
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiTMP-LiBr complex-induced lateral lithiation and cross ester condensation: direct access to isocoumarins from 2-methoxy o-toluate esters†
Mintu
Rehman
,
Sajib
Daimary
,
Rasajna
Madhusudhana
and
Rajendar
Goreti
 *
*
School of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram, Kerala, India-695551. E-mail: rajendar@iisertvm.ac.in
First published on 18th October 2024
Abstract
A LiTMP-LiBr complex facilitates a novel cross-ester coupling of 2-methoxy o-toluate esters to directly yield isocoumarin without the formation of any carbonyl intermediate. Aggregation plays a pivotal role in driving proximity-induced lateral lithiation and expediting acylation. In addition to many natural product precursors, the synthesis of (+)- and (−)-lunatinins is shown.
1H-Isochromen-1-one is a key structural feature of numerous bioactive isocoumarins, which are prevalent in fungi, bacteria, plants, and marine organisms and exhibit diverse biological activities.1 Over the years, extensive research has culminated in the development of diverse synthetic approaches to obtain these privileged structures.2 One well-established, effective, and convergent strategy for isocoumarin synthesis involves lateral lithiation and acylation of 2-methoxy o-toluic acid derivatives followed by subsequent lactonization.3 This approach features wide applicability for introducing diverse substituents at the C3 position by utilizing readily available acylating agents such as acid chlorides, esters and amides.4
The deprotonative metalation of benzylic C–H groups is a valuable method for forming new C–C and C–X bonds.5 Notably, the lateral lithiation of o-toluic acid 1 and its amide 2 produce a stable dianion (I), which selectively attacks various electrophiles, particularly acylating agents, resulting in the formation of the corresponding keto-derivatives (3 or 4).6 Suitable tactics are then required to cyclize the keto derivatives to the desired isocoumarins (Scheme 1a).7 On the other hand, for o-toluate esters 6, lateral lithiation is complicated due to the stability of the arylogous enolate intermediate (II), which leads to self-condensation to give dimerized product 7.8 The Staunton group reported the reaction of o-toluate esters with Wienreb amides through lateral lithiation and acylation to yield keto-esters 8 (Scheme 1b).9 Notwithstanding its good applicability, the approach is somewhat limited in terms of yield, primarily due to the dimerization of the ester. Additionally, a base-mediated enolization is required to convert ketoderivative 8 into isochromenone 9. Additionally, the use of acid chlorides10 produced poor yields along with complex products due to polycondensation. Conversely, esters have remained inert as electrophilic coupling partners in the condensation reactions of ortho-toluate esters to date.11
 | ||
| Scheme 1 Complex-induced lateral lithiation, acylation, enolization and lactonization for isocoumarin synthesis. (III and IV: possible transition states, hypothetical structures). | ||
Esters are abundant and viable starting materials in organic synthesis. However, cross-ester condensation, in general, poses a formidable challenge due to the low electrophilicity of the acceptor ester.12 It is notably susceptible to issues of self-condensation and over-condensation. Self-condensation has been partially alleviated by using excess equivalents of pre-prepared aryl and tert-butyl ester enolates (V),13 or by the use of Ti- or Zr-based enolates.14,15 Nevertheless, cross-ester coupling involving arylogous enolates (VIII) remained an unsolved challenge.
Recently, we found that use of chelating metal ions (LiCl) in lateral lithiation effectively suppresses the self-dimerization of o-toluate esters.16 We anticipated that the excess Li salts promoted complex-induced lateral lithiation via tight chelation (III).17 Additionally, these salts work in tandem with acylating agents to enhance their electrophilicity, thereby facilitating facile nucleophilic addition (IV). This method is only known for Wienreb amides, and they produce only ketone even in the presence of a large excess of base.8,18 Despite extensive efforts, this reaction yielded self-dimerized ketone 7 consistently as the primary byproduct. Building on this finding, we hypothesized that esters might act as electrophilic partners in lateral lithiation, and unlike Wienreb amides,19 they could directly produce isocoumarins via enolization of ketone intermediate in situ.8 However, preventing self-dimerization of the toluate ester is the biggest hurdle. Generating the stable arylogous enolate as well as activating the acceptor ester simultaneously are crucial to achieve this challenging conversion. We speculated that establishing strong chelation is the key to facilitate this challenging cross-ester coupling; however, this strategy has not been used thus far.
The reaction of orsellinate 10 with methyl benzoate in the presence of LDA and excess LiCl provided the desired product 12 in 42% yield along with ketone 11 (22% yield; Table 1, entry 1). The reaction conditions were further optimized as shown in Table 1. Changing the additive to LiBr gave only a 20% yield (entry 2), and LiI and LiClO4 and the use of LiHMDS failed to give any desired product.20 In contrast, the use of LiTMP in combination with LiBr gave a clean conversion with the limitation that the reaction required more than 3 eq. of base to initiate enolate II formation (entries 3 and 4). Increasing the equivalents of base and changing the additives to MgCl2, MgBr2 and ZnCl2 provided the desired product maximum yield of 12 in entries 5–7 as 69%.
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yieldsa,b (%) |
| a Isolated yields. b Conditions A: base was added to a solution of 10, methyl benzoate and LiBr in THF at −78 °C. c Conditions B: methyl benzoate was added to the preprepared enolate of 10 at −78 °C. | ||
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11:12:13 :11:12:13 |
||
| 1 | LDA (3), LiCl (10), THF | 10:22:42:20 |
| 2 | LDA (4), LiBr (10), THF | 50:25:20:0 |
| 3 | LiTMP (4), LiBr (3), THF | 26:30:37:0 |
| 44:0:51:0c |
||
| 4 | LiTMP (8), LiBr (3), THF | 30:0:68:0 |
| 5 | LiTMP (5), MgCl2 (3), THF | 25:0:69:0 |
| 6 | LiTMP (5), MgBr2 (5), THF | 36:0:52:0 |
| 7 | LiTMP (5), ZnCl2 (5), THF | 78:0:12:0 |
| 8 | LiTMP (5), LiBr (3), THF:hexane (2:1) |
0:0:90:0 |
| 9 |
LiTMP (5), LiBr (3), THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :cyclohexane (2:1) :cyclohexane (2:1)
|
0:0:92:0 |
| 10 | LiTMP (5), LiClO4 (3), THF:cyclohexane |
0:0:51:0 |

The LiTMP-LiBr complex, which is known to produce bromide-induced aggregates to establish a strong chelating complex, has been used to control the stereochemistry of enolates.21 Speculating that we could achieve similar effects,22 we added non-polar solvents such as hexane and cyclohexane to produce aggregates, which resulted in excellent yields of 12 (entries 8 and 9). Additives other than LiBr did not produce good yields of 12 (entry 10). More than three equivalents of the LiTMP-LiBr complex were required to generate the enolate, which was indicated by the formation of a red-coloured solution, which underscores the pronounced aggregation effect at play. The intricate aggregate becomes the epicentre for the proximity-induced lateral lithiation through complex III, ultimately leading to the generation of stable arylogous enolate species. Within this aggregate, the acceptor ester also participates through IV, creating a harmonious molecular environment that sets the stage for acylation and subsequent lactonization. Changes in the base, solvent or additive, however, provided unsatisfactory yields of 12.20
Following the establishment of the standard reaction conditions (entry 11, Table 1), we expanded the applicability and versatility of the method as illustrated in Table 2. Aromatic esters with diverse alkyl and aryl substituents produced the corresponding isocoumarins 14, 15, 16 and 24 in >90% yields. The presence of chelating groups on the acceptor ester did not significantly affect the reaction yields, and isocoumarins 17,2318, 2224 and 23 were produced in 83% to 94% yields. Halogen-substituted esters required a change in the addition sequence; the addition of the ester to the prepared enolate gave isocoumarins 19, 20 and 21 in good yields of >80%.

Unfortunately, the method did not work with enolizable esters. Most interestingly, esters having an α- or β-hydroxyl functionality produced the corresponding isocoumarin products 25, 26, 27,252826 and 3829 in fair yields. This was a very interesting result, given that there was no protecting group involved, which paves the way for the synthesis of such hydroxyl-substituted naturally abundant and pharmacologically important 3-substituted isocoumarins.27
Moreover, o-toluate esters bearing 2-OTBS and 2-OTs groups showed no reactivity under the standard conditions. The substrate with a 2-OBn group resulted in a complex mixture, and that with an OMOM group provided 32 in 54% yield. 2-Methoxy toluate ester exhibited good reactivity with various aryl esters, yielding isocoumarins 33, 3428 to 3528 in excellent yields. Similarly, toluate esters with three methoxy substituents showed good reactivity, affording isocoumarins 36 and 3729 in yields exceeding 80%. The presence of a halogen atom in the toluate ester did not affect the reaction outcome, and compound 39 was produced in 89% yield.
Furthermore, role of chelating groups on enolate stability was estimated through control experiments. As shown in Scheme 2a, the absence of the directing group (2-OMe) led to rapid self-dimerization to provide isocoumarin 40 in 65% yield. An unprotected 2-OH group provided complex products, and the reaction of 3-methoxy toluate ester was unsuccessful. These results highlight the significance of the 2-OMe group, which stabilizes the enolate through a tight chelate complex. Conversely, a chelating group at the 5-position, such as that in 2,5-dimethoxy o-toluate ester, interferes with the reaction path, resulting in a complex mixture instead formation of 43.
 | ||
| Scheme 2 Control experiments. | ||
To understand the reaction mechanism, keto-ester 11 was first prepared and then subjected to cyclization. LiOMe (a byproduct in the reaction) was reacted with the ketone at −78 °C, but proved ineffective in promoting cyclization (Scheme 2b). However, at higher temperatures (25 °C), lactonization was promoted effectively to give 12. The LiTMP-LiBr complex effectively promoted enolization and cyclization at −78 °C, resulting in complete conversion to 12. These findings indicate that enolate formation is sequentially facilitated in two stages by the LiTMP-LiBr complex. Isocoumarin is the core structure of many natural products;1 for example 12 serves as a synthetic precursor for montroumarin.31 Similarly, several products obtained in this study can serve as synthetic intermediates for scorzocreticin,23 thumborginols A28 and B,24 cytogenin,26 dimethoxy thumborginol A,29 diaportin, orthosporin,25 and other natural 6,8-dihydroxy-7-methoxy isocoumarins.30
Lunatinin,32 which has been isolated from various sources as both the (+)- and (−)-isomers, possesses intriguing biological activities; notably, it shows strong HMG-CoA reductase inhibitory activity (IC50 128 μg mL−1) comparable to the standard drug Lovastatin (IC50 160 μg mL−1).33 The absolute structure of (−)-lunatinin was established based on ECD studies. We utilized the LiTMP-LiBr complex-induced tandem lateral lithiation, acylation and lactonization of o-toluate esters to give isocoumarin and a shortest possible route to lunatinin.33 The total synthesis commenced with commercial ester 45. It was coupled with commercially available optically pure methyl (S)-3-hydroxybutanoate in the presence of the LiTMP-LiBr complex, producing isocoumarin 45S in 63% yield (Scheme 3). The absolute structure of 45S was confirmed by X-ray analysis. Demethylation of two phenolic groups of 45S using BBr3 afforded (S)-lunatinin (47S) in 93% yield. All the spectral data of synthetic 47S were identical to the reported data; however, the specific rotation α23D= −19.3 (c 0.3, MeOH) showed a deviation in magnitude. Similarly, the cross-ester condensation of 44 with methyl (R)-3-hydroxybutanoate followed by methyl deprotection provided R-lunatinin (46R) in 57% in two steps.
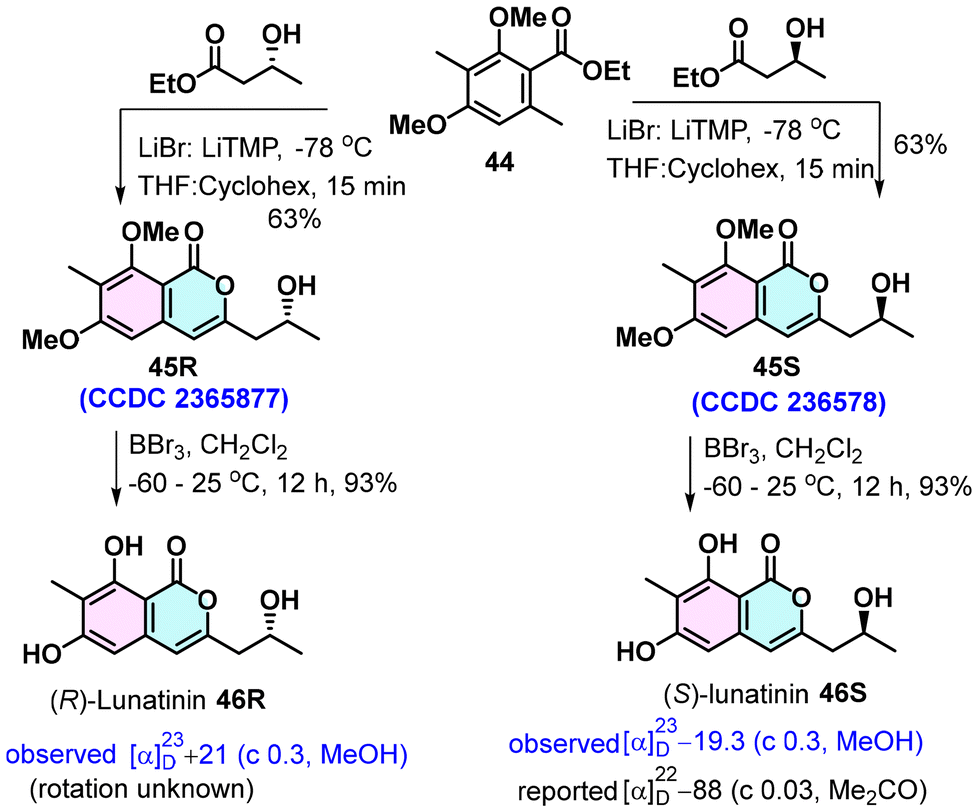 | ||
| Scheme 3 Total synthesis of (+)- and (–)-lunatinins. | ||
In conclusion, our innovative one-step synthetic route to isocoumarins from 2-methoxyl o-toluate esters, employing the LiTMP-LiBr complex, has proven to be a highly efficient and versatile method. This is the first method that shows the influence of aggregates in complex-induced lateral lithiation of toluate esters. This unique feature extends to the first cross-ester coupling of o-toluate ester with other esters. The reaction mechanism involves a seamless tandem process, including proximity-induced lateral lithiation, complex induced acylation, enolization, and lactonization. Furthermore, we demonstrated the utility of our method through a concise total synthesis of (+)- and (−)-lunatinins.
Author contributions
RG developed the concept and written manuscript through the contributions of MR. MR conducted all the experiments with the help of SD and RM. All authors have given approval to the final version of the manuscript.Data availability
All experimental procedures, 1H and 13C NMR data of compounds, and X-ray crystallographic data of compound 45R and 45S are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Alex A for the X-ray analysis. MR thanks IISER-TVM for the research fellowship.References
- (a) A. Saeed, Isocoumarins, miraculous natural products blessed with diverse pharmacological activities E, J. Med. Chem., 2016, 116, 290 CrossRef CAS PubMed; (b) G. Shabir, A. Saeed and H. R. El-Seedi, Natural isocoumarins: Structural styles and biological activities, the revelations carry on…, Phytochemistry, 2021, 181, 112568 CrossRef CAS.
- (a) P. Saikia and S. Gogoi, Isocoumarins: General Aspects and Recent Advances in their Synthesis, Adv. Synth. Catal., 2018, 360, 2063–2075 CrossRef CAS; (b) N. Gogoi, R. Parhi, R. K. P. Tripathi, L. Pachuau and P. P. Kaishap, Recent advances in synthesis of isocoumarins: An overview, Tetrahedron, 2023, 150, 133740 CrossRef.
- (a) F. M. Hauser and R. Rhee, New synthetic strategy for the preparation of linear phenolic natural products, J. Am. Chem. Soc., 1977, 99, 4533–4534 CrossRef CAS; (b) V. Deshpande, B. Rai and R. A. Khan, Simple syntheses of (+)-orthosporin and (−)-semivioxanthin methyl ether, Tetrahedron, 1996, 52, 7159–7162 CrossRef CAS.
- (a) S. Barluenga, P. Lopez, E. Moulin and N. Winssinger, Modular asymmetric synthesis of pochonin C, Angew. Chem., Int. Ed., 2004, 43, 3467–3470 CrossRef CAS; (b) M. A. Marsini, K. M. Gowin and T. R. Pettus, Total synthesis of (±)-mitorubrinic acid, Org. Lett., 2006, 8, 3481–3483 CrossRef CAS PubMed; (c) S. Thayumanavan, A. Basu and P. Beak, Two different pathways of stereoinformation transfer: asymmetric substitutions in the (−)-sparteine mediated reactions of laterally lithiated N, N-diisopropyl-o-ethylbenzamide and N-pivaloyl-o-ethylaniline, J. Am. Chem. Soc., 1997, 119, 8209–8216 CrossRef CAS.
- (a) J. Li, H. Wang, H. Jin, Z. Xiang, L. Chen, P. J. Walsh and G. Liang, Base-Promoted Tandem Synthesis of 3, 4-Dihydroisoquinolones, Org. Lett., 2022, 24, 8125–8129 CrossRef CAS; (b) Y. Gu, Z. Zhang, Y.-E. Wang, Z. Dai, Y. Yuan, D. Xiong, J. Li, P. J. Walsh and J. Mao, Benzylic aroylation of toluenes mediated by a LiN (SiMe3) 2/Cs+ system, J. Org. Chem., 2021, 87, 406–418 CrossRef; (c) D. E. Anderson, A. Tortajada and E. Hevia, Highly reactive hydrocarbon soluble alkylsodium reagents for benzylic aroylation of toluenes using Weinreb amides, Angew. Chem., Int. Ed., 2023, 62, e202218498 CrossRef CAS; (d) C. C. Bao, Y. L. Luo, H. Z. Du and B. T. Guan, Benzylic aroylation of toluenes with unactivated tertiary benzamides promoted by directed ortho-lithiation, Sci. China: Chem., 2021, 64, 1349–1354 CrossRef CAS.
- (a) R. L. Vaulx, W. H. Puterbaugh and C. R. Hauser, Metalation of N-Methyl-o-toluamide with Excess n-Butyllithium. Condensations with Ketones and Aldehydes. Cyclizations, J. Org. Chem., 1964, 29, 3514 CrossRef CAS; (b) P. L. Creger, Metalated carboxylic acids. II. Monoalkylation of metalated toluic acids and dimethylbenzoic acids, J. Am. Chem. Soc., 1970, 92, 1396–1397 CrossRef CAS; (c) F. M. Hauser and R. Rhee, Carboxylation of dilithium ortho-toluates, Synthesis, 1977, 245–246 CrossRef CAS; (d) F. M. Hauser and R. P. Rhee, Syntheses of. alpha.-and. beta.-sorigenin methyl ethers, J. Org. Chem., 1977, 42, 4155–4157 CrossRef CAS.
- S. Ohta, Y. Kamata, T. Inagaki, Y. Masuda and S. Yamamoto, Synthesis of 3-substituted isocoumarins and related natural products, Chem. Pharm. Bull., 1993, 41, 1188–1190 CrossRef CAS.
- F. M. Hauser, R. P. Rhee, S. Prasanna, S. M. Weinreb and J. H. Dodd, ortho-Toluate carbanion chemistry: sulfenylation and selenation, Synthesis, 1980, 72–74 CrossRef CAS.
- C. N. Lewis, P. L. Spargo and J. Staunton, A Convenient Synthesis of 3-Substituted 8-Methoxy- and 6,8-Dimethoxyisocoumarins, Synthesis, 1986, 944–946 CrossRef CAS.
- T. A. Carpenter, G. E. Evans, F. J. Leeper, J. Staunton and M. R. Wilkinson, Reactions of the carbanion from an orsellinate derivative with electrophiles, J. Chem. Soc., Perkin Trans. 1, 1984, 1043–1051 RSC.
- Keto intermediate produced in very poor yields; H. Yaiqing, B. Junker, P. Helquist and R. E. Taylor, Synthesis of anti-angiogenic isocoumarins, Curr. Org. Synth., 2004, 1, 1–9 CrossRef.
- S. Ohta, A. Shimabayashi, S. Hayakawa, M. Sumino and M. Okamoto, Synthesis of t-butyl 3-oxoalkanoates by acylation of t-butyl lithioacetate with alkyl carboxylates, Synthesis, 1985, 45–48 CrossRef CAS.
- K. Yoshizawa, S. Toyota and F. Toda, Solvent-free Claisen and Cannizzaro reactions, Tetrahedron Lett., 2001, 42, 7983–7985 CrossRef CAS.
- (a) T. Zhang, Z. Yang, D. Zhou, F. Meng, Z. Han and H. Huang, Non-metal Lewis acid-catalyzed cross-Claisen condensation for β-keto esters, Org. Biomol. Chem., 2021, 19, 9163–9166 RSC; (b) Y. Yoshida, N. Matsumoto, R. Hamasaki and Y. Tanabe, Catalytic TMSCl promoted powerful aldol addition and Claisen condensation mediated by TiCl4/Bu3N agent: comparison and evaluation with the Mukaiyama aldol addition, Tetrahedron Lett., 1999, 40, 4227 CrossRef CAS; (c) Y. Tanabe, R. Hamasaki and S. Funakoshi, Powerful Claisen condensation and Claisen–aldol tandem reaction of α, α-dialkylated esters promoted by ZrCl4−i,Pr2Net, Chem. Commun., 2001, 1674–1675 RSC.
- (a) A. Iida, K. Takai, T. Okabayashi, T. Misaki and Y. Tanabe, NaOH-Catalyzed crossed Claisen condensation between ketene silyl acetals and methyl esters, Chem. Commun., 2005, 3171–3173 RSC; (b) Y. Nishimoto, A. Okita, M. Yasuda and A. Baba, Indium Tribromide Catalyzed Cross–Claisen Condensation between Carboxylic Acids and Ketene Silyl Acetals Using Alkoxyhydrosilanes, Angew. Chem., Int. Ed., 2011, 37, 8782–8784 CrossRef.
- S. Ranganayakulu, A. Syam and G. Rajendar, A Chiral Pool Approach for Total Synthesis of (+)–Diaportinol,(−)–Peniisocoumarin H and (+) & (−)–Desmethyldiaportinol; Their Structural Revision, Asian J. Org. Chem., 2022, 11, e20210066 CrossRef.
- M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Beyond Thermodynamic Acidity: A Perspective on the Complex–Induced Proximity Effect (CIPE) in Deprotonation Reactions, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRef CAS PubMed.
- P. E. Pfeffer, L. S. Silbert and J. M. Chirinko Jr, alpha. Anions of carboxylic acids. II. Formation and alkylation of. alpha.-metalated aliphatic acids, J. Org. Chem., 1972, 37, 451–458 CrossRef CAS.
- Reaction of arylogous enolates with Wienreb amide produce a stable tetrahedral intermediate through chelation, and it cannot produce ketone in situ until quenching the reaction.
- See ESI† for more optimization details.
- (a) P. L. Hall, J. H. Gilchrist and D. B. Collum, Effects of Lithium Salts on the Stereochemistry of Ketone Enolization by Lithium 2,2,6,6-Tetramethylpiperidide (LiTMP). A Convenient Method for Highly E’-Selective Enolate Formation, J. Am. Chem. Soc., 1991, 113, 9571–9574 CrossRef CAS; (b) P. L. Hall, J. H. Gilchrist, A. T. Harrison, D. J. Fuller and D. B. Collum, Mixed Aggregation of Lithium Enolates and Lithium Halides with Lithium 2,2,6,6-Tetramethylpiperidide (LiTMP), J. Am. Chem. Soc., 1991, 113, 9575–9585 CrossRef CAS.
- The structure of the aggregate is unknown, but its genesis can be deduced from experimental observation. When one equivalent of LiTMP is added to compound 10, an enolate (II) develops, a dark red solution. When LiBr is present in the medium, more than three equivalents of the base are consumed, to produce enolate (red colour solution).
- (a) A. Saeed, Synthesis of 6-O-methyl ether of scorzocreticin and scorzocreticoside I, metabolites from Sorzonera cretica, J. Asian Nat. Prod. Res., 2006, 8, 417–423 CrossRef CAS PubMed; (b) S. Paraschos, P. Migiatis, E. Kalpoutzakis, C. Harvala and A. L. Skaltsounis, Three New Dihydroisocoumarins from the Greek Endemic Species Scorzonera c retica, J. Nat. Prod., 2001, 64, 1585–1587 CrossRef CAS.
- R. Rossi, A. Carpita, F. Bellina, P. Stabile and L. Mannina, Synthesis of 3-arylisocoumarins, including thunberginols A and B, unsymmetrical 3, 4-disubstituted isocoumarins, and 3-ylidenephthalides via iodolactonization of methyl 2-ynylbenzoates or the corresponding carboxylic acids, Tetrahedron, 2003, 59, 2067–2081 CrossRef CAS.
- A. Boller, E. Gäumann, E. Hardegger, F. Kugler, St. Naef-Roth and M. Rosner, Welkstoffe und Antibiotika. 18. Mitteilung Diaporthin, ein Welketoxin aus Kulturen von Endothia parasitica (Murr.) And, Helv. Chim. Acta, 1957, 40, 875–880 CrossRef CAS.
- H. Hiroyuki, M. Tohru, O. Michiyo, H. Seiko, N. Hiroshi, S. Tsutomu, H. Masa, I. Masaaki and T. Tomio, Cytogenin a novel antitumor substance, J. Antibiot., 1990, 43, 1505–1507 CrossRef.
- Y. Honda, S. Katayama, M. Kojima, T. Suzuki and K. Izawa, Synthesis of Optically Active α-Aminoalkyl α’-Halomethyl Ketone: A Cross-Claisen Condensation Approach, Org. Lett., 2002, 4, 447–449 CrossRef CAS.
- K. Sudarshan, M. K. Manna and I. S. Aidhen, Synthesis of 3-arylisocoumarins by using acyl anion chemistry and synthesis of Thunberginol A and Cajanolactone, Eur. J. Org. Chem., 2015, 1797–1803 CrossRef CAS.
- Y.-C. Tzeng, C.-C. Hsieh, M. El-Shazly, S.-Y. Chiang, C.-C. Wu, C.-C. Wu, W.-C. Liu, M.-H. Yang, Y.-C. Wu and F.-R. Chang, Phytochemicals and Estrogen-Receptor Agonists from the Aerial Parts of Liriope platyphylla, Molecules, 2015, 20, 6844–6855 CrossRef PubMed.
- Y. Furutani, H. Naganawa, T. Takeuchi and H. Umezawa, Inhibitors of cyclic nucleotide phosphodiesterases produced by Streptomyces. Part II. Isolation and structure of new isocoumarins, Agric. Biol. Chem., 1977, 41, 1179–1183 CAS.
- S. Jing, Z. Qu, C. Zhao, X. Li, L. Guo, Z. Liu, Y. Zheng and W. Gao, Dihydroisocoumarins and Dihydroisoflavones from the Rhizomes of Dioscorea collettii with Cytotoxic Activity and Structural Revision of 2, 2′-Oxybis (1, 4-di-tert-butylbenzene), Molecules, 2021, 26, 5381 CrossRef CAS PubMed.
- X. Li, C. Liu, Z. Duan and S. Guo, HMG–CoA Reductase Inhibitors from Monascus–Fermented Rice, J. Chem., 2013, 872056 CrossRef.
- (a) Y. Yamaguchi, R. Masuma, Y. P. Kim, R. Uchida, H. Tomoda and S. Omura, Taxonomy and secondary metabolites of Pseudobotrytis sp. FKA-25, Mycoscience, 2004, 45, 9–16 CrossRef CAS; (b) G. Promsuk, P. Chuawong, P. Songjanthuek, S. Thaisri and B. Yongsmith, Absolute configuration of azaphilones from Monascus kaoliang KB9 and solvent effects on their keto and enol forms, Nat. Prod. Res., 2023, 37, 2181–2188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2365878 (for 45S) and 2365877 (for 45R). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01678e |
| This journal is © the Partner Organisations 2025 |