
Unraveling the correlation between biological effects and halogen substituents in cobalt bis(dicarbollide)†
Katarzyna
Zakret-Drozdowska
 a,
Bożena
Szermer-Olearnik
a,
Waldemar
Goldeman
b,
Michalina
Gos
a,
Dawid
Drozdowski
c,
Anna
Gągor
c and
Tomasz M.
Goszczyński
*a
a,
Bożena
Szermer-Olearnik
a,
Waldemar
Goldeman
b,
Michalina
Gos
a,
Dawid
Drozdowski
c,
Anna
Gągor
c and
Tomasz M.
Goszczyński
*a
aLaboratory of Biomedical Chemistry, Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, 53-114 Wrocław, Poland. E-mail: goszczynski@hirszfeld.pl
bDepartment of Organic and Medicinal Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, 50-370 Wrocław, Poland
cInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, 50-422 Wrocław, Poland
First published on 12th November 2024
Abstract
Over the past decade, considerable scientific attention has been given to adapting cobalt bis(dicarbollide) as innovative agent with various biomedical applications. Although the studied compounds show great potential in this field, only a few reports have explored broad, well-thought-out libraries of derivatives to correlate their structure with biological activity. In this study, we investigate a panel of [CoSAN]− derivatives substituted with fluorine, chlorine, bromine and iodine in order to elucidate the impact of the halogen presence on antimicrobial action and selectivity over mammalian cells. We present the first evidence that increasing the atomic mass of a substituent improves the biological activity of a derivative. Our results demonstrate that the addition of a single iodine atom to the [CoSAN]− core results in the most selective antibacterial outcome, especially toward Staphylococcus aureus ATCC 6538. The described correlation between the lipophilicity and the activity of the compounds toward both bacteria and human cell lines highlights the importance of a conscious design method to obtain the most desirable [CoSAN]−-based derivatives.
Introduction
The first reports on metallacarboranes, a class of organometallic compounds, arose nearly 60 years ago in 1965.1 Consisting of centrally localized metal ions and carborane ligands, they form three-dimensional cage-like structures.2 Owing to their structural diversity and varying physicochemical properties, they find application in fields such as materials science,3,4 catalysis5,6 and medicinal chemistry.7–10 Cobalt bis(dicarbollide) [CoSAN]− is one of the most extensively studied representatives within this family of compounds. It consists of two C2B9H112− subunits with a cobalt atom in its 3+ oxidation state and exhibits remarkable physicochemical properties, such as high thermal and chemical stability,11 dispersed negative charge, the ability to form dihydrogen bonds (B–H⋯H–C)12 and superchaotropicity.13,14 Moreover, the ease of chemical modification results in a wide range of [CoSAN]− derivatives.11,15–17 The aforementioned properties make [CoSAN]− an extremely interesting scaffold for the design of new biologically active compounds, especially antibiotics targeting drug-resistant bacteria. Throughout the analysis of the available literature data, we can also conclude that the biological activity of metallacarborane derivatives can be influenced in several ways:(1) The structure of the ligands. Recent studies suggest that the configuration of ligands (ortho [1,2-C2B9H11]2− or meta [1,7-C2B9H11]2−) may impact the antimicrobial activity of [CoSAN]− derivatives.18 Although the activities of both isomers against Gram-positive bacteria are rather similar, the meta-isomer appears to be more effective against Gram-negative strains. The preferential conformation of the [CoSAN]−ortho-isomer is cisoid, leading to its self-assembly and the formation of vesicles and micelles in aqueous solution. Conversely, the preferred transoid conformation of meta[CoSAN]− leads to a 2D lamellar arrangement. This is just one possible explanation for the observed differences in biological activities, but more in-depth research is needed to confirm these observations.
(2) The influence of the central metal. Research on dicarbollides containing metals other than cobalt has been notably limited,19 especially regarding their impact on biological activity.20 Few studies have addressed this aspect with a recent focus on comparing [CoSAN]− and iron-containing ferra bis(dicarbollide) ([FeSAN]−) derivatives in the context of their antimicrobial activity.18 From a chemical perspective, the presence of different metals in metalla bis(dicarbollide) structures imparts significant variations in the redox potential of the M3+/M2+ couple. However, this had a minimal impact on antimicrobial activity. Nevertheless, the latest data suggest that metals (Fe or Co) may play an important role in in vivo experiments in both L4-stage Caenorhabditis elegans nematodes and Caenorhabditis elegans embryos.21
(3) The presence of iodine atoms. Regarding the presence of halogens in the dicarbollide structure, the existing literature conveys a distinct message. Compared with the corresponding parent dicarbollides, compounds containing one or two iodine atoms (B(8)-I or B(8,8′)-I2) consistently exhibit enhanced biological efficacy against bacteria, fungi and eukaryotic organisms.21–26[CoSAN-I2]− is the most extensively studied compound, with a focus on comparing its properties with those of the parent [CoSAN]−. The main differences between them are their conformation (cisoid for [CoSAN]− and transoid for [CoSAN-I2]−) and hydrophobicity. The increased hydrophobicity of [CoSAN-I2]− suggests a higher affinity for lipid membranes22,27 and, consequently, more effective transport across biological barriers. Although it seems to be a logical justification, there is a lack of consideration for the impact of other halogen substituents. This raises the question of whether iodine is the most optimal choice for biological applications.
The goal of this study was to demonstrate a correlation between halogen substitution at the B(8) and/or B(8′)-positions of the [CoSAN]− (ortho isomer) and its broadly understood biological activity. For this purpose, we obtained a series of [CoSAN]− derivatives with various halogens ranging from fluorine to iodine (Fig. 1). These derivatives included both monosubstituted and disubstituted moieties (including homo- and hetero-disubstituted). The biological activity of these compounds was subsequently verified against selected Gram-positive and Gram-negative bacterial strains, along with their antiproliferative efficacy against human normal and cancer cell lines. The selectivity index, a parameter that plays a key role in pharmacology, was calculated from the obtained results. This approach provides a comprehensive insight into the impact of halogenation in [CoSAN]− regarding its potential future applications in medicine.
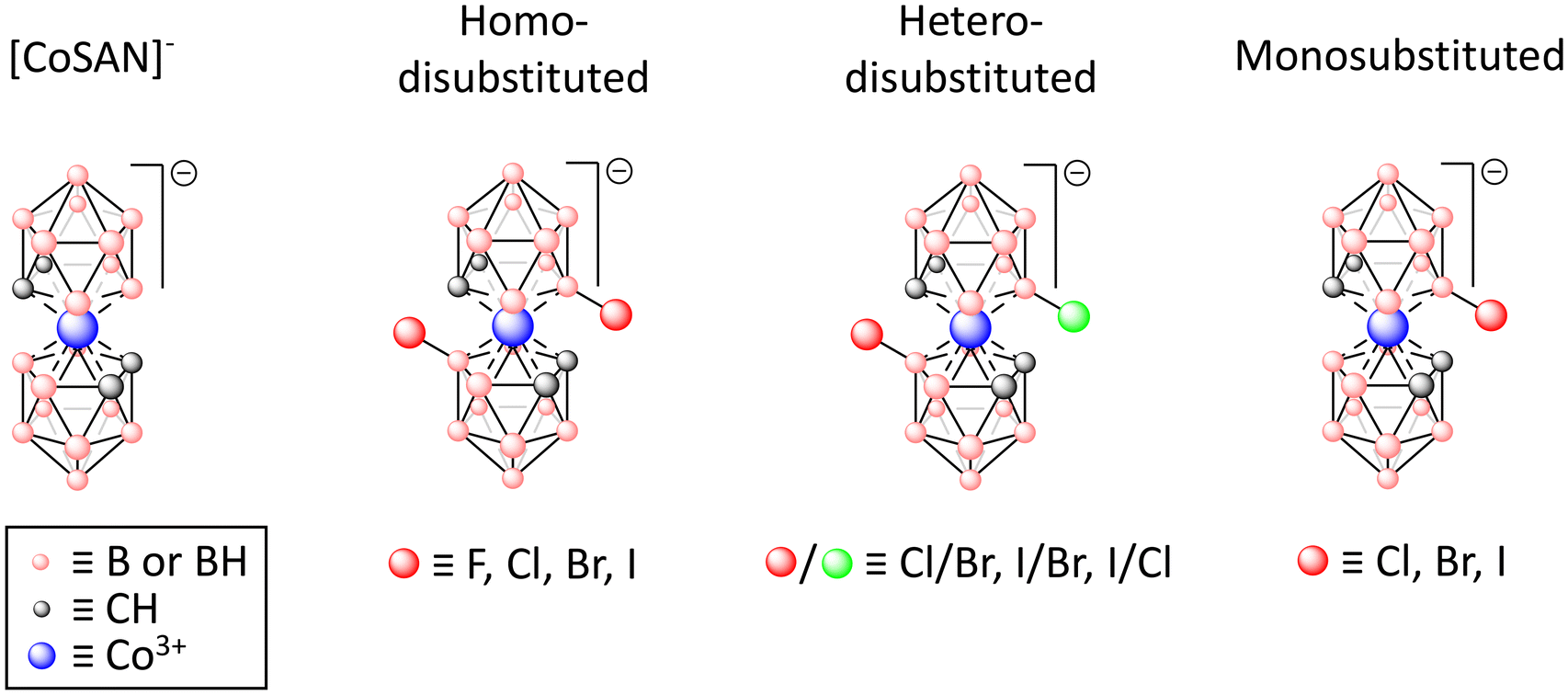 | ||
| Fig. 1 Structures of [CoSAN]− and its halogenated derivatives studied in this work. | ||
Results and discussion
We started our study with the synthesis of B(8,8′)-disubstituted derivatives, incorporating two identical halogen atoms ranging from fluorine to iodine. Based on the initial biological assays on bacterial strains and cell lines, we observed increased activity in all the derivatives, except for [CoSAN-F2]−, compared with the parental [CoSAN]− molecule in all conducted tests. Encouraged by these findings and aiming for a comprehensive understanding of the structure–activity relationships, we decided to broaden the spectrum of the tested compounds. This extension involved the synthesis of monosubstituted and hetero-disubstituted derivatives with halogens ranging from chlorine to iodine (Fig. 1).A unique advantage of anionic metallacarboranes lies in their solubility, which extends to both aqueous and nonaqueous media and is dependent solely on the utilized counterion. The compounds described in this study were synthesized as cesium salts, except for [CoSAN-I,Cl]−, which was obtained as an N,N-diisopropylethylammonium salt. However, for biological assays, we transformed all of these compounds into their sodium salt counterparts, which exhibit excellent solubility in aqueous environments, including biological growth media. The aqueous solubility of [CoSAN]− derivatives is a critical factor influencing their bioavailability.18
Chemistry
To date, synthetic routes for obtaining halogenated [CoSAN]− derivatives have not been widely explored.28–33 Therefore, we find it essential to modify existing methods, revealing new synthetic pathways for these compounds.[CoSAN-F2]− is the least studied of all halogen derivatives, with only a single publication outlining the synthesis of this compound. Fluorination was carried out using 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate), commonly referred to as F-TEDA.34 Considering the limited exploration of [CoSAN-F2]− synthesis, we decided to change the reaction conditions. Starting with the identification of a suitable solvent, we tested acetonitrile (AcN), tetrahydrofuran (THF), dichloromethane (DCM), and methanol (MeOH). Ultimately, the reaction was conducted in methanol under reflux conditions, resulting in the formation of pure [CoSAN-F2]− in the 1-chloromethyl-1,4-diazoniabicyclo[2.2.2]octane salt form, which was achieved through repeated crystallization from hot methanol. Subsequently, when a cation exchange resin was used, the compound was converted into its acidic form – H[CoSAN-F2]. This operation allows to obtain the product with the desired counterion through simple neutralization; for example, using CsOH, a cesium salt can be obtained.
Chlorinated and brominated derivatives of [CoSAN]− can be obtained through two distinct methods: the first of them is the use of elemental halogens or halogen donor reagents (in situ generated hypochlorous acid or N-chlorosuccinimide (NCS) and N-bromosuccinimide (NBS), respectively).29 Another approach for introducing chlorine or bromine into the [CoSAN]− structure involves halogenation induced by γ-radiation.28 We carried out the synthesis of chlorinated [CoSAN]− derivatives following a screening of solvents. THF was ultimately chosen, and through incremental addition of NCS, we optimized both the amount of reagent and the reaction time needed to obtain [CoSAN-Cl2]−. On the basis of these data, we adjusted the reaction conditions required to obtain [CoSAN-Cl]−. Owing to the high reactivity of NCS, the crude reaction mixture consisted of [CoSAN-Cl]− and [CoSAN-Cl2]− (90% and 10%, respectively). Thus, the recrystallization from ethanol was necessary. Using conditions similar to chlorination, we synthesized [CoSAN-Br2]− and [CoSAN-Br]− employing NBS. In the case of [CoSAN-Br]− synthesis, recrystallization of the crude reaction mixture was not required because of the lower reactivity of NBS (and the lower amount of dibromo derivative formed).
For obtaining [COSAN-I2]−, there are two known synthetic methods, using iodine28 or iodine monochloride.35 Due to the highly irritant properties of the iodine monochloride used for the synthesis of [CoSAN-I2]−, we focused our attention on reactions with elemental iodine. Herein, we present an improved synthesis method for [CoSAN-I2]− based on the reaction used for synthesizing [CoSAN-I]−.36 Increasing the amount of iodine and extending reaction time resulted in the substitution of the second iodine. Attempts to iodinate [CoSAN]− using N-iodosuccinimide (NIS) were unsuccessful.
According to the literature data, the hetero-disubstituted derivatives [CoSAN-I,Br]− and [CoSAN-I,Cl]− were synthesized through reactions of [8,8′-μ-I-3,3-Co(1,2-C2B9H10)2] with 1,2-dibromoethane and chloroform, respectively, resulting in moderate yields.37 To obtain these derivatives of [CoSAN]−, we opted for the use of N-halosuccinimides in the halogenation of previously monosubstituted derivatives. Bromination of [CoSAN-Cl]− resulted in [CoSAN-Cl,Br]− in a high yield (85%). To the best of our knowledge, this derivative was obtained for the first time. [CoSAN-I,Br]− was synthesized by brominating [CoSAN-I]−, whereas the reaction of [CoSAN-Br]− with NIS led to the recovery of the substrate. Attempts to obtain [CoSAN-I,Cl]− through the reaction of [CoSAN-Cl]− with NIS or I2 were unsuccessful. Intriguingly, the reaction of [CoSAN-I]− with NCS resulted in a mixture comprising of the starting material, [CoSAN-Cl]− and [CoSAN-I,Cl]−. Therefore, we decided to synthesize [CoSAN-I,Cl]−via opening of the iodonium bridge with CH2Cl2 in the presence of N,N-diisopropylethylamine (yield 74%).
Crystal structures
The crystal structure of Cs[CoSAN] was reported in 1967, but the positions of carbon atoms were selected arbitrarily.38 They were later determined in 1982, but with (C2H5)3NH+ as a counterion.39 To date, little attention has been given to the crystal structure of halogenated derivatives. While parent [CoSAN]− has 27 structures in the CCDC database (with different counterions), one may find only 6 records of derivatives substituted with halogens: Cs[8-I-3,3′-Co(1,2-C2B9H10)(1′,2′-C2B9H11)],40 Cs[8,8′-I2-3,3′-Co(1,2-C2B9H10)2],41 NBu4[8,8′-F2-3,3′-Co(1,2-C2B9H10)2],34 (BEDT-TTF)[8,8′,(7)-Cl2(Cl0,09)-3,3′-Co(1,2-C2B9H9,91)(1′,2′-C2B9H10)], (BEDT-TTF)[8,8′-Br0,75Cl1,25-3,3′-Co(1,2-C2B9H10)2], (BMDT-TTF)4[8,8′-Br1,16(OH)0,72-3,3′-Co(1,2-C2B9H10.06)2] (Refcodes: DEXPOL, DEXPIF, GUPDUQ, BAVBOR, BAVBUX, and BAVCAE respectively). The last three derivatives have nonstoichiometric compositions.42 In addition, there is a niche in terms of crystal structures derived from the room-temperature measurements – conditions corresponding to the natural or application environment of the reported compounds. We examined crystal structures of hetero-disubstituted derivatives, along with those of the parent [CoSAN]− and the most effective antibacterial compounds [CoSAN-I]− and [CoSAN-I2]−.The unit cells of the examined compounds are presented in Fig. S90–S94 (ESI).† The symmetries of [CoSAN]−, [CoSAN-I]− and [CoSAN-I2]− are described in the monoclinic system with P21/c, C2/c, and P21/n space groups, respectively (number of asymmetric units per unit cell Z = 4). Crystal phases of both [CoSAN-I,Br]− and [CoSAN-I,Cl]− are isostructural (P21/n, Z = 2). The [CoSAN-Cl,Br]− is the only among herein reported that adopts the orthorhombic and noncentrosymmetric Pna21 phase (Z = 4). In all hetero-disubstituted compounds, the halogens occupy two sides with equal (50%) probability. As presented in Fig. 2, the incorporation of halides induces changes in conformation. The parent [CoSAN]− is characterized by cisoid, [CoSAN-I]− adopts gauche, while all the examined disubstituted derivatives prefer the transoid conformation.
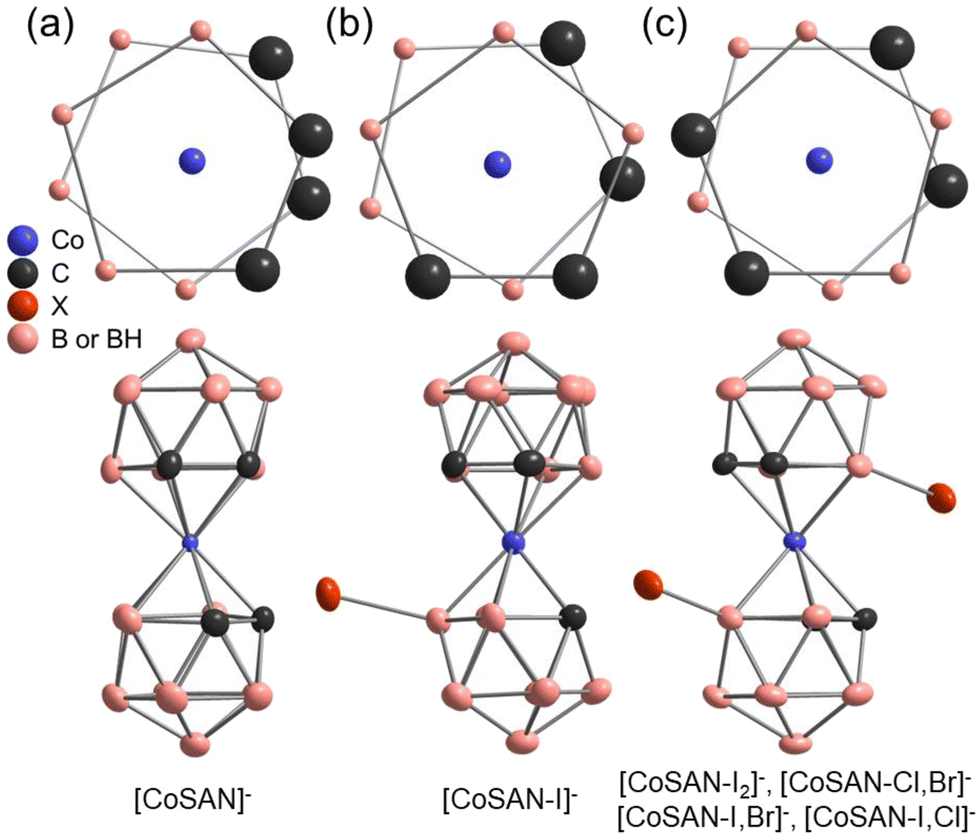 | ||
| Fig. 2 Schematic representation of different conformations of [CoSAN]− and its derivatives: (a) cisoid, (b) gauche, and (c) transoid (different halogens are indicated as X). | ||
Antimicrobial activity
Our investigations of biological activity started with the assessment of the antimicrobial effectiveness of synthesized derivatives and parent [CoSAN]− through the determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) values. This evaluation involved representatives of both Gram-positive and Gram-negative bacteria: Staphylococcus aureus ATCC 6538, Enterococcus faecium PCM 2910, Escherichia coli PCM 1630 and Pseudomonas aeruginosa PCM 2720 (Tables 1 and S2†).| S. aureus ATCC 6538 | E. faecium PCM 2910 | IC50 (95% CI) (μM) | SI S. a. (—) | SI E. f. [=(—) | Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (SE) (—) P (SE) (—) |
Logkw (SE) (—) |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIC (μM) | MBC (μM) | MIC (μM) | MBC (μM) | MCF 10A | HEK293 | |||||
| MIC, minimum inhibitory concentration; MBC, minimum bactericidal concentration; Van, vancomycin; IC50, half maximal inhibitory concentration of proliferation; MCF 10A, human mammary epithelial cell line; HEK293, human embryonic kidney epithelial cell line; SI S. a., selectivity index calculated as IC50 MCF 10A/MICS. aureus; SI E. f., selectivity index calculated as IC50 MCF 10A/MICE. faecium; logP, decimal logarithm of partition coefficient; logkw, decimal logarithm of chromatographic lipophilicity index; SE, standard error; “—” not determined. |
||||||||||
| [CoSAN]− | 50 | 100 | 25 | 50 | 34.94 (—) | 40.59 (33.43–50.87) | 0.70 | 1.40 | 1.59 (0.01) | 3.26 (0.01) |
| [CoSAN-F2]− | 25 | 50 | 50 | 100 | 33.91 (—) | 28.94 (24.49–34.28) | 1.36 | 0.68 | 1.30 (0.01) | 3.06 (0.02) |
| [CoSAN-Cl2]− | 12.5 | 25 | 12.5 | 25 | 25.44 (20.99–29.93) | 22.25 (19.34–25.53) | 2.04 | 2.04 | 3.17 (0.02) | 4.11 (0.02) |
| [CoSAN-Br2]− | 3.1 | 6.2 | 6.2 | 12.5 | 23.93 (19.59–28.42) | 22.56 (18.15–28.06) | 7.72 | 3.86 | 3.65 (0.03) | 4.33 (0.02) |
| [CoSAN-I2]− | 1.6 | 3.1 | 1.6 | 12.5 | 17.47(14.64–20.86) | 17.78 (15.67–20.16) | 10.92 | 10.92 | 4.23 (0.04) | 4.68 (0.03) |
| [CoSAN-Cl]− | 12.5 | 25 | 12.5 | 25 | 32.67 (—) | 29.43 (24.12–37.02) | 2.61 | 2.61 | 2.15 (0.01) | 3.60 (0.02) |
| [CoSAN-Br]− | 3.1 | 6.2 | 6.2 | 25 | 31.66 (—) | 26.59 (22.16–32.28) | 10.21 | 5.11 | 2.40 (0.02) | 3.75 (0.03) |
| [CoSAN-I]− | 0.8 | 1.6 | 1.6 | 12.5 | 31.28 (25.92–35.19) | 20.84 (16.63–26.31) | 39.10 | 19.55 | 2.79 (0.01) | 3.96 (0.02) |
| [CoSAN-Cl,Br]− | 6.2 | 12.5 | 12.5 | 25 | 29.41 (24.27–32.85) | 23.74 (20.48–27.45) | 4.74 | 2.35 | 3.33 (0.02) | 4.23 (0.02) |
| [CoSAN-I,Br]− | 1.6 | 3.1 | 6.2 | 12.5 | 20.65 (16.82–25.06) | 25.67 (21.10–30.60) | 12.91 | 3.33 | 4.12 (0.03) | 4.53 (0.03) |
| [CoSAN-I,Cl]− | 1.6 | 3.1 | 6.2 | 12.5 | 11.15 (—) | 11.66 (10.22–13.26) | 6.97 | 1.80 | 3.78 (0.03) | 4.42 (0.03) |
| Van | <0.4 | 0.4 | <0.4 | 50/100 | — | — | — | — | — | — |
Among all tested compounds, the highest antibacterial activity was observed against Staphylococcus aureus ATCC 6538 (Fig. 3). All synthesized derivatives exhibit better antibacterial effects than parent [CoSAN]−. In general, for mono- and homo-disubstituted derivatives, the antimicrobial activity increases with the mass of halogen present in the structure (MIC ranging from 25 μM for [CoSAN-F2]− to 0.8 μM for [CoSAN-I]−). There is no difference between homo-disubstitution and monosubstitution in MIC and MBC between them, except for iodinated derivatives (MIC for [CoSAN-I]− and [CoSAN-I2]− is 0.8 μM and 1.6 μM, respectively). This finding indicates that the introduction of a second halogen into the structure does not significantly affect the activity against S. aureus ATCC 6538. On the other hand, a significantly greater difference is observed among the hetero-disubstituted derivatives when the presence of iodine is analyzed. The MIC for [CoSAN-I,Br]− and [CoSAN-I,Cl]− is 1.6 μM. However, [CoSAN-Cl,Br]− significantly deviates from the others, with MIC of 6.2 μM. In contrast to this derivative, [CoSAN-I,Br]− and [CoSAN-I,Cl]− have iodine in which their activities are closer to those of [CoSAN-I]− and [CoSAN-I2]−. Thus, it can be inferred that the incorporation of iodine increases the antibacterial activity of the obtained derivative.
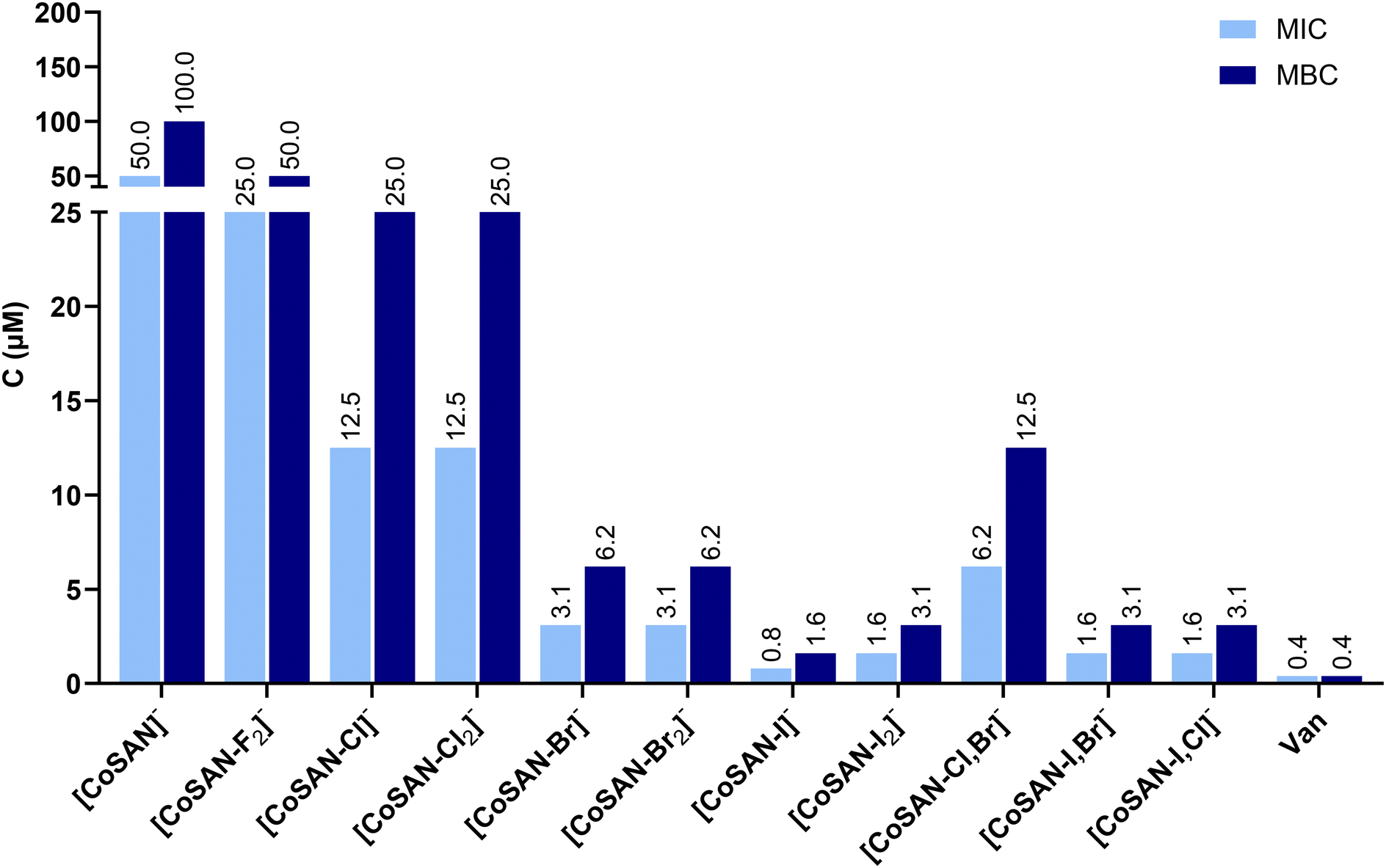 | ||
| Fig. 3 Antibacterial activity of [CoSAN]− and its halogenated derivatives against S. aureus ATCC 6538 (MIC – minimal inhibitory concentration, MBC – minimal bactericidal concentration, C – concentration, Van – vancomycin). | ||
Enterococcus faecium PCM 2910 was the second tested Gram-positive bacteria. Compared with S. aureus, synthesized derivatives showed weaker antibacterial activity. The antibacterial activity of [CoSAN-F2]− was significantly weaker than that of the parent [CoSAN]−. Other trends are consistent with those observed for S. aureus ATCC 6538: the relationship between the mass of the halogen substituent, the weak or no effect of homo-disubstitution compared with that of monosubstituted derivatives and the evident beneficial effect of iodination, i.e., the presence of iodine ensures an enhanced antimicrobial effect.
The obtained derivatives did not show antibacterial activity against Gram-negative Pseudomonas aeruginosa PCM 2720 and Escherichia coli PCM 1630. The MIC and MBC values were above the test threshold of 100 μM, with the exclusion of [CoSAN-I,Cl]− which inhibited the growth of both bacteria at 100 μM.
Time-kill kinetics assay
To further explore the mechanism of action of the most active halo derivatives, a time-kill kinetics assay was used. Considering the Selectivity Index (Table 1), we investigated the antibacterial effects of three iodine-containing compounds: [CoSAN-I]−, [CoSAN-I2]−, and [CoSAN-I,Br]− against S. aureus (Fig. 4A). Among tested compounds, the kinetics of [CoSAN-I2]− action were the quickest. A clear bactericidal effect is observed already after 3 hours of incubation. This may be correlated with the highest lipophilicity of [CoSAN-I2]− among the tested derivatives. After 24 h, all tested compounds had bactericidal effects, with an efficacy of 99.9% for S. aureus reduction. | ||
| Fig. 4 (A) Time-kill kinetics assay of [CoSAN-I2]−, [CoSAN-I]−, and [CoSAN-I,Br]− against S. aureus. (B) Dependency of the logarithmic MIC on logP for the studied derivatives. | ||
Antiproliferative activity
To explore the cytotoxicity of the studied compounds, a sulforhodamine B (SRB) assay was used. The compounds were tested on the normal cell lines: MCF 10A (epithelial human mammary gland cell line) and HEK293 (epithelial human embryonic kidney cell line), as well as the cancer cell lines: A549 (epithelial human lung adenocarcinoma) and MCF-7 (epithelial human breast adenocarcinoma).In the case of the MCF 10A cell line, the monosubstituted derivatives display nearly identical IC50 values, suggesting a similar level of cytotoxicity. However, a noticeable decrease in the IC50 is observed with the introduction of a second halogen. The decreasing IC50 tends to correlate with the increasing atomic mass of the halogen. For the HEK293 cell line, both the monosubstituted and the homo-disubstituted derivatives exhibited a clear trend: the heavier the halogen present in the molecule, the lower the IC50 is. Among the hetero-disubstituted derivatives, [CoSAN-I,Cl]− exhibited the highest cytotoxicity in both MCF 10A and HEK293 cell lines (Table 1).
Tested compounds exhibited the highest cytotoxicity against the A549 cell line. Within monosubstituted and homo-disubstituted derivatives, the IC50 decreases as the halogen present in the structure becomes heavier, with lower concentrations observed for the homo-disubstituted derivatives. [CoSAN-I,Cl]− is the most cytotoxic (8.45 μM). Similar trends were observed for the MCF-7 cell line, with lower cytotoxicity of the compounds than those of the A549 cell line (ESI, Table S2†).
In summary, the IC50 values for all the halo derivatives and the parent [CoSAN]− against both nontumorigenic and cancer cells ranged from 11.15 μM to 75.53 μM. The greatest increase in antiproliferative activity compared to the parent [CoSAN]− was observed for the hetero-disubstituted [CoSAN-I,Cl]−, with a 4.5-fold increase in activity for the MCF-7 cell line and an approximately 3-fold increase in activity for the other cell lines studied.
Selectivity index
The Selectivity Index (SI) is a parameter that plays a crucial role in pharmacology. The safety and efficacy of a compound are assessed by considering its toxic (IC50) and bioactive (MIC) concentrations. The higher the selectivity index is, the better the indication of a more promising antimicrobial agent.43Considering the MIC obtained for S. aureus, all synthesized derivatives have an SI higher than [CoSAN]−, with the highest value characterizing [CoSAN-I]−. As the atomic mass of the halogen present in the structure increases, the SI also increases. However, the introduction of a second halogen leads to a decrease in the SI. The most significant decrease is induced by the introduction of a second iodine atom into the structure (72%, 57%, 56% and 66% for MCF 10A, HEK293, A549 and MCF-7 cell lines, respectively). Among the derivatives substituted with two different halogens, the highest SI characterizes the [CoSAN-I,Br]− compound.
The relationships in the SI obtained for E. faecium (Table S2†) are generally similar to those observed in S. aureus, but with two exceptions. The greatest decrease in the SI occurs with the addition of a second bromine for the A549 cell line, while introducing a second chlorine result in the most significant decrease in the SI for the HEK293 cell line. Overall, the SI values are lower than those obtained for S. aureus, indicating a reduced selectivity toward this microorganism.
Lipophilicity
One of the most crucial physicochemical criteria for novel compound applications in biomedical studies is the lipophilicity parameter. It is characterized as a logarithm of the partition coefficient (logP), typically between n-octanol and water.44,45 The logP values of the tested compounds range from 1.30 to 4.23. Except for [CoSAN-F2]−, the obtained compounds are more lipophilic than [CoSAN]−. The lipophilicities of the derivatives increase with increasing atomic mass of the halogen substituent. The introduction of a second halogen significantly raises the logP, with the heavier halogens causing more pronounced differences in lipophilicity. Another descriptor of lipophilicity, logkw, is determined chromatographically and showcases the compound's interactions with the column's stationary phase in Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC).24 Although logkw differs from logP (values vary from 3.06 to 4.68), the observed relationships are consistent across both parameters.
The graph illustrating the relationship between logP and logMIC (for S. aureus) allows us to advance our understanding of the effects of lipophilicity on antimicrobial activity (Fig. 4B). The most hydrophilic [CoSAN-F2]− stands out from the other derivatives and has the lowest antibacterial activity among all the derivatives. For the monosubstituted and homo-disubstituted compounds, the trend is evident: increasing logP corresponds to enhanced antibacterial activity. Disubstituted compounds with iodine ([CoSAN-I2]−, [CoSAN-I,Br]− and [CoSAN-I,Cl]−) appear to be among the most lipophilic and active. However, [CoSAN-I]−, identified as the most selective antimicrobial agent, is characterized by moderate lipophilicity. Importantly, the lipophilicity of metallacarboranes cannot be used as the primary factor determining their biological activity. In studies on cell lines, [CoSAN-I2]− is often preferred because of its higher lipophilicity and correspondingly higher activity; however, in the antibacterial context, this advantage is lost when compared with that of monosubstituted [CoSAN-I]− due to SI decrease.
To summarize, the biological and physicochemical properties of the studied derivatives strongly depend on the kind of halogen incorporated into [CoSAN]−'s core structure. Synthesizing a range of halogen derivatives revealed that the presence of iodine in the structure provides high activity against Gram-positive strains. The presence of a single iodine atom in [CoSAN]−'s core structure did not significantly affect its antiproliferative activity against the nontumorigenic MCF 10A and HEK293 cell lines, but it increased activity against the S. aureus strain by more than 60-fold and against the E. faecium strain by more than 15-fold. Moreover, the atom substituted at the B(8′)-position notably impacts selectivity toward eukaryotic cells. This phenomenon is particularly evident in the case of the pair [CoSAN-I]−/[CoSAN-I2]−. Introducing the second iodine into the cluster did not improve antibacterial activity, but it reduced the SI nearly by 4-fold for the MCF 10A cell line and by almost 2-fold for the HEK293 cell line.
An essential factor in the biological efficacy of therapeutic compounds is their ability to interact with or permeate cell surfaces. In eukaryotic cells, this barrier is the lipid bilayer, whereas in bacteria it is further reinforced by a cell wall. In Gram-positive bacteria, the cell envelope consists of a thick peptidoglycan layer, while in Gram-negative bacteria, a thinner peptidoglycan layer is surrounded by an additional outer membrane containing lipopolysaccharides. Previous studies utilizing various measurement techniques have provided evidence for the direct membrane permeation of selected [CoSAN]− halogen derivatives.12,22,27,46 Electrophysiological techniques have shown that permeation follows the order [CoSAN-I2]− > [CoSAN-Br2]− > [CoSAN-Cl2]− > [CoSAN]−.12 Unfortunately, these studies compared only the halogen homo-disubstituted derivatives of [CoSAN]−. Similarly, fluorescence displacement assays have demonstrated that the relative translocation rates of the investigated clusters (including the mono-substituted [CoSAN-I]−) follow the order [CoSAN-I2]− > [CoSAN-Cl2]− > [CoSAN-I]− > [CoSAN]−.46 This finding indicates that the mono-substituted [CoSAN-I]− derivative has a lower translocation propensity than the disubstituted [CoSAN-I2]− and [CoSAN-Cl2]− derivatives. These findings again suggest that membrane transport is most efficient for disubstituted derivatives, with the highest efficiency observed for the derivative containing the heaviest halogen, [CoSAN-I2]−. When these results for membrane permeation were compared with our findings on the biological activities of homo-disubstituted derivatives, we observed an identical trend when antimicrobial activity and cytotoxicity were treated separately. Does this, therefore, suggest that the biological activity of [CoSAN]− derivatives is proportional to their membrane permeation rate? This intrinsic membrane affinity can, in part, be rationalized by their polarizabilities and molecular volumes, which depend on the substituents, as well as the resulting lipophilicity. However, our research indicates that traversing the first biological barrier—the lipid membrane—is only the initial step in their biological activity, and the most efficient membrane transport does not necessarily guarantee an optimal biological effect.
From a structural perspective, the presence of halogen atoms in the CoSAN core, each with unique electronegativity, polarizability, and van der Waals radius induces interactions that stabilize specific rotamers.47 In homo- and hetero-disubstituted derivatives, these halogens are expected to enhance the stabilization of cisoid conformations through intramolecular CH⋯X hydrogen bond formation. Among these factors, the van der Waals radius of the halogen has a more significant impact than electronegativity; for example, the highly electronegative fluorine atom does not form intramolecular CH⋯F hydrogen bonds due to its insufficient size. In contrast, for other homo- and hetero-disubstituted derivatives, the strength of intramolecular hydrogen bonds follows the trend I > Br > Cl.47,48 Energetically favorable conformations of [CoSAN]− derivatives, as determined by XRD (solid state) or quantum chemical calculations (typically in the gas phase), may not necessarily be preferred in solution, particularly within biological environments. Therefore, whether specific [CoSAN]− derivative conformations are retained in solution remains an open question. Conformational changes generate a dipole moment that may influence interactions with biological targets, especially given that the presence of a specific biomolecule could induce or stabilize a particular conformation.
Finally, it is worth analyzing the synthetic possibilities that enable the synthesis of [CoSAN-I]− derivatives for biological testing. One such strategy involves the nucleophilic opening of the [8,8′-μ-I-3,3′-Co(1,2-C2B9H10)2] – iodonium bridge, wherein two dicarbollide ligands are linked via an iodine atom.37,49,50 Despite being less common than opening [CoSAN]− cyclic oxonium salts with nucleophiles,11,16,17 this reaction has emerged as an exceedingly valuable tool for expanding the family of [CoSAN-I]− derivatives. The B(8)-I structural motif appears in the group of [CoSAN]− derivatives with noteworthy activity against Gram-positive bacteria.24,51 In addition to iodine, they have an organic substituent at the B(8′)-position. By selecting the appropriate organic substituents, it is possible to synthesize compounds that are effective against pathogenic microorganisms. The results presented in this work emphasize the significant impact of the iodine on biological activity of [CoSAN]− derivatives, which have favorable therapeutic properties and low toxicity to mammalian cells.
Conclusions
The exploration of halo derivatives derived from cobalt bis(dicarbollide) anions commenced in 1968 with the synthesis of [3,3′-Co(8,9,12-Br3-1,2-C2B9H8)2]−.52 Iodo derivatives of metallacarboranes have drawn attention owing to their potential as precursors in boron substitution reactions.36 From a chemical standpoint, this choice is well-founded. However, the biological outlook presents a distinct situation. Although studies comparing the biological properties of the [CoSAN]−/[CoSAN-I2]− pair confirm the positive influence of iodine, the exclusive selection of [CoSAN-I2]− seems to lack a clear rationale.Here, we provide solid scientific evidence justifying the rationale for choosing iodo-derivatives for biological applications. Our findings concerning antibacterial effectiveness are consistent with those of previously published studies, in which iodinated derivatives were shown to have enhanced biological activity.22,24 However, testing various mono- and disubstituted derivatives, with consideration of other biological features, leads to the conclusion that the introduction of only a single iodine at the B(8)-position is beneficial for selective antimicrobial action. Nevertheless, to better understand the effects of iodination on their biological behavior, a more detailed examination of their interactions with other biological compounds and cellular compartments is needed.
Materials and methods
Cs[CoSAN] was purchased from KatChem spol. s r. o. For single-crystal X-ray diffraction measurements, the precipitate was recrystallized from water:ethanol mixture (1:1 v/v). Cs[CoSAN-I] was synthesized according to the procedure published by Rojo et al.36 and recrystallized from water:acetone mixture (1:1 v/v). [8,8′-μ-I-3,3-Co(1,2-C2B9H10)2] was synthesized according to previously published procedures.53
Single-crystal X-ray diffraction
The single-crystal X-ray diffraction experiments of selected compounds were conducted using a conventional four-circle diffractometer (Xcalibur Atlas). The experiments utilized MoKα radiation and a CCD (Atlas) camera. Absorption was corrected by the multi-scan method. The empirical absorption correction using spherical harmonics was done in SCALE3 ABSPACK scaling algorithm implemented in CrysAlis PRO 1.171.42.93a (Rigaku Oxford Diffraction, 2023). For solving and refining the structures, the ShelXT (https://doi.org/10.1107/S2053273314026370) and ShelXL (https://doi.org/10.1107/S2053229614024218) programs were employed, respectively. Hydrogen atoms were introduced at calculated positions and refined as riding atoms. The measurements were conducted at room temperature (295 K). The crystallographic structure of the only noncentrosymmetric phase [CoSAN-Cl,Br]− was refined as an inversion twin. The selected experimental and refinement details are shown in the ESI, Table S1.† CCDC 2298165–2298170.†Nuclear magnetic resonance spectroscopy
The 1H, 13C{1H}, DEPT 135, 11B and 11B{1H} NMR spectra were recorded on a 400 MHz Jeol ECZ 400S (NMR) spectrometer in acetone-d6 (Merck, cat. no. 151793) as the solvent in Wilmad® quartz NMR tubes (Merck, cat. no. Z562262). Chemical shifts (δ) were expressed in parts per million (ppm), while multiplicity was reported as follows: s = singlet, d = dublet, t = triplet, q = quartet, quint = quintet, sext = sextet, hept = heptet, nonet = nonet, m = multiplet (complex pattern), br = broad. For clarity, the set of broad multiplets of B–H protons at ca. 1–4 ppm has been omitted in the 1H NMR description.Ion exchange chromatography
The chromatographic column packed with cation exchange resin (Amberlite® IR120, H form, ion-exchange resin, Merck) was rinsed with 120 mL of 3 M HCl and 500 mL of water to achieve a neutral pH. The mixture was subsequently washed with 120 mL of 3 M NaCl solution, 500 mL of water, and then with 120 mL of acetonitrile (Supelco) and water mixture (50:50 v/v, except for [CoSAN-I,Cl]− – 60:40 v/v). The compound dissolved in an appropriate acetonitrile:water mixture was loaded onto the column. After passing through the column, the desired eluate fraction was collected and lyophilized. The completion of the ion exchange and the concentrations of all compounds, relative to their cesium counterparts, were determined using a quantitative HPLC method described below.
High-performance liquid chromatography (HPLC) and mass spectrometry (MS)
HPLC analyses were carried out using the Ultimate 3000 RS HPLC system (Dionex, Sunnyvale, CA, USA) equipped with a DAD detector using full gradient separation. Column: Hypersil Gold 50 × 2.1 (Thermo Scientific, s/n 0110796A6); mobile phase A: 0.1% trifluoroacetic acid in water, mobile phase B: 0.1% trifluoroacetic acid in CH3CN; gradient: time/% of B: 0/5, 1/5, 16/95, 20/95; flow rate: 0.5 mL min−1; injection 1 μL. The UV-VIS spectra were recorded using acetonitrile–water mixture (55:45 v/v for compound 10, 60:40 v/v for compound 9, 70:30 v/v for remaining compounds) containing 0.1% trifluoroacetic acid. High-resolution mass spectrometry experiments were carried out on a MicrOTOF-Q II spectrometer (Bruker Daltonic, Bremen, Germany) with an electrospray ion source. The instrument was calibrated with a sodium formate solution (10 mM) and operated in the negative-ion mode.
Syntheses of compounds
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H), 50.86 (CH2), 69.47 (CH2Cl); 19F NMR (376 MHz, acetone-d6) δ: −147.00 (q, 1JB–F = 77.1 Hz); note: on 11B, 11B{1H} and 19F NMR spectra additional signals of mono- and trifluoro derivatives are present in trace amounts. The compound was converted into its acidic form using a cation exchange resin (Amberlite® IR120).
H); 19F NMR (376 MHz, acetone-d6) δ: −147.30 (q, 1JB–F = 75.5 Hz); note: on 11B, 11B{1H} and 19F NMR spectra additional signals of mono- and trifluoro-derivatives are present in trace amounts. ESI-MS [M]−m/z (calculated/found) 360.2646/360.2646, HPLC purity: 96.03%.
H), 50.86 (CH2), 69.47 (CH2Cl); 19F NMR (376 MHz, acetone-d6) δ: −147.00 (q, 1JB–F = 77.1 Hz); note: on 11B, 11B{1H} and 19F NMR spectra additional signals of mono- and trifluoro derivatives are present in trace amounts. The compound was converted into its acidic form using a cation exchange resin (Amberlite® IR120).
H); 19F NMR (376 MHz, acetone-d6) δ: −147.30 (q, 1JB–F = 75.5 Hz); note: on 11B, 11B{1H} and 19F NMR spectra additional signals of mono- and trifluoro-derivatives are present in trace amounts. ESI-MS [M]−m/z (calculated/found) 360.2646/360.2646, HPLC purity: 96.03%.
Synthesis of Cs[CoSAN-Cl2]
1.00 g Cs[CoSAN] (2.20 mmol) was dissolved in 40 mL THF. 0.294 g of NCS (Merck, Cat. No. 109681) (2.20 mmol) was added and the reaction mixture darkened. Then, 2.20 mmol of NCS was added three times at 15-minute intervals. Finally, a total amount of 1.18 g (8.80 mmol) of NCS was added. The solution was stirred for 2 hours at room temperature. 1.10 g of Na2SO3 (Acros Organics, code: 424432500) (8.73 mmol) was dissolved in 30 mL of water and added to the reaction mixture. After the evaporation of THF, the product is observed in the form of dark red oil. The product did not solidify after storing for 20 hours at 4 °C, so the evaporation was continued. An orange precipitate formed upon water evaporation. The precipitate was filtered under reduced pressure and washed with 15 mL of water and 15 mL of petroleum ether. The final product was dried under a vacuum. Yield: 0.975 g (84.7%). 1H NMR (400 MHz, acetone-d6) δ: 4.26 (br s, 4 × BCH, 4H); 11B{1H} NMR (128 MHz, acetone-d6) δ: 11.94 (br s, 2B), −0.60 (br s, 2B), −5.98 (br s, 8B), −19.52 (br s, 4B), −25.79 (br s, 2B); 11B NMR (128 MHz, acetone-d6) δ: 11.89 (br s, 2B), −0.57 (br d, J = 135.7 Hz, 2B), −5.98 (br d, J = 140.2 Hz, 8B), −19.52 (br d, J = 159.2 Hz, 4B), 25.81 (br d, J = 169.5 Hz, 2B); 13C{1H} NMR (100 MHz, acetone-d6) δ: 55.77 (br s, 4 × BH). ESI-MS [M]−m/z (calculated/found) 393.2020/393.2040, HPLC purity: 99.9%.
:3 v/v). The Ethanol was partially evaporated, and the resulting orange crystals precipitated. After storing for 20 hours at 4 °C, the precipitate was filtered and washed with 15 mL of water and 15 mL of petroleum ether. The final product was dried under a vacuum. Yield: 1.20 g (89.2%). 1H NMR (400 MHz, acetone-d6) δ: 4.33 (br s, 4 × BCH, 4H); 11B{1H} NMR (128 MHz, acetone-d6) δ: 6.31 (br s, 2B), 0.44 (br s, 2B), −5.47 (br s, 8B), −18.95 (br s, 4B), −24.89 (br s, 2B); 11B NMR (128 MHz, acetone-d6) δ: 6.21 (br s, 2B), 0.44 (br d, J = 112.26 Hz, 2B), −4.31 to −6.05 (m, 8B), −18.95 (br d, J = 159.6 Hz, 4B), −24.87 (br d, J = 180.9 Hz, 2B); 13C{1H} NMR (100 MHz, acetone-d6) δ: 57.61 (br s, 4 × BH). ESI-MS [M]−m/z (calculated/found) 482.1029/482.1023, HPLC purity: 98.7%.
:acetone mixture (2:1 v/v). HPLC purity: 98.5%. All the NMR spectra are in good agreement with the literature data.23,35
H), 56.42 (br s, 2 × BH). ESI-MS [M]−m/z (calculated/found) 358.2452/358.2459, HPLC purity: 98.7%.
H), 58.03 (br s, 2 × BH). ESI-MS [M]−m/z (calculated/found) 403.1949/403.1940, HPLC purity: 98.0%.
H), 57.10 (br s, 2 × BH). For single-crystal X-ray diffraction measurements, the precipitate was recrystallized from water:methanol mixture (1:1 v/v). ESI-MS [M]−m/z (calculated/found) 437.1561/437.1551, HPLC purity: 96.8%.
H), 59.50 (br s, 2 × BH). For single-crystal X-ray diffraction measurements, the precipitate was recrystallized from water:methanol mixture (1:1 v/v). ESI-MS [M]−m/z (calculated/found) 529.0916/529.0906, HPLC purity: 99.0%.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3CH2, J = 7.2 Hz, 3H), 1.67 (d, 2 × (3)2CH, J = 6.8 Hz, 12H), 3.81 (q, CH32, J = 7.2 Hz, 2H), 4.26 (br s, 2 × BCH, 2H), 4.35 (hept, 2 × (CH3)2, J = 7.6 Hz) and 4.38 (br s, 2 × BCH) [total integration 4H]; 11B{1H} NMR (128 MHz, acetone-d6) δ: 12.36 (br s, 1B), 1.09 (br s, 1B), 0.10 (br s, 1B), −4.38 and −5.21 (2 × br s, total integration 8B), −7.02 (br s, 1B), −18.12 (br s, 2B), −19.49 (br s, 2B), −23.59 (br s, 1B), −25.76 (s, 1B); 11B{1H} NMR (128 MHz, acetone-d6) δ: 12.36 (br s, 1B), 1.09 (br s, 1B), 0.10 (br s, 1B), −4.38 and −5.21 (2 × br s, total integration 8B), −7.02 (br s, 1B), −18.12 (br s, 2B), −19.49 (br s, 2B), −23.59 (br s, 1B), −25.76 (br s, 1B); 11B NMR (128 MHz, acetone-d6) δ: 12.36 (br s, 1B), 1.17 (br d, J = 134.8 Hz, 1B), 0.16 (br d, J = 123.6 Hz, 1B), −3.73 to −6.78 (m, 9B), −18.14 (br d, J = 167.7 Hz, 2B), −19.44 (br d, J = 167.7 Hz, 2B), −23.55 (br d, J = 181.6 Hz, 1B), −25.71 (br d, J = 166.9 Hz, 1B); 13C{1H} NMR (100 MHz, acetone-d6) δ: 9.70 (H3CH2), 18.00 ((H3)2CH), 18.13 ((H3)2CH), 57.00 (br s, 2 × BH), 58.98 (br s, 2 × BH), 62.96 (CH3H2), 64.56 ((CH3)2H); note: on 1H NMR and 13C{1H} NMR spectra a peak of residual CH2Cl2 (reaction solvent) is present at 5.54 ppm and 52.93 ppm, respectively. The product was dissolved in 20 mL of ethanol, and a solution of CsCl (0.400 g) in 12 mL of water was added. The mixture was then concentrated until an orange solid, Cs[CoSAN-I,Cl], precipitated. The solid was filtered off, washed with water and petroleum ether, and dried in vacuo. The precipitate was recrystallized from water:acetonitrile mixture (1:2 v/v) to obtain XRD-quality crystals. ESI-MS [M]−m/z (calculated/found) 484.1419/484.1423, HPLC purity: 99.1%.
3CH2, J = 7.2 Hz, 3H), 1.67 (d, 2 × (3)2CH, J = 6.8 Hz, 12H), 3.81 (q, CH32, J = 7.2 Hz, 2H), 4.26 (br s, 2 × BCH, 2H), 4.35 (hept, 2 × (CH3)2, J = 7.6 Hz) and 4.38 (br s, 2 × BCH) [total integration 4H]; 11B{1H} NMR (128 MHz, acetone-d6) δ: 12.36 (br s, 1B), 1.09 (br s, 1B), 0.10 (br s, 1B), −4.38 and −5.21 (2 × br s, total integration 8B), −7.02 (br s, 1B), −18.12 (br s, 2B), −19.49 (br s, 2B), −23.59 (br s, 1B), −25.76 (s, 1B); 11B{1H} NMR (128 MHz, acetone-d6) δ: 12.36 (br s, 1B), 1.09 (br s, 1B), 0.10 (br s, 1B), −4.38 and −5.21 (2 × br s, total integration 8B), −7.02 (br s, 1B), −18.12 (br s, 2B), −19.49 (br s, 2B), −23.59 (br s, 1B), −25.76 (br s, 1B); 11B NMR (128 MHz, acetone-d6) δ: 12.36 (br s, 1B), 1.17 (br d, J = 134.8 Hz, 1B), 0.16 (br d, J = 123.6 Hz, 1B), −3.73 to −6.78 (m, 9B), −18.14 (br d, J = 167.7 Hz, 2B), −19.44 (br d, J = 167.7 Hz, 2B), −23.55 (br d, J = 181.6 Hz, 1B), −25.71 (br d, J = 166.9 Hz, 1B); 13C{1H} NMR (100 MHz, acetone-d6) δ: 9.70 (H3CH2), 18.00 ((H3)2CH), 18.13 ((H3)2CH), 57.00 (br s, 2 × BH), 58.98 (br s, 2 × BH), 62.96 (CH3H2), 64.56 ((CH3)2H); note: on 1H NMR and 13C{1H} NMR spectra a peak of residual CH2Cl2 (reaction solvent) is present at 5.54 ppm and 52.93 ppm, respectively. The product was dissolved in 20 mL of ethanol, and a solution of CsCl (0.400 g) in 12 mL of water was added. The mixture was then concentrated until an orange solid, Cs[CoSAN-I,Cl], precipitated. The solid was filtered off, washed with water and petroleum ether, and dried in vacuo. The precipitate was recrystallized from water:acetonitrile mixture (1:2 v/v) to obtain XRD-quality crystals. ESI-MS [M]−m/z (calculated/found) 484.1419/484.1423, HPLC purity: 99.1%.
Octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
The octanol–water partition coefficient was determined via the shake-flask method.54 To 1.5 mL of an aqueous solution of the selected compound at a concentration of 1.33 mM, 100 μL of octanol was added, and the resulting mixture was shaken for 2 hours at 25 °C. The vials were subsequently centrifuged at 25000g for 10 minutes (25 °C). After phase separation, 5 μL of octanol phase was transferred to another vial and octanol was removed under reduced pressure. After drying, the residue was dissolved in 500 μL of water containing 0.1% TFA and analysed by HPLC. 1 mL of water phase was transferred to another vial and lyophilized. Dry residue was dissolved in 40 μL of water containing 0.1% TFA and analyzed via HPLC. The octanol–water partition coefficient was determined by the ratio of the compound's quantity in the octanol and the water phase. The experiment was performed in three repetitions.
Chromatographic partition coefficient (logkw)
The chromatographic partition coefficient was determined by HPLC according to a previously published procedure.24 The analyses were performed using the Ultimate 3000 RS HPLC system (Dionex, Sunnyvale, CA, USA) with a DAD detector, equipped with a reverse-phase C-18 column (Hypersil Gold 50 × 2.1, Thermo Scientific, s/n 0110796A) using CH3CN/water/0.1% trifluoroacetic acid elution system. The measurements were performed with a flow rate of 0.5 mL min−1 at 25 °C. Retention times (tR) for each compound was obtained via repeated measurements in isocratic elutions in the range of 85–60% of CH3CN with a step of 2.5%. The k value was calculated according to the equation k = (tR − t0)/t0, where tR is the retention time of the substance and t0 is the dead-time. For each compound, a plot of the log capacity factor (logkw) and the mobile phase's composition was generated. The intercept of the plot (logkw) corresponds to the log capacity factor of the compound in 100% water, which is considered the chromatographic partition coefficient between water and the hydrophobic stationary phase.
Bacterial strains
Staphylococcus aureus strain was obtained from ATCC 6538, Enterococcus faecium (PCM 2910), Pseudomonas aeruginosa (PCM 2720), and Escherichia coli (PCM 1630) were obtained from the Polish Collection of Microorganisms HIIET Poland. All bacterial strains were maintained at the Hirszfeld Institute, Wrocław, Poland. Vancomycin (vancomycin, MIP Pharma) and polymyxin B (polymyxin B sulfate salt, Merck) were used as control antibiotics.Minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) assays
The 24-hour inoculum was transferred to a fresh Mueller–Hinton medium (Mueller–Hinton Broth 2, Merck Millipore) and incubated at 0.5 McFarland. The prepared suspension was diluted 100 times with the Mueller–Hinton medium and then 20 μL of the prepared bacterial suspension was added to each well on the plate. A series of dilutions of the tested compounds were made in the range of 100 μM–0.4 μM on a 96-well plate using Mueller–Hinton medium. The wells were then filled with medium and the prepared bacterial inoculum was added to each well to a final volume of 200 μL. The plate was incubated for 24 h at 37 °C, after which the minimum inhibitory concentration (MIC) of the compounds was visually assessed. The minimum bactericidal concentration (MBC) was assessed by plating a suspension from wells in which no bacterial growth was visually observed on agar plates (LAB-AGAR™, BioMaxima S. A.). The plates were incubated for 24 h at 37 °C, and the colonies were counted. The MBC was estimated for the concentrations of compounds causing at least a 99.9% CFU mL−1 reduction. The experiment was performed in three independent repetitions.Time-kill assay
The antimicrobial activity of the compounds was investigated by analyzing the survival of bacteria treated with the compounds at given exposure times. S. aureus ATCC6538 (0.5 McFarland) was treated with the compounds [CoSAN-I]−, [CoSAN-I2]−, and [CoSAN-I,Br]− at the MBC values. At various periods (0.5, 1, 2, 3, and 24 h) equal volumes of samples were diluted and plated on agar plates. Bacterial colonies were counted after 24 h of incubation at 37 °C. The experiment was performed in three independent repetitions.Cell culture lines and culture conditions
A549 (human lung carcinoma) and MCF-7 (human breast adenocarcinoma) cells were purchased from the European Collection of Authenticated Cell Cultures (ECACC; Porton Down, UK), while HEK293 (human embryonic kidney epithelial cell line) and MCF 10A (non-tumorigenic human mammary epithelial cell line) cells were purchased from the American Type Culture Collection (ATCC; Rockville, USA). All the cell lines were maintained at the Hirszfeld Institute of Immunology and Experimental Therapy (HIIET), Wrocław, Poland.The A549 cell line was cultured in OptiMEM (HIIET, PAS, Wroclaw, Poland) supplemented with RPMI 1640 (Thermo Fisher Scientific, Waltham, USA) at a 1:1 ratio and supplemented with 5% (v/v) fetal bovine serum (FBS; GE Healthcare HyClone, Logan, USA) and 2 mM L-glutamine (Merck). The MCF-7 cell line was cultured in Eagle's medium supplemented with 10% (v/v) FBS (Merck), 2 mM L-glutamine (Merck), 1% (v/v) amino acids (Gibco, Thermo Fischer Scientific), and 0.8 μg mL−1 insulin (Merck). The HEK293 cell line was cultured in Eagle's medium (Thermo Fisher Scientific) supplemented with 10% (v/v) FBS (GE Healthcare HyClone) and 2 mM L-glutamine. The MCF 10A cell line was cultured in Ham's F12 medium with glutamine (Corning, New York, USA) supplemented with 5% (v/v) horse serum, 10 μg mL−1 insulin, 0.05 μg mL−1 cholera toxin, 0.5 μg mL−1 hydrocortisone and 20 ng mL−1 hEGF (all from Merck). All culture media were supplemented with the antibiotics – 100 μg mL−1 streptomycin (Polfa Tarchomin, Warsaw, Poland) and 100 U mL−1 penicillin (Merck). The cells were grown at 37 °C in a humid atmosphere saturated with 5% CO2.
Antiproliferative activity assessment by sulforhodamine B assay
The cells were seeded on 384-well plates (Greiner Bio One, Kremsmünster, Austria) at 2 × 103 cells per well density for the HEK293 cell line and 1 × 103 cells per well for the A549, MCF-7 and MCF 10A cell lines. After overnight incubation, the test compounds were applied at various concentrations (ranging from 316 μM to 0.01 μM). After 72 h of incubation, the sulforhodamine B (SRB) assay based on Skehan et al.55 was carried out with slight modifications. In brief, 50 μL of the medium was replaced with 30 μL per well of 25% (w/v) trichloroacetic acid (Avantor). After 40 minutes of incubation at room temperature, the plates were washed three times with deionized water and 20 μL of a 0.1% (w/v) solution of sulforhodamine B (Merck) in 1% (v/v) acetic acid (Avantor) was added to each well. After 30 min of incubation at room temperature, the unbound dye was washed out with 1% (v/v) acetic acid. Bound dye was solubilized with 70 μL of 10 mM unbuffered TRIS (Avantor) solution. The procedure was performed using a BioTek EL-406 washing station (BioTek Instruments). The absorbance was read using a Biotek Hybrid H4 reader at a wavelength of 540 nm. The crude absorbance data were used to calculate the degree of proliferation inhibition using the following formula: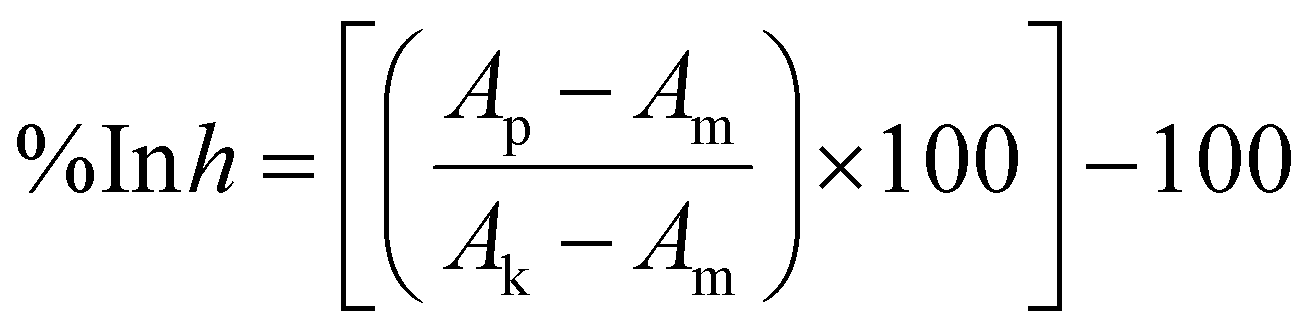 | (1) |
The %Inh was next used for IC50 calculations performed in GraphPad Prism 7.05 (GraphPad Software, Inc.) utilizing the ‘[Inhibitor] vs. response − Variable slope (four parameters)’ model.
The experiment was performed in three independent repetitions.
The selectivity index
The selectivity index was calculated via the following formula: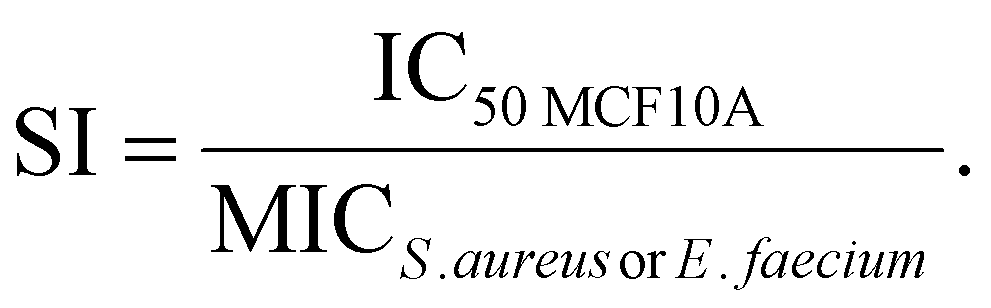 | (2) |
Author contributions
Conceptualization: TMG; data curation: KZ-D, WG, MG, DD, AG; formal analysis: KZ-D, MG, DD, BS-O, WG, TMG; funding acquisition: TMG; investigation: KZ-D, WG, MG, DD, BS-O, TMG; methodology TMG; project administration: TMG; resources TMG, AG, WG; supervision: TMG; validation: TMG, AG, WG; visualization: KZ-D, TMG, DD; writing – original draft: KZ-D; writing – review & editing MG, TMG.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data has been deposited at the CCDC 2298170 – Cs[CoSAN], 2298165 – Cs[CoSAN-I], 2298169 – Cs[CoSAN-I2], 2298168 – Cs[CoSAN-Br,Cl], 2298167 – Cs[CoSAN-I,Br], 298166 – Cs[CoSAN-I,Cl].†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Jakub Cebula for his critical discussion and valuable insights into the final version of this manuscript.References
- M. F. Hawthorne, D. C. Young and P. A. Wegner, Carbametallic Boron Hydride Derivatives. I. Apparent Analogs of Ferrocene and Ferricinium Ion, J. Am. Chem. Soc., 1965, 87, 1818–1819 CrossRef CAS.
- R. N. Grimes, Metallacarboranes in the new millennium, Coord. Chem. Rev., 2000, 200, 773–811 CrossRef.
- P. L. Bora and A. K. Singh, New insights into designing metallacarborane based room temperature hydrogen storage media, J. Chem. Phys., 2013, 139, 164319 CrossRef PubMed.
- J. Brus, J. Czernek, M. Urbanova, J. Rohlíček and T. Plecháček, Transferring Lithium Ions in the Nanochannels of Flexible Metal–Organic Frameworks Featuring Superchaotropic Metallacarborane Guests: Mechanism of Ionic Conductivity at Atomic Resolution, ACS Appl. Mater. Interfaces, 2020, 12, 47447–47456 CrossRef CAS PubMed.
- Z. Yinghuai, S. L. P. Sia, K. Carpenter, F. Kooli and R. A. Kemp, Syntheses and catalytic activities of single-wall carbon nanotubes-supported nickel(II) metallacarboranes for olefin polymerization, J. Phys. Chem. Solids, 2006, 67, 1218–1222 CrossRef.
- I. Guerrero, Z. Kelemen, C. Viñas, I. Romero and F. Teixidor, Metallacarboranes as Photoredox Catalysts in Water, Chem. – Eur. J., 2020, 26, 5027–5036 CrossRef CAS PubMed.
- N. Murphy, E. McCarthy, R. Dwyer and P. Farras, Boron clusters as breast cancer therapeutics, J. Inorg. Biochem., 2021, 218, 111412 CrossRef CAS.
- M. Gozzi, B. Schwarze and E. Hey-Hawkins, Preparing (Metalla)carboranes for Nanomedicine, ChemMedChem, 2021, 16, 1533–1565 CrossRef CAS PubMed.
- K. Fink and M. Uchman, Boron cluster compounds as new chemical leads for antimicrobial therapy, Coord. Chem. Rev., 2021, 431, 213684 CrossRef CAS.
- M. Nuez-Martinez, M. Queralt-Martin, A. Munoz-Juan, V. M. Aguilella, A. Laromaine, F. Teixidor, C. Vinas, C. G. Pinto, T. Pinheiro, J. F. Guerreiro, F. Mendes, C. Roma-Rodrigues, P. V. Baptista, A. R. Fernandes, S. Valic and F. Marques, Boron clusters (ferrabisdicarbollides) shaping the future as radiosensitizers for multimodal (chemo/radio/PBFR) therapy of glioblastoma, J. Mater. Chem. B, 2022, 10, 9794–9815 RSC.
- B. P. Dash, R. Satapathy, B. R. Swain, C. S. Mahanta, B. B. Jena and N. S. Hosmane, Cobalt bis(dicarbollide) anion and its derivatives, J. Organomet. Chem., 2017, 849–850, 170–194 CrossRef CAS.
- T. I. Rokitskaya, I. D. Kosenko, I. B. Sivaev, Y. N. Antonenko and V. I. Bregadze, Fast flip-flop of halogenated cobalt bis(dicarbollide) anion in a lipid bilayer membrane, Phys. Chem. Chem. Phys., 2017, 19, 25122–25128 RSC.
- Y. Chen, A. Barba-Bon, B. Gruner, M. Winterhalter, M. A. Aksoyoglu, S. Pangeni, M. Ashjari, K. Brix, G. Salluce, Y. Folgar-Camean, J. Montenegro and W. M. Nau, Metallacarborane Cluster Anions of the Cobalt Bisdicarbollide-Type as Chaotropic Carriers for Transmembrane and Intracellular Delivery of Cationic Peptides, J. Am. Chem. Soc., 2023, 145, 13089–13098 CrossRef CAS.
- K. I. Assaf and W. M. Nau, The Chaotropic Effect as an Assembly Motif in Chemistry, Angew. Chem., Int. Ed. Engl., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- I. B. Sivaev and V. I. Bregadze, Chemistry of cobalt bis(dicarbollides). A review, Collect. Czech. Chem. Commun., 1999, 64, 783–805 CrossRef CAS.
- L. Pazderova, E. Z. Tuzun, D. Bavol, M. Litecka, L. Fojt and B. Gruner, Chemistry of Carbon-Substituted Derivatives of Cobalt Bis(dicarbollide)(1(-)) Ion and Recent Progress in Boron Substitution, Molecules, 2023, 28, 6971 CrossRef CAS PubMed.
- A. A. Druzina, A. V. Shmalko, I. B. Sivaev and V. I. Bregadze, Cyclic oxonium derivatives of cobalt and iron bis(dicarbollides) and their use in organic synthesis, Russ. Chem. Rev., 2021, 90, 785–830 CrossRef.
- I. Bennour, M. N. Ramos, M. Nuez, J. A. M. Xavier, A. B. Buades, R. Sillanpää, F. Teixidor, D. Choquesillo-Lazarte, I. Romero, M. Martinez-Medina and C. Vinas, Water Soluble Organometallic Small Molecules as Promising Antibacterial Agents: Synthesis, Physical-chemistry properties and Biological Evaluation to Tackling Bacterial Infections, Dalton Trans., 2022, 51, 7188–7209 RSC.
- I. B. Sivaev and V. I. Bregadze, Chemistry of nickel and iron bis(dicarbollides). A review, J. Organomet. Chem., 2000, 614, 27–36 CrossRef.
- N. Murphy, W. J. Tipping, H. J. Braddick, L. T. Wilson, N. C. O. Tomkinson, K. Faulds, D. Graham and P. Farras, Expanding the Range of Bioorthogonal Tags for Multiplex Stimulated Raman Scattering Microscopy, Angew. Chem., Int. Ed., 2023, 62, e202311530 CrossRef CAS PubMed.
- A. Munoz-Juan, M. Nuez-Martinez, A. Laromaine and C. Vinas, Exploring the Role of Metal in the Biointeraction of Metallacarboranes with C. elegans Embryos, Chemistry, 2024, 30, e202302484 CrossRef CAS.
- M. Tarres, E. Canetta, E. Paul, J. Forbes, K. Azzouni, C. Vinas, F. Teixidor and A. J. Harwood, Biological interaction of living cells with COSAN-based synthetic vesicles, Sci. Rep., 2015, 5, 7804 CrossRef CAS PubMed.
- K. Kubinski, M. Maslyk, M. Janeczko, W. Goldeman, A. Nasulewicz-Goldeman, M. Psurski, A. Martyna, A. Boguszewska-Czubara, J. Cebula and T. M. Goszczynski, Metallacarborane Derivatives as Innovative Anti-Candida albicans Agents, J. Med. Chem., 2022, 65, 13935–13945 CrossRef CAS.
- J. Cebula, K. Fink, W. Goldeman, B. Szermer-Olearnik, A. Nasulewicz-Goldeman, M. Psurski, M. Cuprych, A. Kedziora, B. Dudek, G. Bugla-Ploskonska, M. Chaszczewska-Markowska, M. Gos, P. Migdal and T. M. Goszczynski, Structural Patterns Enhancing the Antibacterial Activity of Metallacarborane-Based Antibiotics, J. Med. Chem., 2023, 66, 14948–14962 CrossRef CAS PubMed.
- M. Nuez-Martinez, C. I. G. Pinto, J. F. Guerreiro, F. Mendes, F. Marques, A. Munoz-Juan, J. A. M. Xavier, A. Laromaine, V. Bitonto, N. Protti, S. G. Crich, F. Teixidor and C. Vinas, Cobaltabis(dicarbollide) ([o-COSAN](-)) as Multifunctional Chemotherapeutics: A Prospective Application in Boron Neutron Capture Therapy (BNCT) for Glioblastoma, Cancers, 2021, 13, 6367 CrossRef CAS.
- M. Nuez-Martinez, L. Pedrosa, I. Martinez-Rovira, I. Yousef, D. Diao, F. Teixidor, E. Stanzani, F. Martinez-Soler, A. Tortosa, A. Sierra, J. J. Gonzalez and C. Vinas, Synchrotron-Based Fourier-Transform Infrared Micro-Spectroscopy (SR-FTIRM) Fingerprint of the Small Anionic Molecule Cobaltabis(dicarbollide) Uptake in Glioma Stem Cells, Int. J. Mol. Sci., 2021, 22, 9937 CrossRef CAS PubMed.
- C. Verdia-Baguena, A. Alcaraz, V. M. Aguilella, A. M. Cioran, S. Tachikawa, H. Nakamura, F. Teixidor and C. Vinas, Amphiphilic COSAN and I2-COSAN crossing synthetic lipid membranes: planar bilayers and liposomes, Chem. Commun., 2014, 50, 6700–6703 RSC.
- Ľ. Mátel, F. Macášek, P. Rajec, S. Heřmánek and J. Plešek, B-Halogen derivatives of the bis(1,2-dicarbollyl)cobalt(III) anion, Polyhedron, 1982, 1, 511–519 CrossRef.
- P. K. Hurlburt, R. L. Miller, K. D. Abney, T. M. Foreman, R. J. Butcher and S. A. Kinkead, New Synthetic Routes to B-Halogenated Derivatives of Cobalt Dicarbollide, Inorg. Chem., 1995, 34, 5215–5219 CrossRef CAS.
- A. B. Buades, C. Vinas, X. Fontrodona and F. Teixidor, 1.3 V Inorganic Sequential Redox Chain with an All-Anionic Couple 1-/2-in a Single Framework, Inorg. Chem., 2021, 60, 16168–16177 CrossRef CAS.
- P. Gonzalez-Cardoso, A. I. Stoica, P. Farras, A. Pepiol, C. Vinas and F. Teixidor, Additive Tuning of Redox Potential in Metallacarboranes by Sequential Halogen Substitution, Chem. – Eur. J., 2010, 16, 6660–6665 CrossRef CAS PubMed.
- M. D. Mortimer, C. B. Knobler and M. F. Hawthorne, Methylation of boron vertices of the cobalt dicarbollide anion, Inorg. Chem., 1996, 35, 5750–5751 CrossRef CAS PubMed.
- S. A. Anufriev, M. Y. Stogniy and I. B. Sivaev, A Simple Way to Obtain a Decachloro Derivative of Cobalt Bis(dicarbollide), Reactions, 2023, 4, 148–154 CrossRef CAS.
- A. N. Gashti, J. C. Huffman, A. Edwards, G. Szekeley, A. R. Siedle, J. A. Karty, J. P. Reilly and L. J. Todd, Fluorination studies of the [commo-3,3 ‘-Co(3,1,2-CoC2B9H11)(2)(-1)] ion, J. Organomet. Chem., 2000, 614, 120–124 CrossRef.
- I. Rojo, F. Teixidor, R. Kivekäs, R. Sillanpää and C. Viñas, Methylation and demethylation in cobaltabis(dicarbollide) derivatives, Organometallics, 2003, 22, 4642–4646 CrossRef CAS.
- I. Rojo, F. Teixidor, C. Vinas, R. Kivekas and R. Sillanpaa, Relevance of the electronegativity of boron in eta5-coordinating ligands: regioselective monoalkylation and monoarylation in cobaltabisdicarbollide [3,3′-Co(1,2-C2B9H11)2]- clusters, Chemistry, 2003, 9, 4311–4323 CrossRef CAS PubMed.
- I. D. Kosenko, I. A. Lobanova, I. B. Sivaev, P. V. Petrovskii and V. I. Bregadze, Synthesis of hetero-substituted derivatives of cobalt bis(1,2-dicarbollide), Russ. Chem. Bull., 2011, 60, 2354–2358 CrossRef CAS.
- A. Zalkin, T. E. Hopkins and T. Dh, Crystal Structure of Cs(B9c2h11)2co, Inorg. Chem., 1967, 6, 1911–1915 CrossRef CAS.
- L. Borodinsky, E. Sinn and R. N. Grimes, Crystal and molecular structure of (C2H5)3NH + [Co(1,2-C2B9H11)2], Inorg. Chem., 1982, 21, 1686–1689 CrossRef CAS.
- P. Sivý, A. Preisinger, O. Baumgartner, F. Valach, B. Koreň and L. Mátel, Structure of caesium 8-iodo-3,3′-commo-bis(decahydro-1,2-dicarba-3-cobalta-closo-dodecaborate)(1-), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 30–33 CrossRef.
- P. Sivý, A. Preisinger, O. Baumgartner, F. Valach, B. Koreň and L. Mátel, Structure of caesium 3,3′-commo-bis(decahydro-8-iodo-1,2-dicarba-3-cobalta-closo-dodecaborate)(1-), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 28–30 CrossRef.
- O. N. Kazheva, G. G. Aleksandrov, A. V. Kravchenko, V. A. Starodub, I. A. Lobanova, I. D. Kosenko, I. B. Sivaev, V. I. Bregadze, L. I. Buravov and O. A. Dyachenko, New Fulvalenium Salts of Cobalt Bis(dicarbollide): Crystal Structures and Electrical Conductivities, Crystals, 2012, 2, 43–55 CrossRef CAS.
- G. Indrayanto, G. S. Putra and F. Suhud, Validation of bioassay methods: Application in herbal drug research, Profiles Drug Subst., Excipients, Relat. Methodol., 2021, 46, 273–307 Search PubMed.
- S. Lobo, Is there enough focus on lipophilicity in drug discovery?, Expert Opin. Drug Discovery, 2020, 15, 261–263 CrossRef.
- T. Chmiel, A. Mieszkowska, D. Kempińska-Kupczyk, A. Kot-Wasik, J. Namieśnik and Z. Mazerska, The impact of lipophilicity on environmental processes, drug delivery and bioavailability of food components, Microchem. J., 2019, 146, 393–406 CrossRef CAS.
- K. I. Assaf, B. Begaj, A. Frank, M. Nilam, A. S. Mougharbel, U. Kortz, J. Nekvinda, B. Grüner, D. Gabel and W. M. Nau, High-Affinity Binding of Metallacarborane Cobalt Bis(dicarbollide) Anions to Cyclodextrins and Application to Membrane Translocation, J. Org. Chem., 2019, 84, 11790–11798 CrossRef CAS PubMed.
- I. B. Sivaev and I. D. Kosenko, Rotational conformation of 8,8′-dihalogenated derivatives of cobalt bis(dicarbollide) in solution, Russ. Chem. Bull., 2021, 70, 753–756 CrossRef CAS.
- I. B. Sivaev, Bis(Dicarbollide) Complexes of Transition Metals: How Substituents in Dicarbollide Ligands Affect the Geometry and Properties of the Complexes, Molecules, 2024, 29, 3510 CrossRef CAS PubMed.
- V. I. Bregadze, I. D. Kosenko, I. A. Lobanova, Z. A. Starikova, I. A. Godovikov and I. B. Sivaev, C-H Bond Activation of Arenes by [8,8 ‘-mu-I-3,3 ‘-Co(1,2-C2B9H10)(2)] in the Presence of Sterically Hindered Lewis Bases, Organometallics, 2010, 29, 5366–5372 CrossRef CAS.
- I. D. Kosenko, I. A. Lobanova, I. A. Godovikov, Z. A. Starikova, I. B. Sivaev and V. I. Bregadze, Mild C-H activation of activated aromatics with [8,8 ‘-mu-I-3,3 ‘-Co(1,2-C2B9H10)(2)]: Just mix them, J. Organomet. Chem., 2012, 721, 70–77 CrossRef.
- Y. K. Zheng, W. W. Liu, Y. Chen, H. Jiang, H. Yan, I. Kosenko, L. Chekulaeva, I. Sivaev, V. Bregadze and X. M. Wang, A Highly Potent Antibacterial Agent Targeting Methicillin-Resistant Based on Cobalt Bis(1,2-Dicarbollide) Alkoxy Derivative, Organometallics, 2017, 36, 3484–3490 CrossRef CAS.
- M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, L. F. Warren and P. A. Wegner, Pi-Dicarbollyl Derivatives of Transition Metals. Metallocene Analogs, J. Am. Chem. Soc., 1968, 90, 879–896 CrossRef CAS.
- W. Swietnicki, W. Goldeman, M. Psurski, A. Nasulewicz-Goldeman, A. Boguszewska-Czubara, M. Drab, J. Sycz and T. M. Goszczynski, Metallacarborane Derivatives Effective against Pseudomonas aeruginosa and Yersinia enterocolitica, Int. J. Mol. Sci., 2021, 22, 6762 CrossRef CAS.
- OECD, Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method, 1995 Search PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, New colorimetric cytotoxicity assay for anticancer-drug screening, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2298165–2298170. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02296c |
| This journal is © the Partner Organisations 2025 |