
Photocatalytic arylation/alkylation of olefins/alkynes via halogen-atom transfer mediated by NHC-BH3†
Xinhan
Li‡
,
Yao
Zhong‡
,
Fengsong
Tan
,
Yusong
Fei
,
Xiaohan
Zhao
,
Jianbin
Xu
 * and
Baomin
Fan
*
* and
Baomin
Fan
*
Yunnan Key Laboratory of Chiral Functional Substance Research and Application, & School of Chemistry and Environment, Yunnan Minzu University, Kunming, Yunnan 650504, China. E-mail: xujianbin@ymu.edu.cn; fanbaomin1979@163.com
First published on 29th October 2024
Abstract
A versatile method for radical reductive cross-coupling of iodides and activated olefins under mild conditions, facilitated by NHC-BH3 through photocatalysis, was developed. This method exhibits high efficiency and regioselectivity, achieving yields of up to 98% for aryl, heterocyclic, and alkyl iodides. The reaction employs a minimal amount (1.0 mol%) of photocatalyst and can be performed on a gram scale. Importantly, this approach avoids the use of transition metal co-catalysts, eliminating the need for traditional C–I bond cleavage steps such as oxidative addition, migration insertion, and halogen exchange. This work unveils a novel mechanism for the reductive coupling of iodides in a photocatalytic NHC-BH3 system, providing insights into iodide modification and the functional exploration of NHC-BH3.
Photoredox catalysis, utilizing visible light in organic synthesis, has emerged as a potent strategy for assembling complex molecules under mild conditions.1,2 Operating under mild photocatalytic conditions, this process efficiently generates carbon radicals, providing versatile building blocks for constructing chemical bonds.3,4 Notable advancements over the last decade have predominantly focused on the synergistic catalytic reductive coupling of halides through both photocatalysis and transition metal participation. In this context, transition metal complexes play a crucial role in catalyzing the cleavage of C–X bonds and accepting the radical species formed during the photocatalytic step. Furthermore, the essential function of the photocatalyst involves single electron transfer (SET) in the valence transition of metal complexes (Scheme 1A).5,6
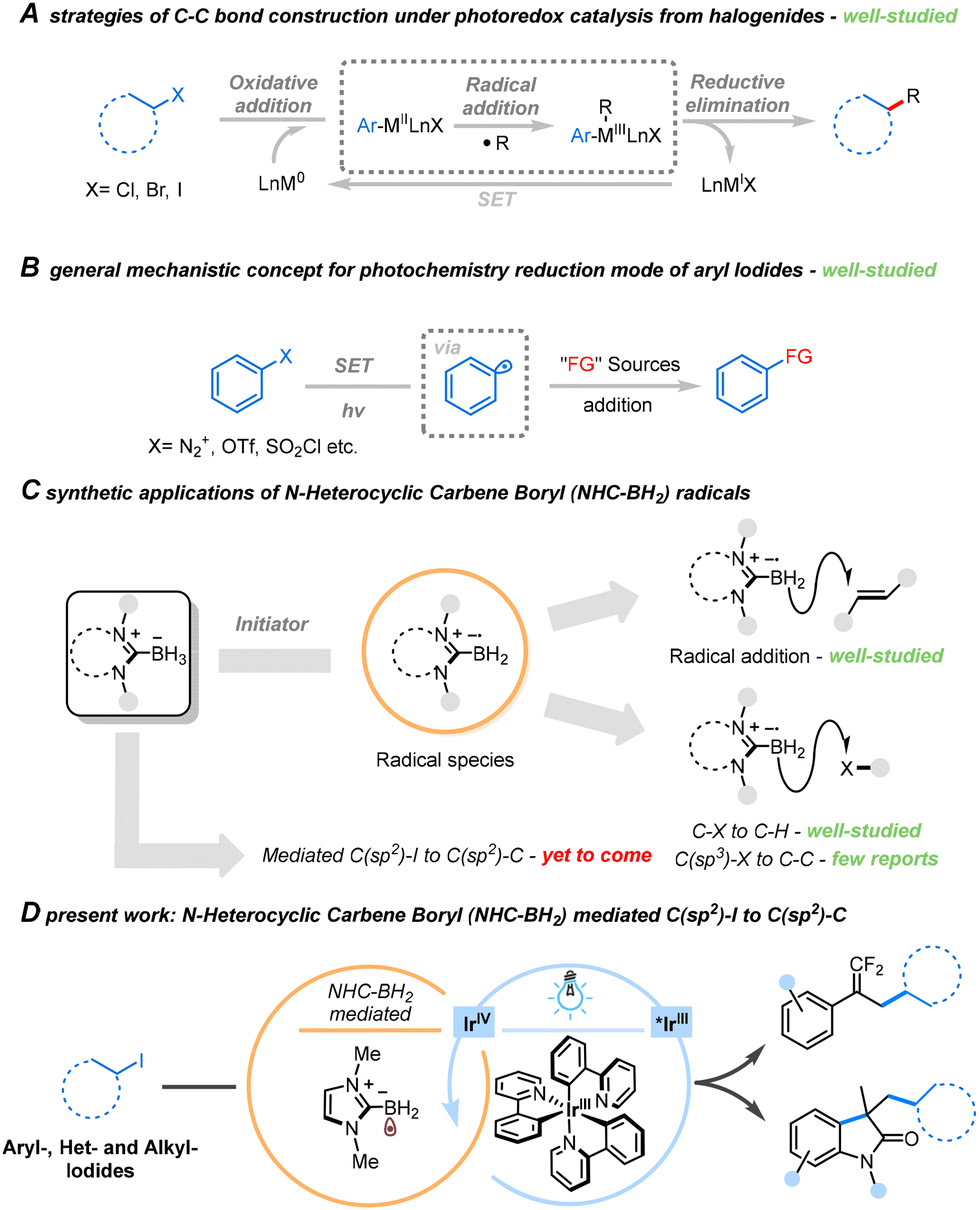 | ||
| Scheme 1 Research contexts of the present work. | ||
Aryl radicals predominantly originate from aryl compounds containing effective leaving groups (Scheme 1B),7–10 such as diazonium salts, iodonium salt and sulfur-containing groups (−OTf or –SO2Cl). However, only diazonium salt substrates of electron-rich aromatic hydrocarbons demonstrate sufficient compatibility for the formation of aryl radicals, restricting the scope of substrates. By contrast, aryl iodides offer an expanded substrate range as a source of aryl radicals, tolerating both electron-poor and complex groups, as well as relatively reactive groups. This extensive range of possible compounds establishes a fundamental basis for subsequent applications in substrate derivatization.
While various transformation strategies of halides by synergistic photocatalysis and metal-based catalysis have been realized, achieving the functional transformation of unactivated halides without the assistance of transition metal complexes during photocatalysis remains a significant challenge. The primary obstacles arise from the hydrogen atom transfer (HAT) induced by high bond dissociation energy (BDE) between the aryl radical and the reducing agent after reductive deiodination.11 This results in the formation of active aryl radical species, leading solely to iodine elimination reduction products with no possibility of further transformation. Furthermore, the low efficiency in generating highly active aryl radicals, difficulties in controlling the reaction, and the challenge of achieving the coupling reaction without transition metal complexes contribute to the overall complexity.
In recent years, our focus has been on N-heterocyclic carbene borane complexes (NHC-BH3). Due to the electron-rich and aromatic nature of the B–H bond, its energy in the borane complex is reduced, making it more susceptible to thermodynamic bond cleavage.12 Consequently, researchers have devised numerous strategies to promote the formation of boron radicals and convert them into borides under the synergistic effect of free radical initiators and proton transfer reagents (Scheme 1C).13–18 The NHC-BH2 radical species generated through the action of free radical initiators also exhibit potential applications as halogen atom transfer (XAT) reagents, presenting an attractive alternative to classical Sn/Si-based derivatives.19–22 Despite the development of such an efficient boron introduction strategy, the general utilization of NHC-BH3 as a promoter for the conversion of iodide-compounds has mostly been overlooked. Hence, there is a need for further exploration of NHC-BH3 in the functionalization of iodides. Based on the aforementioned research, we suggest that NHC-BH3 can serve as an ideal promoter for iodide functionalization because it not only acts as an excellent electron donor but, in the absence of HAT reagent, also prevents the hydrogenation of active carbon free radical species by the protons released during the conversion process, thereby preventing the formation of protonated products.
Inspired by the strategy of halide activation by light-induced and metal-based catalysis synergistic catalytic and the research on NHC-BH3, we have proposed a general strategy for the functionalization of unactivated iodides promoted by photocatalytic NHC-BH3 systems, in which photo-excited Ir complexes transfer energy to the borane compound to generate boryl radicals. The successful development of this strategy is expected to offer new insights into the synthesis methodology for the mild and efficient conversion of iodides, thus providing an important complement to the synergistic photo- and metal-based catalytic conversion strategy of halogenated hydrocarbons. This approach has made significant progress in addressing the challenging tasks of functional conversion and modification of unactivated iodides.
In this work, we present a versatile platform for the functional transformation of unactivated iodides facilitated by NHC-BH3 under blue light irradiation, exemplified in Scheme 1D. Utilizing this protocol, we achieved the radical reductive cross-coupling reaction of aryl, heterocyclic, and alkyl iodides with active olefins (α-(trifluoromethyl)styrenes and N-arylacrylamides). This approach demonstrated a broad substrate scope, high yields, and excellent tolerance towards diverse functional groups. Notably, the resulting gem-difluoroalkenyl functional group serves as an electron isostere of the carbonyl group, while 2-indolone acts as a structural motif in certain drugs and natural products, playing a unique role in the modification of drug molecules.23,24 Compounds synthesized using this protocol expand the library of gem-difluoroalkenes and 2-indolones, offering a potential methodological foundation for screening and applying drug precursors. This successful strategy not only establishes a valuable platform for the trans-formation of inexpensive but also introduces readily available iodides but also introduces a novel avenue for exploring the catalytic functionality of NHC-BH3.
A preliminary study of the novel NHC-BH3-mediated photoredox catalyzed atom transfer radical addition was conducted using iodobenzene (1), α-(trifluoromethyl)styrene (2), Ir(ppy)3 (1 mol%), NHC-BH3, and KHCO3, and exposed to a 30 W blue LED light source in MeCN, achieving a 90% yield of product 4 (Table 1, entry 1). This result validates the successful realization of our proposed concept. Attempts to enhance reaction efficiency by employing borane complexes failed, indicating that these complexes hinder the reaction, further underscoring the necessity of NHC-BH3 for this reaction's success (Table 1, entry 2). Further tests comparing other common halogen atom transfer reagents such as tertiary amines (e.g., TEA) or silanes (e.g., TTMSS) showed inferior results, yielding no detectable product 4, thereby emphasizing the superiority of NHC-BH3 (Table 1, entry 3).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Standard conditions: Ir(ppy)3 (1.0 mol%), B1 (1.5 eq.), KHCO3 (2.0 eq.), iodobenzene 1 (2.2 eq.), α-(trifluoromethyl)styrene 2 (1.0 eq.), MeCN (0.1 M), 30 W LED lamp, room temperature, Ar atmosphere. b Yield determined by 1H NMR spectroscopy using 1,3-benzodioxole as an internal standard. | ||
| 1 | None | 90 |
| 2 | B2–B5 instead of B1 | n.d |
| 3 | TEA or TTMSS instead of B1 | n.d |
| 4 | 4DPAIPN instead of Ir(ppy)3 | 39 |
| 5 | 4CzIPN instead of Ir(ppy)3 | 16 |
| 6 | Na2CO3 instead of KHCO3 | 80 |
| 7 | DABCO instead of KHCO3 | 60 |
| 8 | w/o Ir(ppy)3, light or B1 | n.d |
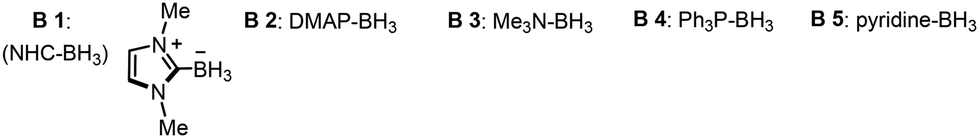
|
||

Evaluations of various photocatalysts revealed that those with lower reduction potentials, such as 4DPAIPN and 4CzIPN, resulted in decreased reaction efficiencies (Table 1, entries 4 and 5). Screening of alkaline additives indicated that both inorganic and organic bases more alkaline than KHCO3 adversely affected the reaction efficiency (Table 1, entries 6 and 7). Control experiments confirmed that the photocatalyst, NHC-BH3, and the light source are all essential components for this reaction, as omitting any of these elements led to no product formation (Table 1, entry 8). Lastly, solvent tests verified that MeCN is the optimal solvent for this reaction (see ESI† for details).
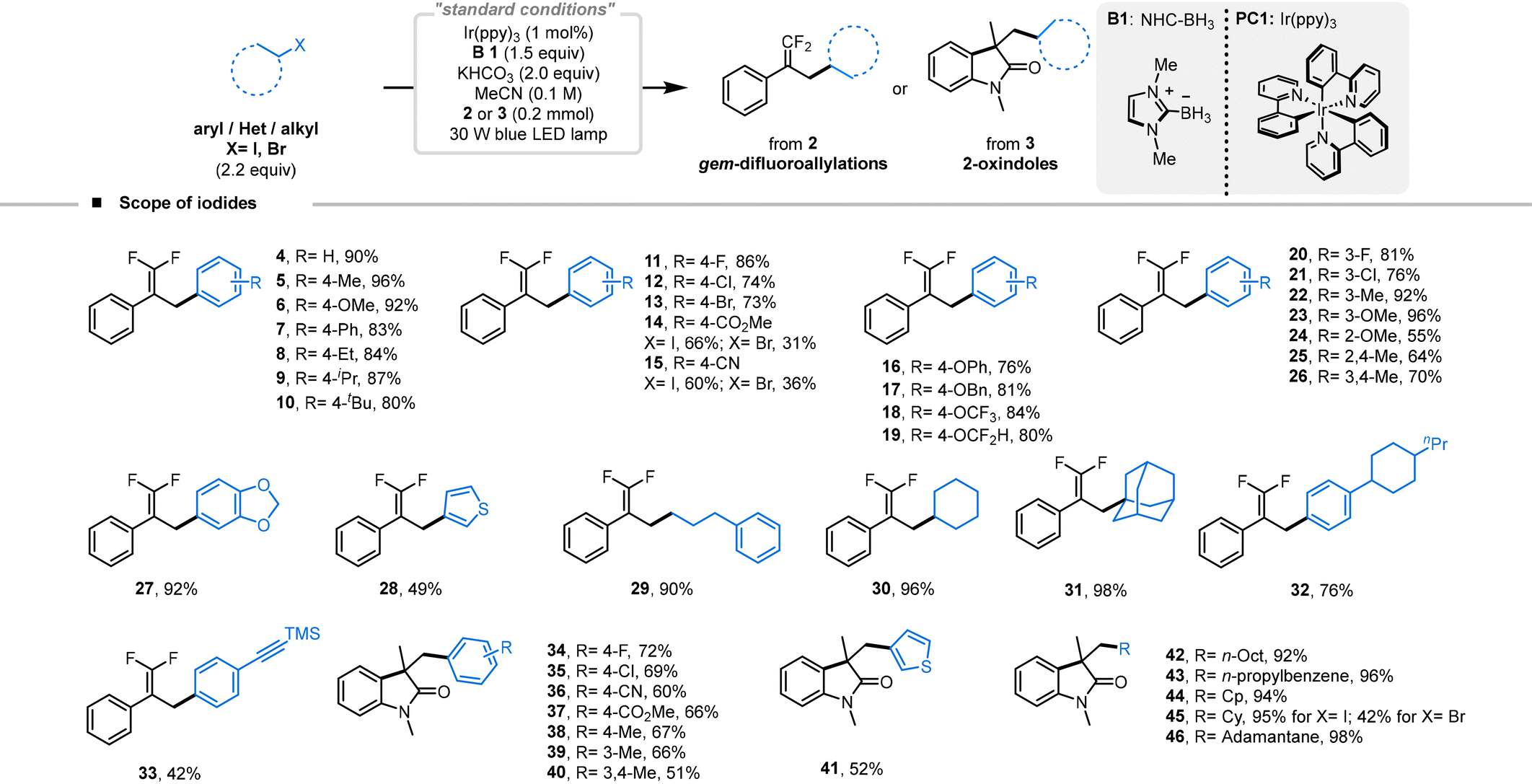
We explored the scope of halide-derived carbon-radical species available for this reaction under optimized conditions. Initially, we found that α-(trifluoromethyl)styrene (2) served as a viable reaction partner (Table 2). Iodobenzenes with electron-donating and sterically hindering groups yielded favorable outcomes (5–10, yields 80–96%). Interestingly, para-substituted iodobenzenes bearing sensitive halogen groups also performed well, and 4-bromoiodobenzene did not participate in any competitive side reactions leading to doubly substituted products (11–13, yields 73–86%). However, electron-withdrawing groups were associated with decreased reaction efficiency, although both electron-withdrawing substituted iodobenzenes and bromobenzenes could be converted, successfully yielding the targeted products (14–15, yields 60–66% and 31–36% for bromobenzene, respectively). para-Substituted iodobenzenes with oxygen-containing groups, prevalent in natural products, were tested and exhibited excellent efficiencies (16–19, yields 76–84%). We also examined the influence of substituents at ortho and meta positions. Electron-donating groups generally enhanced reaction efficiency, but the steric hindrance at the ortho position often resulted in lower yields (20–24, yields 55–96%). Interestingly, di-substituted groups, including those with cyclic ethers, commonly found in biologically active molecules, maintained good reaction efficiencies and typically did not result in C–O bond cleavage (25–27, yields 64–92%). 3-Iodothiophene was successfully converted using this methodology, although alterations in electron cloud density and variances in redox potentials resulted in reduced reaction efficiencies (28, 49%). Remarkably, alkyl iodides demonstrated excellent reactivity; primary, sterically hindered secondary, and bulky tertiary alkyl iodides were all successfully converted to the target products, with iodoadamantane achieving up to 98% yield (29–31, 90–98%). This protocol also proved effective in complex settings: iodobenzenes with large groups or reactive alkynylsilane modifications were converted with relative ease, yielding 76% and 42% for products 32 and 33, respectively. Further use of N-arylacrylamide (3) as a coupling partner with the iodide also yielded excellent results, producing a series of arylated 2-indolone products. para-Substituted iodobenzene with electron-withdrawing groups maintained similar efficiencies, with 4-bromoiodobenzene engaging in the reaction without debromination, producing yields of 60–72% (34–37). Substituents that donate electrons likewise showed comparable efficiencies (38–40, 51–67%). The conversion of 3-iodothiophene proceeded smoothly, resulting in a 52% yield of the target product (41). It was also noted that alkyl iodides consistently gave high yields, with conversions of primary, secondary, and tertiary alkyl iodides occurring without issue, and large sterically hindered iodoadamantanes providing yields up to 98% (42–46, 92–98%). Additionally, the conversion of bromocyclohexane was successfully achieved, with the target product obtained in 42% yield (45).
| a Yield determined by 1H NMR spectroscopy using 1,3-benzodioxole as an internal standard. |
|---|
|
Next, we expanded our investigation to include a broader range of reactive olefins, particularly focusing on α-(trifluoromethyl)styrene and N-arylacrylamide derivatives (Table 3). We observed efficient addition of aryl radicals derived from iodobenzene (1) to these olefins, leading to the formation of the corresponding products. The α-(trifluoromethyl)styrene derivatives, modified para to the vinyl group with various substituents, showed good reactivity. These substituents included electron-donating groups (–Me, –OMe, –SMe), sterically hindering groups (–Et, –iPr, –tBu, –Ph), electron-withdrawing groups (–F, –Cl, –Br), and oxygen-containing groups, all of which successfully yielded the desired gem-difluoroolefins products (47–59, 60–86%). Notably, there were no side reactions such as the elimination of the silyl group in trimethylsilyl (-TMS) substituted substrates, nor was any by-product formation observed in bromine-substituted substrates. Substrates substituted with –Me demonstrated consistent reaction efficiency, whether in meso- or bis-methyl substitution scenarios (60–61, 75%). The cyclic ether structure, commonly found in natural products, also delivered favorable outcomes (62–63, 79–86%), with no observed side reactions involving C–O bond cleavage. The benzothiophene-derived α-(trifluoromethyl)styrene yielded the desired product 64 with an 80% success rate. Exploring the reactivity of N-arylacrylamide derivatives also showed positive outcomes, with a template N-arylacrylamide converting into the arylated 2-indolone product 65 with a 68% yield. Substituting N-arylacrylamide para-positions with electron-donating groups resulted in yields of 70% and 73% for target products 66 and 67, respectively. In contrast, weak electron-withdrawing groups enhanced reaction efficiencies (68–70, 80–81%), while strong electron-withdrawing groups typically lowered them (71–72, 63–65%). Additionally, tetrahydroquinoline-derived arylated 2-indolones 73, which are broadly biologically active, were synthesized with a 60% yield. Furthermore, the versatility of this protocol in complex environments is noteworthy. For instance, adamantane and drug molecules such as flurbiprofen, naproxen, and the ester derivatives of ibuprofen with α-(trifluoromethyl)styrene, all of which feature substantial steric hindrance, successfully underwent arylation of ketodifluoroalkenyls. The resultant target products 74–77 were synthesized with yields of 84%, 87%, 76%, and 85%, respectively, demonstrating the protocol's robustness in accommodating complex molecular frameworks.
| a Yield determined by 1H NMR spectroscopy using 1,3-benzodioxole as an internal standard. |
|---|

|
In the subsequent investigation, we assessed the substrate scope of iodobenzene with non-activated olefins and alkynes as reaction partners. As expected, straightforward aryl olefins, alkyl olefins, and aryl alkynes were all compatible with this protocol, yielding hydrogenated arylated products. Aryl olefins and alkyl olefins exhibited comparable reaction efficiencies, with electron-donating groups yielding better results when the olefin was substituted at the para position (78–81, 40–57%). Of particular interest was the observation that the products derived from aryl alkynes and iodobenzene using this protocol were stereospecific cis-diaryl olefin products (82 and 83, 42 and 49%). This finding presents a novel and efficient avenue for the direct synthesis of cis-olefins, holding promise for future research and production endeavors.
To elucidate the possible reaction mechanism, a series of mechanism validation experiments were performed as outlined in Scheme 2. The introduction of free radical scavengers such as TEMPO and BHT significantly inhibited the reaction, suggesting a radical-mediated process. Notably, the formation of [TEMPO-Ph] and [BHT-Ph] adducts was detected in the GC-MS analysis, confirming radical capture (Scheme 2A; for furtherdetails, see ESI†). UV-Visible analysis further supported that no electron donor–acceptor (EDA) complexes were formed, and the photocatalyst remained stable under reaction conditions (Scheme 2B; for furtherdetails, see ESI†). Additionally, Stern–Volmer fluorescence quenching experiments indicated that the triplet excited state of the photocatalyst (B) actively interacted with iodobenzene, implicating this species in the reaction pathway (Scheme 2C; for furtherdetails, see ESI†). Lastly, light-switching experiments clarified that the visible light redox catalyzed process does not proceed via a chain reaction mechanism, evidenced by a measured quantum yield of 0.01 (Scheme 2D and E). For furtherdetails, see ESI.†
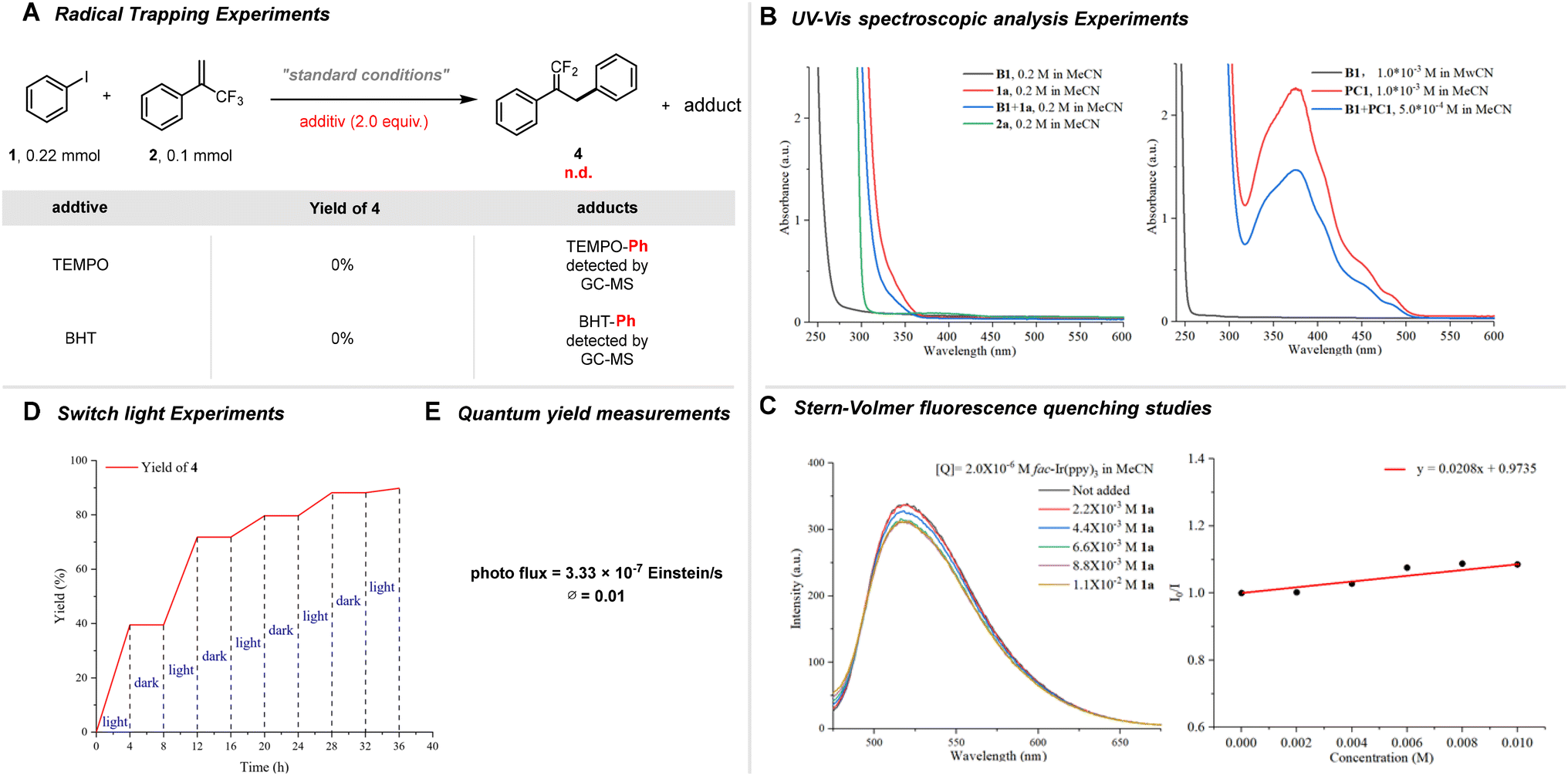 | ||
| Scheme 2 Exploration of the mechanism. | ||
Scheme 3 outlines the proposed mechanism for olefin aryl alkylation using a photoredox catalytic cycle. Initially, an iridium photocatalyst (A) is excited by blue light, achieving a long-lived triplet excited state (B) with a half-reduction potential of Ered1/2[*IrIII/IrIV] = −1.73 V vs. saturated calomel electrode (SCE) in MeCN.25 This excited state photocatalyst (B) is quenched by partial reduction of a halide, generating halide anions and carbon radical species (D), while simultaneously forming a more oxidizing iridium IV species (C) with a potential of Ep = +0.77 V vs. saturated calomel electrode (SCE) in MeCN.25 This oxidized iridium species (C) is then rapidly reduced back to its basal state (A) by NHC-BH3 (B1), which has a potential of Ep = +0.67 V vs. saturated calomel electrode (SCE) in MeCN.26 This reduction not only regenerates the iridium catalyst (A) but also produces an NHC-BH2 radical, demonstrating efficient halogen atom transfer (XAT) capabilities (E). This radical (E) then facilitates further XAT with the halide to generate additional carbon-radical species (D). These radicals can add to olefins and alkynes, forming the corresponding alkyl radicals (G), which then undergo various transformations under different conditions to yield the desired products. Detailed mechanistic studies supporting this atom transfer radical addition mechanism and the transformations of the alkyl radical intermediate (G) could be found in Scheme 3 and the details of the (G) functionalization in (for furtherdetails, see ESI Fig. S11†).
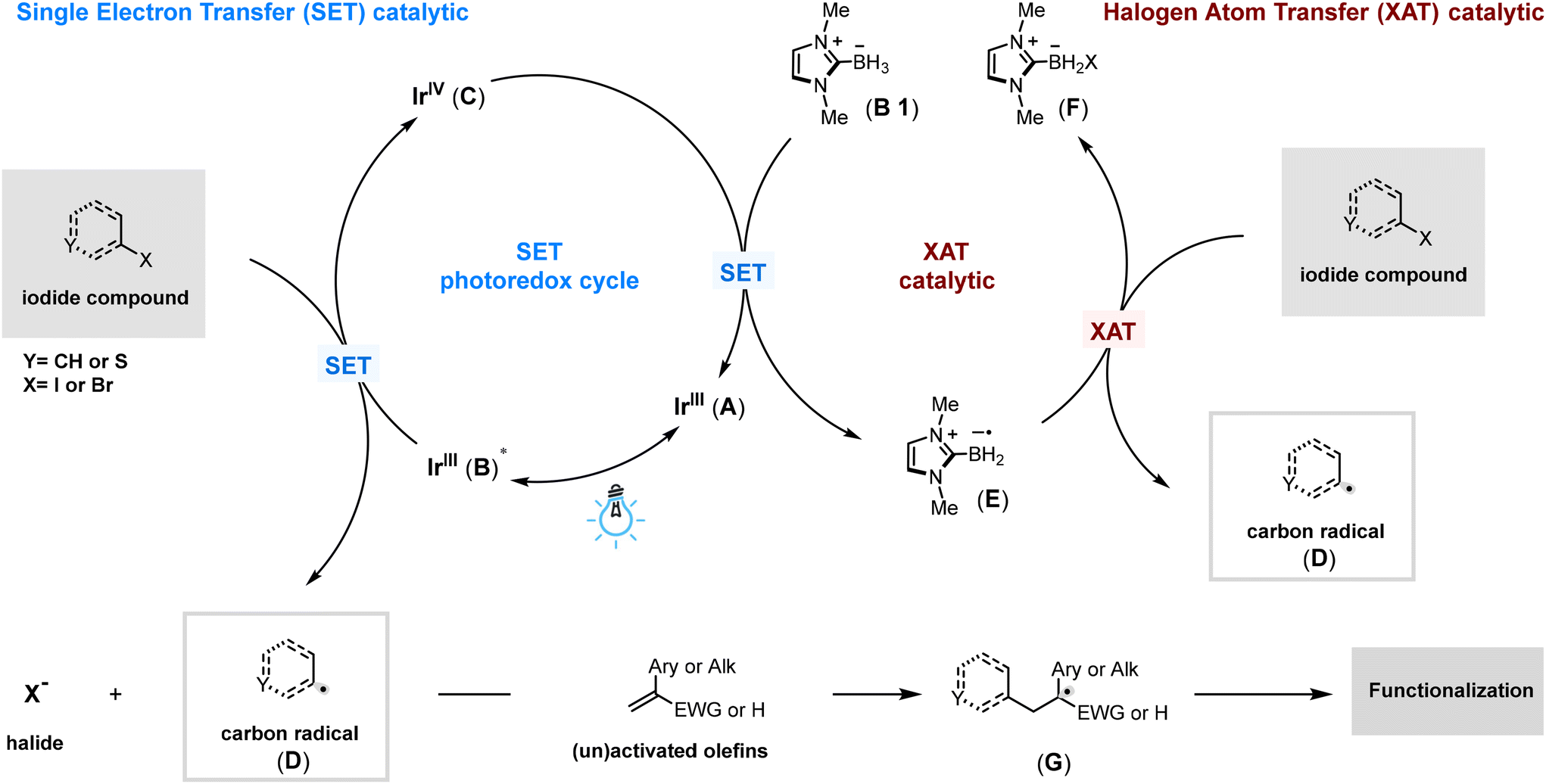 | ||
| Scheme 3 Possible catalytic mechanism. | ||
Conclusions
In summary, we present a novel atom transfer radical addition strategy for the direct aryl/alkylation of (un)activated olefins and alkynes. Our study demonstrates the compatibility of a wide range of olefins, alkynes, iodides, and some bromides as suitable partners, enabling the synthesis of diverse functionalized molecules. Particularly noteworthy is the protocol's versatility in accommodating various radical species, including high-energy aryl radicals, thiophene radicals, primary alkyl radicals, and site-containing alkyl radicals. Moreover, its efficacy in handling complex olefinic substrates suggests potential applicability in late-stage settings. Furthermore, by engaging alkynes as partners, our protocol offers a direct route to generating highly stereoselective cis-conjugated diaryl olefins. We anticipate that the modularity of the protocol, its broad substrate universality, and extensive functional group tolerance will expedite access to pharmaceutically relevant C(sp3)/C(sp2)-rich chemical space.Author contributions
X.H.L. conducted main experiments and prepared several starting materials; Y.Z. optimized the reaction conditions; F.S.T. prepared several starting materials during the revision of the manuscript. X.H.L. conceptualized and directed the project, and drafted the manuscript with the assistance from co-authors. All authors contributed to discussions.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22061048), Yunnan Fundamental Research Projects (202401BC070018, 202301AT070013), Yunnan Key Laboratory of Chiral Functional Substance Research and Application (202402AN360010).References
- C. S. Wang, P. H. Dixneuf and J. F. Soulé, Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Mechanistic Studies in Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- A. L. G. Kanegusuku and J. L. Roizen, Recent Advances in Photoredox-Mediated Radical Conjugate Addition Reactions: An Expanding Toolkit for the Giese Reaction, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef.
- M. D. Kärkäs, B. S. Matsuura and C. R. J. Stephenson, Enchained by visible light–mediated photoredox catalysis, Science, 2015, 349, 1285–1286 CrossRef.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. F. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS.
- D. P. Hari, P. Schroll and B. König, Metal-Free, Visible-Light-Mediated Direct C–H Arylation of Heteroarenes with Aryl Diazonium Salts, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed.
- G. B. Deng, Z. Q. Wang, J. D. Xia, P. C. Qian, R. J. Song, M. Hu, L. B. Gong and J. H. Li, Tandem Cyclizations of 1,6-Enynes with Arylsulfonyl Chlorides by Using Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2013, 52, 1535–1538 CrossRef CAS.
- W. Liu, X. Yang, Y. Gao and C.-J. Li, Simple and Efficient Generation of Aryl Radicals from Aryl Triflates: Synthesis of Aryl Boronates and Aryl Iodides at Room Temperature, J. Am. Chem. Soc., 2017, 139, 8621–8627 CrossRef CAS PubMed.
- N. Kvasovs and V. Gevorgyan, Contemporary methods for generation of aryl radicals, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC.
- S. H. Ueng, M. M. Brahmi, É. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, Complexes of Borane and N-Heterocyclic Carbenes: A New Class of Radical Hydrogen Atom Donor, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS.
- T. Kawamoto, S. J. Geib and D. P. Curran, Radical Reactions of N-Heterocyclic Carbene Boranes with Organic Nitriles: Cyanation of NHC-Boranes and Reductive Decyanation of Malononitriles, J. Am. Chem. Soc., 2015, 137, 8617–8622 CrossRef CAS PubMed.
- N. N. Zhou, X. A. Yuan, Y. Zhao, J. Xie and C. J. Zhu, Synergistic Photoredox Catalysis and Organocatalysis for Inverse Hydroboration of Imines, Angew. Chem., Int. Ed., 2018, 57, 3990–3994 CrossRef CAS PubMed.
- W. Dai, S. J. Geib and D. P. Curran, Facile Synthesis of α-N-Heterocyclic Carbene-Boryl Ketones from N-Heterocyclic Carbene-Boranes and Alkenyl Triflates, J. Am. Chem. Soc., 2019, 141, 12355–12361 CrossRef CAS PubMed.
- P. J. Xia, D. Song, Z. P. Ye, Y. Z. Hu, J. A. Xiao, H. Y. Xiang, X. Q. Chen and H. Yang, Photoredox-Controlled β-Regioselective Radical Hydroboration of Activated Alkenes with NHC-Boranes, Angew. Chem., Int. Ed., 2020, 59, 12817–12821 CrossRef PubMed.
- W. Dai, S. J. Geib and D. P. Curran, 1,4-Hydroboration Reactions of Electron-Poor Aromatic Rings by N-Heterocyclic Carbene Boranes, J. Am. Chem. Soc., 2020, 142, 6261–6267 CrossRef PubMed.
- T. Ye, F. L. Zhang, H. M. Xia, X. Zhou, Z. X. Yu and Y. F. Wang, Stereoselective hydrogen atom transfer to acyclic radicals: a switch enabling diastereodivergent borylative radical cascades, Nat. Commun., 2022, 13, 426 CrossRef CAS PubMed.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Synthesis and Reactions of N-Heterocyclic Carbene Boranes, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS.
- X. C. Pan, E. Lacôte, J. Lalevée and D. P. Curran, Polarity Reversal Catalysis in Radical Reductions of Halides by N-Heterocyclic Carbene Boranes, J. Am. Chem. Soc., 2012, 134, 5669–5674 CrossRef CAS PubMed.
- T. Taniguchi, Advances in chemistry of N-heterocyclic carbene boryl radicals, Chem. Soc. Rev., 2021, 50, 8995–9021 RSC.
- T. Wan, L. Capaldo, D. Ravelli, W. Vitullo, F. J. D. Zwart, B. D. Bruin and T. Noël, Photoinduced Halogen-Atom Transfer by N-Heterocyclic Carbene-Ligated Boryl Radicals for C(sp3)–C(sp3) Bond Formation, J. Am. Chem. Soc., 2023, 145, 991–999 CrossRef CAS PubMed.
- B. Yuan, C. Zhang, H. Y. Dong and C. Wang, Iron-Catalyzed Reductive Ring Opening/gem-Difluoroallylation of Cyclopropyl Ketones, Org. Lett., 2023, 25, 1883–1888 CrossRef CAS.
- X. H. Li, H. Y. Zhao, L. Meng, J. Dong, C. H. Yang, Z. X. Wei, X. M. Wang, J. B. Xu and B. M. Fan, Divergent Radical Cyclization and Hydroaminoalkylation of N-Arylacrylamides Using Photoredox Catalysis, J. Org. Chem., 2023, 88, 5300–5310 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- J. Qi, F. L. Zhang, J. K. Jin, Q. Zhao, B. Li, L. X. Liu and Y. F. Wang, New Radical Borylation Pathways for Organoboron Synthesis Enabled by Photoredox Catalysis, Angew. Chem., Int. Ed., 2020, 59, 12876–12884 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01562b |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |