
Folding of a dynamic macrocyclic system to stabilize its cation radical state†
Arnab
Dutta
 ab,
Krzysztof
Dzieszkowski
a,
Marco
Farinone
a,
Łukasz
Orzeł
a,
Krzysztof
Kruczała
a,
Monika
Kijewska
c and
Miłosz
Pawlicki
*a
ab,
Krzysztof
Dzieszkowski
a,
Marco
Farinone
a,
Łukasz
Orzeł
a,
Krzysztof
Kruczała
a,
Monika
Kijewska
c and
Miłosz
Pawlicki
*a
aFaculty of Chemistry, Jagiellonian University, Functional Organic Materials Team, Gronostajowa 2, 30-387 Kraków, Poland. E-mail: pawlicki@chemia.uj.edu.pl; Web: https://mjplab.org/
bJagiellonian University, Doctoral School of Exact and Natural Sciences, Prof. St Łojasiewicza 11, 30-348, Kraków, Poland
cDepartment of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland
First published on 18th October 2024
Abstract
Precise design of unsaturated systems remains an important factor that determines the quality of formed products based on a specific spatial orientation, but also defines the available π-electron density open for post-synthetic modulation via redox change. Three strictly defined reagents showing different degrees of flexibility, but also introducing two mutual orientations of the reacting ends (parallel and obtuse), determine the quality of the formed products obtained via an intramolecular or an intermolecular reaction. The redox-activated transformation of mono- and double-looped systems results in dissimilar oxidation states of a strongly π-conjugated dication (two-electron process) or a cation radical (one-electron process), derived from the differences in the dynamic skeleton documented for the monomer and dimer, respectively.
Introduction
Design of structural motifs with extended π-delocalisation and based on rational decisions is constantly increasing in importance, as a factor that allows a response to the growing need for specific behaviours and properties, e.g. optical or magnetic.1 In this context, controlled formation of strongly π-conjugated derivatives remains a crucial, highly desirable factor for obtaining strictly defined skeletons with the possibility of stabilizing different oxidation states, which deeply influence optical properties.2 Unsaturated macrocyclic derivatives that merge subunits of different origins (i.e. heterocyclic and/or carbocyclic units) occupy a crucial place in understanding of the π-conjugation phenomenon of 4n + 2 or 4n π-electrons, and the subtle equilibrium between local and global delocalisation(s). On the other hand, cyclic derivatives allow observation of the reactivity, which, in contrast to linear systems, includes stabilization of the open-shell character of neutral3 or cationic derivatives,4 and which is crucial for both magnetic and optical properties. In fact, formation of multicharged systems remains a crucial aspect of the exploration of strongly conjugated systems, because of the structural complexity usually required for the increased total charge. From this perspective, precise planning of the formation of macrocyclic derivatives requires strict control in defining a substrate ready for formation of a cyclic system, usually occurring in the final step5 for either the intramolecular or the intermolecular variant. The two orientations introducing a control factor and lead to either intra- or intermolecular reactivity, and differed by the mutual angle between the active sides were determined. Thus, parallel or obtuse systems based on π-extended acene or heteroacene show dissimilar qualities for their observed products, forming solely monomers (intramolecular, Variant I, Fig. 1)6 or monomers or dimers (intra-/intermolecular, Variant II, Fig. 1), respectively.7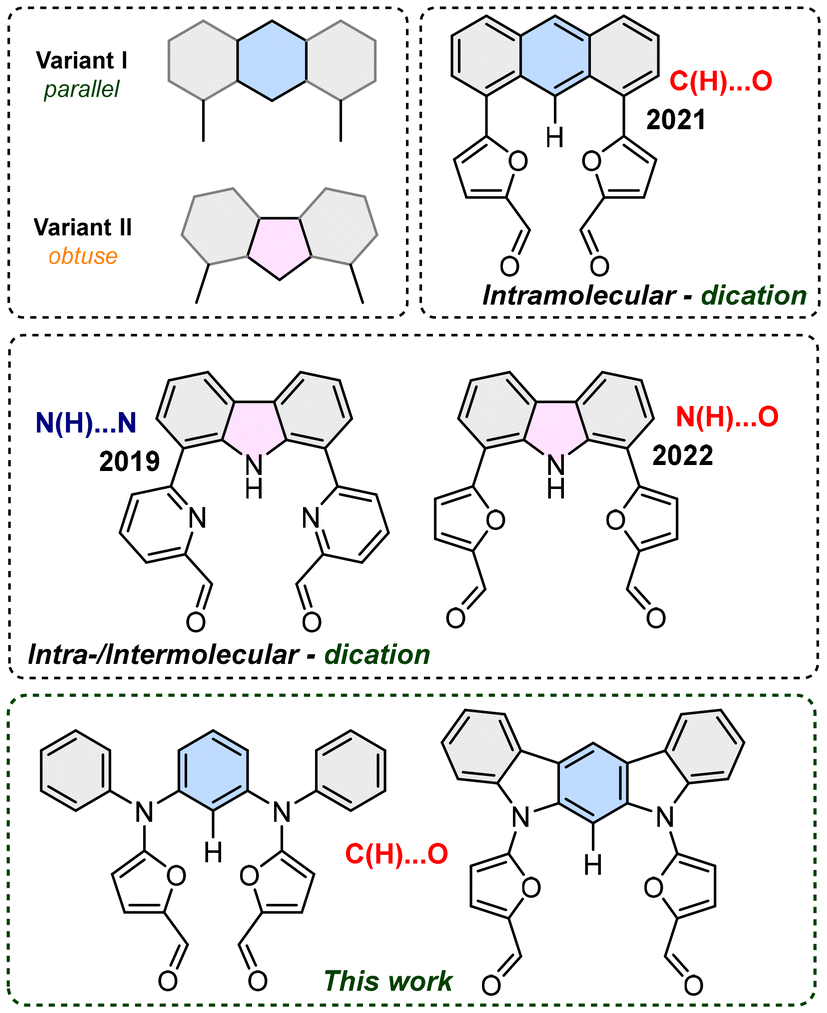 | ||
| Fig. 1 Geometric variants leading to intra- or intermolecular reactions. | ||
In light of these observations we have designed novel substrates that are open for controlled reactivity for the formation of structurally dynamic systems. Incorporation of a bent azaacene and entrapping a five-membered heterocycle unit of furan has been documented as readily undergoing redox changes and stabilizing cationic/radical and globally conjugated derivatives.8 Two structural aspects were considered, namely, the angle/distance between the reacting ends, as well as the overall flexibility, which, as it gradually decreased, was expected to deeply influence the quality of the observed products. All final derivatives were explored with respect to stabilization of globally conjugated systems that potentially entrap unpaired electron(s), as documented for three-dimensional (3D) structure(s) that respond dynamically to the redox trigger forming and taming a cation radical.
Results and discussion
Synthesis and characterisation
The anticipated mutual orientation aspect was based on the m-phenylene diamine core substituted by two phenyls connected to nitrogens (Scheme 1, 1a–1c), which imprints the required geometry, while the reactivity was secured by connecting two furfurals ready for a post-synthetic closure reaction based on the McMurry coupling. The second important factor identified during the analysis was the flexibility of the reagent subjected to the final closing process, which, in our experiments, gradually decreased in the sequence 1a → 1b → 1c. The synthetic approach leading to 1a–1c was focused on the Buchwald–Hartwig reaction, which is widely applied for the formation of a C–N bond in a variety of substrates,9 including π-extended systems.10 The diamino derivatives 2a–2b (Scheme 1) were obtained following previous reports.11,12 The direct reaction between m-dibromobenzene and aniline (Scheme 1, path a) gave 2a in 70% yield.11 The Buchwald–Hartwig coupling for dimethyl 2,4-dibromoisophthalate with aniline (Scheme 1, path b) gave a system that, followed by treatment with methyl lithium, yielded 2b with 74% conversion.122c is a commercially available derivative and was used without any preparation or purification. All diamino derivatives subjected to palladium-catalysed reaction with 2-bromo-furan-5-carboxyaldehyde gave the desired products 1a–1c in good yield (Scheme 1, path c). To examine the exact geometrical factors incorporated into all three derivatives, X-ray analysis of the monocrystals was performed, unambiguously confirming the composition and the quality of the obtained connectivity (Fig. 2A–C). The isolated systems showed a dissimilar spatial arrangement, with the mutual orientation of both furan subunits recalling the parallel orientation (Variant I, Fig. 1) observed for 1a and 1b. The third and the most rigid system, 1c, shows an obtuse angle between the two furans (Variant II, Fig. 1), and in addition stabilizing the C–H⋯O interaction (C⋯O 2.741 Å; H⋯O 2.181 Å).13 In contrast, both units with increased flexibility (1a and 1b) showed a nicely marked sp3 hybridisation of nitrogen bridges, caused by limited global delocalisation and inefficient displacement of a lone pair. Decreased delocalisation of a lone pair changes the geometry, eventually increasing the C(H)–O distance for 1a (C⋯O 2.852 Å; H⋯O 2.557 Å) and for 1b (C⋯O 2.855 Å; H⋯O 2.526 Å), which limits the efficiency of the interaction. Thus, gradually modified flexibility of the substrate for the McMurry reaction is reflected in the documented solid-state geometries.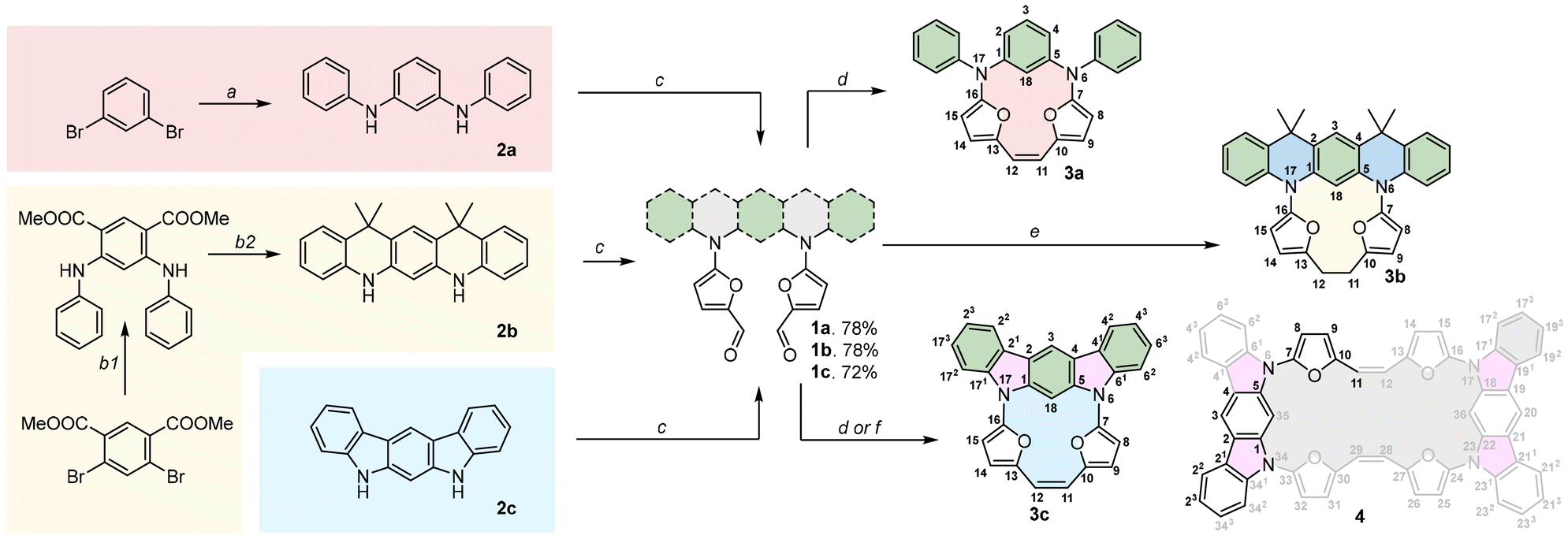 | ||
| Scheme 1 Synthetic approach implemented. Conditions: (a) aniline, Pd2(dba)3, rac-BINAP, NaOtBu, toluene, reflux; (b1) aniline, Pd(OAc)2, Cs2CO3, (t-Bu)3PHBF4, toluene, reflux, 24 h; (b2) (i) CH3Li, THF, −78 °C; (ii) BF3·Et2O, DCM; (c) 5-bromo-2-furaldehyde, Pd2(dba)3, RuPhos, K3PO4, toluene, reflux, 24 h; (d) 1a or 1c, TiCl4, Zn, CuI, THF, reflux; (e) 1b, TiCl4, Zn, CuI, 1,4-dioxane, reflux; (f) 1c, TiCl4, Zn, pyridine, 1,4-dioxane, reflux. | ||
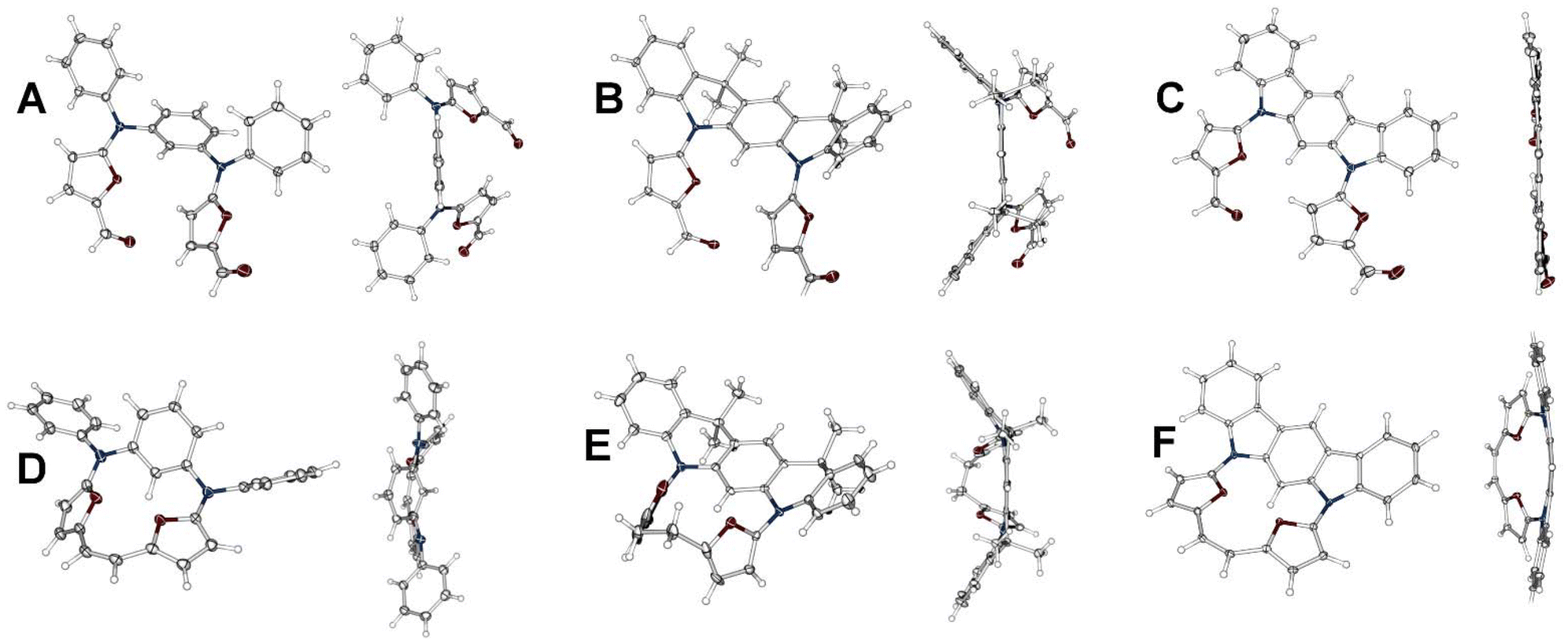 | ||
| Fig. 2 X-ray structures for 1a (A), 1b (B), 1c (C), 3a (D), 3b (E) and 3c (F). Thermal ellipsoids represent 50% probability. | ||
As the cyclisation approach focused on the formation of C–C bonds via McMurry coupling, all three derivatives with carbonyl units were subjected to low-valent titanium activation to form the C–C bond. Based on the above analysis of the geometrical factors, it was expected that a solely intramolecular mode of reaction for 1a would occur, which combines both selection criteria (i.e. parallel orientation of reacting arms and substantial flexibility leading to lack of C–H⋯O interaction). The outcome of this reaction gave 3a, which was isolated with a yield close to unity (92%, Scheme 1, path d), as confirmed by the MS measurements in the ESI positive-ion experiment, where an m/z of 416.1519 was observed (expected 416.1525 for [M+] for C28H20N2O2). Gradually removing flexibility by adding two sp3 bridges, but maintaining the parallel orientation (Variant I) and, as documented in the crystal structure, the lack of C–H⋯O interaction in 1b, also leads to the intramolecular mode of the reaction, giving 3b with 68% yield (Scheme 1, path e) and the saturated C2 bridge (observed 521.2199 for [M + Na]+, expected 521.2205 for [M + Na]+ for C34H30N2O2 + Na). Reduction of the double bond formed under McMurry conditions has been previously reported.7a,14 According to our initial objectives, while subjecting 1c (Variant II) to McMurry conditions an intermolecular mode of reaction accompanied by an intramolecular one can be expected (Scheme 1, path d or f) if the geometrical factors are correctly identified. In addition, we could expect significant support from the decreased flexibility of the reagent. While testing the mixture obtained after classic work-up, two species were observed in the MS spectrum and eventually assigned to 3c (m/z 412.1211, expected 412.1212 [M]+ for C28H16N2O2) and 4 (m/z 824.2573, expected 824.2424 [M]+ for C56H32N4O4). The abundance of both components can be modulated by switching between different solvents: the THF-mediated process gives 40% of 3c and 45% of 4, whereas using dioxane substantially decreases the yield of 4 (24%) while maintaining the same efficiency for the formation of 3c (39%). The decreased yield of 4 on switching between solvents suggests thermodynamic control of a transition state that shows decreased stability at higher temperature, as the yield of 3c does not change significantly.
The X-ray analysis performed for 3a, 3b and 3c unambiguously confirmed all the anticipated structures (Fig. 2D–F), also showing unsaturated (1.355 Å) and saturated (1.552 Å) C2 bridges. The main motifs of 3a and 3b are strongly ruffled from planarity, confirming the expected flexibility. 3a shows a tilt of the m-phenylene that is forced by the efficiency of the p–p overlap between the furan units and the C2 bridge. In contrast to this, 3b adopts a geometry with a strongly disturbed orientation of the heterocyclic subunits caused by the dynamics of a single bond within the C2 bridge.
The bond length analyses in both macrocycles show the maintained character of each subunit incorporated into the final system, consistent with the limited π-electron global delocalisation. The presence of two rigid motifs in 3c leads to a strained structure (Fig. 2F), maintaining planarity within the 5,7-dihydroindolo[2,3-b]carbazole unit and, independently, between the two furans and the C2 bridge. The X-ray analysis for 4 (Fig. 3) confirmed an intermolecular variant of the observed reactivity, as anticipated by the initial geometry of the starting material. In contrast to our previous observations (stabilization of a trans,trans-isomer),7 we have isolated a cis-geometry for the formed double bond bridge. The formation of a cis,cis-isomer suggests a dissimilar transition state with respect to the previously reported intramolecular variant of reactivity.7 In addition, the solid-state analysis showed a condensed/compact geometry, where both 5,7-dihydroindolo[2,3-b]carbazole units are located over each other, with a distance of 3.62 Å. The separation of these subunits suggests that a stabilizing influence is the additional factor responsible for this. The crystal packing (Fig. 2) leads to further stabilization of the cis-isomer in the closed variant through proximity of the furans (∼3.4 Å).
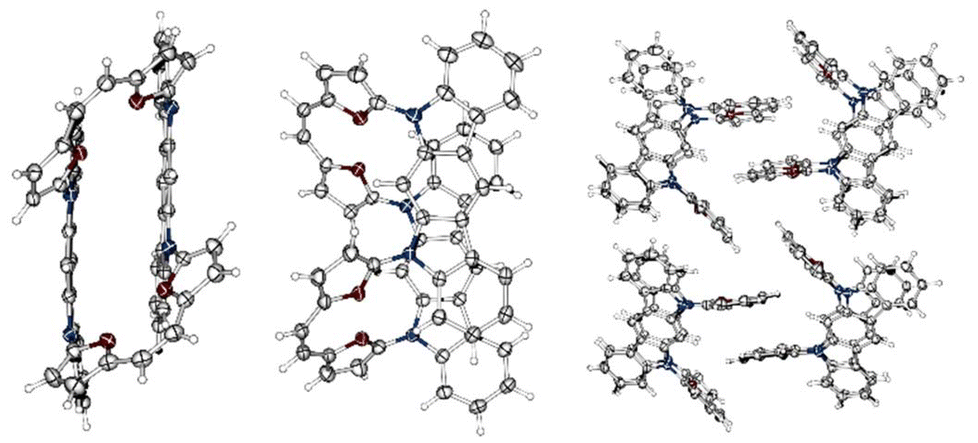 | ||
| Fig. 3 X-ray structures for 4. Thermal ellipsoids represent 50% probability. | ||
Spectroscopic analysis
The spectroscopic analysis, based on a combination of different techniques, showed a picture that was consistent with the solid-state data. The electronic spectra (Fig. 4) showed a transition at λ = 416 nm (3a), λ = 271 nm (3b), λ = 429 nm (3c) and λ = 357 nm (4) as the most bathochromically shifted band(s). The gradual bathochromic shift can be correlated with the increasing number of π-electrons remaining in the limited conjugation, which is least marked for 3b, because of the presence of an sp3–sp3 linker. The dimeric system 4 shows a very efficient emission behaviour, with λem = 459 nm and with a quantum yield of Φ = 0.8 and a lifetime of τ1 = 3.27 ns. By contrast, both conjugated monomeric derivatives 3a and 3c showed substantially lower efficiency of Φ = 0.08 (λem = 479 nm) and Φ = 0.03 (λem = 498 nm), respectively. The time-dependent measurements showed the lifetimes of the excited states as τ1 = 4.40 ns and τ1 = 5.38 ns, respectively.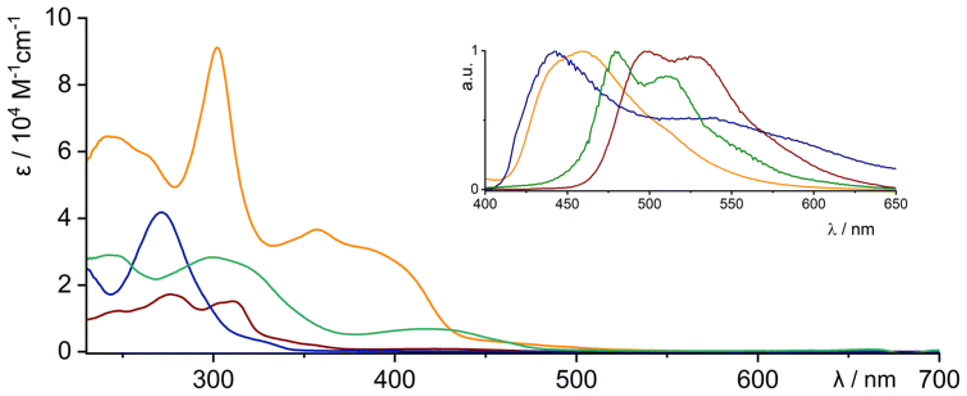 | ||
| Fig. 4 UV-visible experiments for 3a (olive), 3b (navy), 3c (wine) and 4 (orange) systems (conditions: dichloromethane, 295 K). Inset presents emission spectra in the same colour scheme. | ||
The observed hypsochromic shift of absorption and emission in 4 with respect to 3c can be correlated with the modulated conjugation, which has a dissimilar character because of 3D and 2D organisation of the chromophore, respectively. The more rigid geometry in 4, which decreases the strain observed in 3c (Fig. 2F), is potentially also responsible for the significantly higher emission efficiency.
The total number of π-electrons involved in each of the obtained cyclic derivative is 24, for 3a and 3c, while 4 contains 36 (or 48 if one includes both fused C6 rings) π-electrons and follows the 4n rule characteristic of paratropic systems. The 1H NMR spectra for the monomeric systems (3a) (Fig. S5†) and (3c) (Fig. S24†), and the dimeric 4, showed a set of resonances in regions characteristic for each subunit and consistent with the dominance of local effects of delocalisation according to magnetic criterion.15 This behaviour is consistent with the dominating contribution of local diatropic components – carbocyclic or heterocyclic – stabilizing the molecule, with negligible global paratropic current.7 The 1H NMR spectroscopic analyses performed for all the macrocyclic derivatives consistently support the limited global conjugation anticipated from the UV-vis experiments, and support the local effects of conjugation. Due to the shorter C–H⋯O distance following cyclization, the 1H chemical shift of the inside phenyl proton in 1a (7.09 ppm) and 1c (8.44 ppm) moves to 7.92 and 9.42 ppm for 3a and 3c, respectively. The 1H chemical shift ranges for all three monomeric derivatives were recorded in the region characteristic of each subunit incorporated into the final skeleton, with furan resonances at δ = 5.39–6.40 ppm and carbocyclic subunits recorded at δ = 6.00–9.00 ppm. In contrast to 3a and 3c (δ = 5.78 and 5.76 ppm, respectively), the saturated C2 bridge of 3b was observed at δ = 3.10 ppm, consistent with the sp3 hybridized linker. The 1H NMR spectrum recorded for 4 (Fig. 5) showed a number of resonances consistent with the high symmetry of the final derivative, which can be correlated with the geometry observed in the solid state.
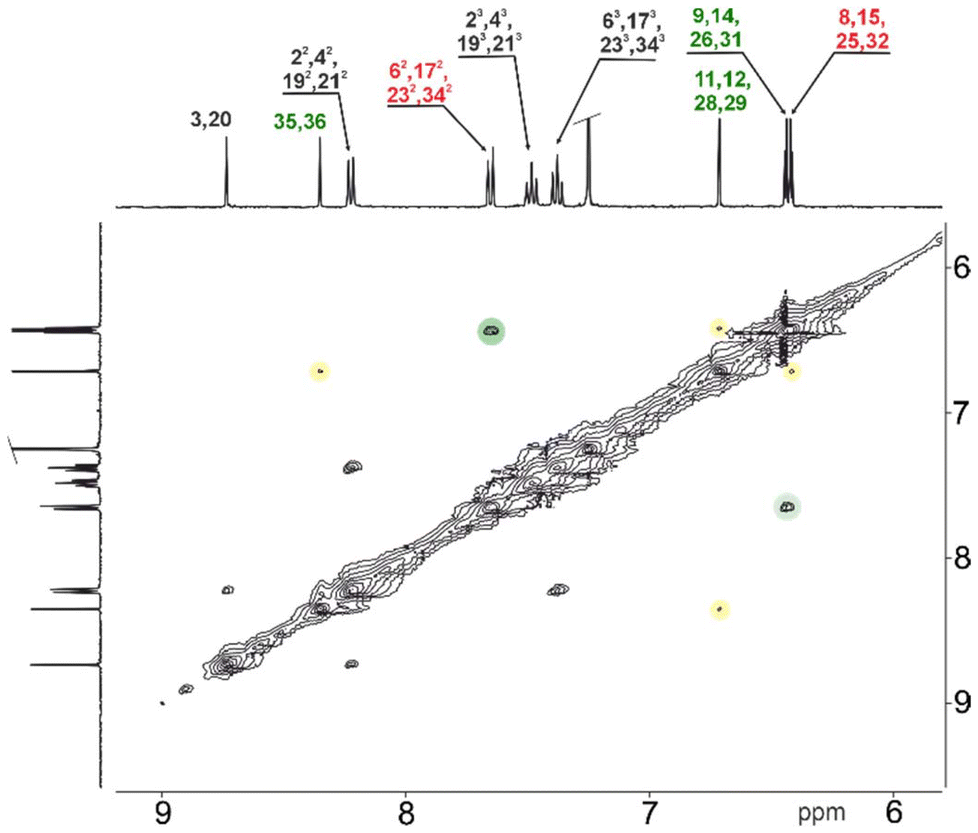 | ||
| Fig. 5 1H–1H NOESY for 4 (400 MHz, CDCl3, 300 K). | ||
Surprisingly, the observed NOESY map (Fig. 5) does not correspond with the expected set of signals for the closed variant of through-space contacts (Scheme 2), but correlates with the planar conformation. Depending on the mutual separation, different intensities of the NOE effect are expected and correlate with the distance. The crucial interactions have been analysed and diagnosed as the correlation between the C2 bridge (H(11/12)) and the central arene (H(35,36)) and β-furan resonance(s) (H(8,15) or H(9,14)) (Scheme 2). Both weak contacts, i.e. H(11)⋯H(35) and H(11)⋯H(9), present on the NOESY map (Fig. 4), do not correlate with the presence of the 4-closed variant in solution. The X-ray analysis of 4-closed (Fig. 3) shows that a strong H(9)⋯H(11) contact should be expected, as the recorded distance does not exceed 2.7 Å. On the other hand, the separation of H(11)⋯H(35) exceeds 5.5 Å and drastically reduces the NOESY contact. These observations suggest the possibility of switching from one geometry to another (open/closed → closed/open) under specific conditions, i.e. while forcing solid-state packing (Fig. 3) with the open variant kept in solution.
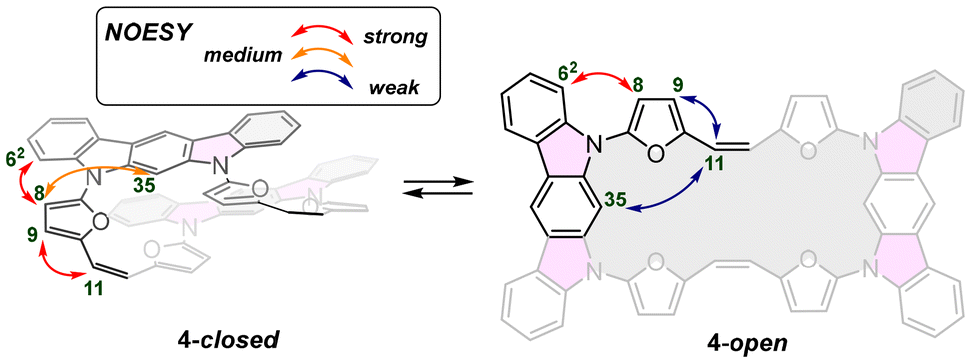 | ||
| Scheme 2 Expected NOE contacts for 4 in closed and open variant. | ||
Reactivity/redox properties
Redox modulation as a fundamental approach for switching between diatropic and paratropic systems has been explored for the formation of neutral, cationic and anionic systems with diatropic or paratropic current(s),16 but also for inducing an open-shell character.17 The electrochemistry experiments performed for all the analysed derivatives (Fig. S50†) showed a dissimilar picture of multistep oxidation. The recorded differences in oxidation potentials can be correlated with the imprinted degree of conjugation, and four reversible waves for 3a, in the sequence 3a → 3˙+ → 3a2+ → 3a3˙+ → 3a4+, which reduce to two waves for 3c (3c → 3c˙+ → 3c2+), were observed (Fig. S50†). The reduced number of oxidations when passing from 3a → 3c correlates nicely with the increased degree of conjugation, allowing efficient delocalisation of the introduced positive charge(s) unavailable for 3a. The significantly decreased degree of conjugation expected for 3b results in a substantially different oxidation potential recorded at 0.87 V, which correlates with the local effects of π-conjugation recorded within each subunit. In contrast to all the monomeric derivatives, 4 shows a broad, quasi-reversible wave at 0.38 V. This behaviour can be correlated with switching between open and closed orientations of the whole molecule, in addition to being activated by oxidation. All systems lack reductions over the whole potential range tested (−1.5 to 0 V; see ESI†).All monomeric derivatives 3a and 3c titrated with NO+SbF6− and monitored by UV-vis absorption experiments showed dissimilar characters while forming positively charged systems (Scheme 2). The oxidation was expected to be a step-by-step process through a cation radical, eventually leading to a double-charged system. The UV-vis monitored experiments for the derivatives (Fig. S40 (3a) and Fig. S42 (3c)† performed at 280 K) showed the oxidation occurring in two-steps, with radical cation(s) in the first step, as documented by low-energy transitions at λ = 1068 nm and λ = 760 nm for 3a and 3c, respectively. In both processes the long wave transitions gradually disappear, shifting hypsochromically, and eventually appearing at λ = 564 nm for 3a and λ = 620 nm for 3c.
As documented experimentally, the 3a → 3˙+ → 3a2+ conversion path goes through a set of isosbestic points, while 3c → 3c˙+ → 3c2+ shows more complex behaviour. This can be correlated with the picture recorded by electrochemistry, with fully reversible oxidations observed for 3a and quasi-reversibility recorded for 3c. A difference in the lowest energy transition location correlates with the potential difference in the observed efficiency of the global conjugation formed after oxidation.7,18 Significantly different behaviour was observed for 3b (Fig. S41†). The observations show the appearance of lower energetic transitions at λ = ∼500 nm, which do not change with further addition of the oxidizing agent (up to 5 eq.). This behaviour can be correlated with a lack of extended conjugation, as secured by the presence of the unsaturated C2 bridge, which is absent in 3b, and which also substantially changes the redox potentials. The UV-vis-near-infrared (NIR) monitored experiment, performed on 4 (Fig. S44†), showed a single transformation that correlated with the electrochemical experiment, and lacked a sharp boarder between the 1st and 2nd oxidation. The recorded transitions reached 1500 nm, fitting within the NIR-II therapeutic window.19
Thus, all analysed derivatives showed redox flexibility with the expected potential for stabilization of the charged systems able to show a different range of delocalisation. In an attempt to better understand the efficiency of the induced changes recorded by UV-vis titration, we repeated these experiments while monitoring with 1H NMR spectroscopy. The oxidation of 3a showed a multistep process that correlated with the number of equivalents of oxidant added. The first equivalent of NO+SbF6− resulted in a disappearance in the spectrum consistent with formation of the cation radical (3˙+), and evolving to a perfectly shaped proton NMR spectrum for 3a2+ after addition of the second equivalent (Fig. S33†). 3c, subjected to the same experiment (Fig. 6), showed a similar transformation sequence, forming a doubly charged system 3c2+ obtained via3c˙+ (a cation radical). Comparing the 1H NMR spectra for both dicationic derivatives, significant dissimilarity eventually correlating with the efficiency of the global conjugation could be observed. A specific construction of 3a limits the global conjugation expected for 3a2+ (Fig. S5 (3a) and Fig. S33 (3a2+)†), while 3c2+ gave a strongly downfield-shifted resonance (Δδ ∼ +2.5 ppm) consistent with very efficient global conjugation (Fig. 6).
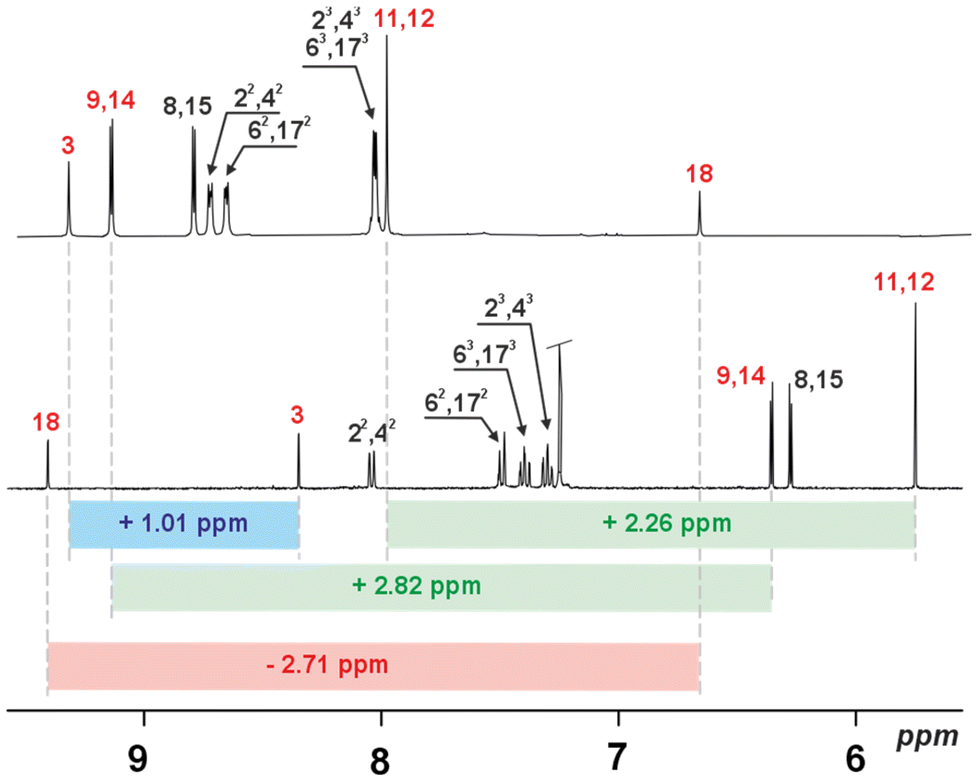 | ||
| Fig. 6 1H NMR spectra of 3c (bottom, CDCl3, 298 K) and 3c2+ (top, CD3CN, 233 K). | ||
At the same time, the internal signal of the CH group has been recorded at δ = 6.73 ppm (Δδ ∼ −3 ppm, Fig. 6, top), which can be correlated with the presence of two competing effects of opposite influence: global conjugation and C(H)⋯O electrostatic interaction.7 In the next step, 4 was subjected to oxidation under the same conditions. In contrast to the monomeric experiments, the proton NMR spectra recorded for the potential sequence of 4 → 4˙+ → 42+ remained silent over the whole range of added oxidant (up to 30 eq.). This correlates with the electrochemical behaviour, where lack of a sharp gap was recorded, which is consistent with a non-discrete process of oxidation. More importantly, lack of a 1H NMR spectrum suggests the constant presence of unpaired electrons in the final molecule. Thus, 4 was subjected to oxidation monitored using EPR spectroscopy, which proved the presence of the open-shell character of 4. The variable temperature experiments (VT, 100 K–200 K) (Fig. 7) showed a constant increase in the intensity of the signal (g = 2.0025 ± 0.0005) with decrease in temperature, following the Curie–Weiss law, which is in opposition to stabilization of the diradicaloid character, where the open-shell singlet state is highly populated at elevated temperature.20 In addition we did not record any signal in the half-field region. Thus, the performed oxidation is reduced to a single-electron process stabilizing 4˙+, and the radical character is entrapped in the flexible skeleton, which dynamically reacts to the oxidation process and leads to a folded geometry.
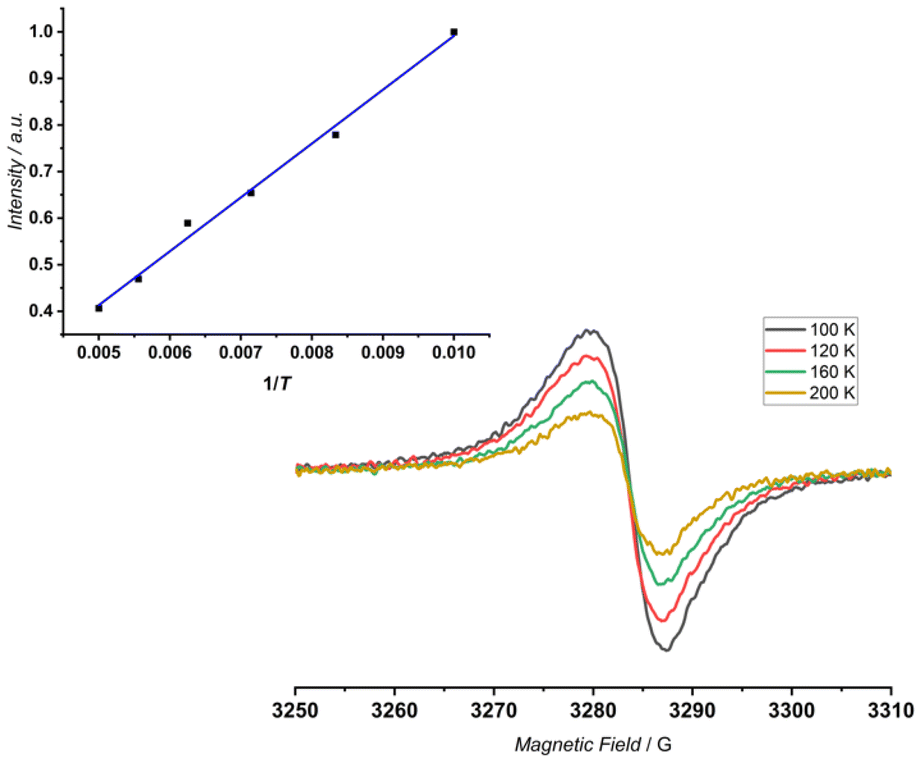 | ||
| Fig. 7 X-band EPR spectra registered at the indicated temperatures: EPR signal intensity vs. 1/T. | ||
Theoretical analysis
To obtain a deeper insight into the observed behaviour we subjected all four derivatives (in neutral and cationic forms) to theoretical analyses using the B3LYP/6-31G(d,p) approach, with chloroform as the solvent (PCM model). All fully optimised geometries were analysed with GIAO, NICS (0)zz21 and AICD22 approaches. In addition, open-shell calculations were made for the 4˙+ system to establish the spin distribution. For all fully optimised geometries of 3a–3c the predicted chemical shifts with the GIAO approach showed a fair correlation to the experimental data, supporting the previous conclusions (Table S2†). Analysis of the changes in current recorded consistently showed the expected correlations with the NMR spectroscopic observations. The AICD visualisation (Fig. S60†) showed local effects for 3a–3c that correlated with the spectroscopic observations.The same consistency between theory and experimental data was recorded for oxidized 3(a–c)2+ forms, eventually leading to the very efficient global conjugation path documented in 3c2+ (Fig. S60†), which is lacking in 3a2+ and 3b2+. The inefficient 4 → 42+ oxidation and formation of an open-shell system (4˙+) comprehensively corresponds with the flexibility of the organic skeleton and the observed differences in geometries recorded in the solid state and in solution. As reported earlier, defects of the neutral, unsaturated system usually lead to a significant increase in the conjugation path, either diatropic or paratropic, which is eventually regarded as a driving force for oxidation.7,23 This adequately correlates with the increased efficiency of the diatropic current in the 3c/3c2+ couple. Nevertheless, the oxidation of 4, while fully performed on the open variant, is expected to form a strongly delocalised derivative stabilized by a diatropic current (see ESI, Fig. S58†). This creates significant problems while switching to the 3D spatial arrangement, as in that observed for 4-closed, which is expected to form a non-obvious conjugation path. As documented in solution, the presence of the solvent stabilizes the open structure, which converts to the closed structure in the solid state. Optimisation of both geometries showed an energy difference of ∼3.7 kcal mol−1 in preference for the closed system (Table 1). Nevertheless, conformational analysis of the 4-closed → 4-open transition showed a conversion barrier reaching ∼10 kcal mol−1 (Fig. 8 and Fig. S61†).
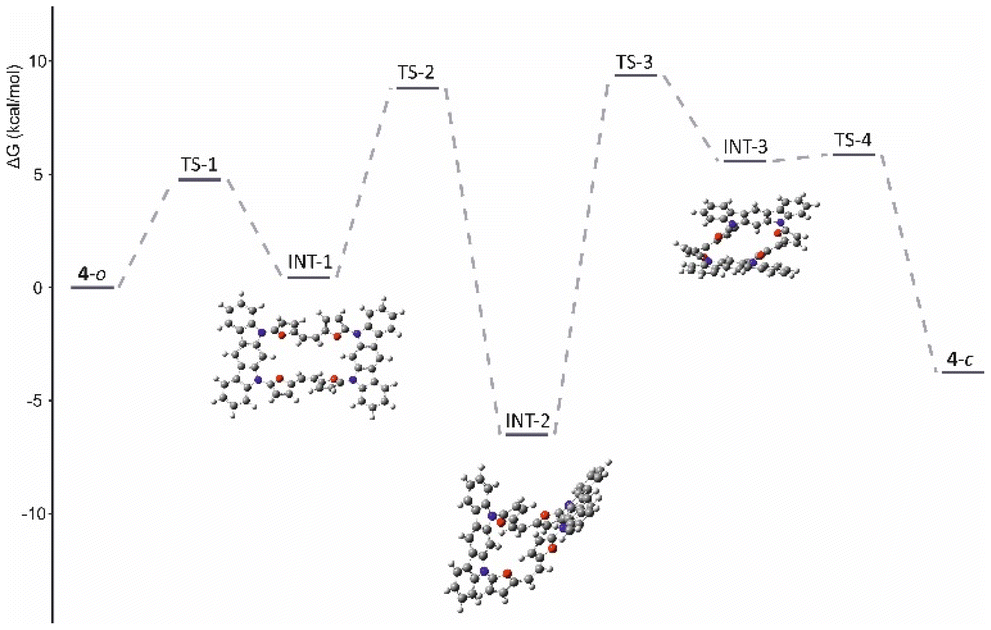 | ||
| Fig. 8 The 4-open → 4-closed folding energy diagram. | ||
| Neutral | Cation radical | |
|---|---|---|
| Open (4-o) | −2673.181394 | −2673.003112 |
| Closed (4-c) | −2673.187377 | −2673.014560 |
| ΔE (Hartree) | 0.005983 | 0.011448 |
| ΔE (kcal mol−1) | 3.75 | 7.18 |
The oxidized 4˙+ significantly increases the stabilization of the 4-closed variant to ∼7.2 kcal mol−1 with respect to 4-open, supporting the possibility of the introduction of a special stress to the molecule by oxidation, which leads to molecular contraction introducing spatial confinements and increasing the observed stability. This relies on the proximity of either furan subunits or the indolocarbazole motif, which can lead to separation of a hole and an electron within the final, closed geometry of 4. The spin-density analysis for both considered geometries, 4˙+-open/4˙+-closed (Fig. 9B), showed a significant difference in distribution over the whole system.
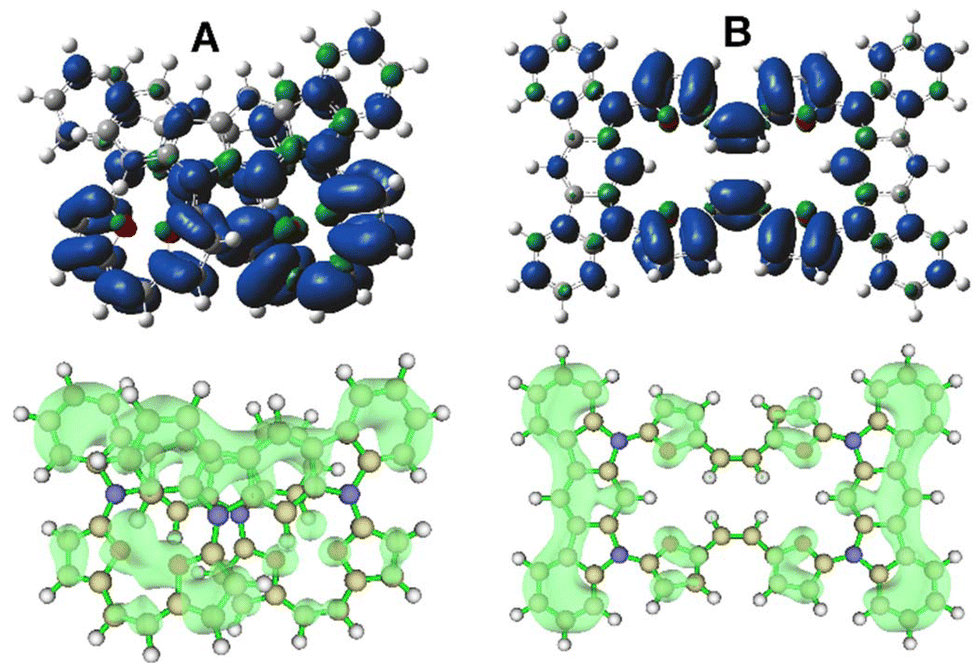 | ||
| Fig. 9 Spin distribution (top, isovalue 0.004) and charge distribution (bottom, isovalue 0.15) of 4˙+ (closed (A) vs. open (B) variant). | ||
The planar geometry of 4˙+-open leads to a fully symmetrical distribution of spin, with substantial location of electron density on the furan units and C2 bridge, and symmetrically distributed over both sides of the macrocyclic system. In contrast to this, 4˙+-closed shows a significant asymmetry in spin distribution (Fig. 9A), which is unequally located on both furan arms. This suggests a potential additional factor influencing the increased stability within the observed system. Spatial constraints documented in the crystal structure and DFT model proximity of the π-extended subunits leads to additional factors responsible for stabilization, which can be deduced from the distribution of positive charge (Fig. 9A, bottom).24 This fills the space unoccupied by electron density and eventually introduces a potential electrostatic interaction between positive and negative zones present in the system, which is impossible in the fully planar 4˙+-open system. Thus, the 4˙+-closed system unequally distributes both spin and positive charge, introducing zones of additional interaction that stabilize the cation radical stage and prevent the system from undergoing further oxidation.
Conclusions
We report on a precisely planned and executed synthetic approach allowing controlled formation of a dimeric system from predefined and easily achievable substrate(s). The detailed analysis of geometric factors imprinted into the initial reagents allows a strict prediction of the quality of the formed product. All the obtained systems showed emission behaviour, with very efficient fluorescence recorded for the dimeric system. All the derivatives maintain the local effect of conjugation, as recorded during the spectroscopic analyses. The redox switching conducted for fully unsaturated systems showed the presence of a global diatropic current, with different efficiencies recorded for monomeric derivatives. The double sized system was also oxidized, eventually stabilizing the open-shell character of a cation radical, which was further analysed experimentally and theoretically. The detailed analysis showed a specific response of the dynamic dimeric system to the oxidation activator, resulting in spatial contraction of the skeleton and leading to a closed geometry. The theoretical analysis conducted for the cation radical showed that the behaviour was consistent with the observed stability and preference in holding the oxidation process at the single-electron step.Author contributions
Conceptualization: MP, AD; methodology: AD, KD, MF, ŁO, KK, MK, MP. Investigation: AD, KD, KK, ŁO, MF, MK. Visualization: MP, AD. Supervision: MP. Writing – original draft: MP. Writing – review & editing: AD, KD, MF, ŁO, KK, MK, MP.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for 1a, 1b, 1c, 3a, 3b, 3c and 4 have been deposited at the CCDC under 2373295–2373301† and can be obtained from: 1a (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373297&Author=Arnab+Dutta), 1b (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373298&Author=Arnab+Dutta), 1c (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373295&Author=Arnab+Dutta), 3a (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373299&Author=Arnab+Dutta), 3b (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373300&Author=Arnab+Dutta), 3c (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373296&Author=Arnab+Dutta), 4 (https://www.ccdc.cam.ac.uk/structures/Search?access=referee&ccdc=2373301&Author=Arnab+Dutta).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Centre, Poland for financial support of the research conducted in project no. 2019/35/B/ST4/00318 (MP) and 2019/35/N/ST4/00393 (MF, synthesis and characterisation of m-phenylenediamine derivatives 1a–3a). We thank Dr Miłosz Siczek for the X-ray measurements. The Wrocław Supercomputer Centre (KDM WCSS) is kindly acknowledged for sharing computation resources necessary for DFT calculations. The study was carried out using the research infrastructure co-funded by the European Union in the framework of the Smart Growth Operational Program, Measure 4.2; grant no. POIR.04.02.00-00-D001/20, “ATOMIN 2.0 – ATOMic scale science for the INnovative economy”.References
- T. He, K. Geng and D. Jiang, All sp2 carbon covalent organic frameworks, Trends Chem., 2021, 3, 431–444 CrossRef CAS.
- (a) W. Stawski, Y. Zhu, I. Rončević, Z. Wei, M. A. Petrukhina and H. L. Anderson, The anti-aromatic dianion and aromatic tetraanion of [18]annulene, Nat. Chem., 2024, 16, 998–1002 CrossRef CAS PubMed; (b) Z. Li, X. Hou, Y. Han, W. Fan, Y. Ni, Q. Zhou, J. Zhu, S. Wu, K.-W. Huang and J. Wu, [8]Cyclo-para-phenylmethine as a Super-Cyclooctatetraene: Dynamic Behavior, Global Aromaticity, and Open-Shell Diradical Character inthe Neutral and Dicationic States, Angew. Chem., Int. Ed., 2022, 61, e202210697 CrossRef CAS; (c) S. Wu, Y. Ni, Y. Han, X. Hou, C. Wang, W. Hu and J. Wu, Hückel- and Baird-Type Global Aromaticity in a 3D Fully Conjugated Molecular Cage, Angew. Chem., Int. Ed., 2022, 61, e202115571 CrossRef CAS PubMed; (d) L. Ren, Y. Han, X. Hou, Y. Ni, Y. Zou, T. Jiao and J. Wu, Aromaticity in Fully π-Conjugated Multicyclic Macrocycles, J. Am. Chem. Soc., 2023, 145, 12398–12406 CrossRef CAS PubMed.
- (a) P. Banachowicz, M. Das, K. Kruczała, M. Siczek, Z. Sojka, M. Kijewska and M. Pawlicki, Breaking Global Diatropic Current to Tame Diradicaloid Character: Thiele's Hydrocarbon Under Macrocyclic Constraints, Angew. Chem., Int. Ed., 2024, 63, e202400780 CrossRef CAS PubMed; (b) Y. Ni, M. E. Sandoval-Salinas, T. Tanaka, H. Pan, T. S. Herng, T. Y. Gopalakrishna, J. Ding, A. Osuka, D. Casanova and J. Wu, [n]Cyclo-para-biphenylmethine Polyradicaloids: [n]Annulene Analogs and Unusual Valence Tautomerization, Chem, 2019, 5, 108–121 CrossRef CAS.
- (a) N. Sprutta, M. Siczek, L. Latos-Grażyński, M. Pawlicki, L. Szterenberg and T. Lis, Dioxadiazuliporphyrin: A Near-IR Redox Switchable Chromophore, J. Org. Chem., 2007, 72, 9501–9509 CrossRef CAS.
- (a) W. Fan, T. M. Fukunaga, S. Wu, Y. Han, Q. Zhou, J. Wang, Z. Li, X. Hou, H. Wei, Y. Ni, H. Isobe and J. Wu, Synthesis and chiral resolution of a triply twisted Möbius carbon nanobelt, Nat. Synth., 2023, 2, 880–887 CrossRef; (b) M. Krzeszewski, H. Ito and K. Itami, Infinitene: A Helically Twisted Figure-Eight [12]Circulene Topoisomer, J. Am. Chem. Soc., 2022, 144, 862–871 CrossRef CAS.
- A. Dutta, W. Stawski, M. Kijewska and M. Pawlicki, Edge Decoration of Anthracene Switches Global Diatropic Current That Controls the Acene Reactivity, Org. Lett., 2021, 23, 9436–9440 CrossRef CAS PubMed.
- (a) J. Klain, W. Stawski, P. J. Chmielewski, J. Cybińska and M. Pawlicki, A route to a cyclobutane-linked double-looped system via a helical macrocycle, Chem. Commun., 2019, 55, 4558–4561 RSC; (b) K. Wypych, M. Dimitrova, D. Sundholm and M. Pawlicki, Diagnosing Ring Current(s) in Figure-Eight Skeletons: A 3D Through-Space Conjugation in the Two-Loops Crossing, Org. Lett., 2022, 24, 4876–4880 CrossRef CAS PubMed.
- (a) E. Vogel, N. Jux, J. Dorr, T. Pelster, T. Berg, H.-S. Bohm, F. Behrens, J. Lex, D. Bremm and G. Hohlneicher, Furan-Based Porphyrins:Tetraoxa[4n+2]porphyrin Dications with 18π-,22π-, or 26π-Electron Systems, Angew. Chem., Int. Ed., 2000, 39, 1101–1105 CrossRef CAS; (b) G. Märkl, J. Stiegler and P. Kreitmeier, Tetraepoxy[32]annulene(4.4.4.4) and tetraoxa-[30]porphyrin(4.4.4.4) dications, Helv. Chim. Acta, 2001, 84, 2022–2036 CrossRef; (c) T. Y. Gopalakrishna and V. G. Anand, Reversible Redox Reaction Between Antiaromatic and Aromatic States of 32p-Expanded Isophlorins, Angew. Chem., Int. Ed., 2014, 53, 6679–6682 Search PubMed.
- P. Riuz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C−N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef.
- (a) M. Pawlicki, K. Hurej, K. Kwiecińska, L. Szterenberg and L. Latos-Grażyński, A fused meso-aminoporphyrin: a switchable near-IR chromophore, Chem. Commun., 2015, 51, 11362–11365 RSC; (b) K. Hurej, W. Stawski, L. Latos-Grażyński and M. Pawlicki, meso-N-Pyrrole as a Versatile Substituent Influencing the Optical Properties of Porphyrin, Chem. – Asian J., 2016, 11, 3329–3333 CrossRef CAS PubMed; (c) K. Urbańska and M. Pawlicki, Porphyrin−Amino Acid Conjugates, J. Org. Chem., 2020, 85, 8196–8202 CrossRef PubMed; (d) A. Nowak-Król and D. T. Gryko, Oxidative Aromatic Coupling of meso-Arylamino-porphyrins, Org. Lett., 2013, 15, 5618–5621 CrossRef; (e) N. Fukui, W.-Y. Cha, S. Lee, S. Tokuji, D. Kim, H. Yorimitsu and A. Osuka, Oxidative Fusion Reactions of meso-(Diarylamino)porphyrins, Angew. Chem., Int. Ed., 2013, 52, 9728–9732 CrossRef CAS.
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Divergent Synthesis of Heteroatom-Centered 4,8,12-Triazatriangulenes, Angew. Chem., Int. Ed., 2017, 56, 5087–5090 CrossRef CAS.
- D. Chai, Y. Zou, Y. Xiang, X. Zeng, Z. Chen, S. Gong and C. Yang, Fused twin-acridine scaffolds as electron donors for thermally activated delayed fluorescence emitters: controllable TADF behavior by methyl substitution, Chem. Commun., 2019, 55, 15125–15128 RSC.
- (a) D. J. Sutor, The C-H⋯O Hydrogen Bond in Crystals, Nature, 1962, 195, 68–69 CrossRef CAS; (b) G. R. Desiraju, The C-H⋯O Hydrogen Bond: Structural Implications and Supramolecular Design, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS; (c) T. Steiner, C–H⋯O Hydrogen Bonding in Crystals, Crystallogr. Rev., 2003, 9, 177–228 CrossRef CAS.
- K. Matsumoto, H. Minami, T. Kawase and M. Oda, [2.2](2,7)-Fluorenophanediene, [2.2.2](2,7)-fluorenophanetriene, and their fluorenide ions, Org. Biomol. Chem., 2004, 2, 2323–2326 RSC.
- M. Pawlicki and L. Latos-Grażyński, Aromaticity Switching in Porphyrinoids, Chem. – Asian J., 2015, 10, 1438–1451 CrossRef CAS PubMed.
- (a) Y. Ni, T. Y. Gopalakrishna, H. Phan, T. Kim, T. S. Herng, Y. Han, T. Tao, J. Ding, D. Kim and J. Wu, 3D global aromaticity in a fully conjugated diradicaloid cage at different oxidation states, Nat. Chem., 2020, 12, 242–248 CrossRef CAS PubMed; (b) Y. Ni, T. Y. Gopalakrishna, S. Wu and J. Wu, A Stable All-Thiophene-Based Core-Modified [38]Octaphyrin Diradicaloid: Conformation and Aromaticity Switch at Different Oxidation States, Angew. Chem., Int. Ed., 2020, 59, 7414–7418 CrossRef CAS PubMed.
- (a) S. Mori, M. Akita, S. Suzuki, M. S. Asano, M. Murata, T. Akiyama, T. Matsumoto, C. Kitamura and S.-i. Kato, Open-shell singlet diradicaloid difluoreno[4,3b:3′,4′-d]furan and its radical cation and dianion, Chem. Commun., 2020, 56, 5881–5884 RSC; (b) N. Tabata, T. Uchino, C. Kitamura, K. Yoshizawa, Y. Shiota and S.-i. Kato, Site-selective radical reactions of kinetically stable open-shell singlet diradicaloid difluorenoheteroles with tributyltin hydride and azo-based radical initiators, Chem. Sci., 2023, 14, 5974–5982 RSC.
- (a) S. P. Panchal, S. C. Gadekar and V. G. Anand, Controlled Core-Modification of a Porphyrin into an Antiaromatic Isophlorin, Angew. Chem., Int. Ed., 2016, 55, 7797–7800 CrossRef CAS; (b) W. Haas, B. Knipp, M. Sicken, J. Lex and E. Vogel, Tetraoxaporphyrinogen (Tetraoxaquaterene): Oxidation to the Tetraoxaporphyrin Dication, Angew. Chem., Int. Ed. Engl., 1988, 27, 409–411 CrossRef; (c) K. Singh, A. Sharma, J. Zhang, W. Xu and D. Zhu, New sulfur bridged neutral annulenes. Structure, physical properties and applications in organic field-effect transistors, Chem. Commun., 2011, 47, 905–907 RSC.
- (a) K. Hurej, W. Oszczędna, E. Opas, S. J. Zalewski, M. Pawlicki, M. J. Białek, Ł. Orzeł and L. Latos-Grażyński, Bispalladium(II) Complexes of di-p-Pyrirubyrin Derivatives as Promising Near-Infrared Photoacoustic Dyes, Angew. Chem., Int. Ed., 2023, 62, e202303394 CrossRef CAS PubMed; (b) K. Shimomura, H. Kai, Y. Nakamura, Y. Hong, S. Mori, K. Miki, K. Ohe, Y. Notsuka, Y. Yamaoka, M. Ishida, D. Kim and H. Furuta, Bis-Metal Complexes of Doubly N-Confused Dioxohexaphyrins as Potential Near-Infrared-II Photoacoustic Dyes, J. Am. Chem. Soc., 2020, 142, 4429–4437 CrossRef CAS PubMed; (c) W. Jiang, L. Lin, P. Wu, H. Lin and J. Sui, Near–Infrared–II Nanomaterials for Activatable Photodiagnosis and Phototherapy, Chem. – Eur. J., 2024, 30, e202400816 CrossRef CAS PubMed.
- (a) S. M. Quintero, M. M. Haley, M. Kertesz and J. Casado, Polycyclic Hydrocarbons from [4n]Annulenes: Correlation versusHybridization Forces in the Formation of Diradicaloids, Angew. Chem., Int. Ed., 2022, 61, e202209138 CrossRef; (b) T. Xu, Y. Han, Z. Shen, X. Hou, Q. Jiang, W. Zeng, P. W. Ng and Ch. Chi, Antiaromatic Dicyclopenta[b,g]/[a,f ]naphthalene Isomers Showing an Open-Shell Singlet Ground State with Tunable Diradical Character, J. Am. Chem. Soc., 2021, 143, 20562–20568 CrossRef CAS PubMed.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simpleand Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef.
- R. Herges and D. Geuenich, Delocalization of Electrons in Molecules, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- (a) Y. Han, J. Zhu, S. Dong, Y. Eng, T. Tao, T. Y. Gopalakrishna and Ch. Chi, Bisazapentalene Dication: Global Aromaticity and Open-Shell Singlet Ground State, Org. Lett., 2023, 25, 3380–3385 CrossRef CAS PubMed; (b) M. D. Peeks, M. Jirásek, T. D. W. Claridge and H. L. Anderson, Global Aromaticity and Antiaromaticity in Porphyrin Nanoring Anions, Angew. Chem., Int. Ed., 2019, 58, 15717–15720 CrossRef CAS; (c) M. D. Peeks, T. D. W. Claridge and H. L. Anderson, Aromatic and antiaromatic ring currents in a molecular nanoring, Nature, 2017, 541, 200–203 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2373295–2373301. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01566e |
| This journal is © the Partner Organisations 2025 |