
Constructing a nickel complex/crystalline carbon nitride hybrid with a built-in electric field for boosting CO2 photoreduction†
Yanrui
Li
 *a,
Linda
Wang
a,
Bozhan
Li
a,
Liangqing
Zhang
a,
Xiaolin
Zhu
c and
Xiang
Gao
*b
*a,
Linda
Wang
a,
Bozhan
Li
a,
Liangqing
Zhang
a,
Xiaolin
Zhu
c and
Xiang
Gao
*b
aCollege of Materials Science and Engineering, Xi'an University of Science and Technology, Xi'an, 710054, China. E-mail: liyanrui91@xust.edu.cn
bCollege of Geology and Environment, Xi'an University of Science and Technology, Xi'an 710054, China. E-mail: gaoxiang@xust.edu.cn
cKey Laboratory of Applied Surface and Colloid Chemistry (Ministry of Education), School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an, 710119, China
First published on 12th November 2024
Abstract
Sluggish charge separation dynamics resulting from the amorphous structure and the lack of driving force for graphitic carbon nitride (GCN) limits its highly effective CO2 photoreduction performance. Herein, a built-in electric field (BEF) was constructed for a well-designed CCN/Ni hybrid composed of crystalline carbon nitride (CCN) and a metal complex, 2,2′-bipyridine-4,4′-dicarboxylic acid NiBr2 (dcabpyNiBr2), to steer charge carrier separation and migration. The CCN/Ni hybrid was synthesized via a solution–dispersion and molten-salt two-step approach, displaying an improved CO2 photoreduction to CO rate of 8.64 μmol g−1 h−1. In situ experimental results and theoretical simulations further investigated the relationships between BEF and photocatalytic activity. This work demonstrates an effective strategy to obtain high-efficiency photocatalytic systems by engineering the crystal structure and constructing a BEF.
Introduction
Global climate warming resulting from excessive carbon dioxide (CO2) caused by the tremendous consumption of fossil fuel has attracted considerable attention worldwide. CO2 photoreduction, converting CO2 and H2O molecules into value-added products driven by solar irradiation energy, is regarded as an effective approach for reducing the concentration of CO2 in the atmosphere, alleviating the greenhouse effect, and reducing the dependence on fossil fuel.1 However, thermodynamically stable CO2 molecules with multiple electrons/protons2 are needed to obtain enough energy and electrons from the photocatalyst to realize photocatalytic CO2 reduction. Unfortunately, fast electron–hole recombination, slow charge carrier migration dynamics and deficiency of surface active sites in photocatalysts inhibit efficient CO2 photoreduction performance. Thus, developing high-efficiency photocatalytic systems for photoreduction of CO2 with desirable efficiency remains highly challenging.3Graphitic carbon nitride (GCN), a two-dimensional organic semiconductor composed of heptazine-based conjugate rings in a plane with interlayer van der Waals forces,4 has been widely utilized as a photocatalyst for CO2 photoreduction owing to its advantages of high chemical and thermal stability, non-toxicity, easy synthesis from nitrogen-rich materials (urea,5 melamine,6 thiourea,7,8etc.) via thermal polymerization,9 and suitable band structure for CO2 reduction. However, GCN suffers from narrow spectral absorption, inhibited charge separation, and limited active sites because of the relatively wide bandgap, low specific surface area and amorphous structure, leading to its poor CO2 photoreduction performance. To overcome the above limitations, modifying the crystal and band structure of GCN has been widely explored to improve its photocatalytic CO2 reduction performance.
Through thermal polymerization, an all-solid-state reaction approach, abundant unpolymerized amino groups (–NH– or –NH2) in amorphous GCN bring about the formation of hydrogen bonds between heptazine-based chains, which disorders the planar arrangement and interlayer stacking order, inhibiting effective charge transfer in the plane and π-stacking direction.4 Usually, molten-salt treatment, providing a high-temperature and liquid-phase condition, has been investigated as a promising approach to improve the crystallinity of semiconductors.10,11 Lotsch et al. utilized a molten salt strategy to synthesize potassium poly(heptazine imide) with high crystallinity.12 It is observed that the high-temperature and liquid-phase condition provide two-dimensional semiconductor error correction to extend crystallites in the lateral dimension, along with metal ions from molten salts introduced into the interlayer arrangement in the π–π stacking direction. Using melamine as the precursor, Wang et al. synthesized a crystalline heptazine-based GCN photocatalyst via the molten salt method.10 Based on the improved crystallinity, the photoexcited charge carrier migration of crystalline heptazine-based GCN was enhanced, which was beneficial to the photocatalytic activity of water splitting.
Beyond these crystal structural properties, broad light absorption, effective charge carrier separation and migration, and the fast catalytic reaction rate at the active site of the photocatalyst are other essential factors for effective CO2 photoreduction performance.13 Constructing a hybrid, combining a different semiconductor,14 metal,15 quantum dot,16etc., has been regarded as a well-studied strategy for improving charge dynamic and reaction kinetics via engineering the band structure of photocatalysts.17 For example, Wang et al. modified CCN with CdS nanoparticles to construct double-junction-involved CCN/CdS hybrids, which promoted the directed migration and spatial separation of photogenerated charge carriers.18 Subsequently, Wang et al. decorated the ultrafine WO3 nanoparticles with matched band structures on the surface of CCN to create an S-scheme CCN/WO3 hybrid for efficient charge transfer and separation performance.19 Zhu et al. fabricated a high-efficiency CCN-based hybrid for photocatalytic CO2 reduction catalyst by anchoring the surface of CCN with sodium 2,5,8-tri(40-pyridyl)-1,3,4,6,7,9-hexaazaphenalenate (TPHAP), featuring a hexaazaphenalenate π-conjugated macrocycle similar to graphitic carbon nitride.20 However, it is difficult to simultaneously enhance light absorption ability, accelerate charge carrier separation and migration, and provide suitable active sites for CO2 reduction reaction in one hybrid based on the traditional inorganic semiconductor. Metal complexes, composed of adjustable central metal ions and the surrounding ligands, are known to possess tailored light absorption ranges via the metal-to-ligand21 or ligand-to-metal22 charge transfer process, MLCT or LMCT, respectively. Given that most photocatalysts have limited visible light response, this advantage makes metal complexes preferential candidates for visible-light-driven CO2 photoreduction by constructing a hybrid with another semiconductor. Depending on the strategy of energy band structural regulation, the units of hybrids possess different conduction bands (CB) and valence bands (VB), thus forming a built-in electric field (BEF) at the interface,23 which drives the photoexcited charge carrier separation and migration to the surface reaction sites. Significantly, as a multielectron reaction process for CO2 photoreduction, the metal ions of metal complexes with variable valence states frequently act as the active centre for the catalytic CO2 reaction.24,25
Considering the above statement of crystal and band structure regulation for an effective photocatalytic system, an improved CO2 photoreduction performance is expected with the “all-in-one” structure modified based on GCN. Herein, with thermally polymerized GCN as a precursor, crystalline carbon nitride (CCN) modified with a metal complex, 2,2′-bipyridine-4,4′-dicarboxylic acid NiBr2 (dcabpyNiBr2), forming CCN/Ni hybrids is obtained via the molten-salt and solution–dispersion two-step approach. The obtained hybrid exhibits improved CO2 photoreduction performance, with the CO and CH4 generation yields reaching 8.64 and 2.54 μmol g−1 h−1, which were about 3.55 times and 15.52 times that of CCN. Further investigations of charge separation dynamics and band structure confirmed the BEF between dcabpyNiBr2 and CCN steering photogenerated charge carrier separation and migration, which was beneficial for improving the CO2 photoreduction performance. This work demonstrates an effective strategy to construct BEF between GCN-based materials and metal complexes with matched energy levels to enhance CO2 photoreduction performance by engineering the crystal and band structure.
Experimental section
Materials
Melamine (C3H6N6, 99%), potassium thiocyanate (KSCN, AR), 4,4′-dimethyl-2,2′-bipyridine, H2SO4 (98%), CrO3 (99%), 4,4′-dimethyl-2,2′-bipyridine (98%), NiBr2, anhydrous ethanol (C2H6O, 99.5%), deionized water. All chemicals were used without further purification.Preparation of photocatalyst
 | ||
| Scheme 1 Molecular structure of 2,2′-bipyridine-4,4′-dicarboxylic acids NiBr2. | ||
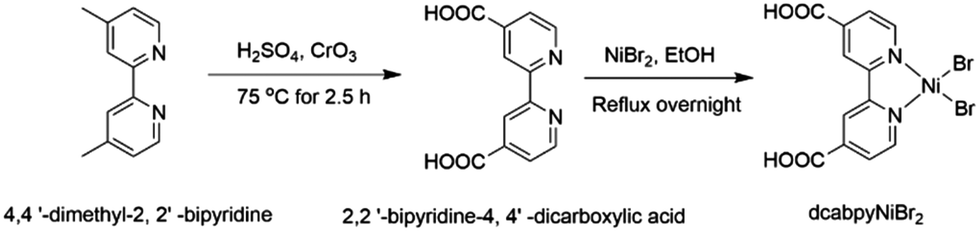 | ||
| Scheme 2 Synthesis of the dcabpyNiBr2 complex. | ||
Results and discussion
Synthesis and characterization of catalyst
Herein, starting from GCN as the precursor, the crystalline carbon nitride (CCN) is synthesized via the molten salt approach, during which the KSCN molten salt provides a liquid condition for engineering the crystal structure of GCN. Based on the CCN with improved crystallinity, the dcabpyNiBr2 metal complex solution is introduced and injected into the CCN suspension to form the hybrid CCN/Nim (m presents the mass percentage of the dcabpyNiBr2 metal complex in the hybrid; m = 1, 5, 10 and 15 meaning 1%, 5%, 10% and 15%, respectively). As shown in Scheme 3, a BEF would be formed between CCN and the dcabpyNiBr2 metal complex with matched energy levels. When CCN is irradiated by photons with equal or higher energy than its band gap (Eg), the photoexcited electron (e−) is excited and translated from VB to CB, leaving photoexcited holes (h+) on VB.3 Driven by the BEF, the photoexcited e− might migrate from CCN to the dcabpyNiBr2 metal complex.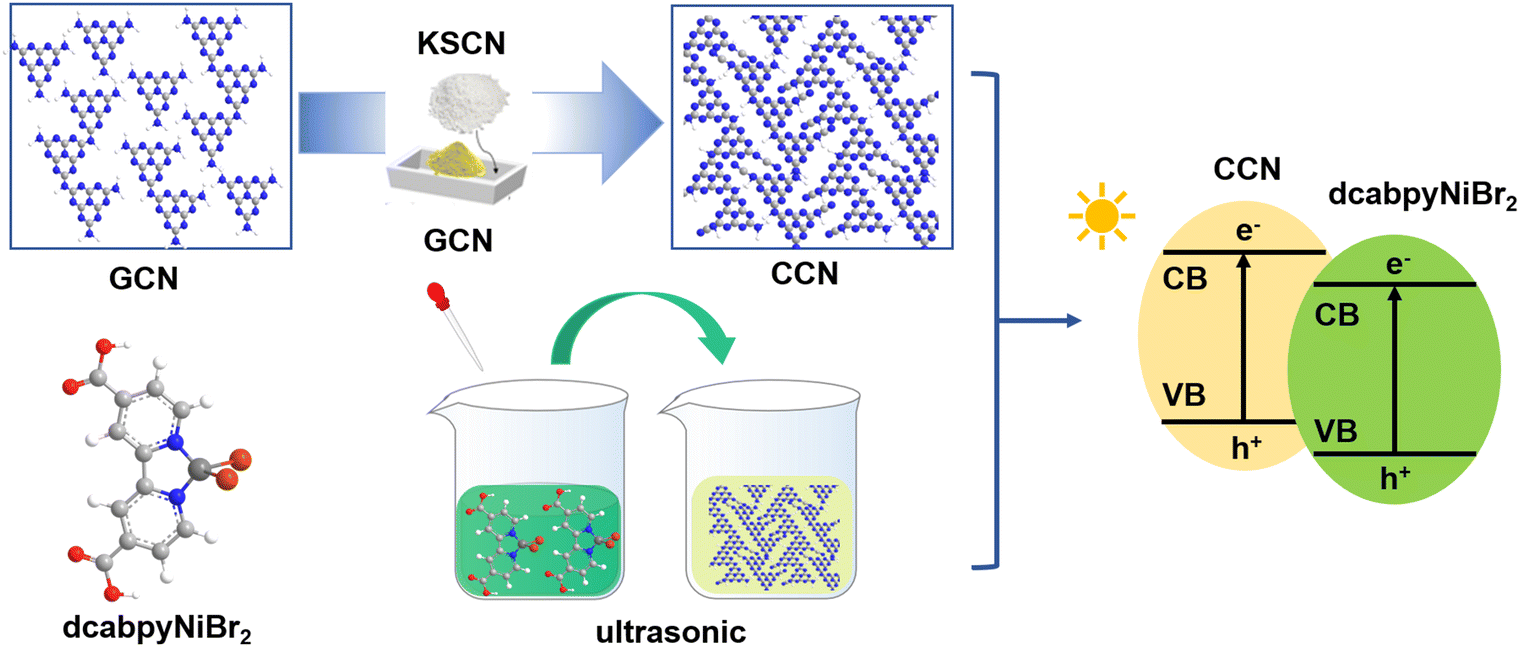 | ||
| Scheme 3 Fabrication process for CCN/Nim hybrids. | ||
The crystal structures of GCN, CCN, and CCN/Nim hybrids were studied by X-ray diffraction (XRD). The characteristic peaks of GCN were located at 13.1° and 27.6° (Fig. 1a), ascribed to the in-plane periodicity and interlayer stacking of the heptazine units, with (100) (d100 = 6.75 Å) and (002) (d002 = 3.23 Å) planes (Table S1†),27 respectively. Compared with amorphous GCN, it was revealed that CCN had an extended in-plane period, with two new peaks 2θ = 10.1° (8.75 Å) and 2θ = 8.0° (11.04 Å) indexed to the in-plane period of (020) and (110) planes,29 respectively. In addition, with the (002) peak of CCN shifted to 27.9° (Fig. 1a inset), CCN obtained a denser interlayer structure with a reduced interlayer distance of 3.20 Å in contrast to 3.23 Å for GCN. Careful observation: the width at half maximum (FWHM) of CCN (1.17) was reduced compared with GCN (1.33) and the clear lattice space of 1.1 nm indexed to the (110) plane in high-resolution TEM (HRTEM) of CCN (Fig. S4†), which synergistically confirmed the improved crystallinity of CCN after the molten salt treatment. As for CCN/Nim hybrids (Fig. 1b), the XRD patterns retained the characteristic CCN peaks of (002), (110), and (020), which suggested the in-plane periodicity and crystallinity remained unchanged after introducing dcabpyNiBr2 metal complexes. No peaks for dcabpyNiBr2 metal complexes were discovered due to the low mass percentage in CCN/Nim hybrids.
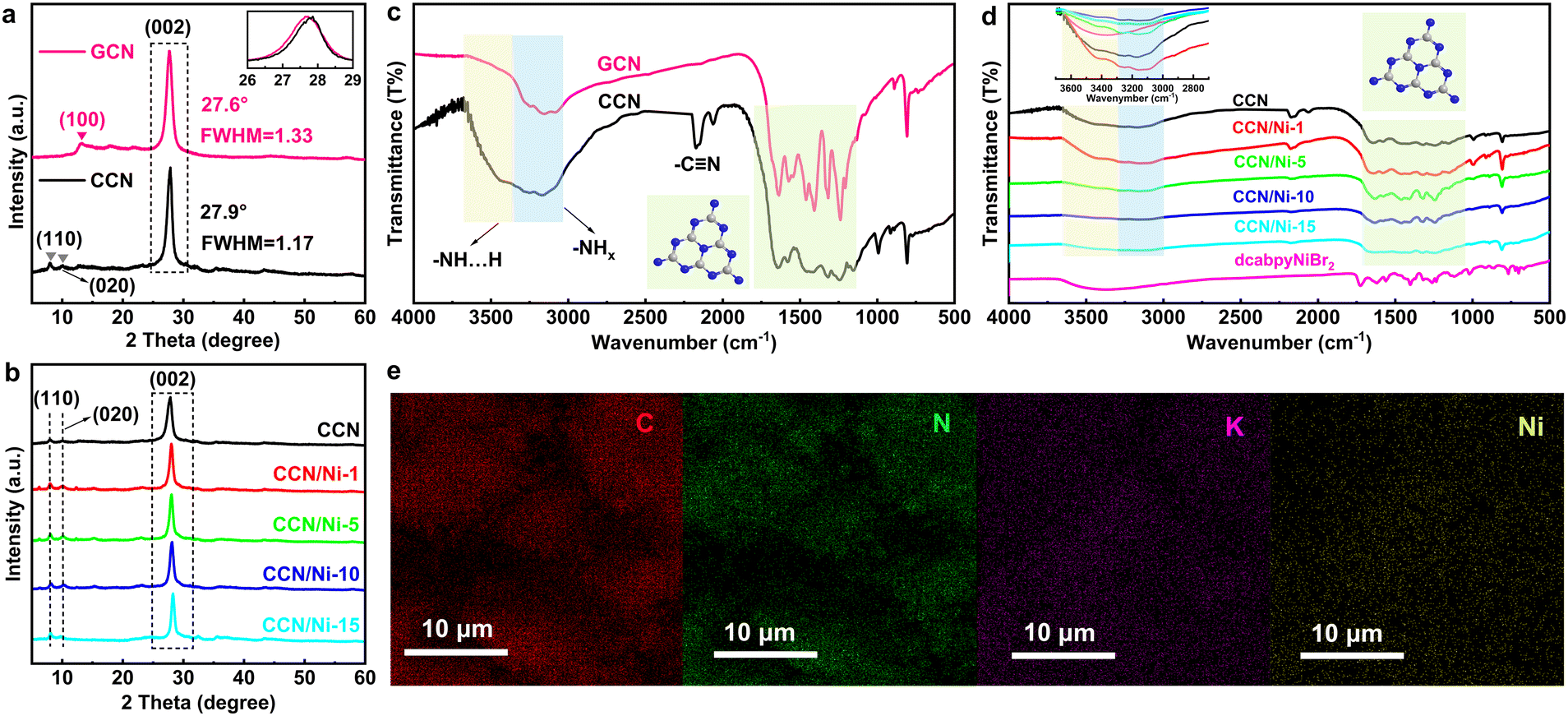 | ||
| Fig. 1 XRD patterns of (a) GCN, CCN, and (b) CCN/Nim hybrids. FTIR patterns of (c) GCN, CCN, and (d) CCN/Nim hybrids. (e) SEM-mapping scan images of C, N, K, and Ni of CCN/Ni-10. | ||
The molecular structure was further investigated by the Fourier transform infrared (FTIR) spectra over the as-prepared photocatalysts. GCN exhibited typical characteristics of out-of-plane bending and telescopic vibration of heptazine heterocycles at the bands of 806 cm−1 and 1200–1400 cm−1 (Fig. 1c).11,30 Similar to GCN, CCN retained the above characteristic bands, revealing that the heptazine-based conjugated rings remained unchanged after the molten salt treatment, which was consistent with the result of XRD patterns. In addition, a band at 994 cm−1 was indexed to the metal–C2N groups. Moreover, the band at 2170 cm−1 assigned to the telescopic vibration of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups (Fig. 1c)31 from the partial translation of –NH– or –NH2 groups to –CN groups after the KSCN molten salt treatment should be attributed to the reduced band intensity of –NH–/–NH2 at 3159 cm−1 in comparison to GCN,27,31,32 as well as the enhanced hydrogen bonds at 3500 cm−1, and due to that the strong electron-withdrawing –CN groups prefer to form hydrogen bonds than –NH–/–NH2 groups.27,31,32 With similar FTIR spectra to CCN (Fig. 1d), CCN/Nim hybrids maintained the molecular structure of CCN after introducing dcabpyNiBr2 to the hybrids, with the –CN groups and heptazine-based units maintained well in hybrids. Careful observations revealed that the band at 3500 cm−1 ascribed to hydrogen bonds displayed an obvious increase with the increasing mass percentage of dcabpyNiBr2, ascribed to the strong electron-withdrawing carboxyl groups of dcabpyNiBr2 preferring to form hydrogen bonds with –NH– or –NH2 groups in CCN.
N groups (Fig. 1c)31 from the partial translation of –NH– or –NH2 groups to –CN groups after the KSCN molten salt treatment should be attributed to the reduced band intensity of –NH–/–NH2 at 3159 cm−1 in comparison to GCN,27,31,32 as well as the enhanced hydrogen bonds at 3500 cm−1, and due to that the strong electron-withdrawing –CN groups prefer to form hydrogen bonds than –NH–/–NH2 groups.27,31,32 With similar FTIR spectra to CCN (Fig. 1d), CCN/Nim hybrids maintained the molecular structure of CCN after introducing dcabpyNiBr2 to the hybrids, with the –CN groups and heptazine-based units maintained well in hybrids. Careful observations revealed that the band at 3500 cm−1 ascribed to hydrogen bonds displayed an obvious increase with the increasing mass percentage of dcabpyNiBr2, ascribed to the strong electron-withdrawing carboxyl groups of dcabpyNiBr2 preferring to form hydrogen bonds with –NH– or –NH2 groups in CCN.
On account of dcabpyNiBr2 just anchored on the surface of CCN via hydrogen bonds, the scanning electron microscopy (SEM) of CCN/Nim hybrids with no obvious change confirmed that the morphological structures of hybrids were not affected by the introduced dcabpyNiBr2 metal complex (Fig. S5†). Except for the uniformly dispersed elements of C, N and K, an SEM-mapping scan with a uniform dispersion of Ni element throughout the surface of CCN, which originated from dcabpyNiBr2 metal complex, suggested the effective introduction of dcabpyNiBr2 on the surface of CCN without any accumulation (Fig. 1e). Significantly, the surficial dispersion of the dcabpyNiBr2 metal complex without accumulation favoured the effective catalytic reaction for CO2 photoreduction.
To further confirm the maintained molecular structure after the molten salt treatment and the chemical state of the metal complex on the surface of CCN, X-ray photoelectron spectroscopy (XPS) was performed. The XPS survey spectrum of CCN was composed of C, O, N, and K elements (Fig. S6d†). It was evident that, except for the graphitic carbon peak at 284.8 eV, a strong peak at 288.1 eV indexed to the sp2-bonded carbon in heptazine unit (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N) could be detected from the C 1s XPS spectrum of CCN (Fig. S6e†), along with a weak peak at 286.1 eV assigned to the –CN groups.33 In addition, two K 2p peaks at 295.5 and 292.8 eV revealed the existence of K+ ions in CCN (Fig. S7†), balancing cation charges for the negatively charged –CN groups. Meanwhile, three characteristic peaks at 401.0, 399.6 and 398.4 eV (Fig. S6f†), ascribed to central tertiary nitrogen, amino groups and triangular edge nitrogen, could be found in the N 1s spectrum of CCN.34 It was clearly recognized from the C 1s and N 1s analysis that the CCN maintained the characteristic molecular structure of the heptazine-based unit and possessed the –CN groups introduced by the molten salt treatment, consistent with the FTIR and XRD results. Given the inevitable graphitic carbon nonparticipating composition of CCN, the surface elemental ratios of carbon to nitrogen (C
C–N) could be detected from the C 1s XPS spectrum of CCN (Fig. S6e†), along with a weak peak at 286.1 eV assigned to the –CN groups.33 In addition, two K 2p peaks at 295.5 and 292.8 eV revealed the existence of K+ ions in CCN (Fig. S7†), balancing cation charges for the negatively charged –CN groups. Meanwhile, three characteristic peaks at 401.0, 399.6 and 398.4 eV (Fig. S6f†), ascribed to central tertiary nitrogen, amino groups and triangular edge nitrogen, could be found in the N 1s spectrum of CCN.34 It was clearly recognized from the C 1s and N 1s analysis that the CCN maintained the characteristic molecular structure of the heptazine-based unit and possessed the –CN groups introduced by the molten salt treatment, consistent with the FTIR and XRD results. Given the inevitable graphitic carbon nonparticipating composition of CCN, the surface elemental ratios of carbon to nitrogen (C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N) in CCN were calculated to be 3:3.643 (Table S2†), which was close to the values of the stoichiometric carbon nitride (3:4). The Ni 2p XPS of the dcabpyNiBr2 metal complex at 855.0 and 872.6 eV were assigned to Ni2+ (Fig. S6g†).35 Significantly, the Ni element could be detected from the XPS survey spectrum of CCN/Ni-10 (Fig. S6h†), verifying the successful introduction of dcabpyNiBr2 in the hybrid. The XPS peaks of C 1s and N 1s of CCN/Ni-10 were similar to that of CCN (Fig. S6j and S6k†), along with the remaining Ni 2p peaks (Fig. S6l†), strongly confirmed that the molecular structure and chemical state of CCN and dcabpyNiBr2 had no change after the self-assembly process in solution.
:N) in CCN were calculated to be 3:3.643 (Table S2†), which was close to the values of the stoichiometric carbon nitride (3:4). The Ni 2p XPS of the dcabpyNiBr2 metal complex at 855.0 and 872.6 eV were assigned to Ni2+ (Fig. S6g†).35 Significantly, the Ni element could be detected from the XPS survey spectrum of CCN/Ni-10 (Fig. S6h†), verifying the successful introduction of dcabpyNiBr2 in the hybrid. The XPS peaks of C 1s and N 1s of CCN/Ni-10 were similar to that of CCN (Fig. S6j and S6k†), along with the remaining Ni 2p peaks (Fig. S6l†), strongly confirmed that the molecular structure and chemical state of CCN and dcabpyNiBr2 had no change after the self-assembly process in solution.
Optical absorption and band structure characteristics
The ultraviolet-visible diffuse reflectance spectroscopy (DRS) and Mott–Schottky (MS) characterizations were conducted to investigate the light absorption ability, CB and VB position of CCN and dcabpyNiBr2. CCN exhibited an adsorption edge at 470 nm with a bandgap of 2.62 eV calculated from the Kubelka–Munk function (Fig. 2a and b). With the adsorption edge at 590 nm (2.10 eV) (Fig. 2c), dcabpyNiBr2 presented a significantly extended optical absorption range to near-infrared region, attributed to the ligand-to-metal charge transfer (MLCT) process in dcabpyNiBr2.36 However, CCN/Nim hybrids presented a weakly enhanced optical absorption range of 450–600 nm, which resulted from the lower mass percentage of dcabpyNiBr2. The flat band potentials detected from the MS plots of CCN and dcabpyNiBr2 were −1.22 V and −0.56 V (vs. Ag/AgCl), i.e., −1.05 V and −0.39 V (vs. NHE, PH = 7) (Fig. 2d and e), respectively. Given the CB possessing more negative potential of 0.2 V than the flat band potentials,37 the CB potentials of CCN and dcabpyNiBr2 were calculated to be −1.25 V and −0.59 V (vs. NHE, PH = 7), respectively. Combined with the bandgap values, the band structure of CCN and dcabpyNiBr2 are outlined in Fig. 2f. It was noticed that the dcabpyNiBr2 possessed a more negative CB potential than CCN, which is beneficial to the formation of BEF between CCN and dcabpyNiBr2, which would drive the photogenerated electrons migrating from CCN to dcabpyNiBr2 for CO2 molecule reduction reaction.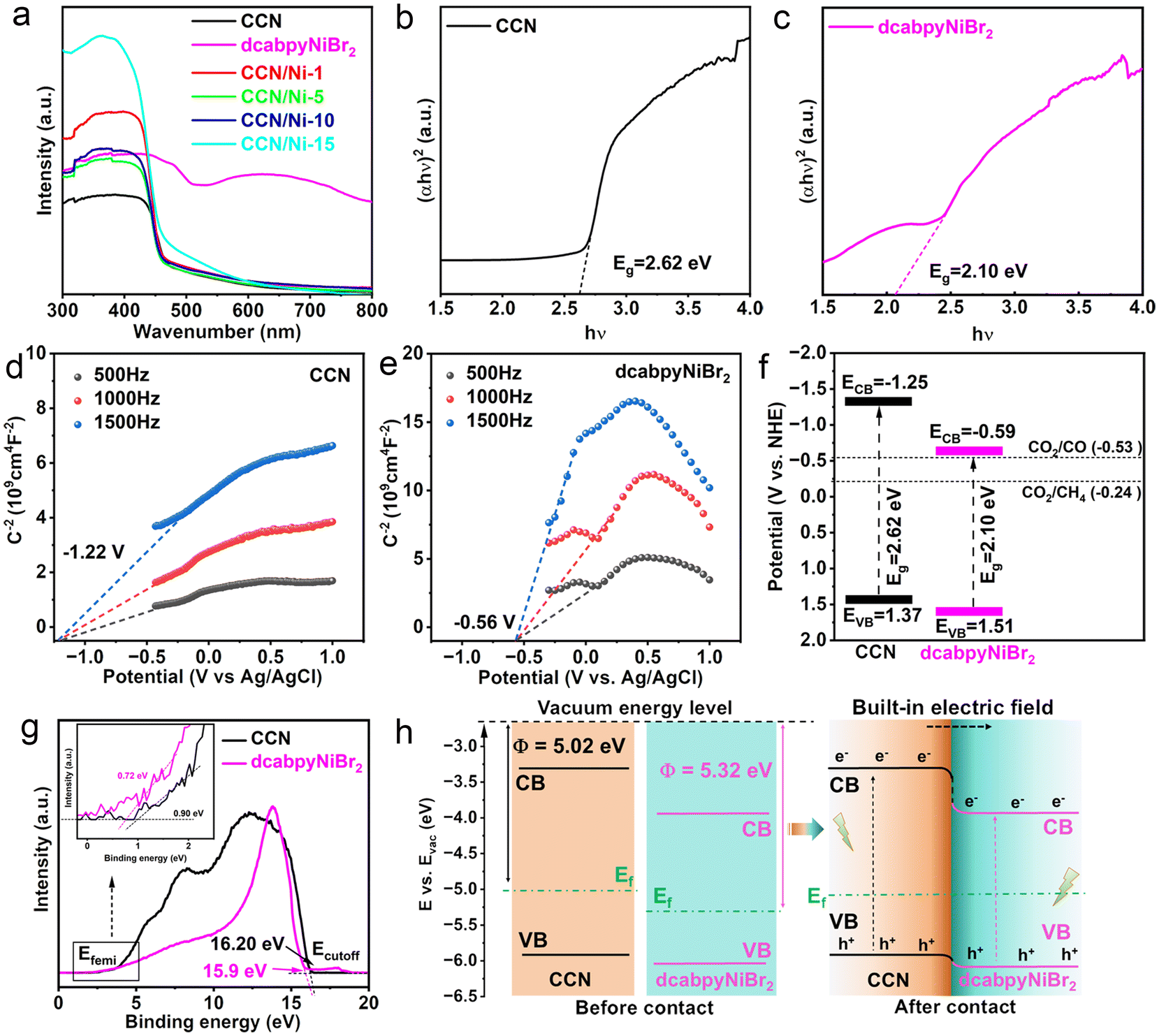 | ||
| Fig. 2 (a) UV-vis patterns of dcabpyNiBr2, CCN, and CCN/Nim hybrids. Tauc plot of (b) CCN and (c) dcabpyNiBr2 calculated from the Kubelka–Munk function. Mott–Schottky curves of (d) CCN and (e) dcabpyNiBr2. (f) The band structures of CCN and dcabpyNiBr2 calculated from the UV-visible DRS and Mott–Schottky plots. (g) UPS spectra of CCN and dcabpyNiBr2. (h) Schematic band structure calculated from UV visible DRS and UPS and a diagram for constructing a BET between CCN and dcabpyNiBr2. | ||
In order to further certify the successful construction of BEF between CCN and dcabpyNiBr2, ultraviolet photoelectron spectroscopy (UPS) and density functional theory (DFT) calculations were carried out. Generally speaking, the BEF originated from two close-contact semiconductors with different work functions (Φ), which induced the electron migration from one unit with a low work function to the other unit with a high work function, thus constructing a potential electric field.38 Therefore, the different work functions or Femi levels could be utilized to confirm the successful construction of a BEF between two materials, as well as judge the orientation of BEF.39,40 As shown in Fig. 2g, the Φ values of CCN and dcabpyNiBr2 were calculated to be 5.02 and 5.32 eV from the UPS spectra. Furthermore, DFT calculations also demonstrated the CCN equipped with a lower Φ than dcabpyNiBr2 (Fig. S8†). Based on the bandgap values obtained from the UV-vis DRS spectra, the band structure of CCN and dcabpyNiBr2 before contact could be outlined (Fig. 2h), which was consistent with the band structure calculated from UV-vis DRS and Motty–Schottky plots. As long as in contact, electron migration would take place from CCN with a high Femi energy (low Φ, Φ = 5.02 eV) to dcabpyNiBr2 with a low Femi energy (high Φ, Φ = 5.32 eV) until both Femi energy levels equilibrated, and then the BEF formed with orientation from CCN to dcabpyNiBr2.
CO2 photoreduction performance
Based on the improved crystallinity of CCN and constructed CCN/Nim hybrids, the photoreduction CO2 performance over the as-prepared catalysts was evaluated under the gas–solid mode. As illustrated in Fig. S9,† it was observed that the CO, as the main reduction product via the two proton and two electron process (CO2 + 2H+ + 2e− → H2O + CO), displayed a higher evolution yield for CCN than GCN, which could be attributed to the accelerated transfer efficiency of photogenerated charge carriers by the enhanced crystallinity. However, pure dcabpyNiBr2 exhibited a low product evolution activity (Fig. 3a and b), which could be resulting from the lack of a driving force for the photogenerated charge carrier. Additionally, all the CCN/Nim hybrids exhibited observably improved photoreduction CO2 activity (Fig. 3a) in contrast to CCN and dcabpyNiBr2, with the CO yields increasing first and then decreasing, depending on the increased mass percentage of the dcabpyNiBr2 metal complex. Among all the as-prepared catalysts, CCN/Ni-10 displayed the highest CO evolution rate, reaching up to 8.64 μmol g−1 h−1 (Fig. 3b), suggesting that this mass percentage of dcabpyNiBr2 was the best suitable for high-efficiency CO2 photoreduction. Unfortunately, the CO evolution rate decreased after a 2 h reaction, due to which the unsaturated CO2 adsorption at the surface resulted in an insufficient CO2 supply and a restricted catalytic reaction rate. The hydrogenation reaction of the CO2-to-CH4 coupling multiple electron and proton transfer was more severely affected by the insufficient CO2 supply, along with low CH4 selectivity, which resulted in an unobvious increase in the CH4 product. In the cyclic reaction measurement (Fig. S10†), CCN/Ni-10 presented good stability for CO2 photoreduction in gas–solid conditions with a slight decay in the CO and CH4 production yields. Additionally, O2 from the H2O oxidized by the photogenerated holes was detected as the oxidation product via the reaction of 2H2O + 4h+ → O2 + 4H+ (Fig. 3c), along with the protons accumulated on the surface of CCN/Nim hybrids, favouring the reduction reaction of CO2-to-CO and CO2-to-CH4. Subsequently, the consumed rate of holes for the oxidation half-reaction with O2 production and consumed rate of electrons for the reduction half-reaction with CO and CH4 were close to the stoichiometric ratio of 1:1 (Fig. 3d), which confirmed that the photogenerated electrons from the conduction band reduced CO2 to CO/CH4, and holes from the valence band oxidized H2O to O2. Significantly, despite eight protons and eight electrons needed for the conversion of CO-to-CH4 (CO2 + 8H+ + 8e− → CH4 + 2H2O), CH4 evolution rates and CH4 selectivity (yield-based and electron-based selectivity) present observably enhanced as long as introducing the dcabpyNiBr2 metal complex (Fig. 3b and d), which may be due to the accumulated protons and accelerated electron–hole separation driven by the BEF for the hydrogenation reaction with multiple electron and proton transfers. In addition, with the increased mass percentage of the dcabpyNiBr2 metal complex, CH4 yield-based and electron-based selectivity over CCN/Nim hybrids showed a continuous increase against the volcano-like activity, implying that the metal complex favours the absorption of an intermediate for CO2-to-CH4 for the subsequent hydrogenation reaction. However, the pure dcabpyNiBr2 displayed a low CH4 yield-based and electron-based selectivity due to a lack of driving force to provide abundant photogenerated electrons and protons for the conversion of CO-to-CH4. Given that the control experiments under the dark (Fig. 3e), Ar atmosphere, no H2O or catalysts have been evaluated and no reduction product detected, the CO2 photoreduction reaction under the gas–solid model was a photo-driven redox reaction of CO2 and H2O by as-synthesized photocatalysts. Furthermore, in the control experiment, the gas–solid reaction was evaluated under the Ar atmosphere without CO/CH4 production, and the 13CO2 isotope tracer experiment with products of 13CH4 (m/z = 17) and 13CO (m/z = 29) verified the carbon source for CO/CH4 originating from CO2 (Fig. 3f).41,42
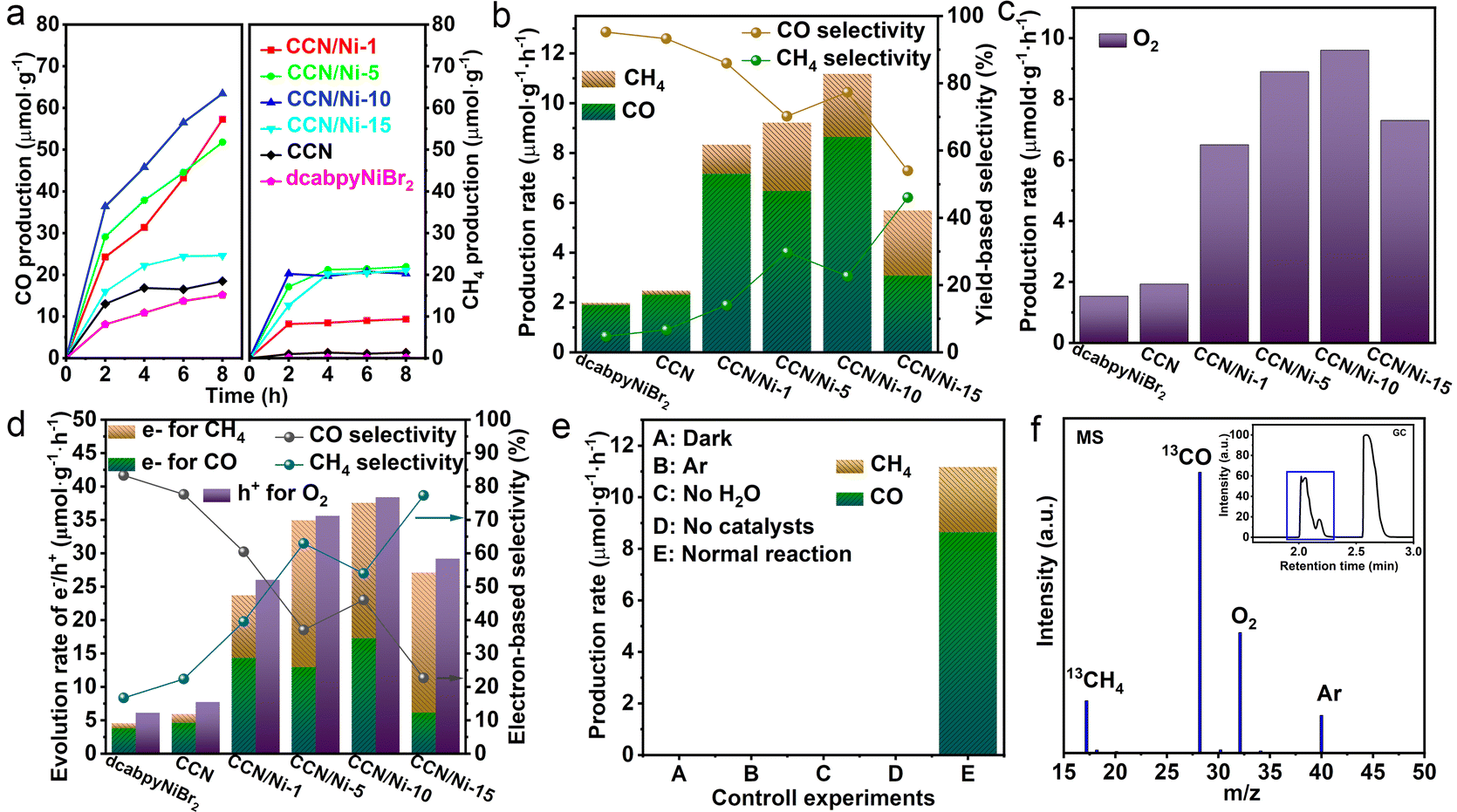 | ||
| Fig. 3 (a) CO and CH4 production with irradiation times for dcabpyNiBr2, CCN and CCN/Nim hybrids. (b) Production rate of CO and CH4 and yield-based selectivity over dcabpyNiBr2, CCN and CCN/Nim hybrids. (c) Evolution rate of O2 over dcabpyNiBr2, CCN and CCN/Nim hybrids. (d) Evolution rate of electrons and holes (e−/h+) and electron-based selectivity for CO2 photoreduction with redox reactions. (e) Control experiments for CCN/Ni-10. (f) Gas chromatography-mass spectrometry (GC-MS) analysis of the product from the gas–solid reaction with 13CO2 over CCN/Ni-10. | ||
Charge separation and transport characteristics
For deep investigation of charge separation efficiency and transfer dynamics steered by the BEF, steady-state and time-resolved photoluminescence (PL) spectra were carried out on CCN and CCN/Nim hybrids. Given the PL emission originates from the recombination of photogenerated e− and h+ pairs, the reduced PL emission intensity confirms the increase in the charge separation efficiency and migration process.20 CCN displayed an observably quenched PL intensity and shortened average lifetime (τave) in contrast to GCN (Fig. S11 and S12†), indicating the improved charge separation efficiency and accelerated transfer process in CCN by the improved crystallinity.43 Furthermore, the PL intensity of CCN/Nim hybrids further weakened in contrast to CCN (Fig. 4a), thanks to the effective charge migration from CCN to dcabpyNiBr2 driven by the BEF, inhibiting the recombination of photogenerated e− and h+ over CCN. With increasing mass percentage of the dcabpyNiBr2 metal complex, the PL intensity of CCN/Nim hybrids weakened first and then strengthened, agreeing with the order in CO2 photoreduction performance. Fig. 4b shows the time-revealed PL spectra with the corresponding τave of photogenerated charges (the inset table). Compared with CCN with a τave of 3.88 ns, CCN/Nim hybrids possessed the prolonged τave of 4.04, 4.32, 4.43, 4.02 ns for CCN/Ni-1, CCN/Ni-5, CCN/Ni-10, and CCN/Ni-15, and the order of prolonged τave was in accordance with the that of quenched PL emission, also consistent with the order in the photocatalytic activity. The prolonged lifetime further confirmed the effective photogenerated charge transfer process in CCN/Nim hybrids driven by the BEF. Obviously, the BEF not only increased the separation efficiency by inducing the photogenerated electrons migrating from CCN to dcabpyNiBr2 but also prolonged the lifetime with more probability of participating in redox reactions at the active sites before recombination. On careful comparison, GCN possessed the longest lifetime with the slowest photocatalytic activity, which resulted from the lack of effective active sites for CO2 photoreduction reaction. Thus, this phenomenon indicated that the introduced dcabpyNiBr2 could act as an active site in CCN/Nim hybrids.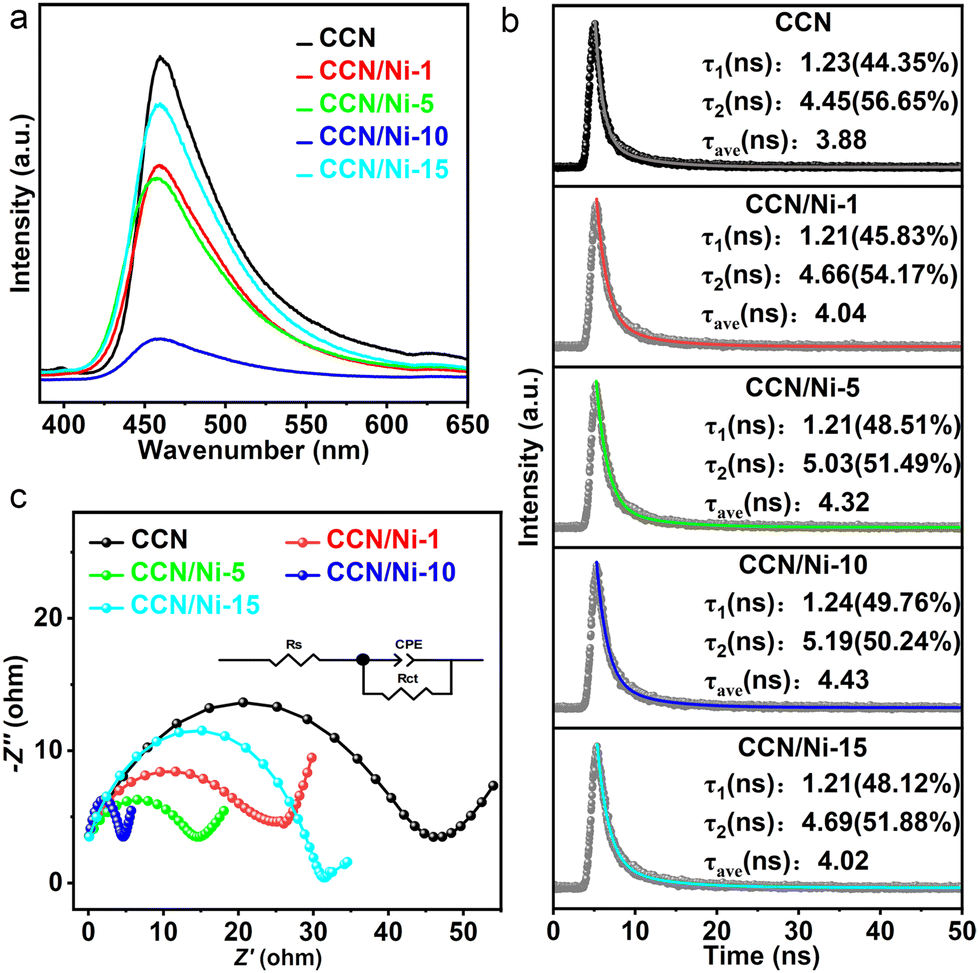 | ||
| Fig. 4 Steady-state (a) and time-resolved (b) photoluminescence (PL) spectra for CCN and CCN/Nim hybrids. (c) EIS Nyquist plots of CCN and CCN/Nim hybrids. | ||
To further confirm the enhanced charge transfer efficiency induced by the BEF, impedance spectra (EIS) and photocurrent measurements were investigated over CCN and CCN/Nim hybrids. It was observable that an equivalent circuit, consistent with solution resistance (Rs), charge transfer resistance through the electrode/electrolyte interface (Rct), and constant phase elements (CPE) (Fig. 4c), was utilized to fit the EIS plots.31 As shown in Table S3,† a pronounced reduction in Rct values occurred for the CCN/Ni-1 (21.18 Ω cm−1), CCN/Ni-5 (6.47 Ω cm−1), CCN/Ni-10 (5.02 Ω cm−1) and CCN/Ni-15 (33.75 Ω cm−1) in contrast to CCN (47.13 Ω cm−1), which verified the enhanced charge transfer process with reduced charge transfer resistance.31,44 Similarly, with the increasing mass percentage of dcabpyNiBr2, the photocurrent intensity of CCN/Nim hybrids distinctly increased compared with CCN (Fig. S13†), which, once more, confirmed the improved charge separation efficiency by the BEF. Significantly, with a suitable mass percentage of dcabpyNiBr2, CCN/Ni-10, possessing the smallest Rct value and highest photocurrent intensity, could be responsible for the best CO2 photoreduction activity.
CO2 photoreduction mechanism
Based on the in situ diffuse reflectance infrared Fourier transformation spectroscopy (DRIFTS), the reaction intermediates of photocatalytic CO2-to-CO reduction could be well explored. Under the dark, the peaks appeared at 1718, 1541 and 1652 cm−1, corresponding to the CO2* asymmetric stretching and surface-adsorption of H2O, demonstrating that the reactant molecule CO2 and H2O for photoreduction CO2 reaction were effectively absorbed on the photocatalyst surface45 (Fig. 5a). Subsequently, the surface-adsorbed H2O and CO2 molecules reacted to form carbon species (HCO3−, b-CO32−) and surface protons through the reaction CO2 + H2O → HCO3− + H+ and CO2 + H2O → CO32− + H+.46–48 Upon illumination, new peaks located at 1594 and 1508 cm−1 appeared and gradually increased with prolonged irradiation time, which could be ascribed to the COOH* intermediate for photocatalytic CO2 reduction into CO (CO2 + H+ + e− → COOH*).49,50 Additionally, one faint band at about 2127 cm−1 was observed and gradually strengthened with prolonged irradiation time, which is generally regarded as the CO* intermediates.51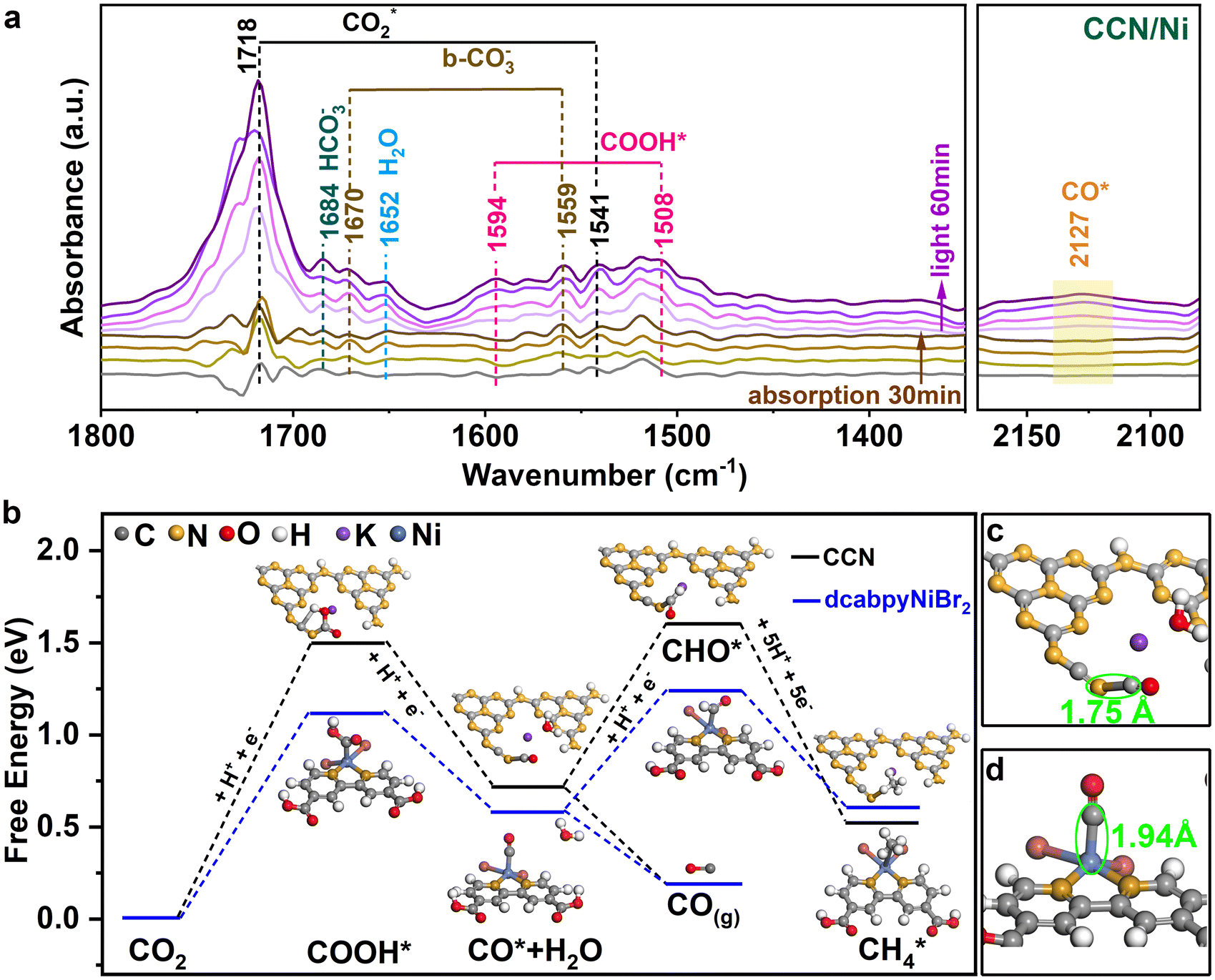 | ||
| Fig. 5 (a) In situ DRIFTS spectra of CCN/Ni hybrids. (b) Calculated free energy diagram for the reaction pathway of photocatalytic CO2 reduction over CCN/Ni and CCN. The distances between the *CO and the active site for (c) dcabpyNiBr2 and (d) CCN. | ||
To further understand the superior catalytic activity based on the photocatalyst, density functional theory (DFT) calculations of the Gibbs free energy of possible reaction pathways for photoreduction CO2-to-CO were performed. Given the photogenerated electrons accumulated on dcabpyNiBr2 driven by the BEF of CCN/Ni hybrid under irradiation, the CO2-to-CO conversion reaction preferred to occur at the dcabpyNiBr2 molecule. With the COOH* and CO* intermediate detected by in situ DRIFTS, the reaction pathways for CO2-to-CO conversion possibly occurred through the hydrogenation pathway:49–51 CO2 + H+ → COOH*, COOH* + H+ → CO* + H2O, CO* → CO(g), CO* + 5H+ → CH4(g). As illustrated in Fig. 5b, the COOH* intermediate formation over CCN and CCN/Ni showed a relatively high free energy difference in contrast to other reaction steps during CO2-to-CO conversion, which acted as the rate-determining step for photoreduction CO2 reaction. The formation of the COOH* intermediate over CCN/Ni possessed a lower free energy difference (1.12 eV) than CCN (1.50 eV), indicating that the introduced dcabpyNiBr2 metal complex favored the hydrogenation reaction of CO2 into COOH*. The formation of CO* revealed an observably reduced free energy difference from 0.72 eV for CCN to 0.58 eV for CCN/Ni, thanks to the accumulated photogenerated e− on the dcabpyNiBr2 metal complex accelerating the conversation of COOH* to CO*. Generally, the formation of COOH* and CO* intermediates over CCN/Ni with the reduced free energy difference in contrast to CCN explained the highly effective performance of CO2 photoreduction on CCN/Ni. The desorption of the CO* intermediate into CO gas for CCN and CCN/Ni was thermodynamically favorable in contrast to forming CHO* as an endothermal process, suggesting that CO2 photoreduction was more likely CO gas formation than a continuous hydrogenation reaction, consistent with high CO selectivity. However, the dcabpyNiBr2 metal complex displayed an improved adsorption ability for CO* with a shorter Ni–C bond length of 1.75 Å than the N–C bond length of 1.94 Å for CCN (Fig. 5c and d), which favored the further hydrogenation reaction from CO* to CH4. Hence, with abundant accumulation of electrons and protons induced by BEF on the dcabpyNiBr2 metal complex, the CH4 selectivity gradually increased with increasing mass percentage of dcabpyNiBr2.
Conclusions
Based on the crystalline CCN, we successfully designed and synthesized CCN/Nim hybrids via the molten-salt and solution–dispersion two-step approach, in which the dcabpyNiBr2 metal complex was anchored on the surface of CCN via the hydrogen bond. It was experimentally displayed that the CCN/Ni-10 hybrid exhibited improved CO2 photoreduction performance, with the CO and CH4 generation rates reaching 8.64 and 2.54 μmol g−1 h−1, which were about 3.55 times and 15.52 times that of CCN. The band structure investigation revealed the BEF was constructed between dcabpyNiBr2 and CCN with matched energy levels. Moreover, the charge carrier dynamics presented that the BEF provided a strong driving force for photogenerated charge migrating from CCN to dcabpyNiBr2 to reduce the CO2 molecule, inhibiting the recombination of photogenerated electron–hole pairs on CCN, which effectively improved the CO2 photoreduction performance. Additionally, the in situ DRIFTS revealed that the COOH* and CO* species acted as the key intermediate for the conversion of CO2-to-CO and continuous hydrogenation reaction for CH4. Furthermore, DFT calculations further investigated the high activity for CCN/Ni with reduced free energy of formation difference for COOH* and CO* intermediates. Accordingly, this study provides a referential strategy for constructing a built-in electric field between GCN-based materials and metal complexes with matched energy levels to target effective CO2 photoreduction performance.Author contributions
Yanrui Li: conceptualization; formal analysis; funding acquisition; resources; writing – review & editing. Linda Wang: investigation; formal analysis; writing – original draft preparation. Xiaolin Zhu: investigation; validation. Bozhan Li: software; validation. Liangqing Zhang: funding acquisition; resources. Xiang Gao: funding acquisition; resources; software.Data availability
The data that support this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22303064, 52103127, 52102232), the Outstanding Youth for Xi'an University of Science and Technology (2023YQ3-07), Xi'an Association for Science and Technology Young Talent Support Program (959202413073), the Natural Science Basic Research Program of Shaanxi (2018JQ2028, 2023-JC-YB-094), the Education Department Fund in Shaanxi Province (2021JK0763).References
- X. Wan, Y. Li, Y. Chen, J. Ma, Y. A. Liu, E. D. Zhao, Y. Gu, Y. Cui, R. Li, D. Liu, R. Long, K. M. Liew and Y. Xiong, Nat. Mater., 2024, 15, 1273 CAS.
- Y. Wang, J. Wang, M. Zhang, S. Zheng, J. Wu, T. Zheng, G. Jiang and Z. Q. Li, Small, 2023, 2300841 CrossRef CAS PubMed.
- S. Roy, ACS Appl. Mater. Interfaces, 2020, 12, 37811–37833 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- M. Liu, S. Wageh, A. A. Al-Ghamdi, P. Xia, B. Cheng, L. Zhang and J. Yu, Chem. Commun., 2019, 55, 14023–14026 RSC.
- Z. Jiang, X. Zhang, H. S. Chen, P. Yang and S. P. Jiang, Small, 2020, 16, 2003910 CrossRef CAS PubMed.
- N. Ojha, A. Bajpai and S. Kumar, Catal. Sci. Technol., 2019, 9, 4598–4613 RSC.
- Q. Wang, Z. Fang, W. Zhang and D. Zhang, Adv. Fiber Mater., 2022, 4, 342–360 CrossRef CAS.
- M. Zhao, J. Feng, W. Yang, S. Song and H. Zhang, ChemCatChem, 2020, 13, 1250–1270 Search PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CAS.
- V. W. H. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CAS.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CAS.
- P. Ganji, R. K. Chowdari and B. Likozar, Energy Fuels, 2023, 37, 7577–7602 CAS.
- C. Yang, Y. Wang, J. Yu and S. Cao, ACS Appl. Energy Mater., 2021, 4, 8734–8738 CrossRef CAS.
- Y. Li, Y. Lei, D. Li, A. Liu, Z. Zheng, H. Liu, J. Guo, S. Liu, C. Hao and D. He, ACS Catal., 2023, 13, 10177–10204 CrossRef CAS.
- Y. Yuan, R. T. Guo, Z. W. Zhang, L. F. Hong, X. Y. Ji, Z. D. Lin and W. G. Pan, Energy Fuels, 2021, 35, 13291–13303 CrossRef CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef.
- G. Chen, F. Wei, Z. Zhou, B. Su, C. Yang, X. F. Lu, S. Wang and X. Wang, Sustainable Energy Fuels, 2023, 7, 381–388 RSC.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, J. Environ. Sci., 2024, 40, 103–112 CrossRef.
- Y. Li, L. Wang, X. Gao, Y. Xue, B. Lia and X. Zhu, J. Mater. Chem. A, 2024, 12, 7807–7816 RSC.
- M. C. Rosko, K. A. Wells, C. E. Hauke and F. N. Castellano, Inorg. Chem., 2021, 60, 8394–8403 CrossRef CAS.
- P. P. Ferreira da Rosa, S. Miyazaki, H. Sakamoto, Y. Kitagawa, K. Miyata, T. Akama, M. Kobayashi, K. Fushimi, K. Onda, T. Taketsugu and Y. Hasegawa, J. Phys. Chem. A, 2021, 125, 209–217 CrossRef CAS PubMed.
- X. Zhu, Y. Jia, Y. Liu, J. Xu, H. He, S. Wang, Y. Shao, Y. Zhai and Y. Zhu, Angew. Chem., Int. Ed., 2024, 63, 202405962 CrossRef PubMed.
- S. Park, H. S. Ahn, C. K. Lee, H. Kim, H. Jin, H. S. Lee, S. Seo, J. Yu and S. Han, Phys. Rev., 2008, 77, 134103 CrossRef.
- W. Zhang, Q. Gu, X. Fu, Y. Wang, Y. Jian, H. Sun and Z. Gao, Inorg. Chem., 2023, 62, 13615–13625 CrossRef CAS PubMed.
- T. Banerjee, A. K. Biswas, U. G. Reddy, T. S. Sahu, A. Das, B. Ganguly and H. N. Ghosh, J. Phys. Chem. C, 2014, 118, 3864–3877 CrossRef CAS.
- D. Gunawan, C. Y. Toe, K. Sun, J. Scott and R. Amal, Photochem. Photobiol. Sci., 2022, 21, 2115–2126 CrossRef CAS.
- D. Gunawan, C. Y. Toe, P. Kumar, J. Scott and R. Amal, ACS Appl. Mater. Interfaces, 2021, 13, 49916–49926 CrossRef CAS.
- M. S. Wu and H. H. Hsieh, Electrochim. Acta, 2008, 53, 3427–3435 CrossRef CAS.
- Y. Wang, S. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 17236–17240 CrossRef CAS PubMed.
- Z. Liao, C. Li, Z. Shu, J. Zhou, T. Li, W. Wang, Z. Zhao, L. Xu, L. Shi and L. Feng, Int. J. Hydrogen Energy, 2021, 46, 26318–26328 CrossRef CAS.
- W. Wang, Z. Shu, J. Zhou, D. Meng, Z. Zhao and T. Li, J. Mater. Chem. A, 2020, 8, 6785–6794 RSC.
- R. Zhang, H. Wang, Y. Li, D. Wang, Y. Lin, Z. Li and T. Xie, ACS Sustainable Chem. Eng., 2021, 9, 7286–7297 CrossRef CAS.
- Z. Jin, R. Hu, H. Wang, J. Hu and T. Ren, Appl. Surf. Sci., 2019, 491, 432–442 CrossRef CAS.
- Y. Li, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huangfu, D. Zhao, L. Gu and S. Shen, Chem. Sci., 2021, 12, 3633–3643 RSC.
- D. A. Cagan, D. Bím, B. Silva, N. P. Kazmierczak, B. J. McNicholas and R. G. Hadt, J. Am. Chem. Soc., 2022, 144, 6516–6531 CrossRef CAS PubMed.
- Q. Liang, Z. Li, Z. H. Huang, F. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS.
- X. Zhao, M. Liu, Y. Wang, Y. Xiong, P. Yang, J. Qin, X. Xiong and Y. Lei, ACS Nano, 2022, 16, 19959–19979 CrossRef CAS PubMed.
- Z. S. Luo, X. Y. Ye, S. J. Zhang, S. K. Xue, C. Yang, Y. D. Hou, W. D. Xing, R. Yu, J. Sun, Z. Y. Yu and X. C. Wang, Nat. Commun., 2022, 13, 2230 CrossRef CAS.
- B. C. Zhu, B. Cheng, L. Y. Zhang and J. G. Yu, Carbon Energy, 2019, 1, 32–56 CrossRef CAS.
- B. Su, Y. Kong, S. Wang, S. Zuo, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415 CrossRef CAS PubMed.
- B. Su, M. Zheng, W. Lin, X. F. Lu, D. Luan, S. Wang and X. W. Lou, Adv. Energy Mater., 2023, 13, 2203290 CrossRef CAS.
- M. Zhou, P. Yang, R. Yuan, A. M. Asiri, M. Wakeel and X. Wang, ChemSusChem, 2017, 10, 4451–4456 Search PubMed.
- H. Zou, B. He, P. Kuang, J. Yu and K. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 22311–22319 CrossRef CAS.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- H. Cao, S. Jiang, J. Xue, X. Zhu, Q. Zhang and J. Bao, J. Phys. Chem. Lett., 2022, 13, 8397–8402 CrossRef CAS.
- Y. Deng, C. Wan, C. Li, Y. Wang, X. Mu, W. Liu, Y. Huang, P. K. Wong and L. Ye, ACS Catal., 2022, 12, 4526–4533 Search PubMed.
- Y. Yu, X. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Zhou and F. Dong, ACS Nano, 2021, 15, 14453–14464 CrossRef CAS.
- D. Sun, J. Li, T. Shen, S. An, B. Qi and Y.-F. Song, ACS Appl. Mater. Interfaces, 2022, 14, 16369–16378 CrossRef CAS PubMed.
- L. Shen, Z. Xie, L. Hou, J. Yang and Q. Li, Energy Fuels, 2022, 36, 11515–11523 CrossRef CAS.
- Y. Mao, M. Zhang, S. Si, G. Zhai, X. Bao, K. Song, L. Zheng, Y. Liu, Z. Wang, Z. Zheng, P. Wang, Y. Dai, H. Cheng and B. Huang, ACS Catal., 2023, 13, 8362–8371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03586k |
| This journal is © The Royal Society of Chemistry 2025 |