
Spiro-[4,5]-cyclohexadiene-8-one polymers: photoactivated crosslinking and switch-on fluorescence for lithography†
Yi
Yuan‡
a,
Mi
Chao‡
a,
Yunyi
Shang
a,
Yujia
Gao
a,
Guangle
Niu
 *b,
Wanggang
Fang
c,
Liqing
He
*c and
Hui
Wang
*a
*b,
Wanggang
Fang
c,
Liqing
He
*c and
Hui
Wang
*a
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, College of Chemistry & Materials Science, Northwest University, Xi’an 710127, China. E-mail: whui210@nwu.edu.cn
bSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: niugl@bit.edu.cn
cNational Engineering & Technical Research Center on Pressure Vessels and Piping Safety, Hefei General Machinery Research Institute Co., Ltd, Hefei 230031, China. E-mail: heli_limao@163.com
First published on 8th November 2024
Abstract
Developing multiple photoresponsive polymers is crucial for creating versatile intelligent materials; however, it poses a significant challenge due to the limited availability of photoactivated moieties. Herein, we present a novel series of dual photoresponsive spiro-[4,5]-cyclohexadiene-8-one polymers exhibiting photoactivated crosslinking and switch-on fluorescence behaviors. These polymers were synthesized through a robust palladium-catalyzed [2+2+1] cycloaddition polymerization reaction of 4-phenol diazonium tetrafluoroborate and diynes. Notably, the single photoreactive spiro-[4,5]-cyclohexadiene-8-one moiety endowed dual photoresponse features to these polymers. Upon UV irradiation, the cyclohexadienone moieties underwent a 2π+2π photocycloaddition reaction to form an insoluble crosslinked polymer network. Concurrently, the photoactivated fluorescence phenomenon of the crosslinked polymers was also observed. To our knowledge, these polymers represent the first examples of merging photocrosslinking and fluorescence turn-on properties into one single functional group. By harnessing the unique photocrosslinking and photoactivated fluorescence properties, we successfully imprinted 2D and 3D photopatterns for lithographic applications. These intriguing results provide an alternative design strategy of multiple photoresponsive polymers for fluorescent labelling and 2D/3D optical security.
Introduction
Stimulated responsive polymers have garnered increasing attention for designing smart materials due to their ability to respond to external stimuli, including electricity, magnetism, heat, and light.1,2 Among various external stimuli, light stands out for its cleanliness, robust remote controllability, and precision in regulating reactions.3 Leveraging these benefits, the integration of specific photosensitive elements into polymers enables the construction of photoresponsive polymers.4–7 Incorporation of fluorochromic groups such as azobenzene and spiropyran into polymers is a general strategy to construct photochromic or photoswitch polymers with reversible color changes or the fluorescence on/off property when exposed to specific wavelengths of light.8 Through the macroscopic fluorescence change, these polymers can effectively encode the information by fluorescence signals. However, the current method for storing information is predominantly confined to two-dimensional (2D) formats.9,10 These 2D encoding approaches suffer from a security risk of information leakage, as the information can be easily decrypted, for instance, through exposure to commercial ultraviolet (UV) light.11 To address this limitation, the integration of a photoresponsive moiety into polymers represents a promising strategy for 3D encoding.12Photocrosslinking polymers facilitate the information programming via mask and wash postprocessing, leading to insoluble 3D images upon illumination.13,14 Inspired by their common photoresponsive characteristics, we envisioned the possibility of designing a photochromic(switch)-integrated-photocrosslinking (PIP) polymer system to realize the 3D-encoding with the aid of mask assistant photocrosslinking technology as well as render the 2D-encoding using fluorochromic dyes. Coumarin15,16 and its derivatives17–19 equipped with carbonyl-activated carbon–carbon double bonds are one of the most widely utilized photocrosslinkable units (Fig. 1a), which are capable of undergoing reversible phototriggered 2π+2π cycloaddition with fast response and excellent thermal stability. The predominant approach for fabricating such systems is focused on integrating fluorochromic dyes into photoclickable polymer chains. However, the complexity of multistep preparation processes hampers their practicality in real samples.20,21 Given these challenges, the exploration and development of a novel polymer backbone containing a single specific functional group that combines both photocrosslinking and fluorochromic characteristics could offer a fresh perspective for composite polymers in the realm of 2D and 3D encoding applications.
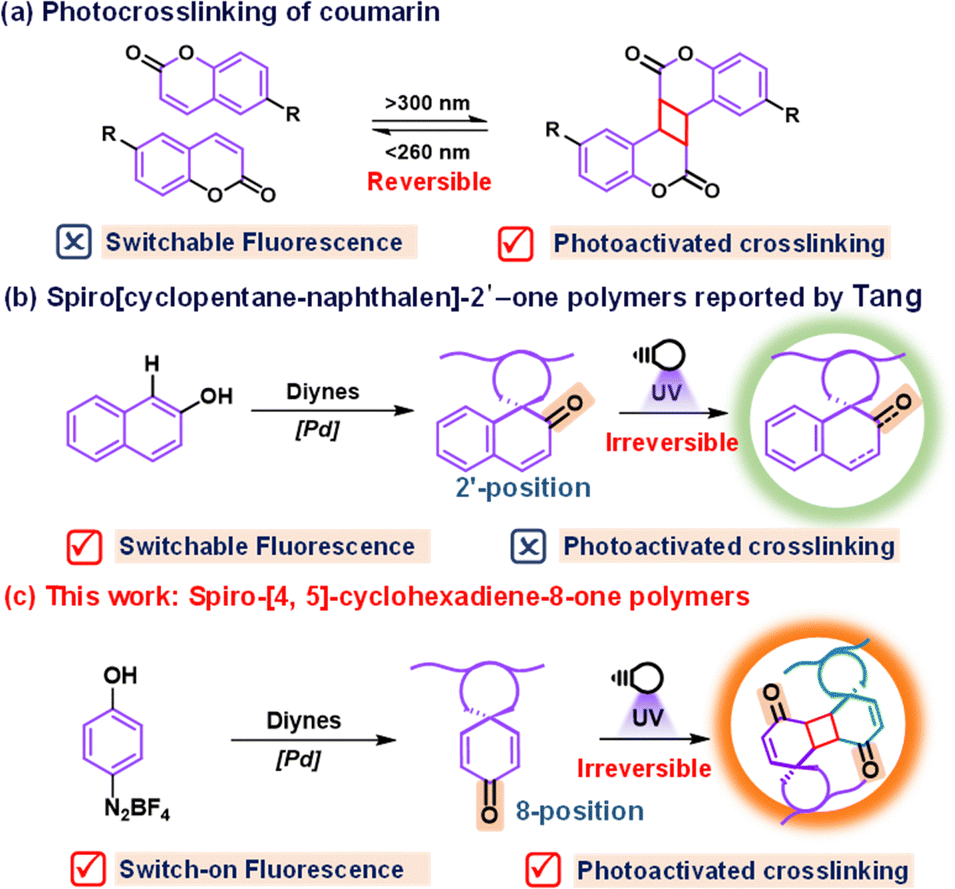 | ||
| Fig. 1 (a) Previous works on photocrosslinking of coumarin; (b) spiro[cyclopentane-naphthalen]-2′-one polymer by Tang; and (c) spiro-[4,5]-cyclohexadiene-8-one polymers in this work. | ||
In 2019, Tang's research group intelligently synthesized a series of spiro[cyclopentane-naphthalen]-2′-one polymers using a simple one-pot C–H activation/dearomatization strategy (Fig. 1b).22 The obtained spiro-polymers can be utilized to fabricate different well-resolved fluorescent photopatterns based on their photoinduced fluorescence switch properties. Moreover, the refractive index undergoes significant changes before and after irradiation, making them suitable for integrated silicon photon technology. What inspires us is that the fluorescence change originated from the photooxidation reaction, instead of the photocrosslinking reaction, between the carbonyl-activated carbon–carbon double bonds, which is typically different from the reported coumarin derivatives. In this respect, we designed and synthesized other spiro-cyclopentane naphthalenone polymer isomers to investigate whether the existence of the spiro-ring or the steric problem impacts the potential photocrosslinking properties and also explore the feasibility of novel PIP polymers through the spirocyclic scaffold.
In this study, we report the synthesis of a novel PIP polymer with the spiro-[4,5]-cyclohexadiene-8-one core through the A1 + B2 step polymerization employing easily accessible 4-phenol diazonium tetrafluoroborate and internal diynes. Notably, the resulting polymers exhibit rapid photocrosslinking capabilities to form dense networks and display photo-activated fluorescent turn-on properties upon exposure to 365 nm light. It is worth noting that these rigid polymers represent the first example of merging photocrosslinking and fluorescence turn-on properties into one single functional group. The unique dual-response behaviour, which is distinct from the 2-carbonyl isomer reported by the Tang group, is attributed to the 2π+2π cycloaddition reaction between spiro-[4,5]-cyclohexadiene-8-one moieties (Fig. 1c). This mechanism was corroborated through ultraviolet fluorescence spectra, 1H-NMR spectra, and infrared spectra. By harnessing the photocrosslinking capabilities and the alteration in the polymer's fluorescence properties upon irradiation, we succeeded in preparing 2D and 3D lithographic patterning using a simple method. Actually, for lithography, the photosensitive polymer development has experienced three different periods: the first one was the oil soluble polymer (also known as a negative resist), which contains many residual double bonds to facilitate crosslinking with other UV sensitive compounds (such as bisaryl azide).23 Upon radiation, the crosslinking took place and images with a resolution of microns became possible. However, due to the non-polar solvent usage for removal of the uncrosslinked regions, such materials suffer from swelling problems of the crosslinked regions and distortion of small features. The second one is the novolac-diazonaphthoquinone (DNQ) system (also known as a positive resist).24 The combined system was insoluble in an aqueous base; however, it became aqueous soluble upon irradiation due to the formation of a carboxylic acid as the photochemical rearrangement of DNQ. More recently, chemically amplified photoresists (CAR) composed of a poly(hydroxystyrene) polymer protected with a t-Boc group and combined with a photo-acid generator were invented by Willson, Fréchet and Ito.25 Upon exposure, the photo-acid generator releases a proton that can deprotect the polymer by release of t-Boc, which generates a high solubility contrast between the unexposed and protected polymers. All the mentioned photolithography techniques focus on the resolution of images and solubility of photo-responsive polymers, but little attention has been paid to the changes in fluorescence signals. Therefore, the proposed dual photoresponsive strategy in this study will provide a new idea for the photolithography techniques related to fluorescence changes.
Results and discussion
Polymerization
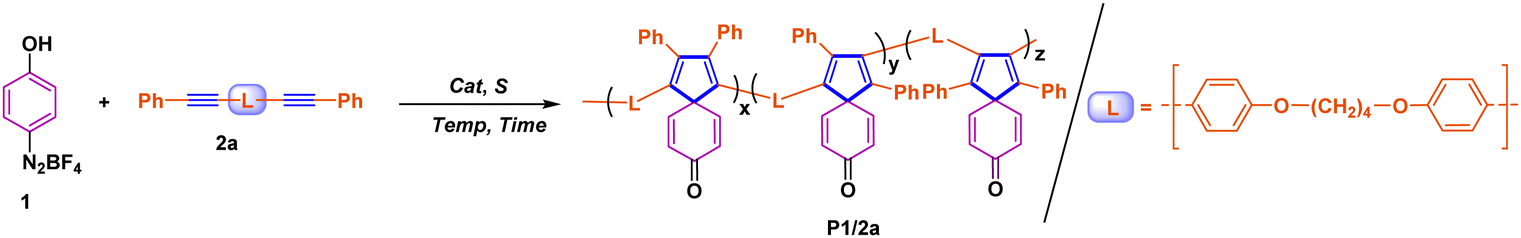
The starting point of this reaction was inspired by the small molecule synthesis from Pd-catalyzed dearomatized coupling of arene diazonium salts and internal alkynes reported by the Schmidt group.26 Considering the robust rigidity of the spiro scaffold, it is necessary to introduce flexible fragments to facilitate polymerization and make the multicoupling much easier. In this regard, the internal diyne 2a featuring flexible four methylene groups was selected as the model monomer to investigate its reactivity with 4-phenol diazonium tetrafluoroborate 1a.In the first experiment, the optimized conditions for the synthesis of small molecule spiro-[4,5]decatetraene-8-one were adopted, that is, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of both monomers, the absence of base, and using 5 mmol% Pd(OAc)2 as a precatalyst in the presence of MeOH. However, only small molecules were obtained without any oligomer or polymer insolubles in the post-solution treatment (see Tables S1 and S2 in the ESI†). Despite temperature adjustments, the situation did not improve. Finally, we found that the poor solubility of diacetylene in methanol hindered the polymerization, prompting us to modify the reaction solvent to a mixed solvent, and successfully promoted the formation of the polymer in the mixed solution of dichloromethane (DCM) and methanol, which are good solvents for diacetylene and diazonium salt, respectively.
:1 ratio of both monomers, the absence of base, and using 5 mmol% Pd(OAc)2 as a precatalyst in the presence of MeOH. However, only small molecules were obtained without any oligomer or polymer insolubles in the post-solution treatment (see Tables S1 and S2 in the ESI†). Despite temperature adjustments, the situation did not improve. Finally, we found that the poor solubility of diacetylene in methanol hindered the polymerization, prompting us to modify the reaction solvent to a mixed solvent, and successfully promoted the formation of the polymer in the mixed solution of dichloromethane (DCM) and methanol, which are good solvents for diacetylene and diazonium salt, respectively.
After identifying the optimal solvent system, we first examined the effects of catalyst and base loading on the polymerization of 1 (0.1 M) and 2a (0.1 M). As shown in entries 1–6 of Table 1, polymeric yellowish-brown solid P1/2a with high molecular weights (Mw = 14000) can be generated in a high yield of 91% when conducting the reaction in 5% mmol PdCl2 without the assistance of any base, which is consistent with the proposed mechanism of oxidative addition, double carbopalladation, aryl insertion and β-hydride elimination cascade process.26 To be noted, due to the asymmetric structure of the internal diyne, the chemical structure of P1/2a shown in Table 1 was supposed to be composed of regioisomers.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | [1]/[2a] (M) | T (h) | Temp. (°C) | Yield (%) | M w (g mol−1) | M n (g mol−1) | Đ | DPc |
|
a Unless otherwise noted, [Pd] = 5 mmol %, DCM/MeOH with a 1/1 vol. ratio was used as the solvent.
b Determined by GPC in THF on the basis of a polymethyl methacrylate calibration. Đ = polydispersity = Mw/Mn.
c Degree of polymerization is determined by dividing the relative molecular weight (Mn) of the polymer by the molecular weight of its repeating unit.
d NaOAc was added, [1]:[NaOAc] = 1:3.
e [Pd] = 10%.
f [Pd] = 20%.
|
||||||||||
| Screening of the catalysts and base | ||||||||||
| 1 | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 24 | 40 | 91 | 14000 |
5100 | 2.7 | 9 |
| 2 | Pd(OAc)2 | DCM:MeOH = 1:1 |
0.1/0.1 | 24 | 40 | 54 | 5300 | 1600 | 3.3 | 3 |
| 3 | Pd(CF3CO2)2 | DCM:MeOH = 1:1 |
0.1/0.1 | 24 | 40 | 44 | 3300 | 2800 | 1.2 | 5 |
| 4d | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 24 | 40 | 85 | 8400 | 4600 | 1.8 | 8 |
| 5d | Pd(OAc)2 | DCM:MeOH = 1:1 |
0.1/0.1 | 24 | 40 | 73 | 8900 | 3600 | 2.5 | 7 |
| 6d | Pd(CF3CO2) | DCM:MeOH = 1:1 |
0.1/0.1 | 24 | 40 | 69 | 7000 | 3700 | 1.9 | 7 |
| Screening of temperature | ||||||||||
| 7 | PdCl2 | DCM:EtOH = 1:1 |
0.1/0.1 | 24 | 70 | 91 | 9500 | 2300 | 4.1 | 4 |
| 8 | PdCl2 | DCM:IPA = 1:1 |
0.1/0.1 | 24 | 70 | — | — | — | — | — |
| 9 | PdCl2 | DCM:TAA = 1:1 |
0.1/0.1 | 24 | 80 | — | — | — | — | — |
| 10 | PdCl2 | DCM:n-BuOH = 1:1 |
0.1/0.1 | 24 | 70 | — | — | — | — | — |
| 11 | PdCl2 | DCM:TFE = 1:1 |
0.1/0.1 | 24 | 40 | — | — | — | — | — |
| 12 | PdCl2 | DCM:HFIP = 1:1 |
0.1/0.1 | 24 | 40 | — | — | — | — | — |
| Screening of monomer concentration | ||||||||||
| 13 | PdCl2 | DCM:MeOH = 1:1 |
0.15/0.15 | 24 | 40 | 76 | 6200 | 4100 | 1.5 | 8 |
| 14 | PdCl2 | DCM:MeOH = 1:1 |
0.05/0.05 | 24 | 40 | 88 | 11800 |
6200 | 1.9 | 12 |
| 15 | PdCl2 | DCM:MeOH = 1:1 |
0.15/0.1 | 24 | 40 | 90 | 12000 |
4500 | 2.7 | 8 |
| 16 | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.15 | 24 | 40 | 84 | 9990 | 4200 | 2.4 | 8 |
| Screening of the reaction time and catalyst loading | ||||||||||
| 17 | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 48 | 40 | 89 | 15400 |
6700 | 2.3 | 13 |
| 18e | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 48 | 40 | 94 | 27400 |
16400 |
1.5 | 31 |
| 19f | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 48 | 40 | 69 | 10300 |
6300 | 1.6 | 12 |
| 20e | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 12 | 40 | 78 | 11600 |
9100 | 1.3 | 17 |
| 21e | PdCl2 | DCM:MeOH = 1:1 |
0.1/0.1 | 72 | 40 | 84 | 13400 |
9900 | 1.4 | 19 |
Next, the effects of temperature on the polymerization were investigated. The results shown in entries 7–12 suggested that variation of methanol to other high-boiling-point alcohols, such as IPA, TAA, and n-BuOH, or other polar protic solvents, including TFE or HFIP, completely halted the reaction. The only exception observed was with the DCM and EtOH mixture, which yielded a reduced polymerization efficiency with a degree of only 4. In this manner, DCM in combination with MeOH was still selected as the best mixture to adjust the temperature to 40 °C. As shown in entries 13–16, the relative proportions and absolute concentrations of the monomers 1 and 2a have a significant effect on the polymerization. Specifically, reducing the concentrations of both monomers 1 and 2a to 0.05 M leads to a moderate Mw of 11800 and a good polydispersity of 1.9. However, the nonstoichiometric proportion of the two monomers, such as 0.15/0.15 or 0.1/0.15, provided inferior results although the reaction can proceed smoothly. After the optimum catalyst, solvent, temperature, and monomer concentration were determined, the effects of catalyst equivalent and reaction time on polymerization were investigated. Satisfactorily, when the catalyst equivalent was increased to 10% and the reaction time was extended to 48 h, the target spiropolymer P1/2a was obtained with 93.7% yield, higher molecular weights (Mw = 27400, Mn = 16400) and a narrow polydispersity (Đ = 1.5).
Under the optimal conditions described above, we then studied the polymerization of diazonium salt with different internal diynes to enrich the polymer structures. As shown in Table 2, in addition to 2a–2c with different numbers (4–8) of flexible alkyl fragments, two rigid and conjugated internal alkynes were screened for the target polymerization, including dibenzothiophene-linked diyne 2d and triphenylamine-linked diyne 2e. Compared with the flexible alkynes with alkyl chains 2a–b, the reaction efficiency was decreased significantly due to the polyaromatic stiff linker of 2d–e as well as the highly rigid and twisted spiro-polymer, leading to oligomer P1/2d–e with a molar weight less than 10000 g mol−1. Interestingly, the efficiency of polymerization did not consistently improve with longer flexible alkyl chains. For example, for compound 2c with m equal to 8, the polymerization efficiency is significantly weakened, which may be related to the reduced probability of collisions between two acetylene molecules.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Polymer | Yield (%) | M w (g mol−1) | M n (g mol−1) | Đ | DPc |
| a Carried out in the presence of PdCl2 at 40 °C in DCM and MeOH under a nitrogen atmosphere for 48 h with [1] = [2] = 0.1 M and PdCl2 = 10 mol%. b Estimated by GPC in THF on the basis of polymethyl methacrylate calibration. Đ = polydispersity = Mw/Mn. c Degree of polymerization is determined by dividing the relative molecular weight (Mn) of the polymer by the molecular weight of its repeating unit. | ||||||
| 1 | P1/2a | 93.7 | 27400 |
16400 |
1.5 | 31 |
| 2 | P1/2b | 89.2 | 24100 |
13300 |
1.8 | 24 |
| 3 | P1/2c | 73.9 | 16000 |
7300 | 2.2 | 12 |
| 4 | P1/2d | 65.5 | 7900 | 3400 | 2.3 | 7 |
| 5 | P1/2e | 75.8 | 4500 | 2600 | 1.7 | 5 |
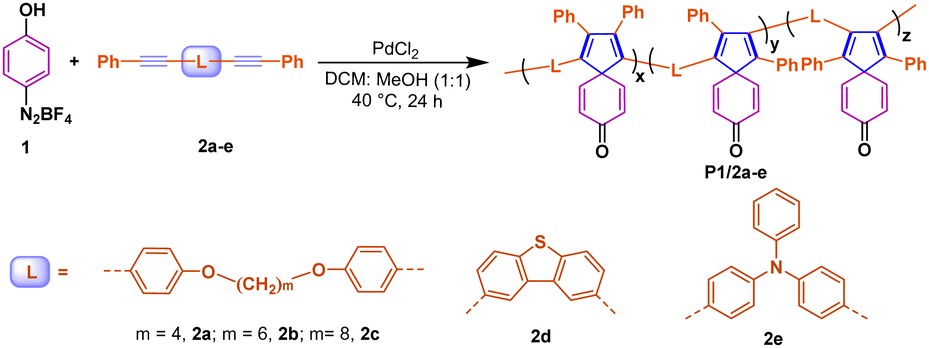
Structural characterization
The model compound 4, synthesized from reactants 4-phenol diazonium tetrafluoroborate 1 and diphenylacetylene 3 under the same reaction conditions, was prepared to confirm the structure of the target spiropolymers. According to reported small-molecule reactions and literature reviews,27,28 we suggested that the Pd–σ-aryl complex formed by the diazonium cation and Pd0 might subsequently undergo double carbopalladation to lead to the formation of target spiropolymers.The structural characterization of model compound 4, spiropolymer P1/2a, and their corresponding monomers 1 and 2a is discussed here as a representative example for comparative analysis. The Fourier-transform infrared (FT-IR) and nuclear magnetic resonance (NMR) spectra were utilized as the major characterization method. As shown in Fig. 2, both the absorption bands related to stretching vibrations of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond located at 2203 cm−1 of 2a in Fig. 2A and the NN bond located at 2230 cm−1 of 1 in Fig. 2B disappeared in Fig. 2C and D, indicating the reaction between the diazonium cation and the internal alkynes for the model reaction and polyannulation. New peaks emerged at 1650 cm−1, attributed to the stretching vibrations of the C
C bond located at 2203 cm−1 of 2a in Fig. 2A and the NN bond located at 2230 cm−1 of 1 in Fig. 2B disappeared in Fig. 2C and D, indicating the reaction between the diazonium cation and the internal alkynes for the model reaction and polyannulation. New peaks emerged at 1650 cm−1, attributed to the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in Fig. 2C and D, demonstrating the occurrence of dearomatized spirocyclization and the presence of spiro-[4,5]-cyclohexadiene-8-one in 4 and P1/2a. Similarly, the other four spiropolymers P1/2b–e displayed similar characteristic variation to P1/2a (Fig. S1–S15 in the ESI†). The structural identity of the generated polymer P1/2a was confirmed by analysis of the 1H NMR spectra of product 4 in comparison with those of monomers 1 and 2a. As illustrated by the low-field region of the 1H-NMR spectra in Fig. 2E–H, the disappearance of broad Ha peaks (4.0–4.4 ppm) corresponding to the phenolic hydroxyl proton in monomer 1 (Fig. 2E) and appearance of downfield double peaks of Hd (6.43 ppm) both in model product 4 and polymer P1/2a (Fig. 2G and H) indicate the formation of the hexadienone moiety in the resulting spiropolymers. Moreover, the sharp peaks corresponding to alkane chains at δ 3.93 (Hb) and δ 1.90 (Hc) in polymer P1/2a remain as sharp as their monomer counterparts in 2a. This indicates the existence of flexible alkyl chains in polymer chains and confirms the occurrence of [2+2+1] cyclization between two equiv. of alkyne and azides to promote the formation of spirorings. The 13C-NMR analysis provides more detailed information on the polymer structure in Fig. 2I–L. The resonance peaks assigned to the acetylene carbon atoms (Ci) at δ 89.4 and 88.0 ppm and the hydroxyl carbon atom (Ch) at δ 175.62 ppm all disappeared in the spectrum of P1/2a. Instead, a new peak at δ 186.2 ppm emerged implying the formation of carbonyl groups in the polymer structure. The combination of 1H NMR and 13C NMR spectra of P1/2a largely resemble those of the model compound 4, confirming the successful synthesis of polymers with the structures shown in Table 1.
O bond in Fig. 2C and D, demonstrating the occurrence of dearomatized spirocyclization and the presence of spiro-[4,5]-cyclohexadiene-8-one in 4 and P1/2a. Similarly, the other four spiropolymers P1/2b–e displayed similar characteristic variation to P1/2a (Fig. S1–S15 in the ESI†). The structural identity of the generated polymer P1/2a was confirmed by analysis of the 1H NMR spectra of product 4 in comparison with those of monomers 1 and 2a. As illustrated by the low-field region of the 1H-NMR spectra in Fig. 2E–H, the disappearance of broad Ha peaks (4.0–4.4 ppm) corresponding to the phenolic hydroxyl proton in monomer 1 (Fig. 2E) and appearance of downfield double peaks of Hd (6.43 ppm) both in model product 4 and polymer P1/2a (Fig. 2G and H) indicate the formation of the hexadienone moiety in the resulting spiropolymers. Moreover, the sharp peaks corresponding to alkane chains at δ 3.93 (Hb) and δ 1.90 (Hc) in polymer P1/2a remain as sharp as their monomer counterparts in 2a. This indicates the existence of flexible alkyl chains in polymer chains and confirms the occurrence of [2+2+1] cyclization between two equiv. of alkyne and azides to promote the formation of spirorings. The 13C-NMR analysis provides more detailed information on the polymer structure in Fig. 2I–L. The resonance peaks assigned to the acetylene carbon atoms (Ci) at δ 89.4 and 88.0 ppm and the hydroxyl carbon atom (Ch) at δ 175.62 ppm all disappeared in the spectrum of P1/2a. Instead, a new peak at δ 186.2 ppm emerged implying the formation of carbonyl groups in the polymer structure. The combination of 1H NMR and 13C NMR spectra of P1/2a largely resemble those of the model compound 4, confirming the successful synthesis of polymers with the structures shown in Table 1.
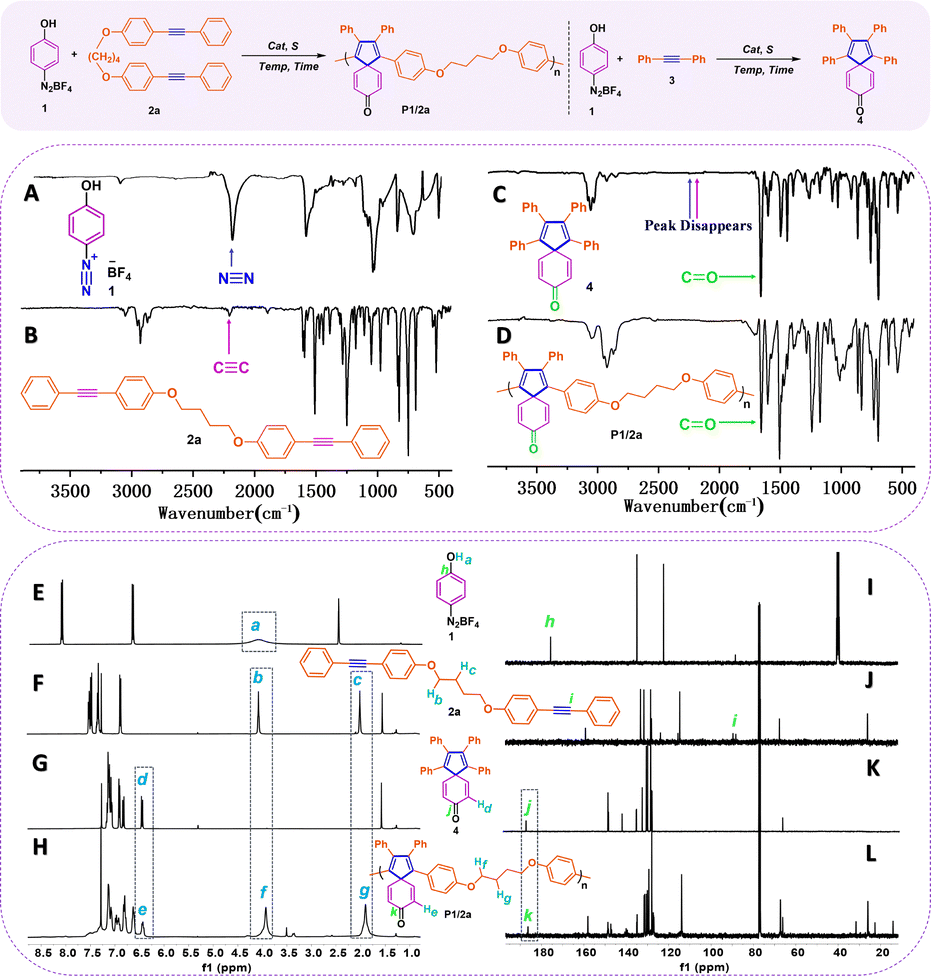 | ||
| Fig. 2 The synthetic route to spiropolymer P1/2a and model compound 4 (top reaction); FT-IR spectra of compound 1 (A), 2a (B), model compound 4 (C) and P1/2a (D) (middle part); and 1H-NMR and 13C-NMR spectra of 1 (E and I), 2a (F and J), model compound 4 (G and K) and P1/2a (H and L). | ||
Thermal stability and optical properties
The obtained spiropolymers P1/2a–2e possess good solubility in common organic solvents, such as chloroform (CHCl3), dioxane, tetrahydrofuran (THF), and dimethyl sulfoxide (DMSO), (Fig. S1–S15 in ESI†) as a result of the synergistic effect of the flexible alkoxyl groups and the suppression of close packing or interchain interactions caused by the rigid spiro-structure.29,30 Next, the thermal properties of the high-weight spiropolymers P1/2a–2c were evaluated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). As shown in Fig. 3A, the decomposition temperature corresponding to 5% weight loss ranged from 334 to 383 °C, and P1/2b can retain almost 60% carbon residues at a high temperature of up to 700 °C. The DSC results (Fig. 3B) indicated that the three polymers have good morphological stability with high glass transition temperatures (Tg) evaluated to be from 147 to 198 °C. Similarly, the Tg exhibited a decreasing trend with longer alkane chain lengths in the internal diynes. This trend was attributed to the plasticizing effect of the longer alkyl chains, which enhanced the rotational freedom of the polymer chains and consequently lowered their Tg.31 Thus, the obtained spiropolymers are promising to be applied in functional devices for their good solution processability and desirable thermal properties.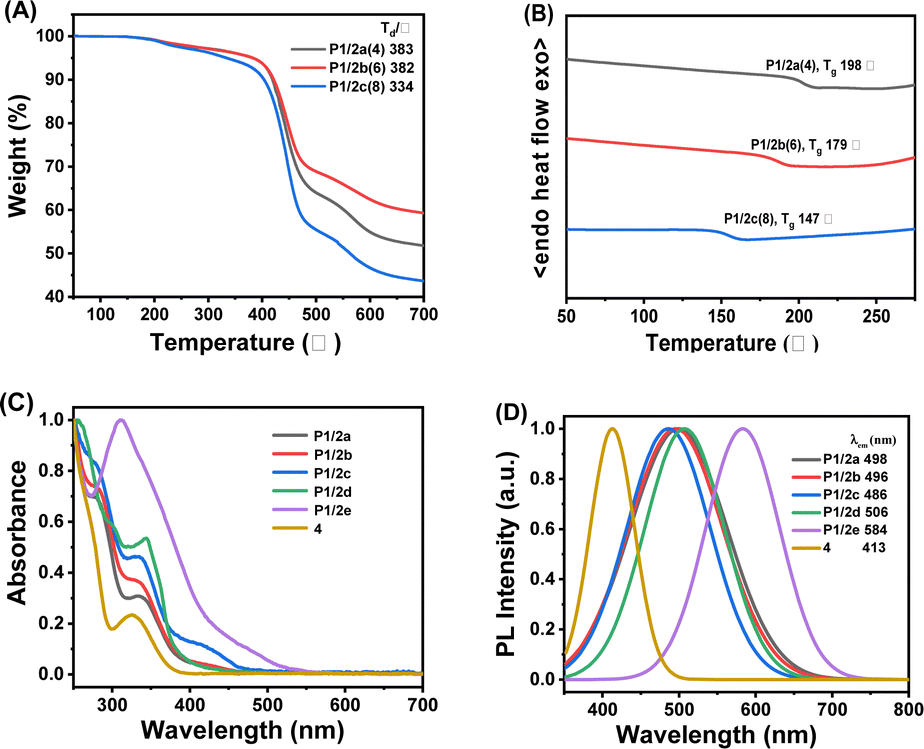 | ||
| Fig. 3 (A) TGA thermograms of P1/2a–c at a heating rate of 10 °C min−1; (B) DSC thermograms of P1/2a–c during the second heating cycle at a scan rate of 10 °C min−1; (C) UV-vis spectra of P1/2a–eand model compound 4 in CHCl3 solutions (1.0 mg mL−1); and (D) PL spectra of P1/2a–e and model compound 4 in CHCl3 solutions (1.0 mg mL−1, λex = 350 nm). | ||
Considering the existence of the conjugated-π moiety and the brown colour of the obtained polymers, the photophysical properties of all the obtained spiropolymers were examined by UV-vis absorption and photoluminescence (PL) spectroscopy (Fig. 3C and D). As shown in Fig. 3C, P1/2a, P1/2b, P1/2c, P1/2d, P1/2e, and model compound 4 in dilute CHCl3 solution exhibited strong UV-vis absorption bands at 334, 332, 330, 343, 311 and 325 nm, respectively, assignable to the π–π* transitions resulting from the conjugation between the alkene moiety and the aromatic rings. Moreover, the energy bandgap evaluated from the tangential line of the long-wavelength absorption spectrum shows a decreasing trend as the π-conjugation length extended, especially for P1/2d and P1/2e. Upon UV irradiation at 345 nm in CHCl3, the maximum fluorescence emission appeared at 486–584 nm covering the blue, green and yellow regions. Compared to the λPL value of 405 nm exhibited by the model compound 4, all polymers displayed varying degrees of redshifted emission, influenced by their conjugation length. Specifically, as the conjugated structure increased in size and the attached aryl groups of the alkyne moiety enhanced its electron-donating capability, a more pronounced spectral bathochromic shift was observed, from P1/2c to P1/2d and further to P1/2e. This indicates the possible occurrence of intramolecular charge transfer, presumably arising from the homoconjugation between spiro-linked donor and acceptor moieties.32 However, when the compound P1/2a is transformed into a solid powder, its fluorescence is completely quenched, indicating that its PET mechanism may be activated in the solid state.33
Photocrosslinking and characterization of organic gels
With the photoactive spirohexadienone-functionalized polymer scaffolds in hand, their potential to undergo photocrosslinking to yield organic gels was then explored using UV spectroscopy. The THF solution of polymer P1/2a was irradiated with a 365 nm portable ultraviolet lamp (10 mW cm−2) at a distance of 3.5 cm in the presence of air. As depicted in Fig. 4A, the absorption peak at 334 nm, attributed to the π–π* transitions of the![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CC
CC![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) bond in the pendent spirohexadienone moiety, underwent a significant reduction during the initial 90 minutes of irradiation, with minimal variation observed thereafter.
bond in the pendent spirohexadienone moiety, underwent a significant reduction during the initial 90 minutes of irradiation, with minimal variation observed thereafter.
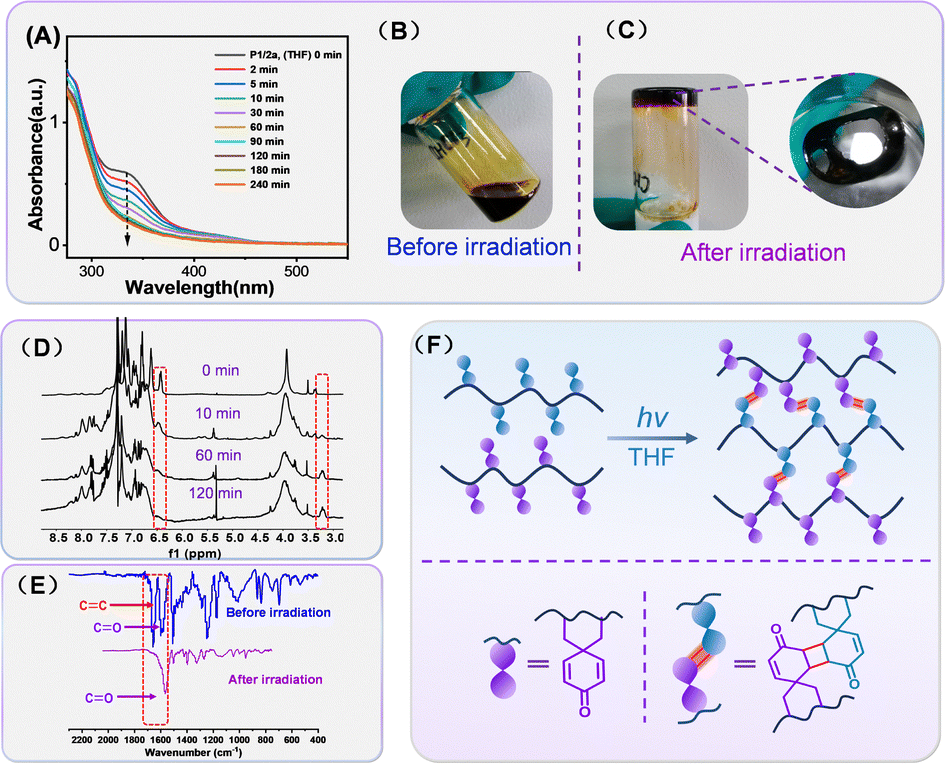 | ||
| Fig. 4 (A) UV-vis spectra of P1/2a in THF irradiated with a 365 nm portable ultraviolet lamp at various irradiation time intervals; pictures of P1/2a before (B) and after (C) 10 h of irradiation at 365 nm in THF; (D) 1H NMR spectra of P1/2a in CDCl3 irradiated with a 365 nm portable ultraviolet lamp at various irradiation time intervals; (E) FT-IR spectra obtained by employing the squashing method of P1/2a before and after irradation; and (F) illustration of spirohexadienone-based-[2+2] photocrosslinking for organo-gel formation. | ||
Moreover, when a slightly higher concentration of P1/2a in THF (10 mg mL−1) was irradiated for 10 h, a black residue that was found to be insoluble in all organic solvents was obtained (Fig. 4B). All the phenomena suggest that the crosslinking of the polymer chains likely occurs via a 2π+2π cycloaddition reaction between the intermolecular carbon–carbon double bonds. Meanwhile, the degree of crosslinking (x) was calculated using the equation x = 1 − (At/A0), where A0 represents the initial absorbance and At represents the absorption value after a certain time of irradiation. Accordingly, the maximum crosslinking degree was estimated to be ∼67%, and this value remained almost unchanged from 60 to 240 minutes. Compared with the reported photoresponsive polymers, the reason for the low crosslinking degree may be related to the large steric hindrance between inter-chains caused by the rigid spiral scaffold. The attempt to enhance the crosslinking efficiency by changing the concentration proved futile. Despite our efforts to raise the concentration and increase the illuminance, the efficiency did not exhibit significant improvement (Fig. S17 in the ESI†). The plots of x against irradiation times for different polymers and solvents were also investigated, and the results are shown in Fig. S16 and S18 (ESI†). In the same solvent – THF, the cross-linking degree gradually declined as the alkyl chain length extended, ultimately reaching merely 45% for P1/2c. This reduction is likely strongly correlated with the decreased collision frequency of the activated carbon–carbon double bonds due to the increased flexibility of the alkyl chains. Furthermore, our findings indicate that changing the solvent had little impact on the maximum achievable crosslinking degree but instead primarily influenced the crosslinking speed, with chloroform solution exhibiting the highest crosslinking rate.
Besides UV-vis monitoring, 1H NMR spectroscopy and FT-IR spectroscopy were used to elucidate the mechanism for the intermolecular 2π+2π cycloaddition reaction. Initially, P1/2a were dissolved in CDCl3 at low concentrations (0.1 mg mL−1) and exposed to UV light (365 nm, 10 mW cm−2) in order to follow the chemical changes of the spirohexadienone moieties under irradiation. As shown in Fig. 4C, the 1H NMR spectra of P1/2a clearly display alkyl, aryl, and olefin proton signals with well-resolved peaks. After 60 minutes of UV irradiation, the resonance signal (δ 6.42 ppm) from the carbonyl-activated olefin protons noticeably weakened, and instead, new high-field signals (δ 3.21 ppm) corresponding to alkyl moieties emerged.34 This phenomenon suggested that the neighbouring alkenes, activated by the carbonyl in spiro-[4,5]-cyclohexadiene-8-one, might undergo intermolecular [2+2] cyclization, leading to the formation of rigid cyclobutane rings after irradiation and thus presenting apparent crosslinking. In addition, changes before and after irradiation were analyzed by FT-IR spectroscopy. As shown in Fig. 4D, the Vc=c at 1657 cm−1 assigned to the absorption of the olefinic bond in the spirohexadienone moiety of P1/2a disappeared after irradiation. The characteristic peak at 1589 cm−1, representing the carbonyl peak in P1/2a, shifted to a higher wave number of 1594 cm−1 due to the loss of conjugation. The combination of FT-IR, 1H NMR and UV-vis spectroscopy further confirmed the exact occurrence of 2π+2π cycloaddition for spiro-[4,5]-cyclohexadiene-8-one functionalized polymers; however, the crosslinking process for these spiro-polymers presented irreversibility nature when compared with traditional photoresponsive coumarin15 derivatives. After the UV-254 nm irradiation experiment [see the ESI†] and literature investigation, the competitive absorption of the spiro-polymer35 and higher energy for rigid crosslinking net restructuring36 are identified as the primary factors contributing to the irreversibility of the crosslinking process.
By successful demonstration of photocrosslinking, the viscoelastic characteristics of the crosslinked photocurable gels were thoroughly analyzed through oscillatory shear rheology conducted at 25 °C to determine their storage modulus (G′) and loss modulus (G′′) (Fig. 5A). The energy storage modulus (G′) values of the gels follow a descending order: P1/2a > P1/2b > P1/2c, indicating that the crosslinking density of P1/2a is the highest. At the same time, P1/2c exhibits a crossover point at a shear strain of 4.9%, demonstrating that the crosslinking network under such stress undergoes a breakdown.37 This observation further underscores the close correlation between the mechanical strength of the gel and the extent of crosslinking, revealing that a lower degree of crosslinking correlates with a more brittle gel. Next, frequency scanning was performed at a constant strain value of 0.1% in the linear viscoelastic region. As depicted in Fig. 5B, the energy storage modulus (G′) of the crosslinked polymer remained consistently higher than the loss modulus (G′′), indicating the presence of distinct gel-like structures as the two moduli remain independent of each other.38
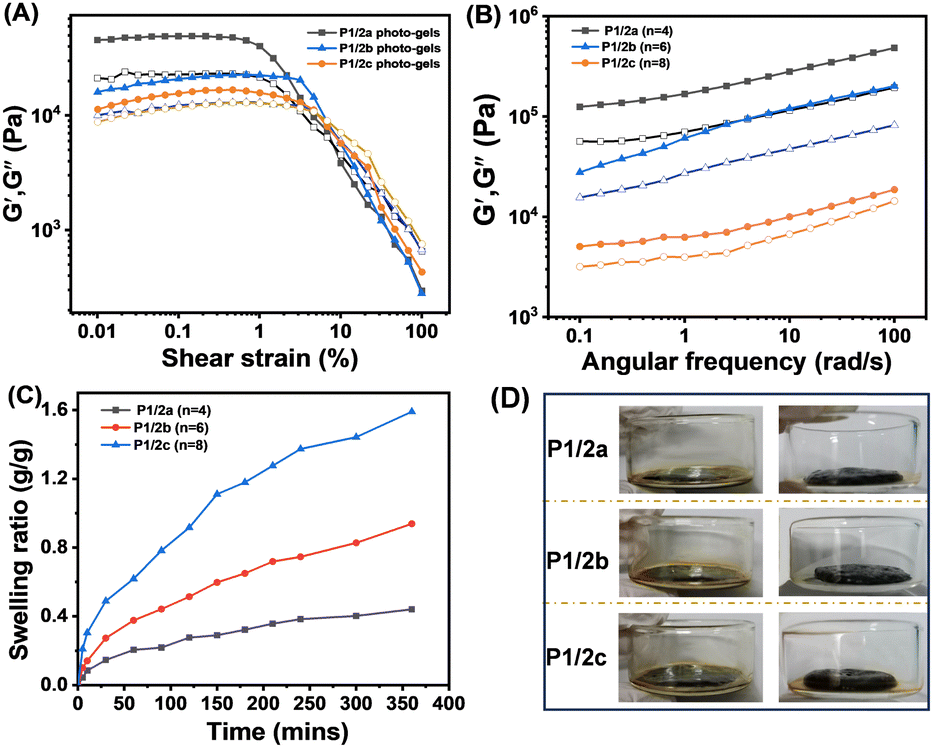 | ||
| Fig. 5 Amplitude sweep measurements of P1/2a–c photo-gels at a constant frequency of ω = 10 rad s−1 at 25 °C (A) and frequency sweep measurements of P1/2a–c photo-gels using a constant strain of γ = 0.1% at 25 °C (B). Swelling ratio (C) and swelling images (D) for P1/2a, P1/2b, and P1/2c xerogels (the y-axis represents the swelling ratio determined from the weight of the swollen gel and xerogel, and the x-axis represents the swelling period from 0 to 360 minutes). | ||
On the basis of these results, the polymer concentration was further increased to study their gelation behavior. In order to select suitable gelation conditions, different concentrations were tested and 25 wt% was selected as the solubility limit for P1/2a–c. Subsequently, 25 wt% of the three polymers were dissolved in chloroform, and the solution was exposed to UV light at 365 nm for 24 hours to ensure complete cross-linking, and the residual solvent was removed using an oven. Next, the swelling test of these three organic gels was carried out in ethyl acetate to study their swelling behavior within 360 minutes (Fig. 5C and D). Calculated from eqn 1, the swelling ratios were 1.44, 0.82, and 0.4 g/g for P1/2c, P1/2b and P1/2a, respectively. The swelling results are in alignment with the principle that polymers with a high crosslinking density absorb a lower amount of solvent as a result of a smaller mesh size, reflecting the anti-swelling property of P1/2a gels.39
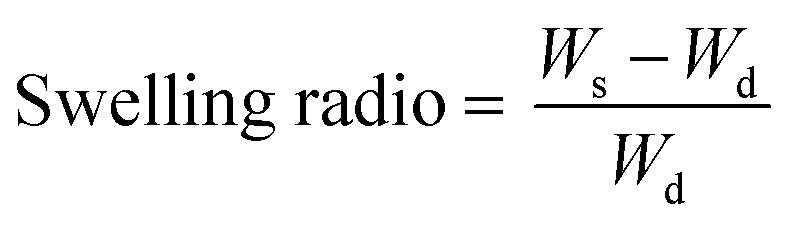 | (1) |
As shown in Fig. 6A, photocrosslinking of the polymer film was conducted in air at room temperature (∼27 °C) for 60 minutes using 365 nm UV light with an intensity of 10 mW cm−2. The polymer film was prepared by dip-coating the polymer solution (30 wt% in CHCl3) onto a silicon wafer. The film was dried in a vacuum oven at room temperature overnight. The fluorescent image was generated using a black paper mask (Fig. 6B and C) or a Cu-negative mask (Fig. 6D) and taken using a digital camera or a fluorescent optical microscope (Olympus BX4) with a 330–385 nm broad range UV source.
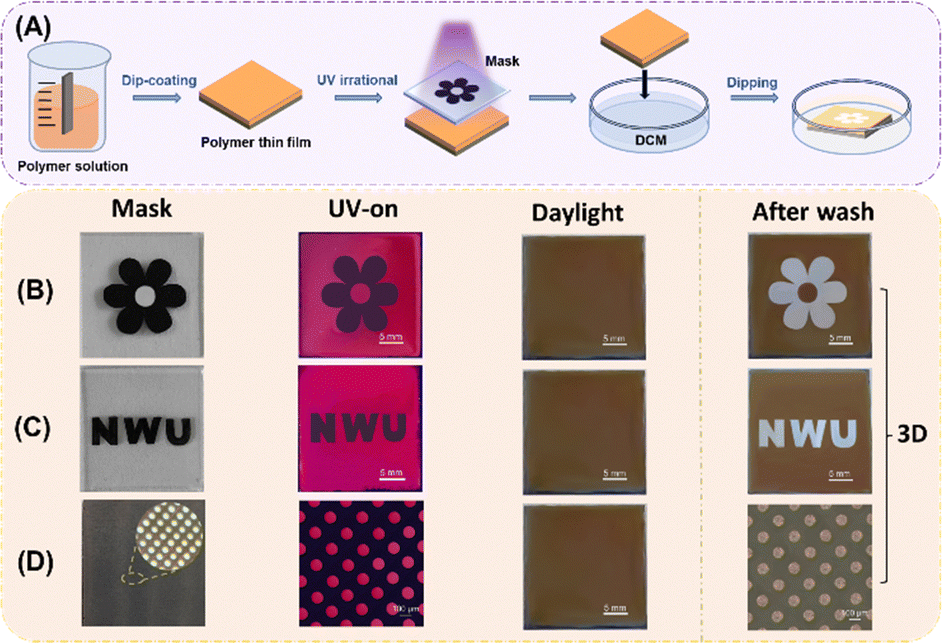 | ||
| Fig. 6 (A) Schematic diagram for photopatterning; the images for fluorescent patterning with a flower mask (B); “NWU” letter mask (C); and copper-negative microscopic mask (D) following the order of mask, UV on, UV off and after wash (notes: the size of all substrates is 2.5 cm × 2.5 cm. In Fig. 6B, the diameter of the flowers is 2.0 cm; each size of the “NMU” letters is 0.6 cm in Fig. 6C, and the diameter of the small circle is 100 microns in Fig. 6D). | ||
Indeed, upon exposure to UV light through black paper masks in the air for 60 minutes, the thin films of P1/2a underwent photocrosslinking, resulting in orange-red fluorescence in the exposed sections due to the blocked PET progress. In contrast, the masked regions, forming special images or letters, remained emission quenched (Fig. S19, ESI†). Consequently, 2D-fluorescent patterns were generated without the need for further development process. Notably, upon turning off the UV light, no mask patterns were observed on the surface of the films, suggesting potential applications of this photolithography process in the encryption field (see the UV-off images).
After the completion of 2D fluorescent patterning, we then immersed the exposed and patterned films into a dichloromethane solution. Since the masked regions were not photocrosslinking and could quickly dissolve in DCM, the masked pattern parts became concave, resulting in the immediate display of 3D-photolithographic patterns. Additionally, the polymers can also be used to fabricate microscopic maps with circular hole as small as 100 microns, typically, the sharp edges of the images can be clearly seen as shown in Fig. 6D. Therefore, the prepared spiro-polymers with exceptional solution processability present promising potential as 2D and 3D-encoding materials for utilization in optical display devices, optical writing and reading systems, as well as anti-counterfeiting applications.
Conclusions
In summary, we have successfully obtained novel photochromic(switch)-integrated-photocrosslinking (PIP) polymers with spiro-[4,5]-cyclohexadiene-8-one as a photoactive moiety through a transition-metal catalyzed [2+2+1] cycloaddition polymerization process. Impressively, the high-molecular-weight functional polymers were synthesized using readily available 4-phenol diazonium tetrafluoroborate and internal diynes as active substrates, catalyzed by palladium under mild reaction conditions. The reaction achieved remarkable yields of up to 93.7%, and the resulting polymers exhibit remarkable solubility, thermal robustness, and morphological stability, coupled with intriguing dual photoresponsive properties. Notably, these polymers, featuring carbon–carbon double bonds activated by the carbonyl group in the 8-position, undergo rapid 2π+2π cycloaddition crosslinking under 365 nm light irradiation, resulting in the formation of a dense crosslinked network. Such a network can form organic gels that possess unique swelling properties towards organic solvents, allowing them to reversibly absorb and release solvent molecules. Furthermore, the photoactivated fluorescence turn-on phenomenon of the crosslinked polymers was also observed during the crosslinking process. Ultimately, by casting the spiro-polymers into films, we realized the 2D and 3D fluorescent patterning based on their dual photoresponsive properties by employing the lithography method, indicating the potential applications of this polymer in fluorescent labelling and 2D/3D optical security fields.Author contributions
H. W. and G. N. directed the project. L. H. assisted with the photopatterning. Y. Y. and M. C. carried out the syntheses and characterization studies of all PIP polymers. Y. S. carried out the model reactions. Y. G. and W. F. performed the 2D and 3D photopatterning. All co-authors contributed to the formal analyses of the results. Y. Y. provided the first draft of the paper that was first corrected by H. W., G. N. and L. H. All co-authors then corrected the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (202030065 and 62105184), the Natural Science Basic Research Program of Shaanxi Province (2022JM-084), the Basic Science Research Program of Shaanxi Institute of Basic Science (23JHQ015), and the Shccig-Qinling Program (SMYJY20220436).Notes and references
- A. Rosario and B. Ma, Stimuli-Responsive Polymer Networks: Application, Design, and Computational Exploration, ACS Appl. Polym. Mater., 2024 DOI:10.1021/acsapm.4c00002.
- M. Mrinalini and S. Prasanthkumar, Recent Advances on Stimuli-Responsive Smart Materials and their Applications, ChemPlusChem, 2019, 84, 1103 CrossRef CAS PubMed.
- O. Bertrand and J. Gohy, Photo-Responsive Polymers: Synthesis and Applications, Polym. Chem., 2017, 8, 52–73 RSC.
- I. Tomatsu, K. Peng and A. Kros, Photoresponsive Hydrogels for Biomedical Applications, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- J. Yang, M. Shankar and H. Zeng, Photochemically Responsive Polymer Films Enable Tunable Gliding Flights, Nat. Commun., 2024, 15, 4684 CrossRef CAS.
- Y. Li, B. Xue, J. Yang, J. Jiang, J. Liu, Y. Zhou, J. Zhang, M. Wu, Y. Yuan, Z. Zhu, Z. Wang, Y. Chen, Y. Harabuchi, T. Nakajima, W. Wang, S. Maeda, J. Gong and Y. Cao, Azobenzene as a Photoswitchable Mechanophore, Nat. Chem., 2023, 16, 446–455 CrossRef PubMed.
- O. Kuksenok and A. Balazs, Stimuli-Responsive Behaviour of Composites Integrating Thermo-Responsive Gels with Photo-Responsive Fibers, Mater. Horiz., 2016, 3, 53–62 RSC.
- A. Abdollahi, H. Roghani-Mamaqani and B. Razavi, Stimuli-Chromism of Photoswitches in Smart Polymers: Recent Advances and Applications as Chemosensors, Prog. Polym. Sci., 2019, 98, 101149 CrossRef CAS.
- Y. Dong, Y. Ling, D. Wang, Y. Liu, X. Chen, S. Zheng, X. Wu, J. Shen, S. Feng, J. Zhang and W. Huang, Harnessing Molecular Isomerization in Polymer Gels for Sequential Logic Encryption and Anticounterfeiting, Sci. Adv., 2022, 8, eadd1980 CrossRef CAS.
- Y. Sun, X. Le, S. Zhou and T. Chen, Recent Progress in Smart Polymeric Gel-Based Information Storage for Anti-Counterfeiting, Adv. Mater., 2022, 34, 2201262 CrossRef CAS.
- H. Wang, X. Ji, Z. Page and J. Sessler, Fluorescent Materials-Based Information Storage, Mater. Chem. Front., 2020, 4, 1024–1039 RSC.
- J. Williams, Photopolymerization and Photocrosslinking of Polymers, Fortschr. Chem. Forsch., 1969, 13, 227–250 CrossRef CAS.
- G. Giustina, S. Giulitti, L. Brigo, M. Zanatta, M. Tromayer, R. Liska, N. Elvassore and G. Brusatin, Hydrogel with Orthogonal Reactive Units: 2D and 3D Cross-Linking Modulation, Macromol. Rapid Commun., 2016, 38, 1600570 CrossRef PubMed.
- M. Lebedevaite, J. Ostrauskaite, E. Skliutas and M. Malinauskas, Photocross-Linked Polymers Based on Plant-Derived Monomers for Potential Application in Optical 3D Printing, J. Appl. Polym. Sci., 2019, 137, 48708 CrossRef.
- M. Maddipatla, D. Wehrung, C. Tang, W. Fan, M. Oyewumi, T. Miyoshi and A. Joy, Photoresponsive Coumarin Polyesters That Exhibit Cross-Linking and Chain Scission Properties, Macromolecules, 2013, 46, 5133–5140 CrossRef CAS.
- J. Chung, K. Lee, C. Neikirk and C. Nelson, Photoresponsive Coumarin-Stabilized Polymeric Nanoparticles as a Detectable Drug Carrier, Small, 2012, 8, 1693–1700 CrossRef CAS PubMed.
- H. Chen, G. Noirbent, S. Liu, D. Brunel, B. Graff, D. Gigmes, Y. Zhang, K. Sun, F. Morlet-Savary, P. Xiao, F. Dumur and D. Lalevée, Bis-Chalcone Derivatives Derived from Natural Products as Near-UV/Visible Light Sensitive Photoinitiators for 3D/4D Printing, Mater. Chem. Front., 2021, 5, 901–916 RSC.
- L. Deng, L. Tang and J. Qu, Novel Chalcone-Based Phenothiazine Derivative Photoinitiators for Visible Light Induced Photopolymerization with Photobleaching and Good Biocompatibility, Prog, Org. Coat., 2022, 167, 106859 CrossRef CAS.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Reversible Cross-Linking of Aliphatic Polyamides Bearing Thermoand Photoresponsive Cinnamoyl Moieties, Macromolecules, 2014, 47, 8247–8254 CrossRef CAS.
- S. Ulrich, X. Wang, M. Rottmar, R. Rossi, B. Nelson, N. Bruns, R. Müller, K. Maniura-Weber, X. Qin and L. Boesel, Nano-3D-Printed Photochromic Micro-Objects, Small, 2021, 17, 2101337 CrossRef CAS.
- B. Xie, G. Wang and X. Zhao, Synthesis and Photochromic Properties of Naphthopyran Polymer Containing Photocrosslinkable Coumarin Moiety, J. Appl. Polym. Sci., 2011, 122, 3377–3382 CrossRef CAS.
- T. Han, Z. Yao, Z. Qiu, Z. Zhao, K. Wu, J. Wang, A. Poon, J. Lam and B. Tang, Photoresponsive Spiro-Polymers Generated in Situ by C–H-activated Polyspiroannulation, Nat. Commun., 2019, 10, 5483 CrossRef CAS.
- L. F. Thompson and R. E. Kerwin, Polymer Resist Systems for Photo- and Electron Lithography, Annu. Rev. Mater. Sci., 1976, 6, 267–301 CrossRef CAS.
- L. F. Thompson, Introduction to Microlithography, American Chemical Society, Washington, DC, 1983 Search PubMed.
- H. Ito, C. G. Willson and J. M. J. Frechet, Positive- and Negative-Working Resist Compositions with Acid Generating Photoinitiator and Polymer with Acid Labile Groups Pendant from Polymer Backbone, US4491628A, 1985 Search PubMed.
- B. Schmidt, R. Berger, A. Kelling and U. Schilde, Pd-Catalyzed [2 + 2 + 1] Coupling of Alkynes and Arenes: Phenol Diazonium Salts as Mechanistic Trapdoors, Chem. Eur. J., 2011, 17, 7032–7040 CrossRef CAS PubMed.
- L. Wang, J. Zhang, M. Lang and J. Wang, Palladium-Catalyzed Ring Contraction Reaction of Naphthoquinones upon Reaction with Alkynes, Org. Chem. Front., 2016, 3, 603–608 RSC.
- B. Song, K. Hu, A. Qin and B. Tang, Oxygen as a Crucial Comonomer in Alkyne-Based Polymerization toward Functional Poly(tetrasubstituted furan)s, Macromolecules, 2018, 51, 7013–7018 CrossRef CAS.
- G. Zhu, N. Lin, X. Wu, J. Shi, B. Tong, Z. Cai, J. Zhi and Y. Dong, Multicomponent Spiropolymerization of Diisocyanides, Activated Alkynes, and Bis-Anhydrides, Macromolecules, 2022, 55, 6150–6159 CrossRef CAS.
- D. Liaw, K. Wang, Y. Huang, K. Lee, J. Lai and C. Ha, Advanced Polyimide Materials: Syntheses, Physical Properties and Applications, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- Y. Yang, J. Huang, R. Zhang and J. Zhu, Designing Bio-Based Plasticizers: Effect of Alkyl Chain Length on Plasticization Properties of Isosorbide Diesters in PVC Blends, Mater. Des., 2017, 126, 29–36 CrossRef CAS.
- H. Hamada, Y. Itabashi, R. Shang and E. Nakamura, Axially Chiral Spiro-conjugated Carbon-bridged p-Phenylenevinylene Congeners: Synthetic Design and Materials Properties, J. Am. Chem. Soc., 2020, 142, 2059–2067 CrossRef CAS PubMed.
- D. Escudero, Revising Intramolecular Photoinduced Electron Transfer (PET) from First-Principles, Acc. Chem. Res., 2016, 49, 1816–1824 CrossRef CAS PubMed.
- J. Yang, M. Dewal, S. Profeta, M. Salvatore, Y. Li and L. Shimizu, Origins of Selectivity for the [2 + 2] Cycloaddition of α,β-unsaturated Ketones within a Porous Self-assembled Organic Framework, J. Am. Chem. Soc., 2008, 130, 612–621 CrossRef CAS.
- L. C. Chambers, C. Barner-Kowollik, L. Barner, L. Michalek and H. Frisch, Photostationary State in Dynamic Covalent Networks, ACS Macro Lett., 2022, 11, 532–536 CrossRef CAS PubMed.
- M. Aljuaid, Y. Chang, D. M. Haddleton, P. Wilson and H. A. Houck, Thermoreversible [2 + 2] Photodimers of Monothiomaleimides and Intrinsically Recyclable Covalent Networks Thereof., J. Am. Chem. Soc., 2024, 146, 19177–19182 CrossRef CAS.
- K. Xu, B. Fan, K. Putera, M. Wawryk, J. Wan, B. Peng, M. Banaszak Holl, A. Patti and S. Thang, Nanoparticle Surface Cross-Linking: A Universal Strategy to Enhance the Mechanical Properties of Latex Films, Macromolecules, 2022, 55, 5301–5313 CrossRef CAS.
- K. Almdal, J. Dyre, S. Hvidt and O. Kramer, Towards a Phenomenological Definition of the Term ‘Gel’, Polym. Gels Networks, 1993, 1, 5–17 CrossRef CAS.
- T. Canal, N. Peppas and J. Biomed, Correlation between Mesh Size and Equilibrium Degree of Swelling of Polymeric Networks, Mater. Res., 1989, 23, 1183–1193 CAS.
- C. Wu, C. Li, X. Yu, L. Chen, C. Gao, X. Zhang, G. Zhang and D. Zhang, An Efficient Diazirine-Based Four-Armed Cross-Linker for Photo-Patterning of Polymeric Semiconductors, Angew. Chem., Inter. Ed., 2021, 60, 21521–21528 CrossRef CAS PubMed.
- X. Liu, X. Liang, Y. Hu, L. Han, Q. Qu, D. Liu, J. Guo, Z. Zeng, B. H. ai, R. Kwok, A. Qin, J. Lam and B. Tang, Catalyst-Free Spontaneous Polymerization with 100% Atom Economy: Facile Synthesis of Photoresponsive Polysulfonates with Multifunctionalities, JACS Au., 2021, 1, 344–353 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qm00688g |
| ‡ Y. Yuan and M. Chao contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |