
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotent EGFR/PARP-1 inhibition by spirooxindole-triazole hybrids for targeted liver cancer therapy†
Mohamed S. Nafie ab,
M. Alic,
Moayad Abdullah Alwehaibic,
Abdulmajeed Abdullah Alayyafc,
Muhanna K. Al-Muhannad,
Naif S. Almuqatie,
Abdullah A. Alghamdie,
Matti Haukkaf,
Syeda Sumayya Tariqg,
Zaheer Ul-Haqg and
Assem Barakat*c
ab,
M. Alic,
Moayad Abdullah Alwehaibic,
Abdulmajeed Abdullah Alayyafc,
Muhanna K. Al-Muhannad,
Naif S. Almuqatie,
Abdullah A. Alghamdie,
Matti Haukkaf,
Syeda Sumayya Tariqg,
Zaheer Ul-Haqg and
Assem Barakat*c
aDepartment of Chemistry, College of Sciences, University of Sharjah, Sharjah, 27272, United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae
bChemistry Department, Faculty of Science, Suez Canal University, Ismailia, 41522, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg
cDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: mayyaf@ksu.edu.sa; Maly.c@ksu.edu.sa; 442105720@student.ksu.edu.sa; ambarakat@ksu.edu.sa
dThe Material Science Research Institute, King Abdulaziz City for Science and Technology (KACST), Riyadh 11442, Saudi Arabia. E-mail: mmuhanna@kacst.edu.sa
eRefining and Petrochemical Technologies Institute (RPTI), KACST, Saudi Arabia
fDepartment of Chemistry, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: matti.o.haukka@jyu.fi
gDr Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan. E-mail: sumayyatariq7@gmail.com; zaheer_qasmi@hotmail.com
First published on 3rd January 2025
Abstract
The search for effective anti-cancer therapies has led to the exploration of dual inhibition strategies targeting multiple key molecular pathways. In this study, we aimed to design a novel candidate capable of dual inhibition targeting both EGFR (Epidermal Growth Factor Receptor) and PARP-1 (poly(ADP-ribose)polymerase-1), two crucial proteins implicated in cancer progression and resistance mechanisms. Through molecular hybridization and structure-based drug design approaches, we synthesized a series of compounds based on spirooxindole with triazole scaffolds with the potential for dual EGFR and PARP-1 inhibition. Interestingly, compounds 4a, 4b and 4f showed potent cytotoxicity against HepG2 cells with IC50 values of 3.6, 4.6, and 1.9 μM compared to doxorubicin (IC50 = 2.78 μM), with poor cytotoxicity against THLE2 cells with IC50 values higher than 40 μM in a selective way. They exhibited potent EGFR and PARP-1 inhibition with IC50 values of 74.6, 85.3, and 116.1 nM compared to Erlotinib (IC50 = 80 nM, 94.7%). Additionally, they exhibited potent PARP-1 inhibition, with IC50 values of 2.01, 3.6, and 18.4 nM compared to Olaparib (IC50 = 1.49 nM, 94.5%). Furthermore, compound 4a-treatment induced increased apoptosis by 6.6-fold, as the effective cell death mechanism. Overall, our study underscores the importance of multi-targeted approaches in developing effective anti-cancer agents.
1 Introduction
Numerous strategies have been approved for cancer treatment.1 These strategies encompass the induction of apoptosis, the inhibition of signal transduction, the expression of gene modulators, and the suppression of angiogenesis.2–4 In the first strategy, apoptosis induction targets and eliminates unwanted cancer cells while leaving healthy cells unaffected. Apoptosis induction can be achieved by inhibiting or activating specific regulatory proteins, such as BAX, BCL-2, caspase-3, caspase-9, tubulin, and various kinases.5 The upregulation of BAX and/or downregulation of BCL-2 have been documented as inducers of apoptotic cell death. Additionally, caspases, a group of protease enzymes, play a crucial role in enhancing apoptotic cell death. Among these, the initiator caspases (e.g., caspase-9) and effector caspases (e.g., caspase-3) are directly involved in the process of apoptosis.5Another valuable strategy in cancer treatment involves the inhibition of the poly(ADP-ribose)polymerase (PARP) family of proteins.6,7 Among the members of this family, PARP-1 is an adenosine diphosphate ribosyl transferase primarily localized within the nucleus and frequently associated with chromatin 8. PARP-1 is an apoptosis-inducing factor that has been implicated in apoptotic cell death triggered by DNA damage. In recent years, significant research attention has been devoted to the development of PARP-1 inhibitors as potential antitumor drugs. PARP-1 plays a significant role in both the DNA repair process and the regulation of cell survival and death.9,10 Numerous PARP-1 inhibitors, including Rucaparib11 (Fig. 1), have been developed and utilized in clinical treatments. Typically, these inhibitors contain an amide fragment crucial for interacting with Ser904 and Gly863 residues, which are considered the key pharmacophore for PARP-1 inhibition. Many other representative examples have been reported, either synthetically or naturally occurring, targeting the PARP-1 inhibition, such as chalcone-based triazole scaffold that exhibited apoptosis inducer and showed PARP-1 degrading activity.12 Compound IV demonstrated potent inhibition of PARP-1 biosynthesis at a nanomolar concentration and exhibited an exceptional selectivity index.13
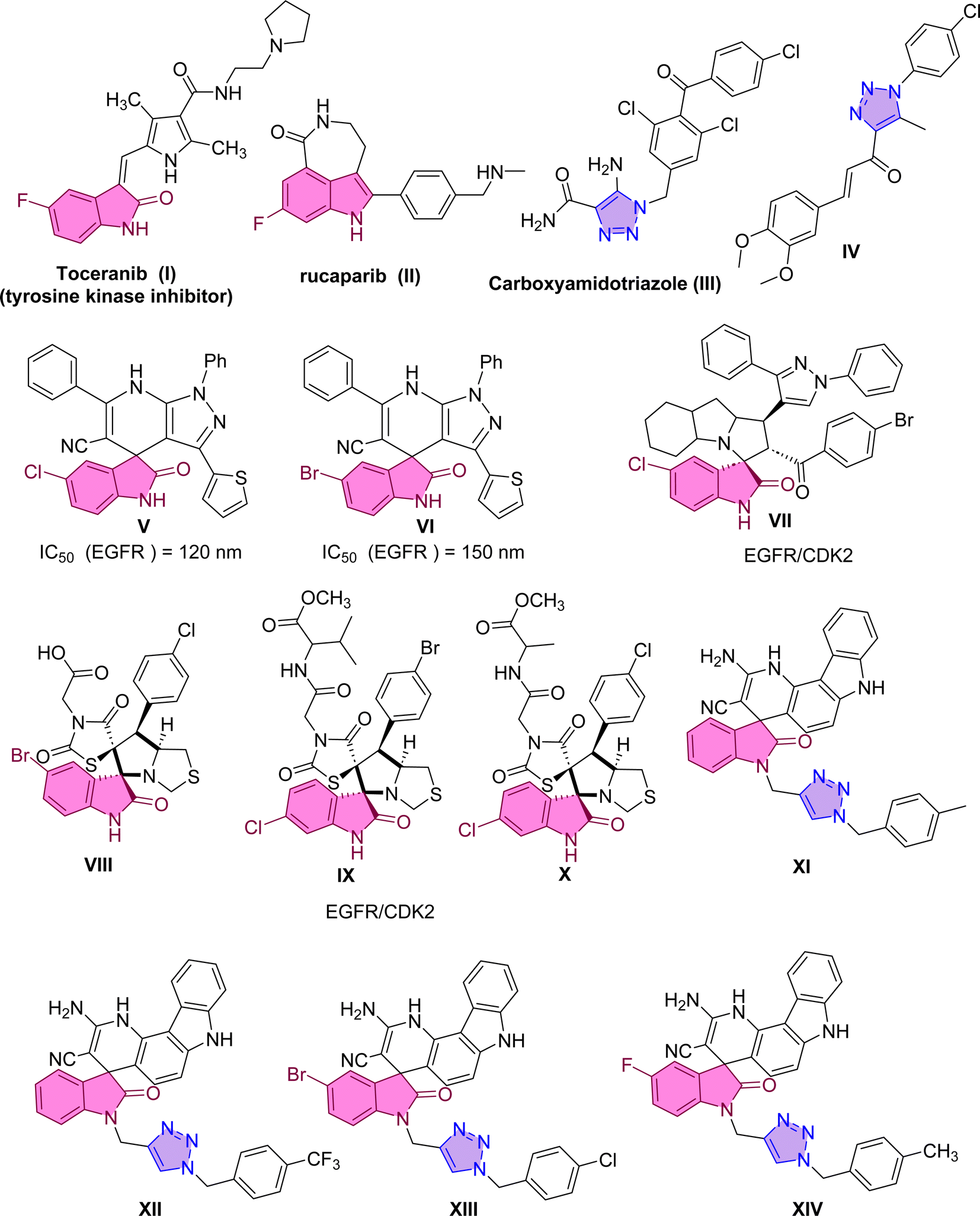 | ||
| Fig. 1 Reported examples targeting EGFR and PARP-1 derived from spiro-heterocycles and triazole scaffold. | ||
Due to its importance as a nitrogen heterocyclic compound, 1,2,3-triazole has found extensive use in constructing drug molecules,14–16 particularly in the realm of anti-cancer agents.17,18 Its chemical properties suggest that 1,2,3-triazole is an effective pharmacophore, facilitating interactions with drug receptors while maintaining favorable chemical and metabolic profiles.19 Therefore, we retained the 1,2,3-triazole moiety to enhance the compound's activity.
Another alternative strategy for cancer treatment involves inhibiting a protein known as EGFR (Epidermal Growth Factor Receptor). The Epidermal Growth Factor Receptor (EGFR) represents a crucial molecular target in growth factor signaling.20 Dysregulated signaling of these factors can result in uncontrolled cell proliferation, suppression of apoptosis, promotion of angiogenesis, facilitation of migration, and metastasis, all hallmark characteristics of cancer cells. The overexpression of EGFR in various human cancer cell types, including liver, bladder, breast, colon, and lung cancer cells, has identified it as a promising molecular target for inhibition. Consequently, researchers have devised strategies to counteract the effects of EGFR activation, aiming to impede tumor growth and invasion. Various agents, such as specific antibodies targeting its ligand-binding domain and small molecules inhibiting its tyrosine kinase activity, are undergoing clinical trials or have already gained approval for clinical treatment. In 2018, Eldehna et al. reported the synthesis of spirooxindole derivatives V and VI through a one-pot three-component reaction.21 These derivatives exhibited potent inhibition of EGFR, with IC50 values of 120 nM and 150 nM, respectively.21
A recent example in 2023, Al-Jassas et al. reported the design, synthesis, and evaluation of a series of spirooxindoles with pyrazole scaffold-targeted EGFR and CDK2 dual inhibitory effects. The spirooxindole hybrid VII displayed significant potential, with IC50 values of 0.189 ± 0.01 μM (MCF-7) and 1.04 ± 0.21 μM (HepG2). Furthermore, it possessed potent CDK-2 inhibition (IC50 = 34.98 nM) and an IC50 of 96.6 nM for EGFR inhibition.22 Compound II also effectively regulated the expression of pro-apoptotic genes (P53, Bax, caspases-3, 8, and 9) while downregulating the anti-apoptotic gene BCL-2. In 2024, pyrrolidinyl-spirooxindole derivatives VIII–X were synthesized and found to possess dual activities against EGFR and CDK-2.23 These compounds exhibited remarkable cytotoxicity against cancer cells, demonstrating notable potency against MCF-7 and MDA-MB-231 cells.
Molecular hybridization represents a novel approach to drug design and development. It involves combining pharmacophoric moieties from different bioactive substances to create a new hybrid compound. This hybridization aims to enhance the affinity and efficacy of the resulting compound compared to its parent drugs. A combination of triazole scaffold with spiro-heterocycles has demonstrated that compounds exhibit potent anti-cancer activities against cervical cancer HeLa cells. In a study by Sarkar et al., a series of chromenocarbazole tethered 1,2,3-triazoles were designed and synthesized using a click chemistry-based one-pot five-component reaction.24 Compounds XI, XII, XIII, and XIV displayed excellent antiproliferative activity (with IC50 values of 4.05, 3.54, 3.83, and 3.35 μM, respectively) in HeLa cells.
Based on these findings and the continuation of our research program about the development of the spirooxindole scaffold,25–27 the aim of this study was to design a novel candidate capable of dual inhibition targeting both EGFR (Epidermal Growth Factor Receptor) and PARP-1 (poly(ADP-ribose)polymerase-1). In this text, we synthesized a series of spirooxindole hybrids with triazole scaffold, the compounds screened against two cancer cell lines (HepG2 and THLE2), and the most active candidate was further explored for mechanism of action targeting EGFR and PARP-1. Molecular docking study and molecular dynamic simulation were also explored.
2 Results and discussion
The synthesis of spirooxindole-triazole hybrids through [3 + 2] cycloaddition reactions offers a promising route for developing potent inhibitors targeting EGFR and PARP-1 for cancer therapy. The unique structure of these hybrids, combining the biological activities of spirooxindoles and 1,2,3-triazoles, is designed to enhance their therapeutic efficacy and specificity (Scheme 1). Isatin derivatives 2a–e, amino acid 3, and chalcone-based triazole scaffolds 1a–d were selected for their ability to form spirooxindole structures 4a–k in a one-step fashion. Azides were utilized to introduce the triazole ring via the [3 + 2] cycloaddition reaction with acetylacetone, forming an acetyl-triazole derivative, which was then employed as a precursor for synthesizing chalcone-based triazoles through aldol condensation with heteroaryl aldehydes followed our previously reported method. 1H and 13C-NMR spectroscopy were employed to confirm the chemical structure of the synthesized hybrids, identifying characteristic peaks corresponding to the oxindole, triazole, and other substituent groups. Single-crystal X-ray diffraction was successfully performed for acetyl-triazole derivative and two chalcone-based triazole compounds 1a and 1b, providing detailed information on their molecular geometry and crystal structure, and offering insights into the spatial arrangement of the hybrid molecules (Fig. 2). Compounds 4c and 4j serve as representative examples of the spirooxindole cycloadducts, whose chemical structures were confirmed through single-crystal X-ray analysis. These results have been published as a CSD Communication in 2024.28,29 The data provide conclusive evidence of the absolute configuration and stereochemistry of the spirooxindole cycloadducts.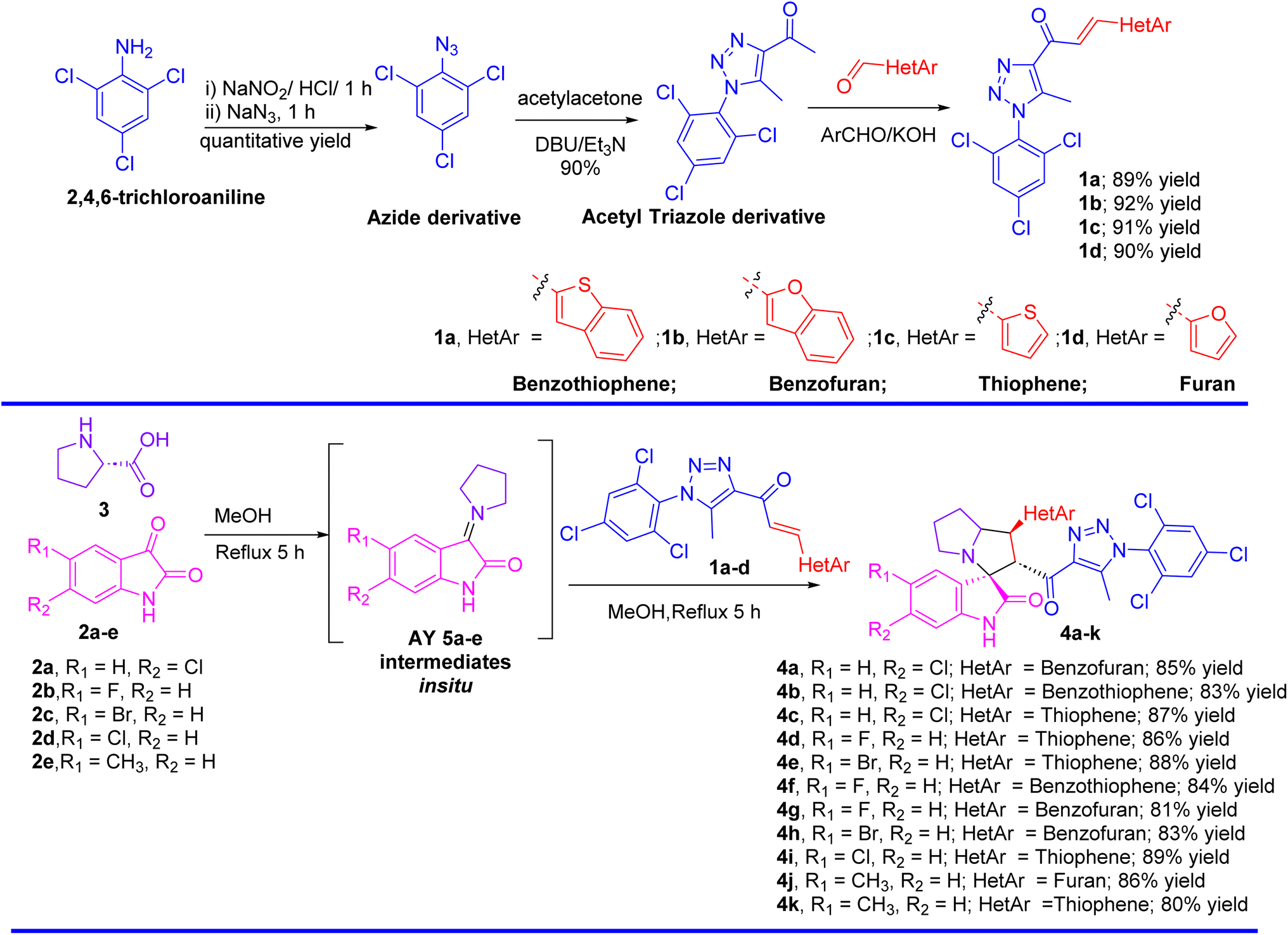 | ||
| Scheme 1 Synthesis of the spirooxindoles 4a–k. | ||
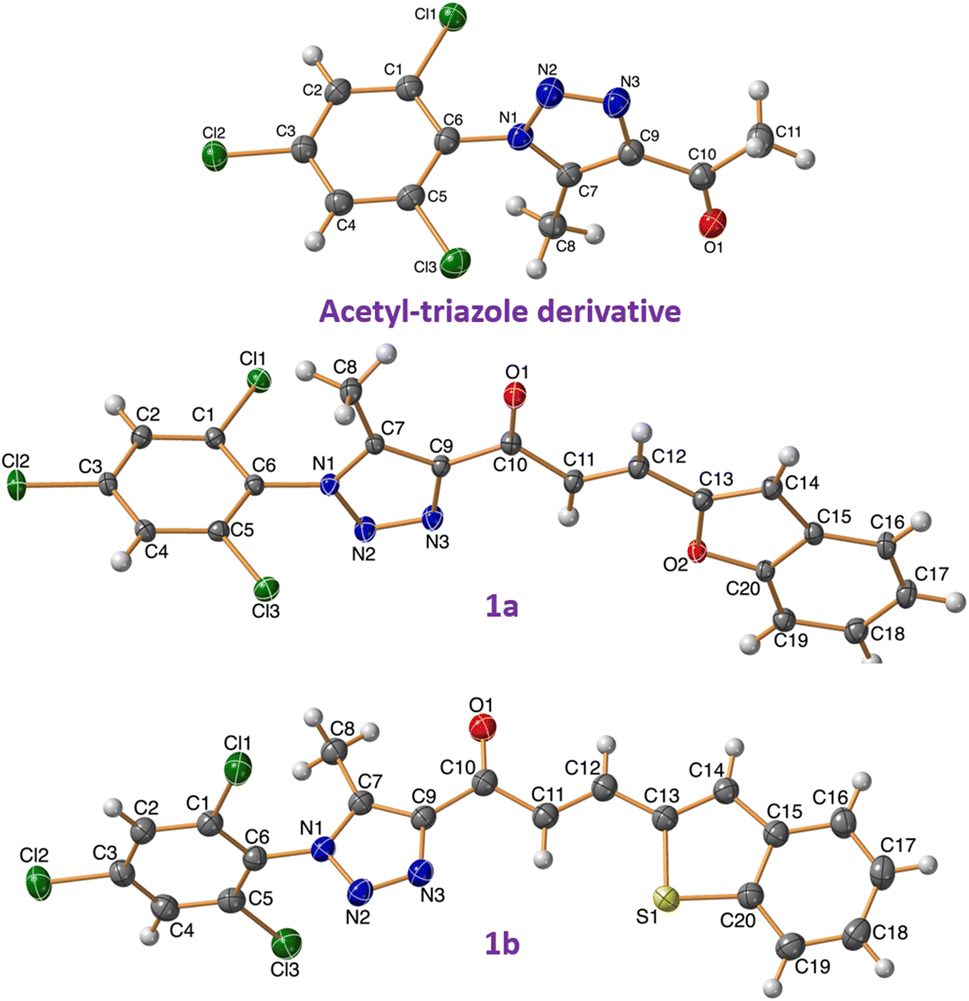 | ||
| Fig. 2 X-ray for the chalcones based triazole scaffold acetyl-triazole derivative and chalcones 1a and 1b. | ||
2.1 Cytotoxicity against cancer cells
The synthesized compounds were screened for their cytotoxicity against HepG2 and THLE2 cells using the MTT assay. As seen in Table 1, compounds 4a, 4b, and 4f showed potent cytotoxicity against HepG2 cells with IC50 values of 3.6, 4.6, and 1.9 μM compared to doxorubicin (IC50 = 2.78 μM), with poor cytotoxicity against THLE2 cells with IC50 values higher than 40 μM in a selective way. While compounds 1b, 1b, 1c, 4g, and 4h showed promising cytotoxicity against HepG2 cells with IC50 values range of 9.7–15.6 μM, with selective cytotoxicity against THLE2 cells with higher IC50 values. In contrast, other compounds weren't cytotoxic against HepG2 cells with higher IC50 values. Hence, compounds 4a, 4b, and 4f with potent cytotoxicity were examined for the EGFR/PARP-1 inhibition as the designed molecular targets.| # | Chemical structures | IC50 ± SD [μM] | |
|---|---|---|---|
| HepG2 | THLE2 | ||
| a Values are expressed as mean ± SD of three independent trials. NT = non-tested. IC50 of doxorubicin (DOX) = 2.78 μM as a reference drug. | |||
| 1a |  |
9.7 ± 0.2 | 45.8 ± 2.1 |
| 1b | 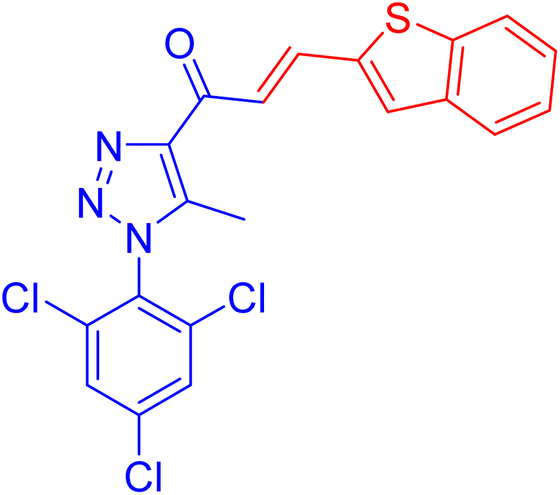 |
8.5 ± 0.4 | 42.9 ± 3.0 |
| 1c | 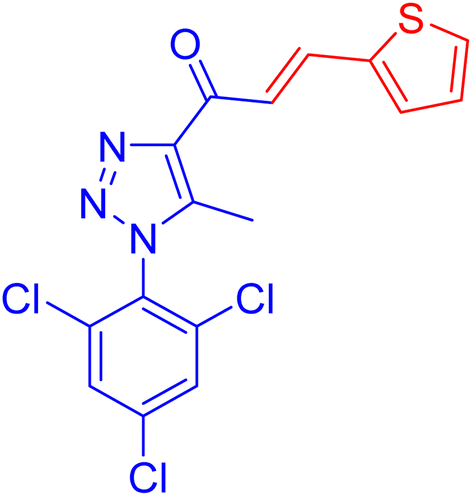 |
16.8 ± 0.5 | 32.5 ± 1.9 |
| 1d | 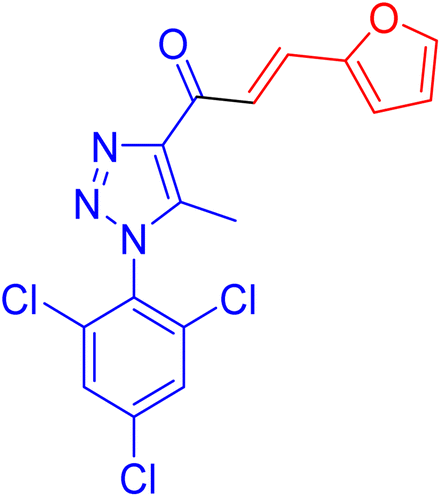 |
17.6 ± 0.4 | 42.3 ± 0.9 |
| 4a |  |
3.6 ± 0.1 | 43.8 ± 2.9 |
| 4b |  |
4.6 ± 0.2 | 39.8 ± 2.0 |
| 4c |  |
27.3 ± 0.9 | NT |
| 4d |  |
31.5 ± 1.5 | NT |
| 4e |  |
28.9 ± 1.1 | NT |
| 4f |  |
1.9 ± 0.1 | 43.7 |
| 4g | 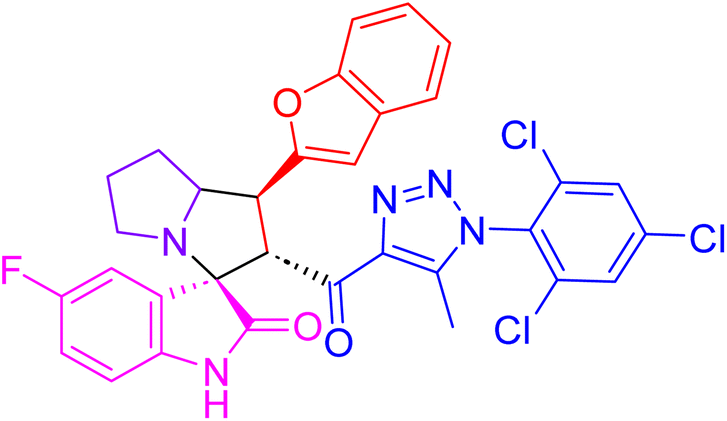 |
8.9 ± 0.3 | 38.4 ± 1.6 |
| 4h | 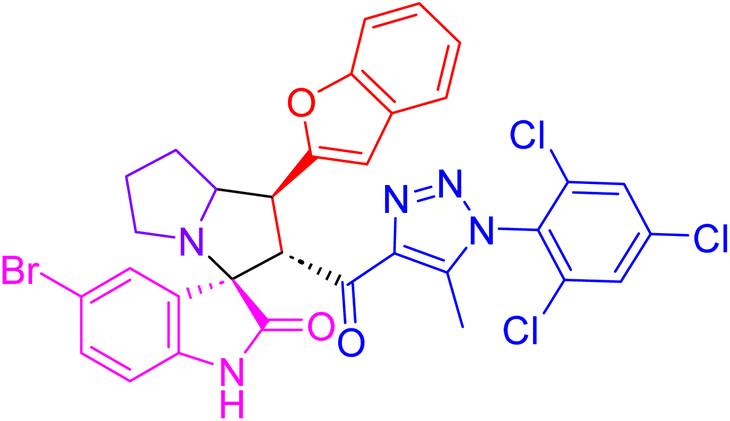 |
15.6 ± 0.4 | 29.7 ± 1.2 |
| 4i |  |
38.9 ± 1.4 | NT |
| 4j |  |
14.3 ± 0.7 | 39.0 ± 2.3 |
| 4k | 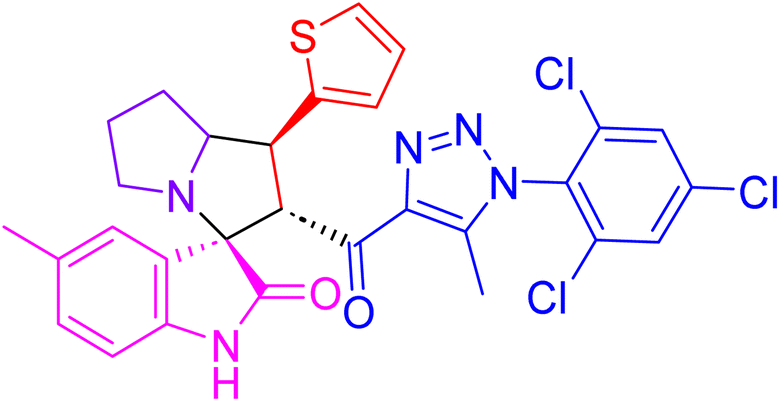 |
37.5 ± 1.7 | NT |
2.2 EGFR/PARP-1 inhibition
Activity against EGFR and PARP-1 was assessed for compounds 4a, 4b, and 4f. It is worth noting, as shown in Table 2, they exhibited potent EGFR inhibition, with IC50 values of 74.6, 85.3, and 116.1 nM with the percentages of inhibition of 94.1%, 90.1%, 84.3% compared to Erlotinib (IC50 = 80 nM, 94.7%). Additionally, they exhibited potent PARP-1 inhibition, with IC50 values of 2.01, 3.6, and 18.4 nM with a percentage of inhibition of 95.2%, 81.2%, 86.6% compared to Olaparib (IC50 = 1.49 nM, 94.5%). The results show that inhibiting the EGFR/PARP-1 enzyme is a potential strategy that could explain the cytotoxic effects.| ≠ | Chemical structure | IC50 ± SD [nM] | |
|---|---|---|---|
| EGFR | PARP-1 | ||
| a “Values are expressed as an average of three independent replicates”. “IC50 values were calculated using sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound”. | |||
| 4a | 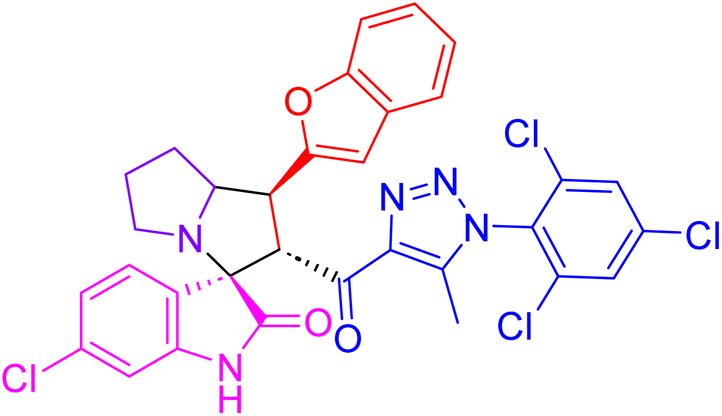 |
74.6 ± 1.8 | 2.01 ± 0.1 |
| 4b | 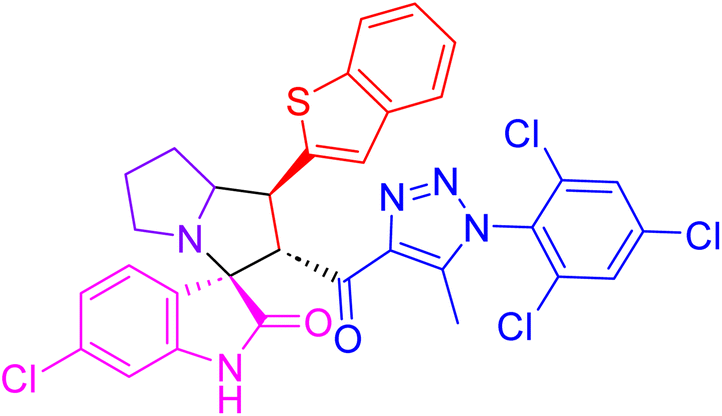 |
85.3 ± 2.9 | 3.6 ± 0.3 |
| 4f | 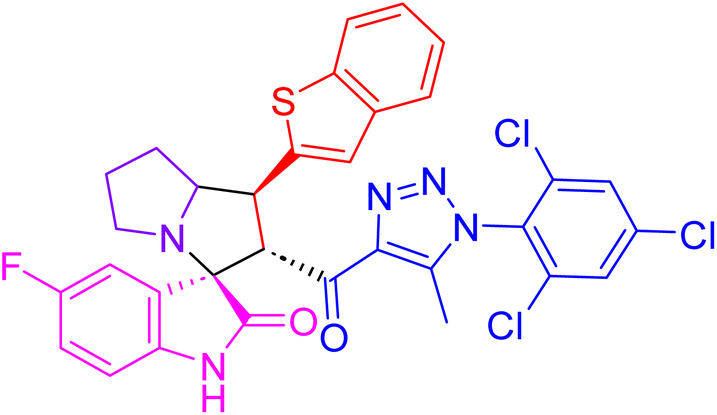 |
116.1 ± 3.1 | 18.4 ± 1.0 |
| Erlotinib | 80.1 ± 1.8 (ref. 30) | — | |
| Olaparib | — | 1.49 ± 0.2 (ref. 31) | |
The spirooxindole triazole compound is an effective treatment for liver cancer due to its ability to block growth factors from activating the EGFR receptor, preventing tumor growth. This bidirectional action targets both the tumor and immune system, enhancing the body's ability to fight cancer. In early experiments, the compound significantly reduced liver cancer tumors and increased survival rates compared to untreated mice. It also increased apoptosis and decreased cell growth, demonstrating its effectiveness against cancer cells in multiple ways.
The spirooxindole triazole compound inhibits the growth of cancer cells. It accomplishes this by focusing on and inhibiting PARP-1, a protein. Cancer cells cannot repair DNA damage if PARP-1 is inhibited, leading to more DNA damage and, ultimately, cell death. The chemical kills cancer cells by targeting PARP-1, which prevents them from repairing themselves. Because it inhibits tumor growth and enhances the immune system's response, the spirooxindole triazole molecule represents an encouraging prospect for cancer treatment. The chemical spirooxindole triazole was utilized in a preclinical investigation; it inhibited PARP-1 and led to DNA damage accumulation in cancer cells. As a result, tumor development in mice was significantly reduced. Additionally, the chemical improved the efficacy of immune checkpoint inhibitors, which accelerated tumor regression and improved immune responses to malignancies.
2.3 Apoptosis-investigation activity
2.3.1.1 Annexin V/PI staining with cell cycle analysis. Flow cytometric examination of Annexin V/PI staining of both untreated and 4a-treated HepG2 cells was used to investigate the apoptotic activity of compound 4a. Fig. 3(A) showed that compound 4a activated apoptotic cell death, increasing the cell population in total apoptosis by 13.05% (0.95% late and 12.1% early apoptosis) compared to the untreated group (1.97%). While, it didn't affect necrotic cell death, as it reduced cells in necrosis by 0.58% compared to 0.61% in the untreated control. Hence, compound 4a-treatment induced apoptosis by 6.62-fold.
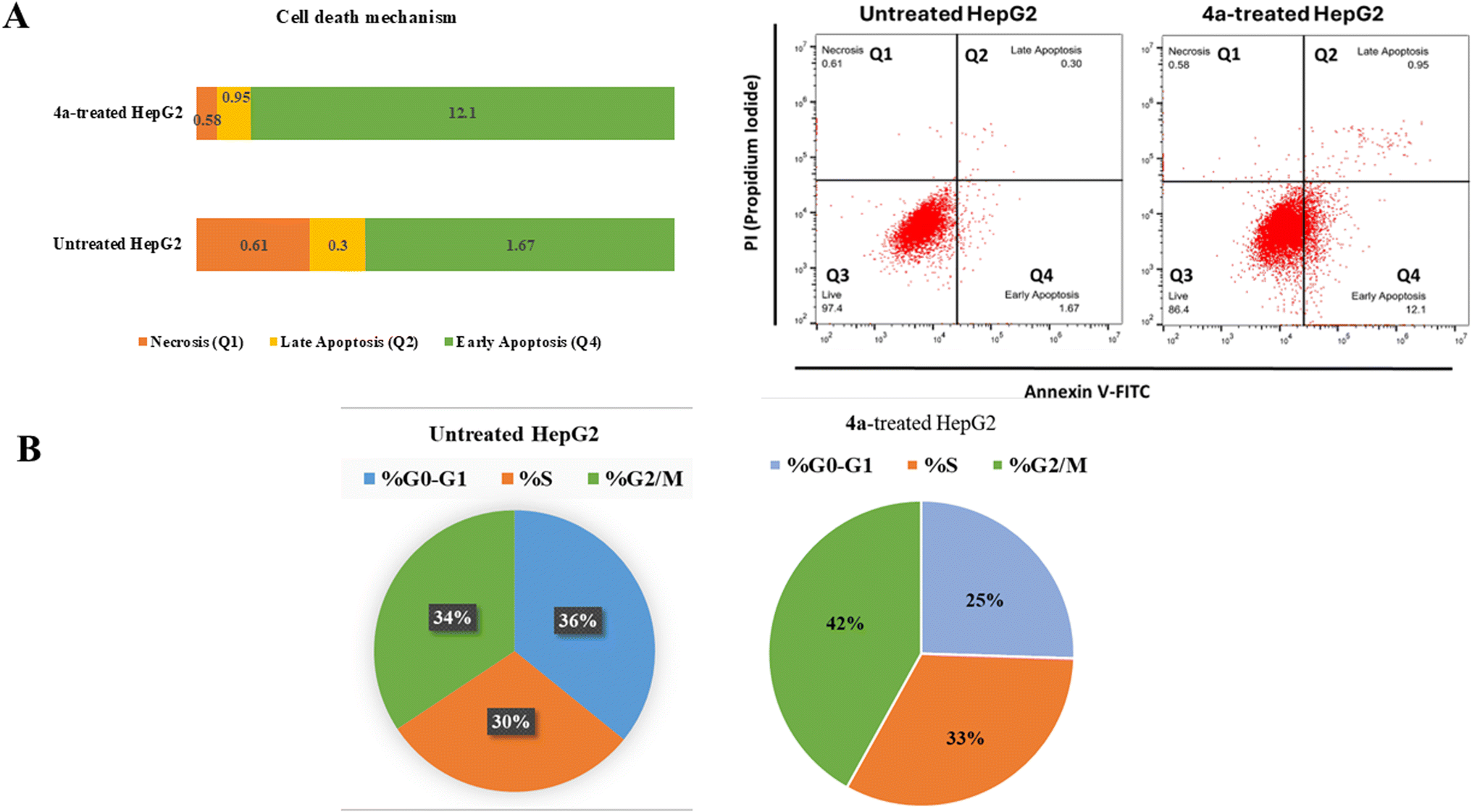 | ||
| Fig. 3 (A) Pie-chart representation of the percentage of apoptosis (early and late apoptotic cell population) using Annexin V/propidium iodide staining. (B) Pie-chart representation of the percentage of cell population at each cell phase in untreated and 4a-treated HepG2 cells, with the IC50 value at 48 h. “The data shown are the average of three independent experimental runs (mean ± SD)”. | ||
Additionally, As can be shown in Fig. 3(B), the cell population in the “G2/M-phase” was significantly increased by 42% after treatment with compound 4a, compared to the control 34%, whereas the cell population in the “S-phase” was non-significantly increased by 33% after treatment compared to the control 30%, hence, in contrast, cells population at “G0–G1-phase” were decreased upon treatment.
2.4 Gene expression analysis of apoptosis-related genes
To confirm apoptotic cell death, RT-PCR was used on both the untreated and treated HepG2 cells (Fig. 4). The expression of pro-apoptotic genes “P53, Bax, and caspases 3, 8, and 9” was upregulated by 4a treatment, corresponding fold changes of 9.3, 4.6, 5.6, 3.5, and 8.6, respectively. Concurrently, it resulted in a 0.37-fold decrease in the gene expression of the anti-apoptotic BCL-2. These findings align with the possibility of triggering cell death by blocking enzymes. The release of “cytochrome c” and loss of mitochondrial potential are the results of activation of the intrinsic pathway of apoptosis. Cell death occurring through “caspase-dependent apoptosis” occurs when the ratio of proteins that promote cell death to those that prevent it increases, setting in motion a cascade reaction involving activations of caspases 3 and 9 proteins. | ||
| Fig. 4 Gene expression levels of apoptosis-related genes of untreated and 4a-treated HepG2 cells with the IC50 value. “Values are expressed as mean ± SD of three independent replicates”. “Data were normalized using β-actin as a housekeeping gene”. “The dashed line represents the gene expression level of untreated control. Fold change was calculated using 2−ΔΔCT”. | ||
In accordance with previous literature,32–34 both molecular targets of EGFR and PARP-1, which target tumor growth and resistance to conventional therapies, hold great promise as a liver cancer treatment option. Researchers aim to inhibit these critical processes, potentially improving the efficacy of radiation therapy and chemotherapy. This approach may be the last resort for patients who have not responded to standard therapies or cannot undergo surgery. A synergistic impact between PARP-1 and EGFR medicines could improve outcomes and survival rates for liver cancer patients. Preclinical studies have shown promising results with the potential to revolutionize future treatments. A mouse model will be further recommended to validate the decrease of tumor size after therapy targeting angiogenesis and immunological checkpoint pathways. This two-pronged approach may circumvent the limitations of current therapies and provide a more tailored a future chance of treatment for liver cancer.
2.5 Molecular interactions
Molecular docking was used for the evaluation of the binding modes of the spirotriazole derivatives against poly(ADP-ribose)polymerase (PARP1) and Epidermal Growth Factor Receptor (EGFR). For this purpose, the crystal structure of PDB IDs 7AAD and 1M17, respectively. The respective controls Olaparib and Erlotinib were also subjected to docking along with the spirotriazole derivatives.Analysis of the PARP1–ligand (4a) complex with the best IC50 (compound 4a) and top-ranked pose via Protein–Ligand Interaction Profiler (PLIP) revealed Hydrogen bonding with Asp766 and Tyr896 residues of the PARP1 at a bond distance of 1.93 and 2.89 Å respectively, while residues Ile872, Leu877, Arg878, Tyr889, Ala898, and Glu988 were found to be involved in hydrophobic interactions. Similarly, the PLIP analysis of the PARP-Olaparib complex revealed Gly863, Ser864, and Ser904 forming hydrogen bonds with Olaparib at a distance of 2.10, 1.76, and 1.85 Å, respectively, while Glu763, Arg878, Tyr896, Tyr907, Glu988 were found involved in hydrophobic bonding. Fig. 5 shows the molecular interactions within (A) PARP1–ligand and (B) PARP1–control.
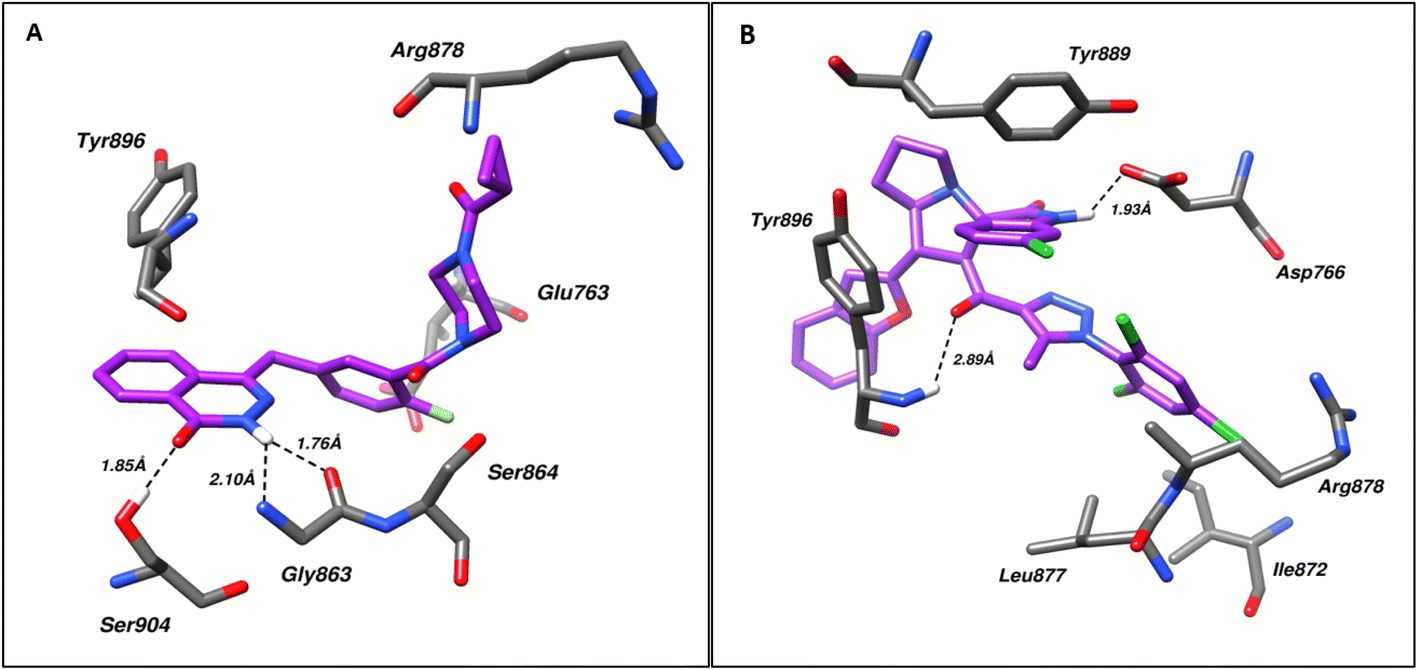 | ||
| Fig. 5 Shows the molecular interactions within (A) PARP1–control and (B) PARP1–ligand (4a). Hydrogen bonds are shown as dotted lines. | ||
In case of EGFR, analysis of the EGFR–ligand (4a) complex with the best IC50 and top-ranked pose via Protein–Ligand Interaction Profiler (PLIP) revealed hydrogen bonding with Arg817 at a distance of 2.25 Å while residues Leu694, and Phe699 took part in hydrophobic bonding with the ligand. On comparison with the EGFR–Erlotinib complex, it was found that this interaction involved hydrogen bonding via Ser696, in addition to Lys721, Thr766, and Met769 at a distance of 3.35, 3.30, 3.16, and 2.63 Å, respectively. Residues Leu694, Val702, Leu768, and Leu820 were found involved in hydrophobic bonding. Fig. 6 shows the molecular interactions within (A) EGFR–ligand and (B) EGFR–control.
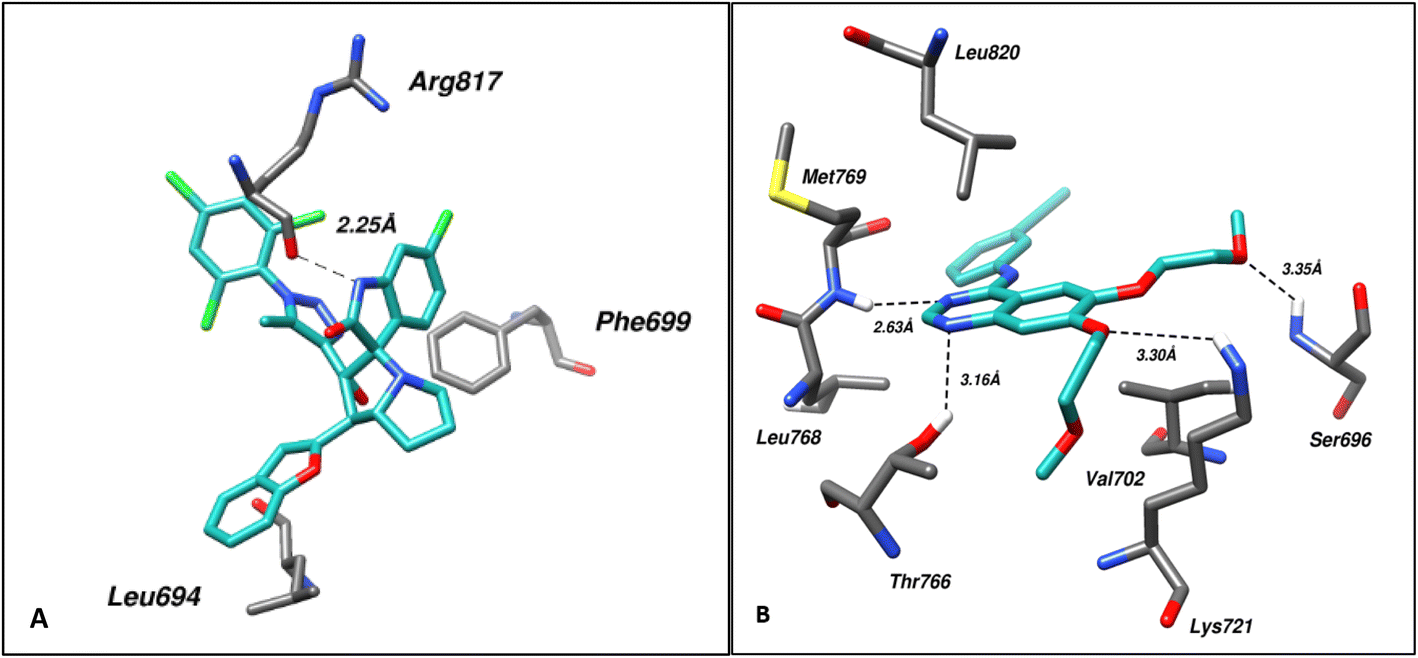 | ||
| Fig. 6 Shows the molecular interactions within (A) EGFR–ligand (4a) and (B) EGFR–control. Hydrogen bonds are shown as dotted lines. | ||
2.6 Molecular dynamics
To assess the structural stability of the protein–ligand complex, RMSDs of the 50 ns simulated trajectories were calculated. The RMSD of any simulated biomolecular system serves as an essential parameter to gauge the system's stability. In the context of structural biology, when studying the atomic coordinates, the lower RMSD values indicate greater system stability, whereas larger RMSD values correspond to less stable complexes.35 Throughout the simulation, both the PARP1 and EGFR exhibited consistent RMSDs with minimal fluctuations, indicating that the complexes remained stable throughout the simulation.In order to assess the inherent flexibility of the protein during the simulation run, Root Mean Square Fluctuations (RMSF) of the simulated trajectories, were computed. The RMSF provides information regarding residue-level perturbations where higher values within a biomolecular system signify flexibility and, consequently a less stable state.36 On the other hand, reduced fluctuation imparts greater stability to the system.37 The average RMSFs recorded for PARP1 and EGFR, indicated that the protein residues remained stable upon interaction.
The structural properties of the simulated ensembles were further assessed via the time-dependent convergence of the radius of gyration (RoG), which is related to the compactness of the protein. A properly folded conformation in general features constant gyration values, while on the other hand, distortion in the folding behavior induces the RoG values to change over time.38 The average RoG recorded for the PARP–ligand and EGFR–ligand complexes demonstrated stability with minimal fluctuations. The RMSD, RMSF, and RoG plots of the PARP1 and EGFR systems are shown in Fig. 7 and 8 respectively.
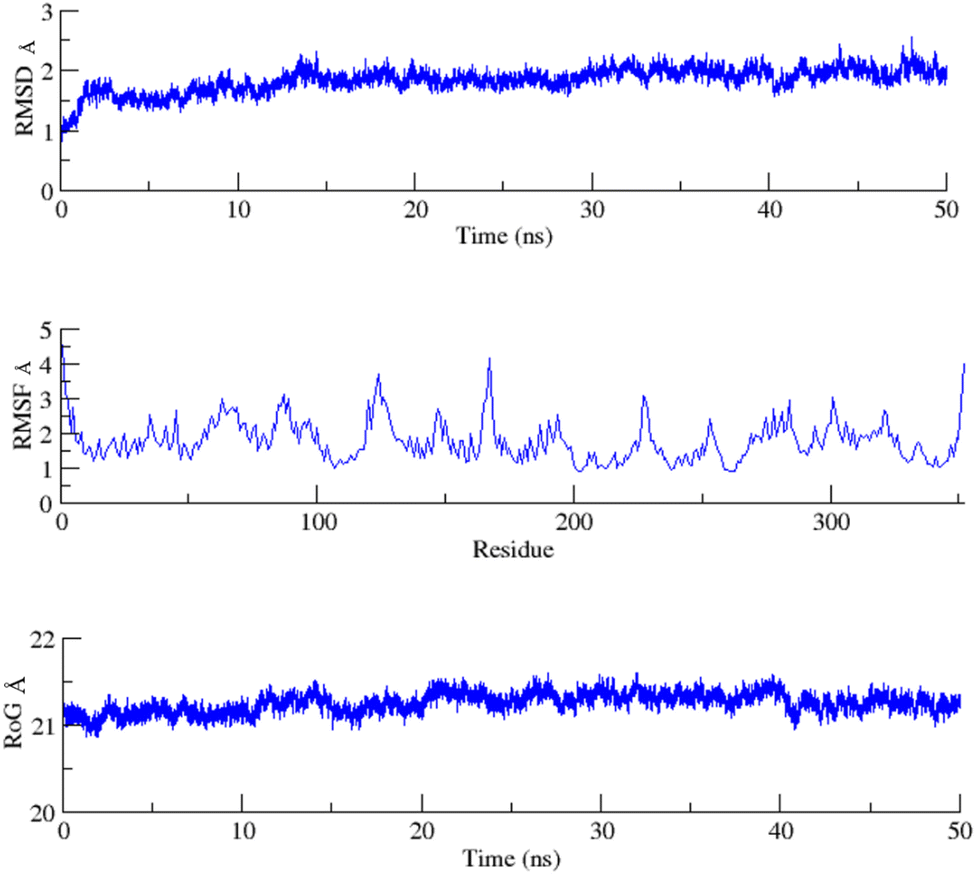 | ||
| Fig. 7 RMSD, RMSF and RoG plots of the PARP1 systems calculated as a function of time. | ||
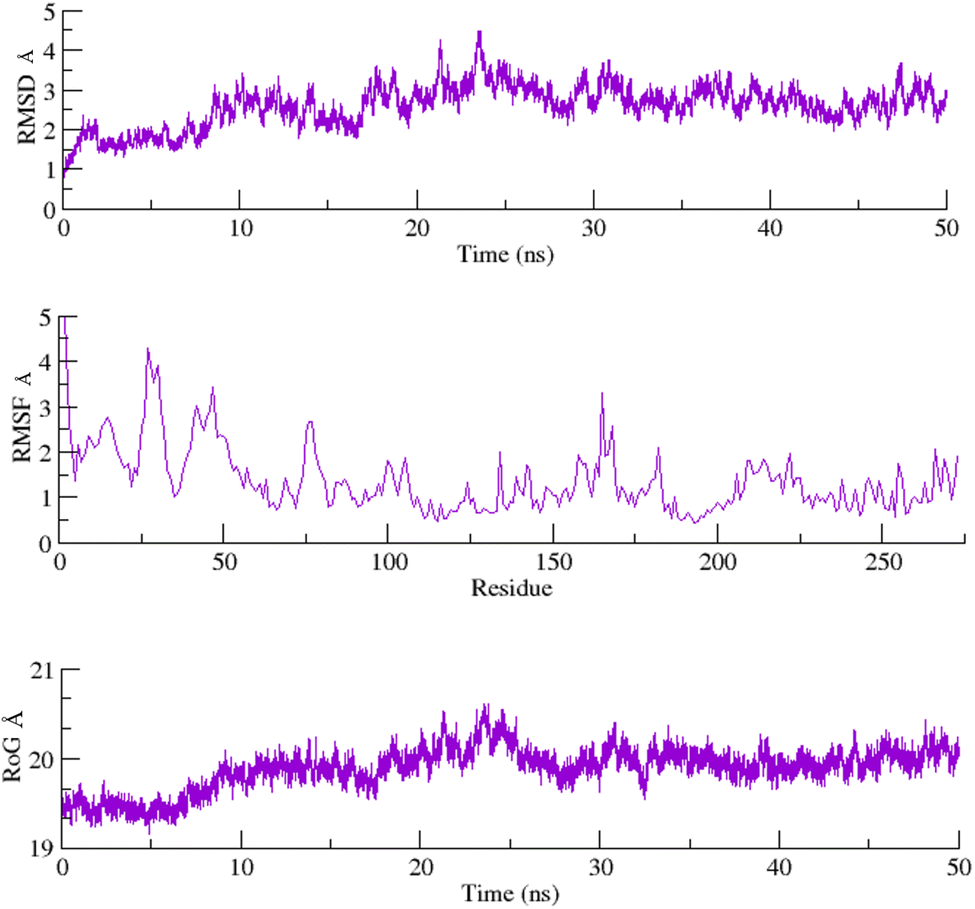 | ||
| Fig. 8 RMSD, RMSF and RoG plots of the EGFR systems calculated as a function of time. | ||
3 Materials and methods
3.1 Synthesis of 1-(5-methyl-1-(2,4,6-trichlorophenyl)-1H-1,2,3-triazol-4-yl)ethan-1-one 1
To a stirring solution of 2,4,6-trichloroaniline (6.05 g, 0.0308 mol) in dil. HCl (40 mL, 3 M). A solution of sodium nitrite (2.34 g, 0.0339 mol, 25 mL, 1 M) is treated at temperature <10 °C. The progress of the reaction is confirmed by liberation of brown gases (1 h). When the brown gases crases a solution of sodium azide (4.006 g, 0.0616 mol) is added to the stirring cold reaction mass. The formation of product is confirmed by TLC (1 h). The reaction mixture was extracted using diethylether. A brown viscous liquid confirmed the product 1.A solution of the appropriate azide (10.0 mmol, 2.22 gm) and acetylacetone (2.0 g, 20.0 mmol) in CHCl3 (20 mL) was treated by adding Et3N (2.02 g, 20.0 mmol) and DBU (0.76 g, 5.00 mmol). The reaction mixture was stirred at room temperature for 1–2 h. The solvent was removed, and water is added then extracted with Et2O, dried with Na2SO4, evaporated and the solid product was washed with the n-hexane to provide pure compound.
1H NMR (500 MHz, DMSO-d6) δ 8.14 (s, 2H), 2.66 (s, 3H), 2.68–2.62 (m, 1H), 2.37 (s, 3H);13C NMR (126 MHz, DMSO-d6) δ 193.55, 142.99, 140.00, 138.09, 134.38, 130.07, 129.66, 28.15, 8.99; m.p: 131 °C; powder.
3.2 General method for the synthesis of the chalcones based triazole 1a–d (GP1)
A mixture of heteroaryle aldehyde (2 mmol) and 1-(5-methyl-1-(2,4,6-trichlorophenyl)-1H-1,2,3-triazol-4-yl)ethan-1-one (0.609 g, 2.0 mmol) dissolved in ethanol (20 mL) was added slowly to an aqueous solution of potassium hydroxide (2.0 mmol, 112 mg) in water (10 mL). The mixture was stirred in crushed-ice bath for 2 h, stirred at 20–25 °C for 4 h. The mixture was filtrated and the residue was washed with cold water and cold alcohol dried to give the titled compound without further purification.Chalcones 1c and 1d was prepared according our prevous published article.39
3.3 General method for the synthesis of spirooxindoles triazole hybrids 4a–k (GP2)
A mixture of the chalcones 1a–d (0.5 mmol), isatin derivatives (0.5 mmol) and L-proline (57.5 mg, 0.5 mmol) in methanol (10 mL) was refluxed on oil bath for appropriate time 5–8 h. After completion of the reaction as evident from TLC, the reaction was kept at room temperature overnight and the solid precipitated upon slow evaporation was filtered off without any further purification.3.4 X-ray structure determination
The crystals of acetyl-triazole derivative, 1a, and 1b were immersed in cryo-oil, mounted in a loop, and measured at a temperature of 120–121 K. The X-ray diffraction data were collected on a Rigaku Oxford Diffraction Supernova diffractometer using Cu Kα radiation (acetyl-triazole derivative and 1b) or Mo Kα radiation (1a). The CrysAlisPro40 software package was used for cell refinements and data reductions. A multi-scan (acetyl-triazole derivative and 1a) or an analytical (1b) absorption correction (CrysAlisPro40) was applied to the intensities before the structure solutions. The structures were solved by the intrinsic phasing (SHELXT41) method. Structural refinements were carried out using SHELXL42 software with SHELXLE43 graphical user interface. All hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–0.99 Å and Uiso = 1.2–1.5 Ueq(parent atom). The crystallographic details are summarized in Table S1.†3.5 Biological investigations
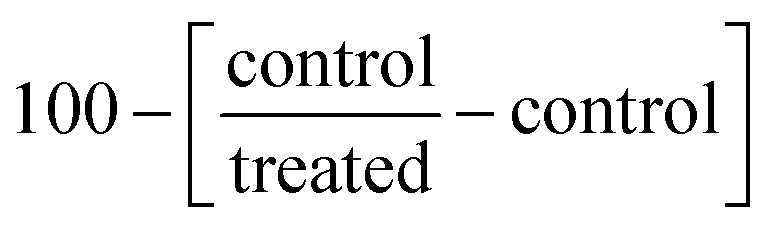 using the curves of percentage inhibition of eight concentrations of each compound, IC50 was calculated using the GraphPad prism7 software.46,47
using the curves of percentage inhibition of eight concentrations of each compound, IC50 was calculated using the GraphPad prism7 software.46,473.6 Flow cytometry using Annexin V/PI staining
HepG2 cells were seeded into 6-well culture plates (3–5 × 105 cells per well) and incubated overnight. Cells were then treated for 48 h with 4a at their IC50 values. Next, media supernatants and cells were collected and rinsed with ice-cold PBS. The cells were then suspended in 100 μL of annexin binding buffer solution “25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4” and incubated with “Annexin V-FITC solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) and propidium iodide (PI)” at a concentration equal to 10 μg mL−1 in the dark for 30 min. Stained cells were then acquired using a Cytoflex flow cytometer (Beckman Coulter C02946) machine. Data were analyzed using Fowjo v10.1 Software (BD life sciences).45,48
:100) and propidium iodide (PI)” at a concentration equal to 10 μg mL−1 in the dark for 30 min. Stained cells were then acquired using a Cytoflex flow cytometer (Beckman Coulter C02946) machine. Data were analyzed using Fowjo v10.1 Software (BD life sciences).45,48
For the purpose of molecular docking, Triangle Matcher method was used as the primary placement method for docking, while LondondG was implemented as the placement scoring method. ‘Induced fit receptor’ algorithm was used for the refinement of the 50 generated poses, out of which, top 10 poses scored via GBVI/WSA dG method were used for further analysis. The binding poses of the top-ranked compounds were then analysed using Protein–Ligand Interaction Profiler (PLIP)54 to visualize, identify, and further analyze protein–ligand interactions. PLIP serves to validate the docking results and allow in-depth study of the protein–ligand interactions, and to visualize the structures and interactions. The 3D interaction figures were made with the help of Chimera.
4 Conclusion
The successful synthesis and characterization of spirooxindole-triazole hybrids through [3 + 2] cycloaddition reactions have resulted in novel compounds with potent inhibitory activity against EGFR and PARP-1. These hybrids represent promising candidates for targeted cancer therapy, combining structural innovation with enhanced biological efficacy. Interestingly, Interestingly, compound 4a showed potent cytotoxicity against HepG2 cells with IC50 values of 3.6 μM compared to doxorubicin (IC50 = 2.78 μM), with poor cytotoxicity against THLE2 cells with IC50 values higher than 40 μM in a selective way. It exhibited potent EGFR and PARP-1 inhibition with IC50 values of 74.6 nM and 2.01 nM, respectively. Furthermore, compound 4a-treatment induced apoptosis by 6.6-fold, arrecting the cell cycle at the G0–G1 phase. Overall, our study underscores the potent activity of compound 4a as dual EGFR/PARP-1 inhibitors against liver cancer. Further in vivo animal model and preclinical studies will focus on optimizing this compound for clinical application, potentially leading to new and effective treatment liver cancer.Data availability
Data supporting this study are openly available from the corresponding author: Prof. Assem Barakat.Author contributions
Assem Barakat, Abdulmajeed Abdullah Alayyaf and Muhanna K. Al-Muhanna: conceptualization and supervision; M. Ali, Moayad Abdullah Alwehaibi, Naif S. Almuqati, and Abdullah A. Alghamdi: investigation, methodology, and formal analysis; Matti Haukka: X-ray diffraction analysis, software; Mohamed S. Nafie: conceptualization of anticancer activity with signaling pathway and biological analysis; Syeda Sumayya Tariq, and Zaheer-Ul Haq: computional study and software; All authors contributed in writing –review & editing in their respective parts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
“The author would like to extend their sincere appreciation to the Researchers Supporting Project (RSP2024R64), King Saud University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Seed Research Project No. (24021440154) granted to Mohamed S. Nafie funded by the Research and Graduate Studies at the University of Sharjah, United Arab Emirates. All biological activity of the tested compounds including cytotoxicity against liver cancer cell line (HepG2), and apoptotic investigations of compound 4a were partially done at “Center of Excellence in molecular medicine”, Suez Canal University.References
- J. L Arias, Drug targeting strategies in cancer treatment: an overview, Mini-Rev. Med. Chem., 2011, 11(1), 1–17 CrossRef PubMed.
- S. H. Kaufmann and W. C. Earnshaw, Induction of apoptosis by cancer chemotherapy, Exp. Cell Res., 2000, 256(1), 42–49 CrossRef PubMed.
- E. K. Rowinsky, Signal events: cell signal transduction and its inhibition in cancer, Oncologist, 2003, 8(S3), 5–17 CrossRef PubMed.
- C. Garnis, T. P. Buys and W. L. Lam, Genetic alteration and gene expression modulation during cancer progression, Mol. Cancer, 2004, 3, 1–23 CrossRef PubMed.
- M. H. Naseri, M. Mahdavi, J. Davoodi, S. H. Tackallou, M. Goudarzvand and S. H. Neishabouri, Up regulation of Bax and down regulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells, Cancer Cell Int., 2015, 15, 1–9 CrossRef PubMed.
- N. J. Curtin, PARP inhibitors for cancer therapy, Expert Rev. Mol. Med., 2005, 7(4), 1–20 CrossRef PubMed.
- M. J. Schiewer and K. E. Knudsen, Transcriptional roles of PARP1 in cancer, Mol. Cancer Res., 2014, 12(8), 1069–1080 CrossRef PubMed.
- M. Rose, J. T. Burgess, K. O'Byrne, D. J. Richard and E. Bolderson, PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance, Front. Cell Dev. Biol., 2020, 8, 564601 CrossRef PubMed.
- L. Wang, W. Cai, W. Zhang, X. Chen, W. Dong, D. Tang, Y. Zhang, C. Ji and M. Zhang, Inhibition of poly (ADP-ribose) polymerase 1 protects against acute myeloid leukemia by suppressing the myeloproliferative leukemia virus oncogene, Oncotarget, 2015, 6(29), 27490 CrossRef PubMed.
- K. Kondo, S. Obitsu, S. Ohta, K. Matsunami, H. Otsuka and R. Teshima, Poly (ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by α-tocopherol and MEK inhibition, J. Biol. Chem., 2010, 285(17), 13079–13091 CrossRef PubMed.
- L. E. Dockery, C. C. Gunderson and K. N. Moore, Rucaparib: the past, present, and future of a newly approved PARP inhibitor for ovarian cancer, OncoTargets Ther., 2017, 3029–3037 CrossRef PubMed.
- S. Li, X. Y. Li, T. J. Zhang, M. O. Kamara, J. W. Liang, J. Zhu and F. H. Meng, Design, synthesis and biological evaluation of homoerythrina alkaloid derivatives bearing a triazole moiety as PARP-1 inhibitors and as potential antitumor drugs, Bioorg. Chem., 2020, 94, 103385 CrossRef CAS PubMed.
- H. F. Ashour, L. A. Abou-Zeid, A. A. Magda and K. B. Selim, 1, 2, 3-Triazole-Chalcone hybrids: Synthesis, in vitro cytotoxic activity and mechanistic investigation of apoptosis induction in multiple myeloma RPMI-8226, Eur. J. Med. Chem., 2020, 189, 112062 CrossRef CAS.
- K. Bozorov, J. Zhao and H. A. Aisa, 1, 2, 3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview, Bioorg. Med. Chem., 2019, 27(16), 3511–3531 CrossRef CAS ..
- D. Dheer, V. Singh and R. Shankar, Medicinal attributes of 1, 2, 3-triazoles: Current developments, Bioorg. Chem., 2017, 71, 30–54 CrossRef CAS ..
- D. Lengerli, K. Ibis, Y. Nural and E. Banoglu, The 1, 2, 3-triazole ‘all-in-one’ring system in drug discovery: A good bioisostere, a good pharmacophore, a good linker, and a versatile synthetic tool, Expert Opin. Drug Delivery, 2022, 17(11), 1209–1236 CrossRef CAS PubMed ..
- I. Shawish, S. Alayoubi, A. El-Faham, A. Aldalbahi, F. Elsenduny, F. Badria, M. Ríos Gutiérrez, H. Hammud, S. Ashraf, Z. Ul-Haq and A. Barakat, Novel Spirooxindole-Triazole Derivatives: Unveiling [3+2] Cycloaddition Reactivity Through Molecular Electron Density Theory and Investigating Their Potential Cytotoxicity Against HepG2 and MDA-MB-231 Cell Lines, Front. Chem., 2024, 12, 1460384, DOI:10.3389/fchem.2024.1460384.
- S. Li, X. Y. Li, T. J. Zhang, J. Zhu, W. H. Xue, X. H. Qian and F. H. Meng, Design, synthesis and biological evaluation of erythrina derivatives bearing a 1, 2, 3-triazole moiety as PARP-1 inhibitors, Bioorg. Chem., 2020, 96, 103575 CrossRef PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Click chemistry: 1, 2, 3-triazoles as pharmacophores, Chem.–Asian J., 2011, 6(10), 2696–2718 CrossRef PubMed.
- N. Normanno, A. De Luca, C. Bianco, L. Strizzi, M. Mancino, M. R. Maiello, A. Carotenuto, G. De Feo, F. Caponigro and D. S. Salomon, Epidermal growth factor receptor (EGFR) signaling in cancer, Gene, 2006, 366(1), 2–16 CrossRef PubMed.
- W. M. Eldehna, D. H. El-Naggar, A. R. Hamed, H. S. Ibrahim, H. A. Ghabbour and H. A. Abdel-Aziz, One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDA-MB-231 cells, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 309–318 CrossRef PubMed.
- R. M. Al-Jassas, M. S. Islam, A. M. Al-Majid, M. S. Nafie, M. Haukka, A. M. Rahman, A. M. A. Alayyaf and A. Barakat, Synthesis and SARs study of novel spiro-oxindoles as potent antiproliferative agents with CDK-2 inhibitory activities, Arch. Pharm., 2023, 356(8), 2300185 CrossRef.
- M. S. Nafie, A. M. Al-Majid, M. Ali, A. A. Alayyaf, M. Haukka, S. Ashraf, Z. Ul-Haq, A. El-Faham and A. Barakat, Exploring pyrrolidinyl-spirooxindole natural products as promising platforms for the synthesis of novel spirooxindoles as EGFR/CDK2 inhibitors for halting breast cancer cells, Front. Chem., 2024, 12, 1364378 CrossRef PubMed.
- P. V. Chavan, U. V. Desai, P. P. Wadgaonkar, S. R. Tapase, K. M. Kodam, A. Choudhari and D. Sarkar, Click chemistry based multicomponent approach in the synthesis of spirochromenocarbazole tethered 1, 2, 3-triazoles as potential anti-cancer agents, Bioorg. Chem., 2019, 85, 475–486 CrossRef PubMed.
- A. Barakat, S. Alshahrani, A. M. Al-Majid, A. S. Alamary, M. Haukka, M. M. Abu-Serie, L. R. Domingo, S. Ashraf, Z. Ul-Haq, M. S. Nafie and M. Teleb, New spiro-indeno [1, 2-b] quinoxalines clubbed with benzimidazole scaffold as CDK2 inhibitors for halting non-small cell lung cancer; stereoselective synthesis, molecular dynamics and structural insights, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2281260 CrossRef PubMed.
- S. Alshahrani, A. M. Al-Majid, A. S. Alamary, M. Ali, M. S. Altowyan, M. Ríos-Gutiérrez, S. Yousuf and A. Barakat, Synthesis and Characterization of New Spirooxindoles Including Triazole and Benzimidazole Pharmacophores via [3+2] Cycloaddition Reaction: An MEDT Study of the Mechanism and Selectivity, Molecules, 2023, 28(19), 6976 CrossRef PubMed.
- A. Barakat, S. Alshahrani, A. M. Al-Majid, A. S. Alamary, M. Haukka, M. M. Abu-Serie, A. Dömling, L. R. Domingo and Y. A. Elshaier, Activation of p53 signaling and regression of breast and prostate carcinoma cells by spirooxindole-benzimidazole small molecules, Front. Pharmacol., 2024, 15, 1358089 CrossRef PubMed.
- M. Ali, M. A. Alwehaibi, A. M. Al-Majid, M. K. Al-Muhanna, S. M. Soliman, M. Ríos-Gutiérrez, M. Haukka, A. A. Alayyaf, A. Barakat, CCDC 2303110: Experimental Crystal Structure Determination, CSD Communication, 2024, DOI:10.5517/ccdc.csd.cc2h9kw8.
- M. Ali, M. A. Alwehaibi, A. M. Al-Majid, M. K. Al-Muhanna, S. M. Soliman, M. Ríos-Gutiérrez, M. Haukka, A. A. Alayyaf, A. Barakat, CCDC 2303111: Experimental Crystal Structure Determination, CSD Communication, 2024, DOI:10.5517/ccdc.csd.cc2h9kx9.
- C. Schettino, M. A. Bareschino, V. Ricci and F. Ciardiello, Erlotinib: an EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment, Expert Rev. Respir. Med., 2008, 2(2), 167–178 CrossRef CAS PubMed.
- C. C. Gunderson and K. N. Moore, Olaparib: an oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer, Future Oncol., 2015, 11(5), 747–757 CrossRef CAS PubMed.
- K. R. Senwar, P. Sharma, T. S. Reddy, M. K. Jeengar, V. L. Nayak, V. G. M. Naidu, A. Kamal and N. Shankaraiah, Spirooxindole-derived morpholine-fused-1, 2, 3-triazoles: Design, synthesis, cytotoxicity and apoptosis inducing studies, Eur. J. Med. Chem., 2015, 102, 413–424 CrossRef CAS.
- M. Rajeswari, S. Kumari and J. M. Khurana, One-pot four component domino strategy for the synthesis of novel spirooxindole pyrrolizine linked 1, 2, 3-triazoles via stereo-and regioselective [3+ 2] cycloaddition reaction in acidic medium, RSC Adv., 2016, 6(11), 9297–9303 RSC.
- R. Bianco, T. Gelardi, V. Damiano, F. Ciardiello and G. Tortora, Rational bases for the development of EGFR inhibitors for cancer treatment, Int. J. Biochem. Cell Biol., 2007, 39(7–8), 1416–1431 CrossRef CAS.
- T. I. Adelusi, A. Q. K. Oyedele, I. D. Boyenle, A. T. Ogunlana, R. O. Adeyemi, C. D. Ukachi, M. O. Idris, O. T. Olaoba, I. O. Adedotun, O. E. Kolawole and Y. Xiaoxing, Molecular modeling in drug discovery, Inform. Med. Unlocked, 2022, 29, 100880 CrossRef.
- M. A. Ashiru, S. O. Ogunyemi, O. R. Temionu, A. C. Ajibare, N. C. Cicero-Mfon, O. A. Ihekuna, M. O. Jagun, L. Abdulmumin, Q. K. Adisa, Y. E. Asibor and C. J. Okorie, Identification of EGFR inhibitors as potential agents for cancer therapy: pharmacophore-based modeling, molecular docking, and molecular dynamics investigations, Mol. Model., 2023, 29(5), 128 CrossRef CAS.
- K. H. Liao, K. B. Chen, W. Y. Lee, M. F. Sun, C. C. Lee and C. Y. C. Chen, Ligand-Based and Structure-Based Investigation for Alzheimer's Disease from Traditional Chinese Medicine, J. Evidence-Based Complementary Altern. Med., 2014, 2014(1), 364819 CrossRef.
- T. E. Ogden, J. C. Yang, M. Schimpl, L. E. Easton, E. Underwood, P. B. Rawlins, M. M. McCauley, M. F. Langelier, J. M. Pascal, K. J. Embrey and D. Neuhaus, Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition, Nucleic Acids Res., 2021, 49(4), 2266–2288 CrossRef CAS.
- A. A. Alayyaf, M. Ali, M. A. Alwehaibi, M. K. Al-Muhanna, S. M. Soliman, M. Ríos-Gutiérrez, M. Haukka and A. Barakat, Utilizing MEDT analysis of [3+ 2] cycloaddition reaction: x-ray crystallography of spirooxindole linked with thiophene/furan heterocycles and triazole framework, BMC Chem., 2024, 18(1), 1–16 CrossRef.
- Rikagu Oxford Diffraction, CrysAlisPro 1.171.43.100a, Rikagu Oxford Diffraction inc., Yarnton, Oxfordshire, England, 2023 Search PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Cryst., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Cryst., 2015, C71, 3–8 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef PubMed.
- A. T. Boraei, E. H. Eltamany, I. A. Ali, S. M. Gebriel and M. S. Nafie, Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: In vitro, in vivo, and in silico integrated approaches, Bioorg. Chem., 2021, 111, 104877 CrossRef PubMed.
- M. Hisham, B. G. Youssif, E. E. A. Osman, A. M. Hayallah and M. Abdel-Aziz, Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents, Eur. J. Med. Chem., 2019, 176, 117–128 CrossRef PubMed.
- I. Shawish, M. S. Nafie, A. Barakat, A. Aldalbahi, H. H. Al-Rasheed, M. Ali, W. Alshaer, M. Al Zoubi, S. Al Ayoubi, B. G. De la Torre and F. Albericio, Pyrazolyl-s-triazine with indole motif as a novel of epidermal growth factor receptor/cyclin-dependent kinase 2 dual inhibitors, Front. Chem., 2022, 10, 1078163 CrossRef PubMed.
- M. S. Nafie, S. M. Kishk, S. Mahgoub and A. M. Amer, Quinoline-based thiazolidinone derivatives as potent cytotoxic and apoptosis-inducing agents through EGFR inhibition, Chem. Biol. Drug Des., 2022, 99(4), 547–560 CrossRef PubMed.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Triaryl dicationic DNA minor-groove binders with antioxidant activity display cytotoxicity and induce apoptosis in breast cancer, Chem.-Biol. Interact., 2020, 324, 109087 CrossRef PubMed.
- A. I. Khodair, M. A. Alsafi and M. S. Nafie, Synthesis, molecular modeling and anti-cancer evaluation of a series of quinazoline derivatives, Carbohydr. Res., 2019, 486, 107832 CrossRef PubMed.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor, J. Biol. Chem., 2002, 277(48), 46265–46272 CrossRef PubMed.
- S. Vilar, G. Cozza and S. Moro, Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery, Curr. Top. Med. Chem., 2008, 8(18), 1555–1572 CrossRef CAS PubMed.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, PLIP: fully automated protein–ligand interaction profiler, Nucleic Acids Res., 2015, 43(W1), W443–W447 CrossRef CAS.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell Jr, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14(1), 71–73 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- R. Fletcher and M. J. Powell, A rapidly convergent descent method for minimization, Comput. J., 1963, 6(2), 163–168 CrossRef.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N log (N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- D. R. Roe and T. E. Cheatham III, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics Modell., 1996, 14(1), 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2360616–2360618. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05966b |
| This journal is © The Royal Society of Chemistry 2025 |