
Mechanistic insight into the dehydrogenation reaction catalyzed by an MLC catalyst with dual ancillary ligand sites†
Gui-Xiang
Zhou
and
Cheng
Hou
 *
*
School of Chemistry and Pharmaceutical Sciences, State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Guangxi Normal University, Guilin, 541004, P. R. China. E-mail: houcheng@gxnu.edu.cn
First published on 29th October 2024
Abstract
Metal–ligand cooperative (MLC) catalysis is central to modern catalytic chemistry, notable for its ability to enhance efficiency. Traditional MLC catalysts with a single auxiliary ligand site have limitations in optimizing catalytic activity, prompting increased interest in employing dual active sites for improved performance. In this study, density functional theory (DFT) is employed to explore the catalytic mechanism of a ruthenium complex featuring a 2-(2-benzimidazolyl)pyridine ligand in the dehydrogenation of benzyl alcohol. This catalyst is distinguished by its ability to use both the O–H group of hydroxy pyridine and the N–H group of benzimidazole as auxiliary sites during the reaction. The research uncovers a dynamic switching mechanism of ligand active sites across different catalytic stages. Specifically, the catalyst utilizes the oxygen site of the pyridone ligand as the proton acceptor during the proton transfer stage, while the N–H group of the benzimidazole ligand serves as the active site during the critical hydride transfer stage. This site-switching mechanism is elucidated through molecular plane parameter (MPP) analysis and Extended Transition State – Natural Orbitals for Chemical Valence (ETS-NOCV) analysis, which reveal that the N-site-assisted pathway during hydrogen transfer is characterized by reduced ligand deformation and enhanced orbital interactions. These factors contribute to the observed mechanistic selectivity. This dynamic site-switching strategy effectively lowers the reaction energy barrier and improves catalytic efficiency. The insights gained from this study not only clarify the roles of the ligand in various catalytic stages but also offer valuable theoretical guidance for the development of novel MLC catalysts with dual active sites.
Introduction
The rational design of catalysts to precisely control their activity, selectivity, and stability for the efficient and green conversion of specific chemical reactions is a shared objective among chemists. Metal–ligand cooperation (MLC),1–3 as a core concept in modern catalytic chemistry, has garnered widespread attention and spurred intense research interest due to its unique catalytic mechanisms and outstanding performance enhancement potential.4–6 The MLC strategy involves the ingenious incorporation of auxiliary ligands into catalysts, achieving synergistic interactions between the metal center and ligands.7–11 This synergy not only enhances catalytic activity but also significantly improves reaction selectivity. Historically, milestone syntheses such as the Noyori–Ikariya12–16 and Milstein catalysts17–20 have successfully utilized the principles of MLC, pushing catalytic efficiency to new heights and bringing revolutionary advancements to fields like fine chemical synthesis, drug development, and energy conversion. However, despite these significant achievements, traditional MLC catalysts are often constrained by the design of a single auxiliary site, which limits the flexibility and scope of tuning catalytic activity. Consequently, exploring new MLC catalysts with multiple auxiliary sites21–23 and high tunability to meet the demands of increasingly complex chemical reactions has become a forefront challenge and direction in current catalytic chemistry research.In recent years, the synthesis of catalysts featuring dual ligand coordination sites has emerged as a pivotal trend within the realm of catalytic chemistry (Fig. 1a). In 2014, Milstein and colleagues achieved a milestone by synthesizing a Ru–PNNH pincer complex24 endowed with dual auxiliary coordination sites. Despite the presence of these dual active sites, no site-switching process25,26 was observed, thereby paving the way for the development of dual-site MLC catalysts. Subsequently, the Khusnutdinova group reported a novel ruthenium complex based on naphthyridinone, which they postulated to possess two potential deprotonation sites: an acidic CH2 group linked to the phosphine arm and the NH moiety of the naphthyridinone.27 This catalyst was employed in the dehydrogenation of primary alcohols. Although harboring two potential reactive sites, the Khusnutdinova team refrained from exploring the potential interplay between these active sites. Ayan Datta and Sreebrata Goswami et al. conducted a study that combines DFT calculations and experimental methods to reveal the ligand-mediated mechanism in the catalytic dehydrogenation of alcohols using a nickel clamp complex.28 The research also highlights the significant role of quantum tunneling in the hydrogen transfer process, demonstrating its potential applications in alcohol oxidation and hydrogen storage.29–33
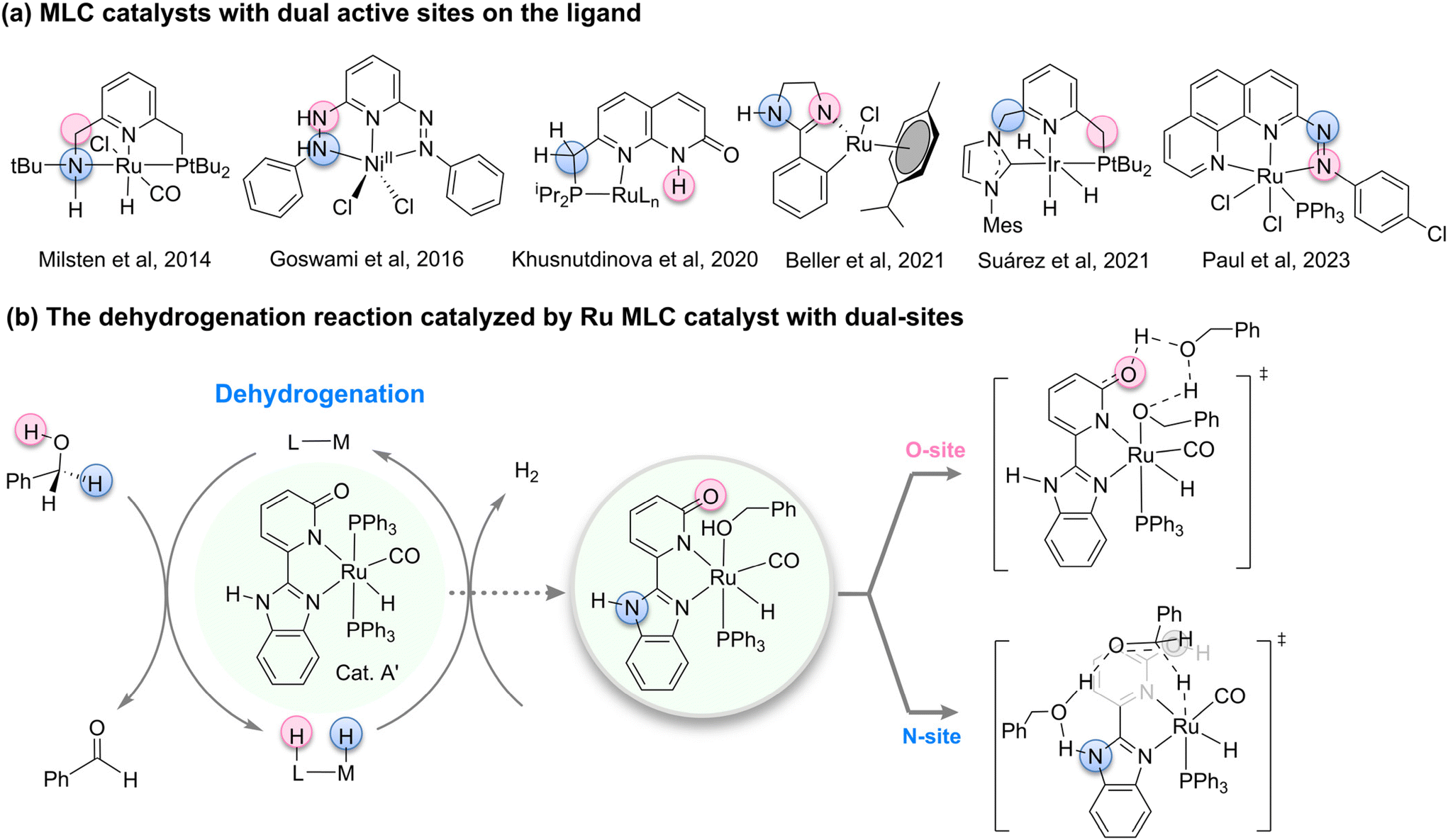 | ||
| Fig. 1 (a) MLC catalysts with dual active sites on the ligand; (b) the dehydrogenation reaction catalyzed by Ru MLC catalyst with dual-sites. | ||
Fast forwarding to 2021, Beller et al. synthesized a ruthenium complex utilizing a phenylimidazoline ligand, which, upon preactivation, presented two potential active sites on the ligand.34 Computational investigations suggested that interconversion between these two active sites was unlikely to occur.26 In the same year, Suárez and colleagues crafted an iridium CNP pincer complex derived from phosphine ligands and N-heterocyclic carbenes.35 Through a combined experimental and theoretical approach, they discovered that during hydrogen activation, the reaction was facilitated by the NHC moiety, whereas during carbonyl hydrogenation, the reaction was predominantly governed by the phosphine ligand side. Subsequently, an intramolecular proton transfer facilitated the switching between these two distinct active sites. In 2022, Suárez et al. successfully applied this dual-site cooperative catalyst to the more intricate reduction of nitrogen oxides, demonstrating the involvement of both ligand active sites in the reaction.36 In 2023, Nanda D. Paul and colleagues synthesized a ruthenium catalyst, postulating that the two N-sites on the ligand played a significant role during catalysis.37 Given this backdrop, it becomes evident that while these dual-active-site MLC catalysts exhibit promising catalytic activities, the presence of two reactive sites on the ligand often leads to intricate and elusive reaction mechanisms.
Recently, Kundu et al. synthesized and characterized a series of ruthenium complexes based on 2-(2-benzimidazolyl)pyridine ligands.38 Notably, Cat. A′, incorporating 2-hydroxypyridine and benzimidazole moieties, demonstrated superior reactivity (Fig. 1b). This Cat. A′, with its dual ligand sites, piqued our research interest: Could there be a possibility of site-switching between these active ligand sites? Which auxiliary ligand site is favored during different stages of catalysis? To address these questions, we conducted density functional theory (DFT) calculations, aiming to elucidate the roles played by different ligand sites at various stages of the catalytic reaction. This theoretical endeavor holds the potential to offer valuable insights for the future design of novel MLC catalysts featuring two active sites.
Computational details
Density functional theory (DFT) calculations were carried out using Gaussian 16 software (version A.03).39 Geometry optimization was performed at the M06-L40/def2SVP41 level of theory. We evaluated a series of functionals (please refer to ESI, Table S1†) and ultimately selected the M06-L functional, striking a balance between computational cost and accuracy. Additionally, frequency analysis was conducted at the M06-L/def2SVP level to ensure the absence of imaginary frequencies in all intermediates and to confirm the presence of exactly one imaginary frequency in each transition state. Intrinsic reaction coordinate (IRC) calculations were executed for all transition states to validate the obtained results. To improve the accuracy of the calculated free energy values, single-point energies of the optimized structures were computed at the M06-L/def2-TZVP level, utilizing the SMD solvent model42 to mimic the reaction environment in 1,4-dioxane. These corrections were facilitated using GoodVibes software (version 2.0.3),43 developed by Funes-Ardoiz and Paton. Furthermore, ETS-NOCV44 and MPP analyses45 were performed using Multiwfn software,46 while the geometries of the crucial transition states were generated and visualized using VMD software47 and CYLview software.48Results and discussion
The inner-sphere mechanism
The inner-sphere mechanism pathway, as illustrated in Fig. 2, starts from the more stable intermediate IM2 (please see the ESI, Fig. S8†).49,50 For this pathway, the benzyl alcohol dehydrogenation reaction is primarily divided into two stages: H2 formation and the regeneration of the metal hydride. In the H2 formation stage, we considered two possible transition states: a four-membered ring transition state TS4a (Ru–H⋯H⋯O–Ru) and a six-membered ring proton-shuttle transition state TS4b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51 (Ru–H⋯H⋯O⋯H⋯O–Ru). Unlike the transfer hydrogenation mechanism, both TS4a and TS4b involve a two-electron transfer process, wherein a proton combines with the metal hydride to generate H2. In TS4a, the proton from benzyl alcohol directly combines with the metal hydride, resulting in H2 formation and producing intermediate IM5. This transition state adopts a compact four-membered ring structure (Ru–H⋯H⋯O–Ru). In contrast, TS4b involves a proton-shuttle mechanism, where the proton on the benzyl alcohol coordinated to the metal is transferred to the oxygen of another benzyl alcohol molecule. Simultaneously, a proton from this second benzyl alcohol combines with the metal hydride to generate H2, leading to the formation of intermediate IM5. An IRC curve analysis further clarifies the proton-shuttle process occurring in TS4b (please see the ESI, Fig. S4 and S5†). Computational results indicate that the proton-shuttle transition state TS4b has an activation barrier of 23.4 kcal mol−1, whereas the four-membered ring transition state TS4a, due to ring strain, has a slightly higher barrier by approximately 1.0 kcal mol−1. Subsequently, the H2-coordinated intermediate IM5 releases H2 in a slightly endothermic process (ΔG = 0.5 kcal mol−1), generating the coordinatively unsaturated intermediate IM6, which provides an open site for the subsequent β-H elimination. The β-H elimination then proceeds viaTS5,27 generating the metal hydride intermediate IM7. IM7 releases a molecule of benzaldehyde, forming IM1. Finally, a molecule of benzyl alcohol coordinates with IM1 to form intermediate IM2, thereby completing the catalytic cycle. The computational results suggest that, in the inner-sphere reaction pathway, the rate-determining step occurs in the H2 formation stage, specifically at the proton-shuttle transition state TS4b, with an activation free energy of 23.4 kcal mol−1.
51 (Ru–H⋯H⋯O⋯H⋯O–Ru). Unlike the transfer hydrogenation mechanism, both TS4a and TS4b involve a two-electron transfer process, wherein a proton combines with the metal hydride to generate H2. In TS4a, the proton from benzyl alcohol directly combines with the metal hydride, resulting in H2 formation and producing intermediate IM5. This transition state adopts a compact four-membered ring structure (Ru–H⋯H⋯O–Ru). In contrast, TS4b involves a proton-shuttle mechanism, where the proton on the benzyl alcohol coordinated to the metal is transferred to the oxygen of another benzyl alcohol molecule. Simultaneously, a proton from this second benzyl alcohol combines with the metal hydride to generate H2, leading to the formation of intermediate IM5. An IRC curve analysis further clarifies the proton-shuttle process occurring in TS4b (please see the ESI, Fig. S4 and S5†). Computational results indicate that the proton-shuttle transition state TS4b has an activation barrier of 23.4 kcal mol−1, whereas the four-membered ring transition state TS4a, due to ring strain, has a slightly higher barrier by approximately 1.0 kcal mol−1. Subsequently, the H2-coordinated intermediate IM5 releases H2 in a slightly endothermic process (ΔG = 0.5 kcal mol−1), generating the coordinatively unsaturated intermediate IM6, which provides an open site for the subsequent β-H elimination. The β-H elimination then proceeds viaTS5,27 generating the metal hydride intermediate IM7. IM7 releases a molecule of benzaldehyde, forming IM1. Finally, a molecule of benzyl alcohol coordinates with IM1 to form intermediate IM2, thereby completing the catalytic cycle. The computational results suggest that, in the inner-sphere reaction pathway, the rate-determining step occurs in the H2 formation stage, specifically at the proton-shuttle transition state TS4b, with an activation free energy of 23.4 kcal mol−1.
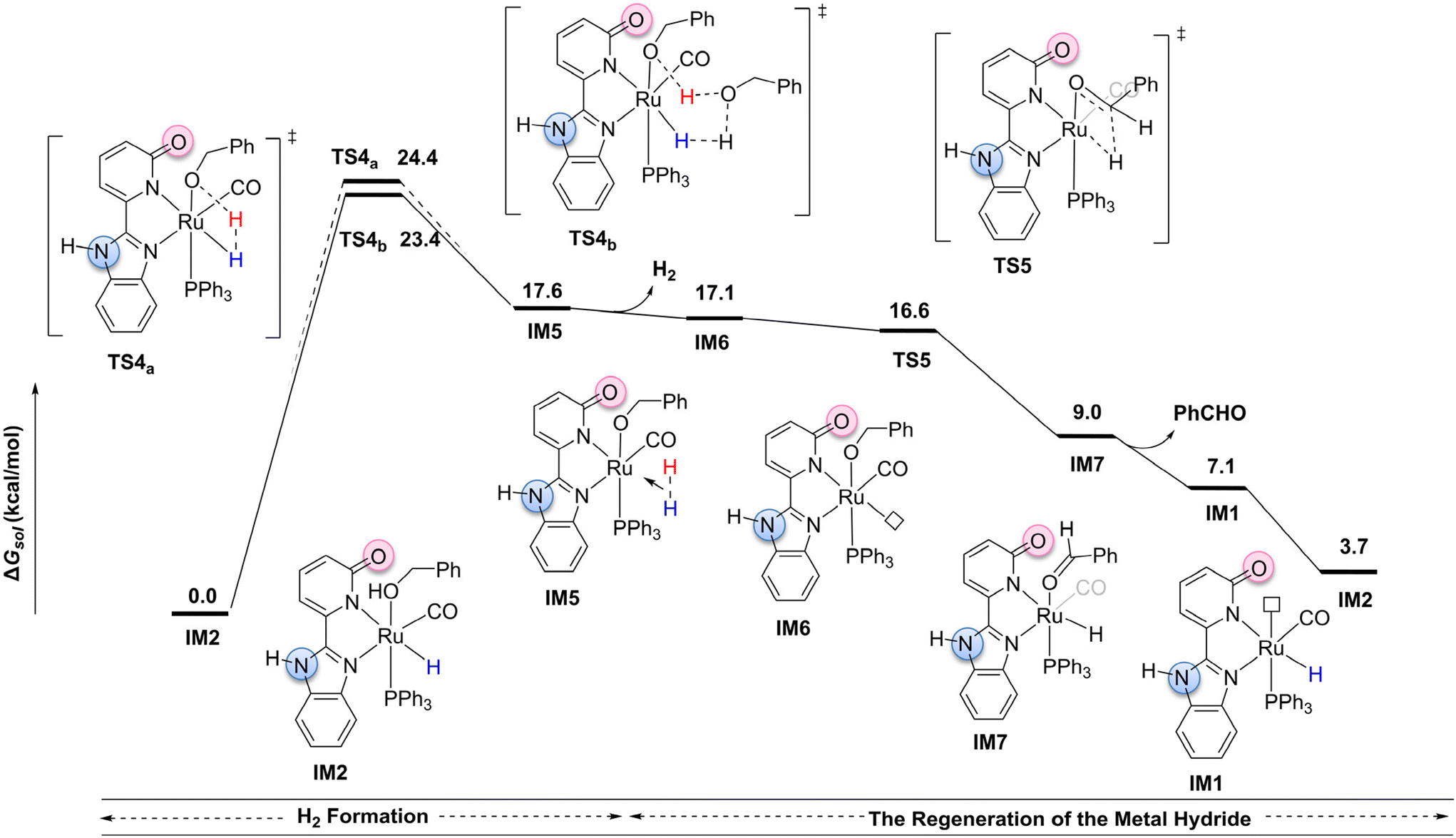 | ||
| Fig. 2 Potential energy diagram for the inner-sphere dehydrogenation mechanism of benzyl alcohol. | ||
The outer-sphere mechanism
As illustrated, the outer mechanism pathway starts from intermediate IM2 as the initiation point of the catalytic cycle. In the context of the outer mechanism, the catalytic cycle can be divided into two main stages: benzyl alcohol dehydrogenation and H2 generation (Fig. 3). The benzyl alcohol dehydrogenation stage comprises two processes: proton transfer and β-H elimination. During the proton transfer process, the ligand carbonyl oxygen site primarily receives the proton. For the β-H elimination process, three possible scenarios were considered: (1) β-H elimination assisted by the ligand O site (TS8); (2) β-H elimination without assistance from any ligand site (TS2); and (3) β-H elimination assisted by the ligand N site (TS6). Firstly, in the reaction process assisted by the ligand O site, there are two possible pathways: a stepwise pathway and a concerted pathway. In the stepwise pathway, the substrate benzyl alcohol first transfers a proton and then undergoes β-H elimination. Intermediate IM2, via transition state TS7a, transfers the proton from benzyl alcohol to the ligand carbonyl, forming intermediate IM8. Intermediate IM8, through transition state TS7b (ΔG‡ = 26.2 kcal mol−1), releases a molecule of benzaldehyde, forming intermediate IM4. In the concerted pathway, intermediate IM2, via transition state TS8 (ΔG‡ = 21.9 kcal mol−1), directly releases benzaldehyde, forming intermediate IM4. The calculation results indicate that in the reaction process assisted by the ligand oxygen site, the concerted pathway is more favorable, with an activation free energy barrier of 21.9 kcal mol−1. | ||
| Fig. 3 Potential energy diagram for the outer-sphere dehydrogenation mechanism of benzyl alcohol. | ||
For the reaction pathways without ligand site assistance and with ligand N site assistance in β-H elimination, first, during the proton transfer from the alcohol in intermediate IM2 to the ligand carbonyl, the proton shuttle transition state TS1b (ΔG‡ = 6.0 kcal mol−1) is lower in energy by 0.3 kcal mol−1 compared to TS1a (ΔG‡ = 6.3 kcal mol−1). Intermediate IM2, through the proton shuttle transition state TS1b, forms intermediate IM9, which is an endothermic process (ΔG = 7.1 kcal mol−1). Intermediate IM9, losing a molecule of benzyl alcohol, forms intermediate IM3. Subsequently, in the β-H elimination process from intermediate IM3 to form intermediate IM4, two possible transition states were considered: transition state TS6, formed by hydrogen bonding between a molecule of alcohol and the ligand N site, and transition state TS2, which does not involve any ligand site assistance. The calculation results indicate that the transition state TS6, with ligand N site assistance, has a lower energy barrier of 19.9 kcal mol−1. In contrast, the transition state TS2, without ligand site assistance, has a significantly higher energy barrier of 49.8 kcal mol−1.
During the H2 generation stage, we also considered two possible transition states: intermediate transition state TS3a and proton shuttle transition state TS3b, with energy barriers of 20.7 kcal mol−1 and 16.6 kcal mol−1, respectively. Intermediate IM4, via the proton shuttle transition state TS3b, releases H2, forming intermediate IM1. Subsequently, a molecule of benzyl alcohol coordinates with intermediate IM1, regenerating intermediate IM2. The above calculation results indicate that in the outer mechanism pathway, the O site is first involved in the proton transfer process during the alcohol dehydrogenation reaction, while the subsequent β-H elimination switches to the N site. During the β-H elimination stage, the transition state TS6 with ligand N site assistance has a significantly lower energy than the transition state TS2 without ligand site assistance and is also lower than the transition states TS7b and TS8 with ligand O site assistance.
Our calculations suggest that this catalyst follows a unique site-switching mechanism. In the proton transfer step of the acceptorless dehydrogenation reaction, the catalyst chooses the oxygen atom site of the pyridinone ligand. However, in the crucial hydride transfer step, the calculations show that the catalyst dynamically switches to the assistance of the N–H group of the benzimidazole ligand. In the final H2 release step, the catalyst reacts again through the oxygen atom site of the hydroxy-pyridine.
To elucidate the phenomenon of site switching in the catalyst and the variations in transition state energy barriers, an ETS-NOCV was subsequently applied to TS6, TS7b, and TS8. As shown in Fig. 4(a), the ETS-NOCV results indicate that the NOCV orbital energies for TS6, TS7b, and TS8 are −53.90 kcal mol−1, −22.39 kcal mol−1, and −53.35 kcal mol−1, respectively. Notably, TS7b (ΔG‡ = 26.2 kcal mol−1) exhibits a relatively small contribution from its NOCV orbitals to the electron density changes. In contrast, TS6 (ΔG‡ = 19.9 kcal mol−1) and TS8 (ΔG‡ = 21.9 kcal mol−1) have larger absolute NOCV orbital energies with similar magnitudes, suggesting a significant contribution to electron density changes in these two transition states.
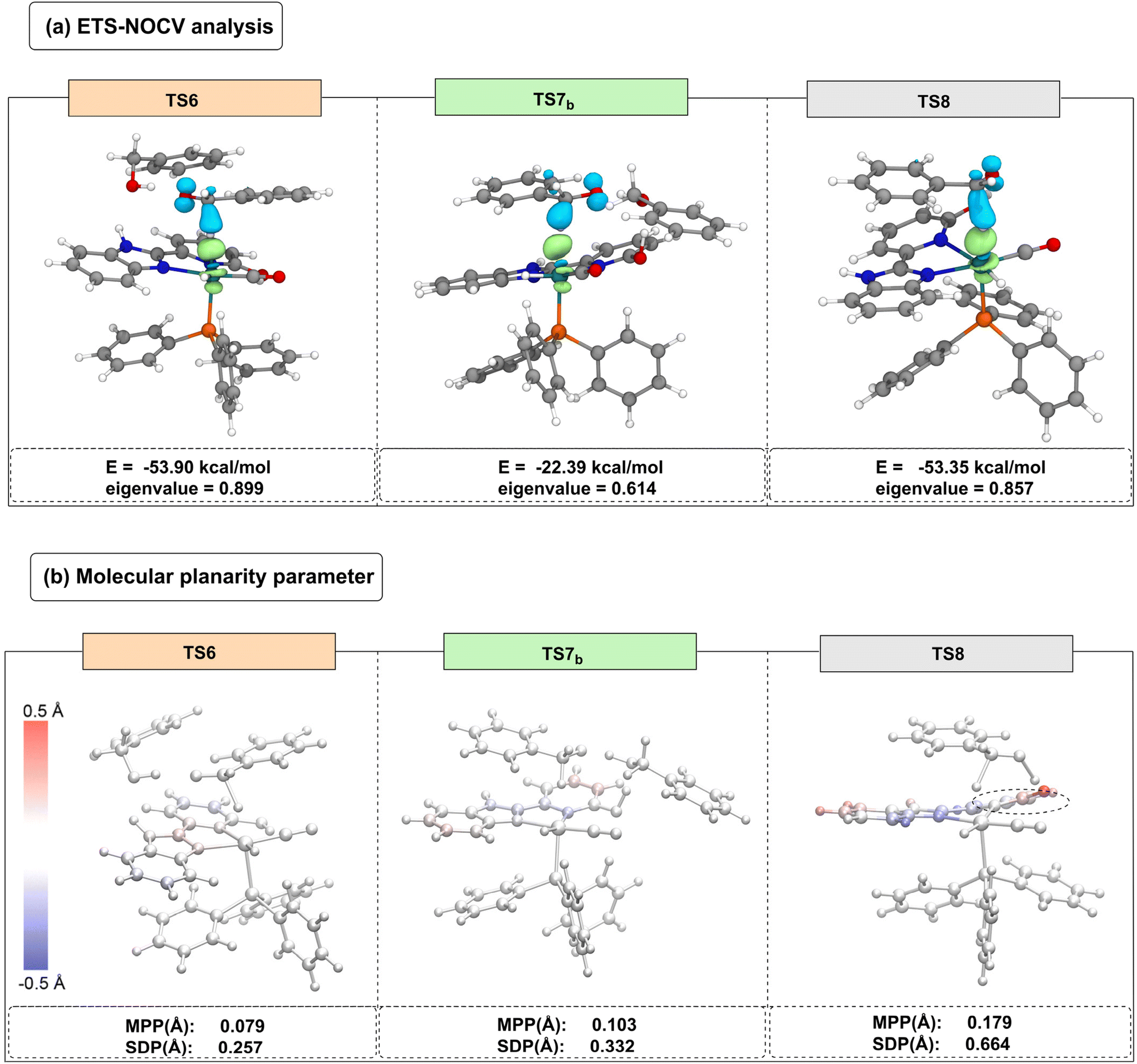 | ||
| Fig. 4 (a) ETS-NOCV analysis; (b) molecular planarity parameter analysis. | ||
To further explain the energy barrier differences between TS6 and TS8, a MPP analysis was performed.52–55 In this analysis, the atoms in the transition state structures are colored based on their standard deviation of planarity (SDP) values, with darker colors indicating greater deviations from the fitted plane. Carbon atoms above the fitted plane are primarily colored red, while those below are blue. As shown in Fig. 4(b), TS6 has an MPP value of 0.079, indicating minimal deviation from planarity in its ligand. In contrast, TS8 has an MPP value of 0.179, with the sp2 hybridized carbonyl carbon atoms appearing red, indicating significant deviation from planarity. This deviation favors the dynamic selection of the N-site on the benzimidazole ligand during the β-H elimination process.
Conclusions
This study utilizes density functional theory (DFT) calculations to elucidate the catalytic mechanism of a ruthenium complex with a 2-(2-benzimidazolyl)pyridine ligand in the benzyl alcohol dehydrogenation reaction. We present a detailed analysis of the dynamic switching mechanism of ligand active sites across various catalytic stages. Our findings indicate that the catalyst predominantly employs the oxygen site of the pyridone ligand during the proton transfer phase, while in the critical hydride transfer stage, it dynamically selects the N–H group of the benzimidazole ligand as the active site. This dynamic site selection mechanism substantially reduces the reaction barrier and enhances catalytic efficiency. These insights offer novel perspectives on the design of traditional MLC catalysts and highlight the capability of dual-site cooperative catalysts to optimize reaction pathways in complex processes. Our study not only advances the understanding of dual-site cooperative catalysis but also provides a robust theoretical foundation for the future development of highly efficient catalysts.Author contributions
Gui-Xiang Zhou: calculation, data curation, writing – original draft. Cheng Hou: conceptualization, writing – review & editing, supervision, funding acquisition.Data availability
Additional data are made available in supplementary tables in ESI† of this manuscript.Conflicts of interest
The authors declared that they have no conflicts of interest to this work.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22163001).Notes and references
- J. R. Khusnutdinova and D. Milstein, Metal-ligand cooperation, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- M. Rauch, S. Kar, A. Kumar, L. Avram, L. J. W. Shimon and D. Milstein, Metal-Ligand Cooperation Facilitates Bond Activation and Catalytic Hydrogenation with Zinc Pincer Complexes, J. Am. Chem. Soc., 2020, 142, 14513–14521 CrossRef CAS.
- M. R. Elsby and R. T. Baker, Strategies and mechanisms of metal-ligand cooperativity in first-row transition metal complex catalysts, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- L. Omann, C. D. F. Konigs, H. F. T. Klare and M. Oestreich, Cooperative Catalysis at Metal-Sulfur Bonds, Acc. Chem. Res., 2017, 50, 1258–1269 CrossRef CAS PubMed.
- J. Luo, M. Montag and D. Milstein, Metal-Ligand Cooperation with Thiols as Transient Cooperative Ligands: Acceleration and Inhibition Effects in (De)Hydrogenation Reactions, Acc. Chem. Res., 2024, 57, 1709–1721 CrossRef CAS PubMed.
- B. Zhao, Z. Han and K. Ding, The N-H functional group in organometallic catalysis, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS.
- X. Wu and L. Zhang, Gold-Catalyzed Homo/Heterodimerization of Terminal Alkynes Facilitated by Metal-Ligand Cooperation, Org. Lett., 2024, 26, 5736–5740 CrossRef CAS PubMed.
- C. Hou, Z. Zhang, C. Zhao and Z. Ke, DFT Study of Acceptorless Alcohol Dehydrogenation Mediated by Ruthenium Pincer Complexes: Ligand Tautomerization Governing Metal Ligand Cooperation, Inorg. Chem., 2016, 55, 6539–6551 CrossRef CAS PubMed.
- N. Nebra, J. Monot, R. Shaw, B. Martin-Vaca and D. Bourissou, Metal–Ligand Cooperation in the Cycloisomerization of Alkynoic Acids with Indenediide Palladium Pincer Complexes, ACS Catal., 2013, 3, 2930–2934 CrossRef CAS.
- A. Scharf, I. Goldberg and A. Vigalok, Evidence for metal-ligand cooperation in a Pd-PNF pincer-catalyzed cross-coupling, J. Am. Chem. Soc., 2013, 135, 967–970 CrossRef CAS PubMed.
- J. M. Perez, R. Postolache, M. Castineira Reis, E. G. Sinnema, D. Vargova, F. de Vries, E. Otten, L. Ge and S. R. Harutyunyan, Manganese(I)-Catalyzed H-P Bond Activation via Metal-Ligand Cooperation, J. Am. Chem. Soc., 2021, 143, 20071–20076 CrossRef CAS PubMed.
- R. Noyori and S. Hashiguchi, Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- R. Noyori, Asymmetric Catalysis: Science and Opportunities (Nobel Lecture), Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- R. Noyori, M. Yamakawa and S. Hashiguchi, Metal–Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS.
- S. H. Akio Fujii, N. Uematsu, T. Ikariya and R. Noyori, Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid-Triethylamine Mixture, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, Asymmetric Transfer Hydrogenation Of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- C. Gunanathan and D. Milstein, Metal–Ligand Cooperation by Aromatization–Dearomatization: A New Paradigm in Bond Activation and “Green” Catalysis, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- E. Ben-Ari, G. Leitus, L. J. W. Shimon and D. Milstein, Metal–Ligand Cooperation in C–H and H2 Activation by an Electron-Rich PNP Ir(I) System: Facile Ligand Dearomatization–Aromatization as Key Steps, J. Am. Chem. Soc., 2006, 128, 15390–15391 CrossRef CAS PubMed.
- C. Gunanathan, B. Gnanaprakasam, M. A. Iron, L. J. W. Shimon and D. Milstein, “Long-Range” Metal–Ligand Cooperation in H2 Activation and Ammonia-Promoted Hydride Transfer with a Ruthenium–Acridine Pincer Complex, J. Am. Chem. Soc., 2010, 132, 14763–14765 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Bond Activation and Catalysis by Ruthenium Pincer Complexes, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS.
- B. Dong, X. Wu, L. Shen, Y. He, X. Chen, S. Zhang and F. Li, Poly(2,2′-Bibenzimidazole)-Supported Iridium Complex: A Recyclable Metal-Polymer Ligand Bifunctional Catalyst for the N-Methylation of Amines with Methanol, Inorg. Chem., 2024, 63, 15072–15080 CrossRef CAS PubMed.
- Y. Zhang, L.-Y. Li and C. Hou, Mechanistic investigation of the transfer hydrogenation of alkynes catalysed by an MLC catalyst with multiple ancillary ligand sites, Org. Chem. Front., 2023, 10, 3766–3775 RSC.
- C. Hou, J. Jiang, Y. Li, C. Zhao and Z. Ke, When Bifunctional Catalyst Encounters Dual MLC Modes: DFT Study on the Mechanistic Preference in Ru-PNNH Pincer Complex Catalyzed Dehydrogenative Coupling Reaction, ACS Catal., 2016, 7, 786–795 CrossRef.
- E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. Shimon and D. Milstein, System with potential dual modes of metal-ligand cooperation: highly catalytically active pyridine-based PNNH-Ru pincer complexes, Chem. – Eur. J., 2014, 20, 15727–15731 CrossRef CAS PubMed.
- Y. Q. Zou, N. von Wolff, M. Rauch, M. Feller, Q. Q. Zhou, A. Anaby, Y. Diskin-Posner, L. J. W. Shimon, L. Avram, Y. Ben-David and D. Milstein, Homogeneous Reforming of Aqueous Ethylene Glycol to Glycolic Acid and Pure Hydrogen Catalyzed by Pincer-Ruthenium Complexes Capable of Metal-Ligand Cooperation, Chem. – Eur. J., 2021, 27, 4715–4722 CrossRef CAS PubMed.
- L. Mei, M. Du, Y. Zhang and C. Hou, Mechanistic insight into borrowing-hydrogen N-alkylation catalyzed by an MLC catalyst with dual proton-responsive sites, Dalton Trans., 2022, 51, 16215–16223 RSC.
- M. Gallardo-Villagran, O. Rivada-Wheelaghan, S. M. W. Rahaman, R. R. Fayzullin and J. R. Khusnutdinova, Proton-responsive naphthyridinone-based Ru(II) complexes and their reactivity with water and alcohols, Dalton Trans., 2020, 49, 12756–12766 RSC.
- D. Sengupta, R. Bhattacharjee, R. Pramanick, S. P. Rath, N. Saha Chowdhury, A. Datta and S. Goswami, Exclusively Ligand-Mediated Catalytic Dehydrogenation of Alcohols, Inorg. Chem., 2016, 55, 9602–9610 CrossRef CAS.
- S. Karmakar and A. Datta, Role of quantum mechanical tunneling on the gamma-effect of silicon on carbenes in 3-trimethylsilylcyclobutylidene, J. Phys. Chem. B, 2014, 118, 2553–2558 CrossRef CAS.
- A. Nijamudheen, S. Karmakar and A. Datta, Understanding the mechanisms of unusually fast H-H, C-H, and C-C bond reductive eliminations from gold(III) complexes, Chem. – Eur. J., 2014, 20, 14650–14658 CrossRef CAS.
- K. Jana and B. Ganguly, DFT studies on quantum mechanical tunneling in tautomerization of three-membered rings, Phys. Chem. Chem. Phys., 2018, 20, 28049–28058 RSC.
- P. R. Schreiner, Quantum Mechanical Tunneling Is Essential to Understanding Chemical Reactivity, Trends Chem., 2020, 2, 980–989 CrossRef CAS.
- S. Kozuch, T. Schleif and A. Karton, Quantum mechanical tunnelling: the missing term to achieve sub-kJ mol−1 barrier heights, Phys. Chem. Chem. Phys., 2021, 23, 10888–10898 RSC.
- P. Piehl, R. Amuso, A. Spannenberg, B. Gabriele, H. Neumann and M. Beller, Efficient methylation of anilines with methanol catalysed by cyclometalated ruthenium complexes, Catal. Sci. Technol., 2021, 11, 2512–2517 RSC.
- P. Sánchez, M. Hernández-Juárez, N. Rendón, J. López-Serrano, E. Álvarez, M. Paneque and A. Suárez, Selective, Base-Free Hydrogenation of Aldehydes Catalyzed by Ir Complexes Based on Proton-Responsive Lutidine-Derived CNP Ligands, Organometallics, 2021, 40, 1314–1327 CrossRef.
- I. Ortega-Lepe, P. Sánchez, L. L. Santos, P. Lara, N. Rendón, J. López-Serrano, V. Salazar-Pereda, E. Álvarez, M. Paneque and A. Suárez, Catalytic Nitrous Oxide Reduction with H2 Mediated by Pincer Ir Complexes, Inorg. Chem., 2022, 61, 18590–18600 CrossRef CAS PubMed.
- A. K. Guin, S. Pal, S. Chakraborty, S. Chakraborty and N. D. Paul, Oxygen Dependent Switchable Selectivity during Ruthenium Catalyzed Selective Synthesis of C3-Alkylated Indoles and Bis(indolyl)methanes, J. Org. Chem., 2023, 88, 16755–16772 CrossRef CAS.
- M. Maji and S. Kundu, Cooperative ruthenium complex catalyzed multicomponent synthesis of pyrimidines, Dalton Trans., 2019, 48, 17479–17487 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2008, 113, 6378–6396 CrossRef.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, GoodVibes: automated thermochemistry for heterogeneous computational chemistry data, F1000Research, 2020, 9, 291 Search PubMed.
- M. P. Mitoraj, M. Parafiniuk, M. Srebro, M. Handzlik, A. Buczek and A. Michalak, Applications of the ETS-NOCV method in descriptions of chemical reactions, J. Mol. Model., 2011, 17, 2337–2352 CrossRef CAS PubMed.
- T. Lu and Q. Chen, A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure, Theor. Chem. Acc., 2020, 139, 25 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 37–38 CrossRef PubMed.
- C. Y. Legault, CYLview20, Université de Sherbrooke, 2020. Available online: https://www.cylview.org Search PubMed.
- J. A. Luque-Urrutia, T. Pèlachs, M. Solà and A. Poater, Double-Carrousel Mechanism for Mn-Catalyzed Dehydrogenative Amide Synthesis from Alcohols and Amines, ACS Catal., 2021, 11, 6155–6161 CrossRef CAS.
- K. Sarkar, P. Behera, L. Roy and B. Maji, Manganese catalyzed chemo-selective synthesis of acyl cyclopentenes: a combined experimental and computational investigation, Chem. Sci., 2024, 15, 14287–14294 RSC.
- N. E. Smith, W. H. Bernskoetter and N. Hazari, The Role of Proton Shuttles in the Reversible Activation of Hydrogen via Metal-Ligand Cooperation, J. Am. Chem. Soc., 2019, 141, 17350–17360 CrossRef CAS.
- T. Lu, Simple, reliable, and universal metrics of molecular planarity, J. Mol. Model., 2021, 27, 263 CrossRef CAS PubMed.
- L. Yu, K. W. Lee, Y. Q. Zhao, Y. Xu, Y. Zhou, M. Li and J. S. Kim, Metal Modulation: An Effortless Tactic for Refining Photoredox Catalysis in Living Cells, Inorg. Chem., 2023, 62, 18767–18778 CrossRef CAS PubMed.
- R. Zaier, A. Martel and T. J. Antosiewicz, Effect of Benzothiadiazole-Based π-Spacers on Fine-Tuning of Optoelectronic Properties of Oligothiophene-Core Donor Materials for Efficient Organic Solar Cells: A DFT Study, J. Phys. Chem. A, 2023, 127, 10555–10569 CrossRef CAS PubMed.
- T. Noor, M. Waqas, M. Shaban, S. Hameed, R. Ateeq Ur, S. B. Ahmed, H. A. Alrafai, S. I. Al-Saeedi, M. A. A. Ibrahim, N. M. A. Hadia, R. A. Khera and A. A. Hassan, Designing Thieno[3,4-c]pyrrole-4,6-dione Core-Based, A2-D-A1-D-A2-Type Acceptor Molecules for Promising Photovoltaic Parameters in Organic Photovoltaic Cells, ACS Omega, 2024, 9, 6403–6422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01767f |
| This journal is © the Partner Organisations 2025 |