
Facile one-pot synthesis of novel imidates as multifunctional organic fluorescent materials†
Feng-Ting
Liu
a,
Shuo
Wang
b,
Yong-Shun
Chen
a,
Jun-Ying
Miao
 b,
Bao-Xiang
Zhao
*a and
Zhao-Min
Lin
*c
b,
Bao-Xiang
Zhao
*a and
Zhao-Min
Lin
*c
aInstitute of Organic Chemistry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P.R. China. E-mail: bxzhao@sdu.edu.cn
bShandong Provincial Key Laboratory of Animal Cells and Developmental Biology, School of Life Science, Shandong University, Qingdao 266237, P.R. China
cInstitute of Medical Sciences, the Second Hospital of Shandong University, Jinan 250033, P.R. China. E-mail: linzhaomin@sdu.edu.cn
First published on 30th October 2024
Abstract
Imidates have enormous applications in the synthesis of natural products and nitrogen-containing heterocyclic compounds, as well as in drug development. However, there is currently a lack of green and efficient synthesis methods for imidate compounds. We present a novel one-pot synthesis of multifunctional organic fluorescent imidates from quinolinium salt derivatives, nitrosoarenes and alcohols. This method has the advantages of operational simplicity, mild reaction conditions, metal-free catalysis, a wide substrate scope (43 examples) and excellent isolated yields (up to 93%). Meanwhile, quinolinium-imidates exhibit good fluorescence intensity and excellent chemical stability, which makes them suitable for application in living cells, and in particular, imidate 4v can target the endoplasmic reticulum with high selectivity.
Introduction
Imidates (also known as imido esters) have attracted much attention from scientists in recent decades due to their great utility in the synthesis of natural products, nitrogen-containing heterocyclic compounds and metal complexes with catalytic effects. For instance, imidates are used as intermediates in the total synthesis of natural products such as (±)-perophoramidine, (±)-communesin F and dehaloperophoramidine.1 Nitrogen-containing molecules, especially nitrogen-containing heterocyclic compounds, are ubiquitous in natural and bioactive products.2 Imidates have been shown to be of particular importance in a diverse range of organic transformations because they exhibit dual reactivity, wherein the nitrogen atom of the imide bond is a nucleophilic center and the corresponding carbon is an electrophilic center.3 The recent application of imidates as building blocks for the synthesis of saturated and unsaturated N-heterocycles like oxazolines, oxazines, quinazolines, isoquinolines, imidazoles, and triazoles, among others, has been a hot topic of discussion.4 For example, N-(dihalophenyl)-imidate can be used as a raw material for the synthesis of benzimidazoles with N-tosylhydrazones and amines via a one-pot process catalyzed by palladium.5 In addition, imidates can not only coordinate with transition metals to form stable complexes,4f,6 but also be converted to amides by the Chapman rearrangement7 or aza-Claisen rearrangement,8 which is of practical significance in pharmaceutical and fine chemical industries. In view of their distinctive structural features and potential applications, the synthesis of imidates has received significant attention from organic chemists and has made good progress.In recent years, various synthetic methods have been reported for constructing imidate derivatives, including cyclic, N-unsubstituted and N-substituted imidates. One of the most common synthetic strategies is the reaction of a nitrile compound with an alcohol to produce imidates upon activation by an acid (Pinner reaction)9 (Fig. 1a). Although this method is simple to operate, the reaction conditions are harsh. It often requires strong acids to activate the cyano group, which are incompatible with acid-sensitive functional groups. Furthermore, this method is generally limited to the synthesis of imidates with unsubstituted NH. Another synthesis method of imidates involves a nucleophilic substitution reaction between imide chloride compounds and sodium alcohols10 (Fig. 1b). The disadvantages of this method are that imine chloride compounds are not easy to synthesize and are unstable. In addition, sodium alcohol is a strong base, making it difficult for some functional groups sensitive to bases to be compatible, and these drawbacks greatly limit the application of this method. In 2015, Wang and co-workers developed a series of trifluoroethoxy-substituted imidate compounds from amides and trifluorodiazoethane in excellent yields under the catalysis of silver salts11 (Fig. 1c). In addition, based on conventional synthesis, a series of new synthetic strategies have been reported in recent years, such as the construction of imidate derivatives using nitriles,12 imines,13 aldehydes14 and arylamines15 as starting materials (Fig. 1d–g). We find that many of the methods have certain shortcomings, such as the need for expensive metal catalysts, initiators, oxidizers and ligands and the involvement of stepwise reactions that reduce overall yield. Therefore, it is important to develop an economical, simple, green and efficient method to synthesize imidate compounds.
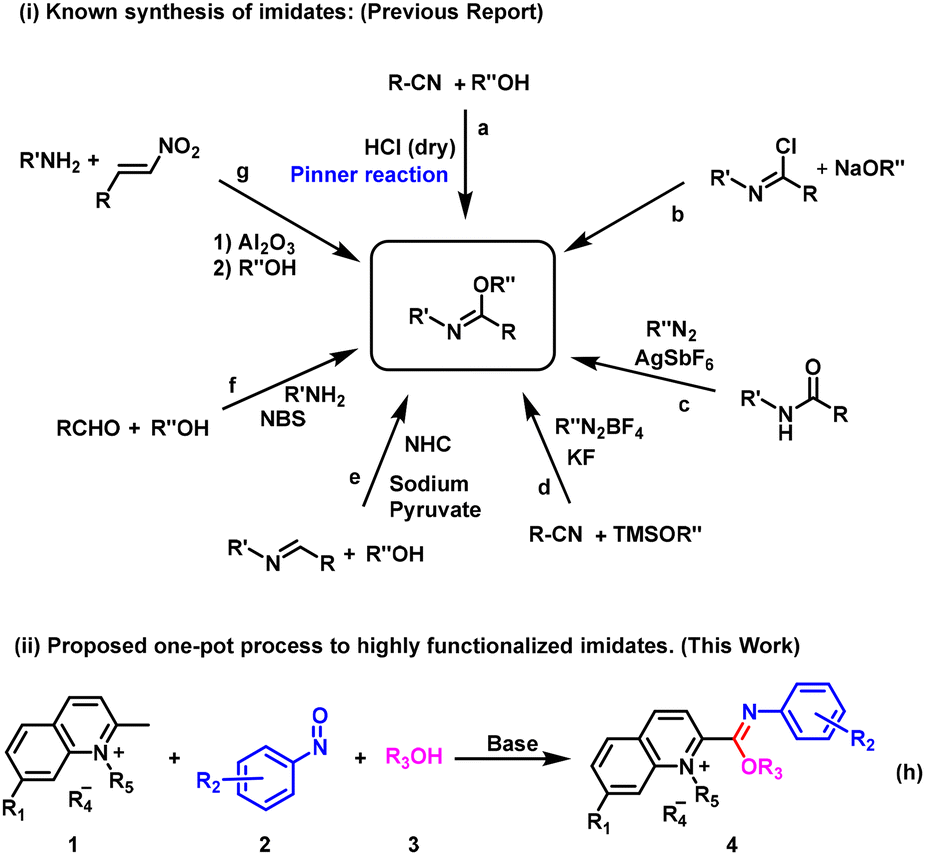 | ||
| Fig. 1 Synthetic strategies towards imidates. | ||
In addition, nitrosoarenes can be regarded as excellent electrophiles and can effectively undergo aldol reaction with carbon nucleophiles.16 Herein, we report an unexpected synthetic route to obtain novel imidate derivatives via a one-pot method utilizing 2-methylquinolinium salts, nitrosoarenes and alcohols in the absence of metal catalysts (Fig. 1h). In our pursuit of developing new organic small fluorescent probes,17 we attempted to synthesize a new probe with a nitrosobenzene structure for the recognition of hydrogen sulfide.18 Therefore, we designed a probe structure (I) and a synthesis route, as shown in Fig. 2. However, we did not obtain the expected compound (I). After carefully characterizing the structure of the product, we identified the structure to be II, an unexpected imidate derivative rather than the anticipated aldol product I or the nitrone derivative III suggested by the literature.19 This finding was exciting because discovery of new reactions is a central topic in organic chemistry. We then continued our exploration of the imidate synthesis reaction using 2-methylquinolinium salts, nitrosoarenes and alcohols as starting materials.
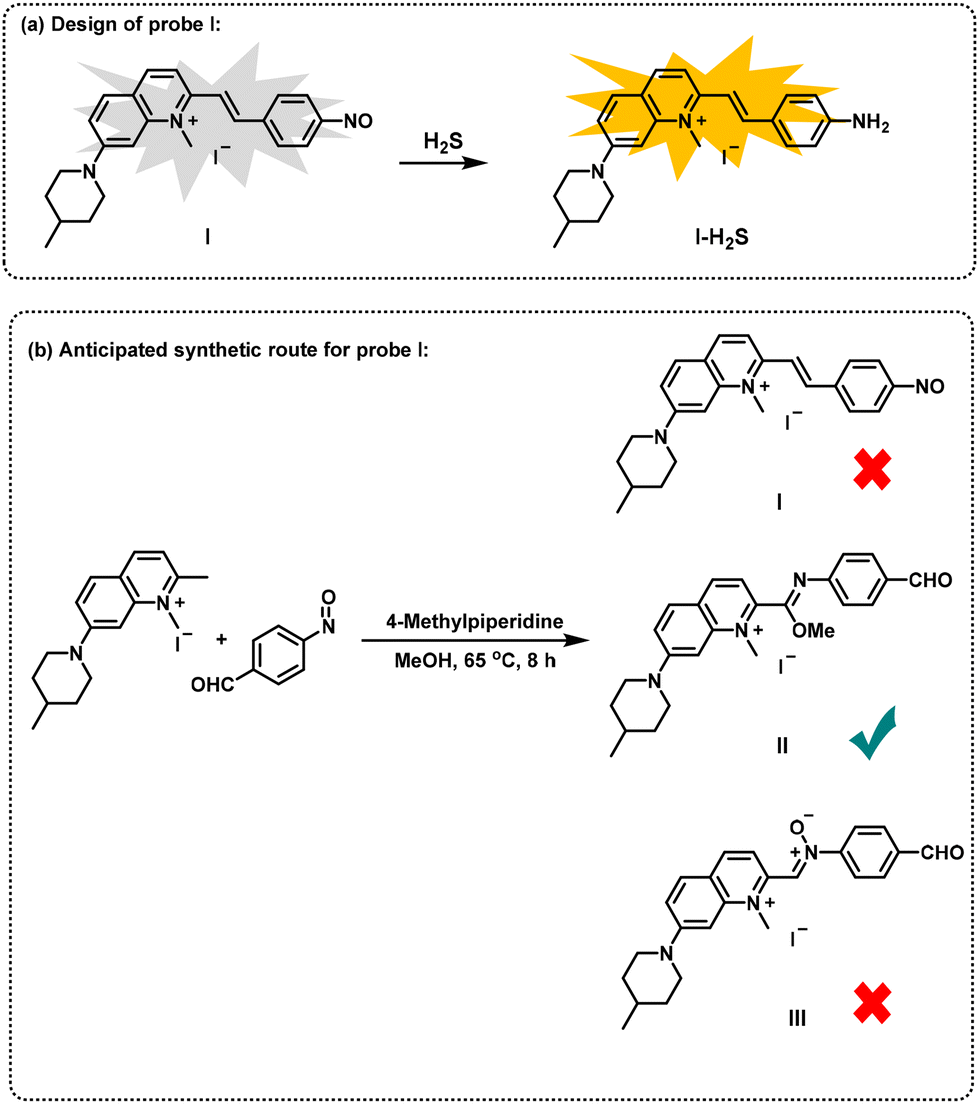 | ||
| Fig. 2 Discovery of an unexpected novel imidate via a synthetic route of designed probe I. | ||
Results and discussion
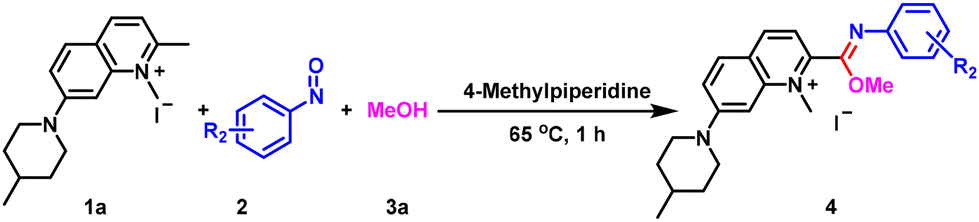
We chose 1,2-dimethyl-7-(4-methylpiperidin-1-yl)quinolin-1-ium iodide (1a), 4-nitrosobenzaldehyde (2a) and methanol (3a) as the model substrates to optimize the reaction conditions (Table 1). When 4-methylpiperidine was used as the base, we found that the reaction could proceed smoothly at 65 °C to give the target product 4a, as determined by NMR analysis. However, when the molar ratio of 1a and 2a was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, the yields were lower than 60%, both when the base dosage was increased or under a nitrogen or an oxygen atmosphere (Table 1, entries 1–3). In order to confirm the reliability of the reaction, we replaced 2a with 1-nitro-4-nitrosobenzene (2d) and methyl 4-nitrosobenzoate (2e) and obtained the target products in 32% and 45% yields, respectively (Table 1, entries 4 and 5). When we increased the dosage of 2a to 2.2 equivalents, the yield of 4a was greatly improved. The reaction was much better (95% yield) under air conditions than under nitrogen protection (76% yield) (Table 1, entries 6 and 7). Next, we examined the effect of the amount of 4-methylpiperidine on the reaction. The target product was not obtained without the base even when the reaction time was extended (Table 1, entry 8). When the equivalents of the base were reduced to 0.2 and 0.5, the yields were 80% and 87%, respectively (Table 1, entries 9 and 10), neither of which was as effective as using 1.0 equivalent. After determining the base amount, we further screened reaction times and temperatures (Table 1, entries 11–15). The reaction at 65 °C for 1 h gave 93% yield of 4a, which not only offered significant time efficiency, but also ensured excellent yield. The reaction could also be carried out smoothly at room temperature, but the yield was not as good as that under the 65 °C conditions. Therefore, we optimized the reaction temperature and time to be 65 °C and 1 h. Finally, we also tested the organic base DBU and the inorganic base K2CO3 (Table 1, entries 16–18), and found that the effect of these bases was inferior to that of 4-methylpiperidine. The finalized optimal reaction conditions were 1a (0.5 mmol), 2a (1.1 mmol) and 4-methylpiperidine (0.5 mmol) in 3a (20 mL) at 65 °C for 1 h (Table 1, entry 11).
:1.2, the yields were lower than 60%, both when the base dosage was increased or under a nitrogen or an oxygen atmosphere (Table 1, entries 1–3). In order to confirm the reliability of the reaction, we replaced 2a with 1-nitro-4-nitrosobenzene (2d) and methyl 4-nitrosobenzoate (2e) and obtained the target products in 32% and 45% yields, respectively (Table 1, entries 4 and 5). When we increased the dosage of 2a to 2.2 equivalents, the yield of 4a was greatly improved. The reaction was much better (95% yield) under air conditions than under nitrogen protection (76% yield) (Table 1, entries 6 and 7). Next, we examined the effect of the amount of 4-methylpiperidine on the reaction. The target product was not obtained without the base even when the reaction time was extended (Table 1, entry 8). When the equivalents of the base were reduced to 0.2 and 0.5, the yields were 80% and 87%, respectively (Table 1, entries 9 and 10), neither of which was as effective as using 1.0 equivalent. After determining the base amount, we further screened reaction times and temperatures (Table 1, entries 11–15). The reaction at 65 °C for 1 h gave 93% yield of 4a, which not only offered significant time efficiency, but also ensured excellent yield. The reaction could also be carried out smoothly at room temperature, but the yield was not as good as that under the 65 °C conditions. Therefore, we optimized the reaction temperature and time to be 65 °C and 1 h. Finally, we also tested the organic base DBU and the inorganic base K2CO3 (Table 1, entries 16–18), and found that the effect of these bases was inferior to that of 4-methylpiperidine. The finalized optimal reaction conditions were 1a (0.5 mmol), 2a (1.1 mmol) and 4-methylpiperidine (0.5 mmol) in 3a (20 mL) at 65 °C for 1 h (Table 1, entry 11).
|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Time (h) | Temp. (°C) | Yieldb (%) |
| a Unless otherwise noted, all reactions were carried out under the following conditions: 1a (0.5 mmol), 4-nitrosobenzaldehyde (2a, 1.1 mmol), 4-methylpiperidine (0.5 mmol), MeOH (20 mL) at 65 °C for 1 h. b Yields of the isolated products. c 2a (0.6 mmol) was employed. d Under N2. e Under O2. f 1-Nitro-4-nitrosobenzene (2d, 0.6 mmol) was employed. g Methyl 4-nitrosobenzoate (2e, 0.6 mmol) was employed. h DBU was employed as a base. i K2CO3 was employed as a base. DBU = 1,8-diazabicyclo-[5.4.0]undec-7-ene, N.P. = no product. | ||||
| 1c | 2 mL | 3 | 65 | 48 |
| 2c,d | 1.0 | 3 | 65 | 41 |
| 3c,e | 1.0 | 3 | 65 | 51 |
| 4e,f | 1.0 | 3 | 65 | 32 |
| 5e,g | 1.0 | 3 | 65 | 45 |
| 6d | 1.0 | 3 | 65 | 76 |
| 7 | 1.0 | 3 | 65 | 95 |
| 8 | 0 | 8 | 65 | N.P. |
| 9 | 0.2 | 3 | 65 | 80 |
| 10 | 0.5 | 3 | 65 | 87 |
| 11 | 1.0 | 1 | 65 | 93 |
| 12 | 1.0 | 0.5 | 65 | 88 |
| 13 | 1.0 | 0.5 | 30 | 77 |
| 14 | 1.0 | 1 | 30 | 80 |
| 15 | 1.0 | 5 | 30 | 85 |
| 16h | 1.0 | 1 | 30 | 69 |
| 17i | 1.0 | 1 | 30 | 71 |
| 18i | 1.0 | 1 | 65 | 43 |
With the optimized conditions in hand, we investigated the substrate scope and limitations of the reaction. To our delight, we found that this method for constructing quinolinium-imidates could tolerate a wide range of functional groups and all the products exhibited excellent fluorescence properties. As alcohol solvents were involved in the reaction, we first investigated the suitability of different alcohol solvents and found that methanol, ethanol, n-propanol, isopropanol and n-butanol were all compatible (Fig. 3, entries 1–5). Notably, when benzyl alcohol and 4-methylbenzyl alcohol were used as solvents at 65 °C, the desired quinolinium-imidates could be obtained in excellent yields (Fig. 3, entries 6 and 7). We then examined the substrate universality of the nitrosoarenes. When 2-nitrosobenzaldehyde was used as the substrate for the reaction, possibly influenced by steric hindrance, we could only detect trace amounts of the target compound (Fig. 3, entry 8). Reassuringly, 3-nitrosobenzaldehyde provided 4i in good yield (Fig. 3, entry 9). In addition, para-substituted nitrosoarenes carrying NO2, CO2Me and CN groups were well tolerated in this transformation, with yields of 83%, 88% and 85%, respectively (Fig. 3, entries 10–12). When halogen (−Cl and −Br) substituted nitrosoarenes were used as substrates, the corresponding imidate products were obtained in high yields under the standard conditions (Fig. 3, entries 13–15). Furthermore, we were pleased to find that the reaction system was not only suitable for nitrosoarenes with electron-withdrawing groups but also for nitrosoarenes with electron-donating groups, providing the corresponding products 4p–4r in 61%, 76% and 61% yields, respectively (Fig. 3, entries 16–18). The results indicated that the yields of reactions involving nitrosoarenes with electron-withdrawing groups were generally higher than those of nitrosoarenes with electron-donating groups. This discrepancy could be attributed to the fact that electron-withdrawing groups enhanced the electrophilicity of the nitrosoarenes, thereby facilitating their reaction with the enamine intermediate produced by the quinolinium salt and potentially increasing the overall reaction rate. Single crystal X-ray diffraction studies were carried out on compounds 4b, 4f, 4i, 4j and 4n to confirm the structures of the products (Fig. 3, entries 6 and 9; Fig. S1†).
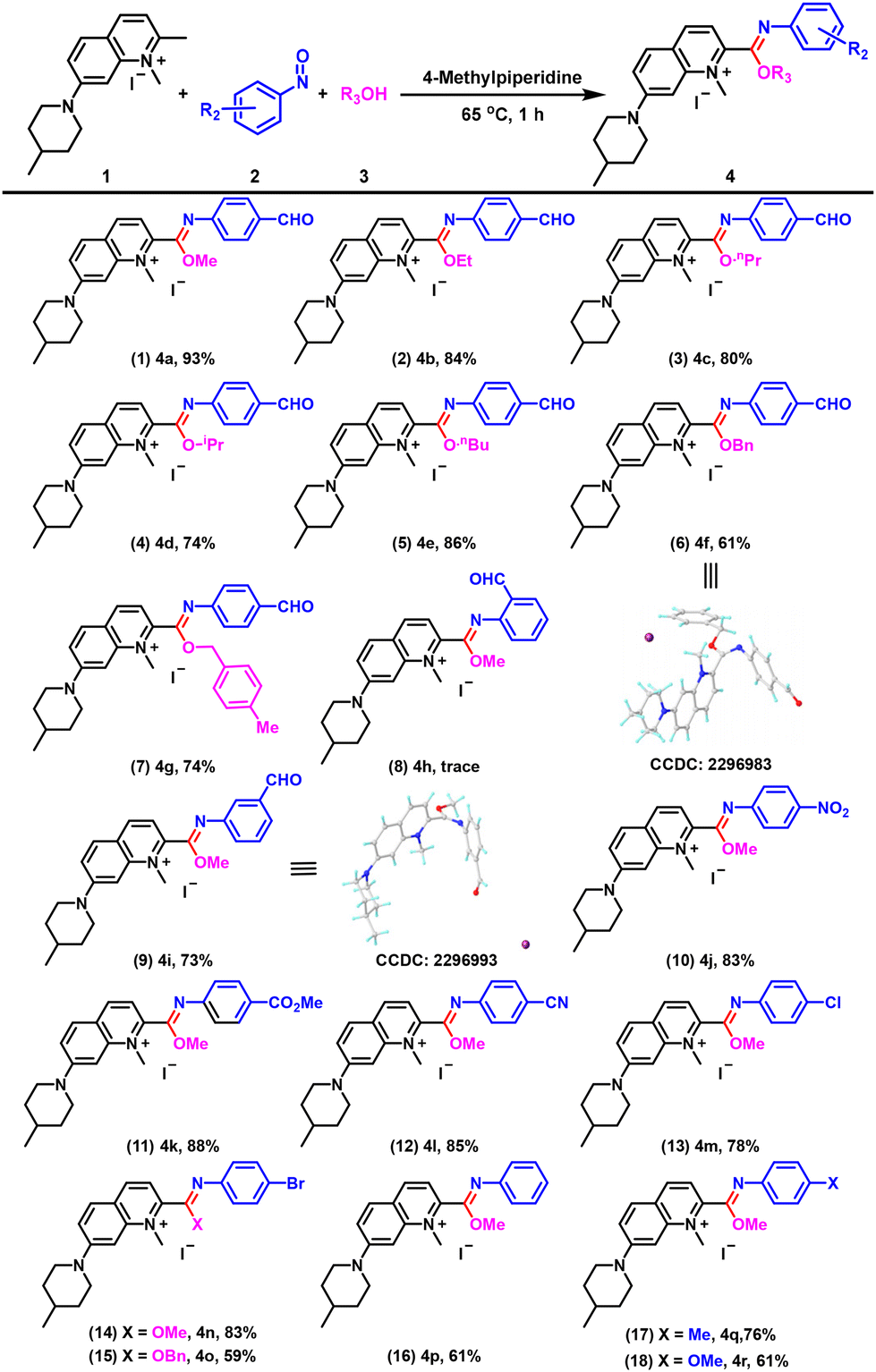 | ||
| Fig. 3 Scopes of alcohols and nitrosobenzene derivatives. Reaction conditions: 1a (0.5 mmol), 2 (1.1 mmol), 4-methylpiperidine (0.5 mmol), and alcohols (20 mL) at 65 °C for 1 h. Yields of isolated products are given. | ||
Next, we investigated the range of quinolinium salts suitable for this reaction. We examined the effect of anions. In addition to I−, −OTf was also compatible with this system and gave 4s in 67% yield (Fig. 4, entry 19). In addition, replacing the methyl group on the nitrogen atom of the quinolinium salt with an ethyl group still afforded the imidate product 4t in excellent yield (Fig. 4, entry 20). Remarkably, a quinolinium salt substituted with morpholine at position 7 was also compatible with this system and could react with nitrosoarenes with different substituents to provide 4u–4ac in good yields (Fig. 4, entry 21–29), which not only increased the structural versatility of the compounds, but also enhanced their ability to target lysosomes for organelle localization, attributed to the morpholine groups. The substituents used at position 7 could be further broadened to include groups such as diethylamino (Fig. 4, entries 30–37) or 4-(tert-butoxycarbonyl)piperazin-1-yl (Fig. 4, entries 38–43), enabling the formation of the desired products. The structures of 4x and 4ae were confirmed by single crystal X-ray diffraction.
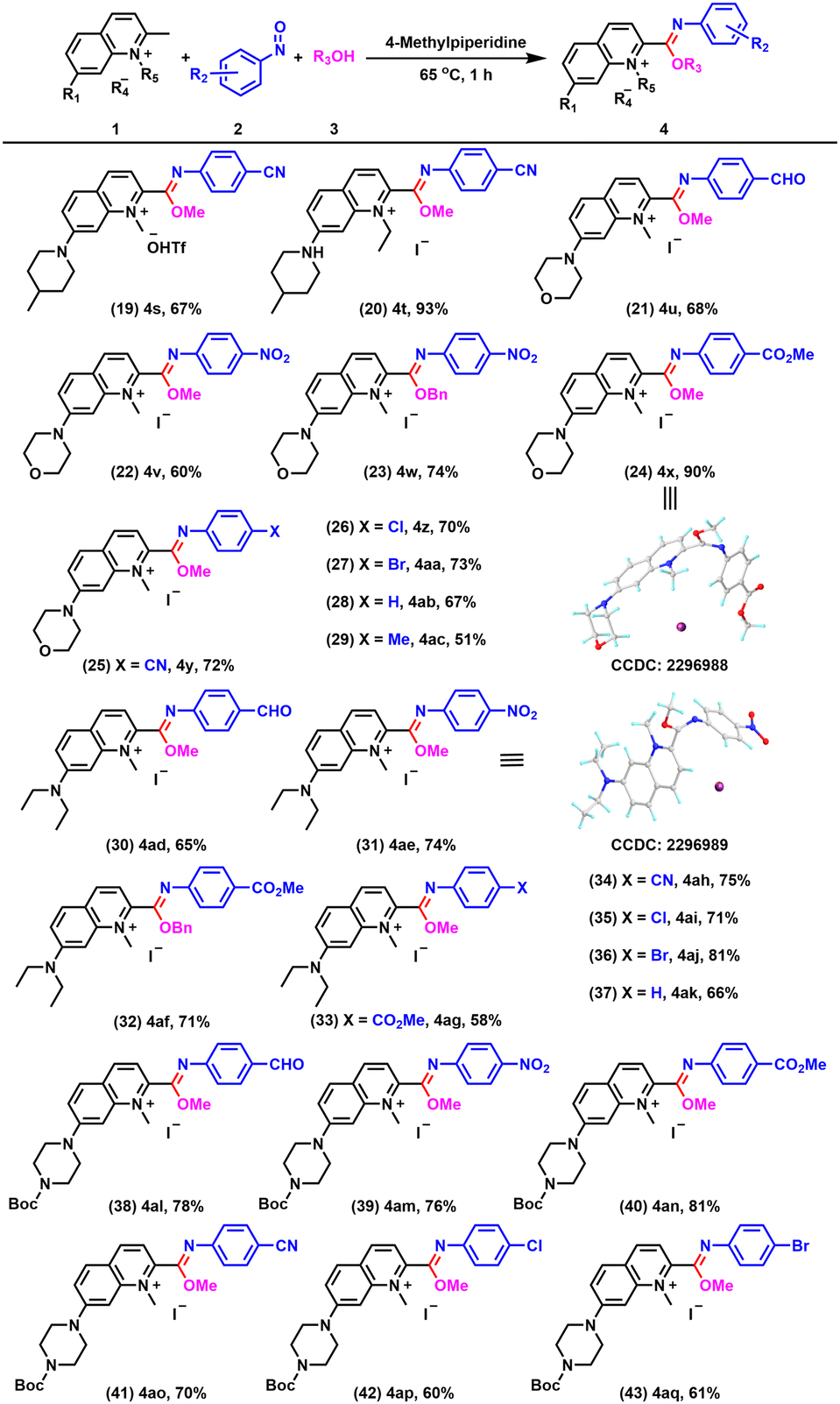 | ||
| Fig. 4 Scopes of alcohols and nitrosobenzene derivatives. Reaction conditions: 1a (0.5 mmol), 2 (1.1 mmol), 4-methylpiperidine (0.5 mmol), and alcohols (20 mL) at 65 °C for 1 h. Yields of isolated products are given. | ||
While exploring the above reactions, we found that when quinolinium salts were reacted with nitrosoarenes using benzyl alcohol as a solvent, the reaction at 100 °C did not give the corresponding imidate product, but gave the amide compound 5a (Scheme 1), which was confirmed by single-crystal X-ray diffraction. In order to verify this reaction outcome, we replaced benzyl alcohol with 4-methylbenzyl alcohol and 4-nitrobenzyl alcohol and still the same amide product was obtained. In addition, the reactions with 1a, 1b and 2e gave the corresponding amide products 5b and 5c when benzyl alcohol was used as the solvent, again confirming the above results. In stark contrast, when n-butanol was used at 100 °C, we did not detect the amide product, instead we obtained the imidate compound 4e in 70% yield. Thus, it was shown that the formation of the amide derivatives was not only related to the reaction temperature, but also to the structure of the alcohols.
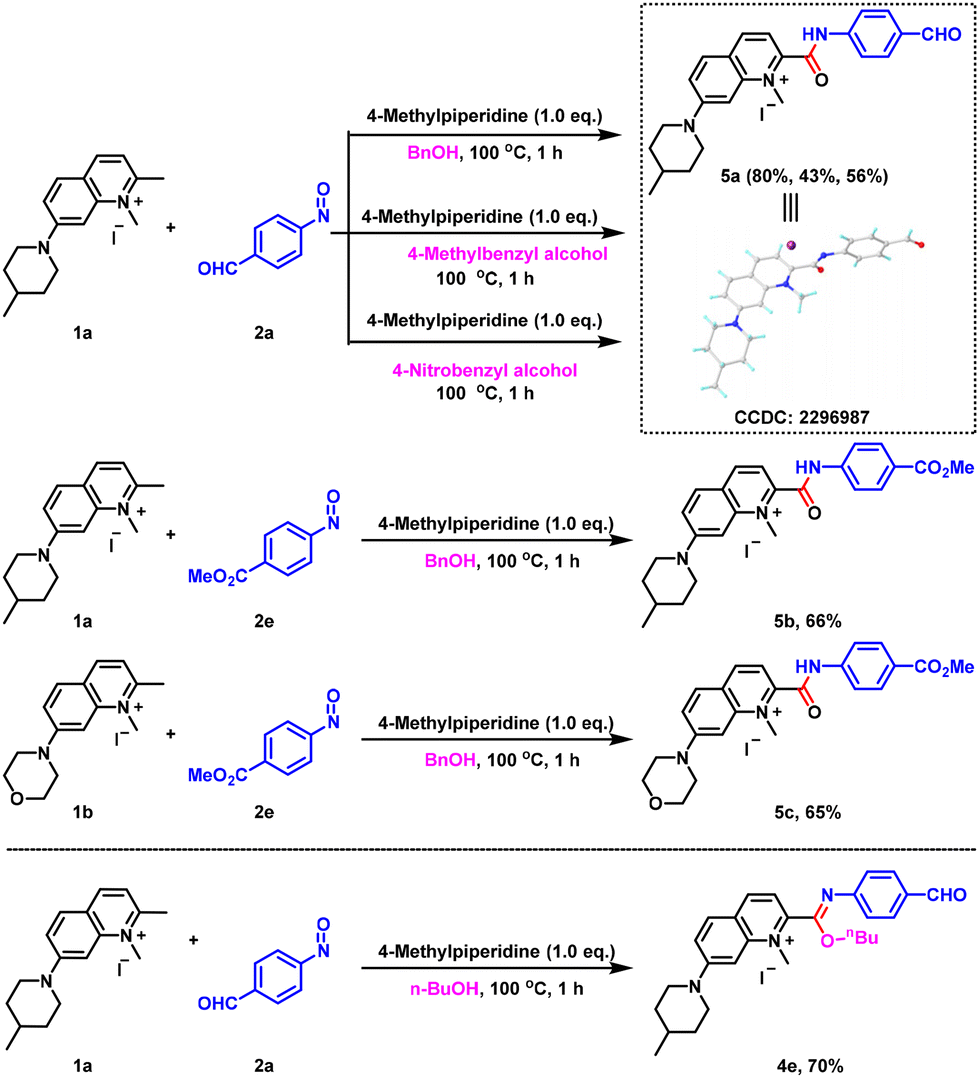 | ||
| Scheme 1 Construction of amide substrates. | ||
In order to further explore the mechanism of the three-component reaction of 2-quinolinium salt derivatives, nitrosoarenes and alcohols, we used 1a, 2a and methanol (or benzyl alcohol) as the model substrates to investigate the reaction mechanism (Scheme 2). The peaks of many intermediates (7, 8, 9, 10, 11, 12, 13 and 4f) could be observed by HRMS. Firstly, 1a reacted with 4-methylpiperidine in an acid–base reaction to produce the enamine intermediate 7,20 which then underwent a nucleophilic addition reaction with the nitroso group of 2a to give intermediate 8.20,21 These two substances were detected by HRMS analysis of the reaction mixture. During condition screening, we found that the yield of the target product could reach more than 50% only when more than 2 equivalents of 2a were involved in the reaction, so we judged that the enamine derivative 7 reacted with 2 equivalents of 2a to generate 10. The detection of intermediate 8 by HRMS suggested that intermediate 7 reacted twice consecutively with the monomer of 2a rather than directly with the dimer of 2a, since the dimer is known to be in equilibrium with the monomeric form in solution.22 Intermediate 8 loses a hydride ion, allowing it to nucleophilically attack another molecule 2a, and this is followed by the deprotonation of the hydroxy group to generate nitrone 10 and the by-product 4-(hydroxyamino)benzaldehyde 9.21 Nitrone 10 underwent a nucleophilic addition reaction with methanol to generate intermediate 11,23 and the subsequent dehydration of intermediate 11 gave imidate 4a. When benzyl alcohol was used at 100 °C, following the mechanism described above, the nitrone compound 10 gave imidate 4f. However, the benzyl positive ion readily dissociated from 4f at elevated temperatures to produce the amide compound 5a, with compound 13 as the major byproduct, providing additional support for this mechanism (Scheme 2).
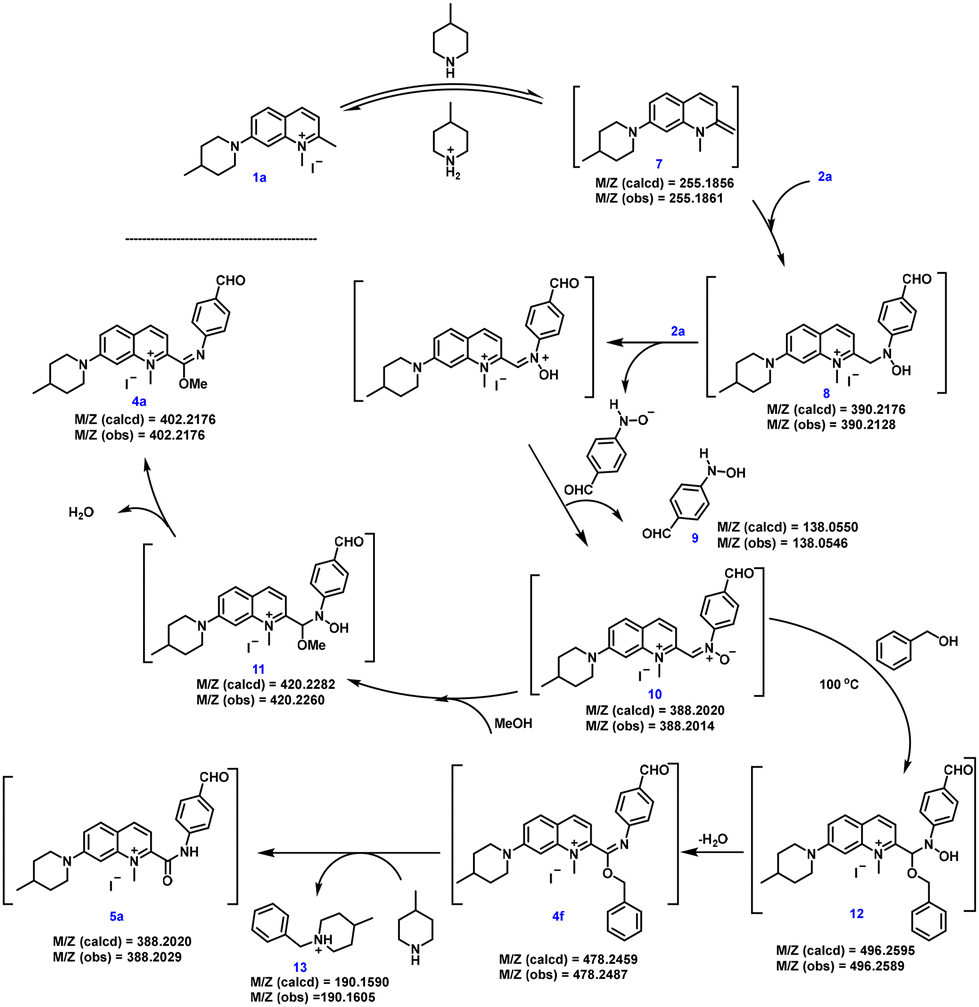 | ||
| Scheme 2 Proposed reaction mechanism. | ||
In order to verify the utility of this simple one-pot reaction, a gram-scale experiment was carried out. A 2 mmol representative reaction of 1f and 2e in methanol proceeded smoothly under the standard reaction conditions to give the corresponding product 4an in 87% yield (Scheme 3a). This indicated that this reaction could be used to prepare imidate compounds on a relatively large scale under mild conditions, which presents potential for future industrial production In the presence of trifluoroacetic acid, imidate 4m could be hydrolyzed efficiently to give amide 6a in 92% yield, confirming its further applicability (Scheme 3b). The structure of 6a was confirmed by single crystal diffraction.
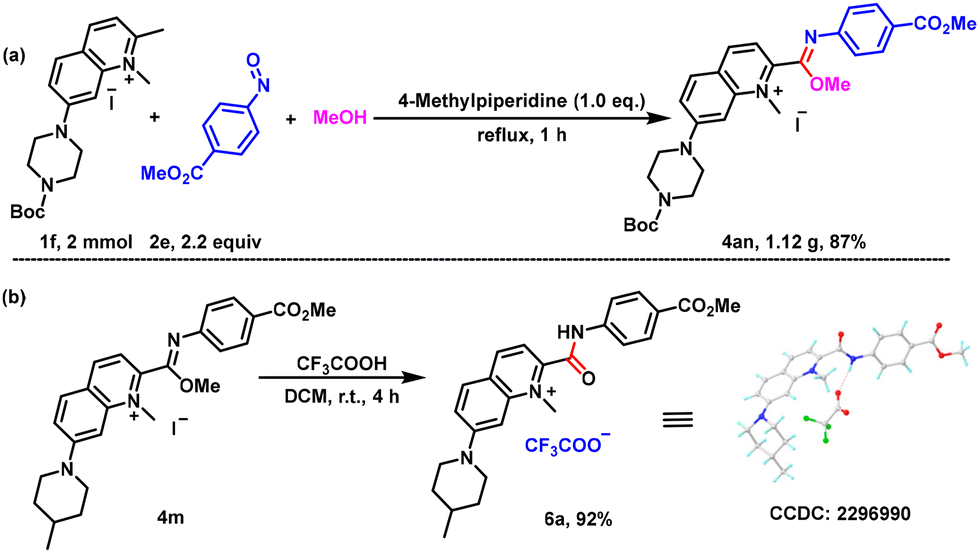 | ||
| Scheme 3 Large scale reaction and synthetic applications. | ||
We then chose ten representative imidate compounds (4a, 4j, 4k, 4m, 4n, 4p, 4q, 4u, 4ad and 4al) for the determination of their photophysical properties. First, the fluorescence spectra of 4a recorded in different solvents showed that DCM was the best solvent, producing a strong emission band centered at 591 nm, and that the maximum emission wavelength underwent a redshift of 27 nm with increasing solvent polarity, which might be caused by the solvent effect (Fig. 5a). Next, we recorded the fluorescence spectra of 4a, 4j, 4k, 4m, 4n, 4p, 4q, 4u, 4ad and 4al in DCM. We observed that compound 4q with an electron-donating group (–Me) exhibited the weakest fluorescence intensity. In contrast, compounds 4a, 4j and 4k, with electron-withdrawing groups, demonstrated stronger fluorescence intensities compared to all other compounds and experienced a redshift of the maximum emission wavelength of 20 nm, probably due to the induced effect of the electron-withdrawing groups (NO2, CHO and CO2Me) (Fig. 5b). Additionally, we determined the UV-vis absorption peaks for each compound in six solvents (DMSO, ACN, EtOH, AC, THF and DCM) (Fig. S2†). The UV-vis absorption peaks of the imidates displayed a redshift of 3–6 nm with increasing solvent polarity, exhibiting a small solvatochromic effect. The absorption intensities of 4a, 4j, 4k, 4m, 4n, 4p, 4q, 4u, 4ad and 4al were comparable in DCM (Fig. 5c), and there was not much difference in their emission wavelengths, a phenomenon which stimulated our interest. Single-crystal X-ray diffraction revealed that the quinoline plane and the aryl ring plane were not coplanar and the electron transfer was suppressed, so there was no significant difference in fluorescence emission wavelengths and UV absorption wavelengths between the different imidates. In addition, we could see that these imidate derivatives showed orange-red fluorescence under 365 nm UV light (Fig. 5d).
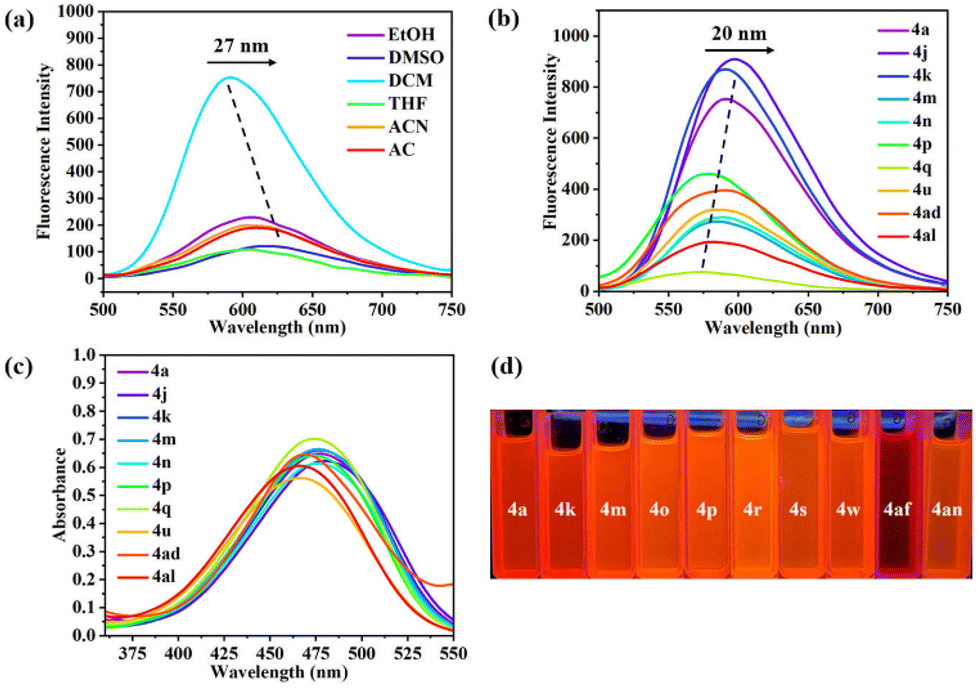 | ||
| Fig. 5 (a) Fluorescence spectra of compound 4a in different solutions (2 × 10−5 M). (b) Fluorescence spectra of 4a, 4j, 4k, 4m, 4n, 4p, 4q, 4u, 4ad and 4al (2 × 10−5 M) in DCM. (c) UV-vis absorption spectra of 4a, 4j, 4k, 4m, 4n, 4p, 4q, 4u, 4ad and 4al (4 × 10−5 M) in DCM. (d) Photos under irradiation of UV light (λ = 365 nm). | ||
Encouraged by these favorable photophysical properties, we chose 4q, 4n and 4v to study their organelle targeting. HeLa cells were pretreated with compounds 4 (5 μM) for 1 h, and then treated with Lyso-Tracker Red DND-99 (1:5000), ER-Tracker Red (1:1000), HCS lipidTox Deep Red (1:400) and Mito-Tracker Deep Red (1:5000) for 20 min, respectively. Some overlaps were observed between the green channel where compound 4 was located and the red channel where the organelle dye was located. It was expected that 4q and 4n would be more inclined to target mitochondria due to their positive charge,24 and co-localization experiments indicated that 4q and 4n indeed exhibited high targeting towards mitochondria (Mito), with Pearson co-localization coefficients of 0.91 and 0.90, respectively (Fig. 6a and b). However, they also showed good targeting towards the endoplasmic reticulum, with Pearson co-localization coefficients of 0.96 and 0.88, respectively, and the Pearson co-localization coefficients of the lipid droplets were not low, suggesting that 4q and 4n did not exclusive target mitochondria. A compound containing a morpholine moiety is usually designed to target lysosomes25 because morpholine can bind to the membrane of the lysosome, allowing the compound to accumulate efficiently in the lysosome. However, 4v, containing the morpholine moiety, exclusively targeted the endoplasmic reticulum with a Pearson co-localization coefficient of 0.85, which was significantly higher than that of the other organelles (Fig. 6c). The results might be due to the fact that the imidate moiety in the compounds counteracted the effect of the morpholine, as 4q and 4n containing the imidate moiety also had high co-localization coefficients to the endoplasmic reticulum. The endoplasmic reticulum (ER) is an indispensable organelle in eukaryotic cells and is involved in protein synthesis and processing, as well as in calcium storage and release.26 Abnormal fluctuations of active substances in the endoplasmic reticulum will lead to an imbalance in homeostasis in the body and further cause endoplasmic reticulum stress, leading to various diseases.27 Therefore, real-time imaging of various active species in the endoplasmic reticulum is important to determine its complex role, which will help uncover the corresponding disturbance mechanisms.
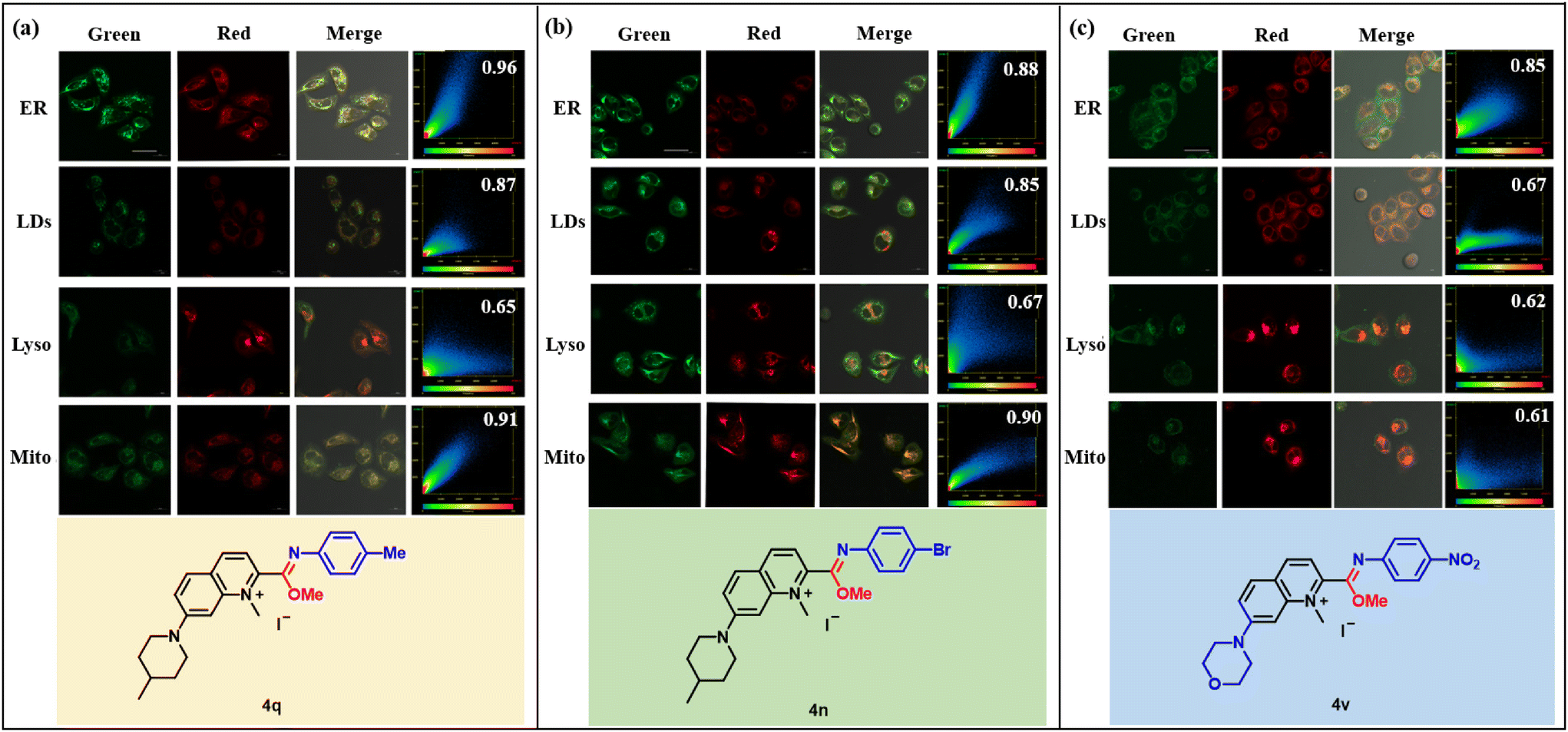 | ||
| Fig. 6 (a–c) Confocal fluorescence images of living HeLa cells co-stained with 4q (5 μM), 4n (5 μM), 4v (5 μM) and organelles. Green channel: 4q, 4n and 4v, λex = 405 nm, λem = 405–530 nm. Red channel: ER-Tracker™ Red (500 nM); HCS Lipid TOXTM Deep Red neutral lipid stain (100 nM); LysoTracker® Deep Red (2.5 mg mL−1) and MitoTracker™ Deep Red (100 nM), λex = 633 nm, λem = 640 nm-700 nm. Scale bar = 20 μm. | ||
Conclusions
In summary, we have developed a fast, efficient and practical one-pot method for constructing imidate derivatives from a three-component approach consisting of 2-methylquinolinium salt derivatives, nitrosoaromatic compounds and alcohols. This synthetic method had the advantages of operational simplicity, mild reaction conditions, metal-free catalysis, gram-scale preparation, a wide substrate scope (43 substrates) and high yields (up to 93%). Notably, the imidate compounds emitted orange-red fluorescence (572–620 nm) and could be applied to cells, especially compound 4v could target endoplasmic reticulum organelles. The protocol provided an experimental basis for further constructing novel fluorescent probes and investigating their organelle function mechanisms.Data availability
The data that support the findings of this study are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFC3602400), the Natural Science Foundation of Shandong Province (ZR2021MB009; ZR2019ZD36), and the National Natural Science Foundation of China (No. 32170744). The authors acknowledge the assistance of Shandong University Structural Constituent and Physical Property Research Facilities. The authors thank Prof. Di Sun at Shandong University for assistance with the data collection of X-ray crystal structures. Technical support provided by the single-crystal X-ray diffraction platform of SDU SC&PP research facilities is acknowledged.References
- (a) J. R. Fuchs and R. L. Funk, Total synthesis of (±)-perophoramidine, J. Am. Chem. Soc., 2004, 126, 5068 CrossRef CAS; (b) J. Yang, H. X. Wu, L. Q. Shen and Y. Qin, Total synthesis of (±)-communesin F, J. Am. Chem. Soc., 2007, 129, 13794 CrossRef CAS; (c) K. Popov, A. Hoang and P. Somfai, Concise total synthesis of dehaloperophoramidine, Angew. Chem., Int. Ed., 2016, 55, 1801 CrossRef CAS.
- (a) J. F. Li, R. F. Zhao, F. Q. Zhou, M. Y. She, J. Zhang, B. Yin, S. Y. Zhang and J. L. Li, Exploring the necessity of an acidic additive for Pd(II)-catalyzed exclusive C4-fluoroalkylation of 3-acetylindole: a detailed DFT study on the mechanism and regioselectivity, Org. Chem. Front., 2019, 6, 2607 RSC; (b) Q. L. Yang, Y. Liu, Y. R. Luo, Z. H. Li, H. W. Jia, Y. B. Fu, G. R. Qu and H. M. Guo, Rhodium(III)-catalyzed synthesis of diverse fluorescent polycyclic purinium salts from 6-arylpurine nucleosides and alkynes, Org. Lett., 2022, 24, 4234 CrossRef CAS; (c) Y. Y. Guo, Z. L. Wang, Y. Zhu, Q. C. Zhang, D. H. Wei, X. Liu and Z. Q. Fu, Access to polyfunctionalized carbazoles through π-extension of 2-methyl-3-oxoacetate indoles, Org. Chem. Front., 2019, 6, 3741 RSC; (d) H. X. Wang, Z. H. Li, W. W. Li, G. R. Qu, Q. L. Yang and H. M. Guo, Electrochemically driven oxidative C–H/N–H cross-coupling reactions of cyclic sulfamidate imines with primary anilines and secondary amines, Green Chem., 2022, 24, 8377 RSC; (e) W. Z. Xiong, Z. Y. Chen, Y. L. Shao, R. H. Li, K. Hu and J. X. Chen, The synthesis of fluorescent benzofuro[2,3-c]pyridines via palladium-catalyzed heteroaromatic C–H addition and sequential tandem cyclization, Org. Chem. Front., 2020, 7, 756 RSC.
- (a) A. G. Chaidali and I. N. Lykakis, Synthetic routes to imidates and their applications in organic transformation: Recent progress, Eur. J. Org. Chem., 2023, 26, e2023004 CrossRef; (b) R. Thakur, Y. Jaiswal and A. Kumar, Imidates: an emerging synthon for N-heterocycles, Org. Biomol. Chem., 2019, 17, 9829 RSC; (c) K. Popov and P. Somfai, Synthesis of imidates: TFA-mediated regioselective amide alkylation using Meerwein's Reagent, J. Org. Chem., 2016, 81, 3470 CrossRef CAS PubMed.
- (a) L. Y. Pan, Z. X. Li, T. Ding, X. M. Fang, W. K. Zhang, H. Xu and Y. Q. Xu, Base-mediated synthesis of unsymmetrical 1,3,5-triazin-2-amines via three-component reaction of imidates, guanidines, and amides or aldehydes, J. Org. Chem., 2017, 82, 10043 CrossRef CAS PubMed; (b) S. J. Shi, K. Xu, C. Jiang and Z. H. Ding, ZnCl2-catalyzed [3 + 2] cycloaddition of benzimidates and 2H-azirines for the synthesis of imidazoles, J. Org. Chem., 2018, 83, 14791 CrossRef CAS PubMed; (c) Y. Zhang, J. L. Zeng, Z. Chen and R. Wang, Base-promoted (3 + 2) cycloaddition of trifluoroacetohydrazonoyl chlorides with imidates en route to trifluoromethyl-1,2,4-triazoles, J. Org. Chem., 2022, 87, 14514 CrossRef CAS PubMed; (d) G. Borah, B. Dam and B. K. Patel, Ortho-functionalization of benzimidates and benzamidines, ChemistrySelect, 2022, 7, e202104583 CrossRef CAS; (e) Y. H. Tian, F. D. Wu, S. Q. Jia, X. N. Gong, H. Mao, P. F. Wang, W. L. Qin and H. L. Yan, Organocatalytic asymmetric construction of tetrasubstituted carbon stereocenters bearing three heteroatoms via intramolecular cyclization of vinylidene ortho-quinone methide with imidates, Org. Lett., 2022, 24, 5073 CrossRef CAS; (f) Q. Q. Lu, S. Gressies, S. Cembellín, F. J. R. Klauck, C. G. Daniliuc and F. Glorius, Redox-neutral manganese(I)-catalyzed C−H activation: Traceless directing group enabled regioselective annulation, Angew. Chem., Int. Ed., 2017, 56, 12778 CrossRef CAS PubMed; (g) E. A. Wappes, K. M. Nakafuku and D. A. Nagib, Directed β C–H amination of alcohols via radical relay chaperones, J. Am. Chem. Soc., 2017, 139, 10204 CrossRef CAS PubMed.
- T. Naret, P. Retailleau, J. Bignon, J. D. Brion, M. Alami and A. Hamze, Palladium-catalyzed one-pot synthesis of 5-(1-arylvinyl)-1H-benzimidazoles: Overcoming the limitation of acetamide partners, Adv. Synth. Catal., 2016, 358, 1833 CrossRef CAS.
- (a) X. G. Li, M. Sun, K. Liu and P. N. Liu, Oxidative alkenylation/annulation of benzimidates via ruthenium(II)-catalyzed C-H activation to generate 3-methyleneisoindolin-1-ones, Adv. Synth. Catal., 2015, 357, 395 CrossRef CAS; (b) S. Gupta, J. Han, Y. Kim, S. W. Lee, Y. H. Rhee and J. Park, C–H activation guided by aromatic N–H ketimines: Synthesis of functionalized isoquinolines using benzyl azides and alkynes, J. Org. Chem., 2014, 79, 9094 CrossRef CAS PubMed.
- (a) J. J. Fang, M. L. Ke, G. X. Huang, Y. Tao, D. Cheng and F. E. Chen, The Chapman rearrangement in a continuous-flow microreactor, RSC Adv., 2019, 9, 9270 RSC; (b) M. A. Epishina, A. S. Kulikov, N. V. Ignat'ev, M. Schulte and N. N. Makhova, Efficient synthesis of tertiary acyclic amides by the Chapman rearrangement of aryl benzimidates in ionic liquids, Mendeleev Commun., 2015, 25, 126 CrossRef CAS; (c) M. Kimura, I. Okabayashi and K. Isogai, The mechanism of the chapman rearrangement of N-arylbenzimidate on the basis of the molecular structure established by X-ray. Part II†, J. Heterocycl. Chem., 1988, 25, 315 CrossRef CAS.
- (a) J. Ma, X. L. Cui, D. J. Yang, J. L. Wu, M. P. Song and Y. J. Wu, Ferrocenylimidazoline palladacycles as efficient catalysts for the aza-Claisen rearrangement reaction of allylic imidates, Appl. Organomet. Chem., 2008, 22, 624 CrossRef CAS; (b) J. Kang, T. H. Kim, K. H. Yew and W. K. Lee, The effect of face-blocking in the enantioselective aza-Claisen rearrangement of allylic imidates, Tetrahedron: Asymmetry, 2003, 14, 415 CrossRef CAS; (c) J. Kang, K. H. Yew, T. H. Kim and D. H. Choi, Preparation of bis[palladacycles] and application to asymmetric aza-Claisen rearrangement of allylic imidates, Tetrahedron Lett., 2002, 43, 9509 CrossRef CAS.
- K. Watanabe, N. Kogoshi, H. Miki and Y. Torisawa, Torisawa, Improved Pinner reaction with CPME as a solvent, Synth. Commun., 2009, 39, 2008 CrossRef CAS.
- H. Liu, F. Wang, C. Han, H. X. Zhang, C. X. Bai, Y. M. Hu and X. Q. Zhang, Cobalt and nickel complexes supported by 2,6-bis(imidate)pyridyl ligands: Synthesis, characterization, and 1,3-butadiene polymerization studies, Inorg. Chim. Acta, 2015, 434, 135 CrossRef CAS.
- H. Q. Luo, G. J. Wu, Y. Zhang and J. B. Wang, Silver(I)-catalyzed N-trifluoroethylation of anilines and O-trifluoroethylation of amides with 2,2,2-trifluorodiazoethane, Angew. Chem., Int. Ed., 2015, 54, 14503 CrossRef CAS.
- (a) K. Zhang, X. H. Xu and F. L. Qing, Copper-promoted Ritter-type trifluoroethoxylation of (hetero)arenediazonium tetrafluoroborates: A method for the preparation of trifluoroethyl imidates, Eur. J. Org. Chem., 2016, 2016, 5088 CrossRef CAS; (b) R. Z. Zhang, W. Q. Huang, R. X. Zhang, C. Xu and M. Wang, Synthesis of N-CF3 amidines/imidates/thioimidates via N-CF3 nitrilium ions, Org. Lett., 2022, 24, 2393 CrossRef CAS.
- G. J. Wang, C. L. Wei, X. F. Hong, Z. Q. Fu and W. Huang, Sodium pyruvate as a peroxide scavenger in aerobic oxidation under carbene catalysis, Green Chem., 2020, 22, 6819 RSC.
- P. Yin, W. B. Ma, Y. Chen, W. C. Huang, Y. Deng and L. He, Highly efficient cyanoimidation of aldehydes, Org. Lett., 2009, 11, 5482 CrossRef CAS PubMed.
- A. G. Chaidali and I. N. Lykakis, Simple synthetic approach to N-(pyridin-2-yl)imidates from nitrostyrenes and 2-aminopyridines via the N-(pyridin-2-yl)iminonitriles as intermediates, Molecules, 2023, 28, 3321 CrossRef CAS PubMed.
- (a) J. N. Payette and H. Yamamoto, Nitrosobenzene-mediated C-C bond cleavage reactions and spectral observation of an oxazetidin-4-one ring system, J. Am. Chem. Soc., 2008, 130, 12276 CrossRef CAS; (b) M. Baidya and H. Yamamoto, Metal nitrite: A powerful oxidizing reagent, J. Am. Chem. Soc., 2011, 133, 13880 CrossRef CAS.
- (a) F. T. Liu, N. Li, Y. S. Chen, H. Y. Yu, J. Y. Miao and B. X. Zhao, A quinoline-coumarin near-infrared ratiometric fluorescent probe for detection of sulfur dioxide derivatives, Anal. Chim. Acta, 2022, 1211, 339908 CrossRef CAS; (b) F. T. Liu, W. W. Han, H. Ren, W. J. Yang, R. N. Wang, J. Y. Miao, B. X. Zhao and Z. M. Lin, A ratiometric lysosome-targeted fluorescent probe for imaging SO2 based on the coumarin-quinoline-julolidine molecular system, Dyes Pigm., 2023, 210, 111017 CrossRef CAS; (c) F. T. Liu, W. W. Han, H. Ren, R. N. Wang, W. J. Yang, J. Y. Miao, B. X. Zhao and Z. M. Lin, A dicyanoisophorone-quinolinium-based near-infrared-emission fluorescent probe for ratiometric sensing of bisulfite/sulfite in living cells, Sens. Actuators, B, 2023, 380, 133305 CrossRef CAS.
- H. H. Chen, X. Y. Gong, X. W. Liu, Z. Li, J. J. Zhang and X. F. Yang, A nitroso-based fluorogenic probe for rapid detection of hydrogen sulfide in living cells, Sens. Actuators, B, 2019, 281, 542 CrossRef CAS.
- P. Zuman and B. Shah, Addition, reduction, and oxidation reactions of nitrosobenzene, Chem. Rev., 1994, 94, 1621 CrossRef CAS.
- X. Gao, F. Zhang, G. J. Deng and L. Yang, Bronsted acid catalyzed benzylic C-H bond functionalization of azaarenes: Nucleophilic addition to nitroso compounds, Org. Lett., 2014, 16, 3664 CrossRef CAS.
- A. J. Fraboni and S. E. Brenner-Moyer, Dienamine-catalyzed nitrone formation via redox reaction, Org. Lett., 2016, 18, 2146 CrossRef CAS PubMed.
- B. G. Gowenlock, Structure and properties of C-nitroso-compounds, Q. Rev., Chem. Soc., 1958, 12, 321 RSC.
- S. I. Murahashi and Y. Imada, Synthesis and transformations of nitrones for organic synthesis, Chem. Rev., 2019, 119, 4684 CrossRef CAS PubMed.
- (a) L. T. Zeng, T. H. Chen, B. Q. Chen, H. Q. Yuan, R. L. Sheng and G. M. Bao, A distinctive mitochondrion-targeting, in situ-activatable near-infrared fluorescent probe for visualizing sulfur dioxide derivatives and their fluctuations in vivo, J. Mater. Chem. B, 2020, 8, 1914 RSC; (b) J. C. Xu, J. Pan, X. M. Jiang, C. Q. Qin, L. T. Zeng, H. Zhang and J. F. Zhang, A mitochondria-targeted ratiometric fluorescent probe for rapid, sensitive and specific detection of biological SO2 derivatives in living cells, Biosens. Bioelectron., 2016, 77, 725 CrossRef CAS.
- (a) W. Q. Zhao, P. Y. Xu, Y. X. Ma, Y. M. Song, Y. H. Wang, P. P. Zhang, B. Li, Y. M. Zhang, J. L. Li and S. P. Wu, Old trees bloom new flowers, lysosome targeted near-infrared fluorescent probe for ratiometric sensing of hypobromous acid in vitro and in vivo based on Nile red skeleton, Bioorg. Chem., 2024, 143, 107031 CrossRef CAS PubMed; (b) K. Q. Liu, J. Zhang, X. X. Li, Y. Y. Xie, Y. Li, X. Wang, X. Y. Jiao, X. L. Xie and B. Tang, Hypochlorous acid-activated two-photon fluorescent probe for evaluation of anticancer drug-induced cardiotoxicity and screening of antioxidant drugs, Org. Chem. Front., 2022, 9, 6795 RSC.
- X. Chen and J. R. Cubillos-Ruiz, Endoplasmic reticulum stress signals in the tumour and its microenvironment, Nat. Rev. Cancer, 2021, 21, 71 CrossRef CAS PubMed.
- (a) I. L. Lemmer, N. Willemsen, N. Hilal and A. Bartelt, A guide to understanding endoplasmic reticulum stress in metabolic disorders, Mol. Metab., 2021, 47, 101169 CrossRef CAS PubMed; (b) L. J. Gui, J. Yan, J. Y. Zhao, S. Y. Wang, Y. Y. Ji, J. Liu, J. S. Wu, K. Yuan, H. Liu, D. W. Deng and Z. W. Yuan, Hypochlorite activatable ratiometric fluorescent probe based on endoplasmic reticulum stress for imaging of atherosclerosis, Biosens. Bioelectron., 2023, 240, 115660 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2296981, 2296983, 2296985–2296990 and 2296993. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01628a |
| This journal is © the Partner Organisations 2025 |