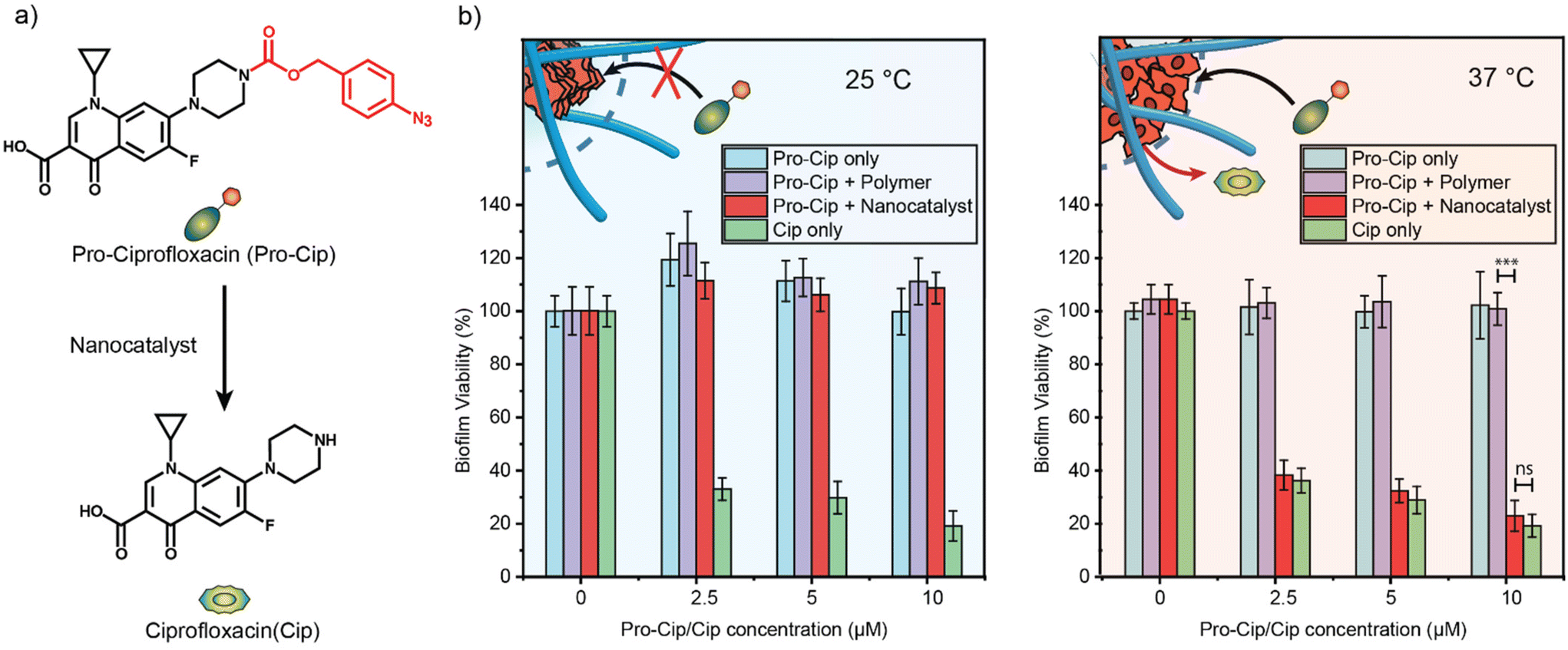 | ||
| Fig. 5 (a) Schematic representation of the activation of pro-Cip, utilizing 1 mM GSH as a reducing agent. (b) Viability of E. coli (CD-2) biofilms after incubation with TMA_70_TEG_30_1:10 (200 nM) and prodrugs for 6 hours at 25 °C and 37 °C. Biofilms treated with prodrug, drug, and prodrug + polymers were used as controls. Each experiment was performed in triplicate, and error bars represent the standard deviation. Statistical analysis was performed using the Student's t-test. ns = not significant; *** = p ≤ 0.001. | ||
In previous work, Cip was strategically modified into a prodrug (pro-Cip) by masking its secondary amino group, efficiently inhibiting the binding to its target enzymes.54,55 This modification involved introducing a bulky aryl-azide carbamate moiety as a caging group, which significantly decreased the antimicrobial activity of Cip by more than two orders of magnitude (Fig. S14†). The efficacy of TMA_70_TEG_30_1:10 in uncaging this prodrug was quantified with a ninhydrin assay (Fig. S15†).
Pathogenic E. coli is a main contributor to bacterial infections in the United States and rapidly acquires antimicrobial resistance, particularly in biofilms.56 We utilized 3-day-old biofilms of pathogenic E. coli (CD-2) to test the effectiveness of pro-Cip activation. E. coli biofilms treated with TMA_70_TEG_30_1:10 and different concentrations of pro-Cip were incubated at two temperatures, 25 °C and 37 °C, over 6 hours. The samples were subsequently washed with PBS, and their viability was assessed using an Alamar Blue assay.57,58 Biofilms treated solely with different concentrations of pro-Cip or TMA_70_TEG_30_1:10 were set as negative controls, whereas biofilms treated with active Cip at varying concentrations served as positive controls. The highest concentration of the pro-drug and drug used during the drug-activation study was 10 μM, respectively, at which the active drug exhibited its maximum bacteria-killing ability.
The drug release experiments were conducted at 25 °C and 37 °C (Fig. 5b). Biofilms exposed to pro-Cip and TMA_70_TEG_30_1:10 at 37 °C demonstrated a significant reduction in viability compared to those treated under the same conditions at 25 °C. However, biofilms treated solely with different concentrations of pro-Cip displayed a negligible reduction in bacterial viability at both temperatures. As positive controls, biofilms treated with active Cip demonstrated a killing effect in a dose-dependent manner, regardless of the incubation temperature. Furthermore, biofilms treated with the pro-drug and the polymer only do not have reduced bacteria viability, emphasizing the necessity of the bioorthogonal catalyst for pro-drug activation. These results demonstrate the capability of TMA_70_TEG_30_1:10 to thermally regulate bioprocesses, facilitating the uncaging of an antimicrobial drug within complex biological environments with an efficiency equivalent to the free drug. Furthermore, our results demonstrate the translatability of the thermal regulation of catalysts to a cellular environment.
Conclusions
In conclusion, this study provides unique insights into the precise control of activation temperature in thermo-responsive nanocatalysts. Our investigation demonstrated that the activation temperature can be finely modulated by tailoring two key parameters: the proportion of TEG-OMe groups in the host polymer structures and the polymer-to-catalyst feed ratios. Our results demonstrated that these parameters synergistically influence the supramolecular interactions within their nanoenvironment. Depending on the catalyst environment and feed ratio, disaggregation of the catalyst can be observed at different activation temperatures. Furthermore, we successfully demonstrated the applicability of this nanocatalyst system in a complex bacterial biofilm environment, where it effectively enabled thermal activation of a pro-drug to eradicate the biofilm. This study provides a robust framework for developing thermo-responsive nanocatalysts with adjustable activation temperatures. The precise control of the polymer interior provides a milestone in the design of supramolecular nanocatalysts, paving the way for applying tailored and targeted catalytic reactions in biomedicine.Author contributions
Conceptualization: R. H., V. M. R.; investigation: R. H., C.-M. H., D. L., C.-H. L., A. N., J. P., A. R., V. M. R.; visualization: R. H.; writing – original draft: R. H., C.-M. H.; writing – review & editing: R. H., C.-M. H., V. M. R.; resources: Y. X., V. M. R.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Institutes of Health R01 EB022641 (V. M. R.) and the National Key Research and Development Program of the International Scientific and Technological Innovation Cooperation Project among Governments of China (2021YFE0100400) (Y. X.). C.-M. H. was partially supported by a fellowship from the University of Massachusetts as part of the Chemistry-Biology Interface Training Program (National Research Service Award T32 GM139789).References
- A. Maleki, Polycycl. Aromat. Compd., 2016, 38, 402–409 CrossRef.
- S. B. Somwanshi and P. B. Kharat, J. Phys.: Conf. Ser., 2020, 1644, 012046 CrossRef.
- Q. Wang, Z. Zhang, E. Wang and S. Dong, Trends Anal. Chem., 2018, 105, 218–224 CrossRef CAS.
- C.-M. Hirschbiegel, X. Zhang, R. Huang, Y. A. Cicek, S. Fedeli and V. M. Rotello, Adv. Drug Delivery Rev., 2023, 195, 114730 CrossRef CAS PubMed.
- X. Zhang, R. Huang, S. Gopalakrishnan, R. Cao-Milán and V. M. Rotello, Trends Chem., 2019, 1, 90–98 CrossRef CAS PubMed.
- R. Ningthoujam, Y. D. Singh, P. J. Babu, A. Tirkey, S. Pradhan and M. Sarma, Chem. Phys., 2022, 4, 100064 Search PubMed.
- F. Lu and D. Astruc, Coord. Chem. Rev., 2020, 408, 213180 CrossRef CAS.
- J. M. Navia-Mendoza, O. A. Filho, L. A. Zambrano-Intriago, M. M. M. B. Duarte, L. S. Quiroz-Fernández, R. J. Baquerizo-Crespo and J. M. Rodríguez-Díaz, Sustainability, 2021, 13, 11676 CrossRef CAS.
- B. Ernest, P. J. Shiny, A. Mukherjee and N. Chandrasekaran, Carbohydr. Res., 2012, 352, 60–64 CrossRef PubMed.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- R. Huang, C.-H. Li, R. Cao-Milán, J. M. Makabenta, X. Zhang, E. Yu and V. M. Rotello, J. Am. Chem. Soc., 2020, 142, 10723–10729 CrossRef CAS PubMed.
- X. Zhang, R. F. Landis, P. Keshir, R. Cao-Milán, D. C. Luther, S. Gopalakrishnan, Y. Liu, R. Huang, G. Li, M. Malassiné, I. Uddin, B. Rondon and V. M. Rotello, Adv. Healthcare Mater., 2020, 10, 2001627 CrossRef.
- M. Jiang, A. N. Chattopadhyay, C.-H. Li, Y. Geng, D. C. Luther, R. Huang and V. M. Rotello, Chem. Sci., 2022, 13, 12899–12905 RSC.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- X. Zhang, Y. Liu, S. Gopalakrishnan, L. Castellanos-Garcia, G. Li, M. Malassiné, I. Uddin, R. Huang, D. C. Luther, R. W. Vachet and V. M. Rotello, ACS Nano, 2020, 14, 4767–4773 CrossRef CAS.
- W. Wang, X. Zhang, R. Huang, C.-M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed.
- C.-H. Li, R. Huang, J. M. Makabenta, S. Schmidt-Malan, R. Patel and V. M. Rotello, Microbiol. Insights, 2021, 14, 117863612199712 Search PubMed.
- J. Hardie, J. M. Makabenta, A. Gupta, R. Huang, R. Cao-Milán, R. Goswami, X. Zhang, P. Abdulpurkar, M. E. Farkas and V. M. Rotello, Mater. Horiz., 2022, 9, 1489–1494 RSC.
- R. Huang, D. C. Luther, X. Zhang, A. Gupta, S. A. Tufts and V. M. Rotello, Nanomaterials, 2021, 11, 1001 CrossRef CAS PubMed.
- C.-M. Hirschbiegel, S. Fedeli, X. Zhang, R. Huang, J. Park, Y. Xu and V. M. Rotello, Materials, 2022, 15, 6487 CrossRef CAS PubMed.
- B. Rubio-Ruiz, A. M. Pérez-López, L. Uson, C. M. Ortega-Liebana, T. Valero, M. Arruebo, J. L. Hueso, V. Sebastian, J. Santamaria and A. Unciti-Broceta, Nano Lett., 2023, 23, 804–811 CAS.
- Y. Wang, W. Wang, Z. Gu, X. Miao, Q. Huang and B. Chang, RSC Adv., 2020, 10, 39954–39966 CAS.
- S. Carregal-Romero, N. J. Buurma, J. Pérez-Juste, L. Liz-Marzán and P. Hervés, Chem. Mater., 2010, 22, 3051–3059 CAS.
- A. A. P. Mansur, H. S. Mansur and S. M. Carvalho, Catal. Today, 2022, 388, 187–198 CrossRef.
- M. Solra, S. Das, A. Srivastava, B. Sen and S. Rana, ACS Appl. Mater. Interfaces, 2022, 14, 45096–45109 CrossRef CAS.
- B. Huang, J. Tian, Z. Cui, S. Weng, W. Wang, X. Jiang and W. Zhang, Chem. Eng. J., 2022, 444, 136164 CrossRef CAS.
- J. Han, H. Gong, X. Ren and X. Yan, Nano Today, 2021, 41, 101295 CrossRef CAS.
- R. Cao-Milán, S. Gopalakrishnan, L. D. He, R. Huang, L.-S. Wang, L. Castellanos, D. C. Luther, R. F. Landis, J. M. V. Makabenta, C.-H. Li, X. Zhang, F. Scaletti, R. W. Vachet and V. M. Rotello, Chem, 2020, 6, 1113–1124 Search PubMed.
- C.-M. Hirschbiegel, R. Goswami, S. Chakraborty, C. Noonan, E. Pham, H. Nagaraj, W. Ndugire, S. Fedeli and V. M. Rotello, J. Polym. Sci., 2024, 62, 3787–3793 CrossRef CAS.
- F. Hapiot, S. Menuel and E. Monflier, ACS Catal., 2013, 3, 1006–1010 CrossRef CAS.
- J.-H. Park, J.-W. Jang, J.-H. Sim, I.-J. Kim, D.-J. Lee, Y.-H. Lee, S.-H. Park and H.-D. Kim, Int. J. Polym. Sci., 2019, 2019, 1–7 CrossRef.
- B. Cheng, Y. Wan, Q. Tang, Y. Du, F. Xu, Z. Huang, W. Qin and X. Chen, Chin. J. Chem., 2021, 40, 806–812 CrossRef.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Koester, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS.
- F. Yang, M. Xu, X. Chen and Y. Luo, Biomed. Pharmacother., 2023, 164, 114933 CrossRef CAS.
- L. Lin, C. Hou, Z. Zhang, Y. Wang, Y. Chen and T. He, Appl. Catal., B, 2018, 221, 312–319 CrossRef CAS.
- C. Wang, R. Yuan, Y. Chai, S. Chen, Y. Zhang, F. Hu and M. Zhang, Electrochim. Acta, 2012, 62, 109–115 CrossRef CAS.
- N. J. Caggiano, B. C. Wilson, R. D. Priestley and R. K. Prud'homme, Mol. Pharm., 2022, 19, 1515–1525 CrossRef CAS.
- W. Ye, G. Zhang, X. Liu, Q. Ren, F. Huang and Y. Yan, J. Drug Delivery Sci. Technol., 2022, 70, 103183 CrossRef CAS.
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B, 2019, 9, 4–18 CrossRef.
- R. Huang, C.-M. Hirschbiegel, X. Zhang, A. Gupta, S. Fedeli, Y. Xu and V. M. Rotello, ACS Appl. Mater. Interfaces, 2022, 14, 31594–31600 CrossRef CAS.
- S. Fedeli, R. Huang, Y. Oz, Z. Zhang, A. Gupta, S. Gopalakrishnan, J. M. V. Makabenta, S. Lamkin, A. Sanyal, Y. Xu and V. M. Rotello, ACS Appl. Mater. Interfaces, 2023, 15, 15260–15268 CrossRef CAS.
- K. M. Pustulka, A. R. Wohl, H.-S. Lee, A. Michel, J. Han, T. R. Hoye, A. V. McCormick, J. Panyam and C. W. Macosko, Mol. Pharm., 2013, 10, 4367–4377 CrossRef CAS PubMed.
- W. S. Saad and R. K. Prud'homme, Nano Today, 2016, 11, 212–227 CrossRef CAS.
- D. N. Hendrickson, M. G. Kinnaird and K. S. Suslick, J. Am. Chem. Soc., 1987, 109, 1243–1244 CrossRef CAS.
- L. Shao, B. Hu, P. Dong, W. Ji and C. Qi, J. Porphyrins Phthalocyanines, 2010, 14, 993–999 CrossRef CAS.
- C.-N. Cao and Y.-T. Long, Acc. Chem. Res., 2018, 51, 331–341 CrossRef CAS.
- A. Satake and Y. Kobuke, Org. Biomol. Chem., 2007, 5, 1679–1679 RSC.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- Y. Chen, W. Li, K. Jiang, C. Y. Wang and X. Yu, J. Magn. Reson. Imaging, 2016, 44, 375–382 CrossRef PubMed.
- S. K. Filippov, A. Bogomolova, L. Kaberov, N. Velychkivska, L. Starovoytova, Z. Cernochova, S. E. Rogers, W. M. Lau, V. V. Khutoryanskiy and M. T. Cook, Langmuir, 2016, 32, 5314–5323 CrossRef CAS PubMed.
- M. Wu, A. M. Vartanian, G. Chong, A. K. Pandiakumar, R. J. Hamers, R. Hernandez and C. J. Murphy, J. Am. Chem. Soc., 2019, 141, 4316–4327 CrossRef CAS.
- A. Shariati, M. Arshadi, M. A. Khosrojerdi, M. Abedinzadeh, M. Ganjalishahi, A. Maleki, M. Heidary and S. Khoshnood, Front. Public Health, 2022, 10, 1025633 CrossRef.
- N. M. Ibrahim, S. H. Fahim, M. Hassan, A. E. Farag and H. H. Georgey, Eur. J. Med. Chem., 2022, 228, 114021 CrossRef CAS.
- A. Nabawy, J. M. Makabenta, S. Schmidt-Malan, J. Park, C.-H. Li, R. Huang, S. Fedeli, A. N. Chattopadhyay, R. Patel and V. M. Rotello, J. Controlled Release, 2022, 347, 379–388 CrossRef CAS PubMed.
- A. Nabawy, R. Huang, D. C. Luther, X. Zhang, C.-H. Li, J. M. Makabenta and V. M. Rotello, Chem. Sci., 2022, 13, 12071–12077 RSC.
- J. M. Rangel, P. H. Sparling, C. Crowe, P. M. Griffin and D. L. Swerdlow, Emerg. Infect. Dis., 2005, 11, 603–609 CrossRef PubMed.
- S. N. Rampersad, Sensors, 2012, 12, 12347–12360 CrossRef CAS PubMed.
- E. M. Longhin, N. El Yamani, E. Rundén-Pran and M. Dusinska, Front. Toxicol., 2022, 4, 981701 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03083d |
| This journal is © The Royal Society of Chemistry 2025 |