
Iridium-catalyzed asymmetric cascade allylation/lactonization of methyl salicylates: enantioselective construction of chiral benzodioxepinones†
Bendu
Pan
 a,
Yunru
Wu
a,
Yaqi
Zhang
a,
Xiaobo
He
a,
Long
Jiang
b and
Liqin
Qiu
*a
a,
Yunru
Wu
a,
Yaqi
Zhang
a,
Xiaobo
He
a,
Long
Jiang
b and
Liqin
Qiu
*a
aSchool of Chemistry, IGCME, Guangdong Provincial Key Laboratory of Chiral Molecules and Drug Discovery, The Key Laboratory of Low-Carbon Chemistry & Energy Conservation of Guangdong Province, Sun Yat-sen University, Guangzhou 510006, China
bInstrumental Analysis and Research Centre, Sun Yat-sen University, Guangzhou 510275, China
First published on 5th November 2024
Abstract
An efficient asymmetric cascade allylation/lactonization of methyl salicylates has been achieved. The utilization of chiral-bridged biphenyl phosphoramidite ligand L3 resulted in good yields (up to 85%) and enantioselectivity (up to 95% ee) for the construction of a wide range of chiral benzodioxepinones with tolerance to diverse substituents. This reaction is featured by low catalyst loading, commercially available substrates and a broad substrate scope. Control experiments indicate that a relay catalytic pathway and kinetic resolution of racemic VEC might occur. In this transformation, the chiral-bridged phosphoramidite ligand L3 shows some advantages in enantioselective control compared to its BINOL-derived counterpart.
Introduction
Lactones are fundamentally important organic molecules that constitute the core of plenty of natural products and many compounds with significant biological activities.1 Therefore, it is always an important research topic to develop simple, efficient and practical methods for the preparation of these cyclic compounds such as chiral benzodioxepinones. However, the scope and synthetic methods of these molecules obtained in previous studies are quite limited,2 for example, Dong1a and co-workers presented an enantioselective approach to benzodioxepinones via a Rh-catalyzed carbonyl hydroacylation strategy in 2008 (Scheme 1a). Zhou1e and colleagues in 2013 developed a silver carboxylate-promoted lactonization for the synthesis of benzodioxepinones. Nevertheless, these methods did not provide any substrates with substituents on the benzene ring, which greatly limited the scope of benzodioxepinones.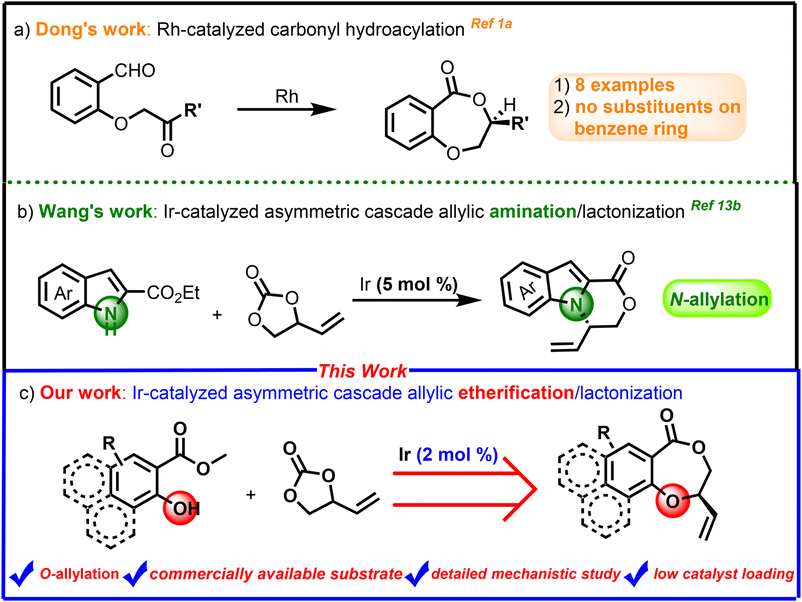 | ||
| Scheme 1 Cascade allylic etherification/lactonization strategy for the construction of chiral benzodioxepinones. | ||
Iridium-catalyzed asymmetric allyl substitution (AAS) reactions have achieved great development in the past decades, and it has evolved as a powerful tool for the construction of carbon–carbon and carbon–heteroatom bonds.3 Outstanding contribution has been made by Helmchen,4 Hartwig,5 Carreira6 and You3d,7 in the formations of oxygen-containing chiral molecules through asymmetric allylic etherification. Several kinds of intramolecular allylic etherification reactions have also been developed in our group using chiral-bridged biphenyl phosphoramidite ligands.8 On the other hand, vinylethylene carbonates (VECs) have emerged as promising building blocks for the synthesis of stereochemically and structurally complex compounds, as they can easily generate zwitterionic π-allyl metal intermediates promoted by transition-metal catalysts,9 which then lead to further diverse transformations to construct structurally divergent five-membered to medium-ring compounds.10,11 Recently, Cui12 and Wang’ groups13 (Scheme 1b) developed several efficient iridium-catalyzed cascade allylic amination/lactonization of VEC with isatoic anhydrides and indole 2-carboxylates, providing facile preparation of a variety of structurally important lactone compounds. However, to the best of our knowledge, the asymmetric version of cascade allylic etherification/lactonization still remains unknown. On the other hand, 5 mol% of Ir catalyst is often required to obtain good results in previous work, which makes the development of more efficient catalytic systems urgent. Further research on the catalytic pathway of such type of cascade reactions is also rare. In view of the potential application value of chiral benzodioxepinones and as an application extension of the chiral-bridged biphenyl phosphoramidite ligands developed by our group, we herein present our studies on Ir-catalyzed asymmetric cascade allylic etherification/lactonization between racemic VEC and various methyl salicylates, thus affording a class of chiral benzodioxepinone derivatives with moderate to good yields and high enantioselectivities (Scheme 1c).
Results and discussion
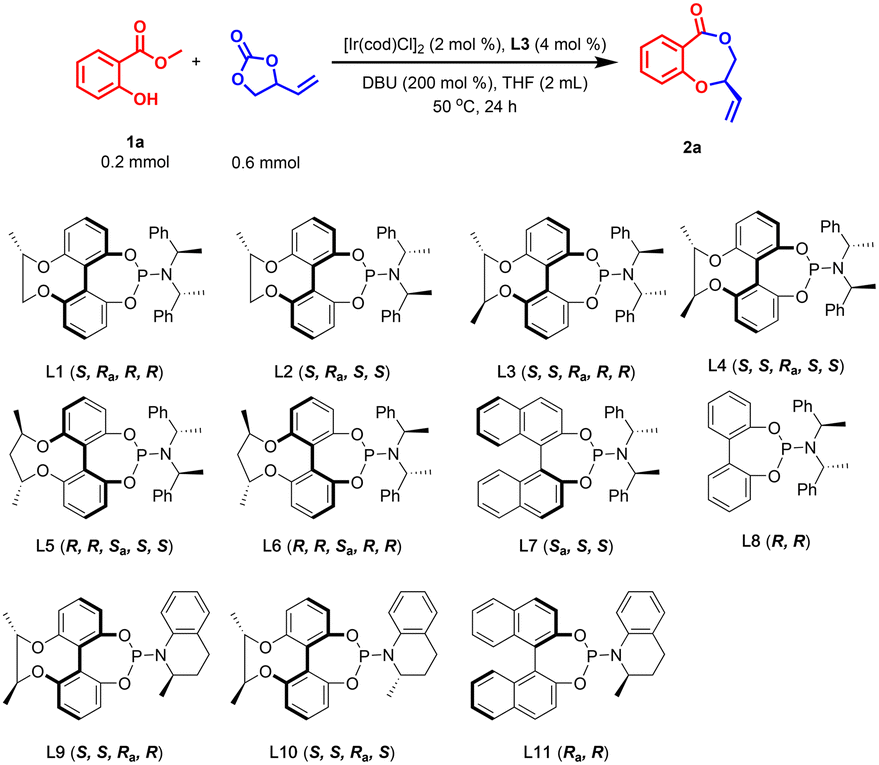
We focused our initial investigation on finding out effective phosphoramidite ligands for this asymmetric cascade allylic etherification/lactonization between racemic VEC and model substrate 1a. To our delight, treatment of 1a and VEC with an Ir catalyst derived from [Ir(cod)Cl]2 and chiral-bridged biphenyl phosphoramidite ligand L3, and DBU as base, in THF at 50 °C for 24 h gave target product 2a in 64% yield with 86% ee (Table 1, entry 1). A variety of other chiral phosphoramidite ligands were then evaluated carefully (Table 1, entries 2–11). Compared with L3, employment of chiral-bridged biphenyl phosphoramidite ligands with a shorter (L1) or longer (L5) chiral chain (Table 1, entries 2 and 5) would lead to similar yields but lower ee values. As shown in Table 1, the ee values of 2a dropped sharply when the diastereomers L2, L4 and L6 of the above ligands were used in this transformation (Table 1, entries 3, 4 and 6). As controls, BINOL-derived ligand L7 and bisphenol-based ligand L8 were also tested (Table 1, entries 7 and 8). However, lower yields and reduced enantioselectivities were observed, indicating that the chiral-bridged phosphoramidite ligand L3 shows some advantages in this reaction. Further screening demonstrates that ligands L9–L11 with chiral tetrahydroquinoline in the amine moiety are also not applicable to this reaction (Table 1, entries 9–11), just delivering 2a in moderate yields and poor ees. Considering the lower reaction yield, we further increased the [Ir(cod)Cl]2 loading to 4 mol% and 6 mol% (Table 1, entries 12 and 13), but the reaction results were still unsatisfactory, with slightly higher yields and moderate ees. Through replacement of DBU with some other bases such as Cs2CO3, K3PO4, DBN, Et3N and DABCO, the reaction efficiency was not improved too (see ESI Table S1† for details). Careful temperature screening proved that the optimal temperature for this reaction is 70 °C concerning both yield and enantioselectivity. These promising results encouraged us to further test different solvents such as PhMe, 1,4-dioxane, DCE, CH3CN, DME, CH3CH2OH, but THF is still the best choice (see ESI Table S1† for details). It was also found that the amount of DBU and VEC had little effect on the reaction results (Table 1, entries 16–18). Based on the improvement of the catalytic activity at 70 °C, we tried to reduce the catalyst loading to 1 mol% of Ir and 2 mol% of L3 (Table 1, entry 19) and the desired 2a was obtained in 82% yield and 90% ee. Substitution of substrate 1a with ethyl 2-hydroxybenzoate also did not provide a better result (Table 1, entry 21), but it was unexpectedly found that the amount of solvent THF was crucial for improving the ee value of 2a (Table 1, entries 22–24). As the dosage of THF increased from 0.5 mL to 5 mL, the ee value of product 2a was heightened sharply from 73% to 95%. This indicates the substrate concentration greatly affects the enantioselective control of the reaction. Further extending the reaction time to 36 h, product 2a was given with an acceptable yield and 95% ee (Table 1, entry 24). Taken together, the optimal conditions were ultimately identified: 1a (0.2 mmol), VEC (0.6 mmol), [Ir(cod)Cl]2 (1 mol%), L3 (2 mol%), and DBU (0.4 mmol) in THF (5 mL) at 70 °C for 36 h.|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | [Ir(cod)Cl]2 loading [mol %] | T [°C] | Yieldb [%] | eec [%] |
| a Conditions: [Ir(cod)Cl]2 (2 mol%), L3 (4 mol%), DBU (0.4 mmol), 1a (0.2 mmol), VEC (0.6 mmol), THF (2.0 mL), 50 °C. b Isolated yields. c Determined by HPLC analysis. d DBU (0.2 mmol). e VEC (0.4 mmol). f VEC (0.8 mmol). g Ethyl 2-hydroxybenzoate was used instead of 1a. h THF (0.5 mL). i THF (4.0 mL). j 36 hours reaction time. k THF (5.0 mL). | |||||
| 1 | L3 | 2 | 50 | 64 | 86 |
| 2 | L1 | 2 | 50 | 60 | 74 |
| 3 | L2 | 2 | 50 | 61 | 58 |
| 4 | L4 | 2 | 50 | 70 | 63 |
| 5 | L5 | 2 | 50 | 69 | −83 |
| 6 | L6 | 2 | 50 | 67 | −80 |
| 7 | L7 | 2 | 50 | 63 | −80 |
| 8 | L8 | 2 | 50 | 51 | 50 |
| 9 | L9 | 2 | 50 | 53 | 8 |
| 10 | L10 | 2 | 50 | 57 | 1 |
| 11 | L11 | 2 | 50 | 54 | 19 |
| 12 | L3 | 4 | 50 | 65 | 72 |
| 13 | L3 | 6 | 50 | 70 | 58 |
| 14 | L3 | 2 | 25 | 39 | 73 |
| 15 | L3 | 2 | 70 | 83 | 86 |
| 16d | L3 | 2 | 70 | 82 | 85 |
| 17e | L3 | 2 | 70 | 70 | 81 |
| 18f | L3 | 2 | 70 | 82 | 86 |
| 19 | L3 | 1 | 70 | 82 | 90 |
| 20 | L3 | 0.5 | 70 | 69 | 80 |
| 21g | L3 | 1 | 70 | 78 | 83 |
| 22h | L3 | 1 | 70 | 84 | 73 |
| 23i,j | L3 | 1 | 70 | 81 | 93 |
| 24j,k | L3 | 1 | 70 | 82 | 95 |
With the optimized reaction conditions in hand, the generality of this newly developed protocol was then evaluated with a variety of substituted methyl salicylates 1 (Scheme 2). Impressively, methyl salicylates with functional groups methyl- (1b) or methoxy-substituents (1c–1d) were well tolerated under optimized conditions, furnishing the corresponding chiral benzodioxepinones 2b–2d in moderate to high yields (52%–85%) with excellent enantioselectivities (90%–94% ees). Although slight enantioselectivity erosion was observed for substrates bearing halogens (1e–1j), the desired products 2e–2j were produced in acceptable yields (68%–83%) with good enantioselectivities (81%–85% ees). Through Suzuki coupling reaction, different substrates bearing aromatic substituents were prepared conveniently. Delightfully, the asymmetric cascade reactions also underwent smoothly without erosion of yield (63%–81%) or enantioselectivity (88%–93% ees) when various aromatic rings such as substituted phenyl (1k–1o, 1t, 1w), 1-naphthyl (1p), 2-thiophenyl (1q, 1x), 2-benzofuryl (1r) and 2-furyl (1s) were introduced into the benzene ring of methyl salicylate. Moreover, the corresponding reactions could still proceed (48%–84% yields, 83%–92% ees) even if the parent benzene ring of methyl salicylate was replaced by a naphthalene ring (1u–1v). Unfortunately, either 6-position substituted substrate 1A or 3-position substituted substrate 1B failed to yield the desired product perhaps due to the steric influence on the nucleophilic attack of the hydroxyl oxygen atom on the intermediate π-allyl iridium electrophile. 2C, 2D and 2E could not been obtained, either probably due to active proton competitition between their functional groups and the hydroxyl group. Replacing L3 with classical ligand L7 as a control, interestingly, both the ee values and yields of 2j and 2u descended. This further demonstrates the necessity and effectiveness of the chiral bridge to regulate the asymmetric catalytic performance of ligand. The configuration of 2p was successfully determined as R by single-crystal X-ray diffraction (CCDC: 2294598†).
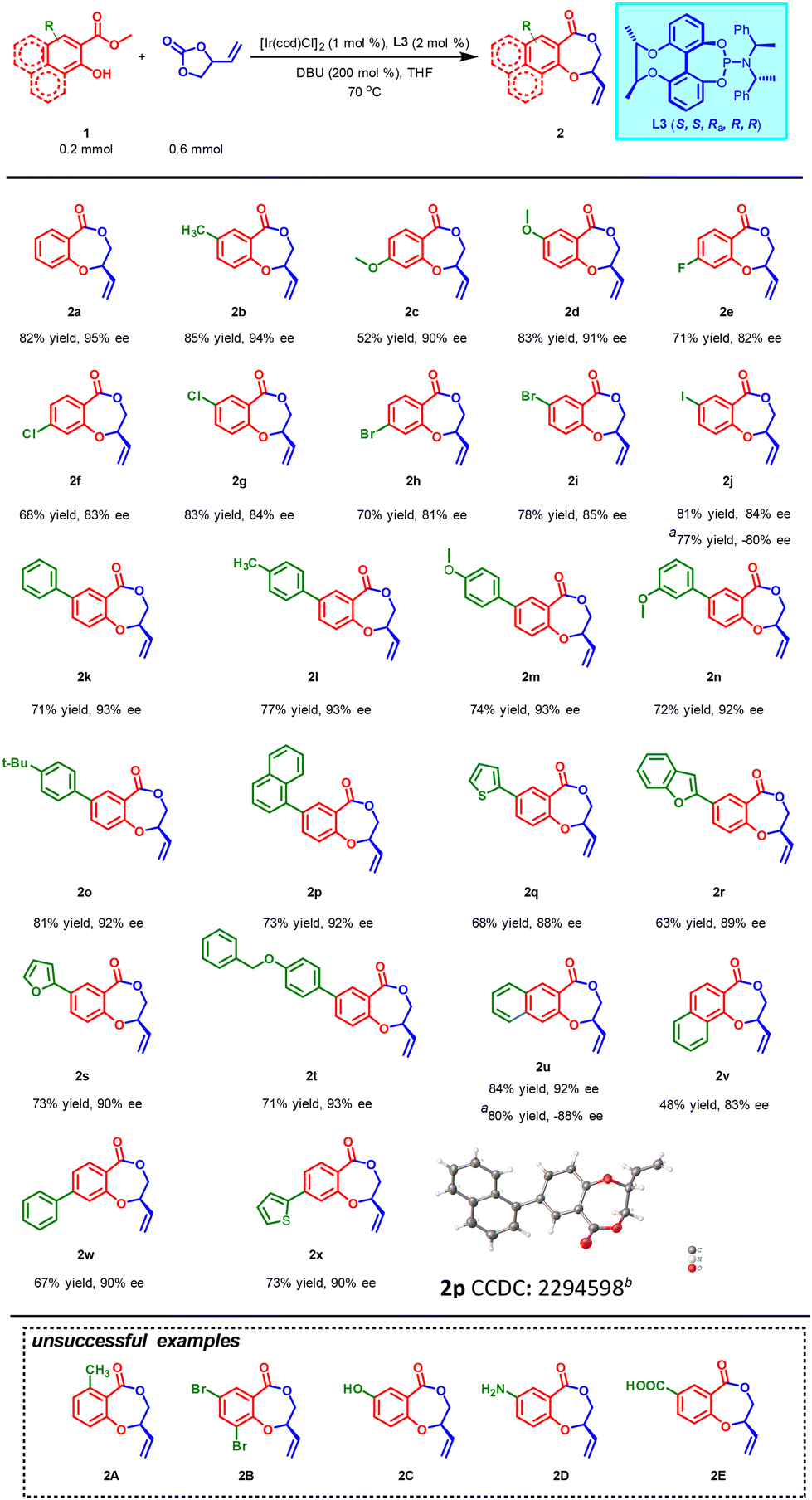 | ||
| Scheme 2 Substrate scope of methyl salicylates.a–c Conditions: a[Ir(cod)Cl]2 (1 mol%), L3 (2 mol%), DBU (0.4 mmol), 1a (0.2 mmol), VEC (0.6 mmol), THF (5.0 mL), 70 °C, 36 h. bIsolated yields. cDetermined by HPLC analysis. dL7 was used instead of L3. eProb = 50. | ||
To examine the synthetic utility of this protocol, a gram-scale reaction was performed with 1a and racemic VEC to obtain 2a in 70% yield and 94% ee (Scheme 3).
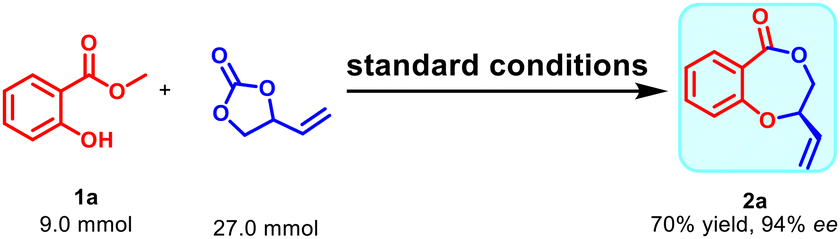 | ||
| Scheme 3 Gram-scale experiment. | ||
After completing the investigation on the substrate scope, a preliminary mechanistic study was carried out to explore the possible catalytic pathway (Scheme 4). Inspired by Wang's work13 of catalytic cascade allylation/macrolactonization, we wonder if our Ir-catalyzed asymmetric cascade allylic etherification/lactonization also went through a similar kinetic resolution. As shown in Scheme 4, interestingly, both the catalyst and substrate concentrations had a great impact on the ee value of product 2a (Scheme 4a), and an increase in the concentration of substrates or iridium-phosphoramidite ligand complex in the system would lead to a decrease in enantioselectivity of 2a in these control experiments. Therefore, it is speculated that the reaction rate of the more reactive (R)-VECs is already fast enough so that an increase in concentration will not have a significant impact on its reaction rate. On the contrary, the increase in concentration will sharply accelerate the reaction rate of the less reactive (S)-VECs, resulting in a drop in selectivity factor and difficulty in kinetic resolution, which in turn brings about a decrease in enantioselectivity.14 This is in line with the perception of kinetic resolution. Further investigation of the cascade reaction between 1a (0.2 mmol) and excess racemic VEC (0.6 mmol) provided 2a in 82% yield with 95% ee, accompanied by 40% recovery of VEC with 90% ee (Scheme 4b). These results indicate that the kinetic resolution of racemic VEC occurred in this transformation. Moreover, allylic etherification intermediate 2a′ could be isolated in 87% yield and 86% ee under condition A (Scheme 4c), and when intermediate 2a′ was exposed to condition B, the corresponding product 2a could also be obtained from a one-pot reaction (condition C) in 82% yield and 83% ee. Further control experiments showed that when intermediate 2a′ reacted under condition E (room temperature) and condition F (no DBU added), the corresponding product 2a could not be obtained. However, 2a could be conveniently given when intermediate 2a′ was exposed to condition D (without Ir complex). From the results obtained in the one-pot reaction (condition C) and the stepwise experiments, it can be concluded that the reaction proceeds through a relay catalytic pathway and DBU catalyzes the lactonization step at 70 °C.
 | ||
| Scheme 4 Mechanistic studies. | ||
Based on the experimental results of the above mechanistic studies and literature report,13 a plausible pathway was depicted in Scheme 5 to rationalize this Ir-catalyzed asymmetric cascade transformation. First, the coordination of racemic VEC with chiral Ir(I) catalyst initiates the formation of diastereomeric complexes [Ir(I)*]·(R)-VEC and [Ir(I)*]·(S)-VEC (Scheme 5). Because of the significant difference in diastereomeric reactivity, decarboxylative oxidative addition of [Ir(I)*]·(R)-VEC preferentially generates Ir(III)-π-allyl species A with configuration inversion, while (S)-VEC is recovered through the kinetic resolution process. However, according to the results of control experiments, high concentrations of catalyst and substrates would increase the reactivity of the less reactive [Ir(I)*] (S)-VEC, thereby naturally weakening the kinetic resolution ability. Subsequent nucleophilic attack of 1a in the presence of DBU produces anionic intermediate B, which finally undergoes lactonization to form the target product 2a and regenerates the catalyst for the next catalytic cycle.
 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In summary, for the first time, we have established an efficient Ir-catalyzed asymmetric cascade allylic etherification/lactonization using commercially available methyl salicylates, which allows for the convenient synthesis of a class of highly enantioenriched benzodioxepinones with a broad substrate scope in moderate to good yields. Low catalyst loading demonstrates the practicability of this newly developed strategy. The mechanistic investigations support that this reaction involves a relay catalytic pathway and kinetic resolution. Moreover, the chiral-bridged phosphoramidite ligand L3 shows some advantages in enantioselective control compared to BINOL-derived counterpart in this transformation. Given the potential pharmacological activity of these multiple substituted chiral benzodioxepinones prepared, we expect that the target molecules will find applications in new drug development industry.Author contributions
Bendu Pan conceived the idea and designed and performed most of the experiment, Yunru Wu, Yaqi Zhang and Xiaobo He synthesized some of the starting materials, Long Jiang helped to analyze single crystal X-ray diffraction and Liqin Qiu conceived the idea and supervised the research.Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant 21772238), the Key-Area Research and Development Program of Guangdong Province (2020B010188001), Science and Technology Program of Guangzhou (20220602JBGS02). We appreciate Prof. Qing-Hua Fan (Institute of Chemistry, Chinese Academy of Sciences) and Prof. Yong-Gui Zhou (Dalian Institute of Chemical Physics, Chinese Academy of Sciences) for their generous support and providing enantiopure 2-methyl-1,2,3,4-tetrahydroquinoline. We also appreciate Prof. Chun-Jiang Wang (Wuhan University) for his generous support in chiral GC test of the recovered VEC.References
- (a) Z. Shen, H. A. Khan and V. M. Dong, Rh-Catalyzed Carbonyl Hydroacylation: An Enantioselective Approach to Lactones, J. Am. Chem. Soc., 2008, 130, 2916 CrossRef CAS PubMed; (b) Z. Shen, P. K. Donan, H. A. Khan, T. K. Woo and V. M. Dong, Mechanistic Insights into the Rhodium-Catalyzed Intramolecular Ketone Hydroacylation, J. Am. Chem. Soc., 2009, 131, 1077 CrossRef CAS PubMed; (c) C. A. Rose and K. Zeitler, Org. Lett., 2010, 12, 4552 CrossRef CAS; (d) H. Li, A. Spannenberg, H. Neumann, M. Beller and X.-F. Wu, Regioselective Synthesis of 2,3-Dihydrobenzodioxepinones from Epoxides and 2-Bromophenols via Palladium-Catalyzed Carbonylation, Chem. Commun., 2014, 50, 2114 RSC; (e) L. Liu, S. Xu and H. Zhou, Silver Carboxylate Promoted Lactonization: A General Method Applicable to Prepare Medium and Large-sized Lactones without High Dilution or Slow Addition, Tetrahedron, 2013, 69, 8386 CrossRef CAS; (f) I. Shiina, H. Fukui and A. Sasaki, Synthesis of Lactones Using Substituted Benzoic Anhydride as A Coupling Reagent, Nat. Protoc., 2007, 2, 2312 CrossRef CAS PubMed.
- L. Ding, H. Song, C. Zheng and S.-L. You, Enantioselective Synthesis of Medium-Sized-Ring Lactones via Iridium-Catalyzed Z–Retentive Asymmetric Allylic Substitution Reaction, J. Am. Chem. Soc., 2022, 144, 4770 CrossRef CAS.
- (a) J. F. Hartwig and L. M. Stanley, Mechanistically Driven Development of Iridium Catalysts for Asymmetric Allylic Substitution, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS; (b) J. Qu and G. Helmchen, Applications of Iridium-Catalyzed Asymmetric Allylic Substitution Reactions in Target-Oriented Synthesis, Acc. Chem. Res., 2017, 50, 2539 CrossRef CAS PubMed; (c) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Iridium-Catalyzed Asymmetric Allylic Substitution Reactions, Chem. Rev., 2019, 119, 1855 CrossRef CAS PubMed; (d) Y. Wang, W.-Y. Zhang and S.-L. You, Ketones and Aldehydes as O-Nucleophiles in Iridium-Catalyzed Intramolecular Asymmetric Allylic Substitution Reaction, J. Am. Chem. Soc., 2019, 141, 2228 CrossRef CAS; (e) C. M. Pearson, J. W. B. Fyfe and T. N. Snaddon, A Regio- and Stereodivergent Synthesis of Homoallylic Amines by a One-Pot Cooperative-Catalysis-Based Allylic Alkylation/Hofmann Rearrangement Strategy, Angew. Chem., Int. Ed., 2019, 58, 10521 CrossRef CAS; (f) L. Huang, Y. Cai, H.-J. Zhang, C. Zheng, L.-X. Dai and S.-L. You, Highly Diastereoand Enantioselective Synthesis of Quinuclidine Derivatives by an Iridium-Catalyzed Intramolecular Allylic Dearomatization Reaction, CCS Chem., 2019, 1, 106 CrossRef CAS; (g) X.-J. Liu, S. Jin, W.-Y. Zhang, Q.-Q. Liu, C. Zheng and S.-L. You, Sequence-Dependent Stereodivergent Allylic Alkylation/Fluorination Reaction of Acyclic Ketones, Angew. Chem., Int. Ed., 2020, 59, 2039 CrossRef CAS PubMed; (h) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Diastereodivergent Synthesis of Enantioenriched α,β-Disubstituted γ-Butyrolactones via Cooperative N-Heterocyclic Carbene and Ir Catalysis, Nat. Catal., 2020, 3, 48 CrossRef CAS; (i) H.-F. Tu, P. Yang, Z.-H. Lin, C. Zheng and S.-L. You, Time-Dependent Enantiodivergent Synthesis via Sequential Kinetic Sesolution, Nat. Chem., 2020, 12, 838 CrossRef CAS; (j) R. Jiang, Q.-R. Zhao, C. Zheng and S.-L. You, Structurally Defined Anti-π-Allyliridium Complexes Catalyse Z-Retentive Asymmetric Allylic Alkylation of Oxindoles, Nat. Catal., 2022, 5, 1089 CrossRef CAS; (k) B. Pan, C. V. Subba Reddy, Y. Wu and L. Qiu, Recent Advances in Cyclization Reactions via Catalytic Allylic Substitution for the Asymmetric Synthesis of Heterocyclic Compounds, Tetrahedron Lett., 2024, 141, 155071 CrossRef CAS.
- C. Welter, A. Dahnz, B. Brunner, S. Streiff, P. Dübon and G. Helmchen, Highly Enantioselective Syntheses of Heterocycles via Intramolecular Ir-Catalyzed Allylic Amination and Etherification, Org. Lett., 2005, 7, 1239 CrossRef CAS PubMed.
- (a) L. M. Stanley, C. Bai, M. Ueda and J. F. Hartwig, Iridium-Catalyzed Kinetic Asymmetric Transformations of Racemic Allylic Benzoates, J. Am. Chem. Soc., 2010, 132, 8918 CrossRef CAS; (b) C. Shu and J. F. Hartwig, Iridium-Catalyzed Intermolecular Allylic Etherification with Aliphatic Alkoxides: Asymmetric Synthesis of Dihydropyrans and Dihydrofurans, Angew. Chem., Int. Ed., 2004, 43, 4794 CrossRef CAS; (c) S. Ueno and J. F. Hartwig, Direct, Iridium-Catalyzed Enantioselective and Regioselective Allylic Etherification with Aliphatic Alcohols, Angew. Chem., Int. Ed., 2008, 47, 1928 CrossRef CAS.
- (a) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, Readily Available [2.2.2]-Bicyclooctadienes as New Chiral Ligands for Ir(I): Catalytic, Kinetic Resolution of Allyl Carbonates, J. Am. Chem. Soc., 2004, 126, 1628 CrossRef CAS; (b) M. Roggen and E. M. Carreira, Enantioselective Allylic Etherification: Selective Coupling of Two Unactivated Alcohols, Angew. Chem., Int. Ed., 2011, 50, 5568 CrossRef CAS PubMed.
- (a) H. He, K. Ye, Q. Wu, L. Dai and S.-L. You, Iridium-Catalyzed Asymmetric Allylic Etherification and Ring-Closing Metathesis Reaction for Enantioselective Synthesis of Chromene and 2,5-Dihydrobenzo[b]oxepine Derivatives, Adv. Synth. Catal., 2012, 354, 1084 CrossRef CAS; (b) Q. Cheng, Y. Wang and S.-L. You, Chemo-, Diastereo-, and Enantioselective Iridium-Catalyzed Allylic Intramolecular Dearomatization Reaction of Naphthol Derivatives, Angew. Chem., Int. Ed., 2016, 55, 3496 CrossRef CAS PubMed.
- (a) B. Pan, J.-S. Ouyang, Y. Zhang, H. Liang, Q. Ni, B. Chen, X. Pu, L. Jiang, R. Cao and L. Qiu, Iridium-Catalyzed Intramolecular Asymmetric Allylic Etherification of Salicylic Acid Derivatives with Chiral-Bridged Biphenyl Phosphoramidite Ligands, Org. Chem. Front., 2021, 8, 4514 RSC; (b) B. Pan, X. Qian, Y. Zhang, L. Jiang, R. Cao and L. Qiu, Iridium-Catalyzed Intramolecular Asymmetric Allylic Etherification of Pyrimidinemethanols: Enantioselective Construction of Multifunctionalized Pyrimidine-Fused Oxazepines, Org. Lett., 2023, 25, 5498 CrossRef CAS.
- (a) W. S. Guo, J. E. Gomez, A. Cristofol, J. N. Xie and A. W. Kleij, Catalytic Transformations of Functionalized Cyclic Organic Carbonates, Angew. Chem., Int. Ed., 2018, 57, 13735 CrossRef CAS PubMed; (b) Q.-Z. Li, Y. Liu, M.-Z. Li, X. Zhang, T. Qi and J.-L. Li, Palladium-Catalysed Decarboxylative Annulations of Vinylethylene Carbonates Leading to Diverse Functionalised Heterocycles, Org. Biomol. Chem., 2020, 18, 3638 RSC; (c) Z.-K. Liu, Y. Gao and X.-Q. Hu, Recent Advances in Catalytic Synthesis of Medium-Ring Lactones and their Derivatives, Catal. Sci. Technol., 2021, 11, 6931 RSC.
- (a) A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y. J. Zhang, Palladium-Catalyzed Decarboxylative Cycloaddition of Vinylethylene Carbonates with Formaldehyde: Enantioselective Construction of Tertiary Vinylglycols, Angew. Chem., Int. Ed., 2014, 53, 6439 CrossRef CAS; (b) L. Yang, A. Khan, R. Zheng, L. Y. Jin and L. Y. Zhang, Pd-Catalyzed Asymmetric Decarboxylative Cycloaddition of Vinylethylene Carbonates with Imines, Org. Lett., 2015, 17, 6230 CrossRef CAS PubMed; (c) K. Liu, I. Khan, J. Cheng, Y. J. Hsueh and Y. J. Zhang, Asymmetric Decarboxylative Cycloaddition of Vinylethylene Carbonates with β-Nitroolefins by Cooperative Catalysis of Palladium Complex and Squaramide, ACS Catal., 2018, 8, 11600 CrossRef CAS; (d) Y. Xia, Q.-F. Bao, Y. Li, L.-J. Wang, B.-S. Zhang, H.-C. Liu and Y.-M. Liang, Ligand-Controlled Regiodivergent π-Allyl Palladium Catalysis Enables a Switch Between [3 + 2] and [3 + 3] Cycloadditions, Chem. Commun., 2019, 55, 4675 RSC; (e) Y. Xu, L. Chen, Y.-W. Yang, Z. Zhang and W. Yang, Vinylethylene Carbonates as α,β-Unsaturated Aldehyde Surrogates for Regioselective [3 + 3] Cycloaddition, Org. Lett., 2019, 21, 6674 CrossRef CAS; (f) Y. Peng, X. Huo, Y. Luo, L. Wu and W. Zhang, Enantio- and Diastereodivergent Synthesis of Spirocycles through Dual-Metal-Catalyzed [3 + 2] Annulation of 2-Vinyloxiranes with Nucleophilic Dipoles, Angew. Chem., Int. Ed., 2021, 60, 24941 CrossRef CAS.
- (a) Z.-Q. Rong, L.-C. Yang, S. Liu, Z. Yu, Y.-N. Wang, Z. Y. Tan, R.-Z. Huang, Y. Lan and Y. Zhao, Nine-Membered Benzofuran-Fused Heterocycles: Enantioselective Synthesis by Pd-Catalysis and Rearrangement via Transannular Bond Formation, J. Am. Chem. Soc., 2017, 139, 15304 CrossRef CAS PubMed; (b) P. Das, S. Gondo, P. Nagender, H. Uno, E. Tokunaga and N. Shibata, Access to Benzo-Fused Nine-Membered Heterocyclic Alkenes with a Trifluoromethyl Carbinol Moiety via a Double Decarboxylative Formal Ringexpansion Process under Palladium Catalysis, Chem. Sci., 2018, 9, 3276 RSC; (c) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, Enantioselective Trapping of Pd-Containing 1,5-Dipoles by Photogenerated Ketenes: Access to 7-Membered Lactones Bearing Chiral Quaternary Stereocenters, J. Am. Chem. Soc., 2019, 141, 133 CrossRef CAS PubMed; (d) R. Zeng, J.-L. Li, X. Zhang, Y.-Q. Liu, Z.-Q. Jia, H.-J. Leng, Q.-W. Huang, Y. Liu and Q.-Z. Li, Diastereoselective Construction of 6,8-Dioxabicyclo[3.2.1]octane Frameworks from Vinylethylene Carbonates via Palladium-Organo Relay Catalysis, ACS Catal., 2019, 9, 8256 CrossRef CAS; (e) H.-H. Wu, X.-Z. Fan, Z. Tang, H. Zhang, L.-Y. Cai, X.-F. Bi and H.-W. Zhao, Palladium-Catalyzed Formal (5 + 6) Cycloaddition of Vinylethylene Carbonates with Isatoic Anhydrides for the Synthesis of Medium-Sized N,O-Containing Heterocycles, Org. Lett., 2021, 23, 2802 CrossRef CAS.
- W. Fu, L. Wang, Z. Yang, J.-S. Shen, F. Tang, J. Zhang and X. Cui, Facile Access to Versatile Aza-macrolides through Iridium-Catalysed Cascade Allyl-Amination/Macrolactonization, Chem. Commun., 2020, 56, 960 RSC.
- (a) L. Xiao, L. Wei and C.-J. Wang, Stereodivergent Synthesis of Enantioenriched γ-Butyrolactones Bearing Two Vicinal Stereocenters Enabled by Synergistic Copper and Iridium Catalysis, Angew. Chem., Int. Ed., 2021, 60, 24930 CrossRef CAS; (b) Q. Xiong, L. Xiao, X.-Q. Dong and C.-J. Wang, Asymmetric Synthesis of Chiral Aza-macrodiolides via Iridium-Catalyzed Cascade Allylation/Macrolactonization, Org. Lett., 2022, 24, 2579 CrossRef CAS PubMed; (c) F. Xiao, P. Liao, X. Lu, J. Wang, X.-Q. Dong and C.-J. Wang, Iridium-Catalyzed Asymmetric Cascade Allylation/Lactonization of Indole Esters: Access to Chiral Tricyclic Oxazinoindolones, Org. Lett., 2022, 24, 8592 CrossRef CAS; (d) W.-Y. Wang, Z.-Y. Yi, Z.-F. Wang, X.-Q. Dong and C.-J. Wang, Iridium-Catalyzed Asymmetric Cascade Dearomative Allylation/Acyl Transfer Rearrangement: Access to Chiral N-Substituted 2-Pyridones, Chem. Commun., 2024, 60, 5086 RSC; (e) K. Tian, X. Chang, L. Xiao, X.-Q. Dong and C.-J. Wang, Stereodivergent Synthesis of α-Fluoro α-Azaaryl γ-Butyrolactones via Cooperative Copper and Iridium Catalysis, Fundam. Res., 2024, 4, 77 CrossRef CAS PubMed; (f) Z.-Y. Yi, H. Xu, X. Chang, Y. Dang, X.-Q. Dong and C.-J. Wang, Iridium-Catalyzed Asymmetric Cascade Allylation/[1,4]-Phospha-Brook Rearrangement Reaction, ACS Catal., 2024, 14, 14058 CrossRef CAS.
- M. Lopez-Iglesias, A. Arizpe, F. J. Sayago, V. Gotor, C. Cativiela and V. Gotor-Fernandez, Lipase-Catalyzed Dynamic Kinetic Resolution of Dimethyl (1,3-Dihydro-2H-isoindol-1-yl)phosphonate, Tetrahedron, 2016, 72, 7311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2294598. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01771d |
| This journal is © the Partner Organisations 2025 |