
Photoinduced cobaloxime catalysis for allylic mono- and diphosphinylation of alkenes with hydrogen evolution†
Aijun
Zhang
a,
Miao-Miao
Li
*a,
Lei
Guo
b,
Huaixiang
Yang
a,
Jiefei
Guo
a,
Da
Xu
b and
Wei
Ding
 *a
*a
aDivision of Molecular Catalysis and Synthesis, Henan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: miaomiaoli116@zzu.edu.cn; ding_w@zzu.edu.cn
bTechnology Center, China Tobacco Jiangxi Industrial Co. Ltd, Nanchang 330096, P. R. China
First published on 29th October 2024
Abstract
The visible-light-induced cobalt-catalyzed hydrogen evolution radical coupling reaction has emerged as a powerful strategy for the synthesis of structurally diverse compounds, whereas transformations with phosphorus-centered radicals remain largely unexplored. Herein, a photoinduced allylic radical phosphinylation of alkenes with secondary phosphine oxides by cobaloxime catalysis is described. The reaction tolerates a wide range of alkenes, including activated electron-deficient alkenes and nonactivated aryl- and alkylsubstituted alkenes, affording allylic phosphine oxides in good yields with high site-selectivities. Furthermore, this photoinduced cobaloxime catalytic system can be extended to the radical diphosphinylation of simple alkenes to construct DPPP-dioxide analogues. The new phosphine–olefin and diphosphine ligands are generated smoothly by reduction of mono- and diphosphinylation products.
Introduction
Organophosphorus compounds are widely used in many fields including medicine and drug discovery, agriculture and crop protection, and materials science, owing to their unique properties.1 Moreover, phosphorus-containing compounds play significant roles in metal-catalyzed and organocatalytic reactions.2 Among these, allylic phosphine oxides are key structural units in some bioactive molecules (Scheme 1a) and serve as versatile building blocks to access a variety of functionalized organophosphorus compounds.3 Consequently, the construction of these scaffolds has attracted substantial interest, and notable progress has been made. Conventional approaches for accessing allylic phosphine oxides typically include transition metal-catalyzed Tsuji–Trost allylic substitution with phosphine oxides as nucleophiles and hydrophosphinylation of 1,3-dienens or allenes (Scheme 1b).4,5 Despite remarkable advancements in this field, the need for the utilization of highly prefunctionalized and specialized starting materials and a relatively narrow substrate scope have limited their general application. Therefore, the development of a novel and efficient strategy for constructing allylic organophosphorus compounds from simple materials remains a highly desirable yet challenging goal.6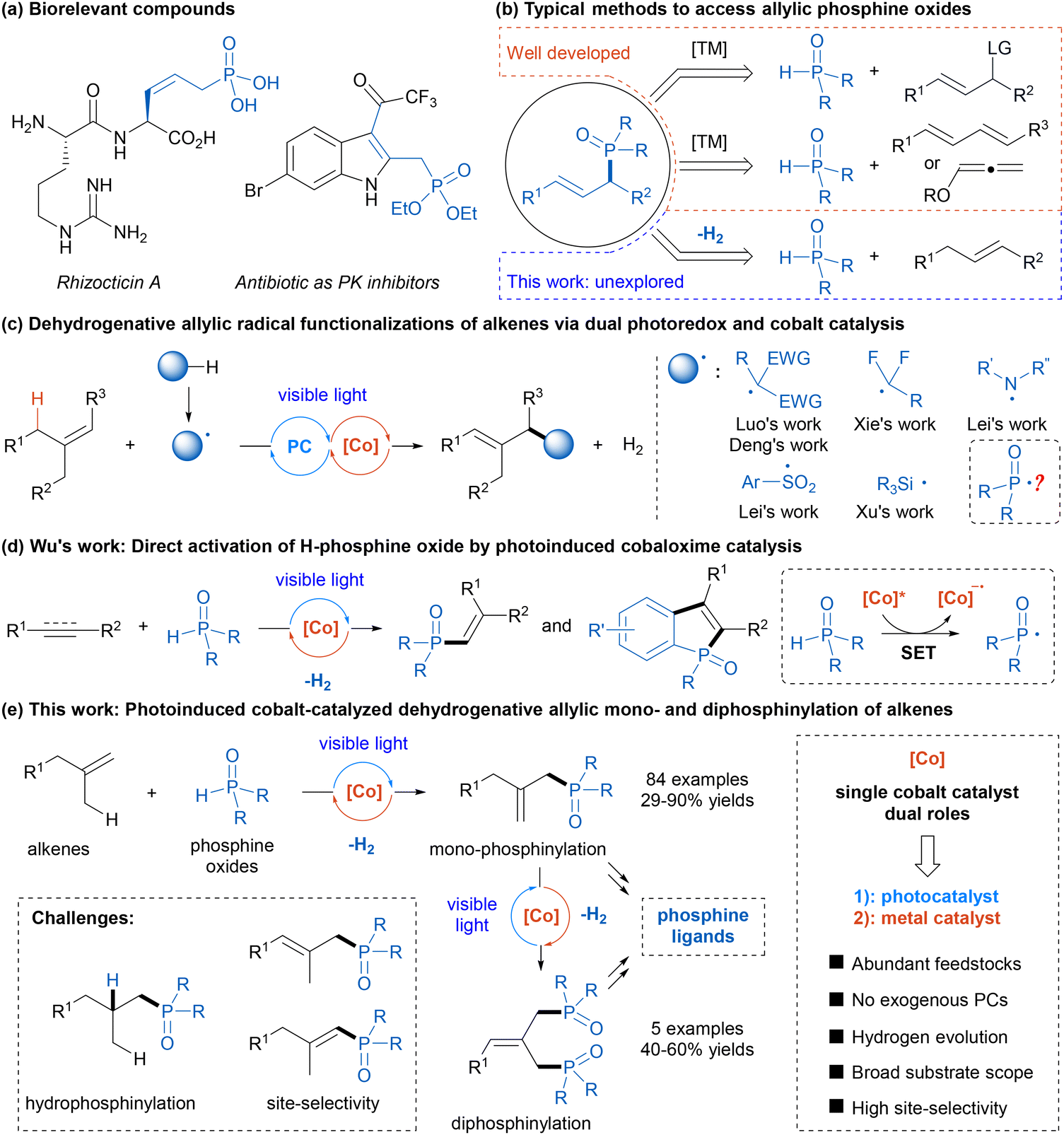 | ||
| Scheme 1 Outline of this work. | ||
With rapid advancements in photochemistry over the past decade, metallaphotoredox catalysis, which combines visible-light photoredox catalysis with transition-metal catalysis, has emerged as a powerful platform for constructing complex molecules under mild conditions.7 In this context, a photoredox/cobalt dual catalytic strategy helps accomplish many new synthetic transformations, which are either not feasible or difficult to perform through single catalytic systems.8 In particular, cobaloxime complexes, initially synthesized to mimic the structure of vitamin B12,9 have been utilized as powerful proton reduction catalysts in conjunction with photocatalysis to achieve various hydrogen-evolution reactions,10,11 avoiding the use of external hydrogen acceptors or chemical oxidants. In recent years, dehydrogenative allylic radical functionalization of alkenes via dual photoredox and cobaloxime catalysis has been exploited as a straightforward and atom-economical strategy to synthesize allylic compounds (Scheme 1c),12 wherein photoredox catalysis can generate radical intermediates through single-electron transfer (SET) or hydrogen atom transfer (HAT), and cobaloxime catalysis facilitates hydrogen evolution. Many kinds of radicals such as alkyl radicals,12a–c nitrogen-centered radicals,12d–f sulfonyl radicals,12g and silyl radicals12h have been applied in this transformation by the Luo, Deng, Xie, Lei, and Xu groups. To our knowledge, the analogous reaction with phosphorus-centered radicals, which may offer a route to allylic organophosphorus compounds, remains largely underexplored. In 2019, Wu and co-workers elegantly developed a direct activation of H-phosphine oxide by photoinduced cobaloxime catalysis to afford a reactive phosphinoyl radical (Scheme 1d).13a Since then, Wu and other groups have used this strategy to achieve radical phosphorylation reactions of unsaturated compounds to afford various alkenylphosphine oxides and phosphorylated heteroaromatics.13 Critical to this success is the dual role of cobaloxime as both a photoredox catalyst and a hydrogen evolution metal catalyst.14 Inspired by these remarkable studies and in continuation of our recent focus on photochemical synthesis,15 we hypothesized that this visible-light-induced cobaloxime catalysis could enable dehydrogenative allylic radical phosphinylation of simple alkenes with secondary phosphine oxides, providing efficient and general access to allylic phosphine oxides (Scheme 1e). And this strategy could also be extended to the dehydrogenative diphosphinylation of alkenes to produce diphosphine oxides. Potential challenges of this strategy include (1) competitive hydrophosphinylation process16 and (2) control of site-selectivity for the regeneration of the double bond.
Results and discussion
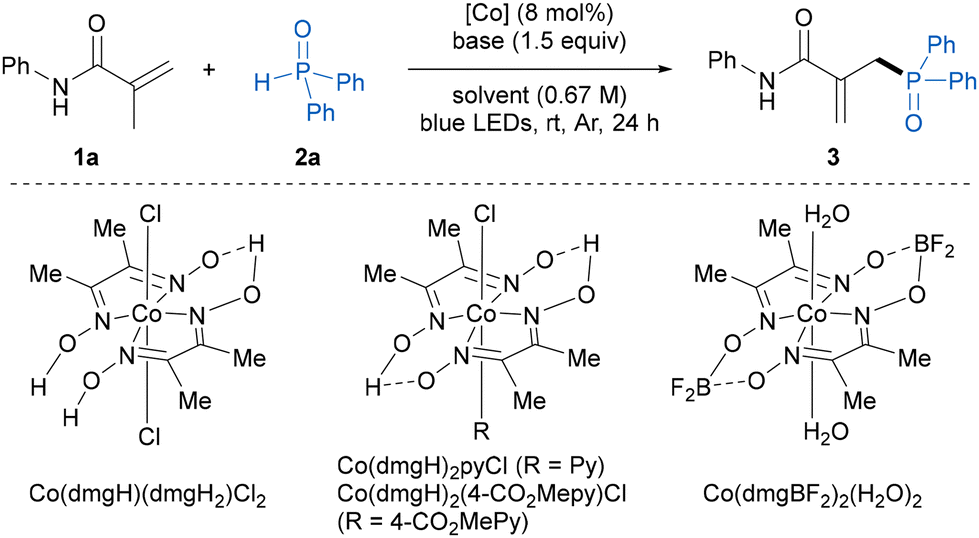
Herein, we began our investigation by evaluating dehydrogenative phosphinylation using N-phenylmethacrylamide 1a and diphenylphosphine oxide 2a as the model substrates (Table 1). To our delight, with the combination of Co(dmgH)(dmgH2)Cl2 (10 mol%) as catalyst and pyridine (1.5 equiv.) as base, the allylic phosphinylation reaction proceeded smoothly in DCM under blue LED irradiation at ambient temperature, affording the expected allylic phosphine oxide product 3 in 79% NMR yield with exclusive site-selectivity (entry 1). Then, a variety of cobaloxime catalysts were investigated. The Co(III) complexes were capable of catalyzing this transformation and Co(dmgH)2(4-CO2Mepy)Cl showed the best catalytic activity with 84% NMR yield, whereas the use of the Co(II)-catalyst Co(dmgBF2)2(H2O)2 led to a significant drop in the yield of 3 (entries 2–4). Notably, decreasing the cobaloxime catalyst loading to 8 mol% gave the same NMR yield and an isolated yield of 78% of 3 (entry 5). Regarding the base, DMAP could be used in place of pyridine and resulted in a decreased yield, while other inorganic and organic bases such as Et3N and K3PO4 completely shut down the reaction (entries 6–8). In addition, the use of other solvents instead of DCM, such as DCE, toluene, and MeCN, resulted in diminished yields (entries 9–11). Control experiments demonstrated that a cobalt catalyst, base and visible light were indispensable in this transformation (entries 12–14). It was found that an excess of the alkene substrate was beneficial for this transformation, and 2 equivalents of 1a gave the highest yield (entry 15 and Table S5†). In fact, some portion of alkene 1a was recovered and no alkene reductive hydrogenation byproduct was observed in the reaction mixture.|
|
||||
|---|---|---|---|---|
| Entry | [Co] | Base | Solvent | Yieldb (%) |
| a Unless otherwise noted, the reaction conditions were as follows: 1a (0.2 mmol, 2.0 equiv.), 2a (0.1 mmol), [Co] (8 mol%), base (1.5 equiv.), solvent (1.5 mL), irradiation via 40 W blue LEDs under Ar at room temperature for 24 h. b Yields are determined by 1H NMR using dibromomethane as an internal standard. c [Co] (10 mol%) was used. d Isolated yield is given in parentheses. e The reaction was performed in the dark. f 1 (0.1 mmol) and 2a (0.1 mmol) were used. | ||||
| 1c | Co(dmgH)(dmgH2)Cl2 | Pyridine | DCM | 79 |
| 2c | Co(dmgH)2pyCl | Pyridine | DCM | 80 |
| 3c | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | DCM | 84 |
| 4c | Co(dmgBF2)2(H2O)2 | Pyridine | DCM | 25 |
| 5 | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | DCM | 84 (78)d |
| 6 | Co(dmgH)2(4-CO2Mepy)Cl | DMAP | DCM | 67 |
| 7 | Co(dmgH)2(4-CO2Mepy)Cl | Et3N | DCM | 0 |
| 8 | Co(dmgH)2(4-CO2Mepy)Cl | K3PO4 | DCM | 0 |
| 9 | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | DCE | 51 |
| 10 | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | Toluene | 48 |
| 11 | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | MeCN | 44 |
| 12 | — | Pyridine | DCM | 0 |
| 13 | Co(dmgH)2(4-CO2Mepy)Cl | — | DCM | 0 |
| 14e | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | DCM | 0 |
| 15f | Co(dmgH)2(4-CO2Mepy)Cl | Pyridine | DCM | 48 |
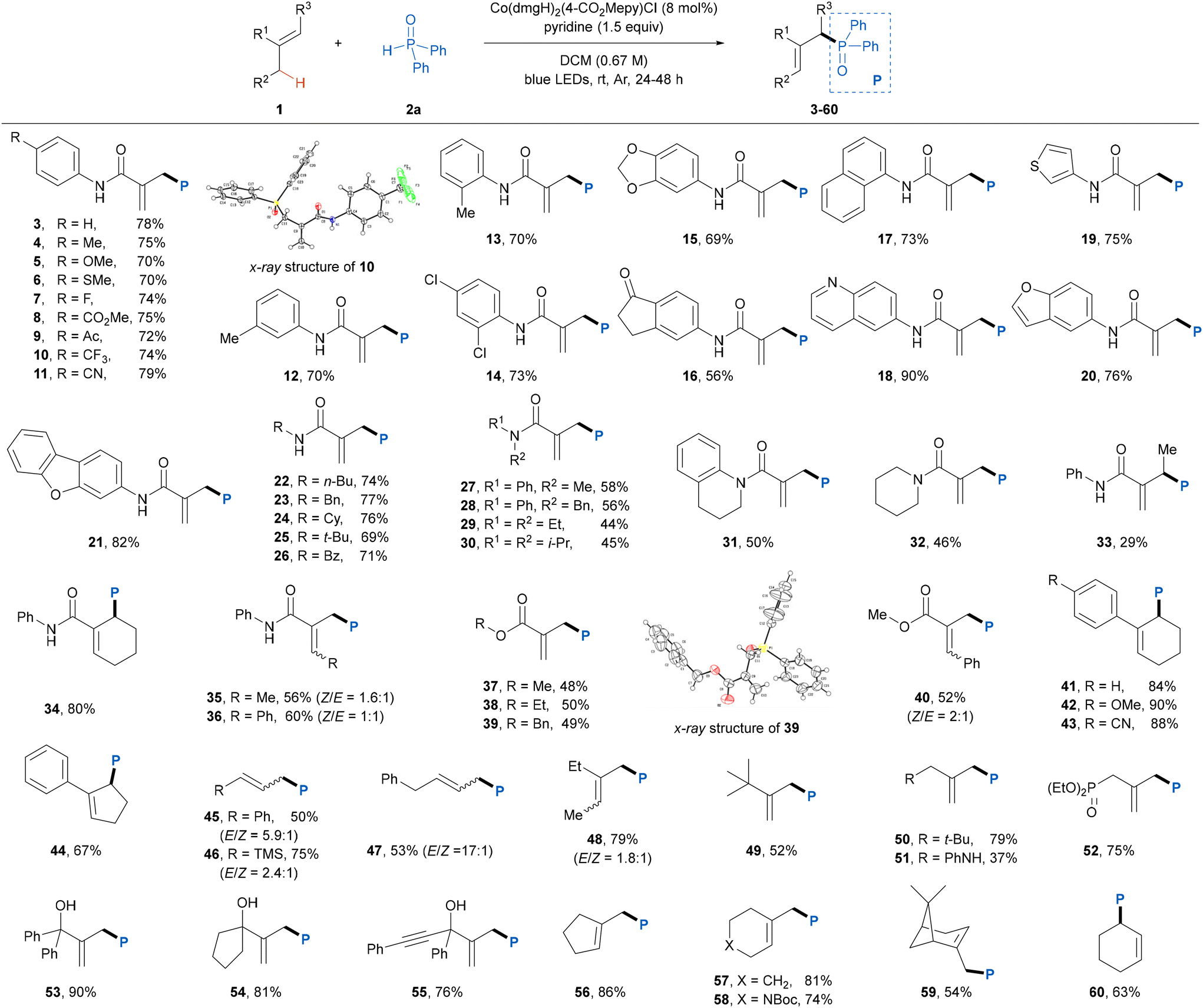
After establishing the optimal reaction conditions, the alkene scope of this photoinduced dehydrogenative phosphinylation reaction was explored (Table 2). Firstly, we investigated the generality of activated electron-deficient alkenes. A diverse range of N-aryl methacrylamides bearing both an electron-donating and an electron-withdrawing group or two substituents at different positions of the benzene ring smoothly participated in this protocol to give the desired products 3–16 in good yields with great site-selectivities. Polycyclic- and heterocyclic-substituted methacrylamides were also suitable for this reaction, affording the corresponding products 17–21 in 73–90% yields. In addition to N-aryl, the methacrylamides featuring N-alkyl or benzoyl groups were also compatible with the present reaction conditions, providing products 22–26 in good yields. It was found that the presence of the N–H bond in methacrylamide substrates was beneficial for this protocol as the unsaturated disubstituted amides afforded relatively low reaction efficiencies (27–32). When an acyclic internal alkene was used in this reaction, a decreased yield (29%) of product 33 was obtained, perhaps because the high steric hindrance led to relatively slower addition rate of the phosphinoyl radical to the double bond, whereas a cyclic internal alkene showed good compatibility with this reaction, delivering the corresponding product 34 in 80% yield. Note that the reactions of ethyl and benzyl acrylamides also proceeded smoothly to produce products 35 and 36 in 56% and 64% yields, respectively, albeit with a low E/Z ratio. We next explored the scope of α,β-unsaturated esters in this transformation. A variety of 2-methyl acrylates proved to be amenable to this transformation, delivering the desired products 37–40 in moderate yields. Moreover, cyclic aromatic alkenes were found to be well tolerated in this protocol, affording the desired products 41–44 in good yields. To further expand the generality of this reaction, we tested more challenging nonactivated aliphatic alkenes, which have usually been shown to be incompatible in some previous studies.12 We were pleased to find that a series of monosubstituted and 1,1-disubstituted aliphatic alkenes gave the corresponding products 45–55 in moderate to good yields with exclusive site-selectivities. Exocyclic alkenes and cyclohexene were also proven to be suitable substrates, giving the desired cyclic alkene derivatives (56–60) in good yields. The precise structures of products 10 and 39 were unambiguously determined by X-ray crystallographic analysis (CCDC 2372952 and 2372953†).
| a Reaction conditions: 1 (0.4 mmol, 2.0 equiv.), 2a (0.2 mmol), Co(dmgH)2(4-CO2Mepy)Cl (8 mol%), pyridine (0.3 mmol, 1.5 equiv.), DCM (3.0 mL), irradiation via 40 W blue LEDs under Ar at room temperature for 24–48 h. |
|---|
|
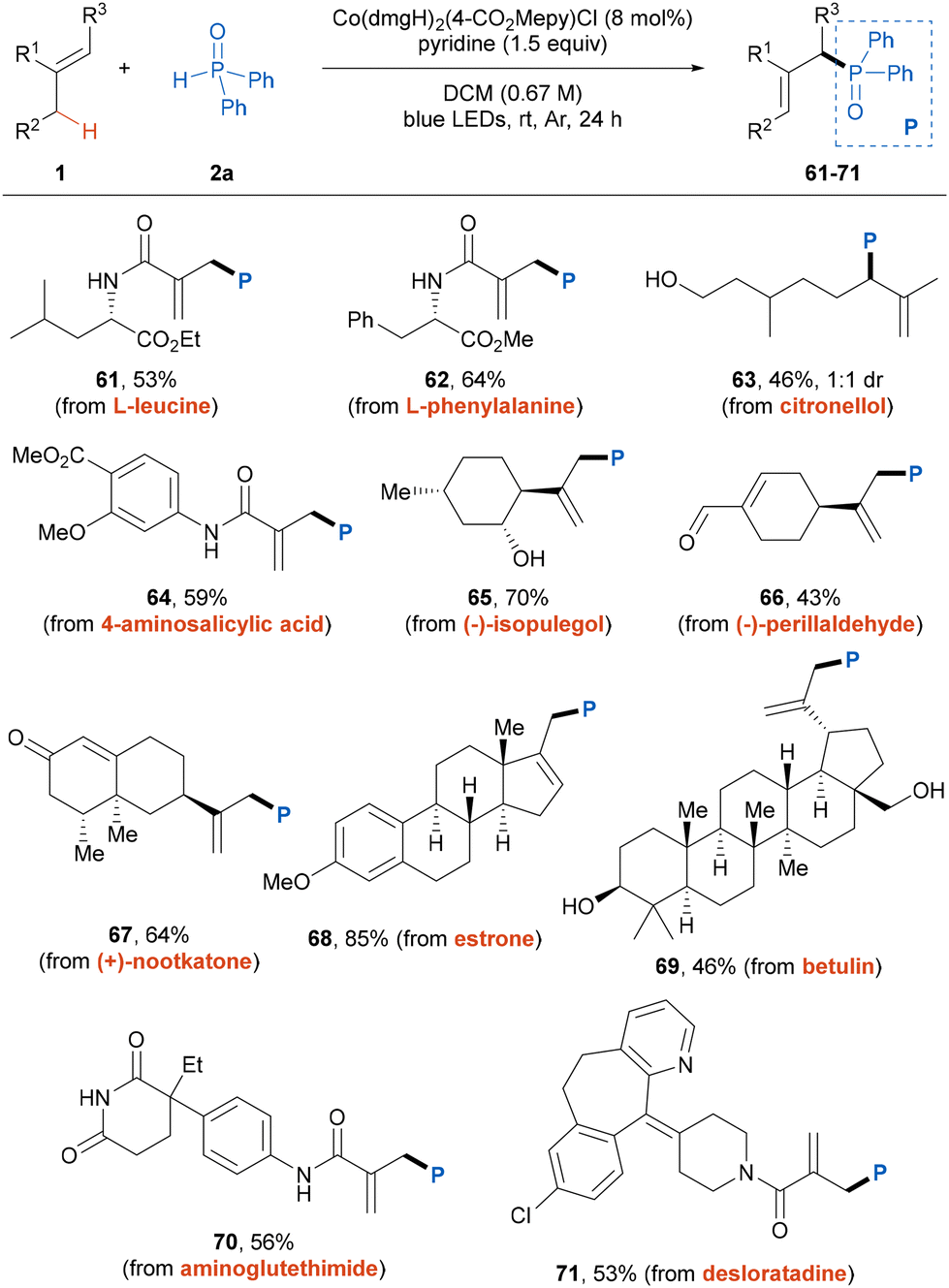
To further demonstrate the versatility and practicality of this photoinduced dehydrogenative phosphinylation reaction, we extended the protocol to the late-stage functionalization of bioactive compounds and drug molecules (Table 3). A variety of alkene-contained natural-products and derivatives participated in the reactions smoothly to afford the corresponding products 61–69 in moderate to good yields with high site-selectivities. And a series of functional groups were found to be well tolerated. Moreover, the alkenes derived from pharmaceutical drugs, including aminoglutethimide and desloratadine, were also compatible with this transformation, providing the desired products smoothly.
| a Reaction conditions: 1 (0.4 mmol, 2.0 equiv.), 2a (0.2 mmol), Co(dmgH)2(4-CO2Mepy)Cl (8 mol%), pyridine (0.3 mmol, 1.5 equiv.), DCM (3.0 mL), irradiation via 40 W blue LEDs under Ar at room temperature for 24 h. |
|---|
|
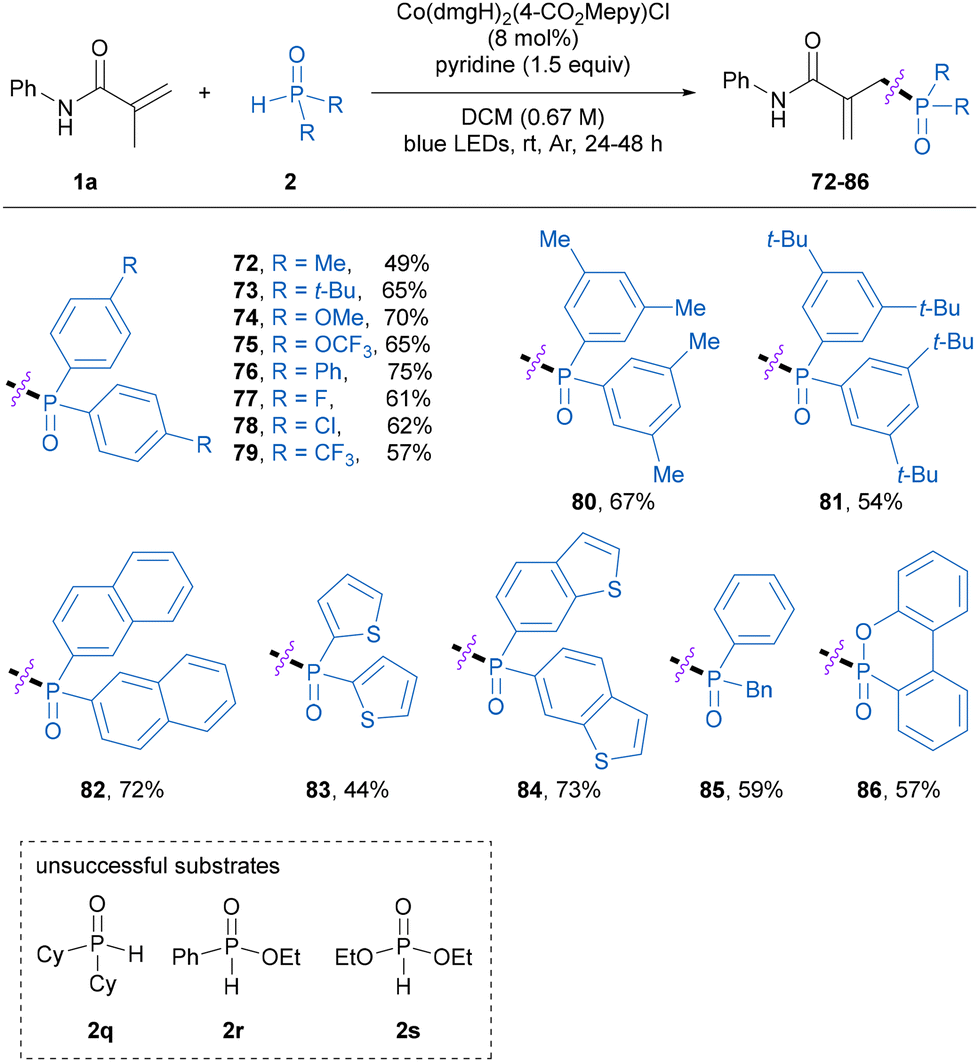
Next, the scope of secondary phosphine oxide was investigated in the phosphinylation of methacrylamide 1a. As shown in Table 4, a variety of monosubstituted and disubstituted diarylphosphine oxides smoothly participated in the reaction to afford the corresponding allylic phosphine oxide products 72–81 in 49–75% yields. Moreover, naphthalene- and heterocycle-substituted diarylphosphine oxides were found to be compatible with this approach, producing the corresponding products 82–84 in 44–73% yields. The asymmetric benzylphenylphosphine oxide was also a suitable substrate and delivered the desired product 85 in 59% yield. Note that 6H-dibenz[c,e][1,2]oxaphosphorin 6-oxide (DOPO) participated in this reaction smoothly to provide the desired product 86 in a moderate yield, whereas dicyclohexylphosphine oxide, ethyl phenylphosphinate, and diethyl phosphite failed to participate in the present allylic phosphinylation reaction at the current stage.
| a Reaction conditions: 1a (0.4 mmol, 2.0 equiv.), 2 (0.2 mmol), Co(dmgH)2(4-CO2Mepy)Cl (8 mol%), pyridine (0.3 mmol, 1.5 equiv.), DCM (3.0 mL), irradiation via 40 W blue LEDs under Ar at room temperature for 24–48 h. |
|---|
|
Diphosphines and their dioxide analogues play significant roles in organic synthesis, particularly as prevalent bidentate ligands for transition metal catalysis.17 In the traditional synthetic methods for obtaining these scaffolds, the use of highly reactive organometallic reagents and strong bases leads to poor functional group compatibility and hampers broader applications.18 We found that the dehydrogenative diphosphinylation of simple alkenes with 2 equivalents of diphenylphosphine oxide 2a proceeded smoothly using the present photoinduced cobaloxime catalysis (Scheme 2),19 which provides a straightforward and atom-economical route to access DPPP-dioxide analogues. 1,1-Disubstituted alkenes, isobutylene and 2,3-dimethyl-1-butene participated in the diphosphinylation process to produce the symmetric diphosphine oxide products 88 and 89 in 62% and 41% yields, respectively. In addition, the structure of 89 was further confirmed by X-ray crystallographic analysis (CCDC 2372954†). Moreover, trisubstituted alkenes, 2-methyl-2-butene and 3-methyl-2-buten-1-ol were also suitable substrates to obtain the asymmetric allylic diphosphine oxide compounds 90 and 91 in moderate yields. It is worth noting that the reaction of the tetrasubstituted alkene 2,3-dimethyl-2-butene gave the desired product 92, and the concomitant formation of compound 89 was observed, which was probably generated from the alkene isomerization enabled by [CoIII]–H through a reversible addition–elimination process (vide infra).
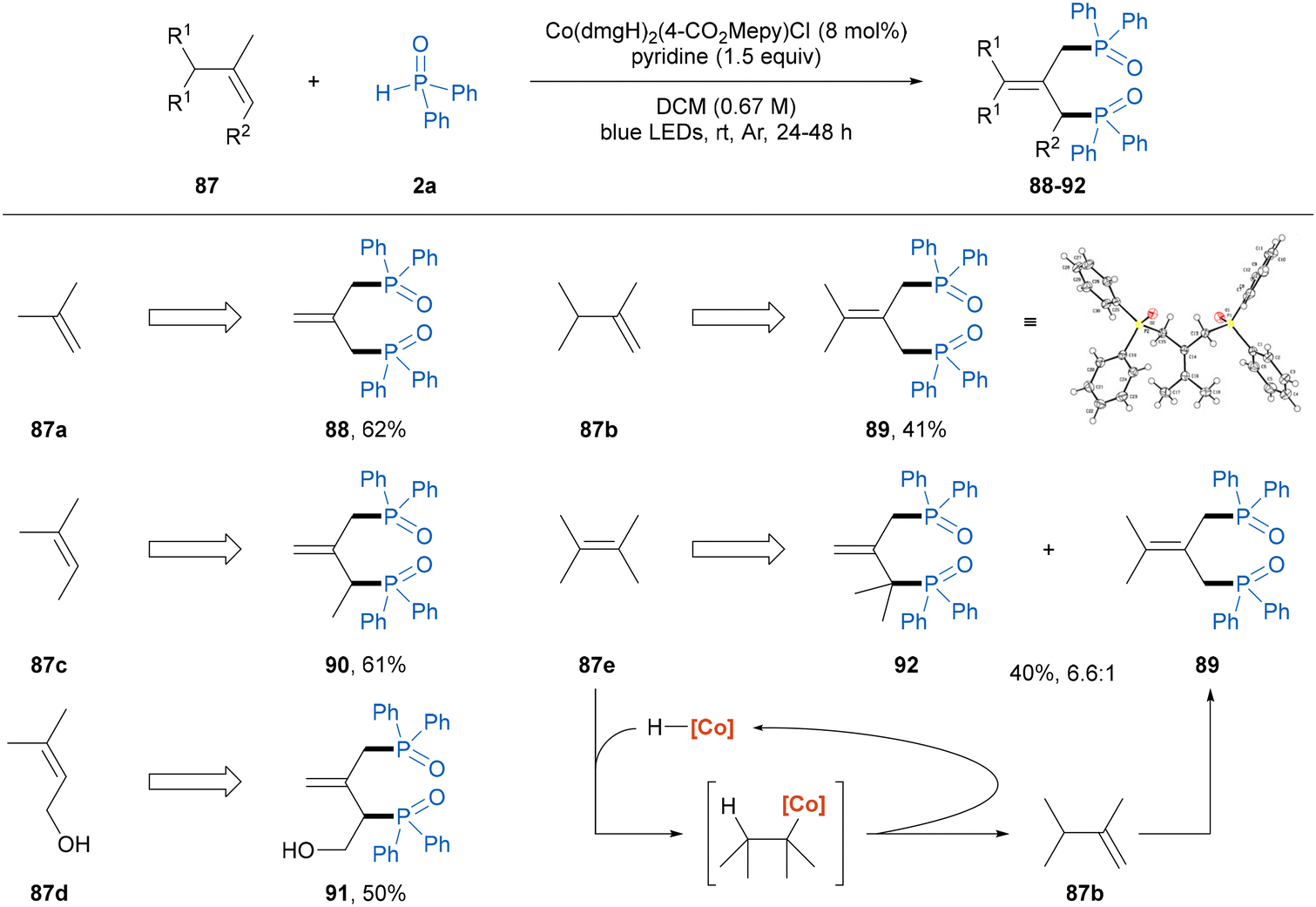 | ||
| Scheme 2 Diphosphinylation of alkenes. Reaction conditions: 87 (0.2 mmol), 2a (0.4 mmol, 2.0 equiv.), Co(dmgH)2(4-CO2Mepy)Cl (8 mol%), pyridine (0.3 mmol, 1.5 equiv.), DCM (3.0 mL), irradiation via 40 W blue LEDs under Ar at room temperature for 24–48 h. | ||
To demonstrate the practical application of the current protocol, we conducted large-scale reactions and explored further transformations of allylic phosphine oxide products (Scheme 3). A 5.2 mmol scale reaction of 1a and a 3.0 mmol scale reaction of 1bb were carried out under the standard conditions, yielding products 3 (1.27 g) and 56 (0.51 g) in 68% and 60% yields, respectively (Scheme 3a). Then, the reduction of the C–C double bond of allylic phosphine oxide 56 with a catalytic amount of Pd/C under a hydrogen gas atmosphere afforded the diphenyl alkyl phosphine oxide 93 in a quantitative yield. In addition, epoxidation and cyclopropanation of the olefin moiety in 56 with mCPBA and dichlorocarbene could be achieved, affording phosphinoyl substituted fused-cyclic compounds 94 and 95 in good yields.20 The phosphine oxide group in 56 could not only undergo the oxygen–sulfur exchange process with the Lawesson reagent to generate the phosphine sulfide 96 in 69% yield, but could also be readily reduced by the Si–H reagent to afford the allylic phosphine compound 97 in 72% yield as a potential bidentate phosphine–olefin ligand.21 Furthermore, a new diphosphine ligand 98 was obtained by the reduction of the diphosphine oxide compound 89 in good yield.
 | ||
| Scheme 3 Large-scale reactions and synthetic applications. | ||
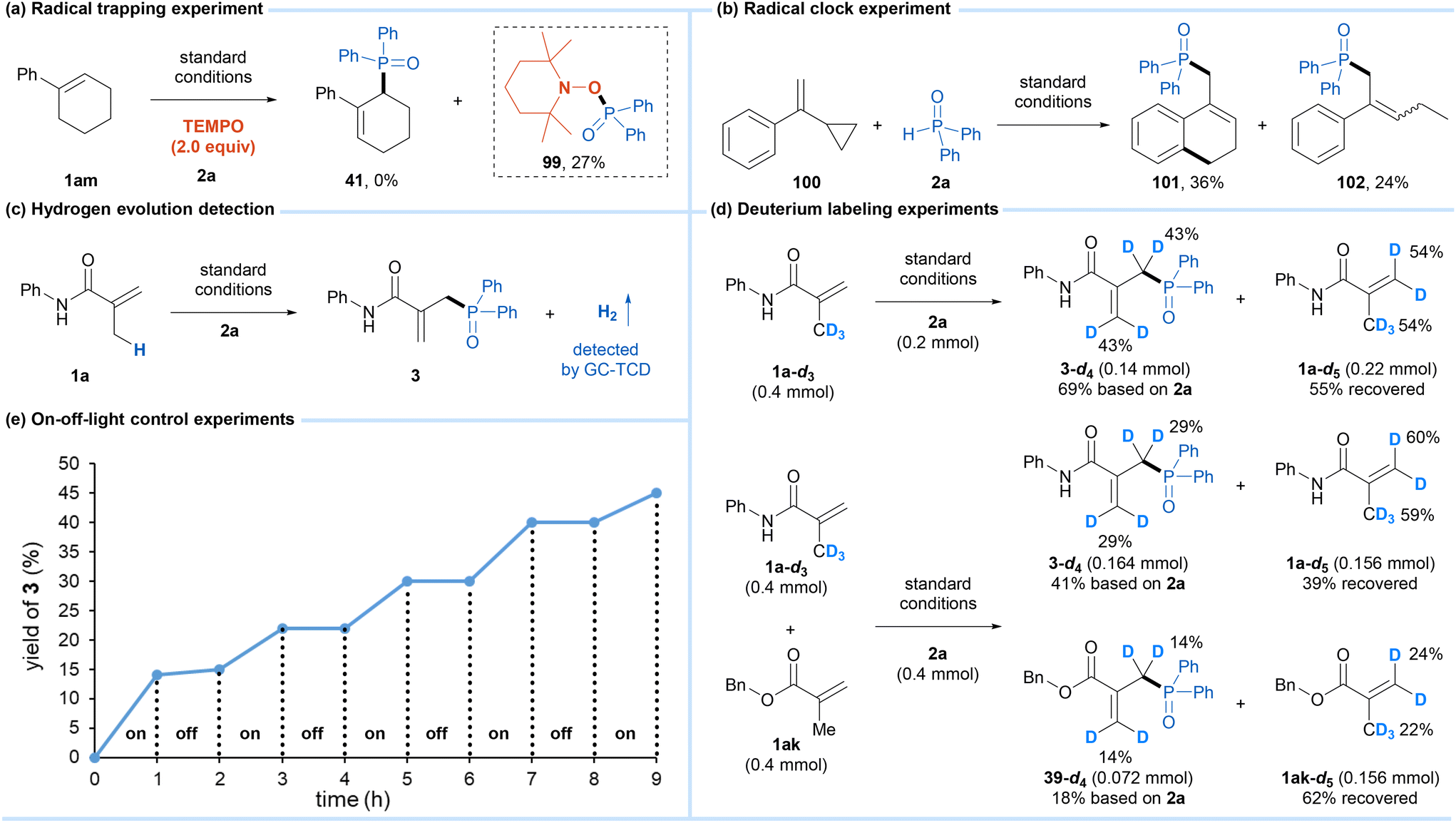
To gain some insights into this reaction mechanism, mechanistic studies were conducted. First, when 2 equivalents of the radical scavenger, 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) was added to the reaction with 1am, the formation of product 41 was inhibited completely, and the radical trapping product 99 formed by the reaction of the phosphinoyl radical and TEMPO was obtained in 27% yield (Scheme 4a). Then, a radical clock experiment with cyclopropylstyrene 100 provided the cyclization product 101 and the ring-opening product 102 in 36% and 24% yields, respectively (Scheme 4b). The results of these experiments indicated that the phosphinoyl radical was involved in this reaction. We noted that the yellow color of the reaction mixture quickly turned light green during the reaction process. Meanwhile, upon irradiation using blue LEDs for 20 minutes, the same color alteration was observed for the soultion of cobaloxime catalyst, diphenylphosphine oxide 2a, and pyridine in DCM (Fig. S6†). Based on Wu's studies13 and these observations, the activation of phosphine oxide by photoexcited cobaloxime in the presence of a base to generate the phosphinoyl radical is reasonable. Moreover, the hydrogen evolution during the reaction was detected by GC-TCD (Scheme 4c). In addition, the reaction of the deuterated alkene substrate 1a-d3 and 2a under the standard conditions produced the tetra-deuterated product 3-d4 in 69% yield with 43% deuterium content at the allyl-carbon and vinyl-carbon positions, and the recovered alkene substrate 1a-d5 also showed the H/D exchange between the allyl-carbon and vinyl-carbon positions (Scheme 4d). The H/D exchange could be explained by the [CoIII]–H promoted isomerization of the alkene through a reversible addition–elimination process,12a,b which was further supported by a competitive reaction between 1a-d3 and 1ak. Finally, light on/off experiments demonstrated that continuous light was an essential condition for the progress of this protocol (Scheme 4e).
 | ||
| Scheme 4 Mechanistic investigations. | ||
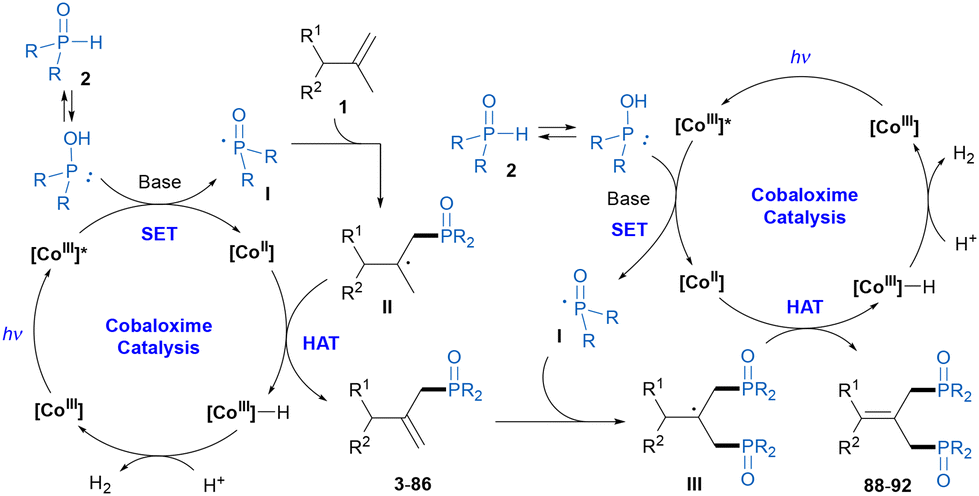
On the basis of the above mechanistic studies and related literature reports,11–13 a catalytic cycle was proposed, as shown in Scheme 5. First, under visible-light irradiation, cobaloxime as a photocatalyst was excited. Then, the photoexcited [CoIII] complex oxidized the secondary phosphine oxide 2 with the assistance of a base to generate the phosphinoyl radical I and [CoII] species. The subsequent addition of the phosphinoyl radical I to alkene 1 gave the carbon radical intermediate II. Hydrogen atom abstraction by the [CoII] species produced the phosphinylation product and [CoIII]–H complex, which was the site-selectivity determining step through preferential cleavage of the allylic C–H bond with less steric hindrance.12 According to the experimental results, the [CoIII]–H complex could promote alkene isomerization and could also react with the proton to release H2 with the regeneration of the [CoIII] catalyst to complete the cobalt catalytic cycle. At this point, we posited that the phosphinylation product might participate as the starting material in a second dehydrogenative phosphinylation process to produce the diphosphinylation product.
 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed an allylic radical phosphinylation of alkenes with secondary phosphine oxides by visible-light-induced cobaloxime catalysis. This reaction features photocatalyst- and oxidant-free conditions, a wide substrate scope, and good functional group tolerance, producing a diverse range of allylic phosphine oxides in good yields with high site-selectivities accompanied by hydrogen evolution. Meanwhile, the dehydrogenative radical diphosphinylation of simple alkenes also occurred smoothly to efficiently construct DPPP-dioxide analogues. In addition, the ease of scale up and versatile transformations of products demonstrate the great potential applications of this approach for the synthesis of functional organophosphorus molecules. Detailed mechanism studies were performed to elucidate the reaction mechanism. We anticipate that this visible-light-induced cobaloxime catalysis will offer a novel approach for the rapid construction of organophosphorus compounds.Author contributions
W. D. and M.-M. L. conceived the idea and supervised the project. A. Z., H. Y. and J. G. designed and conducted all experiments and analysed the data. W. D., L. G., and D. X. wrote the manuscript with the input from all authors.Data availability
The data supporting this article have been included as part of the ESI.†CCDC 2372952, 2372953 and 2372954† contain the supplementary crystallographic data for this paper.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22201265 and 22201264), the China Postdoctoral Science Foundation (2022M710133 and 2022TQ0287), and the Technology Project of Jiangxi Tobacco Industrial Co., Ltd (2022-02).References
- (a) T. Baumgartner and R. Réau, Organophosphorus π-Conjugated Materials, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (b) H. W. He, The Use of OP and Development of New Organophosphorus Agrochemicals in China, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 266–279 CrossRef CAS; (c) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Surface Modification Using Phosphonic Acids and Esters, Chem. Rev., 2012, 112, 3777–3807 CrossRef; (d) J. B. Rodriguez and C. Gallo-Rodriguez, The Role of the Phosphorus Atom in Drug Design, ChemMedChem, 2019, 14, 190–216 CrossRef CAS PubMed; (e) P. Finkbeiner, J. P. Hehn and C. Gnamm, Phosphine Oxides from a Medicinal Chemist's Perspective: Physicochemical and in Vitro Parameters Relevant for Drug Discovery, J. Med. Chem., 2020, 63, 7081–7107 CrossRef CAS; (f) H. Yu, H. Yang, E. Shi and W. Tang, Development and Clinical Application of Phosphorus-Containing Drugs, Med. Drug Discovery, 2020, 8, 100063 CrossRef CAS.
- (a) P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS; (b) P. W. N. M. v. Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Phosphite-Containing Ligands for Asymmetric Catalysis, Chem. Rev., 2011, 111, 2077–2118 CrossRef PubMed; (c) H. Ni, W.-L. Chan and Y. Lu, Phosphine-Catalyzed Asymmetric Organic Reactions, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS; (d) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed; (e) A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Trends in the Usage of Bidentate Phosphines as Ligands in Nickel Catalysis, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- (a) J. M. Gil, J. H. Hah, K. Y. Park and D. Y. Oh, A Facile Synthesis of Diethyl 1-Formylalkane PhosphonatesviaOzonolysis of 1-Alkyl Allylic Phosphonates, Synth. Commun., 2000, 30, 789–794 CrossRef CAS; (b) M. Gahungu, A. Arguelles-Arias, P. Fickers, A. Zervosen, B. Joris, C. Damblon and A. Luxen, Synthesis and biological evaluation of potential threonine synthase inhibitors: Rhizocticin A and Plumbemycin A, Bioorg. Med. Chem., 2013, 21, 4958–4967 CrossRef CAS; (c) Y. Niu, P.-B. Bai, Q.-X. Lou and S.-D. Yang, Generation of a Key Synthon of Indole Alkaloid Synthesis by Palladium(II)–Catalyzed Indole 2−Methylenephosphorylation, ChemCatChem, 2020, 12, 3644–3649 CrossRef CAS; (d) N. M. Eren, S. A. Orr, C. D. Thompson, E. C. Border, M. A. Stevens and V. L. Blair, Synthesis, Structure, and Solution Studies of Lithiated Allylic Phosphines and Phosphine Oxides, Organometallics, 2020, 39, 2080–2090 CrossRef CAS; (e) H. Wang, X. Li and T. Xu, Synthesis of chiral allylic phosphonates via asymmetric reductive cross-coupling of α-bromophosphonates and vinyl bromides, Sci. China: Chem., 2023, 66, 2621–2625 CrossRef CAS; (f) X.-H. Wei, Y.-W. Xue, C.-Y. Bai, X. Liu, X.-H. Wang, Y.-B. Wang and Q. Su, Lewis acid-catalyzed phosphinoylation and halogenation of α,β-unsaturated ketones: access to γ-halo allylic phosphonates, Org. Chem. Front., 2024, 11, 4385–4390 RSC.
- (a) P. Butti, R. Rochat, A. D. Sadow and A. Togni, Palladium–Catalyzed Enantioselective Allylic Phosphination, Angew. Chem., Int. Ed., 2008, 47, 4878–4881 CrossRef CAS; (b) L. Zhang, W. Liu and X. Zhao, Carbon–Phosphorus Bond Formation by Enantioselective Palladium–Catalyzed Allylation of Diphenylphosphine Oxide, Eur. J. Org. Chem., 2014, 6846–6849 CrossRef CAS; (c) X. Wo, P. Xie, W. Fu, C. Gao, Y. Liu, Z. Sun and T.-P. Loh, Barium-catalyzed C–OH/P–H dehydrative cross-coupling for C–P bond construction, Chem. Commun., 2018, 54, 11132–11135 RSC; (d) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, Ni-Catalyzed Asymmetric Allylation of Secondary Phosphine Oxides, J. Am. Chem. Soc., 2019, 141, 16584–16589 CrossRef CAS; (e) H. Qiu, Q. Dai, J. He, W. Li and J. Zhang, Access to P-chiral sec- and tert-phosphine oxides enabled by Le-Phos-catalyzed asymmetric kinetic resolution, Chem. Sci., 2020, 11, 9983–9988 RSC; (f) X. Yang, B. Li, H. Xing, J. Qiu, T.-P. Loh and P. Xie, Dehydrative allylation of P–H species under metal-free conditions, Green Chem., 2021, 23, 1633–1637 RSC; (g) Z.-H. Wu, H.-Y. Wang, H.-L. Yang, L.-H. Wei, T. Hayashi and W.-L. Duan, Secondary Phosphine Sulfide–Enabled Iridium–Catalyzed Asymmetric Allylic Substitution, Angew. Chem., Int. Ed., 2022, 61, e202213904 CrossRef CAS PubMed; (h) B. Li, M. Liu, S. U. Rehman and C. Li, Rh-Catalyzed Regio- and Enantioselective Allylic Phosphinylation, J. Am. Chem. Soc., 2022, 144, 2893–2898 CrossRef CAS; (i) H. Huang, Y.-Q. Wu, L.-Y. Han, L. Jiang, Z.-Z. Zhang, X. Zhang, B. Han, W. Huang and J.-L. Li, Palladium-catalyzed (Z)-selective allylation of phosphine oxides with vinylethylene carbonates to construct phosphorus allyl alcohols, Org. Biomol. Chem., 2024, 22, 3068–3072 RSC.
- (a) S. Z. Nie, R. T. Davison and V. M. Dong, Enantioselective Coupling of Dienes and Phosphine Oxides, J. Am. Chem. Soc., 2018, 140, 16450–16454 CrossRef CAS PubMed; (b) Z. Yang and J. Wang, Enantioselective Palladium–Catalyzed Hydrophosphinylation of Allenes with Phosphine Oxides: Access to Chiral Allylic Phosphine Oxides, Angew. Chem., Int. Ed., 2021, 60, 27288–27292 CrossRef CAS; (c) J. Long, Y. Li, W. Zhao and G. Yin, Nickel/Brønsted acid dual-catalyzed regio- and enantioselective hydrophosphinylation of 1,3-dienes: access to chiral allylic phosphine oxides, Chem. Sci., 2022, 13, 1390–1397 RSC.
- (a) B. Yang, H.-Y. Zhang and S.-D. Yang, Copper-catalyzed allylic C–H phosphonation, Org. Biomol. Chem., 2015, 13, 3561–3565 RSC; (b) M.-X. Cheng, R.-S. Ma, Q. Yang and S.-D. Yang, Chiral Brønsted Acid Catalyzed Enantioselective Phosphonylation of Allylamine via Oxidative Dehydrogenation Coupling, Org. Lett., 2016, 18, 3262–3265 CrossRef CAS PubMed; (c) Q. Chen, C. Wen, X. Wang, G. Yu, Y. Ou, Y. Huo and K. Zhang, DDQ–Promoted Allylic C−H Phosphorylation of 1,3−Diarylpropenes, Adv. Synth. Catal., 2018, 360, 3590–3594 CrossRef CAS.
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (b) X. Lang, J. Zhao and X. Chen, Cooperative photoredox catalysis, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC; (c) A. Lipp, S. O. Badir and G. A. Molander, Stereoinduction in Metallaphotoredox Catalysis, Angew. Chem., Int. Ed., 2020, 60, 1714–1726 CrossRef; (d) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, A review of enantioselective dual transition metal/photoredox catalysis, Sci. China: Chem., 2020, 63, 637–647 CrossRef CAS; (e) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2021, 122, 1485–1542 CrossRef; (f) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Recent advances in transition-metal-catalysed asymmetric coupling reactions with light intervention, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC; (g) F.-D. Lu, G.-F. He, L.-Q. Lu and W.-J. Xiao, Metallaphotoredox catalysis for multicomponent coupling reactions, Green Chem., 2021, 23, 5379–5393 RSC; (h) Q.-Y. Huang and M. Shi, Recent advancements in Ni/photoredox dual catalysis for Csp3−Csp3 cross-coupling reactions, Org. Chem. Front., 2024, 11, 4913–4925 RSC.
- (a) M. Kojima and S. Matsunaga, The Merger of Photoredox and Cobalt Catalysis, Trends Chem., 2020, 2, 410–426 CrossRef CAS; (b) K. R. Bajya and S. Selvakumar, Dual Photoredox and Cobalt Catalysis Enabled Transformations, Eur. J. Org. Chem., 2022, e202200229 CrossRef; (c) T. Yasui and Y. Yamamoto, Cobalt/Photoredox Cooperative Catalysis–Enabled Cycloaddition Reactions of 1,6−Diynes and Related Compounds, ChemCatChem, 2024, 16, e202400488 CrossRef CAS.
- (a) A. J. Moser, B. E. Funk and J. G. West, Vitamin B12 in Photocatalysis – An Underexplored Frontier in Cooperative Catalysis, ChemCatChem, 2024, 16, e202301231 CrossRef CAS PubMed; (b) P. Dam, K. Zuo, L. M. Azofra and O. El-Sepelgy, Biomimetic Photoexcited Cobaloxime Catalysis in Organic Synthesis, Angew. Chem., 2024, 64, e202405775 Search PubMed.
- (a) B. Chen, L.-Z. Wu and C.-H. Tung, Photocatalytic Activation of Less Reactive Bonds and Their Functionalization via Hydrogen-Evolution Cross-Couplings, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS; (b) K. C. Cartwright, A. M. Davies and J. A. Tunge, Cobaloxime–Catalyzed Hydrogen Evolution in Photoredox–Facilitated Small–Molecule Functionalization, Eur. J. Org. Chem., 2020, 1245–1258 CrossRef CAS; (c) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Recent Advances in Oxidative R1-H/R2-H Cross-Coupling with Hydrogen Evolution via Photo-/Electrochemistry, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed.
- (a) J.-J. Zhong, Q.-Y. Meng, B. Liu, X.-B. Li, X.-W. Gao, T. Lei, C.-J. Wu, Z.-J. Li, C.-H. Tung and L.-Z. Wu, Cross-Coupling Hydrogen Evolution Reaction in Homogeneous Solution without Noble Metals, Org. Lett., 2014, 16, 1988–1991 CrossRef CAS PubMed; (b) G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C–H Thiolation for Constructing C–S Bonds, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed; (c) Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, Photocatalytic Hydrogen-Evolution Cross-Couplings: Benzene C–H Amination and Hydroxylation, J. Am. Chem. Soc., 2016, 138, 10080–10083 CrossRef CAS; (d) Q.-Q. Zhao, X.-Q. Hu, M.-N. Yang, J.-R. Chen and W.-J. Xiao, A visible-light photocatalytic N-radical cascade of hydrazones for the synthesis of dihydropyrazole-fused benzosultams, Chem. Commun., 2016, 52, 12749–12752 RSC; (e) K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X.-L. Yang and Y. Li, Acceptorless Dehydrogenation of N–Heterocycles by Merging Visible–Light Photoredox Catalysis and Cobalt Catalysis, Angew. Chem., Int. Ed., 2017, 56, 3080–3084 CrossRef CAS; (f) X. Sun, J. Chen and T. Ritter, Catalytic dehydrogenative decarboxyolefination of carboxylic acids, Nat. Chem., 2018, 10, 1229–1233 CrossRef CAS; (g) H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes, Nat. Commun., 2020, 11, 1956 CrossRef CAS; (h) J. B. McManus, J. D. Griffin, A. R. White and D. A. Nicewicz, Homobenzylic Oxygenation Enabled by Dual Organic Photoredox and Cobalt Catalysis, J. Am. Chem. Soc., 2020, 142, 10325–10330 CrossRef CAS; (i) S. U. Dighe, F. Juliá, A. Luridiana, J. J. Douglas and D. Leonori, A photochemical dehydrogenative strategy for aniline synthesis, Nature, 2020, 584, 75–81 CrossRef; (j) M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, Site-Selective Acceptorless Dehydrogenation of Aliphatics Enabled by Organophotoredox/Cobalt Dual Catalysis, J. Am. Chem. Soc., 2021, 143, 16470–16485 CrossRef CAS PubMed; (k) C.-Y. Huang, J. Li and C.-J. Li, A cross-dehydrogenative C(sp3)−H heteroarylation via photo-induced catalytic chlorine radical generation, Nat. Commun., 2021, 12, 4010 CrossRef CAS; (l) E. Bergamaschi, C. Weike, V. J. Mayerhofer, I. Funes-Ardoiz and C. J. Teskey, Dual Photoredox/Cobaloxime Catalysis for Cross-Dehydrogenative α-Heteroarylation of Amines, Org. Lett., 2021, 23, 5378–5382 CrossRef CAS PubMed; (m) L. Huang, T. F. Ji, C. Zhu, H. F. Yue, N. Zhumabay and M. Rueping, Bioinspired desaturation of alcohols enabled by photoredox proton-coupled electron transfer and cobalt dual catalysis, Nat. Commun., 2022, 13, 809 CrossRef CAS PubMed; (n) A. F. Prusinowski, H. C. Sise, T. N. Bednar and D. A. Nagib, Radical Aza-Heck Cyclization of Imidates via Energy Transfer, Electron Transfer, and Cobalt Catalysis, ACS Catal., 2022, 12, 4327–4332 CrossRef CAS; (o) Z. Jia, L. Cheng, L. Zhang and S. Luo, Asymmetric C–H Dehydrogenative Alkenylation via a Photo-induced Chiral α-Imino Radical Intermediate, Nat. Commun., 2024, 15, 4044 CrossRef CAS PubMed; (p) J. Corpas, H. P. Caldora, E. M. Di Tommaso, A. C. Hernandez-Perez, O. Turner, L. M. Azofra, A. Ruffoni and D. Leonori, A general strategy for the amination of electron-rich and electron-poor heteroaromatics by desaturative catalysis, Nat. Catal., 2024, 7, 593–603 CrossRef CAS.
- (a) Z. Jia, L. Zhang and S. Luo, Asymmetric C–H Dehydrogenative Allylic Alkylation by Ternary Photoredox-Cobalt-Chiral Primary Amine Catalysis under Visible Light, J. Am. Chem. Soc., 2022, 144, 10705–10710 CrossRef CAS; (b) M.-Y. Dong, C.-Y. Han, D.-S. Li, Y. Hong, F. Liu and H.-P. Deng, Hydrogen-Evolution Allylic C(sp3)–H Alkylation with Protic C(sp3)–H Bonds via Triplet Synergistic Brønsted Base/Cobalt/Photoredox Catalysis, ACS Catal., 2022, 12, 9533–9539 CrossRef CAS; (c) T. Zhong, C. Gu, Y. Li, J. Huang, J. Han, C. Zhu, J. Han and J. Xie, Manganese/Cobalt Bimetallic Relay Catalysis for Divergent Dehydrogenative Difluoroalkylation of Alkenes, Angew. Chem., Int. Ed., 2023, 62, e202310762 CrossRef CAS; (d) S. Wang, Y. Gao, Z. Liu, D. Ren, H. Sun, L. Niu, D. Yang, D. Zhang, X. a. Liang, R. Shi, X. Qi and A. Lei, Site-selective amination towards tertiary aliphatic allylamines, Nat. Catal., 2022, 5, 642–651 CrossRef CAS; (e) G. Li and J. Chen, Dual photoredox and cobalt catalysis enpowers site-selective allylic amination, Green Synth. Catal., 2022, 3, 306–308 CrossRef CAS; (f) W.-L. Yu, Z.-G. Ren, W. Ma, H. Zheng, W. Wu and P.-F. Xu, Intramolecular dehydrogenative amination of alkenes via dual organic photoredox and cobalt catalysis without a hydrogen acceptor, Green Chem., 2022, 24, 6131–6137 RSC; (g) G. Zhang, L. Zhang, H. Yi, Y. Luo, X. Qi, C.-H. Tung, L.-Z. Wu and A. Lei, Visible-light induced oxidant-free oxidative cross-coupling for constructing allylic sulfones from olefins and sulfinic acids, Chem. Commun., 2016, 52, 10407–10410 RSC; (h) W.-L. Yu, Y.-C. Luo, L. Yan, D. Liu, Z.-Y. Wang and P.-F. Xu, Dehydrogenative Silylation of Alkenes for the Synthesis of Substituted Allylsilanes by Photoredox, Hydrogen–Atom Transfer, and Cobalt Catalysis, Angew. Chem., Int. Ed., 2019, 58, 10941–10945 CrossRef CAS; (i) M. Chen, Z.-J. Wu, J. Song and H.-C. Xu, Electrocatalytic Allylic C−H Alkylation Enabled by a Dual–Function Cobalt Catalyst, Angew. Chem., Int. Ed., 2022, 61, e202115954 CrossRef CAS; (j) J.-L. Liu, J.-L. Tu and F. Liu, Visible-Light-Promoted Intramolecular α-Allylation of Aldehydes in the Absence of Sacrificial Hydrogen Acceptors, Org. Lett., 2020, 22, 7369–7372 CrossRef CAS.
- (a) W.-Q. Liu, T. Lei, S. Zhou, X.-L. Yang, J. Li, B. Chen, J. Sivaguru, C.-H. Tung and L.-Z. Wu, Cobaloxime Catalysis: Selective Synthesis of Alkenylphosphine Oxides under Visible Light, J. Am. Chem. Soc., 2019, 141, 13941–13947 CrossRef CAS PubMed; (b) T. Lei, G. Liang, Y.-Y. Cheng, B. Chen, C.-H. Tung and L.-Z. Wu, Cobaloxime Catalysis for Enamine Phosphorylation with Hydrogen Evolution, Org. Lett., 2020, 22, 5385–5389 CrossRef CAS; (c) A. Shao, J. Chen, L. Wang, M. Yi, H. Yang, Y. Zhang, S. Fan, S. Chen, H. Wu and R. Shi, Excited-state cobaloxime catalysis enabled scalable oxidant-free dehydrogenative C–H phosphinoylation of undirected heterocycles, Org. Chem. Front., 2022, 9, 4379–4387 RSC; (d) J.-X. Yu, Y.-Y. Cheng, B. Chen, C.-H. Tung and L.-Z. Wu, Cobaloxime Photocatalysis for the Synthesis of Phosphorylated Heteroaromatics, Angew. Chem., Int. Ed., 2022, 61, e202209293 CrossRef CAS PubMed.
- (a) M. Parasram and V. Gevorgyan, Visible light-induced transition metal-catalyzed transformations: beyond conventional photosensitizers, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC; (b) W.-M. Cheng and R. Shang, Transition Metal-Catalyzed Organic Reactions under Visible Light: Recent Developments and Future Perspectives, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (c) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Visible Light-Induced Transition Metal Catalysis, Chem. Rev., 2021, 122, 1543–1625 CrossRef.
- (a) X. Mao, M.-M. Li, P. Wang, Q. Cao, W. Zhou and W. Ding, Transition-Metal-Free Anti-Markovnikov Hydroarylation of Alkenes with Aryl Chlorides through Consecutive Photoinduced Electron Transfer, Org. Lett., 2024, 26, 1265–1270 CrossRef CAS; (b) Q. Cao, M.-M. Li, X. Mao, Q.-Q. Zhou and W. Ding, Visible-Light-Induced Regioselective Radical-Polar Crossover 1,4-Hydrophosphinylation of 1,3-Enynes: Access to Trisubstituted Allenes Bearing a Phosphine Oxide Group, Org. Lett., 2024, 26, 4678–4683 CrossRef CAS PubMed.
- (a) W. J. Yoo and S. Kobayashi, Hydrophosphinylation of unactivated alkenes with secondary phosphine oxides under visible-light photocatalysis, Green Chem., 2013, 15, 1844–1848 RSC; (b) Z. Yuan, S. Wang, M. Li, T. Chen, J. Fan, F. Xiong, Q. Li, P. Hu, B.-Q. Wang, P. Cao and Y. Li, Visible light induced hydrophosphinylation of unactivated alkenes catalyzed by salicylaldehyde, Green Chem., 2021, 23, 3600–3606 RSC.
- (a) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Ligand Bite Angle Effects in Metal-catalyzed C−C Bond Formation, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS; (b) W. Tang and X. Zhang, New Chiral Phosphorus Ligands for Enantioselective Hydrogenation, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS; (c) T. Kuribara, A. Kaneki, Y. Matsuda and T. Nemoto, Visible-Light-Antenna Ligand-Enabled Samarium-Catalyzed Reductive Transformations, J. Am. Chem. Soc., 2024, 146, 20904–20912 CrossRef CAS.
- (a) A. M. Aguiar and D. Daigle, A Stereospecific Route to trans- and cis-1,2-Vinylenebis(diphenylphosphine), J. Am. Chem. Soc., 1964, 86, 2299–2300 CrossRef CAS; (b) M. T. Honaker, B. J. Sandefur, J. L. Hargett, A. L. McDaniel and R. N. Salvatore, CsOH-promoted P-alkylation: a convenient and highly efficient synthesis of tertiary phosphines, Tetrahedron Lett., 2003, 44, 8373–8377 CrossRef CAS; (c) A. Yoshimura, Y. Saga, Y. Sato, A. Ogawa, T. Chen and L.-B. Han, An efficient base-catalyzed double addition of H-phosphine oxides to alkynes, Tetrahedron Lett., 2016, 57, 3382–3384 CrossRef CAS.
- (a) Y. Sato, S. i. Kawaguchi, A. Nomoto and A. Ogawa, Highly Selective Phosphinylphosphination of Alkenes with Tetraphenyldiphosphine Monoxide, Angew. Chem., Int. Ed., 2016, 55, 9700–9703 CrossRef CAS PubMed; (b) Y. Okugawa, K. Hirano and M. Miura, Copper–Catalyzed Vicinal Diphosphination of Styrenes: Access to 1,2−Bis(diphenylphosphino)ethane–Type Bidentate Ligands from Olefins, Angew. Chem., Int. Ed., 2016, 55, 13558–13561 CrossRef CAS; (c) K. Hirano and M. Miura, Recent advances in diphosphination of alkynes and alkenes, Tetrahedron Lett., 2017, 58, 4317–4322 CrossRef CAS.
- A. Bell, A. H. Davidson, C. Earnshaw, H. K. Norrish, R. S. Torr, D. B. Trowbridge and S. Warren, Synthesis of β-(diphenylphosphinoyl) ketones, J. Chem. Soc., Perkin Trans. 1, 1983, 2879–2891 RSC.
- (a) E. Piras, F. Läng, H. Rüegger, D. Stein, M. Wörle and H. Grützmacher, Chiral Phosphane Alkenes (PALs): Simple Synthesis, Applications in Catalysis, and Functional Hemilability, Chem. – Eur. J., 2006, 12, 5849–5858 CrossRef CAS; (b) W.-L. Duan, H. Iwamura, R. Shintani and T. Hayashi, Chiral Phosphine−Olefin Ligands in the Rhodium-Catalyzed Asymmetric 1,4-Addition Reactions, J. Am. Chem. Soc., 2007, 129, 2130–2138 CrossRef CAS; (c) Z. Cao, Y. Liu, Z. Liu, X. Feng, M. Zhuang and H. Du, Pd-Catalyzed Asymmetric Allylic Alkylation of Indoles and Pyrroles by Chiral Alkene-Phosphine Ligands, Org. Lett., 2011, 13, 2164–2167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, characterization of compounds, crystallographic data. CCDC 2372952–2372954. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01638f |
| This journal is © the Partner Organisations 2025 |