
General ppm-level Pd-catalysed asymmetric diarylalkyne hydrosilylation to access structurally diverse Si-stereogenic vinylsilanes†
Heng
Xu‡
a,
Han-Xiao
Yu
a,
An-Jiu
Wen
a,
Fang-Ying
Ling
a,
Fei
Ye
a,
Li
Li
a,
Fuk Yee
Kwong
 b and
Li-Wen
Xu
*a
b and
Li-Wen
Xu
*a
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, and Key Laboratory of Organosilicon Material Technology of Zhejiang Province, Hangzhou Normal University, No. 2318, Yuhangtang Road, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn
bDepartment of Chemistry and State Key Laboratory of Synthetic Chemistry and Shenzhen Municipal Key Laboratory of Chemical Synthesis of Medicinal Organic Molecules, Shenzhen Research Institute, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China
First published on 17th October 2024
Abstract
Alkyne hydrosilylation represents a straightforward method to synthesize vinylsilanes, which have found numerous applications in synthetic chemistry and materials science. However, it is challenging to produce silicon-stereogenic hydrosilanes through catalytic asymmetric hydrosilylation of prochiral dihydrosilanes with internal diarylalkynes. Herein, we present a highly efficient palladium-catalyzed asymmetric hydrosilylation of general internal diarylalkynes with different symmetric or asymmetric aryl groups, each proceeding through the desymmetrization of prochiral dihydrosilanes. The efficiency of this approach is systematically demonstrated for the first time in the synthesis of chiral-at-silicon organosilicon compounds bearing various aromatic groups with high enantioselectivity (up to 97% ee), which would be challenging to produce by means of traditional synthetic methods. Unique efficiency and reaction results for the chiral ligand-controllable hydrosilylation reaction of alkynes were also described using a ppm-level Pd catalyst. To date, this is the most efficient palladium-based catalyst system for the catalytic asymmetric hydrosilylation of alkynes.
Introduction
Catalytic hydrosilylation involving the addition of the silicon-hydrogen bond of hydrosilanes to an unsaturated bond of alkenes or alkynes is one of the most important Si–C bond-forming reactions in organosilicon chemistry.1 Since the first hydrosilylation of alkenes was reported in 1947,2a new catalytic strategies for hydrosilylation of various unsaturated compounds, especially alkyne hydrosilylation, have been realized by structurally diverse transition-metal complexes, such as platinum,2 palladium,3 rhodium,4 cobalt,5 iron,6 zinc,7 rare-earth metals,8 and other molecular catalysts,9 presenting a straightforward strategy to afford vinylsilanes and other organosilicon products with a high atom economy. Furthermore, the catalytic hydrosilylation of alkynes and their synthetic application in the synthesis of functionalized vinylsilanes have attracted increasing attention.10 Nevertheless, the development of enantioselective alkyne hydrosilylation for the synthesis of optically pure silicon-stereogenic vinylsilanes is still a challenging task since an effective ligand and chiral catalyst systems are limited and unpredictable owing to the difficulty in desymmetric activation of one of the Si–H bonds on dihydrosilanes.During the past years, different ligands and transition-metal catalyst systems have been developed for the enantioselective hydrosilylation of alkynes to afford chiral silanes or other silacycles.11–14 For example, Tomooka and co-workers reported a pioneering example of asymmetric hydrosilylation of alkynes in 2012,12 in which they found that the enantioselective hydrosilylation of dihydrosilanes with symmetric internal dialkylalkynes but not aromatic alkynes could be completed by [Pt(dba)3] with the aid of TADDOL-derived phosphonite ligands (Fig. 1a). Although the enantioselective induction of the platinum catalyst system was not good enough (up to 86% ee), this method provided a facile process for the desymmetrization of dihydrosilanes as starting materials to afford Si-stereogenic alkenylhydrosilanes. Subsequently, Lu and co-workers developed co-catalyzed one-pot hydrosilylation/hydrogenation of aromatic terminal alkynes to give structurally diverse chiral silanes (>99% ee) (Fig. 1b).13 In 2018, Huang and co-workers reported an efficient method for catalytic asymmetric hydrosilylation of prochiral dihydrosilanes with alkyne, which was completed by a new cobalt/PyBox complex to obtain Si-stereogenic vinylhydrosilanes with excellent enantioselectivities (up to 91% ee) (Fig. 1b).14 Although this is an elegant example of an enantioselective hydrosilylation reaction with terminal alkynes that occurs with high regio- and enantio-selectivity, this catalyst system is not suitable for the enantioselective hydrosilylation of internal diarylalkynes with symmetric aromatic groups because of its inferior enantioselectivity. For example, when diphenylacetylene was used in this reaction, only 63% ee of the corresponding silicon-stereogenic vinylsilane was achieved. Therefore, the development of new catalyst systems for the enantioselective hydrosilylation of simple and internal diarylalkynes with dihydrosilanes is still an underexplored and continuous task.15
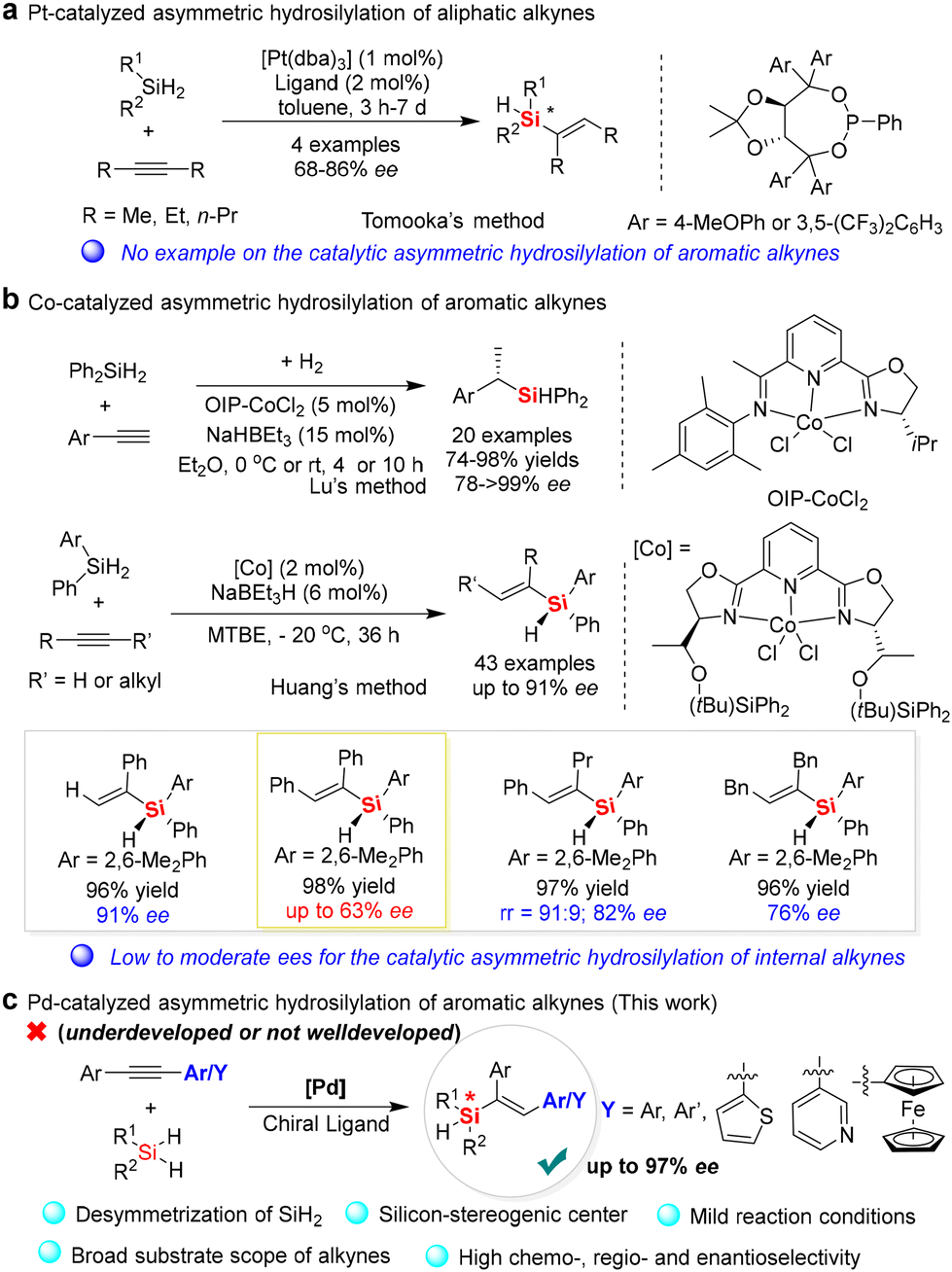 | ||
| Fig. 1 Asymmetric hydrosilylation of alkyne reactions. (a) Pt-catalyzed asymmetric hydrosilylation of aliphatic alkynes. (b) Co-catalyzed asymmetric hydrosilylation of aromatic alkynes. (c) Development of Pd-catalyzed asymmetric hydrosilylation of aromatic alkynes (this work). | ||
In this work, we realize the aim of the efficient synthesis of silicon-stereogenic vinylsilanes through the enantioselective hydrosilylation of internal diarylalkynes and describe a powerful ligand-enabled desymmetrization platform using a chiral P-ligand to control the enantioselective palladium-catalyzed hydrosilylation of dihydrosilanes with general aromatic internal alkynes (Fig. 1c), resulting in high enantioselectivity through desymmetric Si–H bond activation and new Si–C bond formation. By exploiting the palladium catalysis enhanced by the chiral P-ligand, a broad scope of aromatic or heterocyclic internal alkynes can be efficiently converted into silicon-stereogenic organosilicon compounds with compatible functional groups.
Results and discussion
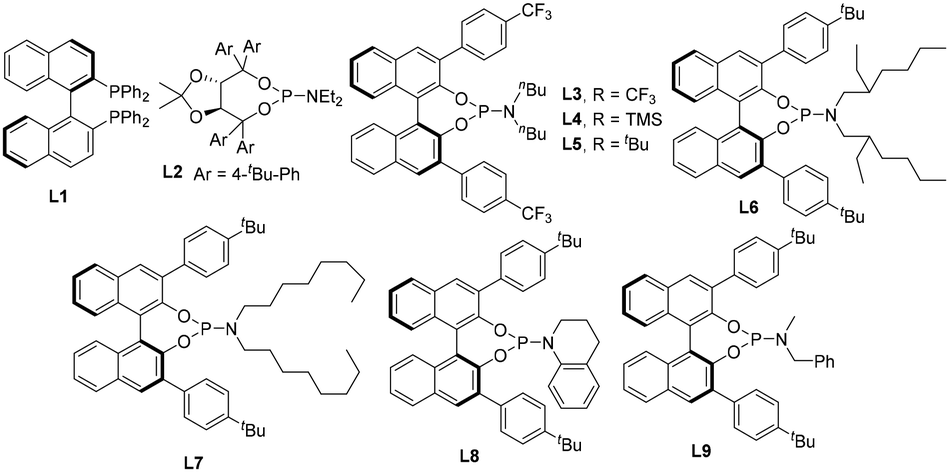
Initially, we investigated the enantioselective palladium-catalyzed hydrosilylation of diphenylacetylene, a symmetric and aromatic internal alkyne, 1a, with hydrosilane 2a. The model reaction was performed using Pd2(dba)3 as a precatalyst and in the presence of a series of chiral P-ligands to establish the optimized reaction conditions. It was found that the chiral P-ligands played varied roles in this reaction, for example, when BINAP (L1) was used as the chiral ligand in dichloromethane (DCM) at 0 °C, poor enantioselectivity (10% ee) and low yield (10%) were obtained. Also, the TADDOL-derived phosphoramidite ligand (L2) exhibited no enantioselective induction in this reaction (entries 1 and 2, Table 1). Thus, inspired by previous findings on the palladium-catalyzed hydrosilylation of 1,3-diynes and ynones,16 we focused on investigating BINOL-derived phosphoramidite ligands for this reaction with cyclohexane as the solvent at 0 °C. As expected, some exciting findings in the Pd-catalyzed enantioselective hydrosilylation of diphenylacetylene 1a were observed, as shown in Table 1. By examining a set of chiral BINOL-derived P-ligands and their analogues, we were pleased to find that most of the BINOL-derived P-ligands enabled the enantioselective synthesis of silicon-stereogenic vinylsilane 3a, albeit with different enantioselective induction and conversion of dihydrosilane 2a. For example, BINOL-derived phosphoramidite ligand L8 exhibited inferior enantioselectivity in comparison to that of L3–L7. Further investigation revealed that BINOL-derived phosphoramidite ligand L9 would be an ideal P-ligand, which was obtained by precise modification of the phenyl substituents on the binaphthyl backbone or nitrogen atom, for the enhancement of enantioselective induction in this asymmetric Pd-catalyzed hydrosilylation (entry 9, 90% yield and 92% ee).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.2 mmol) and 2a (0.2 mmol), with Pd2(dba)3·CHCl3 (2 mol%), chiral P-ligand (8 mol%), solvent (2 mL), at 0 °C for 6 h and the data of yield is determined by NMR analysis. b Isolated yield. c The ee values were determined through HPLC using chiral columns. | ||||
| 1 | L1 | DCM | 10 | 10 |
| 2 | L2 | DCM | 30 | 0 |
| 3 | L3 | c-Hex | 85 | 82 |
| 4 | L4 | c-Hex | 65 | 85 |
| 5 | L5 | c-Hex | 75 | 72 |
| 6 | L6 | c-Hex | 88 | 87 |
| 7 | L7 | c-Hex | 89 | 83 |
| 8 | L8 | c-Hex | 88 | 87 |
| 9 | L9 | c-Hex | 90 | 92 |
|
||||
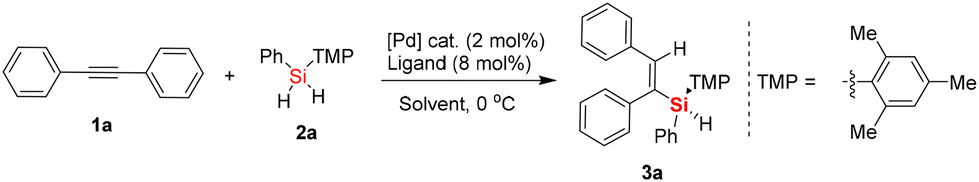
With the BINOL-derived phosphoramidite ligand L9 in hand, and after successfully determining the optimized reaction conditions, subsequently we investigated the substrate scope of the asymmetric Pd-catalyzed hydrosilylation of aromatic internal alkynes 1 and dihydrosilanes 2. Firstly, a series of symmetric aromatic alkynes bearing various electron-withdrawing, electron-neutral, and electron-donating groups on the phenyl ring was tested (Scheme 1). We found that the Pd-catalyzed asymmetric hydrosilylation of aromatic internal alkynes proceeded smoothly to give the desired silicon-stereogenic vinylsilanes 3a–3q with excellent enantioselectivity (up to 94% ee). Also, the enantioselectivity was generally high between 90% ee and 94% ee when aromatic alkynes with substituted groups (Me, iPr, Et, F, CF3, Cl, Br, etc.) on the para-position were used in this reaction, which revealed that the reactivity of the aromatic internal alkynes was not affected by the electrical properties of the substituents on the internal alkynes. We also checked other aromatic internal alkynes with substituted groups on the meta-position of the phenyl ring, in which the corresponding products could be achieved in good yields and enantiomeric excess. For example, product 3k bearing a meta-substituted fluorine was obtained in 90% yield and 91% ee. However, methoxy-substituted aromatic internal alkynes resulted in inferior enantioselectivity in this reaction (80% ee). Notably, this reaction could not tolerate ortho-substituted aromatic alkynes, which always resulted in poor transformations. For example, when 1,2-di-o-tolylethyne was used the substrate, poor yield of the desired product 3u was obtained in this reaction. Fortunately, the use of 1,2-bis(2-fluorophenyl)ethyne as the substrate led to the formation of silicon-stereogenic vinylsilane 3l with excellent yield and ee (98% yield, 92% ee). In addition, we also evaluated the asymmetric Pd-catalyzed hydrosilylation of symmetric internal alkynes with heterocyclic rings (1,2-di(thiophen-2-yl)ethyne) and found that good yield and enantioselectivity could be achieved in this case (3o, 88% yield and 84% ee). This result indicates that the sulfur atom on the thiophene ring did not have a significant effect on the chiral palladium catalyst system, leading to a slight decrease in enantioselectivity. Similarly, 1,2-di(pyridin-3-yl)ethyne was also an effective substrate in the Pd-catalyzed hydrosilylation, giving the corresponding silicon-stereogenic vinylsilane 3q with 87% ee. Similar to previous work,16,17 we also found that steric repulsion on aromatic dihydrosilanes is crucial for the enhancement in enantioselectivity in this reaction. For example, when phenyl(o-tolyl)silane was used as the substrate in this hydrosilylation, only 34% ee of the corresponding product 3r was achieved. Unfortunately, the hydrosilylation of diphenylacetylene with methyl(phenyl)silane or tert-butyl(phenyl)silane resulted in product 3s or 3t without the exact ee value because it could not be determined by chiral HPLC analysis. Although there is no report on the catalytic hydrosilylation of 1,2-bis(4-nitrophenyl)ethyne to date, we checked this reaction with dihydrosilane 2a and found only trace product 3v was detected.
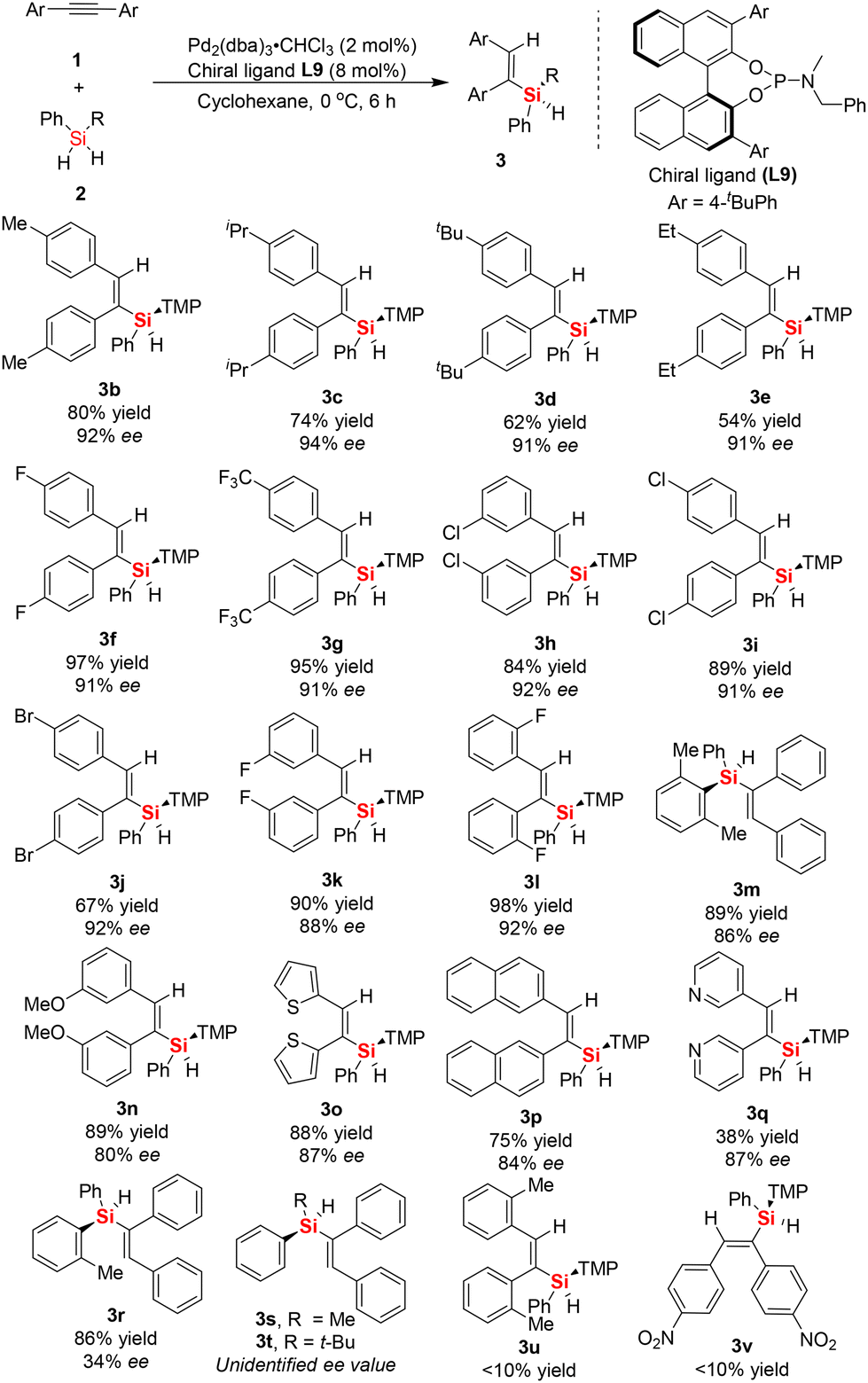 | ||
| Scheme 1 Substrate scope of enantioselective Pd-catalyzed hydrosilylation of aromatic internal alkynes to access silicon-stereogenic vinylsilanes. | ||
Subsequently, the gram-scale synthesis of silicon-stereogenic vinylsilane 3a was investigated to realize the synthetic practicality (Scheme 2). It should be noted that this year, Profs. Luescher, Gallou and Lipshutz jointly published an important perspective about the impact of earth-abundant metals as a replacement for palladium catalysts in cross-coupling reactions, which provides comprehensive and objective support for the synthetic value of palladium catalyst systems.18 Thus, the price of palladium catalysts should not be the main consideration in the development of atom-economic catalytic transformations. In this case, to realize the powerful potential of the Pd-catalyzed hydrosilylation of aromatic alkynes, it is necessary to significantly reduce the catalytic amount of Pd catalyst in this reaction. We evaluated the model reaction of 1a with 2a on a 20 mmol scale to support the synthetic potential of this enantioselective Pd-catalyzed hydrosilylation. As shown in eqn (1) of Scheme 2, the use of a low loading of Pd catalyst (0.03 mol% of Pd2(dba)3) resulted in the corresponding silicon-stereogenic vinylsilane in the same level of excellent enantioselectivity (87% ee), albeit the yield of 3a decreased slightly to 78%. In this case, the yield of the desired product was obtained to reach a record-high turnover number (TON, 1300) in catalytic hydrosilylation. To the best of our knowledge, besides the Pd-catalyzed cross-coupling reaction that enabled the use of ppm-level palladium catalyst systems in the past,19 this is the first example of using ppm-level palladium catalysts to achieve homogenous catalytic hydrosilylation reactions of alkynes20 and realize good enantioselective control in this process.
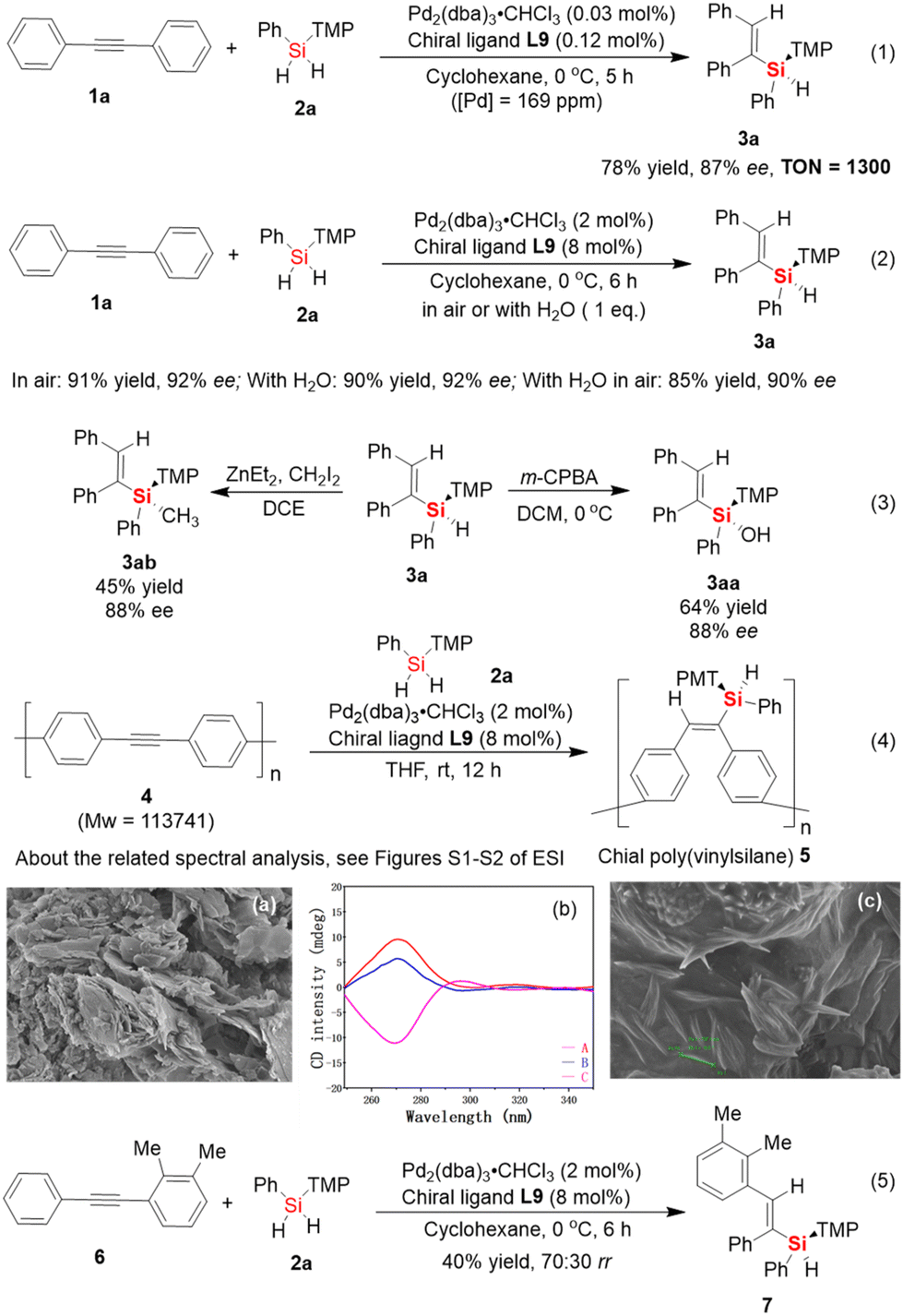 | ||
| Scheme 2 Gram-scale synthesis of silicon-stereogenic vinylsilane 3a with the ppm level of palladium catalyst, the determination of new reaction systems that are insensitive to water or air, and a preliminary result for enantioselective Pd-catalyzed hydrosilylation of polyalkyne and asymmetric diarylalkyne. | ||
Moreover, evidence that the palladium catalysts are not easily deactivated was also obtained from other control experiments (eqn (2) of Scheme 2). For example, the Pd-catalyzed hydrosilylation reaction could be carried out in air without protection of the reaction system under a special nitrogen or argon atmosphere. Also, the addition of a trace amount of water (1 eq.) to the solvent had almost no impact on the catalytic performance, giving the corresponding silicon-stereogenic vinylsilane 3a without any loss of yield and enantioselectivity (90% yield, 92% ee). When this reaction was carried out in the presence of 1 eq. H2O under an air atmosphere, the desired product was obtained with slightly inferior results of 85% yield and 90% ee. In addition, silicon-stereogenic silane 3a could be transformed into Si-stereogenic silanol 3aa and methyl-substituted derivatives 3ab with retention of the configuration of the chirality-at-silicon (eqn (3)).16b,21 These experimental results are sufficient to support that the enantioselective Pd-catalyzed hydrosilylation reaction was unaffected by moisture and oxygen, which may exhibit powerful potential in industrial application and has synthetic value in large-scale preparation.
Next, the enantioselective hydrosilylation of aromatic alkynes inspired us to expand it as a benchtop protocol for the synthesis of a chiral organosilicon polymer bearing a silicon-stereogenic pendant. Although the newly synthesized polyalkyne 4 was prepared as an insoluble and solid material in cyclohexane, we also established the possibility for the catalytic synthesis of a chiral organosilicon polymer bearing silicon-stereogenic centers. In this content, we checked the palladium-catalyzed hydrosilylation of polyalkyne 4 (Mw > 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) with the aid of chiral P-ligand L9 (eqn (4) of Scheme 2). As expected, a high yield of the corresponding poly(vinylsilane) 5 with an Si-stereogenic pendant, as confirmed by IR, SEM, TGA, and circular dichroism (CD) spectra, could be achieved (see eqn (4) and (5) of Scheme 2 and Fig. S1–S4 of the ESI†). For example, during the circular dichroism (CD) analysis, chiral poly(vinylsilane) 5 with a reverse chiral-at-silicon configuration for a pendant Si-stereogenic center exhibited different and corresponding to positive and negative Cotton effect, which realized the CD spectrum of chiral poly(vinylsilane) caused by the fragments of silicon-stereogenic silanes.
000) with the aid of chiral P-ligand L9 (eqn (4) of Scheme 2). As expected, a high yield of the corresponding poly(vinylsilane) 5 with an Si-stereogenic pendant, as confirmed by IR, SEM, TGA, and circular dichroism (CD) spectra, could be achieved (see eqn (4) and (5) of Scheme 2 and Fig. S1–S4 of the ESI†). For example, during the circular dichroism (CD) analysis, chiral poly(vinylsilane) 5 with a reverse chiral-at-silicon configuration for a pendant Si-stereogenic center exhibited different and corresponding to positive and negative Cotton effect, which realized the CD spectrum of chiral poly(vinylsilane) caused by the fragments of silicon-stereogenic silanes.
Notably, the development of the catalytic asymmetric hydrosilylation reaction of asymmetric internal diarylalkynes is one of the most challenging reaction systems in synthetic chemistry, given that the substituents at both ends of aromatic internal alkynes rarely have significant differences in their electrical properties. Thus, two possible products are always produced because of the poor regioselectivity in hydrosilylation reaction. Based on the exploration of the scope of the Pd-catalyzed hydrosilylation with symmetric and aromatic internal alkynes, the poor reactivity of ortho-substituted aromatic internal alkynes provided useful information to design effective asymmetric internal alkynes for regioselective hydrosilylation. To test the hypothesis that an asymmetric internal alkyne bearing an ortho-substituted aromatic ring may be a suitable substrate for the regioselective formation of an Si–C bond, which occurs on the side of the internal alkyne with lower steric hindrance, we checked the Pd-catalyzed hydrosilylation of asymmetric and aromatic internal alkyne 6 with dihydrosilane 2a. Interestingly, the desired product 7 was obtained in only moderate yield and regioselectivity (40% yield of mixed isomers with 70:30 rr). This result indicates that it is difficult to achieve the regioselective Si–C bond-forming hydrosilylation of asymmetric and aromatic alkynes through the precise tuning of the steric hindrance effect. Therefore, it is expected to achieve high regioselective hydrosilylation of asymmetric internal alkynes by controlling the different electronic effects at both ends of alkynes.
As is well known, ferrocene displays high stability, exceptionally well-defined redox features, and interesting geometries with two cyclopentadienyl rings.22 In particular, the special electronic properties of ferrocene and its derivatives make them ideal candidates in the field of organometallic chemistry and materials science.22,23 Accordingly, there are significant differences in the aromaticity or electronic effects between ferrocene and benzene rings.24 Thus, to expand the scope of this protocol, we investigated the catalytic asymmetric hydrosilylation of asymmetric internal alkynes with a ferrocenyl group and aromatic ring at both ends of the internal alkyne (ferrocenyl phenylacetylenes), respectively (Scheme 3). Interestingly, when asymmetric ferrocenyl phenylacetylene 8a was employed as the model substrate to react with dihydrosilane 2a under the standard conditions, only one isomer of vinylsilane (9a) was achieved with excellent regioselectivity (>20:1) and good enantioselectivity (97% ee). Then, we conducted substrate-scope studies to determine the range of substituted ferrocenyl arylacetylenes that can be used in the enantioselective hydrosilylation reaction with dihydrosilane, in which a series of ferrocenyl-substituted silicon-stereogenic vinylsilanes 9 was obtained in good yields (up to 94%) and excellent regio- and enantio-selectivity (up to 20:1 rr and 90–97% ee). As shown in Scheme 3, our results showed that the hydrosilylation reaction was effective with a variety of asymmetric ferrocenyl arylacetylenes, regardless of the substituents on the benzene ring, such as Me, t-Bu, Cl, Br, F, CF3, and OMe. Additionally, the reaction proceeded well with ferrocenyl arylacetylenes bearing a heterocyclic substituent in good yield and enantioselectivity (for example, 92% yield and 90% ee for 9m, 65% yield and 90% ee for product 9n, respectively). The excellent regioselectivity and enantioselectivity of the ferrocenyl arylacetylenes can be attributed to the different electronic properties on the two alkynyl carbons and steric hindrance of dihydrosilane, which provided suitable coordination for Si–Pd–H insertion into the alkynyl substrate.
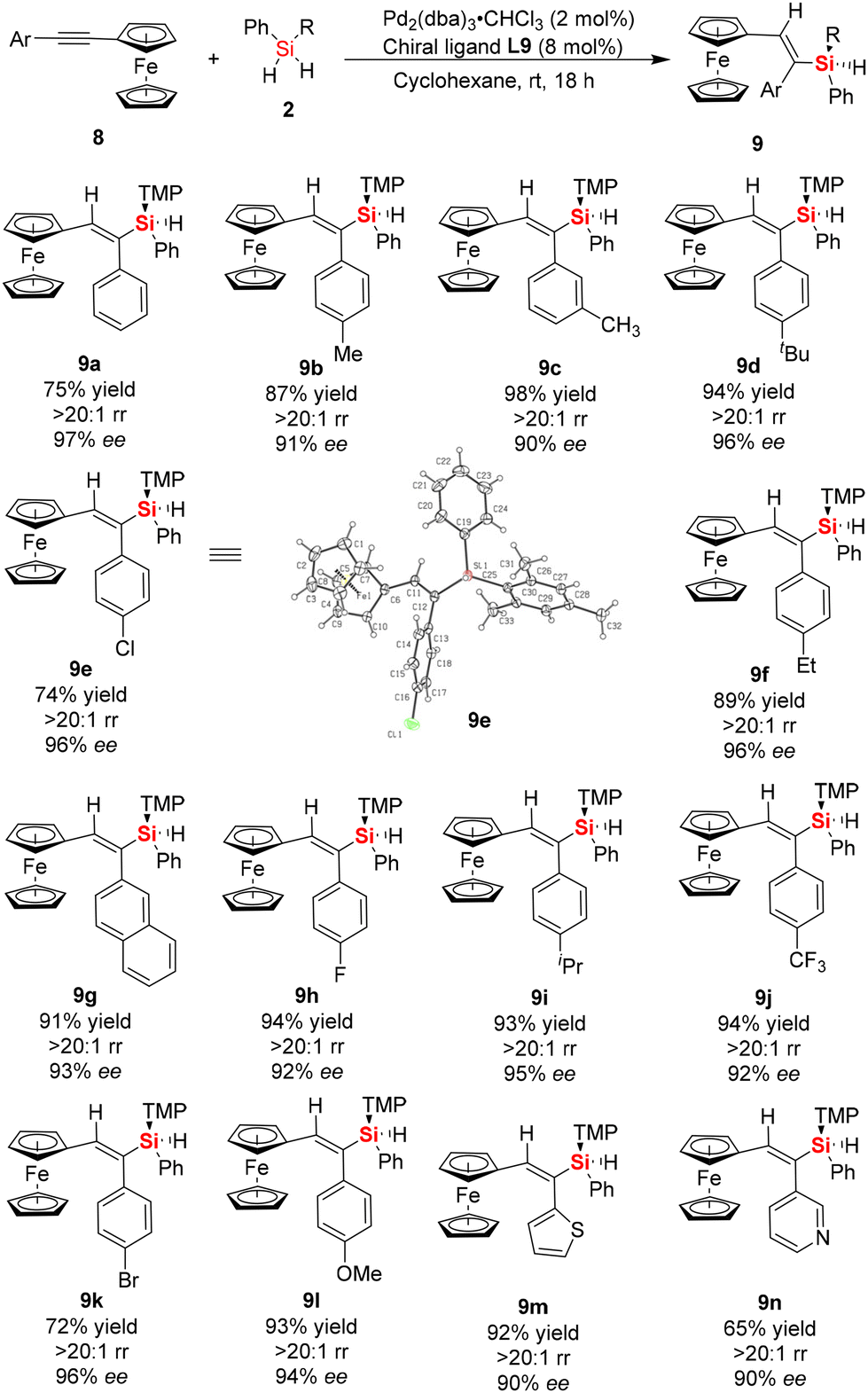 | ||
| Scheme 3 Synthesis of ferrocene-derived chiral Si-stereogenic vinylsilanes 9 from enantioselective Pd-catalyzed hydrosilylation of asymmetric ferrocene-based aromatic alkynes. | ||
To gain insight into the reaction mechanism of the Pd-catalyzed hydrosilylation of diarylalkyne with dihydrosilane, we performed a kinetic study to reveal the effect of product 3a on the reaction. Similar to previous work,16b the Pd-catalyzed hydrosilylation could be accelerated by the addition of a catalytic amount of product 3a (see Fig. 2a). Although almost no product was obtained in the absence of the chiral P-ligand when 3a was used as an additive in this Pd-catalyzed hydrosilylation, these experimental results also support the possibility of product-involved autocatalysis because of the existence of catalyst aggregation in this reaction. Moreover, the non-linear effect (NLE)25 was investigated in the presence of ligand L9 (Fig. 2b), and it was found to be a positive model of NLE under the optimized reaction conditions. Notably, this reaction could be successfully carried out in an air atmosphere or aqueous solvent to achieve the desired products with a high ee value (90%–92% ee, see Scheme 2). These experimental results revealed that the catalyst aggregation would be possibly formed by the product-involved molecular interaction (aromatic interaction) between the catalyst and substrate in this reaction.
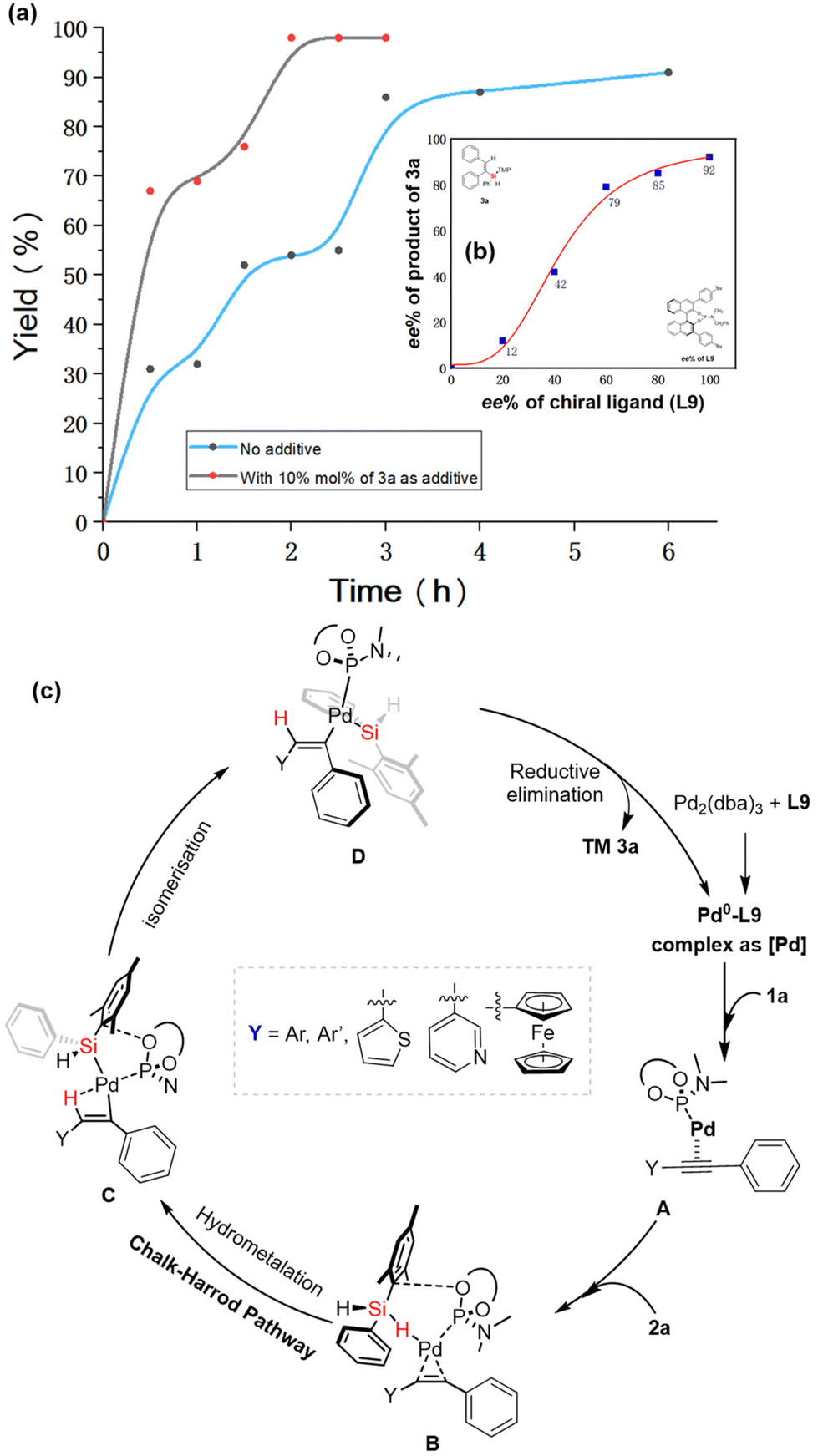 | ||
| Fig. 2 Controlled experiment for the proposed mechanism for Pd-catalyzed hydrosilylation of aromatic alkynes. (a) Kinetic analysis for the determination of the additive effect of product 3a on the Pd-catalyzed hydrosilylation of 1a and 2a. (b) Non-linear effect (NLE) study for Pd-catalyzed hydrosilylation of 1a with 2a. (c) Proposed reaction mechanism. | ||
Based on the previously reported mechanism of palladium-catalyzed hydrosilylation of alkynes,16 we proposed the reaction mechanism in Fig. 2c. Initially, the alkyne-coordinated Pd species A is generated from the Pd2(dba)3/L9 complex with ligand exchange and substrate coordination with diarylalkyne. Then, the Si–H activation of intermediate B and subsequent hydrometallation accelerated by the Pd catalyst occur to give intermediate C through the α-selective alkyne insertion of a hydrogen atom. Finally, the reductive elimination of Pd species D gives the desired silicon-stereogenic vinylsilanes, and the corresponding palladium species is re-generated for the next catalytic cycles.
Conclusions
In summary, we reported the first Pd-catalyzed asymmetric hydrosilylation of diarylalkynes with dihydrosilanes using a chiral phosphine ligand. The catalytic asymmetric hydrosilylation of symmetric diarylalkynes and asymmetric ferrocenyl arylacetylenes was characterized by a wide substrate scope and excellent regioselectivity under mild conditions, in which the enantioselective synthesis of the corresponding silicon-stereogenic vinylsilanes with high enantioselectivities (up to 97% ee) was achieved. In addition, this method could be expanded to the facile synthesis of chiral polyvinylsilane bearing a silicon-stereogenic pendant. Furthermore, we found that a ppm-level of Pd catalyst could be used for the asymmetric hydrosilylation of diarylalkynes with dihydrosilanes. With the BINOL-derived phosphoramidite ligand, a palladium catalyst loading as low as 169 ppm could efficiently catalyze this hydrosilylation with a high TON of 1300.Author contributions
L. W. X. conceived the concept. H. X. developed the enantioselective Pd-catalyzed hydrosilylation of aromatic alkynes. H. X. Y. carried out some hydrosilylation reaction, A. J. W. carried out the DFT calculation and spectral analysis of absolute configuration of new products. F. Y. L., F. Y., L. L. and F. Y. K. assisted with structural analysis of the new silicon-stereogenic product and theoretical analysis of reaction pathway. All authors discussed the results and participated in writing the manuscript. L. W. X. supervised the project and wrote this paper.Data availability
All experimental procedures, details of the optimization, and additional reaction data can be found in the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the grants of National Natural Science Foundation of China (NSFC No. 22371060, 22072035, and 22361162606), NSFC/RGC Joint Research Scheme (N_CUHK426/23), Special Support Program for High-level Talents of Zhejiang Province (2021R51005), Zhejiang Provincial Natural Science Foundation of China (No. LZ23B020002, LR22B020002 and LY22B020006) are gratefully acknowledged. The authors also thank Dr Y. M. Cui, Dr K. Z. Jiang, Dr K. F. Yang and Dr Z. Xu for their assistance on the spectral analysis.References
- For recent reviews, see: (a) J. V. Obligacion and P. J. Chirik, Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed; (b) R. Y. Shi, Z. K. Zhang and X. L. Hu, Nickamine and Analogous Nickel Pincer Catalysts for Cross-Coupling of Alkyl Halides and Hydrosilylation of Alkenes, Acc. Chem. Res., 2019, 52, 1471–1483 CrossRef CAS; (c) K. Kucinski and G. Hreczycho, Hydrosilylation and hydroboration in a sustainable manner: from Earth-abundant catalysts to catalyst-free solutions, Green Chem., 2020, 22, 5210–5224 RSC; (d) K. M. Lewis and S. Couderc, 1946 and the Early History of Hydrosilylation, Molecules, 2022, 27, 4341 CrossRef CAS; (e) E. S. Gulyaeva, E. S. Osipova, R. Buhaibeh, Y. Canac, J.-B. Sortais and D. A. Valyaev, Towards ligand simplification in manganese-catalyzed hydrogenation and hydrosilylation processes, Coord. Chem. Rev., 2022, 458, 214421 CrossRef CAS; (f) L. Li, W. S. Huang, Z. Xu and L. W. Xu, Catalytic asymmetric silicon-carbon bond-forming transformations based on Si-H functionalization, Sci. China: Chem., 2023, 66, 1654–1687 CrossRef CAS; (g) W. Sun and S. F. Zhu, Hydrosilylation Reactions of Alkene with Tertiary Silanes Catalyzed by Iron-Series Metals, Chin. J. Org. Chem., 2023, 43, 3339–3351 CrossRef CAS; (h) J. Jose and T. V. Mathew, Cobalt-Catalyzed Hydrosilylation across Carbon-Carbon, Carbon-Oxygen, and Carbon-Nitrogen Multiple Bonds - A Comprehensive Review, ChemCatChem, 2024, 16, e202301626 CrossRef CAS; (i) A. Vignesh, J. Liu, Z. Wang, Y. Liu and Z. Ke, Nascent developments in main group element-catalyzed hydrosilylation and dehydrogenative silylation of alkenes and alkynes, Org. Chem. Front., 2024, 11, 576–596 RSC.
- (a) L. H. Sommer, E. W. Pietrusza and F. C. Whitmore, Peroxide-Catalyzed Addition of Trichlorosilane to 1-Octene, J. Am. Chem. Soc., 1947, 69, 188 CrossRef CAS; (b) Z. Lamprecht, F. P. Malan, S. Lotz and D. I. Bezuidenhout, Synthesis and structure of thienyl Fischer carbene complexes of PtII for application in alkyne hydrosilylation, New J. Chem., 2021, 45, 6220–6230 RSC; (c) E. E. Ondar, J. V. Burykina and V. P. Ananikov, Evidence for the “cocktail” nature of platinum-catalyzed alkyne and alkene hydrosilylation reactions, Catal. Sci. Technol., 2022, 12, 1173–1186 RSC; (d) L. Ibanez-Ibanez, A. Lazaro, C. Mejuto, M. Crespo, C. Vicent, L. Rodríguez and J. A. Mata, Visible light harvesting alkyne hydrosilylation mediated by pincer platinum complexes, J. Catal., 2023, 428, 115155 CrossRef CAS; (e) Z.-L. Wang, Y. Wang, Y.-C. Sun, J.-B. Zhao and Y.-H. Xu, Regiodivergent Hydrosilylation of Polar Enynes to Synthesize Site-Specific Silyl-Substituted Dienes, Angew. Chem., Int. Ed., 2024, 63, e202405791 CrossRef CAS.
- H. Zhou and C. Moberg, Regio- and stereoselective hydrosilylation of 1,3-enynes catalyzed by palladium, Org. Lett., 2013, 15, 1444–1447 CrossRef CAS PubMed.
- (a) A. Mori, E. Takahisa, Y. Yamamura, T. Kato, A. P. Mudalige, H. Kajiro, K. Hirabayashi, Y. Nishihara and T. Hiyama, Stereodivergent syntheses of (Z)- and (E)-alkenylsilanes via hydrosilylation of terminal alkynes catalyzed by rhodium(I) iodide complexes and application to silicon-containing polymer syntheses, Organometallics, 2004, 23, 1755–1765 CrossRef CAS; (b) B. Sanchez-Page, J. Munarriz, M. V. Jimenez, J. J. Perez-Torrente, J. Blasco, G. Subias, V. Passarelli and P. Alvarez, β-(Z) Selectivity Control by Cyclometalated Rhodium(III)-Triazolylidene Homogeneous and Heterogeneous Terminal Alkyne Hydrosilylation Catalysts, ACS Catal., 2020, 10, 13334–13351 CrossRef CAS; (c) M. González-Lainez, M. V. Jiménez, V. Passarelli and J. J. Pérez-Torrente, β-(Z)-Selective alkyne hydrosilylation by a N,O-functionalized NHC-based rhodium(I) catalyst, Dalton Trans., 2023, 52, 11503–11517 RSC; (d) W. Li, R. Wu, H. Ruan, B. Xiao, X. Gao, H. Jiang, K. Chen, T.-Y. Sun and S. F. Zhu, Axial Ligand Enables Synthesis of Allenylsilane through Dirhodium(II) Catalysis, Angew. Chem., Int. Ed., 2024, 63, e202409332 CrossRef CAS.
- (a) Z. Cheng, J. Guo, Y. Sun, Y. Zheng, Z. Zhou and Z. Lu, Regio-controllable Cobalt-Catalyzed Sequential Hydrosilylation/Hydroboration of Arylacetylenes, Angew. Chem., Int. Ed., 2021, 60, 22454–22460 CrossRef CAS; (b) M. Skrodzki, V. O. Garrido, A. G. Csáky and P. Pawluc, Searching for highly active cobalt catalysts bearing Schiff base ligands for Markovnikov-selective hydrosilylation of alkynes with tertiary silanes, J. Catal., 2022, 411, 116–121 CrossRef CAS; (c) J. W. Park, Cobalt-catalyzed alkyne hydrosilylation as a new frontier to selectively access silyl-hydrocarbons, Chem. Commun., 2022, 58, 491–504 RSC; (d) J. S. Jia, Y. Cao, T. X. Wu, Y. Tao, Y. M. Pan, F. P. Huang and H. T. Tang, Highly regio- and stereoselective Markovinkov hydrosilylation of alkynes catalyzed by high-nuclearity {Co14} clusters, ACS Catal., 2021, 11, 6944–6950 CrossRef CAS; (e) Z. Mo, J. Xiao, Y. Gao and L. Deng, Regio- and Stereoselective Hydrosilylation of Alkynes Catalyzed by Three-Coordinate Cobalt(I) Alkyl and Silyl Complexes, J. Am. Chem. Soc., 2014, 136, 17414–17417 CrossRef CAS PubMed.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, Preparation and molecular and electronic structures of iron(0) dinitrogen and silane complexes and their application to catalytic hydrogenation and hydrosilation, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed; (b) A. M. Tondreau, C. C. Atienza, H. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Iron Catalysts for Selective Anti-Markovnikov Alkene Hydrosilylation Using Tertiary Silanes, Science, 2012, 335, 567–570 CrossRef CAS; (c) M.-Y. Hu, Q. He, S.-J. Fan, Z.-C. Wang, L.-Y. Liu, Y.-J. Mu, Q. Peng and S.-F. Zhu, Ligands with 1,10-phenanthroline scaffold for highly regioselective iron-catalyzed alkene hydrosilylation, Nat. Commun., 2018, 9, 221 CrossRef PubMed; (d) M. Y. Hu, P. He, T. Z. Qiao, W. Sun, W. T. Li, J. Lian, J. H. Li and S. F. Zhu, Iron-Catalyzed Regiodivergent Alkyne Hydrosilylation, J. Am. Chem. Soc., 2020, 142, 16894–16902 CrossRef CAS PubMed; (e) Z. Guo, H. Wen, G. Liu and Z. Huang, Iron-Catalyzed Regio- and Stereoselective Hydrosilylation of 1,3- Enynes To Access 1,3-Dienylsilanes, Org. Lett., 2021, 23, 2375–2379 CrossRef CAS; (f) P. He, M.-Y. Hu, J.-H. Li, T.-Z. Qiao, Y.-L. Lu and S.-F. Zhu, Spin effect on redox acceleration and regioselectivity in Fe-catalyzed alkyne hydrosilylation, Natl. Sci. Rev., 2024, 11, nwad324 CrossRef CAS PubMed.
- I. Raz and R. Dobrovetsky, Highly chemoselective Zn+2-catalyzed hydrosilylation of alkynes, Chem. – Eur. J., 2023, 29, e202300798 CrossRef CAS PubMed.
- (a) X. Xu, A. Gao, W. Chen, J. Li and C. Cui, Lanthanum Ate Amide-catalyzed regio- and stereoselective hydrosilylation of internal alkynes, ACS Catal., 2023, 13, 3743–3748 CrossRef CAS; (b) E. E. Ondar, A. Yu, J. V. Burykina, A. S. Galushko and V. P. Ananikov, Examination of Pt2dba3 as a “cocktail”-type catalytic system for alkene and alkyne hydrosilylation reactions, Catal. Sci. Technol., 2023, 13, 6022–6040 RSC.
- (a) Q. Li, S. Huo, L. Meng and X. Li, Mechanism and origin of the stereoselectivity of manganese-catalyzed hydrosilylation of alkynes: A DFT study, Catal. Sci. Technol., 2022, 12, 2649–2658 RSC; (b) R. Liu, X. Liu, T. Cheng and Y. Chen, Organocalcium complex-catalyzed dehydrogenative coupling of hydrosilanes ith terminal alkynes, Eur. J. Org. Chem., 2022, e202101218 CrossRef CAS; (c) C. Chen, Q. Mo, Y. Wang and L. Zhang, Cooperative catalytic alkyne hydrosilylation by a Porphyrinic metal-organic framework composite, Inorg. Chem., 2023, 62, 16882–16889 CrossRef CAS.
- (a) X. Du, W. Hou, Y. Zhang and Z. Huang, Pincer cobalt complex-catalyzed Z-selective hydrosilylation of terminal alkynes, Org. Chem. Front., 2017, 4, 1517–1521 RSC; (b) W. J. Teo, C. Wang, Y. W. Tan and S. Ge, Cobalt-catalyzed Z-selective hydrosilylation of terminal alkynes, Angew. Chem., Int. Ed., 2017, 56, 4328–4332 CrossRef CAS; (c) Z. Zuo, J. Yang and Z. Huang, Cobalt-catalyzed alkyne hydrosilylation and sequential vinylsilane hydroboration with Markovnikov slectviity, Angew. Chem., Int. Ed., 2016, 55, 10839–10843 CrossRef CAS; (d) Z. Cheng, M. Li, X. Y. Zhang, Y. Sun, Q. L. Yu, X. H. Zhang and Z. Lu, Cobalt-catalyzed regiodivergent double hydrosilylation of arylacetylenes, Angew. Chem., Int. Ed., 2023, 62, e202215029 CrossRef CAS; (e) D. Wang, Y. Lai, P. Wang, X. Leng, J. Xiao and L. Deng, Markovnikov hydrosilylation of alkynes with tertiary silanes catalyzed by dinuclear cobalt carbonyl complexes with NHC ligation, J. Am. Chem. Soc., 2021, 143, 12847–12856 CrossRef CAS PubMed; (f) K. Itami, T. Nokami and J.-i. Yoshida, Facile generation of [bis(2-pyridyldimethylsilyl)methyl]lithium and its reaction with carbonyl compounds. New method for the stereoselective syntheis of vinylsilanes, Tetrahedron, 2001, 57, 5045–5054 CrossRef CAS; (g) J. L. Han, Y. Qin, C. W. Ju and D. Zhao, Divergent Synthesis of Vinyl-, Benzyl-, and Borylsilanes: Aryl to Alkyl 1,5-Palladium Migration/Coupling Sequences, Angew. Chem., Int. Ed., 2020, 59, 6555–6560 CrossRef CAS; (h) J. Szudkowska-Frątczak, G. Hreczycho and P. Pawluć, Silylative coupling of olefins with vinylsilanes in the synthesis of functionalized alkenes, Org. Front. Chem., 2015, 2, 730–738 RSC.
- For recent review, see: (a) Y. Wu and P. Wang, Silicon-Stereogenic Monohydrosilane: Synthesis and Applications, Angew. Chem., Int. Ed., 2022, 61, e202205382 CrossRef CAS; (b) J. Guo and Z. Lu, Highly Chemo-, Regio-, and Stereoselective Cobalt-Catalyzed Markovnikov Hydrosilylation of Alkynes, Angew. Chem., Int. Ed., 2016, 55, 10835–10838 CrossRef CAS; (c) J. Guo, H. Wang, S. Xing, X. Hong and Z. Lu, Cobalt-catalyzed asymmetric synthesis of gen-bis(silyl)alkanes by double hdyrosilylation of aliphatic terminal alkynes, Chem, 2019, 5, 881–895 CrossRef CAS; (d) J. L. Xu, Z. L. Wang, J. B. Zhao and Y. H. Xu, Enantioselective construction of Si-stereogenic linear alkenylhydrosilanes via copper-catalyzed hydrosilylation of alkynes, Chem Catal., 2024, 4, 100887 CrossRef CAS; (e) W. Lu, Y. Zhao and F. Meng, Cobalt-catalyzed sequential site- and stereoselective hydrosilylation of 1,3- and 1,4-enynes, J. Am. Chem. Soc., 2022, 144, 5233–5524 CrossRef CAS PubMed.
- K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Catalytic Enantioselective Synthesis of Alkenylhydrosilanes, Angew. Chem., Int. Ed., 2012, 51, 12745–12748 CrossRef CAS PubMed.
- J. Guo, X. Shen and Z. Lu, Regio- and Enantioselective Cobalt-Catalyzed Sequential Hydrosilylation/Hydrogenation of Terminal Alkynes, Angew. Chem., Int. Ed., 2017, 56, 615–618 CrossRef CAS PubMed.
- H. Wen, X. Wan and Z. Huang, Asymmetric Synthesis of Silicon-Stereogenic Vinylhydrosilanes by Cobalt-Catalyzed Regio- and Enantioselective Alkyne Hydrosilylation with Dihydrosilanes, Angew. Chem., Int. Ed., 2018, 57, 6319–6323 CrossRef CAS.
- P. He, M. Y. Hu, X. Y. Zhang and S. F. Zhu, Transition-metal-catalyzed stereo- and regioselective hydrosilylation of unsymmetric alkynes, Synthesis, 2022, 49–66 CAS.
- (a) J. L. Xie, Z. Xu, H. Q. Zhou, Y. X. Nie, J. Cao, G. W. Yin, J. P. Bouillon and L. W. Xu, Palladium-Catalyzed Hydrosilylation of Ynones to Access Silicon-Stereogenic Silylenones by Stereospecific Aromatic Interaction-assisted Si-H Activation, Sci. China: Chem., 2021, 64, 761–769 CrossRef CAS; (b) F.-Y. Ling, F. Ye, X.-J. Fang, X.-H. Zhou, W.-S. Huang, Z. Xu and L.-W. Xu, An unusual autocatalysis with an air-stable Pd complex to promote enantioselective synthesis of Si-stereogenic enynes, Chem. Sci., 2023, 14, 1123–1131 RSC.
- (a) S. Rendler, G. Auer, M. Keller and M. Oestreich, Preparation of a Privileged Silicon-Stereogenic Silane: Classical versus Kinetic Resolution, Adv. Synth. Catal., 2006, 348, 1171–1182 CrossRef CAS; (b) L. Chen, J.-B. Huang, Z. Xu, Z.-J. Zheng, K.-F. Yang, Y.-M. Cui, J. Cao and L.-W. Xu, Palladium-catalyzed Si–C bond-forming silylation of aryl iodides with hydrosilanes: an enhanced enantioselective synthesis of silicon-stereogenic silanes by desymmetrization, RSC Adv., 2016, 6, 67113–67117 RSC; (c) S. Chen, J. Zhu, J. Ke, Y. Li and C. He, Enantioselective Intermolecular C−H Silylation of Heteroarenes for the Synthesis of Acyclic Si-Stereogenic Silanes, Angew. Chem., Int. Ed., 2022, 61, e202117820 CrossRef CAS PubMed; (d) D. Mu, S. Pan, X. Wang, X. Liao, Y. Huang and J. Chen, Enantioselective synthesis of acyclic monohydrosilanes by steric hindrance assisted C–H silylation, Chem. Commun., 2022, 58, 7388–7391 RSC; (e) W. Yuan, X. Zhu, Y. Xu and C. He, Synthesis of Si-Stereogenic Silanols by Catalytic Asymmetric Hydrolytic Oxidation, Angew. Chem., Int. Ed., 2022, 61, e202204912 CrossRef CAS; (f) W. Yang, L. Liu, J. Guo, S.-G. Wang, J.-Y. Zhang, L.-W. Fan, Y. Tian, L.-L. Wang, C. Luan, Z.-L. Li, C. He, X. Wang, Q.-S. Gu and X.-Y. Liu, Enantioselective Hydroxylation of Dihydrosilanes to Si-Chiral Silanols Catalyzed by In Situ Generated Copper(II) Species, Angew. Chem., Int. Ed., 2022, 61, e202205743 CrossRef CAS PubMed; (g) M.-M. Liu, Y. Xu and C. He, Catalytic Asymmetric Dehydrogenative Si−H/N−H Coupling: Synthesis of Silicon-Stereogenic Silazanes, J. Am. Chem. Soc., 2023, 145, 11727–11734 CrossRef CAS PubMed; (h) J.-L. Xu, Z.-L. Wang, J.-B. Zhao and Y.-H. Xu, Enantioselective construction of Si- stereogenic linear alkenylhydrosilanes via copper-catalyzed hydrosilylation of alkynes, Chem Catal., 2024, 4, 100887 CrossRef CAS.
- M. U. Luescher, F. Gallou and B. H. Lipshutz, The impact of earth-abundant metals as a replacement for Pd in cross coupling reactions, Chem. Sci., 2024, 15, 9016–9025 RSC.
- (a) D. Roy and Y. Uozumi, Recent Advances in Palladium-Catalyzed Cross-Coupling Reactions at ppm to ppb Molar Catalyst Loadings, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef CAS; (b) S. Handa, M. P. Andersson, F. Gallou, J. Reilly and B. H. Lipshutz, HandaPhos: A General Ligand Enabling Sustainable ppm Levels of Palladium-Catalyzed Cross-Couplings in Water at Room Temperature, Angew. Chem., Int. Ed., 2016, 55, 4914–4918 CrossRef CAS PubMed; (c) Y. Zhang, B. S. Takale, F. Gallou, J. Reillyc and B. H. Lipshutz, Sustainable ppm level palladium-catalyzed aminations in nanoreactors under mild, aqueous conditions, Chem. Sci., 2019, 10, 10556–10561 RSC; (d) W. Lin, L. Zhang, Y. Ma, T. Liang and W. H. Sun, Sterically enhanced 2-iminopyridylpalladium chlorides as recyclable ppm-palladium catalyst for Suzuki–Miyaura coupling in aqueous solution, Appl. Organomet. Chem., 2022, 36, e6474 CrossRef CAS.
- J.-J. Chen, J.-H. Zeng, Y. Yang, Z.-K. Liu, Y.-N. Jiang, M.-R. Li, L. Chen and Z.-P. Zhan, A Bithiophene-Promoted ppm Levels of Palladium-Catalyzed Regioselective Hydrosilylation of Terminal Allenes, Adv. Synth. Catal., 2020, 362, 2360–2366 CrossRef CAS.
- It was found that the epoxidation of alkene moiety did not occur and only oxidation of Si–H bond with m-CPBA occurred smoothly to give the product 3aa. This product 3a was quite stable in the presence of alcohol and transition-metal catalysts.
- (a) G. Wilkinson, M. Rosenblum, C. M. Whiting and R. B. Woodward, The structure of iron bis-cyclopentadienyl, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS; (b) T. J. Kealy and P. L. Pauson, A New Type of Organo-Iron Compound, Nature, 1951, 168, 1039–1040 CrossRef CAS; (c) K. Heinze and H. Lang, Ferrocene—Beauty and Function, Organometallics, 2013, 32, 5623–5625 CrossRef CAS; (d) D. Astruc, Why is Ferrocene so Exceptional?, Eur. J. Inorg. Chem., 2017, 2017, 6–29 CrossRef CAS.
- (a) G. Roy, R. Gupta, S. R. Sahoo, S. Saha, D. Asthana and P. C. Mondal, Ferrocene as an iconic redox marker: From solution chemistry to molecular electronic devices, Coord. Chem. Rev., 2022, 473, 214816 CrossRef CAS; (b) S. Giri and I. Dash, Ferrocene to functionalized ferrocene: a versatile redox-active electrolyte for high-performance aqueous and non-aqueous organic redox flow batteries, J. Mater. Chem. A, 2023, 11, 16458–16493 RSC.
- (a) Z. S. Liu, N. Zhu, L. M. Han, R. J. Xie, H. L. Hong and Q. L. Suo, Detecting the electron density of benzene using ferrocene as an indicator, J. Coord. Chem., 2012, 65, 2804–2810 CrossRef CAS; (b) T. L. R. Bennett, L. A. Wilkinson, J. M. A. Lok, R. C. P. O'Toole and N. J. Long, Synthesis, Electrochemistry, and Optical Properties of Highly Conjugated Alkynyl-Ferrocenes and -Biferrocenes, Organometallics, 2021, 40, 1156–1162 CrossRef CAS; (c) T. L. R. Bennett and N. J. Long, A convenient synthesis of ferrocene- (ethynylphenyl)thioacetates, Dalton Trans., 2023, 52, 16465–16471 RSC.
- (a) Y. Geiger and S. Bellemin-Laponnaz, Non-Linear Effects in Asymmetric Catalysis: Impact of Catalyst Precipitation, ChemCatChem, 2022, 14, e202200165 CrossRef CAS; (b) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS PubMed; (c) C. Girard and H. B. Kagan, Nonlinear Effects In Asymmetric Catalysis, Angew. Chem., Int. Ed., 1998, 37, 2922–2959 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2380481. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01698j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |