
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomerization of pirazolopyrimidines to pyrazolopyridines by ring-opening/closing reaction in aqueous NaOH†
Carlos Cifuentes,
Nestor Bravo,
Daniel Restrepo,
Mario Macías and
Jaime Portilla*
and
Jaime Portilla*
Department of Chemistry, Universidad de Los Andes, Carrera 1 No. 18A-10, Bogotá 111711, Colombia. E-mail: jportill@uniandes.edu.co
First published on 22nd January 2025
Abstract
An isomerization reaction of 7-aryl-3-formylpyrazolo[1,5-a]pyrimidines to 5-aroyl-NH-pyrazolo[3,4-b]pyridines proceeding with high yields in aqueous NaOH under microwave conditions is reported. This unprecedented transformation occurs by adding and eliminating a water molecule via an ANRORC mechanism (adding the nucleophile, ring-opening, and ring-closing) studied using DFT calculations. The product's utility was proved as they have aroyl and NH groups that simple methods and readily available reagents easily modified; likewise, their optical properties were studied, highlighting their high potential as highly emissive modular dyes (φF up to 99%). NMR, HRMS, and X-ray diffraction analysis resolved the products' structures.
Introduction
Exploring efficient methods to yield aza-heterocyclic compounds (N–HCs) is essential in organic and medicinal chemistry; this focus is inspired by the high potential to incorporate diverse functionalities into these compounds to expand their scope and pertinency.1–3 Along these lines, heteroaromatic rings with three or more nitrogen atoms in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 fused rings have shown particular interest as they are structural analogs of purines found in various biological and photophysically relevant compounds (Fig. 1a).4–7 Especially pyrazolo[3,4-b]pyridines (PPys), such as the anxiolytic drug tracazolate (I), the 5-benzoyl derivative (II, inhibitor of CDK2 protein), and the antihypertensive agent riociguat (III) are N–HCs that have been explored for their therapeutic potential.8–11 Likewise, some PPys have been highlighted for their fluorescent properties (typical property of 5:6 fused ring12,13) in chemosensors discovery (e.g., probe IV),14–16 (Fig. 1b).
:6 fused rings have shown particular interest as they are structural analogs of purines found in various biological and photophysically relevant compounds (Fig. 1a).4–7 Especially pyrazolo[3,4-b]pyridines (PPys), such as the anxiolytic drug tracazolate (I), the 5-benzoyl derivative (II, inhibitor of CDK2 protein), and the antihypertensive agent riociguat (III) are N–HCs that have been explored for their therapeutic potential.8–11 Likewise, some PPys have been highlighted for their fluorescent properties (typical property of 5:6 fused ring12,13) in chemosensors discovery (e.g., probe IV),14–16 (Fig. 1b).
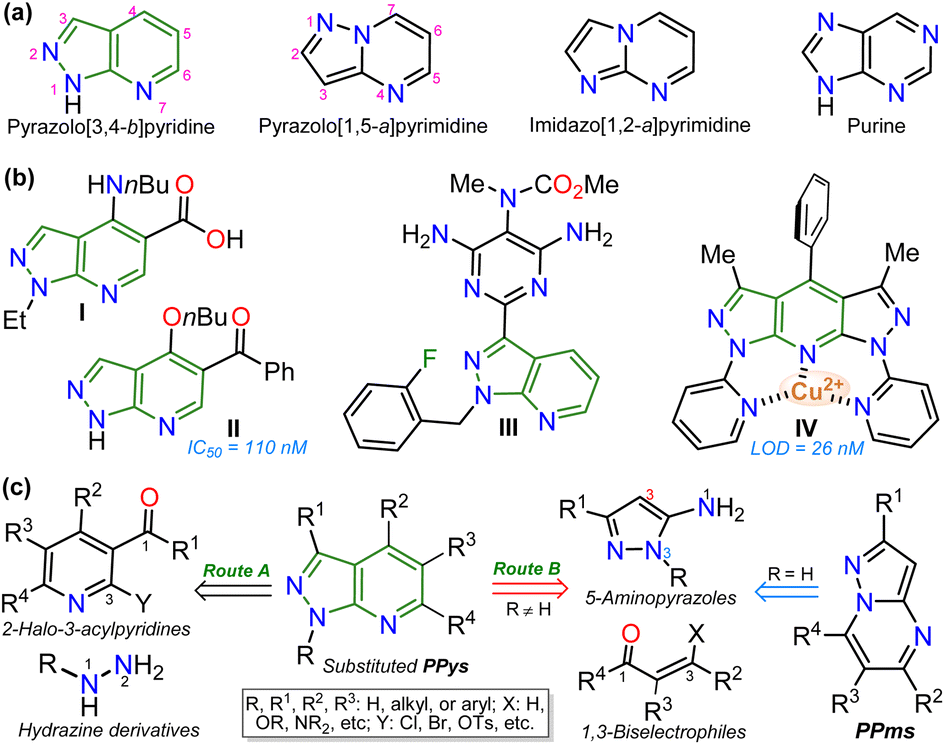 | ||
| Fig. 1 (a) Structure of aza-heteroaromatic 5:6 fused rings with heteroaromatic character. (b) Examples of biological and physically relevant pyrazolo[3,4-b]pyridines. (c) Synthesis general of pyrazolo[3,4-b]pyridines. | ||
The pyrazolo[3,4-b]pyridine (PPy) ring formation is achieved through cyclization reactions of 1,3-biselectrophilic pyridines (e.g., 3-acyl-2-halopyridines, route A) with hydrazine derivatives and, to a greater extent, from N-substituted aminopyrazoles with 1,3-biselectrophilic reagents (e.g., β-alkoxyenones, route B).6,15–19 However, by using NH-5-aminopyrazoles in route B, the reaction mainly yields pyrazolo[1,5-a]pyrimidines,5,20 and route A is limited by the poor availability of substrates6,21 (Fig. 1c). As a result, access to N-unsubstituted PPys is limited since most existing examples are route A-based,6,20,21 and rarer are PPys substituted with an aroyl group at position 5.8,22–24 Most reports on obtaining 5-aroyl-NH-pyrazolo[3,4-b]pyridines are found in pharmaceutical patents,25 and due to this, finding one article with one derivative was only possible.20 In that work, the authors react 3-formylchromones V with 5-amino-3-methylpyrazole (VI) to access the 5-aroyl group by a reaction proceeding with the opening of the pyranic ring in V; however, this method has limitations such as the poor availability of substrate, the recurrence of a phenolic moiety in products, and the competence with pyrazolo[1,5-a]pyrimidines (Scheme 1a).20 Importantly, 5-benzoyl-NH-pyrazolo[3,4-b]pyridines (similar to II in Fig. 1) were obtained in poor global yields (up to ∼5%) using synthetic strategies implying more than seven steps with a deprotection protocol for the NH-pyrazolic group.11
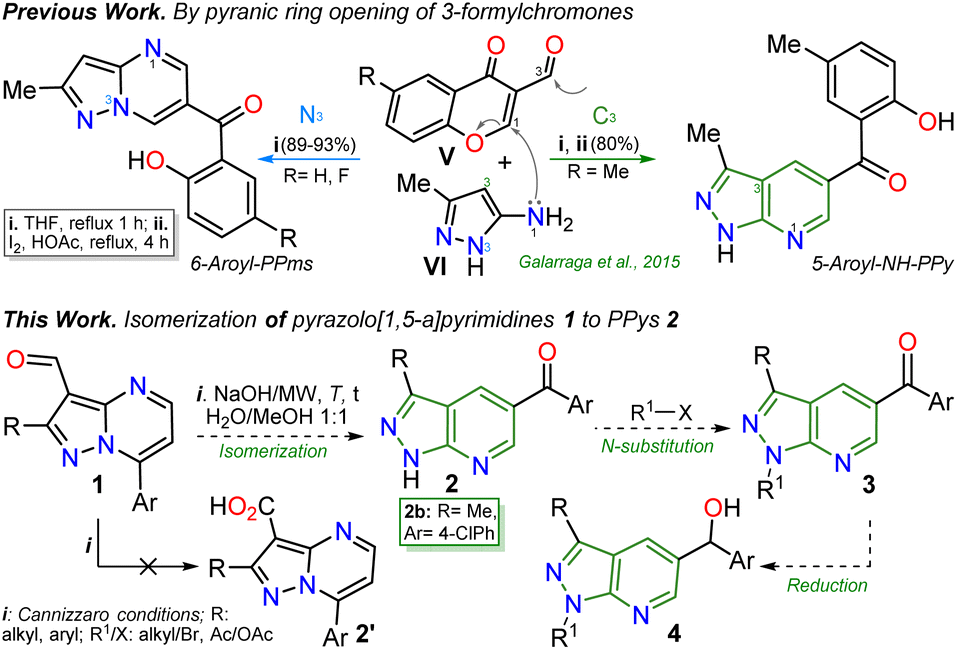 | ||
| Scheme 1 Synthesis of 5-aroyl-NH-pyrazolopyridines studied in previous work (at the top) and in this work (at the bottom). | ||
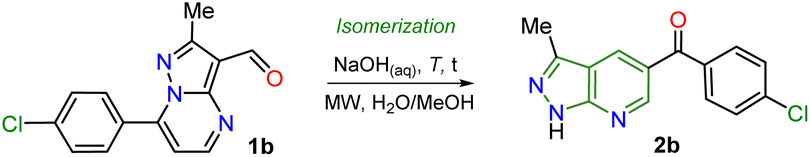
Inspired by the synthetic applications of pyrazolo[1,5-a]pyrimidines under the microwave conditions (MWC) and our investigations in this area,5,26–28 the Cannizzaro reaction26–28 on the 3-formyl derivative 1b was utilized to access the respective carboxylic acid 2′. This acid type could be used in further studies as an N,O-donor ligand in recognition or coordination chemistry, an unusual line in these fused pyrazoles.5 However, an unexpected but fascinating result was found in the used reaction conditions (excess NaOH) as the isomerization product 2b was obtained instead of the desired carboxylic acid 2′. Therefore, we aimed to thoroughly study this isomerization reaction to get a novel family of pyrazolo[3,4-b]pyridines employing an optimized synthetic approach; we also proposed to examine the practical applicability of this protocol using functionalization strategies and the appropriate photophysical study of some representative derivatives (Scheme 2b). These investigations were proposed as isomerization products would be functionalized with easily modifiable aroyl and NH groups, and 5:6 fused rings have evidenced relevant fluorescent properties.12–16
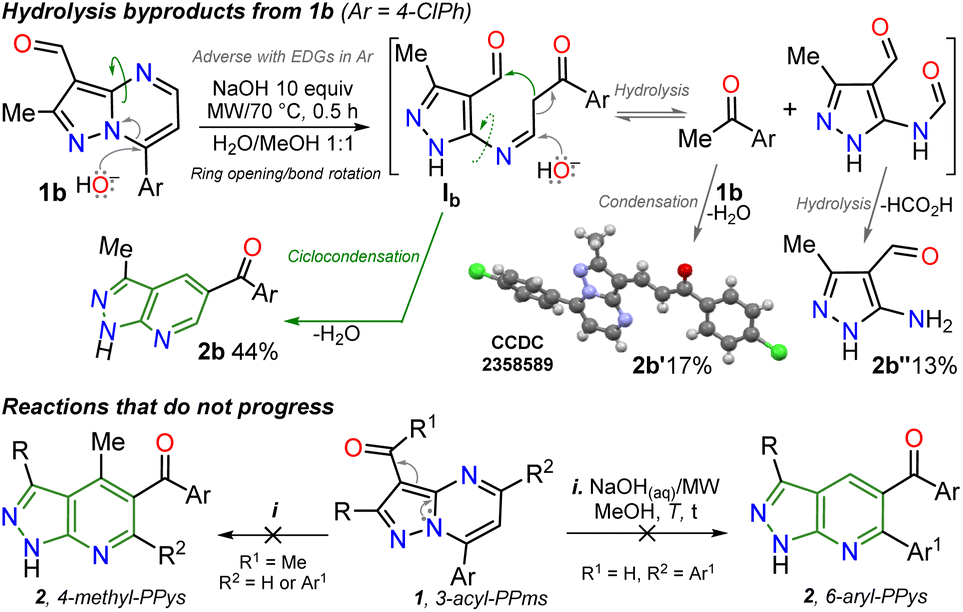 | ||
| Scheme 2 Highlighted results for the studied isomerization reaction of 1b. | ||
Results and discussion
Synthesis
It is important to note that this is the first report on this isomerization reaction class, and due to the strange nature of the transformation, it wasn't easy to establish the structure of 2b; we achieved it by X-ray diffraction (XRD) analysis (Scheme 2, see ESI† for more details). From these exciting findings, we wonder whether the isomerization could be optimized using the 3-formylpyraolo[1,5-a]pyrimidine 1b (R/Ar = Me/4-ClPh, Scheme 1b) as a model substrate and a high excess NaOH (10 equiv.) at 70 °C for 30 minutes as initial conditions.The reaction does not progress when developed in water as 1b is insoluble; thus, the reaction was carried out in water/methanol to improve the substrate solubility under microwave irradiation, obtaining 2b in 44% yield (Table 1, entry 1 versus 2). However, the excess NaOH used also favours the formation of enone 2b′ and the pyrazole derivative 2b′′. This result could be evidence of the ring-opening intermediate Ib that also delivered 2b, also suffers total hydrolysis – e.g., Ab is converted to 4-chloroacetophenone that reacts with a second molecule of 1b – (Scheme 2a). It should be noted that the reaction (i.e., NaOH with substrate 2b) only works well in H2O/MeOH due to its better solubility behavior; indeed, the reaction worsens in H2O/EtOH and does not advance in H2O/MeCN.
|
|||||
|---|---|---|---|---|---|
| Entry | NaOH (equiv.) | H2O/MeOH | t (min) | T (°C) | 2b, yield (%) |
| a Reaction conditions: 52 mg of 1b (0.19 mmol) and NaOH (7.6–76 mg) in 0.7 mL of solvent under microwave conditions; reactions run in a 10 mL sealed tube.b Reaction under conventional heating at reflux with a heating mantle in 2 mL of solvent. | |||||
| 1 | 10 | 1:0 |
30 | 70 | NR |
| 2 | 10 | 1:1 |
30 | 70 | 44 |
| 3 | 10 | 1:1 |
30 | 100 | 68 |
| 4 | 10 | 1:1 |
30 | 120 | 75 |
| 5 | 5 | 1:1 |
30 | 100 | 85 |
| 6 | 5 | 1:1 |
10 | 100 | 87 |
| 7 | 2 | 1:1 |
5 | 100 | 90 |
| 8 | 1 | 1:1 |
5 | 100 | 52 |
| 9 | 1.5 | 1:1 |
5 | 100 | 63 |
| 10 | 2 | 5:1 |
5 | 100 | Traces |
| 11 | 2 | 3:1 |
5 | 100 | 44 |
| 12 | 2 | 2:1 |
5 | 100 | 91 |
| 13b | 2 | 2:1 |
150 | Reflux | 61 |
We continued the isomerization reaction study by increasing the reaction temperature, and good yields were obtained despite the slight presence of 2b′ and 2b′′. Thus, the amount of base and the reaction time were decreased (entries 3–4 versus 5–9), finding excellent results with 2 equivalents of NaOH at 100 °C for 5 minutes as 2b was obtained in 90% yield (Table 1, entry 7). To achieve a greener protocol, we tried to reduce the MeOH amount, noticing that 1b was wholly dissolved in a mixture with up to a 2:1 ratio of H2O/MeOH (entries 10–12). Finally, obtaining the pyrazolo[1,5-a]pyrimidine 2b under conventional heating at reflux was also possible but in moderate yield (Table 1, entry 13).
With the optimal conditions established (Table 1, entry 12), we next explored the scope of the isomerization reaction using substrates 1a–l. Remarkably, excellent tolerance for substituent at positions 2 (Me, tBu, and aryl) and 7 (aryl and hetaryl), with different stereoelectronic natures, was observed. However, substrates bearing electron-donating groups (EDGs) at position C7 (i.e., 1d, 1g, 1i, and 1l) increase the electron density at this carbon atom5 (please see 13C NMR data of 1a–l (ref. 26–28) and LUMO orbital of 1a in ESI†) to an adverse initial nucleophilic attack decreasing reaction yields (Scheme 2a). For example, the conversion from 1l to 2l was only possible under reflux conditions for 48 hours, obtaining the lowest yield (50%) as the substrate's triphenylamine group is highly electron-donating. Curiously, although there was complete conversion, the reaction also offered moderate yields (62–65%) with highly electron-withdrawing groups (EWGs) at position C7 of substrate (as the pyridine ring in 1e and 1f); this result is due to the extraction process since 2e and 2f are slightly more soluble in aqueous medium than the other PPys (Scheme 3). Unfortunately, highly insoluble substrates (i.e., 7-Ar = 4-O2NPh, 4-NCPh, or 4-tolyl) or containing another aryl group at position 5 (i.e., 5-Ar = Ph or 4-ClPh) or an acetyl group instead of formyl are resistant to any change under the optimized reaction conditions; this result is probably as they are very polar or more stable molecules by a better π-conjugation5,28 (Scheme 2b).
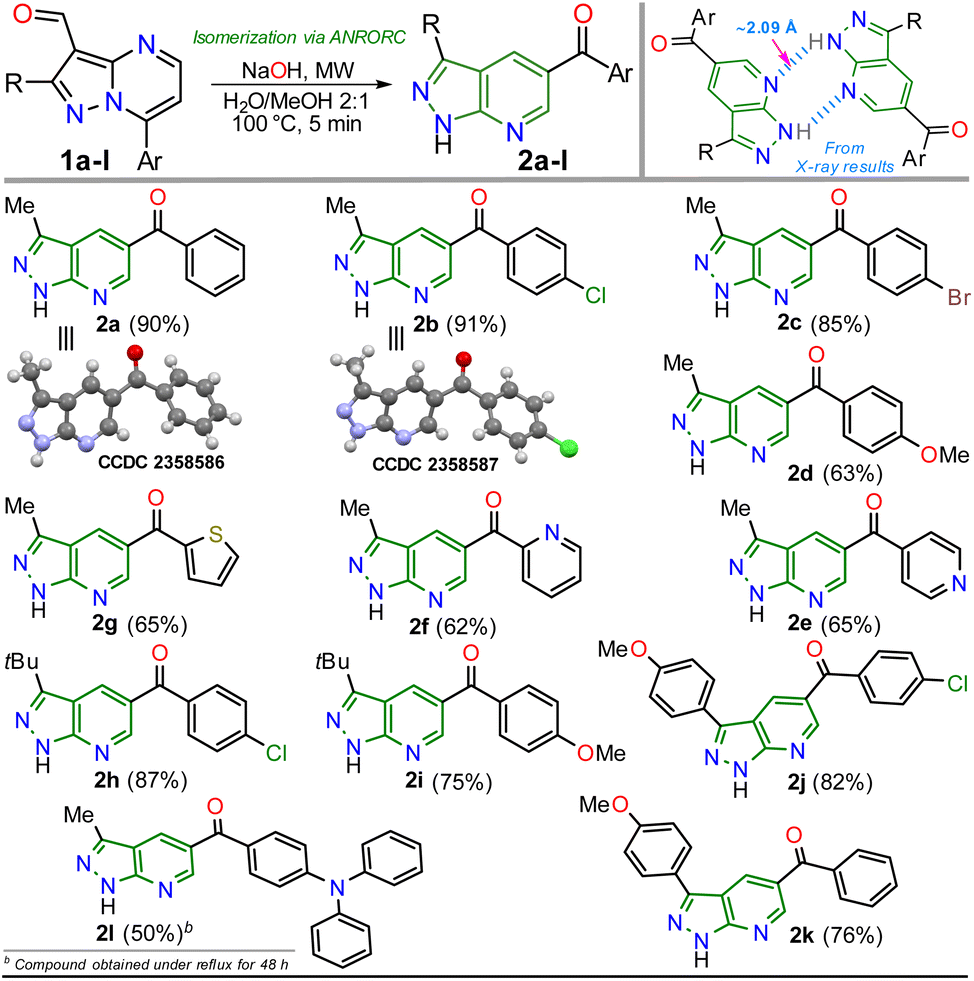 | ||
| Scheme 3 Scope of the isomerization reaction using substrates 1a–l. | ||
Once the 5-aroyl-NH-pyrazolo[3,4-b]pyridines 2a–l were obtained, we developed functionalization reactions to access new derivatives with this relevant heterocyclic core, obtaining strategic compounds with a high potential in medicinal and organic chemistry.20–24 In this way, the N-alkylation reaction on the pyrazole moiety was conducted using alkyl halides (2 equiv.) and caesium carbonate (2 equiv.) in MeCN (2 mL) at 80 °C for 15 minutes under MWC, obtaining 3a–f in high yields (85–90%); the amide ion was previously generated at 50 °C for 3 minutes also under microwave conditions (Scheme 4).
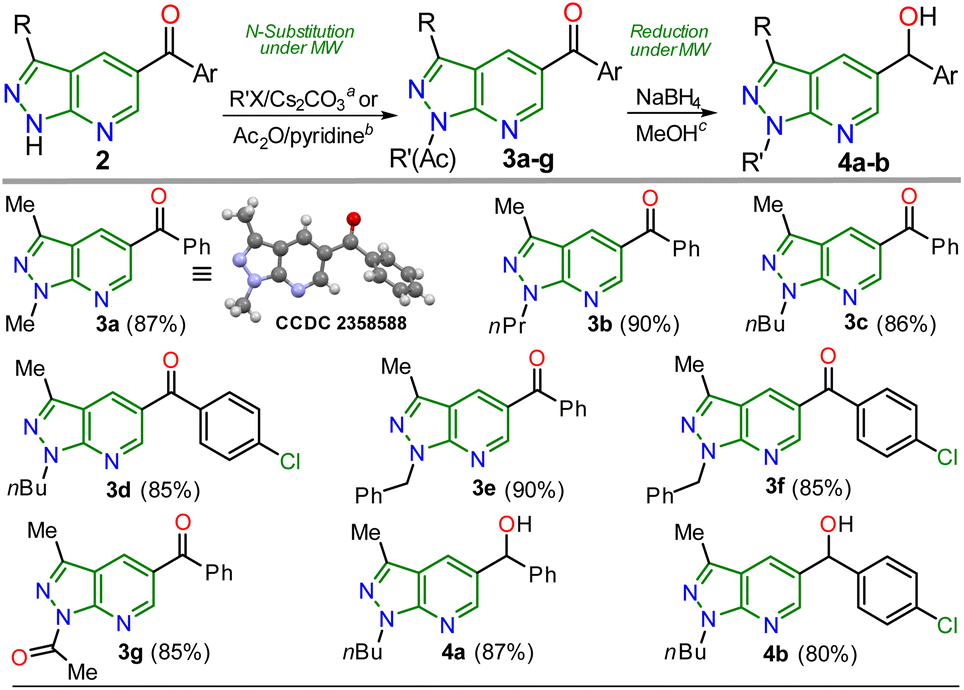 | ||
| Scheme 4 Synthesis of N-substituted 5-aroylpyrazolo[3,4-b]pyridines 3a–g and reduction products (alcohols) 4a–b. | ||
As expected, the acetylation reaction of 2a (0.25 mmol) with acetic anhydride (2 equiv.) in pyridine (2 mL) at 120 °C for 30 minutes in MWC efficiently afforded the N-acetyl derivate 3g in 85% yield. Ultimately, the reduction reaction of the carbonyl group in 3c–d was successfully developed using NaBH4 (2 equiv.) in methanol at 100 °C for 5 minutes under MWC, forming alcohols 4a–b in high yields (80–87%, Scheme 4). Pleasantly, all the obtained pyrazolo[3,4-b]pyridines are structurally novel and functional, so they are potentially helpful for new synthetic procedures, allowing their application in various fields of chemistry; for example, compounds 2a–l could be used as N,N-donor ligands to access coordination complexes, as observed in the hydrogen bonding interactions found by XRD studies (Scheme 3 and Fig. S48†). Likewise, structures of enone 2b′ (Scheme 2), 2a–b (Scheme 3), and N-methyl derivative 3a (Scheme 4) were solved using single-crystal XRD analysis.29 All the obtained compounds (2b′, 2b′′, 2a–l, 3a–g, and 4a–b) were characterized through NMR and high-resolution mass spectra (HRMS) analysis (see ESI† for details).
Photophysical studies
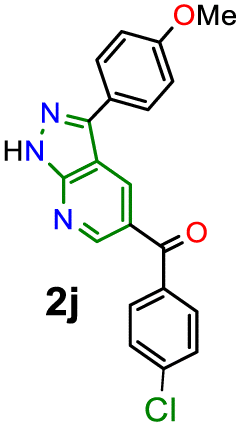
During synthetic experiments, we noticed that some products exhibit luminescent properties in solution and solid state under exposure to UV light, mainly the two derivatives with 4-anisyl group (4-MeOPh) at position 3; thus, the photophysical study of 2j–k was conducted to establish the scope of their donor–π–acceptor molecular architecture (D–π–A or MeO–π–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in organic fluorophores development. Due to their synthetic potential, these results would allow us to document PPys 2j–k as strategic intermediates of new functional fluorophores, considering some fluorescent dyes bearing pyrazolo[1,5-a]pyridine ring.15,16,30 In addition to the initially observed luminescence, probes 2j–k were photophysically studied since the 4-anisyl group, bonded to diverse chromophores, has proven helpful in this area, mainly by intramolecular charge transfer (ICT) phenomena using 5:6 fused aza-heterocyclic rings.31–34 In this manner, the absorption and emission spectra of fluorophores 2j–k were recorded in solvents of different polarity (solvent, Δf): toluene (MePh, 0.013), chloroform (CHCl3, 0.149), tetrahydrofuran (THF, 0.210), dimethylsulfoxide (DMSO, 0.265), N,N-dimethylformamide (DMF, 0.275), acetonitrile (ACN, 0.306), and methanol (MeOH, 0.309) (Fig. 2 and Table 2).
O) in organic fluorophores development. Due to their synthetic potential, these results would allow us to document PPys 2j–k as strategic intermediates of new functional fluorophores, considering some fluorescent dyes bearing pyrazolo[1,5-a]pyridine ring.15,16,30 In addition to the initially observed luminescence, probes 2j–k were photophysically studied since the 4-anisyl group, bonded to diverse chromophores, has proven helpful in this area, mainly by intramolecular charge transfer (ICT) phenomena using 5:6 fused aza-heterocyclic rings.31–34 In this manner, the absorption and emission spectra of fluorophores 2j–k were recorded in solvents of different polarity (solvent, Δf): toluene (MePh, 0.013), chloroform (CHCl3, 0.149), tetrahydrofuran (THF, 0.210), dimethylsulfoxide (DMSO, 0.265), N,N-dimethylformamide (DMF, 0.275), acetonitrile (ACN, 0.306), and methanol (MeOH, 0.309) (Fig. 2 and Table 2).
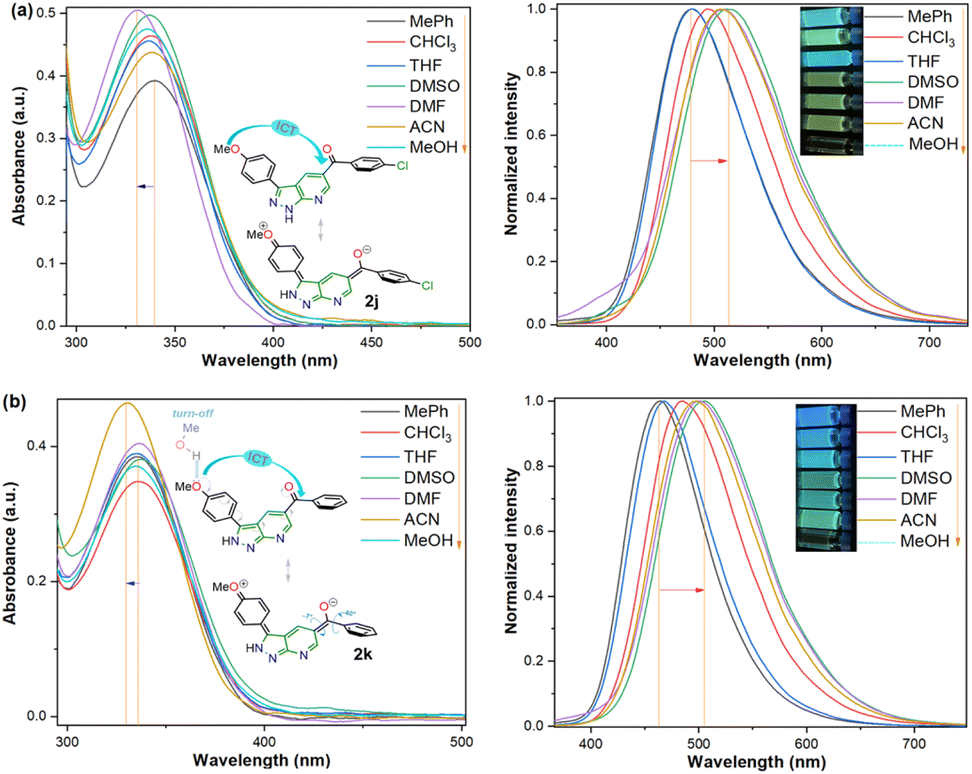 | ||
| Fig. 2 Absorption (left) and emission (right, λex = 335 nm) spectra and photographs of probes (a) 2j and (b) 2k in different solvents (50 μM) at 20 °C. | ||
| Probe | Solventb | λabs (nm) | ε (M−1 cm−1) | λem (nm) | SS (cm−1) | ϕF | B (ε × ϕF) |
|---|---|---|---|---|---|---|---|
| a Quantum yield (ϕF) values were determined using Prodan as a standard. Absorbance (ab), fluorescence emission (em), molar absorption coefficient (ε), Stokes shift (SS), and calculated bright (B) data are also shown.b Variation of orientation polarizability (Δf): 0.013, 0.149, 0.210, 0.265, 0.275, 0.306, and 0.309, respectively. | |||||||
 |
MePh | 339 | 7840 | 478 | 8578 | 0.99 | 7762 |
| CHCl3 | 337 | 9260 | 493 | 9389 | 0.78 | 7223 | |
| THF | 336 | 9100 | 479 | 8885 | 0.78 | 7098 | |
| DMSO | 338 | 8740 | 512 | 10054 |
0.53 | 4632 | |
| DMF | 337 | 9940 | 508 | 9988 | 0.31 | 3082 | |
| ACN | 331 | 10100 |
507 | 10487 |
0.30 | 3030 | |
| MeOH | 335 | 9500 | 519 | 10582 |
0.0069 | 66 | |
 |
MePh | 335 | 7700 | 464 | 8299 | 0.93 | 7161 |
| CHCl3 | 335 | 6940 | 484 | 9189 | 0.88 | 6107 | |
| THF | 334 | 7760 | 468 | 8572 | 0.84 | 6519 | |
| DMSO | 337 | 7560 | 503 | 9792 | 0.96 | 7258 | |
| DMF | 336 | 8080 | 499 | 9721 | 0.77 | 6222 | |
| ACN | 330 | 9280 | 498 | 10222 |
0.74 | 6867 | |
| MeOH | 334 | 7400 | 524 | 10856 |
0.01 | 74 | |
Compounds 2j–k displayed absorption spectra with the typical band of S0 → ICT transitions (i.e., MeO → CO) at around 335 nm with extinction coefficient (ε) values of 7840–10100 M−1 cm−1 for 2j (4-ClBz) and 6940–9280 M−1 cm−1 for 2k (Bz). These results revealed a slight blue-shifted and best absorptivity by increasing solvent polarity, with 2j as the best chromophore due to its higher number of electrons (Fig. 2, left). Both probes exhibited high fluorescence quantum yield (ϕF) and brightness (B = ε × ϕF) values, mainly in less polar solvents (e.g., ϕF = 99% and B = 7762 for 2j in MePh), with a sharp decrease in the emission intensity of 2j in DMF or ACN (Fig. 2, right). This is owing to the stability of non-emitting excited states, meaning that the energy received by solute is dissipated in non-radiative ways rather than released as light. Polar solvents can help with nonradiative relaxing by creating an environment where vibrational energy can be more efficiently transmitted to the solvent; this reduces the quantity of energy emitted as light, decreasing the fluorescence quantum yield.35–40 Moreover, both probes displayed significant Stokes shifts (8578–10582 cm−1 for 2j and 8299–10856 cm−1 for 2k) with a marked red-shifted by increasing solvent polarity (Fig. 2 and S49,† Table 2).
Importantly, the photophysical properties 2j–k were dramatically affected in methanol due to strong hydrogen bonding interactions of their anisyl group with solvent molecules (ϕF ≤ 1%). This solvent acts as a fluorescence quencher in 2j–k, blocking the ICT process to afford energy transfer without light emission; however, this property could be relevant in determining water or ethanol in some organic solvents or distilled spirits (Fig. 2).36,37 In addition, the Lippert–Mataga plots for 2j–k illustrate their typical behaviour in the excited states with the variation of orientation polarizability (Δf) of the solvent; as expected, they evidenced different interactions with the evaluated solvents, leading to two unique straight-line correlations with positive slopes (Fig. 3). This can be explained as a general solvation effect in low polarity solvents; in contrast, specific interactions like dipole–dipole in polar solvents stabilise the system; it would not be dependable to compute the change in the dipole moment using these numbers due to the low R2 value, suggesting no strong association polarity factor/Stokes shift. However, from the positive slopes in the graphs, it is possible to see the defined trends according to the slope direction for each probe, demonstrating positive solvatofluorochromism.35–40
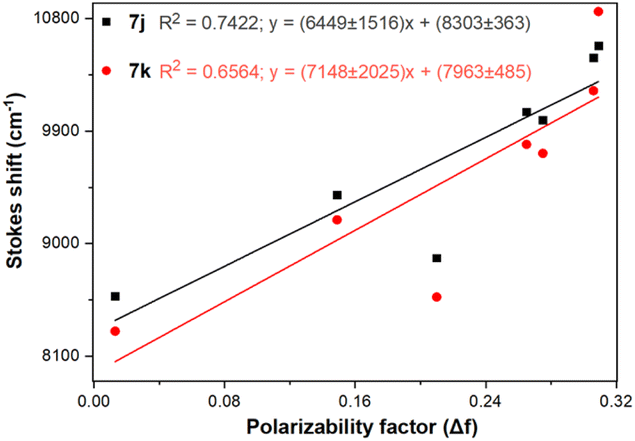 | ||
| Fig. 3 Lippert–Mataga plots (Δν vs. Δf) for probes 7j–k in different solvents. | ||
Accordingly, it is proved that the ICT phenomenon in fluorescent probes 2j–k favours a good charge separation within these molecules through resonance from the anisyl group towards the carbonyl group (i.e., MeO → CO). It should be noted that the benzene ring in the aroyl group at position 5 of the aza-heterocyclic core does not greatly affect the ICT process as this ring is not coplanar with the D–π–A system of fluorophores 2j–k (Fig. 2); this structural characteristic could be determined through the dihedral angles found from crystallographic X-ray diffraction analyses for similar structures (i.e., 2a and 2b, Fig. S48 in ESI†). Finally, from the significant photophysical properties found for probes 2j–k and the good synthetic results involving compounds 2a–l, 3a–g, and 4a–b, we can establish that the heterocyclic core of pyrazolo[3,4-b]pyridine is a promisor functional fluorophore; specifically for developing applications in coordination chemistry, diagnostic bioimaging in living systems, chemosensors discovery, photosensitizers, or organic materials science.13,31–40
DFT calculations
The primary reaction was studied using density functional theory (DFT) B3LYP/6-311+(d,p) calculations to understand better the mechanism of the discovered reaction with the associated relative free energies (ΔG) by substrate 1a.41,42 The relative free energy calculated for the overall reaction is −11.79 kcal mol−1, indicating greater product 2a stability than substrate 1a due to the high aromatic character of the pyridine ring in 2a.5,6 It is important to note that a negative charge is induced within structures during the reaction due to the medium nature and the initial attack of the hydroxyl anion; however, the isomerization involves the addition and elimination of a water molecule through an ANRORC (adding the nucleophile, ring-opening, and ring-closing) mechanism. Implicit water was initially modelled, and explicit modelling of a single water molecule proved crucial in stabilizing transitional states by clustering negative charges within acceptor hydrogen groups along structures. Incorporating a hydroxyl group at position 7 on substrate 1a exhibits lower energy than the initial structure, implying greater stabilization for intermediate I1, allowing the pyrimidine ring-opening to find metastable state cause of possible intermediates I2 and I3. This study supports that rotation in the imine group of I2 is favourable, leading to the pyridine ring-closing to afford I4 by reacting the stabilized carbanion with the C-carbonyl in I3 (Scheme 5).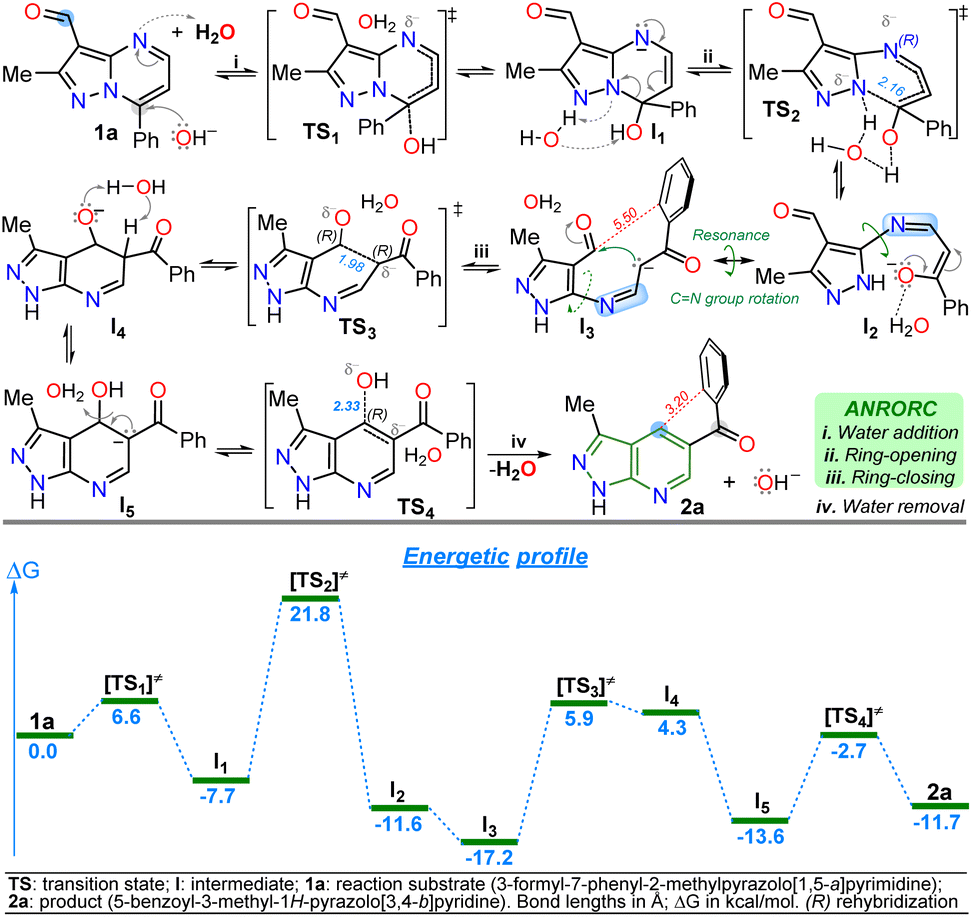 | ||
| Scheme 5 Proposed mechanism (at the top) for the isomerization reaction of 1a to 2a with its respective energetic profile (at the bottom). | ||
The explored mechanism reveals four crucial transition states involving the bonding of the hydroxyl group TS1, its detachment TS4, and the isomerization process via ring-opening TS2 and ring-zar13 closing TS3. The free energies in TS2/TS3 are higher than in TS1/TS4, indicating that the most critical reaction steps involve opening the pyrimidine ring and closing the pyridine ring under MWC. The energy levels of structures TS3 and I4 are closely matched. Deprotonation of the acidic hydrogen at position 5 of the dihydro-pyridine ring in I4 reduces the relative free energy, resulting in the discovery of a metastable state I5 that finally facilitates the formation of a hydroxyl group (base regeneration) to yield the heteroaromatic product 2a (Scheme 5). Notably, the anionic intermediate I3 has the lowest energy in the energetic profile, even lower than 2a, due to the implicit water solvation model used in the presence of the hydroxyl anion; indeed, 2a has an electronic repulsion between the two rings of the molecule at 3.20 Å in different planes. In contrast, I3 has larger distances (5.50 Å) between the ring of the aroyl group and the nearest carbon atom that would be part of the fused ring, reducing intramolecular electronic repulsions (Scheme 5 and Fig. S51†).
Conclusions
In summary, a NaOH-mediated isomerization reaction of 7-aryl-3-formylpyrazolo[1,5-a]pyrimidines to 5-aroylpyrazolo[3,4-b]pyridines was successfully developed under microwave conditions at 100 °C. Eleven isomerization products were obtained in high yields (up to 91%) employing mild reaction conditions in an aqueous medium and cheap reagents. Notably, a mechanistic route for this transformation was also proposed based on DFT calculations and the experimental results; this implies an ANRORC route that started with the hydroxy anion addition at the C7 of the aza-heterocyclic core of the substrate. Additionally, the obtained ketoamines have strategic functional groups (aroyl and NH-heteroaryl) that were easily modified by simple approaches and readily available reagents. The structure of all the obtained compounds was established based on NMR, HRMS, and X-ray diffraction studies. Likewise, the two more luminescent obtained ketoamines were photophysically studied, indicating the utility of the 4-anisyl group and 5:6 fused rings in the optoelectronic properties of fluorophores acting through ICT phenomena. Fluorescence quantum yields of up to 99% were obtained with dyes having positive (redshift) solvatofluorochromism, highlighting the high potential of these two compounds as functional fluorophores; for example, as methanol acted as a solvent fluorescence quenching blocking the ICT process in the two tested ketoamines, new applications in sensing water or ethanol in different mediums could be possible.
Data availability
The experimental data, NMR and HRSM spectra, crystallographic and photophysical details (CCDC: 2a 2358586, 2b 2358587, 2b′ 2358589, and 3a 2358588), and computational details have been included in the ESI.Author contributions
The individuals listed as authors have contributed to developing this manuscript, and no other person was involved. The authors' contributions included: C. C. and N. B. carried out experiments and literature review; D. R. and M. M. carried out computational and XRD studies, respectively; and J. P. the composition of the original draft, supervision, and sources. All authors have read and agreed to the published version of this manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the Chemistry Department and Vicerrectoría de Investigaciones at the Universidad de Los Andes for financial support (project INV-2023-162-2810). N.B. acknowledges to Ministerio de Ciencia Tecnología e Innovación (MINCIENCIAS) for a PhD fellowship (Con. 1 to FCTel of SGR). We also recognize S. Ortiz and the High-Performance Computing (HPC) services of Universidad de Los Andes to acquire the mass spectra and access the computational facilities.Notes and references
- N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, More sustainable approaches for the synthesis of n-based heterocycles, Chem. Rev., 2009, 109, 2703–2802 CrossRef CAS.
- S. Kuwata and F. E. Hahn, Complexes Bearing Protic N-Heterocyclic Carbene Ligands, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS.
- Y. Wang, W.-X. Zhang and Z. Xi, Carbodiimide-based synthesis of N-heterocycles: moving from two classical reactive sites to chemical bond breaking/forming reaction, Chem. Soc. Rev., 2020, 49, 5810–5849 RSC.
- R. Z. U. Kobak and B. Akkurt, Formation and Uses of Imidazo[1,2-a]pyrimidines and Related Compounds: A Review Comprising Years 2000-2021, J. Turk. Chem. Soc., Sect. A, 2022, 9, 1335–1386 CrossRef CAS.
- J. Portilla, in Adv. Heterocycl. Chem., Elsevier Inc., 2024, 1st edn, pp. 71–138 Search PubMed.
- A. Donaire-Arias, A. M. Montagut, R. P. de la Bellacasa, R. Estrada-Tejedor, J. Teixidó and J. I. Borrell, 1H-Pyrazolo[3,4-b]pyridines: Biomedical Applications Synthesis and Biomedical Applications, Molecules, 2022, 27, 2237 CrossRef CAS PubMed.
- S. M. Hoy, Pimitespib: First Approval, Drugs, 2022, 82, 1413–1418 CrossRef CAS.
- S. B. Bharate, T. R. Mahajan, Y. R. Gole, M. Nambiar, T. T. Matan, A. Kulkarni-Almeida, S. Balachandran, H. Junjappa, A. Balakrishnan and R. A. Vishwakarma, Synthesis and evaluation of pyrazolo[3,4-b]pyridines and its structural analogues as TNF-α and IL-6 inhibitors, Bioorg. Med. Chem., 2008, 16, 7167–7176 CrossRef CAS PubMed.
- J. Mittendorf, S. Weigand, C. Alonso-Alija, E. Bischoff, A. Feurer, M. Gerisch, A. Kern, A. Knorr, D. Lang, K. Muenter, M. Radtke, H. Schirok, K. H. Schlemmer, E. Stahl, A. Straub, F. Wunder and J. P. Stasch, Discovery of riociguat (BAY 63-2521): A potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension, ChemMedChem, 2009, 4, 853–865 CrossRef CAS PubMed.
- M. C. Ríos and J. Portilla, Recent Advances in Synthesis and Properties of Pyrazoles, Chemistry, 2022, 4, 940–968 CrossRef.
- R. N. Misra, D. B. Rawlins, H. Y. Xiao, W. Shan, I. Bursuker, K. A. Kellar, J. G. Mulheron, J. S. Sack, J. S. Tokarski, S. D. Kimball and K. R. Webster, 1H-Pyrazolo[3,4-b]pyridine inhibitors of cyclin-dependent kinases, Bioorg. Med. Chem. Lett., 2003, 13, 1133–1136 CrossRef CAS PubMed.
- M. L. S. O. Lima, C. B. Braga, T. B. Becher, M. Odriozola-Gimeno, M. Torrent-Sucarrat, I. Rivilla, F. P. Cossío, A. J. Marsaioli and C. Ornelas, Fluorescent Imidazo[1,2-a]pyrimidine Compounds as Biocompatible Organic Photosensitizers that Generate Singlet Oxygen: A Potential Tool for Phototheranostics, Chem.–Eur. J., 2021, 27, 6213–6222 CrossRef CAS.
- J. T. Sarmiento and J. Portilla, Current advances in chemosensors diazoles-based for CN– and F– detection, Curr. Org. Synth., 2023, 20, 61–79 CrossRef.
- P. Moskwa, A. Kolbus, T. Uchacz, A. Danel and K. Gałczy, Fluorescent Sensor Based on 1H-Pyrazolo[3,4-b]quinoline Derivative for Detecting Zn2+ Cations, Molecules, 2024, 29, 823 CrossRef.
- J. Orrego-Hernández, C. Lizarazo, J. Cobo and J. Portilla, Pyrazolo-fused 4-azafluorenones as key reagents for the synthesis of fluorescent dicyanovinylidene-substituted derivatives, RSC Adv., 2019, 9, 27318–27323 RSC.
- M. García, I. Romero and J. Portilla, Synthesis of Fluorescent 1,7-Dipyridyl-bis-pyrazolo[3,4-b:4′,3′-e]pyridines: Design of Reversible Chemosensors for Nanomolar Detection of Cu2+, ACS Omega, 2019, 4, 6757–6768 CrossRef PubMed.
- N. Medishetti, A. Kale, J. B. Nanubolu and K. Atmakur, Iron(III)chloride induced synthesis of pyrazolopyridines & quinolines, Synth. Commun., 2020, 50, 3642–3651 CrossRef CAS.
- X. Y. Miao, Y. J. Hu, F. R. Liu, Y. Y. Sun, D. Sun, A. X. Wu and Y. P. Zhu, Synthesis of Diversified Pyrazolo[3,4-b]pyridine Frameworks from 5-Aminopyrazoles and Alkynyl Aldehydes via Switchable C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C Bond Activation Approaches, Molecules, 2022, 27, 6381 CrossRef CAS PubMed.
C Bond Activation Approaches, Molecules, 2022, 27, 6381 CrossRef CAS PubMed. - R. F. Barghash, W. M. Eldehna, M. Kovalová, V. Vojáčková, V. Kryštof and H. A. Abdel-Aziz, One-pot three-component synthesis of novel pyrazolo[3,4-b]pyridines as potent antileukemic agents, Eur. J. Med. Chem., 2022, 227, 113952 CrossRef CAS PubMed.
- E. Galarraga, N. Urdaneta, K. J. Gutierrez and J. C. Herrera, In vitro cytotoxicity of hydrazones, pyrazoles, pyrazolo-pyrimidines, and pyrazolo-pyridine synthesized from 6-substituted 3-formylchromones, Z. Naturforsch., B: Chem. Sci., 2015, 70, 305–310 CrossRef CAS.
- G. L. Beutner, J. T. Kuethe, M. M. Kim and N. Yasuda, Expedient synthesis of 3-alkoxymethyl- And 3-aminomethyl-pyrazolo[3,4-b]pyridines, J. Org. Chem., 2009, 74, 789–794 CrossRef CAS PubMed.
- T. Denzel and H. Höhn, Synthese von 1H-Pyrazolo[3,4-b]pyridin-5-ketonen, Arch. Pharm., 1975, 1106, 486–503 Search PubMed.
- M. Lácová, A. Puchala, E. Solčanyova, J. Lac, P. Koiš, J. Chovancová and D. Rasala, 3-formylchromones IV. The rearrangement of 3-formylchromone enamines as a simple, facile route to novel pyrazolo[3,4-b]pyridines and the synthetic utility of the latter, Molecules, 2005, 10, 809–821 CrossRef PubMed.
- S. V. Ryabukhin, A. S. Plaskon, D. M. Volochnyuk and A. A. Tolmachev, Chlorotrimethylsilane-mediated synthesis of functionalized fused pyridines: Reaction of 3-formylchromones with electron-rich aminoheterocycles, Synthesis, 2007, 1861–1871 CAS.
- N. R. Misra, S. D. Kimball, D. B. Rawlins, K. R. Webster and I. Bursuker, Use of pyrazolo[3,4-b]pyridine as cyclin dependant kinase inhibitors, WO1999030710, 2007.
- J. C. Castillo, A. Tigreros and J. Portilla, 3-Formylpyrazolo[1,5-a]pyrimidines as Key Intermediates for the Preparation of Functional Fluorophores, J. Org. Chem., 2018, 83, 10887–10897 CrossRef CAS PubMed.
- J. C. Castillo, H. A. Rosero and J. Portilla, Simple access toward 3-halo- and 3-nitro-pyrazolo[1,5-a]pyrimidines through a one-pot sequence, RSC Adv., 2017, 7, 28483–28488 RSC.
- S. Aranzazu, A. Tigreros, A. Arias-G, J. Zapata-Rivera and J. Portilla, BF3-Mediated Acetylation of Pyrazolo[1,5-a]pyrimidines and Other π-Excedent (N-Hetero)arenes, J. Org. Chem., 2022, 87, 9839–9850 CrossRef CAS PubMed.
- CCDC 2358586 (2a), 2358587 (2b), 2358589 (2b′), and 2358588 (3a) contain the supplementary crystallographic data for this paper.
- U. Krishnan, S. Manickam and S. K. Iyer, BF3 detection by pyrazolo-pyridine based fluorescent probe and applications in bioimaging and paper strip analysis, J. Mol. Liq., 2023, 385, 122413 CrossRef CAS.
- M. L. S. O. Lima, C. B. Braga, T. B. Becher, M. Odriozola-Gimeno, M. Torrent-Sucarrat, I. Rivilla, F. P. Cossío, A. J. Marsaioli and C. Ornelas, Fluorescent Imidazo[1,2-a]pyrimidine Compounds as Biocompatible Organic Photosensitizers that Generate Singlet Oxygen: A Potential Tool for Phototheranostics, Chem.–Eur. J., 2021, 27, 6213–6222 CrossRef CAS PubMed.
- S. Velázquez-Olvera, H. Salgado-Zamora, M. Velázquez-Ponce, E. Campos-Aldrete, A. Reyes-Arellano and C. Pérez-González, Fluorescent property of 3-hydroxymethyl imidazo[1,2-a]pyridine and pyrimidine derivatives, Chem. Cent. J., 2012, 6, 1–9 CrossRef.
- V. V. Fedotov, M. I. Valieva, O. S. Taniya, S. V. Aminov, M. A. Kharitonov, A. S. Novikov, D. S. Kopchuk, P. A. Slepukhin, G. V. Zyryanov, E. N. Ulomsky, V. L. Rusinov and V. N. Charushin, 4-(Aryl)-Benzo[4,5]imidazo[1,2-a]pyrimidine-3-Carbonitrile-Based Fluorophores: Povarov Reaction-Based Synthesis, Photophysical Studies, and DFT Calculations, Molecules, 2022, 27, 8029 CrossRef CAS.
- A. Tigreros, J. C. Castillo and J. Portilla, Cyanide chemosensors based on 3-dicyanovinylpyrazolo[1,5-a]pyrimidines: Effects of peripheral 4-anisyl group substitution on the photophysical properties, Talanta, 2020, 215, 120905 CrossRef CAS.
- M. Homocianu, Exploring solvatochromism: A comprehensive analysis of research data, Microchem. J., 2024, 198, 110166 CrossRef CAS.
- A. Tigreros, M. Macías and J. Portilla, Structural, Photophysical, and Water Sensing Properties of Pyrazolo[1,5-a]pyrimidine-Triphenylamine Hybrid Systems, ChemPhotoChem, 2022, 6, e202200133 CrossRef CAS.
- A. Tigreros, M. Macías and J. Portilla, Expeditious ethanol quantification present in hydrocarbons and distilled spirits: Extending photophysical usages of the pyrazolo[1,5-a]pyrimidines, Dyes Pigm., 2022, 202, 110299 CrossRef CAS.
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2001, vol. 1 Search PubMed.
- O. A. Kucherak, P. Didier, M. Yves and A. S. Klymchenko, Fluorene Analogues of Prodan with Superior Fluorescence Brightness and Solvatochromism, J. Phys. Chem. Lett., 2010, 1, 616–620 CrossRef CAS.
- E. Benedetti, L. S. Kocsis and K. M. Brummond, Synthesis and photophysical properties of a series of cyclopenta[b]naphthalene solvatochromic fluorophores, J. Am. Chem. Soc., 2012, 134, 12418–12421 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, NMR and HRMS charts, crystallographic and theoretical details, and photophysical studies. CCDC: 2a 2358586, 2b 2358587, 2b′ 2358589, and 3a 2358588. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06345g |
| This journal is © The Royal Society of Chemistry 2025 |