
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticomponent reactions (MCRs) yielding medicinally relevant rings: a recent update and chemical space analysis of the scaffolds†
Mukesh Tandi
a,
Vaibhav Sharma
a,
Balasubramanian Gopal
 b and
Sandeep Sundriyal
*a
b and
Sandeep Sundriyal
*a
aDepartment of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Rajasthan 333031, India. E-mail: sandeep.sundriyal@pilani.bits-pilani.ac.in
bMolecular Biophysics Unit, Indian Institute of Science, Bangalore, 560012, India
First published on 16th January 2025
Abstract
In this review we have compiled multicomponent reactions (MCRs) that produce cyclic structures. We have covered articles reported since 2019 to showcase the recent advances in this area. In contrast to other available reviews on this topic, we focus specifically on MCRs with strong prospects in medicinal chemistry. Consequently, the reactions operating in a single-pot and yielding novel rings or new substitution patterns under mild conditions are highlighted. Moreover, MCRs that do not require special reagents or catalysts and yield diverse products from commercially available building blocks are reviewed. The synthetic schemes, substrate scope, and other key aspects such as regio- and stereoselectivity are discussed for each MCR. Using cheminformatic tools, we have also attempted to characterize the chemical space of the scaffolds obtained from these MCRs. We show that the MCR scaffolds are novel, more complex, and globular in shape compared to the approved drugs and clinical candidates. Thus, our review represents a step towards identifying and characterizing the novel ring space that can be accessed efficiently through MCRs in a short timeframe.
 Mukesh Tandi | Mukesh Tandi received his MS (Pharm.) in Medicinal Chemistry from the National Institute of Pharmaceutical Education and Research (NIPER), Raebareli, Uttar Pradesh. He is pursuing his doctoral research under the supervision of Prof. Sandeep Sundriyal. His focus includes exploiting MCRs for designing small-molecule inhibitors of key enzymes in pathogens. |
 Vaibhav Sharma | Vaibhav Sharma holds a Master's degree in Pharmaceutical Chemistry from BITS Pilani. His research in Sundriyal's lab focused on the design and development of novel polymers using MCRs. Vaibhav recently completed an internship at Biophore India Pharmaceuticals in their Intellectual Property Management department, where he gained valuable experience as an IP analyst focused on Active Pharmaceutical Ingredients (APIs), further strengthening his understanding of drug discovery and intellectual property. |
 Balasubramanian Gopal | B. Gopal is a Professor at the Division of Biological Sciences, Indian Institute of Science, Bengaluru. He is structural biologist by training. His current research interests include gene expression analysis and synthetic microbiology to produce active pharmaceutical ingredients. |
 Sandeep Sundriyal | Sandeep Sundriyal is an Associate Professor at the Department of Pharmacy, BITS Pilani. After obtaining PhD in Medicinal Chemistry in 2008 he worked as postdoctoral fellow at various international institutes including Imperial College London. His current research interests include antimicrobial and anticancer drug design using computational and synthetic chemistry tools. |
1 Introduction and background
Multicomponent reactions (MCRs) utilize three or more building blocks that react sequentially to afford the final product with multiple C–C and C–X bond formations (Fig. 1). MCRs have garnered considerable significance in recent times, as evidenced by the increasing number of publications on this topic (Fig. 2). MCRs find significant applications in multiple research areas including medicinal chemistry,1–5 diversity-oriented synthesis,6 heterocyclic chemistry,7 natural product chemistry,8 and material chemistry.9 | ||
| Fig. 1 MCR approach in synthetic chemistry. | ||
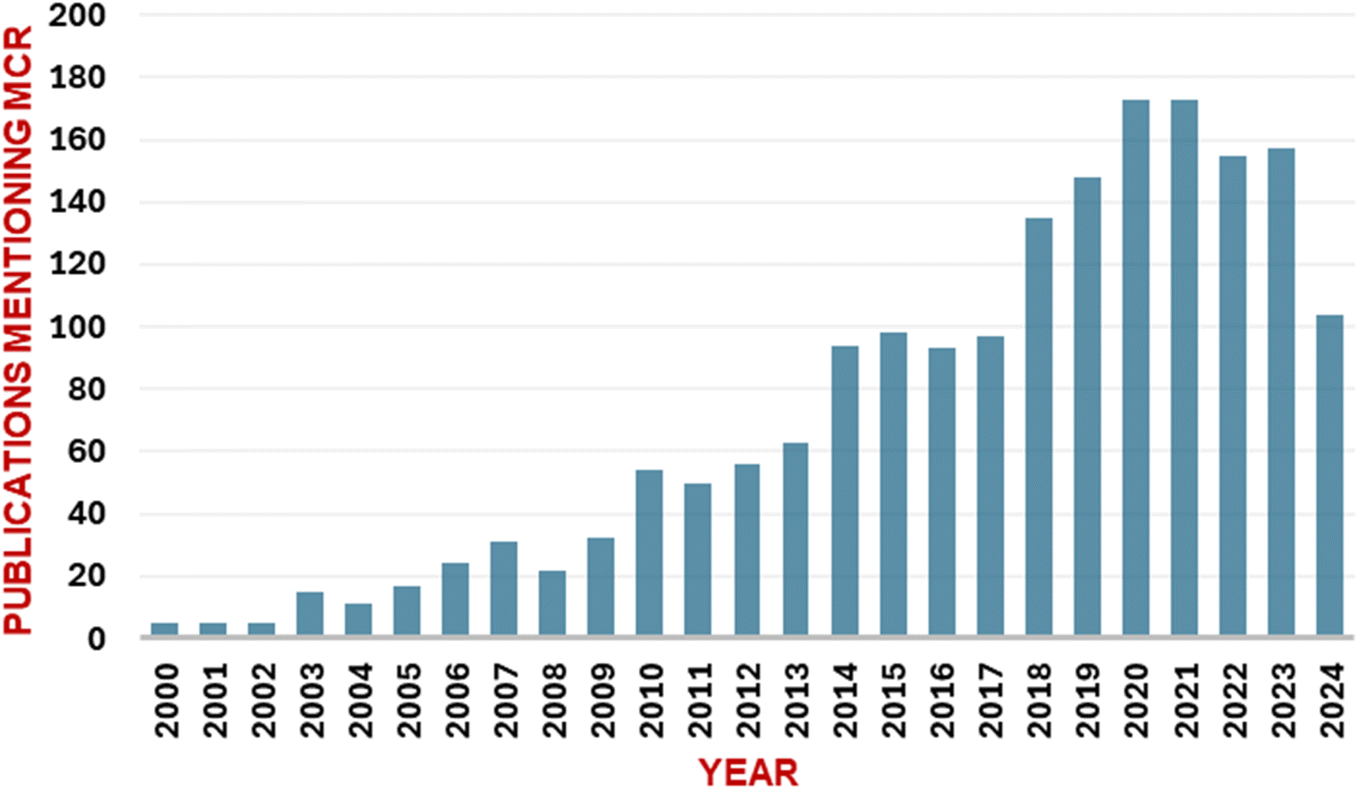 | ||
| Fig. 2 A pubMed search reveals the rising number of publications mentioning ‘MCR’ in the ‘Title’ or ‘Abstract’ in recent years (2000–2024). The terms used for the search are “multiple-component reaction”, “multicomponent reaction”, or “multicomponent reaction.” | ||
MCRs offer several advantages over conventional multistep synthetic strategies. These include (i) the possibility of convergent synthesis as multiple starting materials are used in the single step,10 (ii) high atom-economy and pot-economy,11 (iii) construction of complex molecules from simpler starting materials,3 (iv) access to a diverse compound library by varying building blocks,6,12–14 (v) possibility of post-MCR modifications to construct novel ring systems and substitution patterns,7,15,16 (vi) convenient and mild reaction conditions leading to greener approach.17–20 Due to these advantages, MCRs are particularly suitable in the drug discovery scenario where access to large compound libraries can often be a bottleneck.1,2,21 MCRs can be used to generate multiple analogues of a hit molecule in a short time frame, thus lowering the costs of early medicinal chemistry efforts. The design of MCR-based synthetic processes for approved drugs may also lead to overall lower production costs and has the potential to offer cost-effective medicines.22–25
In drug discovery, the importance of ring systems cannot be overstated.26–30 However, out of the vast theoretical possibilities,28 a few rings and substitution patterns reappear in most drugs and clinical candidates.30 MCR-derived new ring systems with diverse three-dimensional (3-D) topologies offer novel chemical space for drug design.2,15,16,31
Given the significance of MCRs, several classical MCRs, such as Hantzsch, Strecker, Ugi, Passerini, Gewald, Biginelli, and Mannich (Fig. 3), are named after their discoverers. Most of these classical reactions have been exploited in drug discovery and other fields, and multiple reviews are available on these old MCRs.2,4,5,9,21,32 Therefore, to avoid overlap, we focused on reviewing relatively newer MCRs (2019 onwards). Given the enormity of the literature, we have used the following criteria for compiling information in this review:
 | ||
| Fig. 3 Examples of classical MCRs. | ||
(i) Only novel MCRs with the potential to have a significant impact on medicinal chemistry are included. Consequently, minor modifications or variations of the previously known MCRs are not considered.
(ii) MCRs leading to the construction of medicinally relevant rings or previously inaccessible substitution patterns that may benefit drug discovery projects are incorporated.
(iii) In general, a conscious effort was made to include MCRs operating under mild conditions from readily available building blocks and common reagents.
(iv) Only those MCRs were selected in which the product is obtained in a single pot, thus avoiding the isolation of intermediates and high potential for scale-up.
Thus, the current review is not meant to be comprehensive but representative of the literature on MCRs. The text is divided into sections based on the type of heterocycle being formed in the MCR. General synthetic schemes and substrate scope are discussed briefly, followed by the cheminformatic analysis of the scaffolds obtained from the representative molecules.
2 Nitrogen heterocycles
Nitrogen-containing heterocycles frequently occur in structural motifs in several drug molecules and natural drugs.27,33–37 Among diverse N-heterocycles, tetrahydropyridines (THPs) and piperidines are especially important38 since these increase the three-dimensionality of the molecules and help them escape the ‘flatland’.39 Usually, THPs and piperidines are obtained from the reduction of corresponding pyridine rings and intramolecular cyclization, which often generates waste.40–42 Thus, green and convenient methods for synthesizing these aza-heterocyclic rings are highly desirable.Wei et al. reported a mild method to synthesize poly-substituted 1,2,5,6-THPs using a copper-catalyzed MCR.43 The reaction involves a cascade radical cyclization reaction between three components, an F-masked benzene-sulfonamide allene (1), an alkene (2), and trimethylsilyl cyanide (TMSCN) (3), yielding THPs with rare C3 or C5 substituents (e.g. 4a, Fig. 4). The screening of reaction conditions revealed tetrakis(acetonitrile)copper(I) hexafluorophosphate [Cu(CH3CN)4PF6] in conjunction with a bisoxazoline (BOX) ligand and fluorobenzene as solvent to be optimal (Fig. 4). Thus, the model compound 4a was obtained in 65% yield under these optimized conditions. Although the BOX ligand provided good regioselectivity, installing the cyanide group at the primary carbon with the least steric hindrance, stereoselectivity was not observed despite using a chiral ligand.
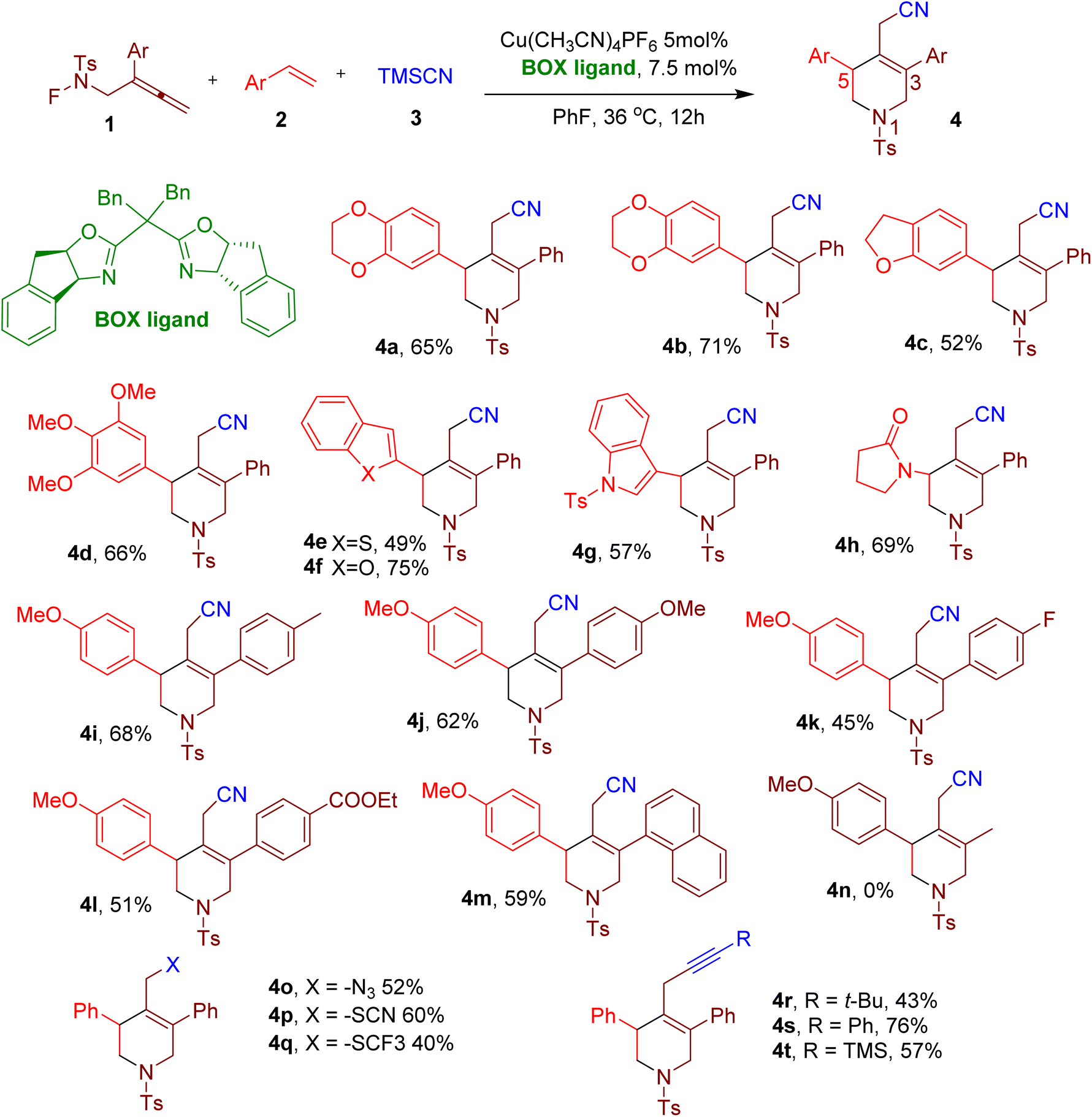 | ||
| Fig. 4 MCR for the synthesis of diversely substituted tetrahydropyridine derivatives. | ||
A variety of alkenes bearing either single or multiple aromatic ring substituents reacted successfully under the optimized conditions, yielding diverse C5-substituted THPs (e.g., 4b–4h). The heterocyclic rings and heteroatom substituents could also be coupled at C5. Similarly, a variety of electron-donating groups (EDGs) and electron-withdrawing groups (EWGs) were tolerated in the aryl ring of the F-masked sulfonamides, thus yielding a variety of C3 substituents (e.g., 4i–4m). However, alkyl groups (e.g., 4n) could not be installed at C3, highlighting this MCR's key limitation. In addition to TMSCN, other nucleophiles such as azidotrimethylsilane (TMSN3), TMSSCN, and silver(I) trifluoromethanethiolate (AgSCF3) also yielded the corresponding products (e.g. 4o–4q) in good yields. The reaction also proceeded smoothly with various alkynyltrimethoxysilanes as nucleophiles to furnish the corresponding THPs (e.g. 4r–4t), displaying the versatility of the reaction. The synthetic utility of this reaction was further demonstrated by making various derivatives of the THP products and by scaling the reaction to a gram scale.
Cui and coworkers designed an MCR strategy to obtain benzofuran-fused piperidines employing an electron-rich benzofuran ring (5), primary amine (6), and formaldehyde (7, Fig. 5).44 The reaction utilizes the unactivated benzylic C(sp3)–H and position-2 C(sp2)–H bonds of the benzofuran ring as nucleophilic sites in a double Mannich reaction. Thus, the newly formed compounds consist of N-substituted piperidine fused with the positions 2 and 3 of the benzofuran. The model reaction between 6-methoxy-3-methylbenzofuran, formaldehyde (4 equiv.), and glycine methyl ester hydrochloride (2 equiv.) yielded the model compound 8a most efficiently when acetic acid was used as solvent.
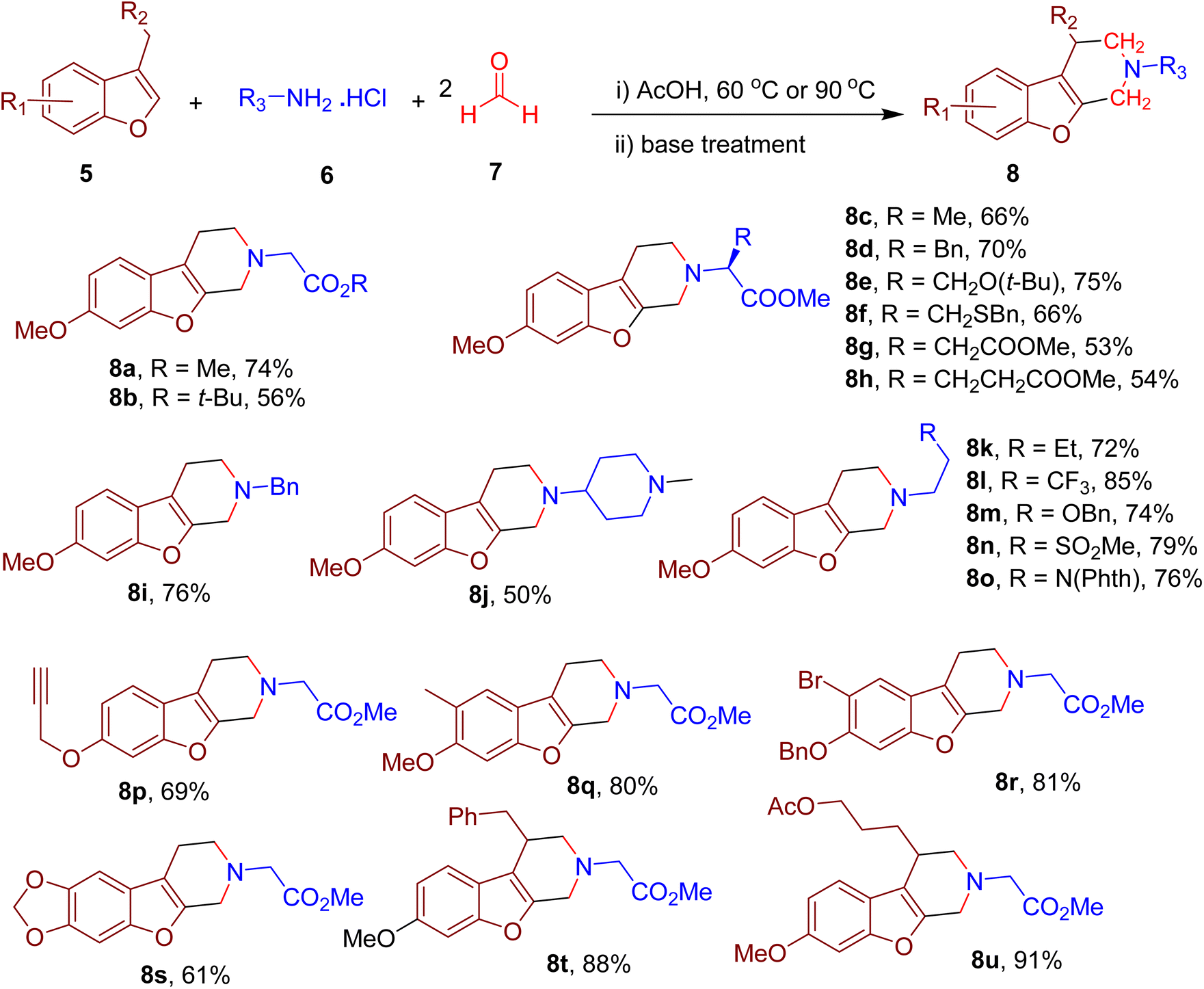 | ||
| Fig. 5 MCR for the synthesis of diverse benzofuran-fused piperidines. | ||
Under the optimized conditions, various amino acids reacted effectively and furnished the corresponding benzofuran-fused piperidines (e.g., 8b–8h) in good yields with the retention of configuration on α-carbons. In addition, other primary amines with diverse functionalities (e.g., 8i–8o) were also well-tolerated, suggesting the MCR's versatility. Late-stage modification of several drug derivatives with amine functionalities could also be achieved, indicating the potential usage of this one-pot MCR in medicinal chemistry. The benzofuran derivatives with diverse EDGs also afforded the corresponding piperidine products (e.g., 8p–8s) in good yields. Various multi-substituted piperidines could also be obtained by employing benzofurans with substituents on the benzyl carbon, thus providing compounds (e.g., 8t and 8u) with diverse physicochemical properties. The presence of boronate and alkyne functionalities in some of the final products also offers an opportunity for further diversification of piperidine-containing compound libraries for medicinal chemistry projects. Mechanistic investigations and density functional theory (DFT) calculations suggested the involvement of several intermediates formed involving Mannich, retro-Mannich, and nucleophilic substitution reactions.
By replacing benzofuran with 2-methyl indole, the same group also reported the synthesis of a piperidine-fused indole chemotype resembling natural alkaloids, which generally requires multistep synthesis. Notably, indole is among the most important heterocycles of numerous drugs and clinical candidates.26,27,45–47 Performing the model reaction in dimethylformamide (DMF) solvent, with N-benzyl-2-methyl indole (9), methyl glycine ester, and formaldehyde yielded γ-tetrahydrocarboline 10a in excellent yield (Fig. 6).48
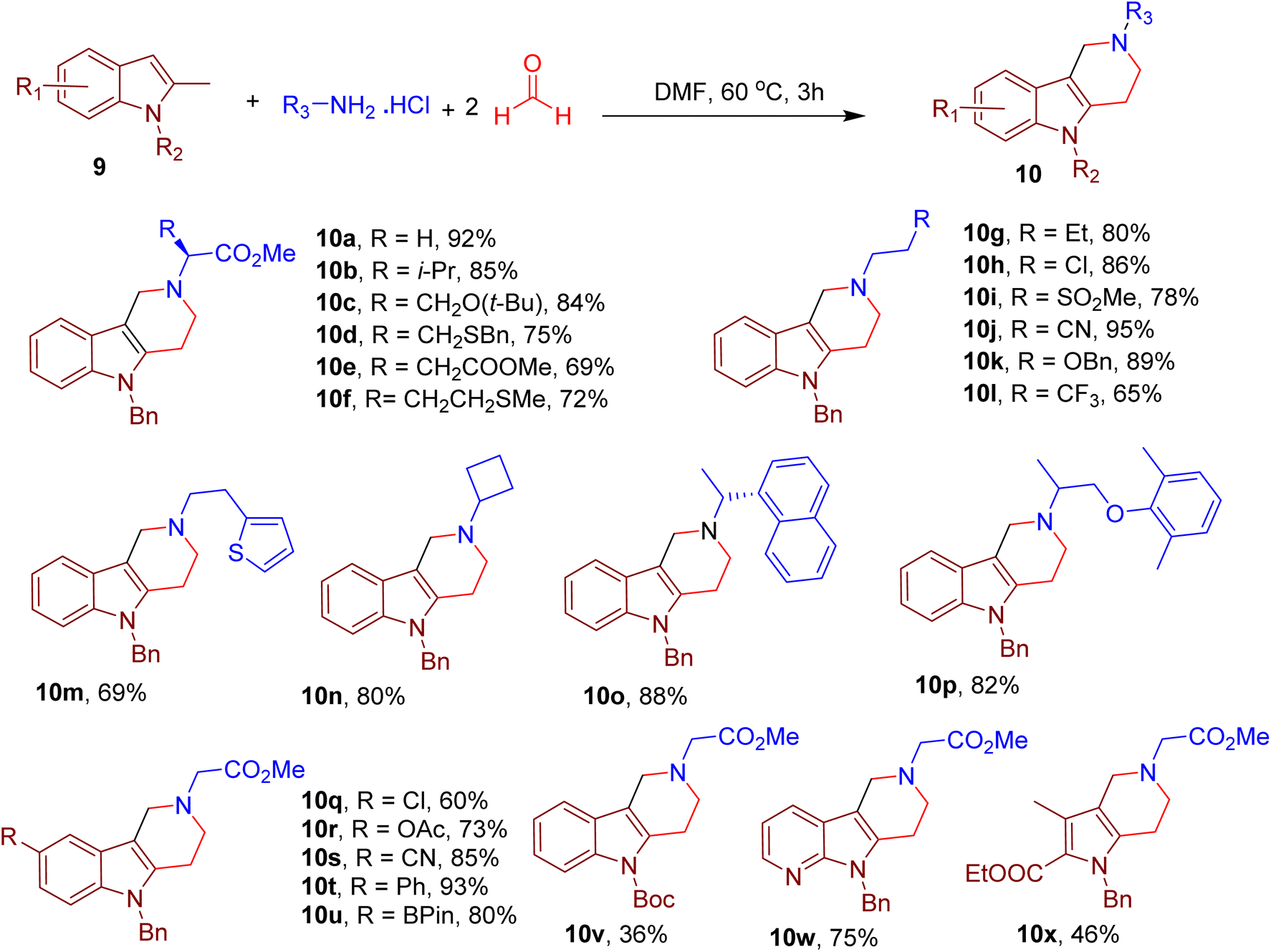 | ||
| Fig. 6 MCR for the synthesis of diverse γ-tetrahydrocarbolines. | ||
The substrate scope revealed the compatibility of various amino acids (e.g., 10a–10f) and aliphatic amines bearing diverse functional groups and rings (e.g., 10g–10p). Many substitutions in the indole ring were also tolerated (e.g., 10q–10u). Nevertheless, replacing the N-alkyl substitution with H-atom or EWGs (such as Boc, e.g., 10v) reduced yields, suggesting the importance of electron density on indole nitrogen. Interestingly, this MCR could also obtain azaindole (e.g. 10w) and pyrrole fused piperidines (e.g., 10x) by selecting corresponding starting materials.
The authors further expanded the scope of the reaction and reported the synthesis of related β-tetrahydrocarboline chemotype by replacing 2-methyl indoles (9) with 3-methyl indoles (13, Fig. 7). In this case, the N-tert-butoxycarbonyl (N-Boc) group and acetonitrile (ACN) as solvent were found to be ideal for the high reaction yields. Again, diverse amines were tolerated under the reported conditions (e.g., 14a–14j). However, perturbing electron density in indole by changing the dimethoxy substitution pattern resulted in poor yields of the resulting products (e.g., 14k–14u). Similarly, N-substitutions other than the Boc group also resulted in diminished yields (e.g., 14v). The MCR's utility was demonstrated by synthesizing a few indole-based drugs and clinical candidates.
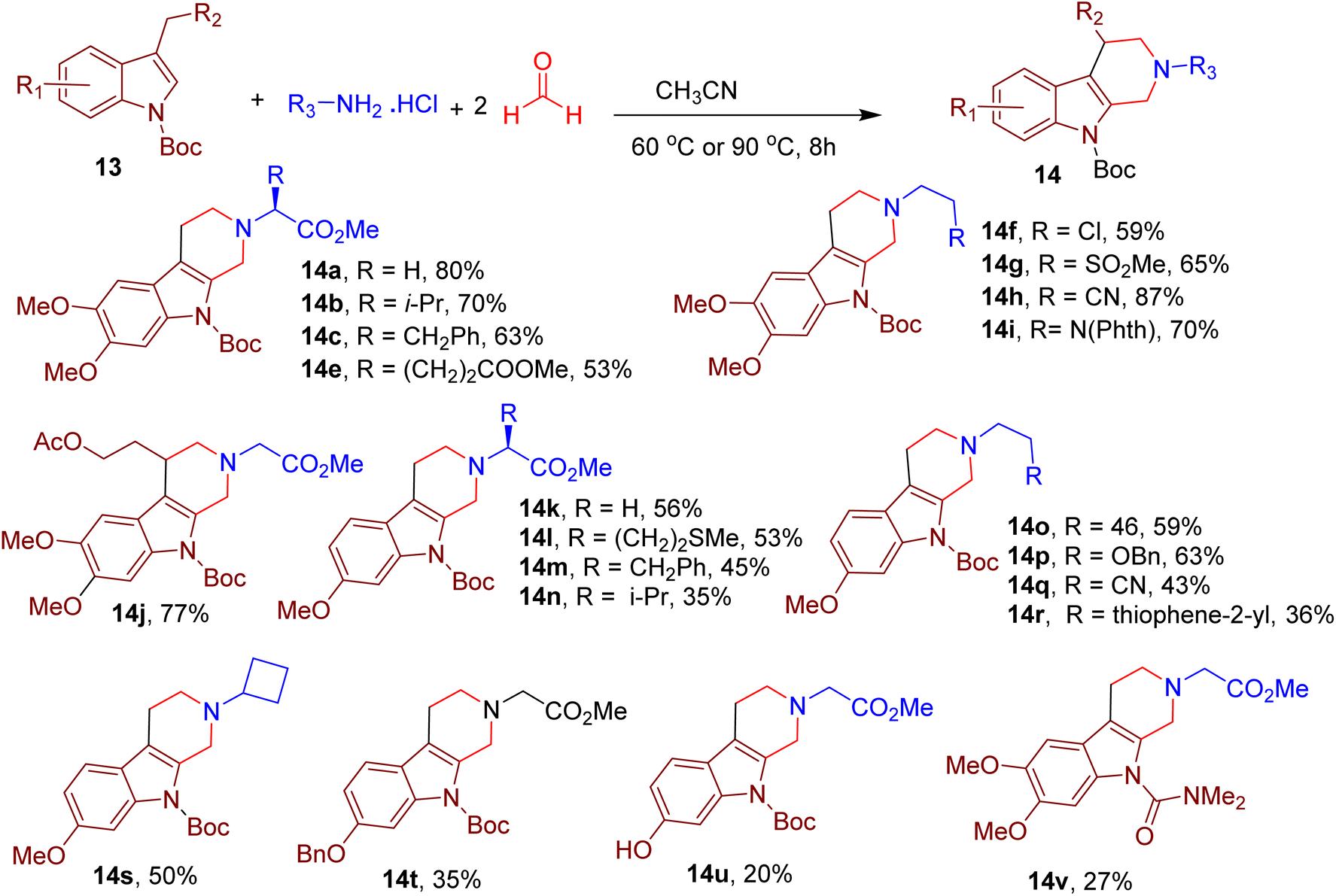 | ||
| Fig. 7 MCR for the synthesis of diverse β-tetrahydrocarbolines. | ||
Continuing with their indole-based MCRs, Cui and coworkers also reported the construction of indole-fused seven-membered heterocycles. The reaction involved the participation of the indole–NH and carbon at position 2. The MCR involves a reaction between 3-methylindole, excess formaldehyde, and amine hydrochloride salt in tetrahydrofuran (THF), resulting in an indole-fused oxadiazepine derivative (Fig. 8).49 Different amino acids (e.g., 15a–15c) and other aliphatic amines (e.g., 15d–15k) furnished the corresponding oxadiazepines in moderate to excellent yields. The integrity of the chiral centre of the amino acids was also maintained under the optimized conditions. Various EWGs and EDGs, such as halogens, methoxy, alkene, and alkyne, were also tolerated on the indole ring (e.g., 15l–15r), indicating the generality of the MCR.
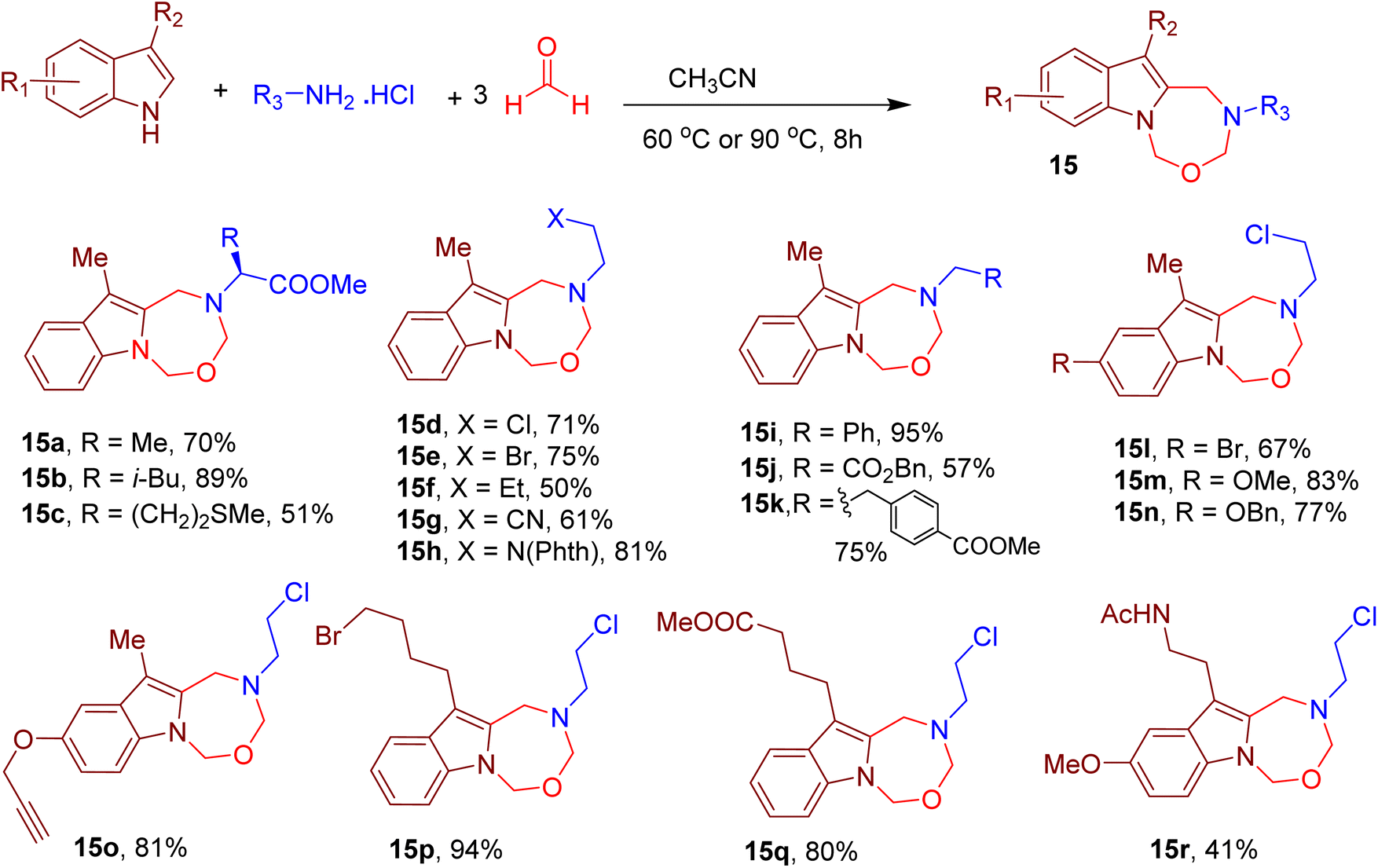 | ||
| Fig. 8 MCR for the synthesis of indole-fused oxadiazepines. | ||
Interestingly, the inclusion of sodium thiosulfate (16) in the same reaction and the use of aqueous hydrochloric acid (HCl) afforded indole-fused thiadiazepine analogues (e.g., 17a–17n), further expanding the scope of this MCR (Fig. 9).
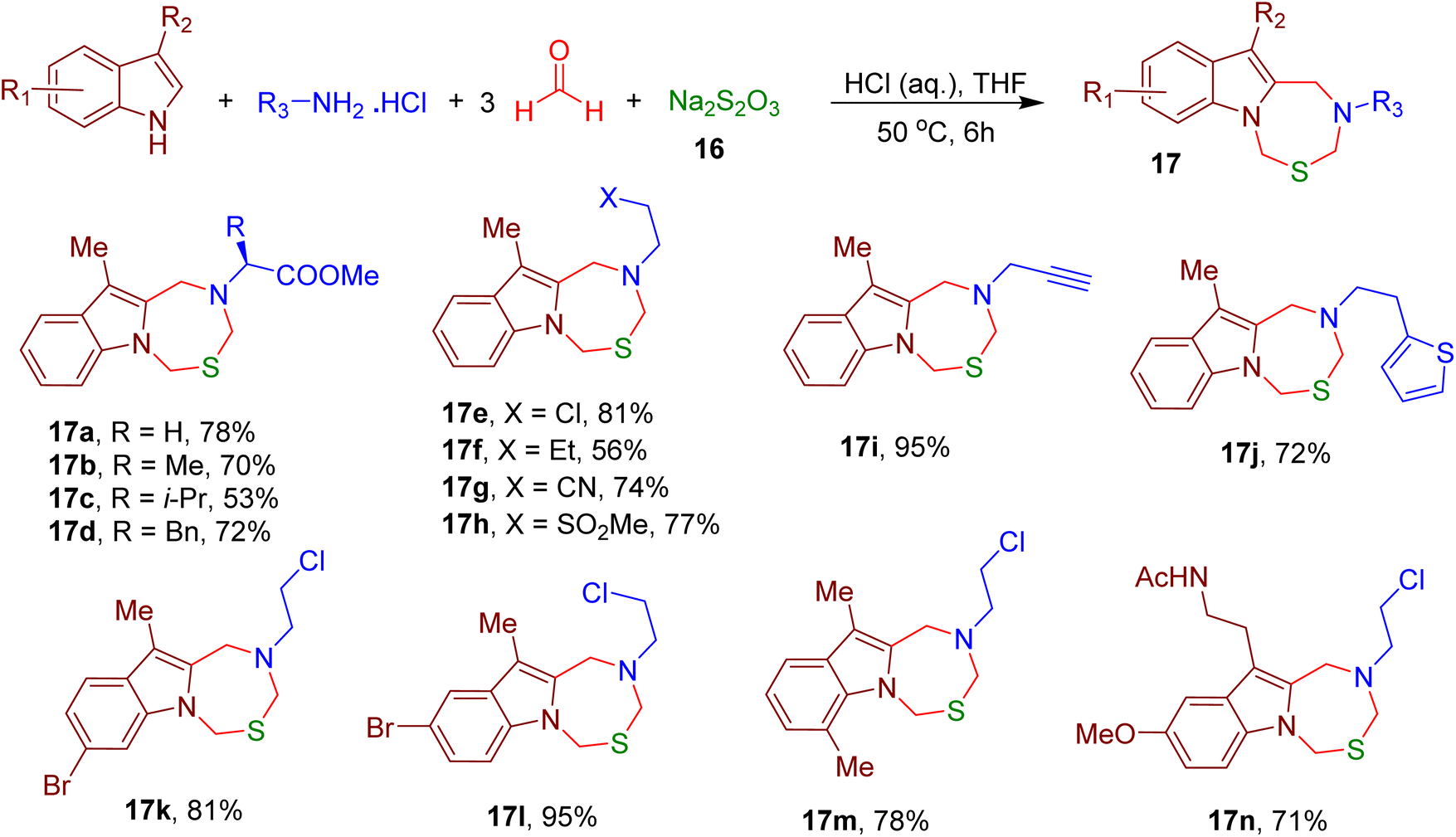 | ||
| Fig. 9 MCR for the synthesis of indole-fused thiadiazepines. | ||
Several newly synthesized oxadiazepine and thiadiazepine analogues displayed inhibition of the liver (BEL7402) and breast cancer (MDA-MB-231) cell lines in the micromolar (μM) range. Further, the indole ring of tryptophan (Trp) and Trp-bearing peptides was also shown to participate in this reaction, suggesting the potential usage of this MCR for peptide and protein modification.
The indole ring is commonly synthesized using Fischer indole synthesis, performed under strong acidic conditions and employing hazardous phenylhydrazine. Despite the plethora of research based on indole compounds, very few MCR-based methodologies exist for synthesizing indole and its derivatives.
Domling and coworkers reported the facile synthesis of tetrazole indoles by exploiting the Ugi-tetrazole four-component reaction (Fig. 10). In this two-step, MCR, a substituted arylamine (18), dimethoxyacetaldehyde (19), isocyanide (20), and TMSN3 (21) were reacted at room temperature to obtain the Ugi-tetrazole adducts (22) which are further cyclized to indole derivatives (23) under acidic conditions.50 The developed methodology displayed a broad scope tolerating a variety of EWGs and EDGs on isocyanide and aniline building blocks (e.g., 23a–23h). The best yields were obtained with methane sulfonic acid (MSA) for cyclization. In the case of unsymmetrical amines, the authors optimized temperature for the selective precipitation of the major regioisomer. This two-step MCR was ultimately translated to a one-pot procedure where 23b was obtained in 49% overall yield. The reaction could be carried out at a gram scale without requiring chromatographic separation, demonstrating the scalability and convenience of the synthetic protocol. The end products could be further functionalized to expand the diversity of the indole derivatives.
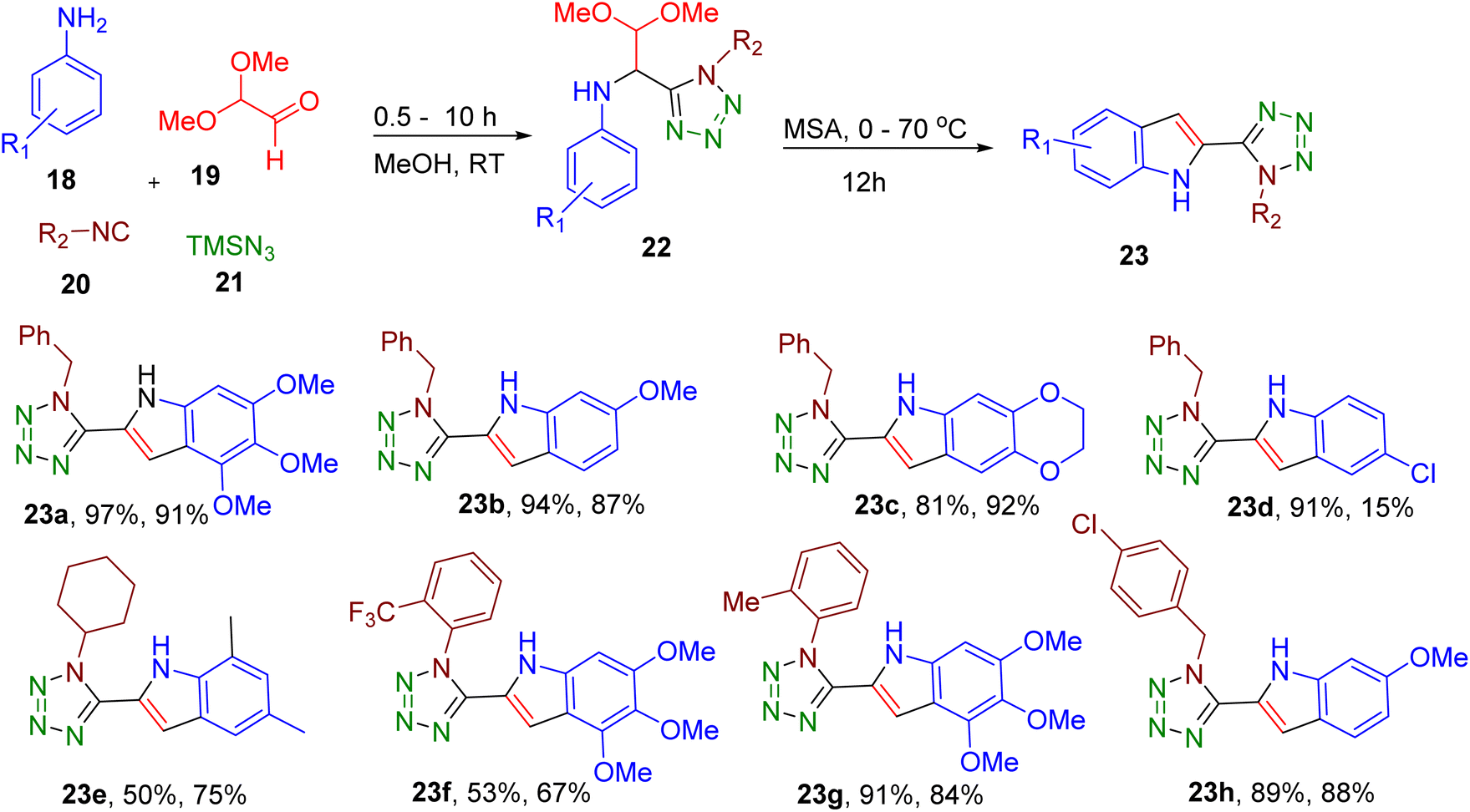 | ||
| Fig. 10 MCR reported for the synthesis of tetrazole-indole derivatives. The yields of the first and second steps are reported, respectively. In cases where a mixture of regioisomers is obtained, the structure of the major isomer is provided. | ||
Thus, this MCR represents mild and facile synthesis of privileged indole core in a one-pot operation.
The group also reported a similar approach to obtain highly substituted indole-2-carboxamide derivatives (Fig. 11). Initially, Ugi adducts were obtained from substituted aniline, glyoxal dimethyl acetal, isocyanide, and formic acid (24) and further cyclized using MSA to get the indole carboxamides (26).51 This two-step approach worked well with anilines with EDGs (e.g., 26a–26f), while electron-deficient anilines either did not undergo cyclization or reacted inefficiently (e.g., 26g–26i). In contrast, various isocyanides afforded the corresponding products in good yields, producing a library of indole carboxamides with diverse physiochemical properties. The authors developed the single-pot-two-step procedure and displayed the scalability of the methodology to the gram scale. To further highlight the usefulness of their designed protocol, the authors demonstrated the synthesis of two anti-tuberculosis agents (e.g., 26j and 26k) and a SET domain containing 2 (SETD2) inhibitor (e.g. 26l) in a short timeframe using this convenient and mild MCR approach.
 | ||
| Fig. 11 MCR for the synthesis of indole-2-carboxamide derivatives. The yields of the first and second steps are reported, respectively. Single yields are reported for the compounds synthesized in one-pot operation. | ||
Quinazolinone derivatives display multiple pharmacological properties, including antimicrobial, anticancer, antidiabetic, and antihypertensive properties.52,53 Several methods for the synthesis of N1,N3-disubstituted 2,3-dihydroquinazolin-4(1H)-ones are known. However, most of these multistep methodologies limit the diversity of N1-substituents and might require specialized catalysts and tedious conditions.54,55
Zhang et al. reported the synthesis of N-substituted quinazolinones employing a Pd(II)-catalyzed cascade reaction between an o-aminobenzoic acid (27), amine (28), aldehyde (29), and carbon monoxide (Fig. 12).56 This one-pot procedure involved the Pd-catalyzed carbonylation of o-aminobenzoic acid followed by the reaction of the resultant isatoic anhydrides with the amine and aldehydes components to afford N1,N3-disubstituted 2,3-dihydroquinazolin-4(1H)-ones. The model reaction was favoured by the usage of copper(II) acetate [Cu(OAc)2] as the oxidant, additives potassium iodide (KI), acetic acid (AcOH), and ACN solvent at 65 °C.
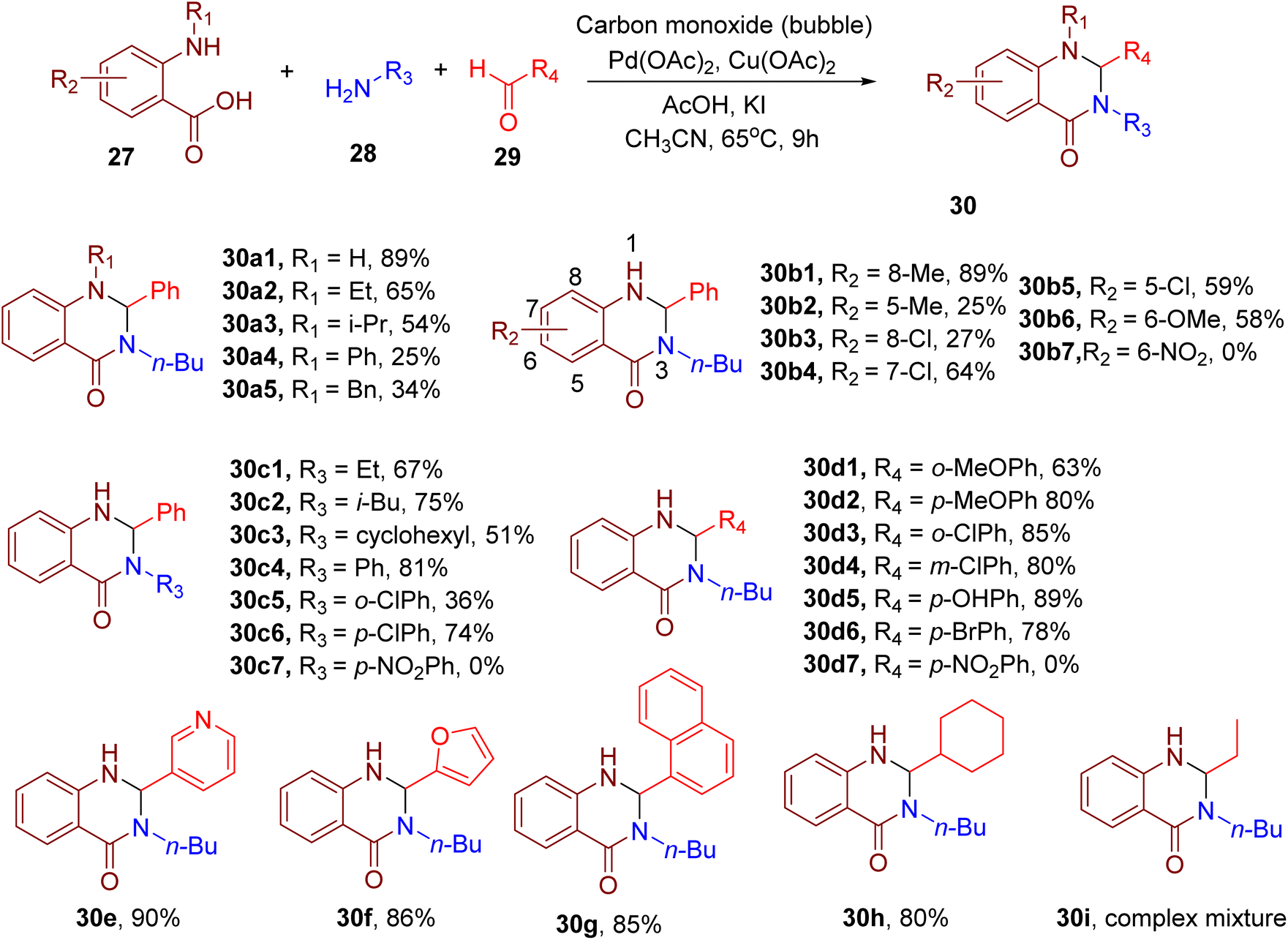 | ||
| Fig. 12 MCR for the synthesis of N1,N3-substituted quinazolinones. | ||
Different o-aminobenzoic acids with or without N-substituents generally reacted smoothly under the optimized conditions to furnish the target products (e.g., 30a1–30a5). However, sterically hindered substrates with bulky N-substituents furnished lower yields than those with smaller N-substituents (e.g., 30a3–30a5). In general, different substituents on the phenyl ring of o-aminobenzoic acids yielded moderate to good yields (e.g., 30b1–30b6), except for the nitro group-bearing aminobenzoic acid (e.g., 30b7) for which no product was obtained.
The authors demonstrated the generality of the reaction with a variety of aliphatic and aromatic amines (e.g., 30c1–30c7). However, bulky amines led to lower product yields (e.g., 30c3). The detrimental effect of a steric factor was also observed with arylamines having ortho-substituents (e.g., 30c5), except for those with EDGs. Conversely, the presence of strong EWGs like nitro on the arylamine (e.g., 30c7) led to the failure of the reaction.
The reaction also worked well with a diverse set of aromatic aldehydes, all of which reacted efficiently regardless of the electronic or steric nature of the substituents (e.g., 30d1–30d6). However, the reaction with 4-nitrobenzaldehyde, bearing a strong EWG like NO2, did not afford the corresponding product (e.g., 30d7). A variety of aromatic heterocyclic (e.g., 30e, 30f) and polycyclic aldehydes (e.g., 30g) also furnished the products in good yields, displaying the wide substrate scope of this MCR. Interestingly, in the case of aliphatic aldehydes, those with branched chains afforded higher yields (e.g., 30h), while an intractable mixture was obtained with the linear ones (e.g., 30i).
In medicinal chemistry, fluorine and trifluoromethyl groups are frequently used to improve various drug properties, such as target potency, metabolic stability, and bioavailability.57 The trifluorodiazoethane reagent has emerged as a key reagent to access diverse trifluoromethylated heterocycles.58–61
Building on previous reports,60 Chandrasekharan et al. designed an MCR between o-alkynyl benzaldehyde (31), amine (32), and trifluorodiazoethane (33) or diazoacetonitrile (34) for the construction of pharmaceutically important benzo[d]azepine motif (35, Fig. 13).62 During the reaction optimization, various catalysts were screened, and silver trifluoroacetic acid (AgTFA) was found to afford the best yield of the model compound (e.g., 35a1). The use of 1,4-dioxane solvent, 4 Å molecular sieves, and moderate temperature was also found to improve the yields.
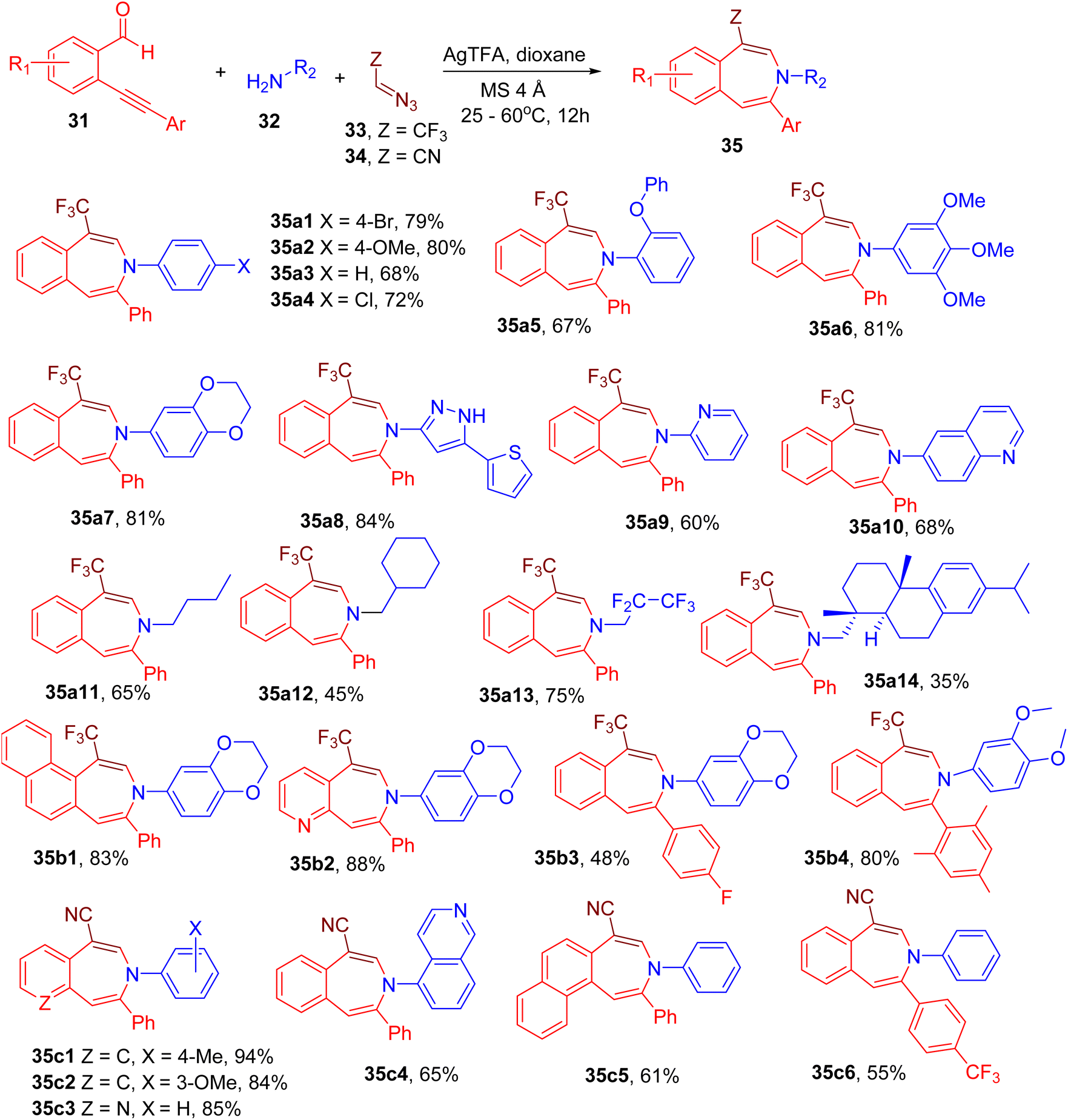 | ||
| Fig. 13 MCR for the synthesis of trifluoromethyl- or cyano-functionalized benzo[d]azepines. | ||
A variety of arylamines (35a1–35a7), heteroarylamines (35a8–35a10), and alkylamines (35a12–35a14) were found to yield the corresponding azepine products indicating the broad scope of amine substrates. Different o-akynylarylaldehydes were also tolerated under the given conditions to yield interesting azepine derivatives (35b1–35b4).
Interestingly, the replacement of trifluorodiazoethane (33) by diazoacetonitrile (34) resulted in analogous cyano-substituted azepine products instead of trifluoromethylated ones. Thus, a variety of amines and akynylarylaldehydes were shown to furnish the cyano-functionalized azepines (35c1–35c6), extending the scope of the reaction. Since nitrile function is amenable to further synthetic modifications, this MCR may provide access to a diverse library of azepine derivatives for drug discovery.
Highly substituted pyrrole rings are of great significance in medicinal chemistry. Several pyrrole-based compounds have been shown to possess anticancer, antibiotic, antifungal, and antiviral properties.63
Afratis et al. recently reported MCRs between a thioamide-enol (36), an aldehyde (37), and an ammonia source to obtain an intermediate (38) which, upon extrusion of sulfur, yielded the substituted pyrrole product (39, Fig. 14).64 Initially, by-product enamine was also observed in appreciable amounts, and reaction conditions were optimized to reduce its formation and improve the overall yield of the reaction. In general, the optimized conditions worked well with diverse heteroaromatic and aromatic aldehydes (e.g., 39a–39i) except for the electron-neutral/-rich aryl aldehydes or alkyl aldehydes. Diverse substitutions on the aromatic ring of thioamide were also tolerated (e.g., 39j–39l) with no loss in yield. Moreover, modifications to the lactam moiety were also tolerated (e.g., 39m), and the lack of lactam moiety also yielded the cyclic alkyl fused pyrroles in good yields (e.g., 39n and 39o).
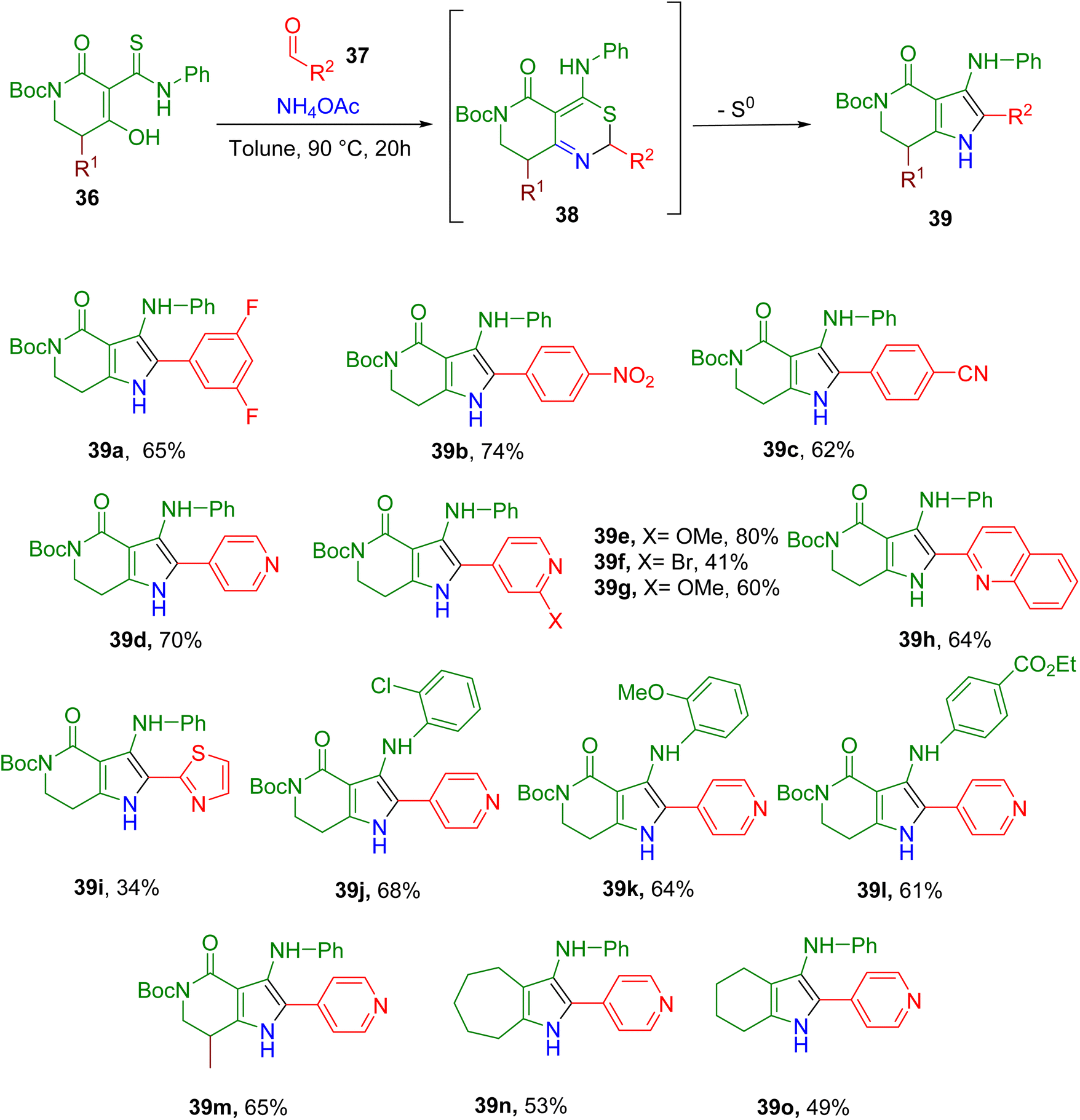 | ||
| Fig. 14 Synthesis of fully substituted fused pyrroles through an oxidant-free MCR. | ||
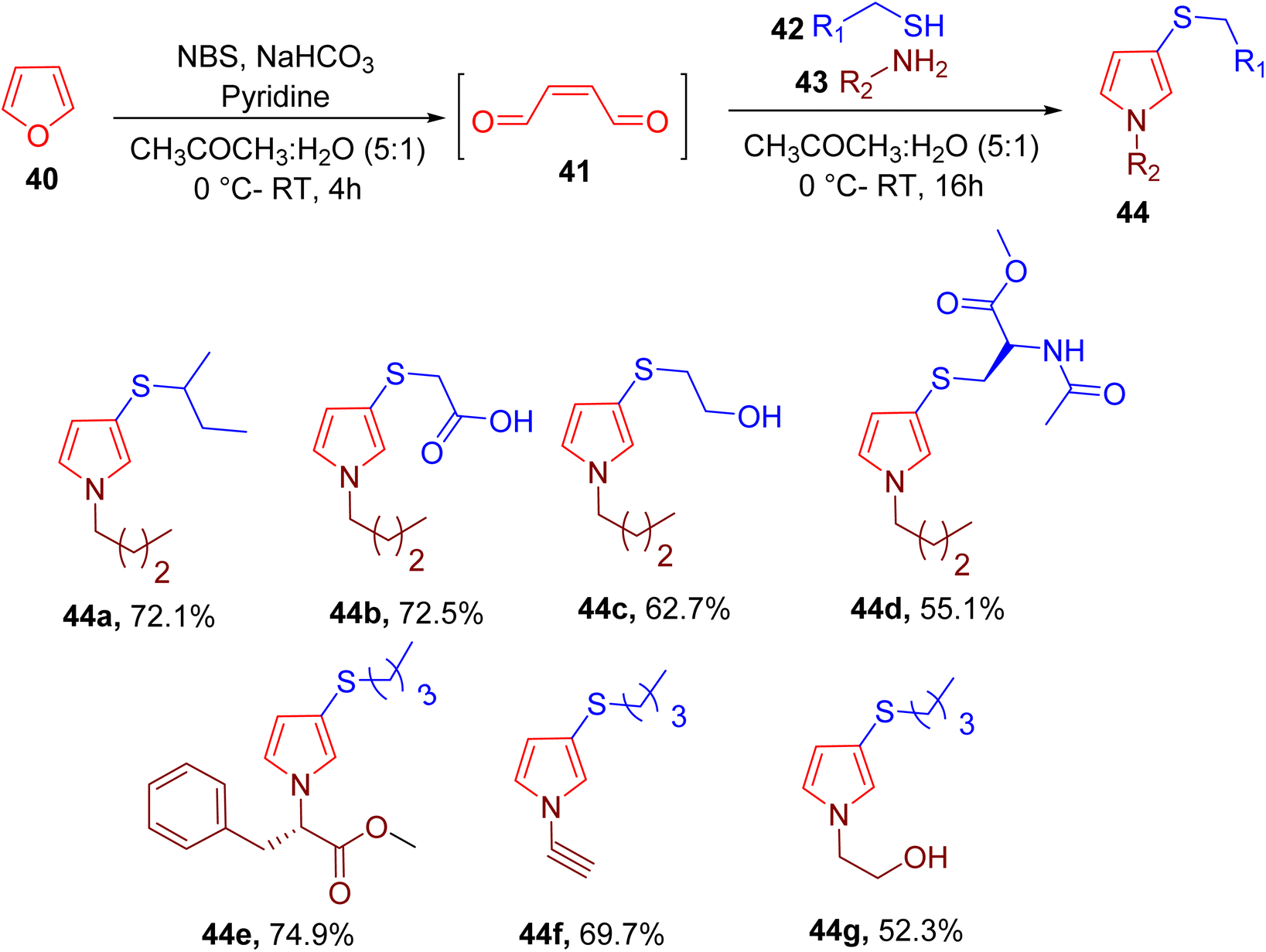
Wang et al. reported a convenient synthesis of 3-thio N-pyrrole ring (44) from thiol (42), amine (43), and cis-2-butene-1,4-dial (BDA) (41). The latter was obtained in situ from the oxidation of pyrrole, leading to an overall two-step single-vessel process (Fig. 15).65 A mixture of acetone and water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was found to be an optimum condition for this MCR. A variety of thiols and amines were used to generate multiple N-pyrrole derivatives bearing varied functional groups (e.g., 44a–44g). Less reactive aniline and different amino acids were also used to obtain the corresponding products in good yields. However, the substrate scope of heteroaromatic amines and thiols is not reported.
:1) was found to be an optimum condition for this MCR. A variety of thiols and amines were used to generate multiple N-pyrrole derivatives bearing varied functional groups (e.g., 44a–44g). Less reactive aniline and different amino acids were also used to obtain the corresponding products in good yields. However, the substrate scope of heteroaromatic amines and thiols is not reported.
 | ||
| Fig. 15 Development of one-pot furan-thiol-amine via MCR approach. | ||
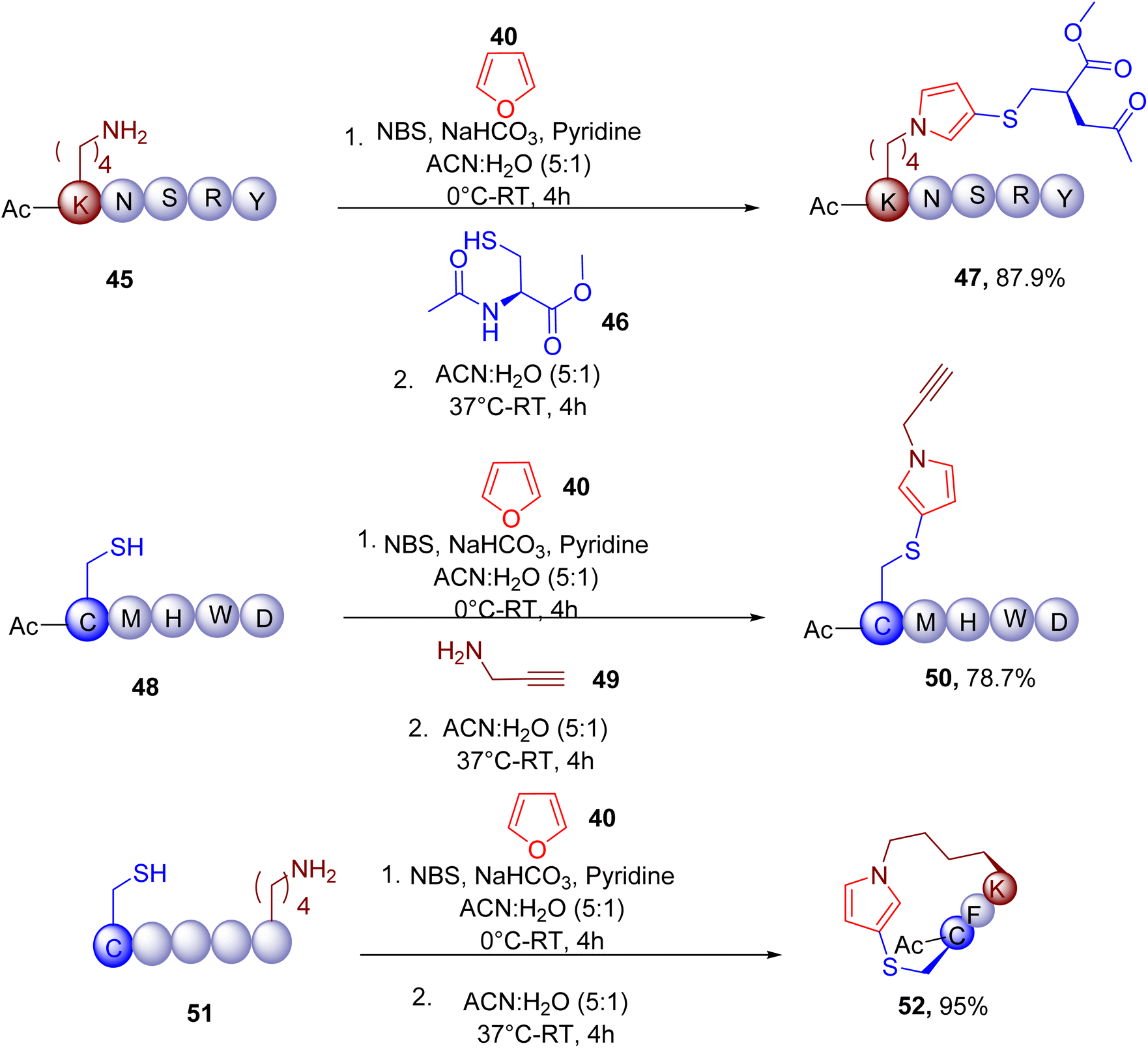
Given the chemoselectivity and robustness of the reaction in aqueous conditions, the MCR was also used for the late-stage modification (Fig. 16) of the lysine (e.g., 47 from 45) and cysteine residues (e.g., 50 from 48) of peptides. Similarly, the macrocyclization of peptides (e.g., 52 from 51) containing both free thiols and amino groups was also demonstrated. Moreover, the amine and thiol groups of several proteins were also shown to participate in this reaction in a complex cell lysate mixture, demonstrating the application of this MCR for amino acid profiling and protein labelling.
 | ||
| Fig. 16 Chemoselective modification of lysine and cysteine residues by one-pot furan-thiol-amine MCR. | ||
In recent years, several organoselenium compounds have demonstrated biological properties.66 Hence, the convenient synthesis of organoselenium compounds, is of great importance in medicinal chemistry.
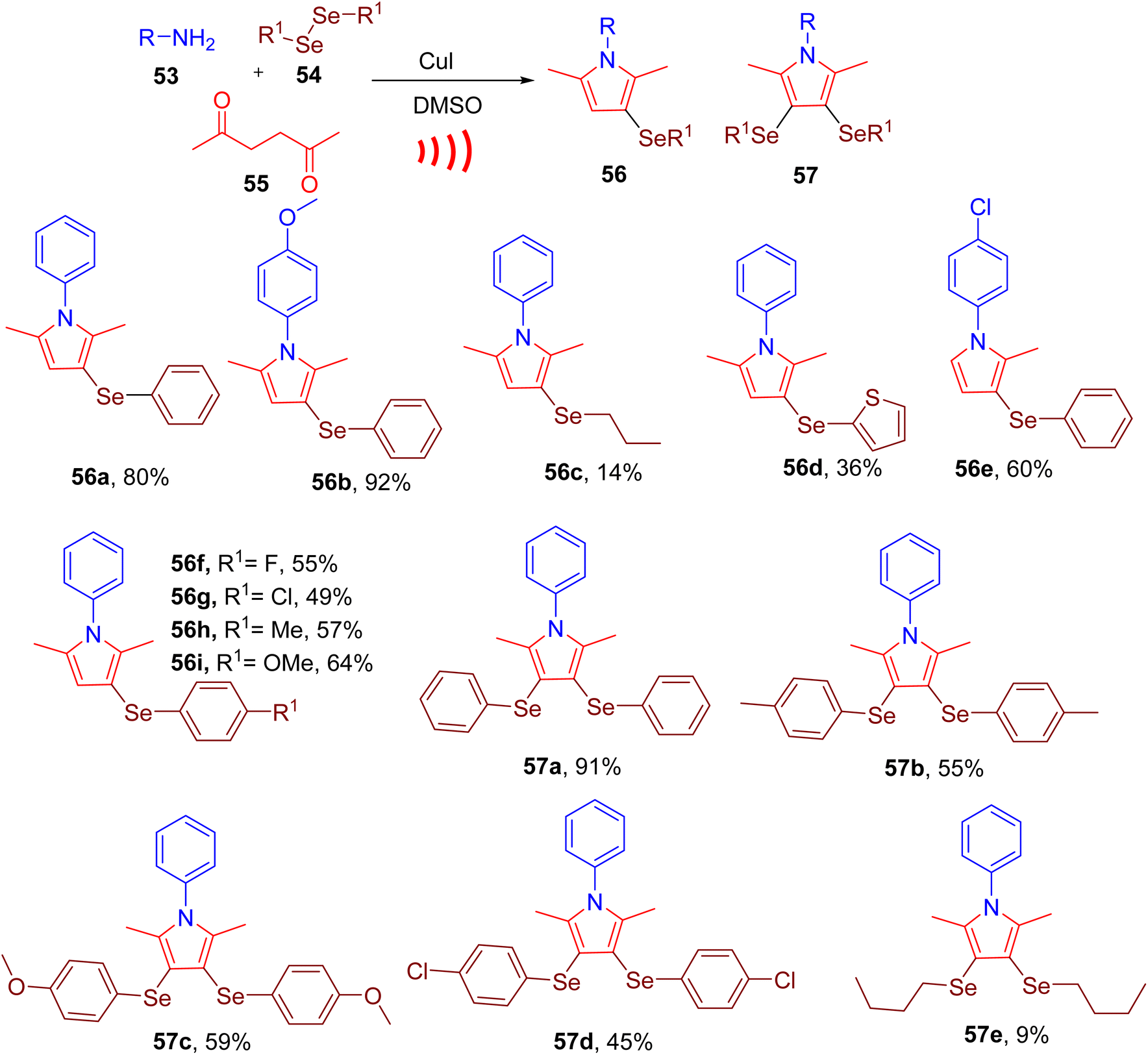
Peglow et al. recently achieved synthesis of pyrrole ring with mono- or di-substituted selanyl groups through MCR (Fig. 17). Building on their previous work, the authors attempted the design of a one-pot methodology using amine (53, 0.5 mmol), diselenide (54, 0.25 mmol), and 2,5-hexanedione (55, 0.5 mmol) to afford selenium-substituted pyrroles (e.g. 56 or 57).67 After screening different conditions, the best yields of monoselenylated pyrrole were achieved using CuI catalyst in dimethysulfoxide (DMSO) using ultrasound (US) (60% amplitude) as the source of energy. The molar ratio of the starting materials and diphenyl diselenide was also found to affect the final yields. The reaction worked effectively with a variety of alkyl, aryl, and heteroaryl diorganyl diselenides and amines to yield pyrroles in moderate to good yields (e.g., 56a–56i). However, diaryl diselenides with EWG (e.g., 56f and 56g) required a longer time to obtain moderate yields of the products compared to those with EDG (e.g., 56h and 56i). The reaction did not perform well with dibutyl diselenide, affording only a 14% yield for the corresponding product (e.g. 56c).
 | ||
| Fig. 17 US-assisted one-pot synthesis of mono- or bis-substituted organylselanyl pyrroles. | ||
Using an excess of diphenyl diselenide (0.5 mmol) and copper iodide (CuI) (5–20 mol%) bis-selenylated pyrroles could be obtained as major products (e.g., 57a–57e). The reaction was found to be efficient with a variety of amines and organoselenides. However, di(thiophen-2-yl)diselenide and dibutyl diselenide (e.g., 57e) produced unsatisfactory results; furnishing lower yields of corresponding products.
Pyrimidine rings occur in several natural products and biomolecules, including deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) bases. Several pyrimidine-based compounds with broad biological properties are reported in the literature.68,69 Many synthetic strategies are available for the synthesis of pyrimidines and their derivatives. However, very few methods exist for the synthesis of asymmetric bipyrimidines, such as 4,5′- and 3,5′-bipyrimidines.
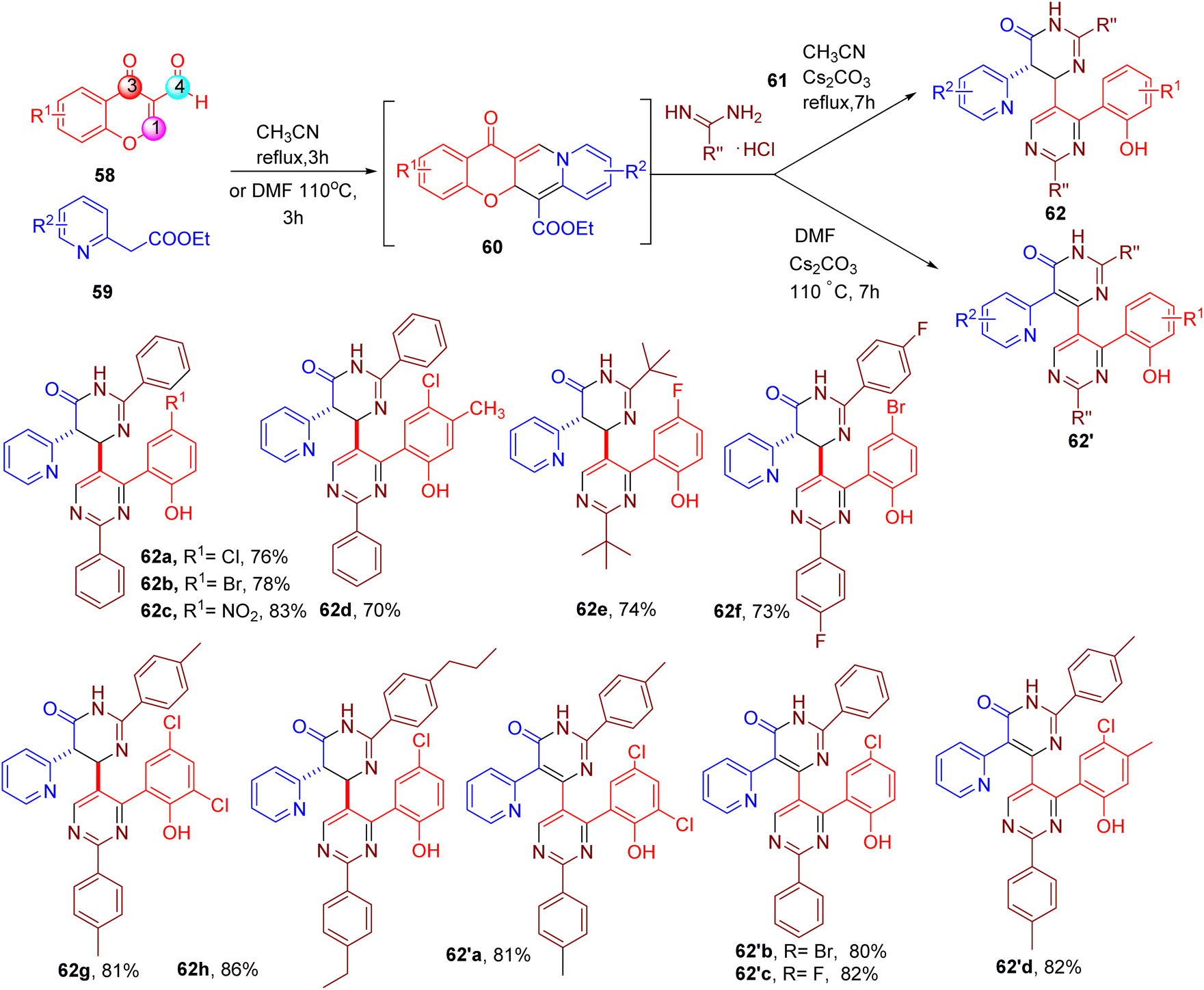
Chen et al. serendipitously discovered an MCR cascade leading to the synthesis of [4,5′-bipyrimidin]-6(1H)-one, a highly functionalized asymmetric bipyrimidine scaffold (62, Fig. 18).70 The authors explored the condensation of 3-formylchromone (58) in the presence of dinucleophiles such as 2-(pyridine-2-yl) acetate (59) and amidine hydrochlorides (61), which yielded (62 or 62′), via intermediate (60), depending on the solvent and temperature. Both EWGs and EDGs groups on the chromone ring were well-tolerated (e.g., 62a–62h and 62′a–62′d). In addition, various benzamidine with differently substituted aromatic rings or alkyl groups could also be employed to obtain the desired products (e.g., 62g and 62h).
 | ||
| Fig. 18 MCR for the synthesis of asymmetric 4,5′-bipyrimidine. | ||
The bipyridines were proposed to form through the cascade of Michael addition, cyclization, intra- and intermolecular ring opening, and dehydrogenative aromatization reactions with the overall formation of five bonds and cleavage of one bond.
Lyapustin et al. reported the unexpected synthesis of a unique azolopyrimidine scaffold (66, Fig. 19) from MCR involving nitroenamine (63), aminoazoles (64), and aromatic aldehyde (65).71 The reaction optimization suggested the use of butanol solvent and boron trifluoride etherate (1.5 equiv.) to obtain the model compound 66a in good yield. While different azolopyrimidines were obtained by varying building blocks (e.g., 66a–66f, Fig. 19), the scope of the reaction seems limited since most of the derivatives consist of only p-nitro and p-methoxybenzaldehydes. Instead of aminoazole (64), a few compounds with other aminotriazole are also reported (66g–66j) to show the generality of the reaction. The nitro and ester groups can be exploited to diversify this scaffold for further drug discovery application.
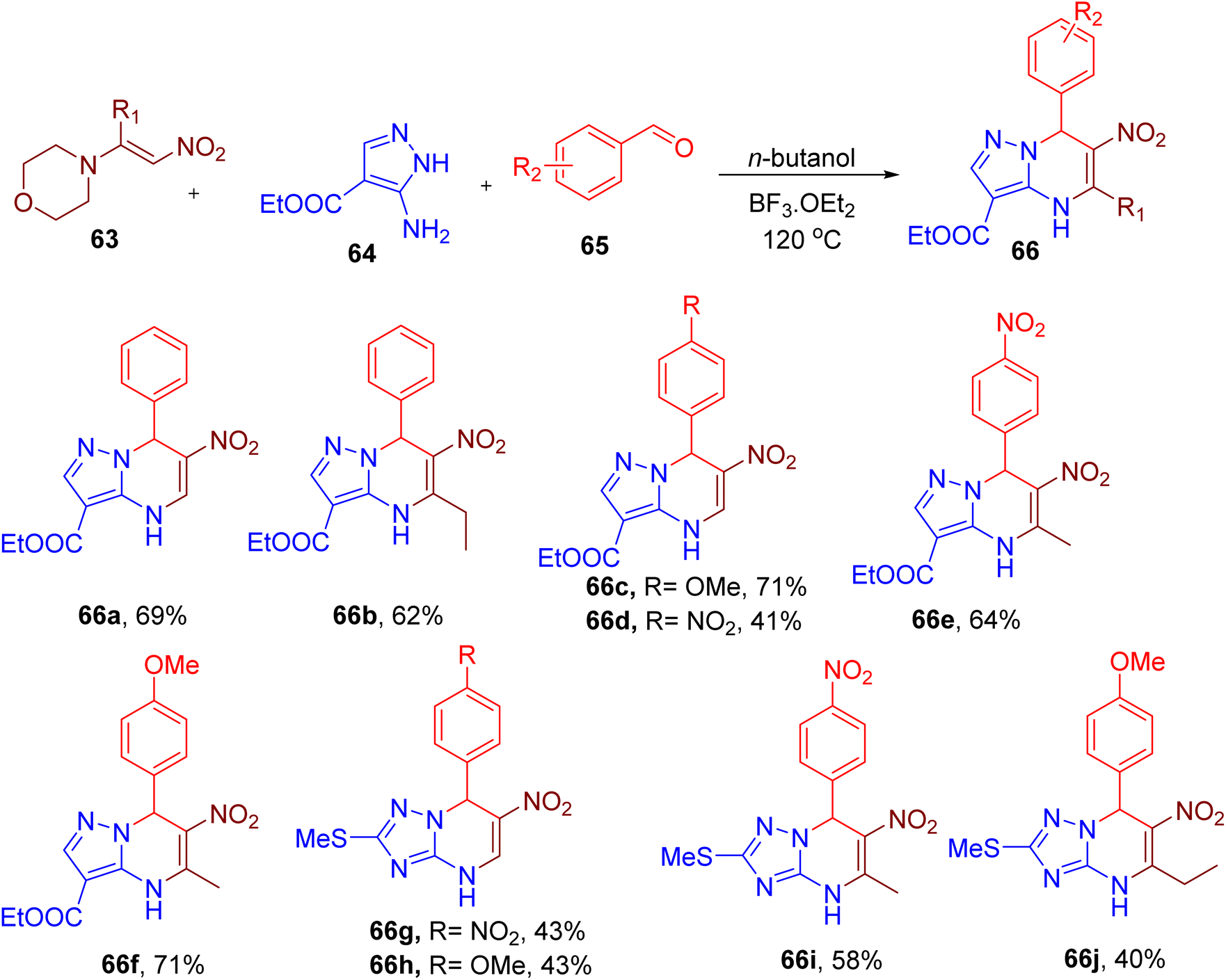 | ||
| Fig. 19 MCR for the synthesis of azolopyrimidine scaffold. | ||
Zhang et al. also reported a facile synthesis of chromone-fused pyrimidine derivatives using a three-component reaction. The highest yield (98%) of the model compound 5H-chromeno[2,3-d]pyrimidin-5-one (e.g. 70) was obtained by heating 3-formylchromone (67), p-toluidine (68), paraformaldehyde (69) under microwave (MW) irradiation without solvent (Fig. 20).72
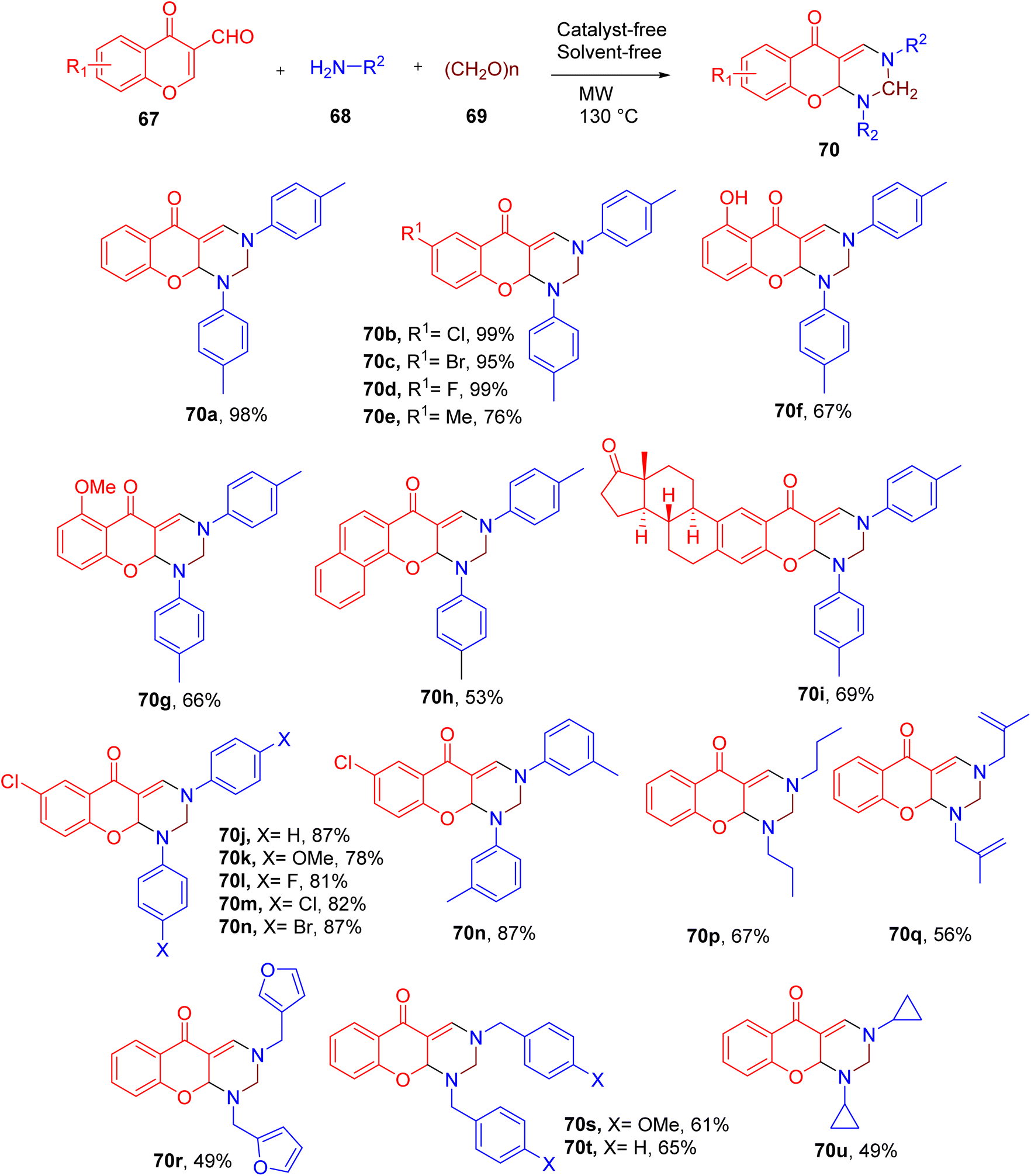 | ||
| Fig. 20 MCR for the synthesis of chromone-fused pyrimidines. | ||
Substrate scope studies reveal that the chromones substituted with halides afforded higher yield of the end products compared to the methyl, hydroxy, or methoxy EDGs (e.g., 70a–70g). Similarly, naphthalene-fused chromone (70h) also displayed lower conversion to the corresponding product. The estrone-based chromone also yielded the desired pyrimidine-fused derivative (70i) in good yield, supporting the late-stage derivatization potential of this MCR.
A variety of EWGs and EDGs were also tolerated on the aryl amines without significant change in the yields of the final products (e.g. 70j–70n). However, alkyl amines furnished the corresponding products in relatively lower yields, especially the ones offering steric hindrance around the amino group (e.g., 70u).
Overall, this MCR represents an important one-pot strategy to construct a chromone-fused pyrimidine scaffold under solvent-free and catalyst-free conditions.
Tan and Wang reported one-pot MCR involving readily available ethylenediamine (71), cinnamaldehydes (72), and 1,3-dicarbonyl compounds (73), resulting in the construction of tetra-substituted hexahydroimidazo[1,2-a]pyridine ring system (74, Fig. 21).73
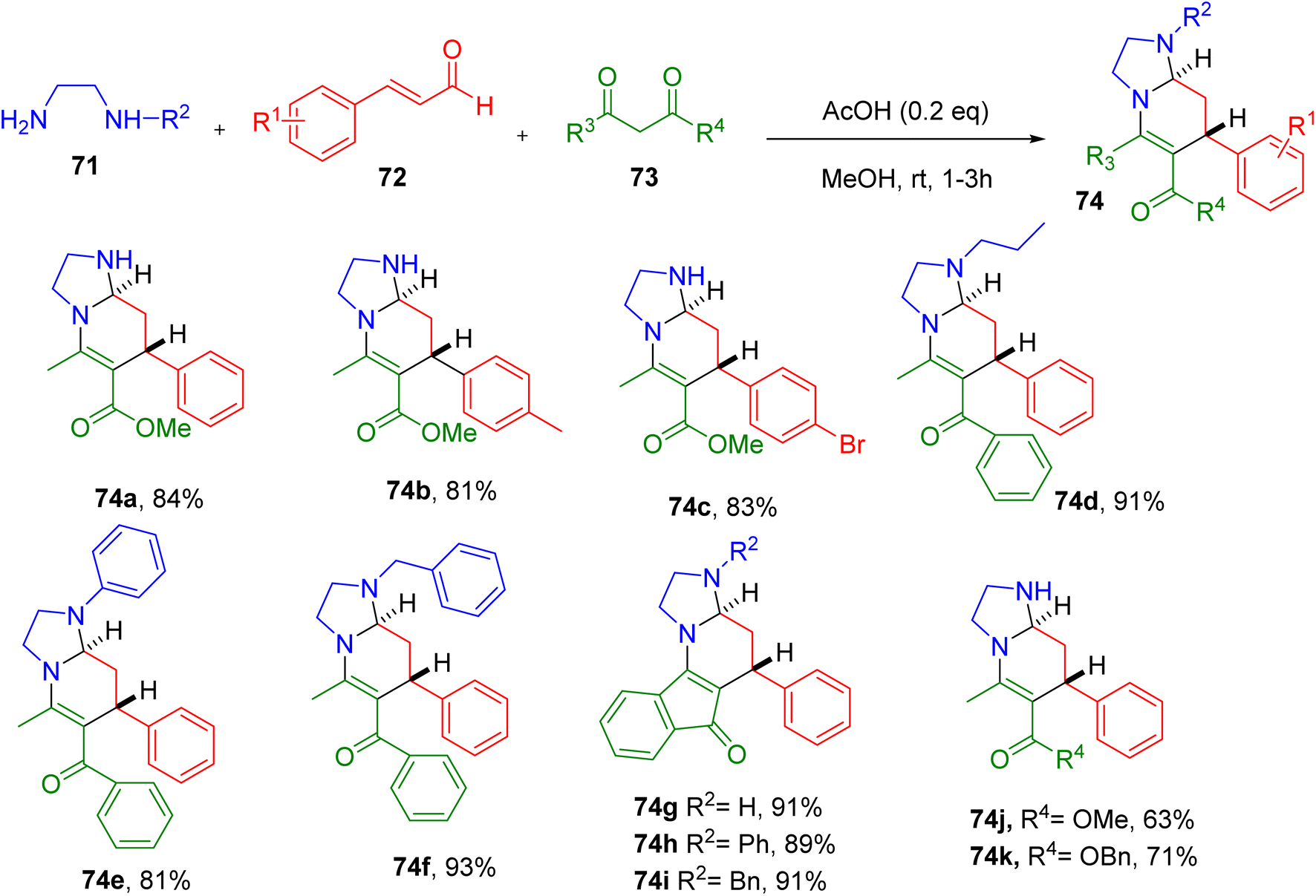 | ||
| Fig. 21 MCR for the synthesis of novel hexahydroimidazo[1,2-a]pyridine derivatives. | ||
The screening of different reaction conditions suggested the use of an AcOH catalyst (0.2 equiv.) in methanol solvent to be optimum for the synthesis of the model compound 74a. The MCR efficiently generated 74a at 84% at room temperature with excellent trans-selectivity (dr > 99:1), which was confirmed by single-crystal X-ray analysis.
A diverse set of cinnamaldehydes with different EWGs and EDGs afforded good yields of the corresponding products (e.g., 74a–74c), except those with the ortho-substituents. Ethylenediamine substituted at one end with alkyl, phenyl, or benzyl groups also furnished the desired compounds in good yields (e.g., 74d–74f). Similarly, a variety of 1,3-dicarbonyl starting materials, including indanedione, afforded the target compounds good yields, suggesting the generality of the reaction (e.g., 74g–74i). Interestingly, employing aminoethanol instead of ethylenediamine furnished oxazolo[3,2-a]pyridine (e.g., 74j and 74k), a natural product-like fragment.
Overall, this MCR is a key tool for generating natural product-like molecules, starting with the commercially available building blocks. The key features of this MCR include high atom economy, mild conditions, high diastereoselectivity, and the use of cheap catalysts.
The heterocyclic ring 1,3,5-triazines and its derivatives are part of many naturally occurring and synthetic molecules displaying important biological applications.74 In particular, 2,4-diamino-1,3,5-triazines display potent antimicrobial and anticancer activities. This heterocyclic chemotype is usually synthesized from biguanide and one-carbon synthons under harsh conditions.
Zhao et al. recently developed a one-pot MCR method for the synthesis of 2,4-diamino-1,3,5-triazines (78) from methyl ketones (75), anilines (76), and cyanamides (77) (Fig. 22).75 The molecular iodine (2 equiv.) in combination with TFA (1 equiv.) as an additive provided the best yields of the model product 78a substituted with an iodine atom at the ortho-position of the aromatic amine.
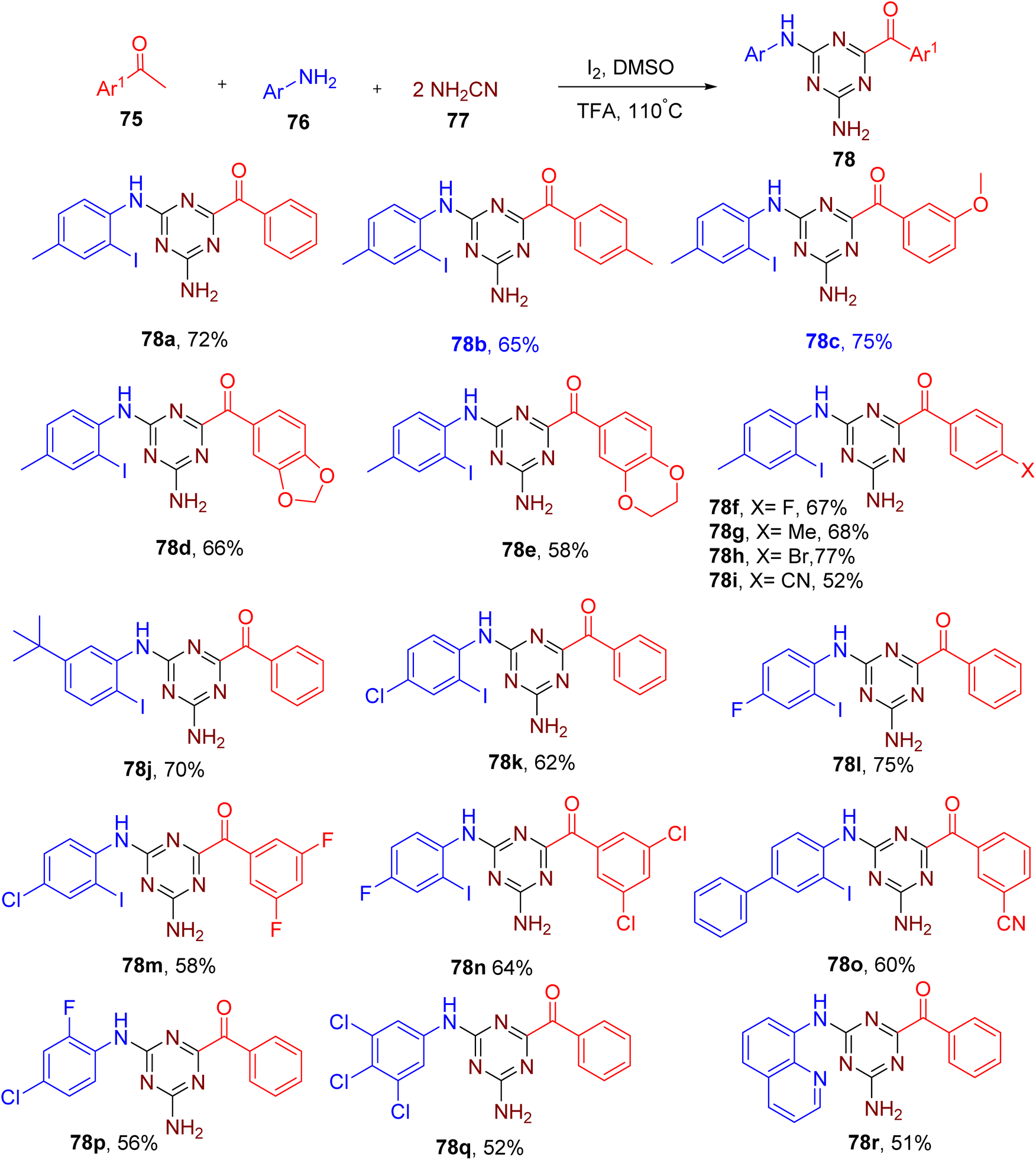 | ||
| Fig. 22 Synthesis of 2,4-diamino-1,3,5-triazines via MCR. | ||
Methyl ketones with different EWGs and EDGs were found to be compatible with the optimized conditions (e.g., 78b–78i). Electron-rich and halogen-substituted aniline substrates also yielded the corresponding products in good yields (e.g., 78j–78o). Electron-deficient anilines with multiple halogens yielded the products without an iodo-substituent at the ortho position (e.g., 78p and 78q), which is in accordance with the poor nucleophilicity of such anilines at the ortho position. Medicinally relevant bi-heterocyclic molecules (e.g. 78r) were also obtained using quinoline-based amine or ketone.
Phthalimide heterocycle is an important pharmacophore in many clinically used drugs, biologically active molecules,76 natural products,77–79 and in the emerging field of PROteolysis.
Targeting chimeras (PROTACs).80,81 The existing synthesis of phthalimides and their analogues requires multistep synthesis using metal catalysts and special building blocks and reagents.82–84
Alizadeh et al. reported the US-mediated MCR reaction for the construction of a phthalimide skeleton. Initially, they synthesized the intermediate methyl 2-(3-benzyl-4-oxo-2-thioxothiazolidin-5-ylidene)acetate (82) from an amine (79), CS2 (80), and dimethyl acetylenedicarboxylate (81, Fig. 23).85 The latter was further reacted with the malononitrile derivative (83 and 84) in the same pot to afford the desired functionalized phthalimide skeleton (85 and 86). The US irradiation with 80% amplitude and the use of triethylamine (Et3N) as base provided the best results in terms of reaction time and final yields. The reaction afforded products in good yields with a variety of 84 derivatives (e.g., 85a–85d) except for those with EWGs like nitro. Switching to the 2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile (83) also resulted in the corresponding phthalimide products (86) with the fused bicyclic indene ring in good yields (e.g., 86a–86c), suggesting the generality of the reaction.
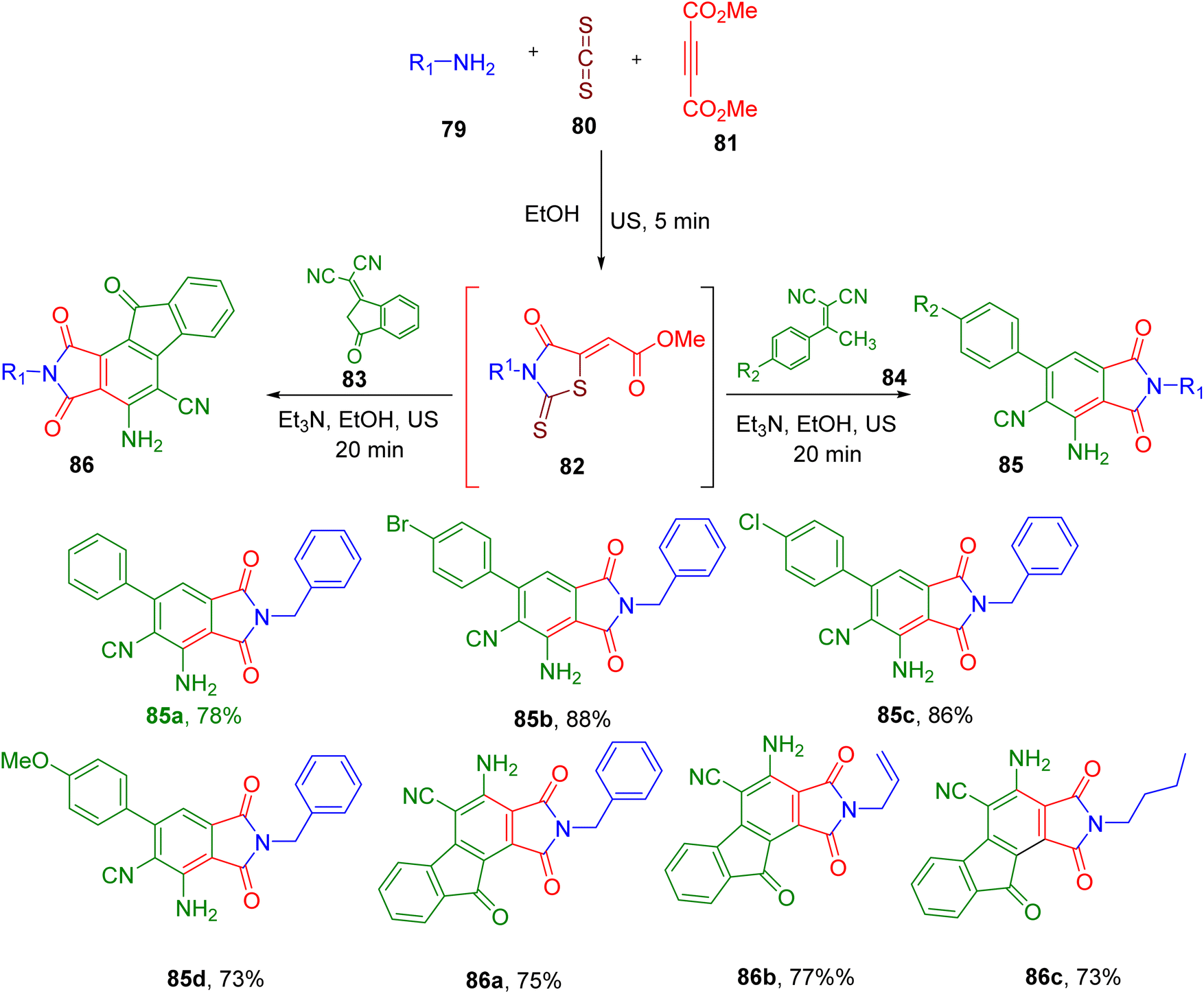 | ||
| Fig. 23 US-mediated MCR for the synthesis of substituted phthalimides via one-pot MCR. | ||
Like podophyllotoxin, 4-azapodophylolotoxins also exhibit potent anticancer activity and excellent tubulin polymerization inhibitory properties.86 Thi et al. reported an efficient synthesis of hybrid molecules by combining 4-aza-podophylolotoxins pharmacophore with aza-anthraquinones. The latter are also known to display cytotoxicity by intercalating DNA. The hybrid molecules were obtained by MCR between 2-hydroxy-1,4-naphthoquinone (87), tetronic acid (88), benzaldehyde derivative (89), and ammonium acetate (90) under MW irradiation (Fig. 24).87 The use of glacial acetic acid as solvent and molecular sieves and heating at 120 °C provided the best yield of the model compound 91a. Various aldehyde derivatives possessing diverse EDGs and EWGs were found to afford the corresponding compounds with good to excellent isolated yields (e.g., 91a–91k). As expected, some of these new compounds also displayed potent cytotoxicity against different cancer cell lines.
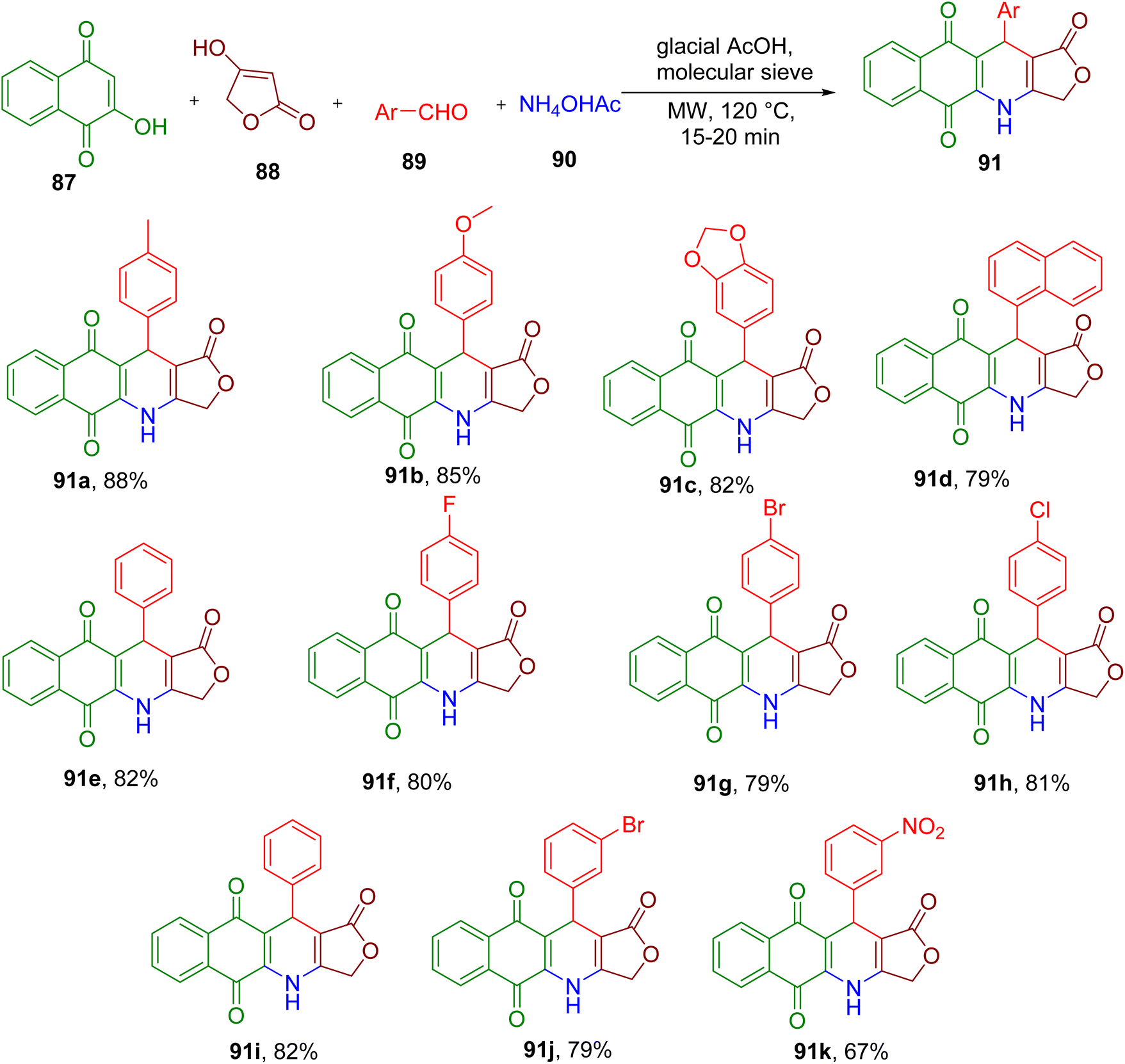 | ||
| Fig. 24 Synthesis of dihydrobenzo[g]furo[3,4-b]quinoline-1,5,10(3H)-triones derivatives. | ||
Aminopyrazoles are important heterocycles in medicinal chemistry and key building blocks for the synthesis of other fused heterocycles.88,89
Annes et al. reported a metal-free synthesis of amino pyrazole derivatives (94, Fig. 25) with a thioether substitution using an MCR between phenylhydrazine (92), aryl thiol (93), and aminocrotononitrile (Fig. 25).90 The model product (95a) obtained a good yield when a solventless mixture of all components was heated in the presence of iodine (1 equiv.).
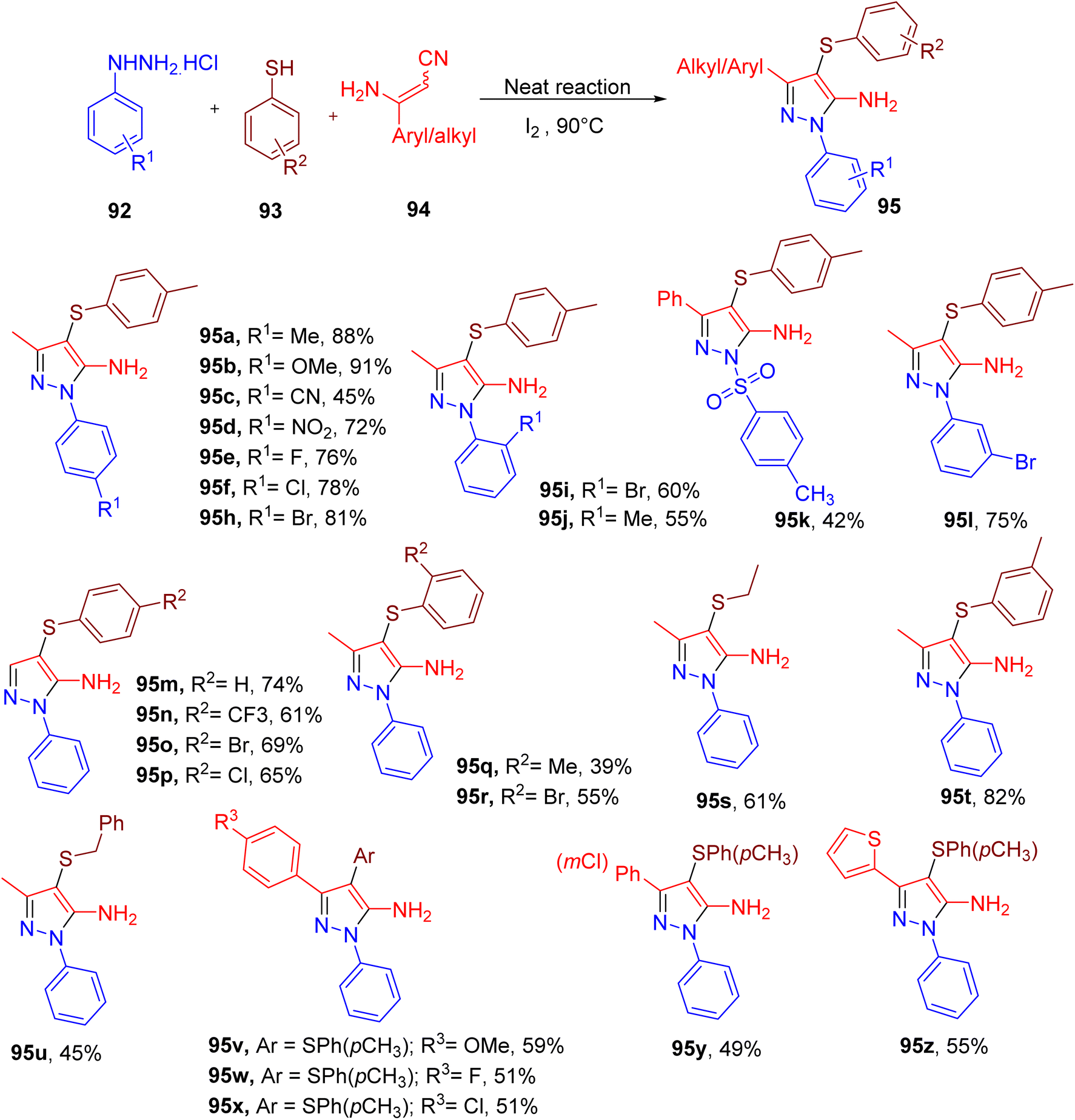 | ||
| Fig. 25 Synthesis of amino pyrazole thioether derivatives via solvent-free cascade reaction. | ||
A variety of phenylhydrazines and thiols were used to afford corresponding pyrazole products (95a–95z). Overall, both phenylhydrazines and thiols showed similarities in terms of the steric and electronic effects. In both cases, EWGs or halide substitutions furnished relatively lower yields of the products (e.g., 95c–95h; 95m–95p) compared to those with EDGs. Also, ortho-substitution afforded lower yields of the corresponding pyrazoles (e.g., 95i and 95j), potentially due to the steric effect. Interestingly, in the case of tosyl hydrazide, the tosyl group remained intact in the corresponding product (95k), albeit with a lower yield. The benzyl and aliphatic thiols (e.g., 95s and 95u) reacted less efficiently than the phenylthiols under the optimized conditions. The scope of β-enaminonitriles was found to be lower as replacing aminocrotononitrile with different aryl-substituted aminonitrile furnished the products in lower yields (e.g. 95v–95z).
Overall, a diverse range of aminopyrazoles could be obtained in this one-pot MCR. The end products with halide substituents are particularly useful for the potential of further modifications.
Pyrazole ring is commonly observed in medicinally relevant molecules and other areas of chemical research.91–93 Building on their earlier work,94 Barroso et al. developed a three-component reaction between alkyne (96), hydrazone (97), and N-heterocycle (98) to obtain multi-substituted pyrazole scaffold (99) (Fig. 26).95 The highest yield of the model compound (99a) was attained by heating the building blocks in ACN in the presence of excess potassium carbonate (K2CO3) (5 equiv.).
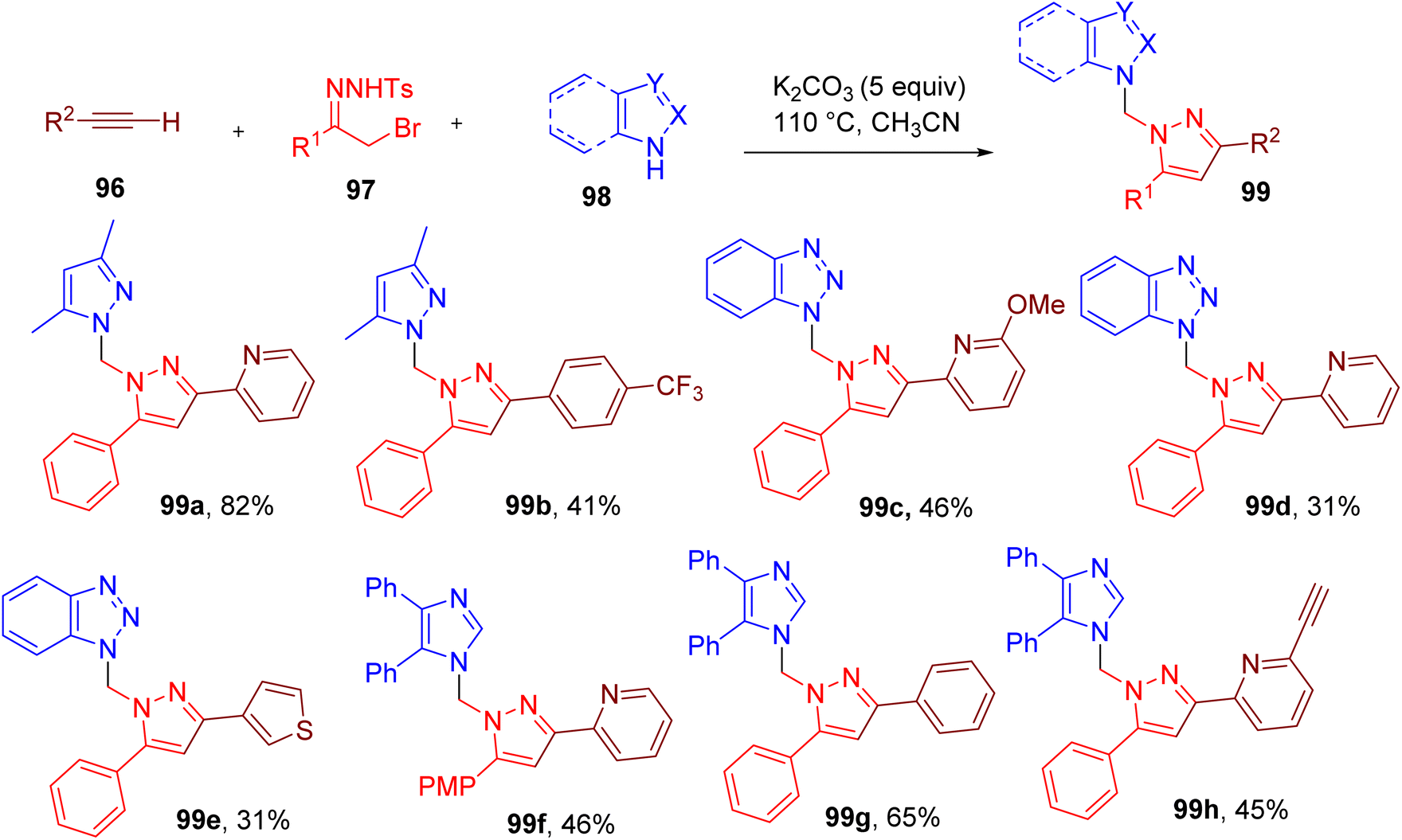 | ||
| Fig. 26 MCR approach for the synthesis of the trisubstituted pyrazoles. | ||
A variety of terminal alkynes attached to aryl and heteroaryl rings in combination with nucleophilic N-heterocycles such as benzotriazole and imidazole were used to furnish diverse trisubstituted pyrazole products in good to moderate yield (99b–99h).
The authors further designed a two-step, four-component methodology for this MCR where N-tosyl hydrazones were obtained from α-bromoketones and tosylhydrazide in the first step, followed by the addition of alkyne and azole components. This process resulted in the simplification of the MCR, thus improving its potential in medicinal chemistry.
The pyridine ring, is an important fragment in drugs, catalyst ligands, and other molecules.27,96 However, metal-free, economical, and environmentally friendly methods to construct 2-substituted pyridines are lacking.97–99
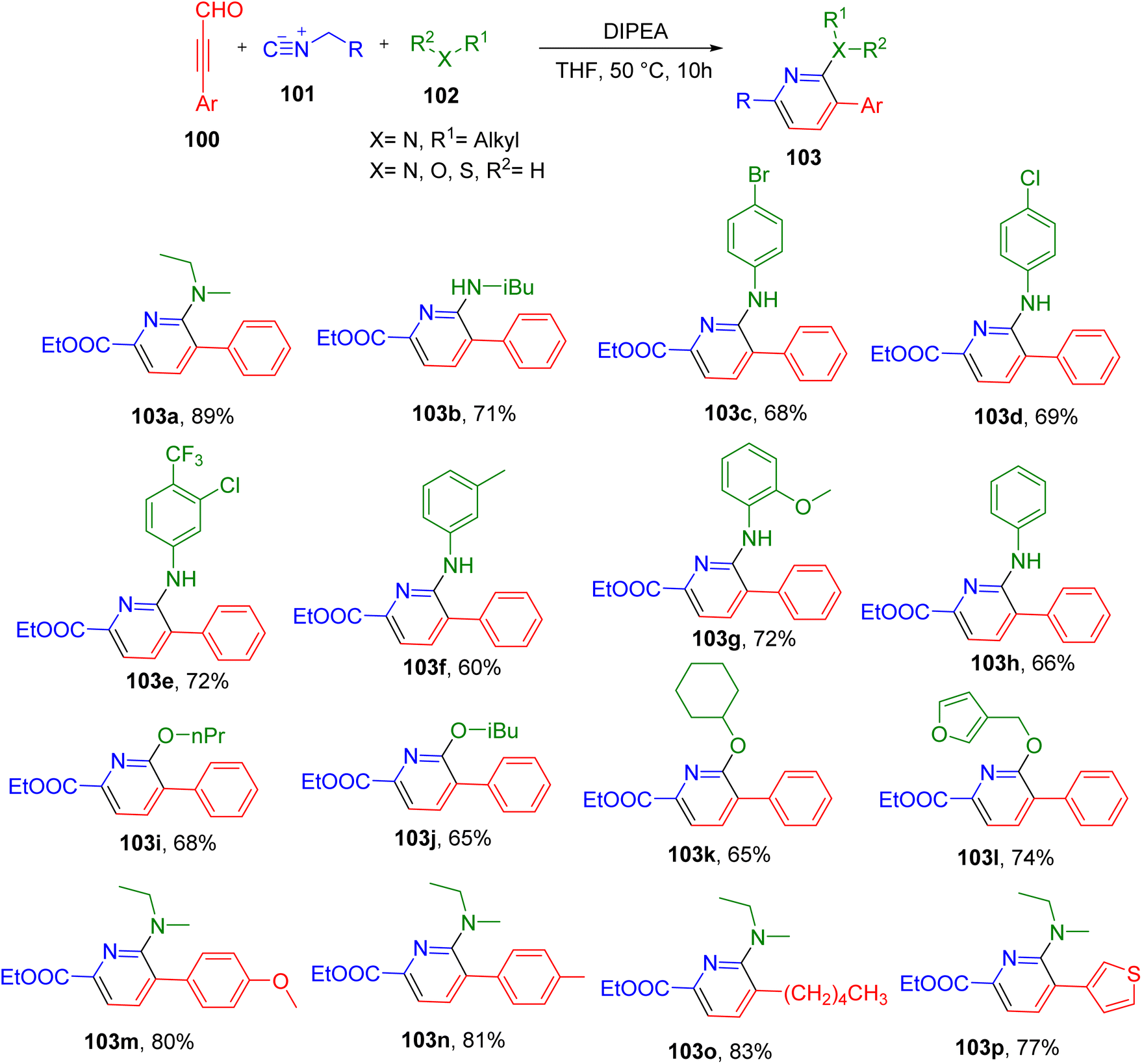
Zhu et al. reported a MCR between ynals (100), isocyanides (101), and amines/alcohols (102) for the synthesis of pyridine derivatives under mild conditions (Fig. 27).100 The optimum reaction conditions included the use of THF solvent and diisopropylethylamine (DIPEA) base at 50 °C (Fig. 27). Under these conditions secondary and primary amines afforded the targeted pyridine derivatives in good yields (e.g., 103a and 103b). The aniline derivatives with different EDGs and EWGs also resulted in the formation of corresponding desired products with good to moderate yields (e.g., 103c–103h). The use of diverse alcohols in place of an amine also yielded the corresponding 2-O-substituted pyridine derivatives (e.g., 103i–103l), showing the broad application of this MCR.
 | ||
| Fig. 27 MCR for the synthesis of tri-substituted pyridines. | ||
Although most of the analogues were obtained from phenylpropiolaldehyde as a ynal component, the substitution of methoxy and methyl groups was also tolerated on the phenyl ring (e.g., 103m and 103n). The reaction also progressed efficiently with oct-2-ynal (103o) and 3-(thiophen-3-yl)propionaldehyde and (103p). In the case of isocyanides, the substrate scope was found to be limited as moving away from the model substrate ethyl isocyanoacetate did not yield the corresponding pyridines.
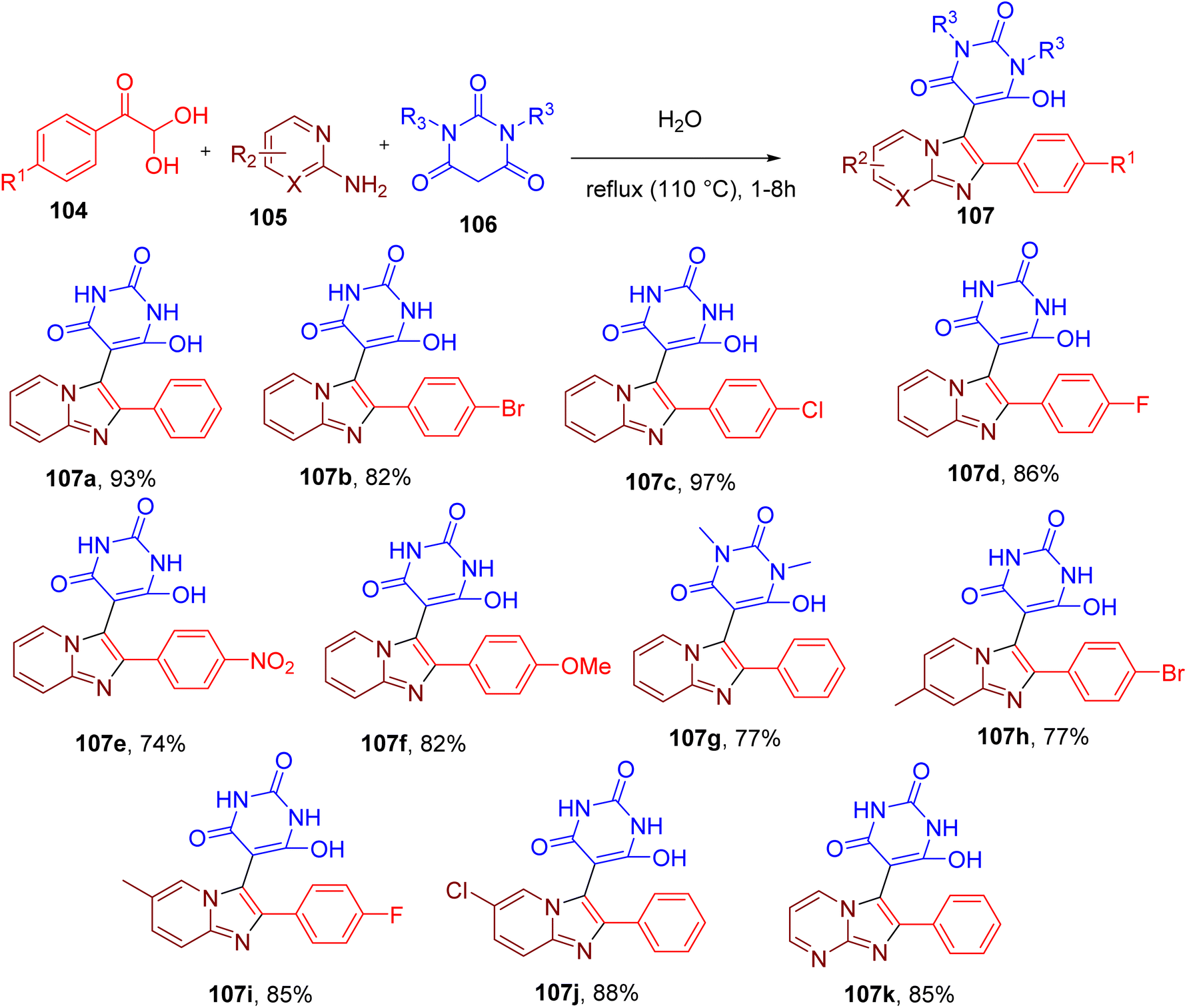
The imidazolopyridine scaffold is known to possess important therapeutic properties.101–105 Previously reported methodologies for the construction of this scaffold involve either multistep synthesis or the use of metal catalysts and hazardous organic solvents.101,102,104 Brahmachari et al. reported a reaction between equimolar quantities of phenylglyoxal (104), 2-aminopyridine (105), and barbituric acid (106) to obtain imidazolopyridine functionalized at C-3 with pyrimidine ring (107, Fig. 28).106 Water at reflux condition was found to be the best solvent for this MCR during reaction optimization studies, yielding 93% of the model compound (107a). These catalyst-free conditions also worked efficiently with other phenylglyoxals substituted with halides, methoxy, and nitro groups, providing the target molecules with good yields (e.g., 107b–107f). Replacing barbituric acid with N,N-dimethyl barbituric acid also produced the desired products (e.g., 107g) in good yields. Similarly, substitutions on the aminopyridine component, such as methyl and halides, were also tolerated (e.g., 107h–107j). Replacing aminopyridine with 2-aminopyrimidine also afforded the corresponding products (e.g., 107k) in good yields suggesting the broad scope of this MCR.
 | ||
| Fig. 28 MCR for the synthesis of substituted imidazolopyridines. | ||
All the products precipitated from the reaction mixture and could be obtained in high purity upon filtration. Overall, this MCR represents a facile catalyst-free green methodology with a high atom economy and very low E-factor.
3 Oxygen heterocycles
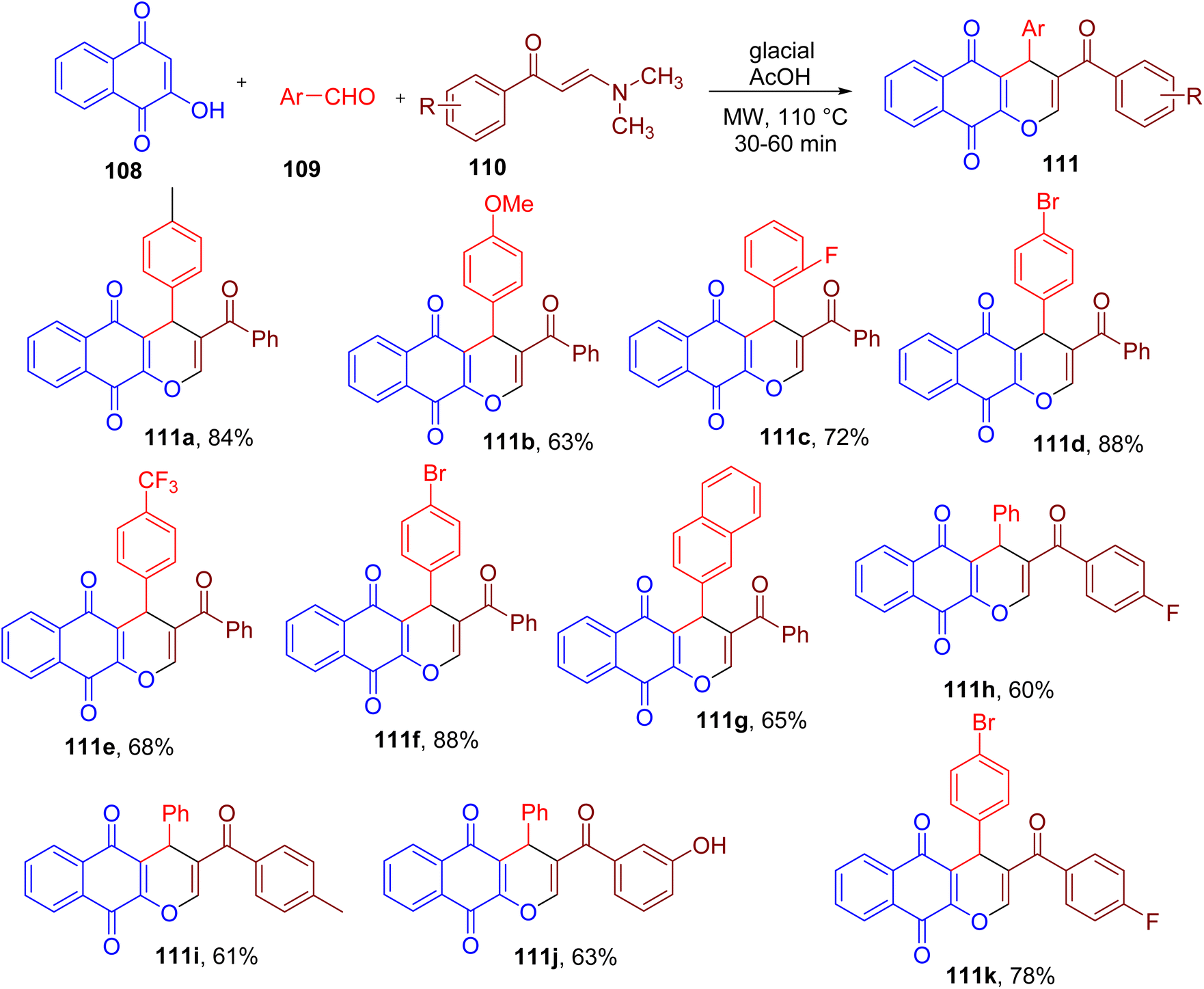
A variety of oxygen heterocycles play an important role in medicinal chemistry.107 For instance, pyranonaphthoquinone scaffold exhibits a variety of biological activities and constitutes several natural products.108,109Nguyen and coworkers reported a one-pot synthesis of novel 3-benzoyl-4H-benzo[g]chromene-5,10-diones (111) from 2-hydroxy-1,4-naphthoquinone (108), aromatic aldehydes (109) and arylenaminones (110) under MW irradiation (Fig. 29).110 Among various reaction conditions, the use of glacial acetic acid as a solvent was found to result in the highest yield of the model compound 111a. The optimized methodology yielded products with aromatic aldehydes and arylenaminones substituted with different EDGs and EWGs in moderate to high yields (e.g., 111a–111k).
 | ||
| Fig. 29 MCR for the synthesis of 3-benzoyl-4H-benzo[g]chromene-5,10-diones. | ||
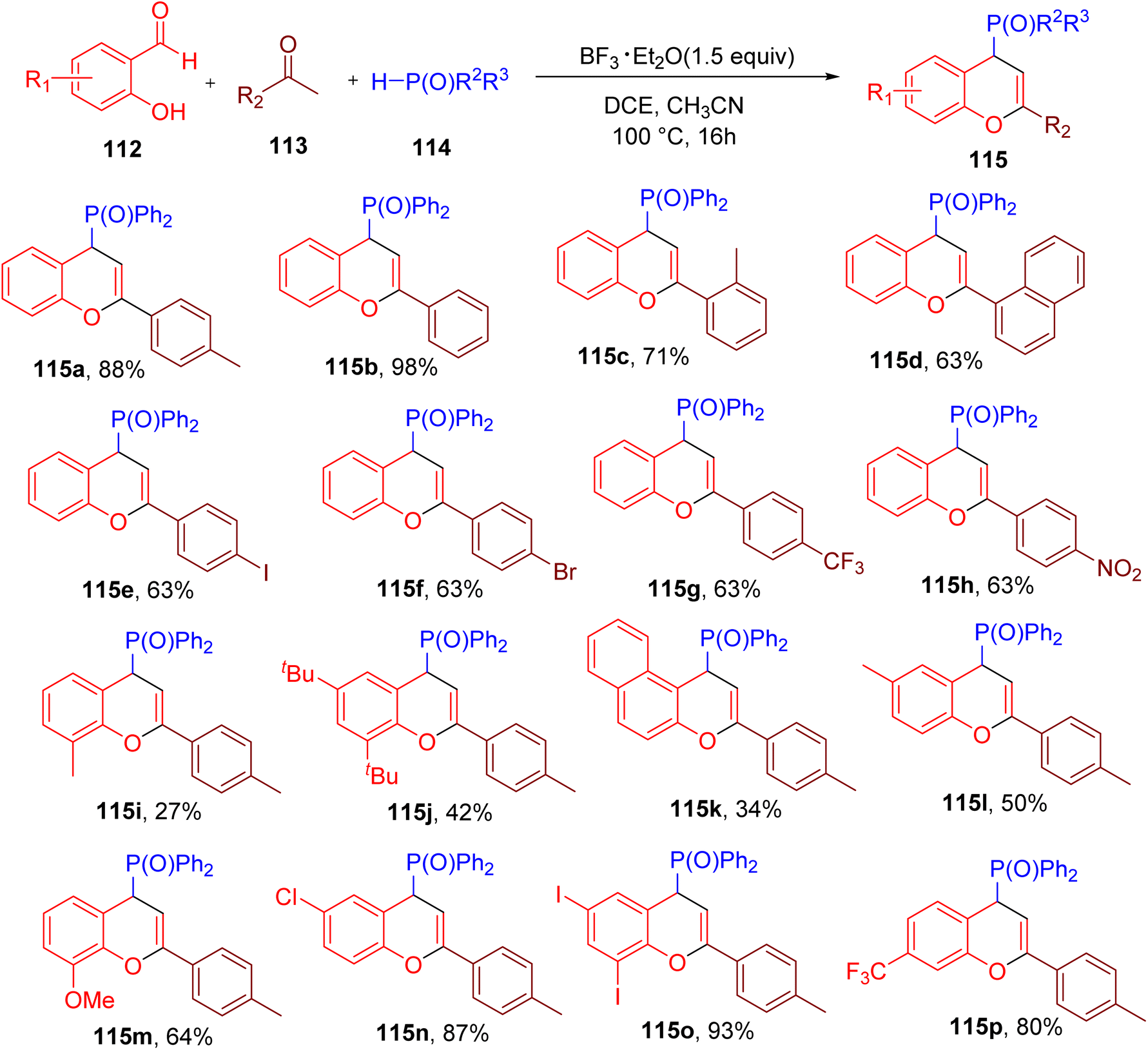
Building on their previous work,111 Su and colleagues reported the MCR-based synthesis of 4-phosphorylated 4H-chromenes (115) from 2-hydroxy benzaldehyde (112), ketone (113), and a diphenylphosphine oxide (114, Fig. 30).112
 | ||
| Fig. 30 MCR approach for the synthesis of 4H-phosphorylated-4H-chromenes. | ||
The optimization of reaction conditions with model substrates revealed that among various tested acids, boron trifluoride etherate (BF3·Et2O) in a mixture of solvents dichloroethane (DCE) and ACN afforded the best yield of 115a. Substrate scope studies showed that a variety of ketones with diverse EWGs and EDGs were tolerated under the optimized conditions (e.g., 115b–115h). However, certain ketones with ortho and meta substituents and alkyl ketones afforded lower yields of the chromene products (e.g., 115c and 115d). In general, various substitutions on 2-hydroxy benzaldehydes were also compatible under the optimized conditions (e.g. 115i–115p). However, substrates with substituents ortho to the hydroxy group reacted less efficiently (e.g., 115i and 115j).
Huang et al. achieved the synthesis of polysubstituted dihydronaphthofuran motif113 with a quaternary carbon observed in some medicinally relevant natural products.114 The convenient synthesis of dihydronaphthofuran (119) was achieved via one-pot MCR between readily available components, aromatic amine (116), α-hydroxyl ketone (117), and naphthol (118, Fig. 31).113 The best yield and diastereoselectivity of the model compound 119a were achieved using chloroform as solvent and benzoic acid as the catalyst.
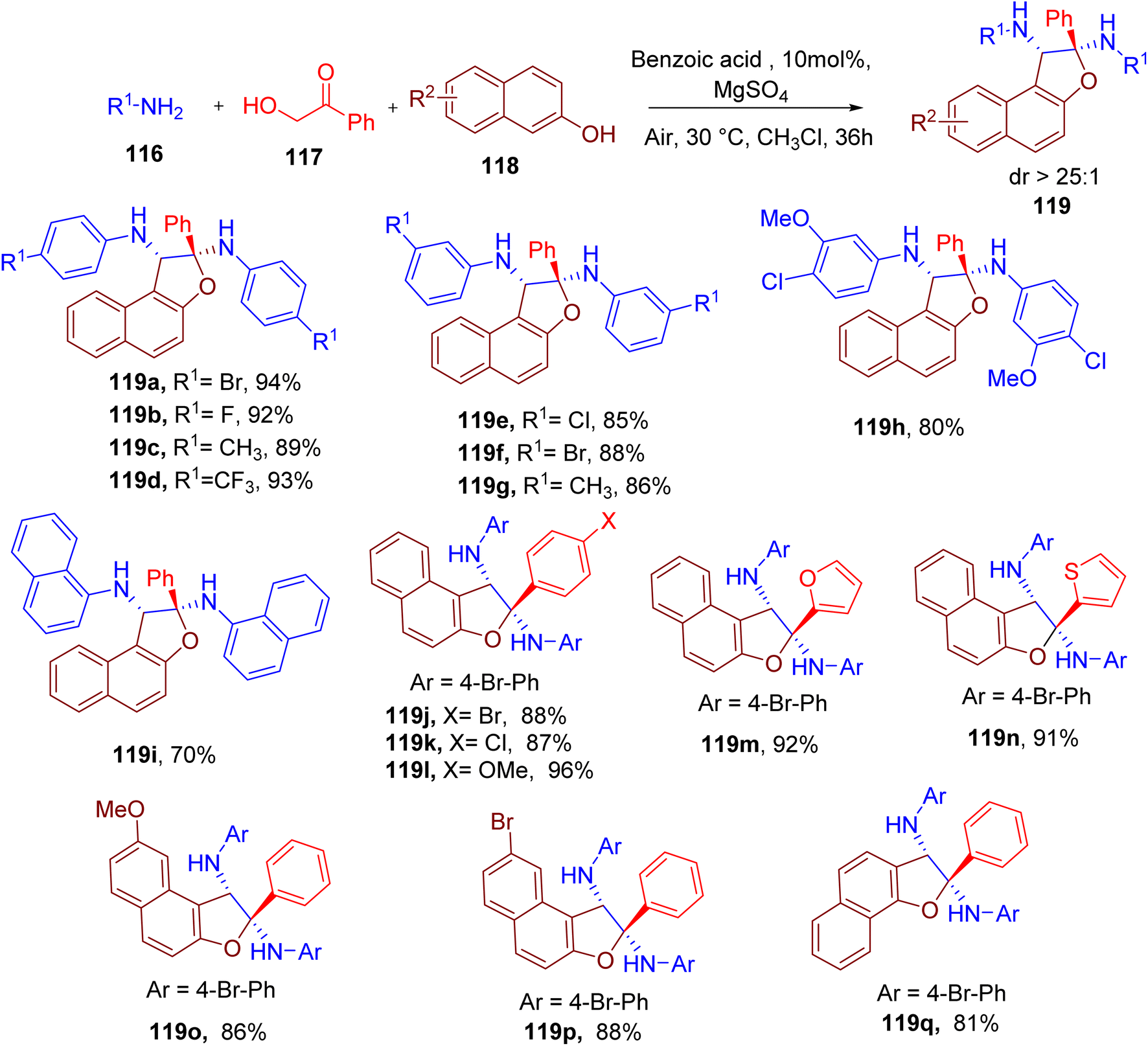 | ||
| Fig. 31 MCR for the synthesis of poly-substituted dihydronaphthofurans. | ||
Aryl amines with single or multiple EWGs and EDGs at the para or meta position (e.g., 119a–119h) were tolerated in the reaction but not at the ortho position due to the steric hindrance. Nonetheless, bulky naphthylamine afforded the corresponding product 119i in good yield. However, the use of alkylamines instead of arylamines in this MCR was not successful. The reaction also worked efficiently with other α-hydroxy ketones, including those bearing a heterocyclic ring (e.g., 119j–119n). A few other naphthols, including α-naphthol, were also found to be compatible under optimized conditions yielding corresponding products (e.g., 119o–119q). The reaction was also scaled to the gram scale, yielding 119a with high diastereoselectivity, which was confirmed by the single-crystal X-ray structure.
4 Sulphur heterocycles
Thiazole ring is present in diverse natural products, drugs, dyes, chemosensing agents, and organic semiconductors.115–122 Recently, Banerjee et al. reported an interesting MCR approach to construct pyrazole-linked thiazole derivatives.123In this straightforward synthesis, an equimolar mixture of model substrates, phenylglyoxal monohydrate (120), thiobenzamide (121), and 3-methyl-1-phenyl-1H-pyrazol-5(4H)-one (122), was stirred to obtain the thiazole derivatives substituted with the pyrazolyl ring at position 5 (Fig. 32). Among various solvents, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) provided the best yields of the model product 123a. Notably, HFIP is emerging as a valuable highly polar solvent in organic synthesis with a strong tendency to stabilize polar intermediates.124 Moreover, owing to its high recyclability, HFIP is considered as a safe and green solvent.125
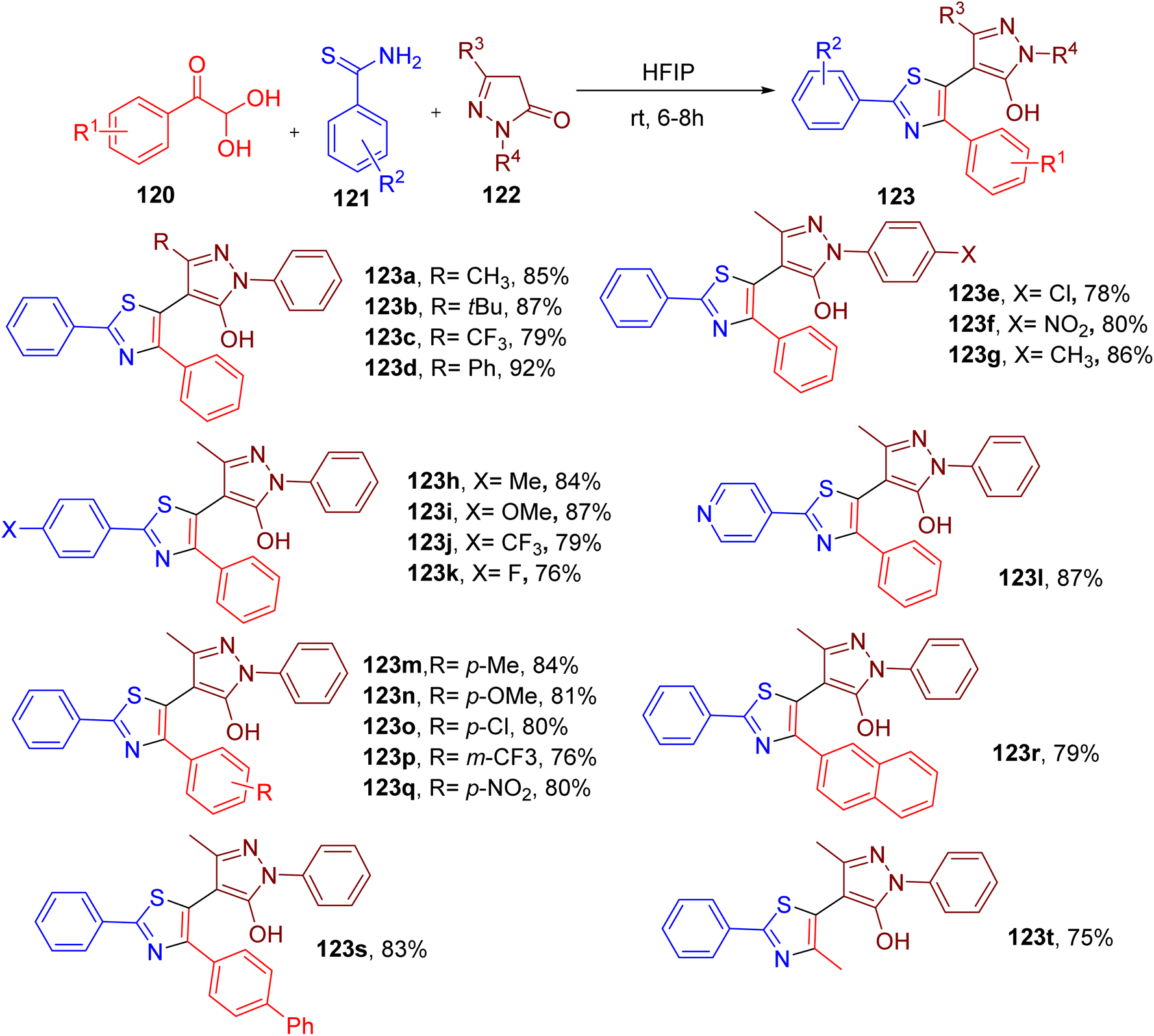 | ||
| Fig. 32 MCR for the synthesis of pyrazole-linked thiazole scaffold. | ||
The substrate scope studies demonstrated that the differently substituted pyrazolone component was well tolerated to yield the desired thiazole products (e.g., 123a–123g). In general, thiobenzamides with different para-substitutions also afforded the resultant pyrazole derivatives in good yield (e.g., 123h–123k). However, a relatively lower yield of the product 123l was obtained with heterocyclic 4-pyridinethioamide. A diverse set of arylglyoxals with variable EDGs and EWGs also reacted smoothly under the reaction conditions to afford the corresponding thiazole compounds in excellent yields (e.g., 123m–123q). The bulky substrates, such as naphthyl glyoxal (e.g., 137r) and biphenyl glyoxal (e.g., 123s), and smaller-sized methyl glyoxal (e.g., 123t) also reacted effectively in the MCR to yield the targeted thiazole analogues.
Notably, like thiazole, the pyrazolone ring is also an important heterocycle in medicinal chemistry.126,127 Thus, the linking of two pharmacophores through this mild MCR methodology may be of great importance in the field of drug design.
5 Carbocyclic rings
Indanone ring is an important structural feature of numerous bioactive compounds,128,129 and thus, methodologies for developing substituted indazoles are of great interest. Hazra and coworkers developed a three-component MCR for the synthesis of 3-aryl-1-indanones, employing benzaldehydes, silyl enolates, and an arene as a nucleophile (Fig. 33).130 This two-step single-pot strategy avoided the use of metal catalysts employed in previous approaches of indanone synthesis.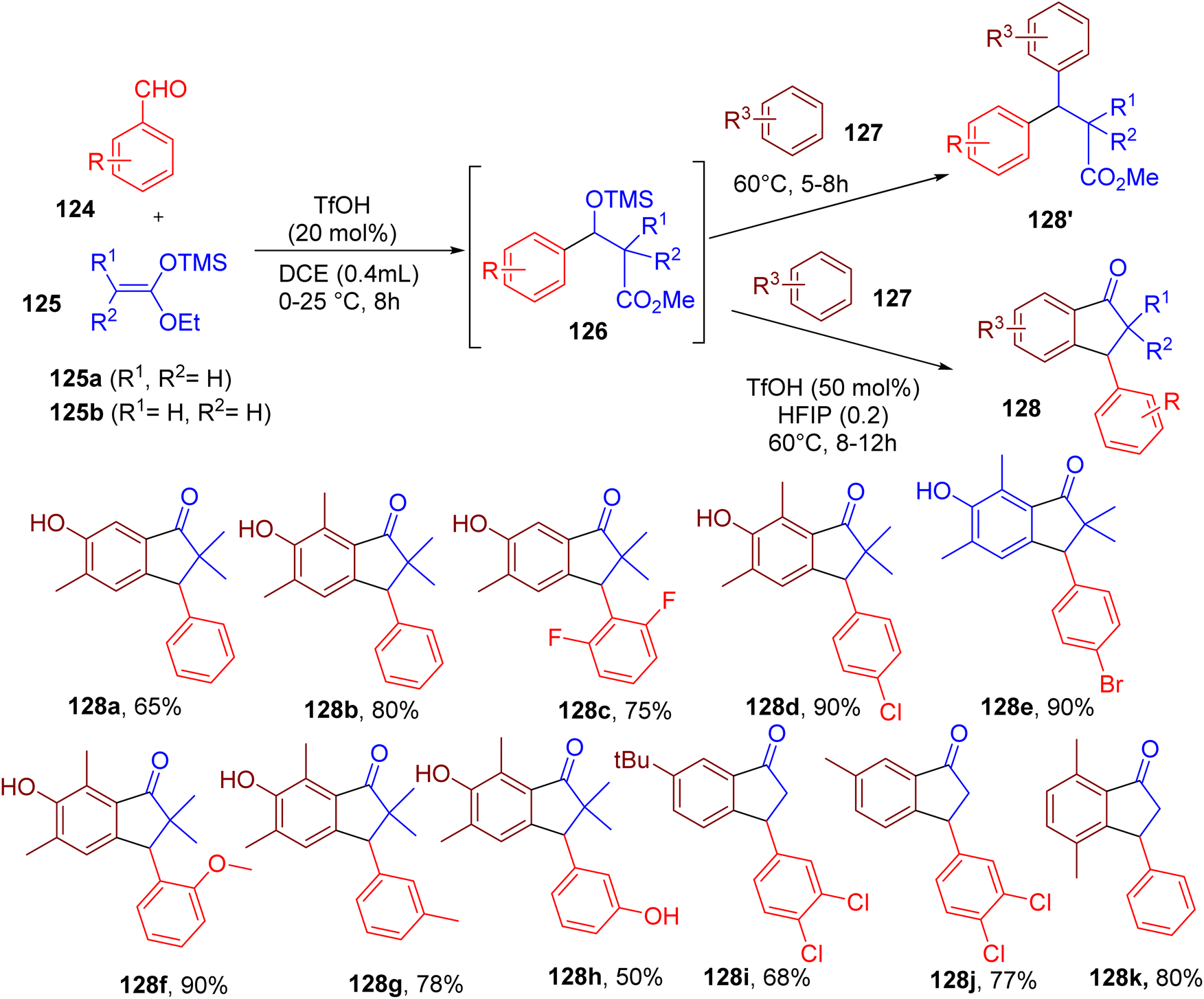 | ||
| Fig. 33 MCR for the one-pot synthesis of 3-aryl-1-indanones. | ||
In the first step, a benzaldehyde derivative (124) and dimethylketene methyl trimethylsilyl acetal (125) was reacted via Mukaiyama aldol reaction to produce the beta-O-silyl ether (126) at room temperature using trimethylsilyl trifluoromethanesulfonate (TMS-OTf) (20 mol%) as a catalyst. The intermediate (126) was used in the second step to generate a benzylic carbocation, which is captured by a nucleophilic arene (127), resulting in a variety of β,β-diaryl esters (128′). The authors later developed a one-pot process to convert 128′ into different indanones (128) through Friedle–Crafts acylation using a combination of triflic acid (TfOH) and HFIP. The reaction worked well when different strong and weak arene nucleophiles were reacted with diverse aldehydes (e.g., 128a–128k). The utility of this MCR strategy was shown by synthesizing an antidepressant drug, indatraline, and a muscle relaxant drug, tolterodine.
Tallarida et al. reported MCR to obtain spirocyclopropyl oxindole scaffold synthesis, which is quite challenging due to the ring strain. In this reaction, a new highly substituted cyclopropyl ring is formed, which is fused to position 3 of the oxindole building block (Fig. 34).131
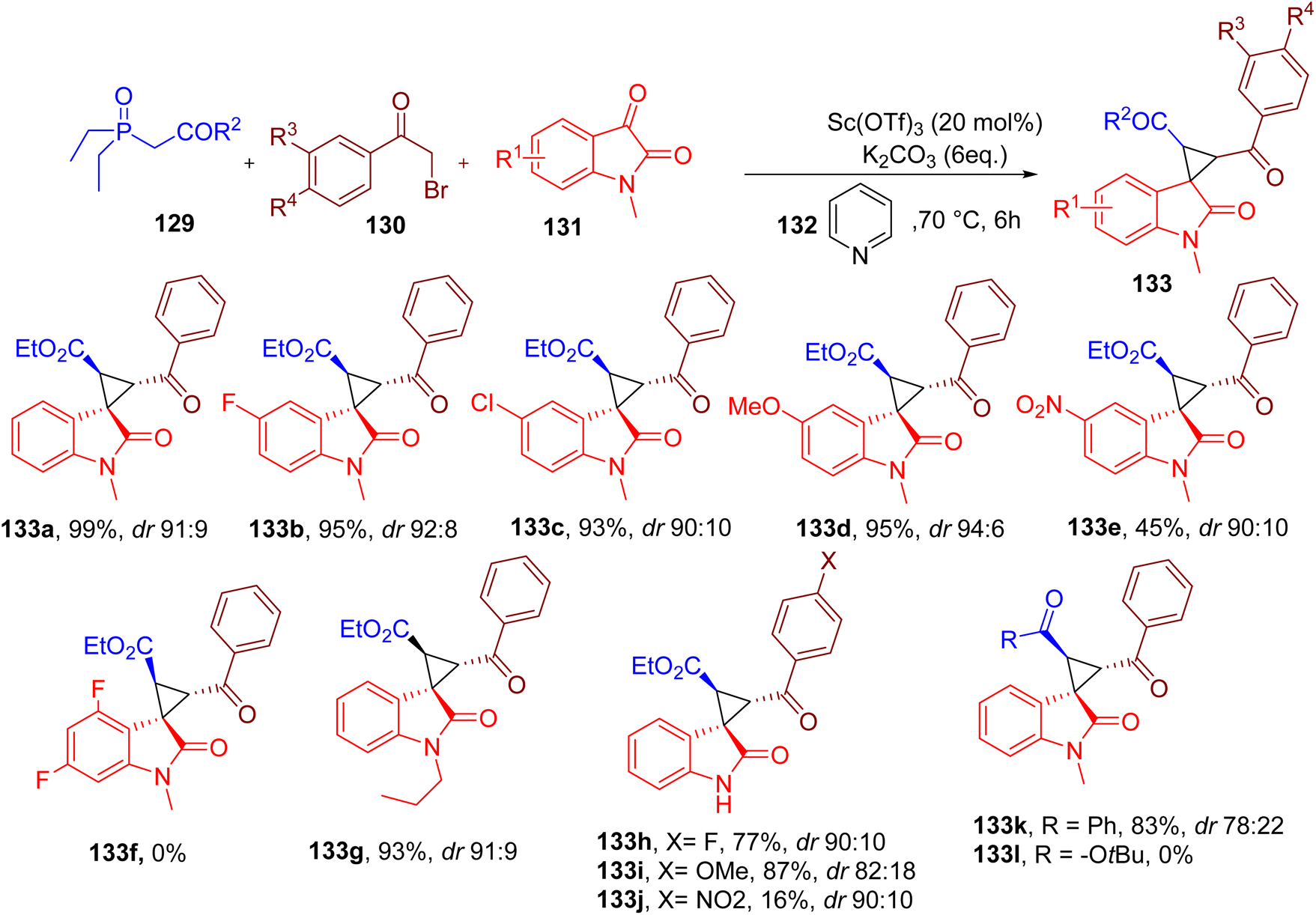 | ||
| Fig. 34 MCR for the synthesis of spirocyclopropyl oxindoles. | ||
The reaction involves triethyl phosphono-acetate (129), 2-bromoacetophenone (130), N-methylisatin (131), and pyridine (132) components to yield a multi-substituted cyclopropyl scaffold (133). Dry pyridine was used as both a reagent and solvent, and it was proposed to form pyridinium ylide, thus playing a key role in ring formation.
The four components were heated in the presence of potassium carbonate and a rare earth metal (REM) salt as a Lewis acid to obtain a single diastereomer as the major product. The best result for the model product 133a was obtained with scandium trifluoromethanesulfonate [Sc(OTf)3] as REM catalyst in pyridine as a solvent, although DMF and ACN also gave satisfactory results.
A variety of commercially available isatins with different EDGs and halides on the benzene afforded excellent yields under the optimized reaction conditions with high diastereoselectivity (e.g. 133a–133d). However, electron-deficient isatins with a nitro group (133e) or two fluorine substituents (133f) reacted less efficiently. Isatins with longer N-alkyl chains (133g) also afforded the corresponding spirocyclopropyl oxindole products in good yields.
A diverse set of substituents at the aromatic ring of 2-bromoacetophenone were also compatible in the MCR, generating the target products with high yields (e.g., 133h and 133i). However, the nitro-substituted bromoacetophenone reacted inefficiently, with low yields of the corresponding product 133j.
Two other phosphonate derivatives were also screened in the reaction, one of which worked well with the phenyl substituent (133k). In contrast, phosphonate with tert-butyl group (133l) did not yield the corresponding product, presumably due to the steric hindrance offered by the bulky tert-butyl group. One of the major disadvantages of this MCR is the requirement of strictly anhydrous conditions for the metal catalyst to furnish high yields and diastereoselectivity.
6 Rings with two different heteroatoms
Benzoxazole heterocycle is an important structural motif present in diverse natural and synthetic molecules with multiple applications.132–135 Several synthetic strategies are available to access 2-substituted benzoxazoles by reacting o-aminophenols with different carbonyl functional groups. However, these methods mostly require harsh conditions and hazardous reagents.136Sharghi et al. recently reported an MCR-based milder synthesis of benzoxazoles from catechols (134), ammonium acetate (135), and aldehydes (136) in the presence of air as an oxidant (Fig. 35).137 Among various catalysts, the use of Fe(III)–salen complex (137) in ethanol at 50 °C afforded the highest yields of the model benzoxazole product 138a. Interestingly, the expected corresponding benzimidazole side product was not obtained under these conditions. Substrate scope studies revealed that the yield of benzoxazoles was not much affected by the nature of the substituents on the aryl aldehydes, all of which afforded final products in excellent yields (e.g., 138a–138f). In addition, sterically hindered 1-naphthaldehyde (138g) and different N, O, and S-containing heteroaryl aldehydes also afforded the benzoxazoles (e.g., 138h–138j) in excellent yields. However, aliphatic aldehydes did not afford the corresponding products in reasonable yields (e.g., 138k). The major limitation of this MCR methodology was the limited number of substitutions tolerated on the catechol component, which were limited to t-butyl or methyl groups (e.g., 138l–138o). A few other catechol derivatives with nitro and halogen substituents were also screened under the optimized MCR conditions, but they did not yield the desired products. Notably, the authors also achieved gram-scale synthesis of 138a with no loss of yield, thus affirming the robustness of the methodology.
 | ||
| Fig. 35 MCR for the synthesis of benzoxazole derivatives. | ||
The thienopyrimidinone ring is known to exhibit antimicrobial, anticancer, and other biological activities.138–141 Generally, the synthesis of this bicyclic heterocycle requires multistep sequential synthesis.139 Shi et al. devised a single-vessel methodology for the synthesis of thieno[2,3-d]pyrimidin-4(3H)-ones (142) involving MCR between a ketone (139), ethyl cyanoacetate (141), formamide (140), and sulfur (S8) (Fig. 36).142 The reaction optimization suggested that the catalytic system consisting of diethylamine and L-proline (20 mol% each) at high temperature provided the best yield of the model thienopyrimidinone derivative 142a. Several ketones with varied EWGs and EDGs on the phenyl ring furnished the corresponding products in high yields (e.g., 142b–142f). The heterocyclic ketones (e.g. 142g–142i), cyclic ketone (142j), phenylacetaldehyde (142k), and phenyl-substituted phenylpropan-2-one (142l) substrates also yielded the desired products under the optimized conditions. The reaction could be scaled to a 100 mmol scale without requiring chromatographic purification, which was the key advantage of this MCR. Due to its catalytic and single-pot procedure, the MCR was found to have an E-factor of 1.5, suggesting it is a green and environmentally friendly methodology.
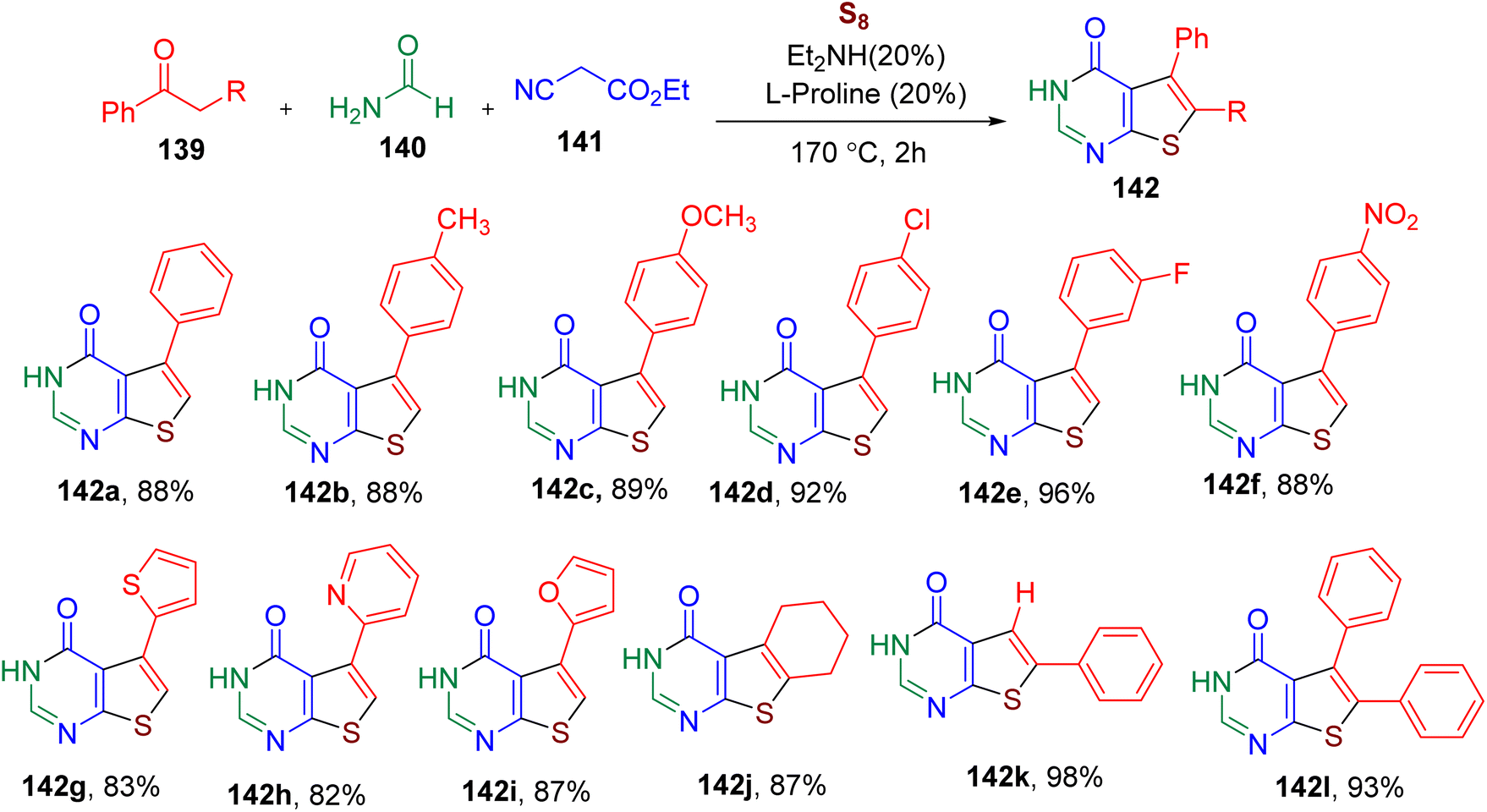 | ||
| Fig. 36 MCR for the synthesis of thieno[2,3-d]pyrimidin-4(3H)-ones. | ||
7 Miscellaneous poly-heterocycles
Polycyclic heterocycles are very often seen in natural products and often pose synthetic challenges. These frameworks impart diversity and complexity to a molecule and, hence are relevant to medicinal chemistry research.143 Therefore, convenient one-pot synthetic methodologies to obtain poly-heterocycles are warranted.Cao et al. reported an interesting MCR between isatin, proline, and alkyl propiolate to construct several new polyheterocyclic systems. A reaction between L-proline (143), N-benzylisatin (144), and alkyl propiolate (145) in chloroform yielded an unprecedented azocino[1,2-a]benzo[c][1,5]diazocines ring system (146a1–146a4, Fig. 37A).144 However, an intractable mixture was obtained when isatins with free –NH or those with N-methyl and N-butyl groups were used in this reaction. Switching to the alcoholic solvent resulted in the products based on the spiro-indoline-3,3′-pyrrolizine scaffold (146b1–146b8, Fig. 37B) thus confirming the mechanism of the reaction involving [3 + 2] cycloaddition of azomethine ylide intermediate with alkyl propiolate. Overall, isatins with different N-substituents were found to react differently in this MCR depending on the reaction conditions, yielding novel ring systems. The chemical structures and stereochemistry of several of these derivatives were confirmed by solving single-crystal X-ray structures.
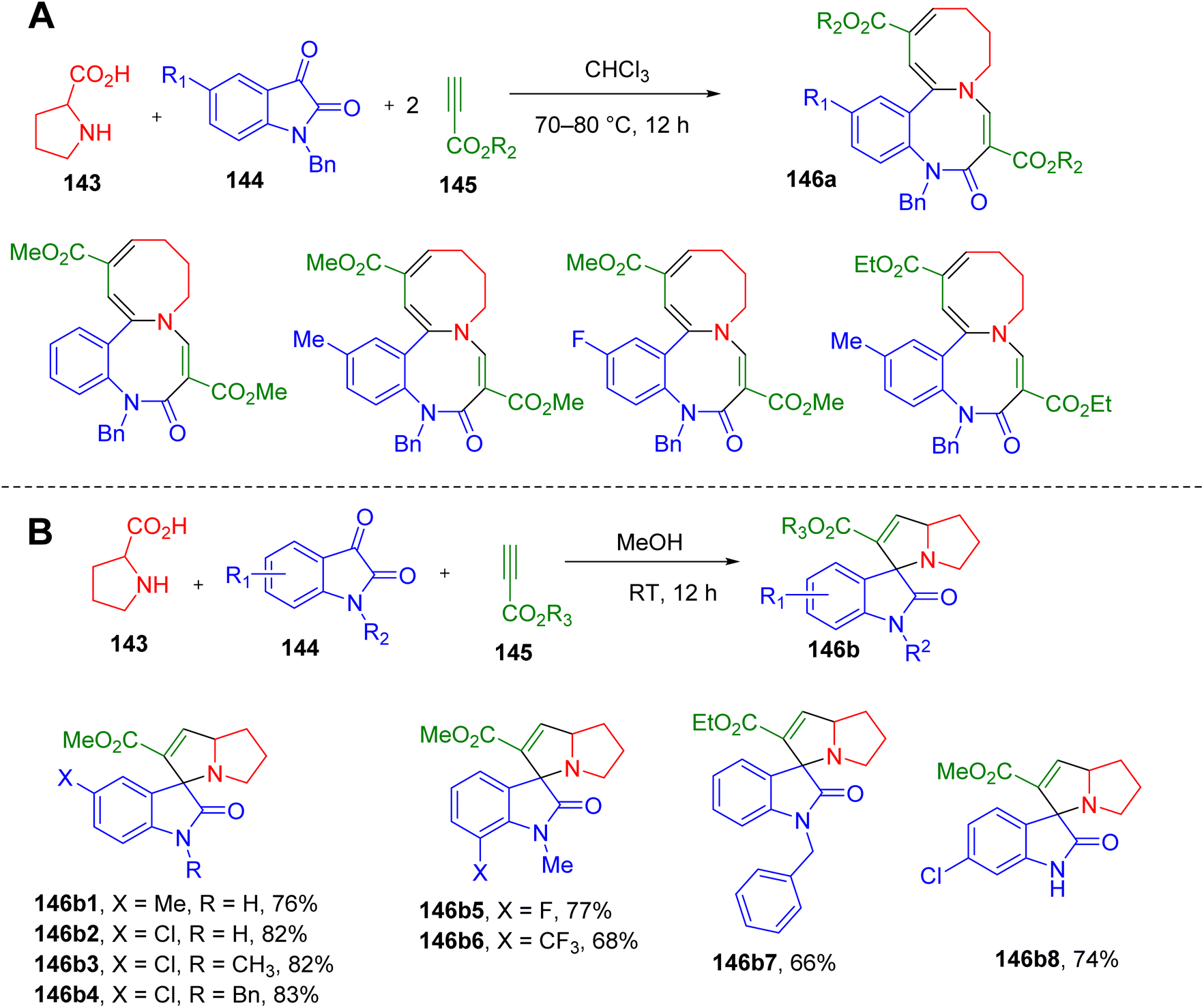 | ||
| Fig. 37 MCR for the synthesis of (A) azocino[1,2-a]benzo[c][1,5]diazocines, and (B) spiro-indoline-3,3′-pyrrolizines. | ||
Zamani et al. reported a three-step single-pot MCR to afford chromeno[3,2-d]oxazole derivatives representing a unique fusion of two heterocycles (Fig. 38). The novel heterocyclic system (151) was obtained from an aldehyde (148), hippuric acid derivative (149), and a cyclic diketone (150) involving a sequence of three steps in single-pot; azalactone formation, Michael addition, and Vilsmeier reactions.145 The optimized conditions included the use of propylene carbonate (PC) solvent at 110 °C and an excess of phosphoryl chloride (POCl3) (1.6 equiv.).
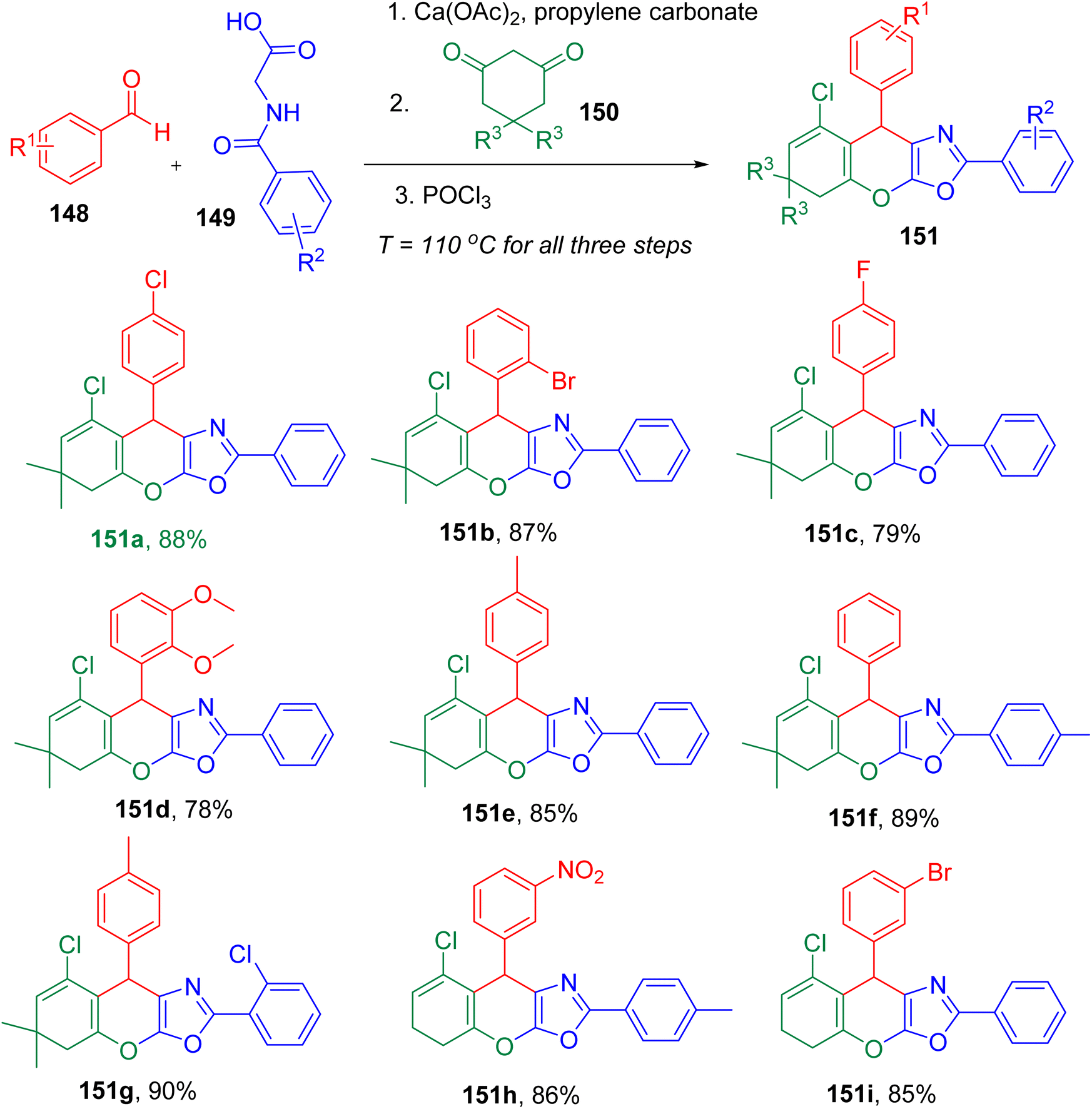 | ||
| Fig. 38 MCR approach for the synthesis of diverse chromeno[3,2-d]oxazoles derivative. | ||
After obtaining the model product (151a) with 88% yield, the authors studied the substrate scope for the optimized conditions. A variety of aldehydes, three different hippuric acid derivatives, and 1,3-cyclohexanedione or dimedone as cyclic ketone were found to furnish corresponding products (e.g., 151a–151h) efficiently. This broad compatibility of substrates suggested no influence of the electronic effects on the reaction yield and established generality of the reaction.
Palanivel and Gnanasambandam reported one-pot MCR for the construction of natural product-like poly-fused heterocycles (Fig. 39).12 Firstly, the authors reported the synthesis of aza-cyclopenta(cd)diindene core (156) from a three-component reaction involving an equimolar mixture of phenylglyoxal (152), benzimidazole acetonitrile (153), malononitrile (154, Fig. 39). The use of ACN solvent and TfOH (2 equiv.) at room temperature was found to furnish the highest yields of the product (156a1). The same product was obtained when cyanoacetamide (155) was used in place of malononitrile under slightly different reaction conditions (Fig. 39). A variety of substituents on the para-position of phenylglyoxal were found to be compatible with providing the desired products with good yields (e.g., 156a1–156a9). Naphthylglyoxal (158a7), heteroarylglyoxals (e.g. 156a8), and methylglyoxals (156a9) also furnished the corresponding products in excellent yields.
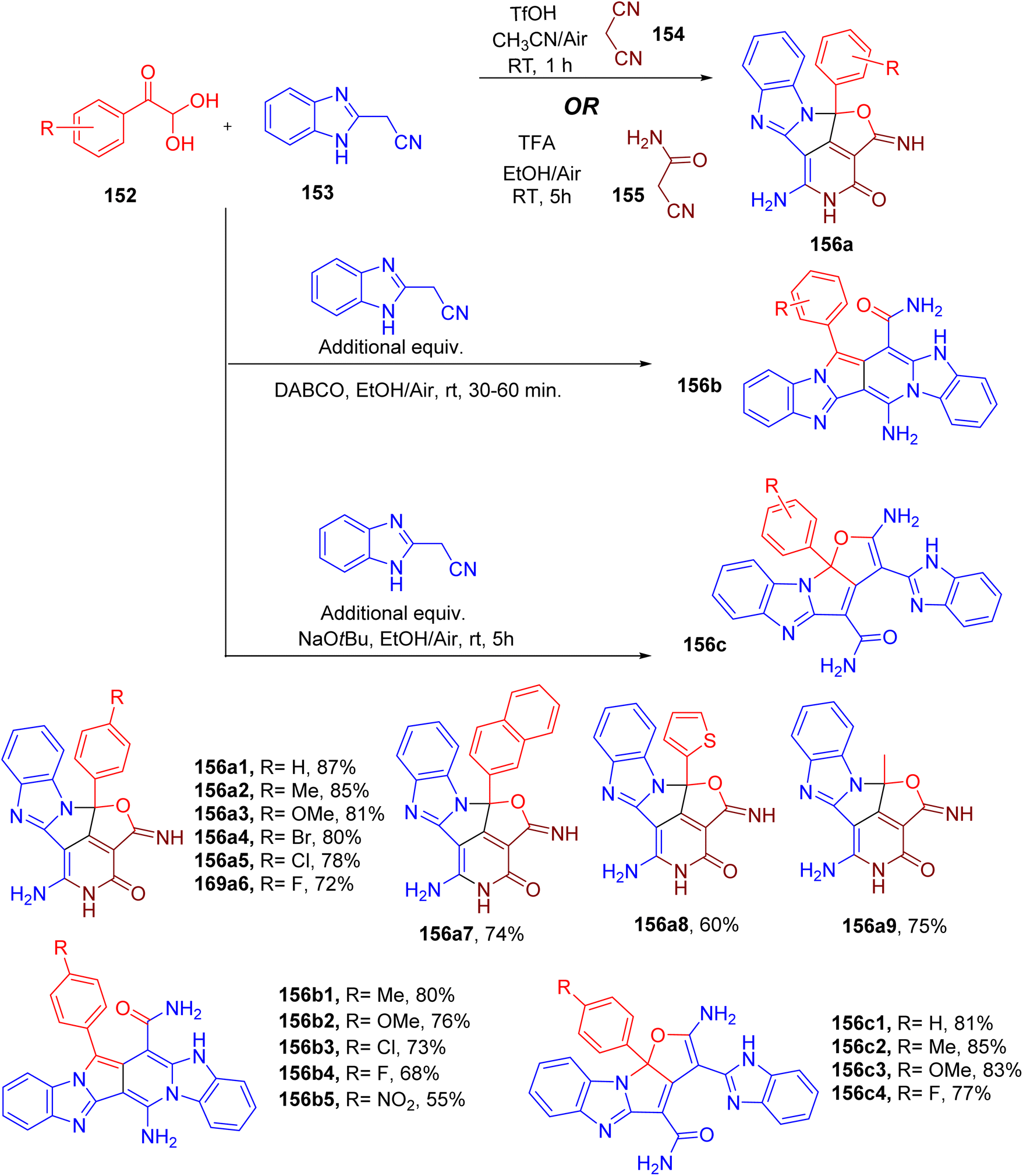 | ||
| Fig. 39 MCR for the synthesis of aza-cyclopenta(cd)diindene (156a), pyrrolo(3,4-d)pyridine-13-carboxamide (156b), and furo-pyrrolo(1,2-a)imidazole-4-carboxamide (156c) scaffolds. | ||
Using an excess of benzimidazole acetonitrile (153, 2 equiv.), however, resulted in the formation of two new polycyclic scaffolds under slightly different conditions (Fig. 39). The optimization of both reactions suggested using 1,4-diazabicyclo[2.2.2]octane (DABCO) base for the synthesis of pyrrolo(3,4-d)pyridine-13-carboxamide derivatives (e.g., 156b1–156b5). On the other hand, sodium tert-butoxide (t-BuONa) base resulted in the formation of pyrrolo(1,2-a)imidazole-4-carboxamides in good yield (e.g., 156c1–156c4, Fig. 39). In both cases, a limited number of substituted phenylglyoxals were exploited to obtain the corresponding target products.
Interestingly, the authors also achieved the synthesis of rare furo(2,3-b)furan derivatives (160) by using methylglyoxal (157), benzothiazole acetonitrile (158), and benzoyl acetonitrile (159) with an additional electrophilic carbonyl centre (Fig. 40). A number of substitutions on benzimidazole acetonitrile and benzoyl acetonitrile were found to be compatible, yielding the corresponding furo(2,3-b)furan end products (e.g. 160a–160h). However, only methylglyoxal seemed to be compatible as the third component in this MCR.
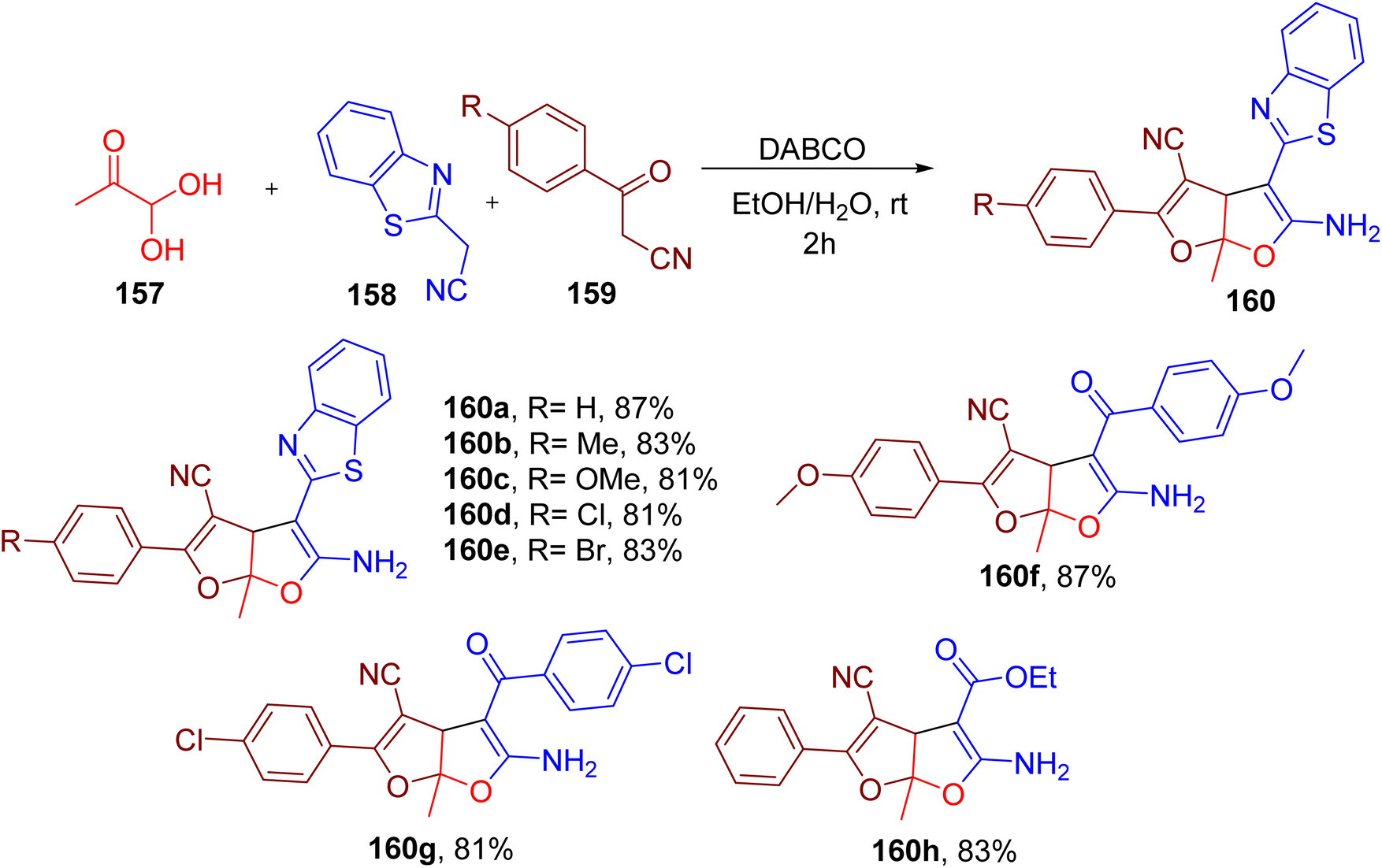 | ||
| Fig. 40 MCR for the synthesis of furo(2,3-b)furan derivatives. | ||
Cao et al. reported146 interesting MCR results in the simultaneous synthesis of triazole and oxazole rings, both of which are important heterocycles in medicinal chemistry.147–149 The optimized reaction is catalyzed by cerium(III) trifluoromethanesulfonate [Ce(OTf)3] in toluene at elevated temperature and involves alkynyl carboxylic acids (161), excess tert-butyl isocyanide (162) and organic azides components (163, Fig. 41).146 The mechanistic studies confirm the participation of three molecules of tert-butyl isocyanide embodying the N-atom of the oxazole ring and substituents at positions 4 and 5. The optimized conditions were found to work well with a broad set of alkynyl carboxylic acids that included a phenyl ring and different EWGs and EDGs substituted-phenyl rings (e.g. 164a–164o). The carboxylic acids with thiophene (e.g., 164h), trimethylsilyl (e.g., 164i), and ethyl groups (e.g., 164l) also reacted efficiently under these conditions. Likewise, various benzyl (e.g., 164j, 164k, 164m, and 164n) and aliphatic azides (e.g. 164o) also afforded the target products in good yields.
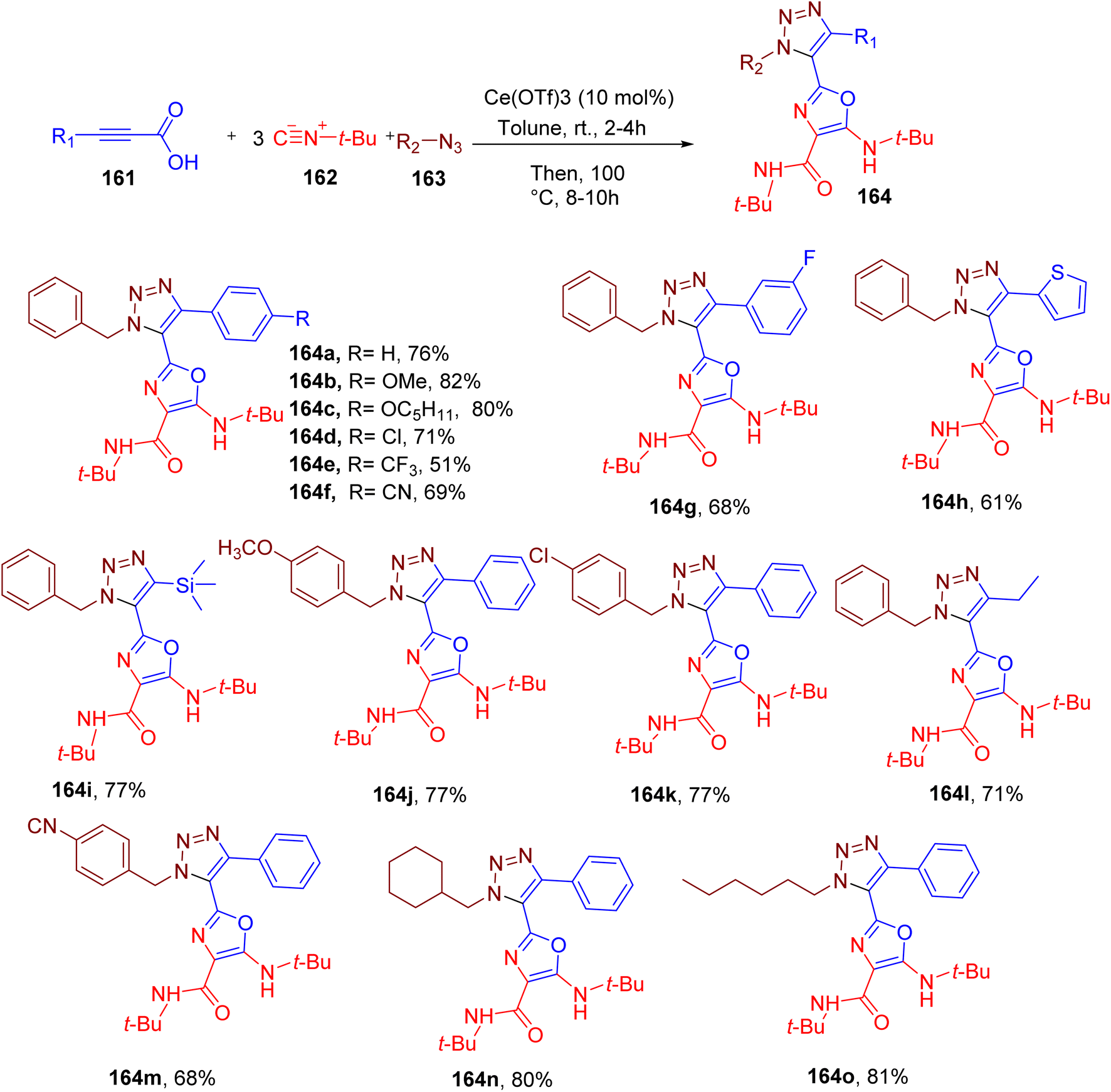 | ||
| Fig. 41 MCR for the simultaneous synthesis of triazole-oxazole-based derivatives. | ||
An important feature of this MCR is its high atom economy and significant chemo and regioselectivity.
8 Ring analysis and comparison with drugs
To evaluate the novelty of the ring systems generated from MCRs, we compared MCR ring space with the rings of approved drugs and clinical candidates. To this end, we selected a set of 41 unique molecules obtained from the MCRs reviewed in previous sections. A library of 5968 drugs and investigational agents (henceforth referred to as ‘drugs’) was obtained from Chemical Database of Bioactive Molecules (ChEMBL) after removing duplicates, metals, and gaseous molecules. The Murcko scaffolds30 of drugs and MCR products were derived and plotted on a similarity chart in Datawarrior. The 2D-Rubber Band Scaling (2D-RBS) algorithm150 is implemented in Datawarrior, which places a compound closer to its similarity neighbourhood and connects the similar molecules above a predefined threshold (80%) with a line. In general, molecules with more similar neighbours tend to occupy the centre of the plot, while those with fewer neighbours show on the edges. The similarity chart demonstrates that most of the MCR scaffolds are not connected to the drug scaffolds and appear at the plot borders, thus indicating the uniqueness of these scaffolds (Fig. 42). | ||
| Fig. 42 Chemical space visualization of Murcko scaffolds derived from MCR products and approved drugs/clinical candidates. The MCR scaffolds are not connected to drug scaffolds, indicating low or no similarity among most of the scaffolds of the two classes. Also, most of the MCR scaffolds have zero neighbours, as depicted by the background colour, which represents the ‘Neighbour Count’. The skelsphere descriptors150 was used for the similarity calculation. | ||
Next, we carried out a direct comparison of Murcko scaffolds and Murcko skeletons of the MCR library and drug library. Eight of the 41 MCR scaffolds had at least one similar scaffolds (≥80% threshold) in the drug library (Table 1), but no identical match was found. Some of the MCR scaffolds shown in Table 1 display interesting similarities and variations with respect to the existing drugs. Therefore, the MCRs corresponding to these eight scaffolds represent an interesting opportunity to generate new analogue libraries of the corresponding drugs in one-pot. Such a comparison between MCR and drug scaffolds may enable the cost-effective discovery of ‘me-too’ drugs.151
| MCR product ID | MCR Murcko scaffold | Corresponding similar drug scaffold (80% skelsphere) | Corresponding drug(s) name |
|---|---|---|---|
| 14a |  |
 |
Frovatriptan |
| 103g | 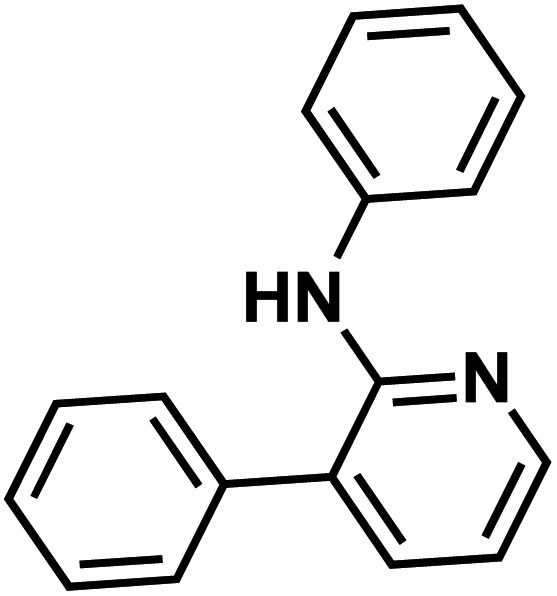 |
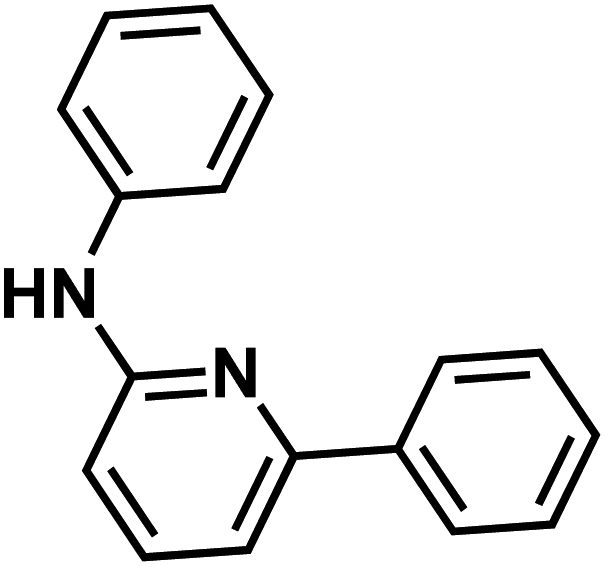 |
VX-702 |
| 44e | 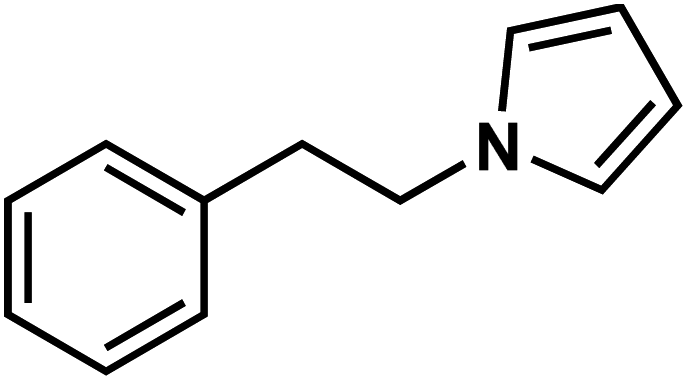 |
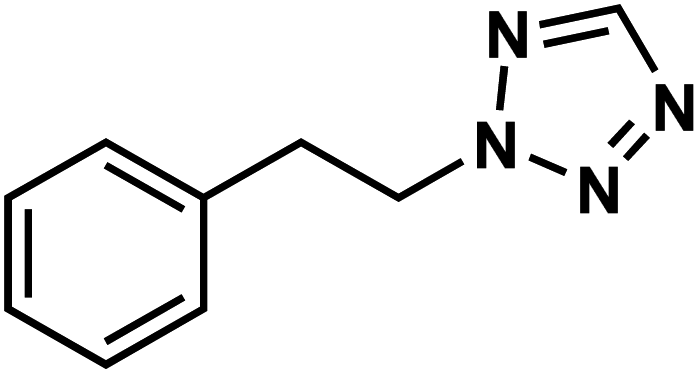 |
Cenobamate |
| 85b | 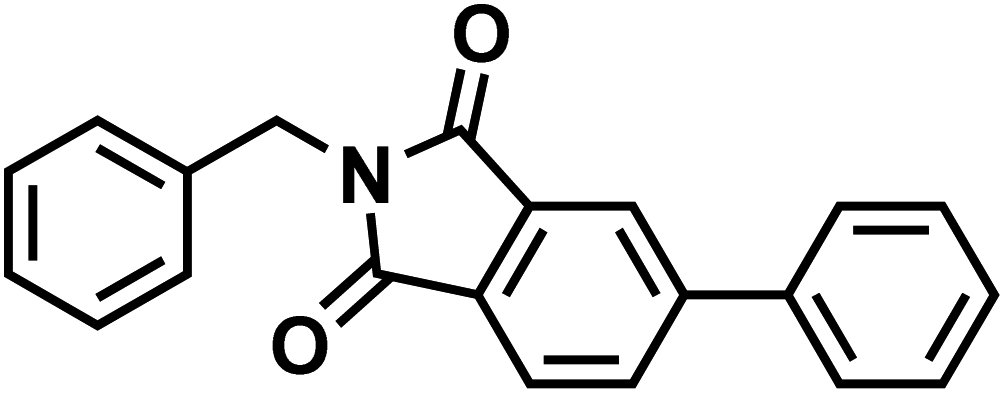 |
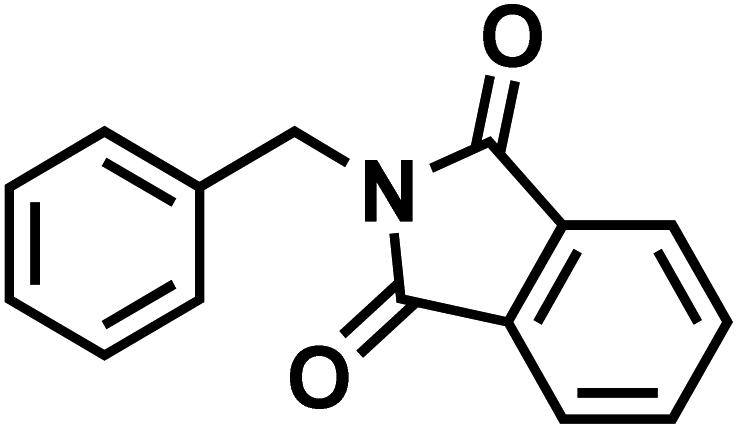 |
CDC-801 |
| Apremilast | |||
| 26l | 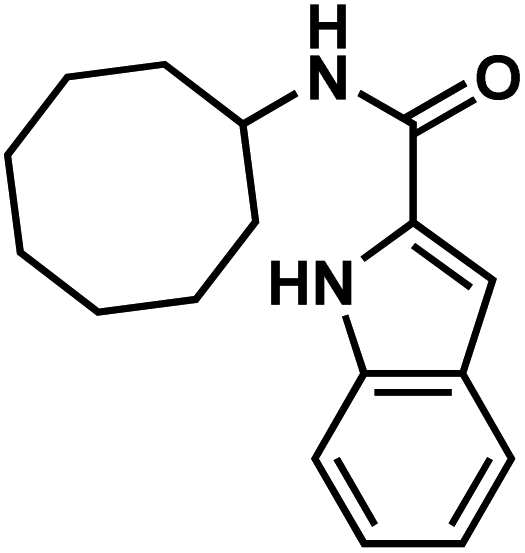 |
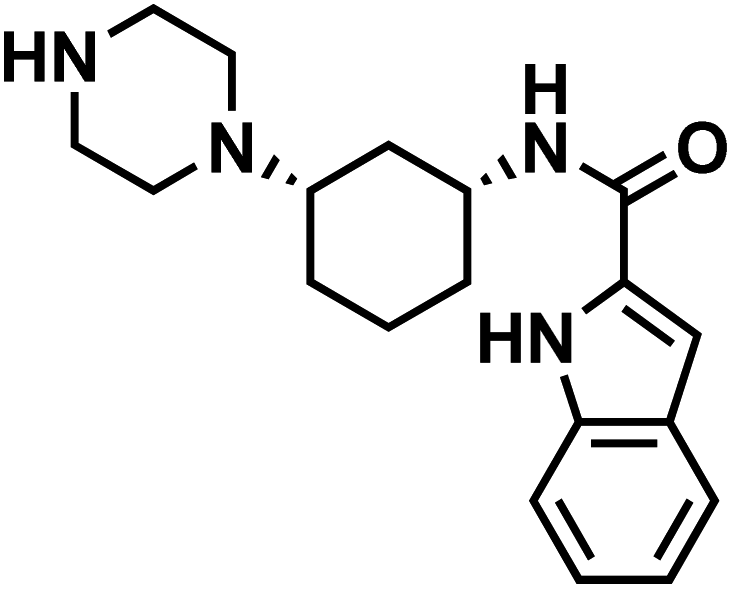 |
EZM-0414 |
| 138g | 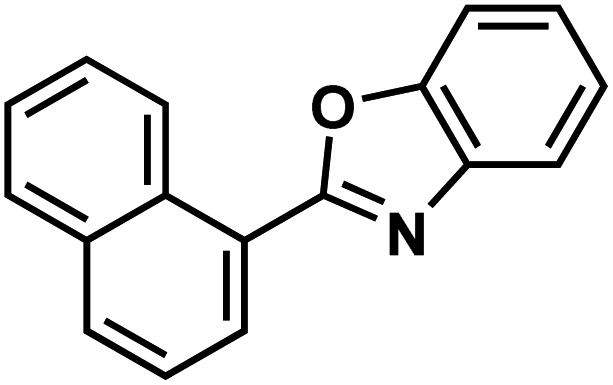 |
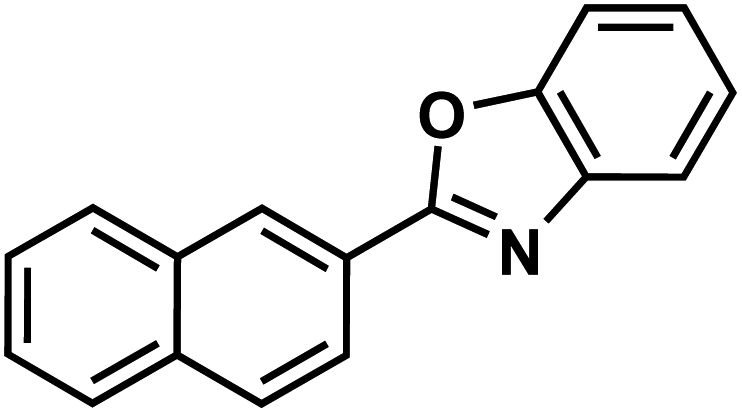 |
Ezutromid |
| 95q | 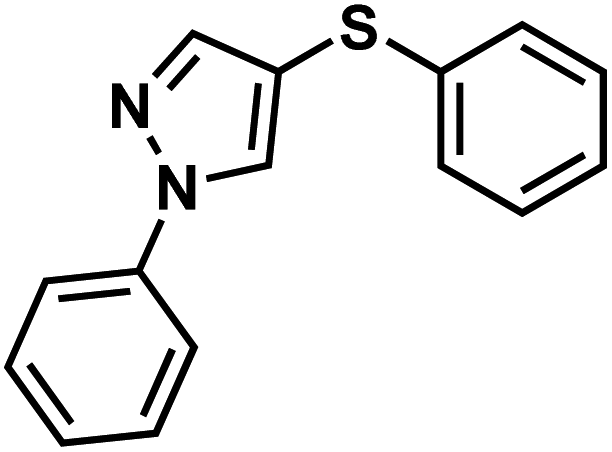 |
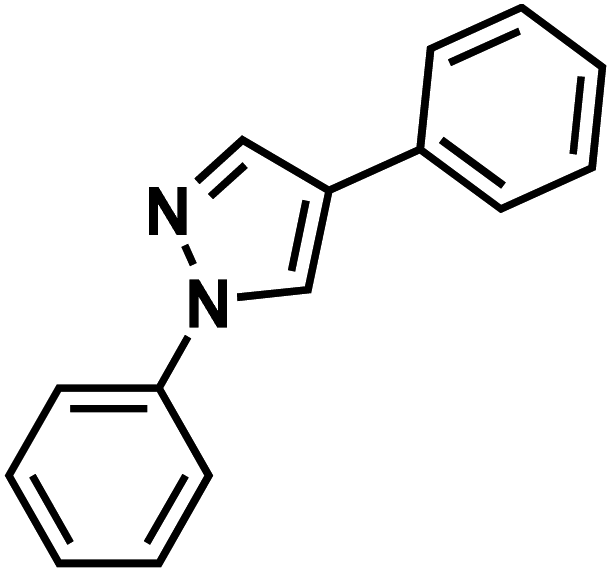 |
Lonazolac |
| 128i |  |
 |
Anisindione |
| Fluindione | |||
| Phenindione |
A comparison of Murcko skeletons30 shows only seven exact matches of skeletons between the two sets of molecules (data not shown). However, considering the similarity (≥80% threshold) between the Murcko skeletons reveals that 37 of the 41 MCR Murcko skeletons had at least one match with the skeletons of the drugs. This is expected since the Murcko skeleton does not consider the atom types or bond orders, and thus, a similarity comparison offers more flexibility.
Subsequently, we evaluated the shape and complexity of the scaffolds from MCR products and drugs. Since the Murcko scaffolds consist of multiple rings, we calculated all small rings (7-membered or lesser size) in both classes for comparison. In general, scaffolds from MCR consist of more rings, as indicated by the high mean and median values (Fig. 43A). This is expected since MCR reactions often yield bicyclic or polycyclic systems. In Datawarrior, the ‘Shape Index’ of the molecules can be calculated based on the 2D topology of the molecules.150 The values higher than 0.5 indicate a linear shape, while smaller values indicate the presence of rings or bridges. On average, MCR scaffolds are significantly less linear (mean 0.504) than drug scaffolds (mean 0.578) (Fig. 43B). In addition to the shape based on 2D structure, we also compared the 3D globularity, which was calculated based on the randomly generated first 16 conformers in the Datawarrior. A value of 1.0 indicates perfect spherical molecules, while flat or linear molecules have values close to 0.0. The MCR scaffolds display a statistically significant increase in the mean and median values than the drug scaffolds, suggesting that the former have more 3D character (Fig. 43C).
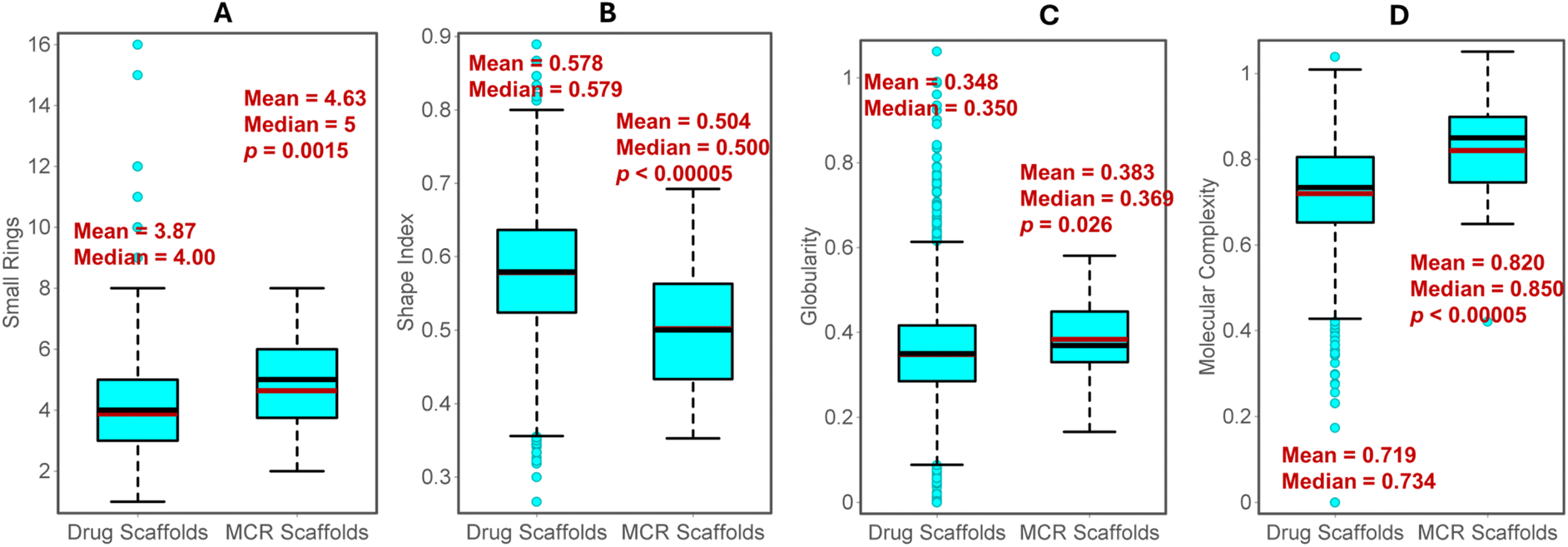 | ||
| Fig. 43 The comparison of average properties of MCR and drug scaffolds. (A) Small rings, (B) shape index, (C) globularity, (D) molecular complexity. The red and black lines in the box represents mean and median, respectively. | ||
Several efforts are reported to mathematically quantify the complexity of a molecule and its correlation with other pharmaceutical traits.143 MCRs are known to yield complex rings with multiple connectivity and substitutions.3,4 As per the Datawarrior measure of complexity, MCR scaffolds possess higher complexity than drug scaffolds (Fig. 43D), indicating the prevalence of branched structures and bridgeheads. Thus, the ligands based on the MCR scaffolds offer an advantage as these may display better target selectivity and safety profiles due to higher molecular complexity.
9 Toxicity assessment of representative scaffolds
To further assess the suitability of these scaffolds in early drug discovery we assessed their toxicity potential using Datawarrior. The latter program predicts the potential of a compound to have mutagenic, tumorigenic, reproductive toxicity, and irritant properties. These predictions are based on the structural fragments present in molecules recorded by Registry of Toxic Effects of Chemical Substances (RTECS) database.152,153 The Datawarrior provides qualitative assessment, and a compound is predicted to belong to either ‘high’, ‘low’, or ‘none’ class for a particular toxicity potential (Fig. 44).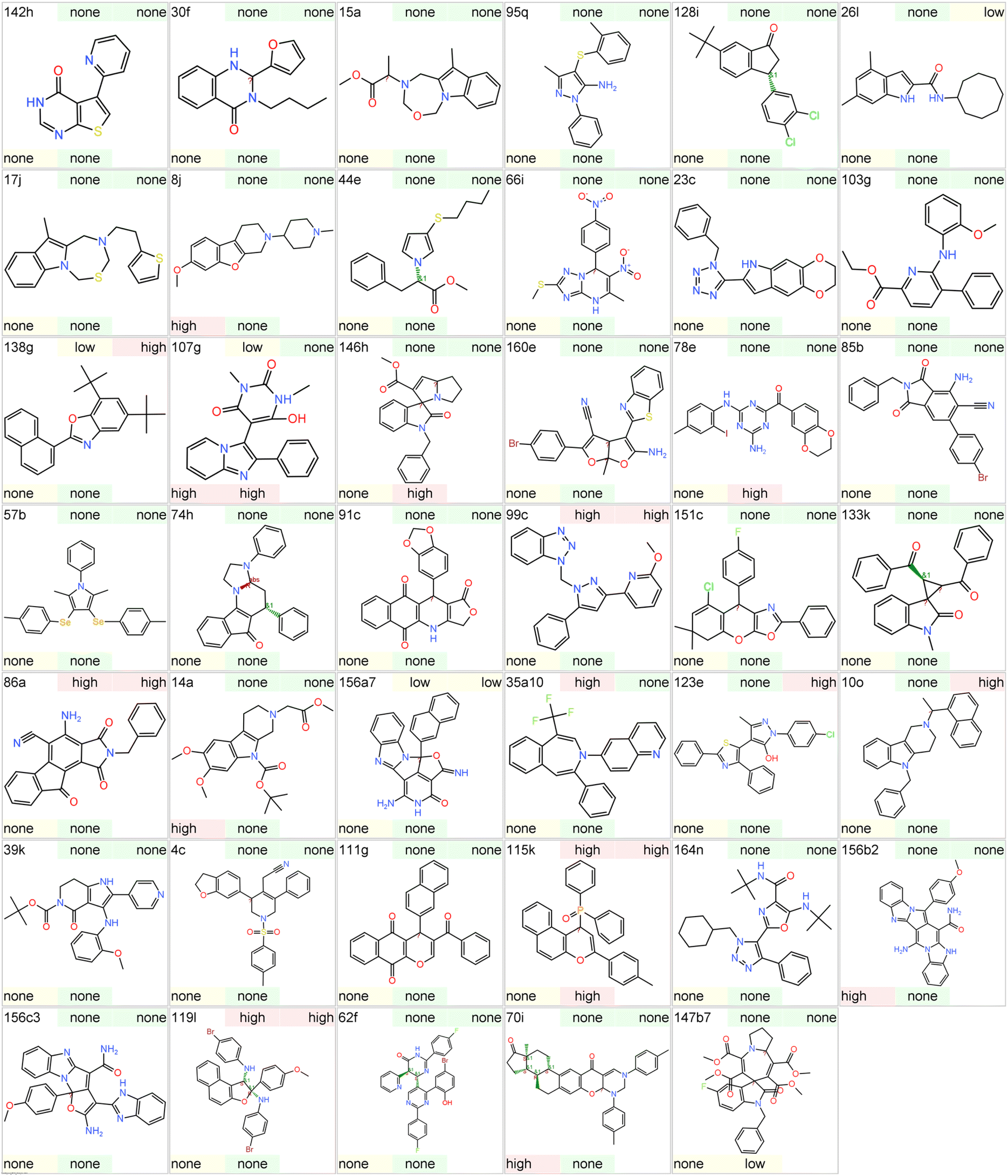 | ||
| Fig. 44 Toxicity risk assessment for compounds representing different scaffolds. The compound ID is given in the top left corner. The toxicity potential is predicted by Datawarrior as high, low or none for mutagenicity (top middle), tumorigenicity (top right), reproductive toxicity (bottom left), and irritant (bottom middle). | ||
Amongst 41 scaffolds, only 5 were predicted to have ‘high’ potential for mutagenic effect which is considered to the most serious toxicity in drug discovery. Similarly, for tumorigenic, reproductive effect, and irritants, the number was 8, 5, and 4, respectively. Overall, 26 scaffolds were computed to have either ‘low’ or ‘none’ potential for all the four toxicity categories. However, it should be noted that only representative examples of each scaffold were selected for this prediction. The change in substituents around the scaffold may give varying results. In early drug discovery it is common to synthesize different analogues of a hit molecule thus, predictions need to be repeated for the newly synthesized derivatives of a particular scaffold. Therefore, these computational results should only be used to flag a particular scaffold and not for excluding it. Similarly, the absence of any alert does not imply that the scaffold is free from toxicity and standard experiments need to be performed to assess toxicity potential during lead optimization. Nonetheless, most of the scaffolds represent a good starting point for their application in medicinal chemistry projects.
Formation of N-nitrosamines during chemical synthesis is another source of mutagenic impurities.154 The N-nitrosamines are formed by reaction of a nitrosating agent with a secondary or tertiary amine, under acidic conditions.155,156 In many cases, degradation of the drug molecules or residual solvent DMF, results in the formation of secondary amines that can result in nitrosamines. Similarly, use of many acids, catalysts, and other reagents can participate in nitrosamine formation. The degradation pathways and structural features can be predicted in silico to assess the formation of nitrosamines during the final synthesis of molecule.157–159 Thus, the synthetic schemes of these scaffolds can be screened using such tools to identify the structural alerts contributing to the formation of nitrosamine generation. This will enable the selection of scaffolds that can be produced without compromising the toxicity profile of the final products, especially during large scale synthesis for preclinical studies.
10 Conclusion
MCRs are known to provide several advantages over traditional multi-step organic synthesis. Here we review recently reported novel MCRs generating medicinally relevant ring frameworks in a single pot operation. Most of these MCRs operate under mild conditions and employ common building blocks such as aldehydes, amines, carboxylic acids, nitriles, and isocyanides. The commercial availability of these building blocks ensures the diversity of chemical space that can be accessed with these MCRs. The rings produced by these MCRs display unique substitution patterns, novel connectivity, and natural product-like architectures. High regioselectivity, diastereoselectivity, broad scope, and robustness are some of the key hallmarks of these MCRs. Most of the examples are related to N-containing heterocycles, which are known to play an important role in drug discovery. Cheminformatic analysis suggests that these MCRs reviewed here generate unique Murcko scaffolds that are not present in clinically used drugs and investigational small molecules. We also show that some of the MCRs may be exploited to access analogues of existing drugs in a one-pot fashion, thus, assisting the discovery of me-too drugs. In addition, with the more complex topology, globular shapes, and favourable computed toxicity profile, MCR scaffolds offer a valuable opportunity to access diverse and unique chemical space for drug discovery.List of abbreviations
| MCR | Multicomponent reaction |
| THPs | Tetrahydropyridines |
| TMSCN | Trimethylsilyl cyanide |
| Cu(CH3CN)4PF6 | Tetrakis(acetonitrile)copper(I) hexafluorophosphate |
| BOX | Bisoxazoline |
| EDGs | Electron donation groups |
| EWGs | Electron withdrawing groups |
| TMSN3 | Azidotrimethylsilane |
| AgSCF3 | Silver(I) trifluoromethanethiolate |
| Equiv. | Equivalent |
| DFT | Density functional theory |
| DMF | Dimethylformamide |
| Boc | Tert-butyloxycarbonyl |
| ACN | Acetonitrile |
| THF | Tetrahydrofuran |
| HCl | Hydrochloric acid |
| μM | Micromolar |
| Trp | Tryptophan |
| MSA | Methanesulfonic acid |
| SETD2 | SET domain containing 2 |
| KI | Potassium iodide |
| AcOH | Acetic acid |
| Cu(OAc)2 | Copper(II) acetate |
| TFA | Trifluoroacetic acid |
| BDA | Cis-2-butene-1,4-dial |
| DMSO | Dimethylsulfoxide |
| US | Ultrasonic |
| CuI | Copper iodide |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| MW | Microwave |
| PROTACs | Proteolysis targeting chimeras |
| Et3N | Triethylamine |
| K2CO3 | Potassium carbonate |
| DIPEA | Diisopropylethylamine |
| BF3·Et2O | Boron trifluoride etherate |
| DCE | Dichloroethane |
| HFIP | Hexafluoroisopropanol |
| TMS-OTf | Trimethylsilyl trifluoromethanesulfonate |
| POCl3 | Phosphoryl chloride |
| TfOH | Triflic acid |
| PC | Propylene carbonate |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| t-BuONa | Sodium tert-butoxide |
| Py | Pyridine |
| REM | Rare earth metal |
| Sc(OTf)3 | Scandium trifluoromethanesulfonate |
| Ce(OTf)3 | Cerium(III) trifluoromethanesulfonate |
| 2D-RBS | 2D-Rubber band scaling |
Data availability
The data supporting this article have been included in the manuscript and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Indian Council of Medical Research (ICMR), New Delhi, India, for the funding support (Grant No. 67/3/2020-DDI/BMS).References
- P. Slobbe, E. Ruijter and R. V. A. Orru, MedChemComm, 2012, 3, 1189–1218 RSC.
- A. Dömling, W. Wang, K. Wang, A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- M. J. Buskes, A. Coffin, D. M. Troast, R. Stein and M. J. Blanco, ACS Med. Chem. Lett., 2023, 14, 376–385 CrossRef CAS PubMed.
- H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Expert Opin. Ther. Pat., 2021, 31, 267–289 CrossRef CAS PubMed.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed.
- B. Voigt, M. Linke and R. Mahrwald, Org. Lett., 2015, 17, 2606–2609 CrossRef CAS PubMed.
- B. Yang, Y. Zhao, Y. Wei, C. Fu and L. Tao, Polym. Chem., 2015, 6, 8233–8239 RSC.
- L. Reguera, Y. Méndez, A. R. Humpierre, O. Valdés and D. G. Rivera, Acc. Chem. Res., 2018, 51, 1475–1486 CrossRef CAS PubMed.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- L. Palanivel and V. Gnanasambandam, Org. Biomol. Chem., 2020, 18, 3082–3092 RSC.
- C. Hulme and J. Dietrich, Mol. Diversity, 2009, 13, 195–207 CrossRef CAS PubMed.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- M. Tandi, N. Tripathi, A. Gaur, B. Gopal and S. Sundriyal, Mol. Diversity, 2024, 28, 37–50 CrossRef CAS PubMed.
- D. J. Boruah, L. Borkotoky, U. D. Newar, R. A. Maurya and P. Yuvaraj, Asian J. Org. Chem., 2023, 12, e202300297 CrossRef CAS.
- S. Pelliccia, I. A. Alfano, U. Galli, E. Novellino, M. Giustiniano and G. C. Tron, Symmetry, 2019, 11, 798 CrossRef CAS.
- M. A. Khanfar, L. Quinti, H. Wang, S. H. Choi, A. G. Kazantsev and R. B. Silverman, Eur. J. Med. Chem., 2014, 76, 414–426 CrossRef CAS PubMed.
- J. Lei, J. P. Meng, D. Y. Tang, B. Frett, Z. Z. Chen and Z. G. Xu, Mol. Diversity, 2018, 22, 503–516 CrossRef CAS PubMed.
- W. Zhang, S. Zhi and X. Ma, Org. Biomol. Chem., 2019, 17, 7632–7650 RSC.
- L. Weber, Curr. Med. Chem., 2012, 9, 2085–2093 CrossRef PubMed.
- H. Cao, H. Liu and A. Dömling, Chem.–Eur. J., 2010, 16, 12296–12298 CrossRef CAS PubMed.
- M. Jida and S. Ballet, ChemistrySelect, 2018, 3, 1027–1031 CrossRef CAS.
- J. Zhang, Y. Y. Wang, H. Sun, S. Y. Li, S. H. Xiang and B. Tan, Sci. China: Chem., 2020, 63, 47–54 CrossRef CAS.
- H. D. Preschel, R. T. Otte, Y. Zhuo, R. E. Ruscoe, A. J. Burke, R. Kellerhals, B. Horst, S. Hennig, E. Janssen, A. P. Green, N. J. Turner and E. Ruijter, J. Org. Chem., 2023, 88, 12565–12571 CrossRef CAS PubMed.
- J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- R. Visini, J. Arús-Pous, M. Awale and J. L. Reymond, J. Chem. Inf. Model., 2017, 57, 2707–2718 CrossRef CAS PubMed.
- P. Ertl, S. Jelfs, J. Mühlbacher, A. Schuffenhauer and P. Selzer, J. Med. Chem., 2006, 49, 4568–4573 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
- E. M. M. Abdelraheem, R. Madhavachary, A. Rossetti, K. Kurpiewska, J. Kalinowska-Tluścik, S. Shaabani and A. Dömling, Org. Lett., 2017, 19, 6176–6179 CrossRef CAS PubMed.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25, 505 CrossRef PubMed.
- H. Valluri, A. Bhanot, S. Shah, N. Bhandaru and S. Sundriyal, J. Med. Chem., 2023, 66, 8382–8406 CrossRef CAS PubMed.
- A. Bhanot and S. Sundriyal, ACS Omega, 2021, 6, 6424–6437 CrossRef CAS PubMed.
- L. D. Pennington and D. T. Moustakas, J. Med. Chem., 2017, 60, 3552–3579 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- S. Sundriyal, Future Med. Chem., 2024, 16, 2069–2071 Search PubMed.
- N. Mateeva, L. Winfield and K. Redda, Curr. Med. Chem., 2012, 12, 551–571 Search PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- S. Dongbang, D. N. Confair and J. A. Ellman, Acc. Chem. Res., 2021, 54, 1766–1778 CrossRef CAS PubMed.
- Z. S. Ye, M. W. Chen, Q. A. Chen, L. Shi, Y. Duan and Y. G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10181–10184 CrossRef CAS PubMed.
- F. Glorius, N. Spielkamp, S. Holle, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2004, 43, 2850–2852 CrossRef CAS PubMed.
- S. Wei, G. Zhang, Y. Wang, M. You, Y. Wang, L. Zhou and Z. Zhang, iScience, 2023, 26, 106137 CrossRef CAS PubMed.
- Z. Lai, R. Wu, J. Li, X. Chen, L. Zeng, X. Wang, J. Guo, Z. Zhao, H. Sajiki and S. Cui, Nat. Commun., 2022, 13, 435 CrossRef CAS PubMed.
- N. Teraiya, K. Agrawal, T. M. Patel, A. Patel, S. Patel, U. Shah, S. Shah, K. Rathod and K. Patel, Curr. Drug Discovery Technol., 2023, 20, 9–37 Search PubMed.
- A. Dorababu, RSC Med. Chem., 2020, 11, 1335–1353 RSC.
- A. Bendi, Versha, Rajni, L. Singh and Taruna, ChemistrySelect, 2023, 8, e202303872 CrossRef CAS.
- J. Li, Z. Lai, W. Zhang, L. Zeng and S. Cui, Nat. Commun., 2023, 14, 4806 CrossRef CAS PubMed.
- J. Li, H. Ni, W. Zhang, Z. Lai, H. Jin, L. Zeng and S. Cui, Chem. Sci., 2024, 15, 5211–5217 RSC.
- X. Lei, P. Lampiri, P. Patil, G. Angeli, C. G. Neochoritis and A. Dömling, Chem. Commun., 2021, 57, 6652–6655 RSC.
- X. Lei, G. K. Angeli, C. G. Neochoritis and A. Dömling, Green Chem., 2022, 24, 6168–6171 RSC.
- I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193–244 CrossRef CAS PubMed.
- A. Hameed, M. Al-Rashida, M. Uroos, S. A. Ali, Arshia, M. Ishtiaq and K. M. Khan, Expert Opin. Ther. Pat., 2018, 28, 281–297 CrossRef CAS PubMed.
- C. L. Yoo, J. C. Fettinger and M. J. Kurth, J. Org. Chem., 2005, 70, 6941–6943 CrossRef CAS PubMed.
- D. Shi, L. Rong, J. Wang, Q. Zhuang, X. Wang and H. Hu, Tetrahedron Lett., 2003, 44, 3199–3201 CrossRef CAS.
- X. Zhang, J. Wang, J. Xu, Q. Pang, D. Liu and G. Zhang, J. Org. Chem., 2023, 88, 10266–10276 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- Z. Chen, Y. Zheng and J. Ma, Angew. Chem., 2017, 129, 4640–4645 CrossRef.
- F.-G. Zhang, Y. Wei, Y.-P. Yi, J. Nie and J.-A. Ma, Org. Lett., 2014, 16, 3122–3125 CrossRef CAS PubMed.
- Z. Chen, N. Ren, X. Ma, J. Nie, F. G. Zhang and J. A. Ma, ACS Catal., 2019, 9, 4600–4608 CrossRef CAS.
- S. A. Künzi, B. Morandi and E. M. Carreira, Org. Lett., 2012, 14, 1900–1901 CrossRef PubMed.
- S. P. Chandrasekharan, A. Dhami and K. Mohanan, Org. Lett., 2023, 25, 5806–5811 CrossRef CAS PubMed.
- G. Li Petri, V. Spanò, R. Spatola, R. Holl, M. V. Raimondi, P. Barraja and A. Montalbano, Eur. J. Med. Chem., 2020, 208, 112783 CrossRef CAS PubMed.
- K. Afratis, J. M. Bateman, B. F. Rahemtulla, O. Hughes, B. C. Milgram, T. A. Mulhern and E. P. A. Talbot, Org. Lett., 2023, 25, 461–465 CrossRef CAS PubMed.
- Y. Wang, P. Czabala and M. Raj, Nat. Commun., 2023, 14, 4086 CrossRef CAS PubMed.
- C. Gallo-Rodriguez and J. B. Rodriguez, ChemMedChem, 2024, 19, e202400063 CrossRef CAS PubMed.
- T. J. Peglow, G. P. Da Costa, L. F. B. Duarte, M. S. Silva, T. Barcellos, G. Perin and D. Alves, J. Org. Chem., 2019, 84, 5471–5482 CrossRef CAS PubMed.
- B. Nammalwar and R. A. Bunce, Pharmaceuticals, 2024, 17, 104 CrossRef CAS PubMed.
- X.-J. Dai, L.-P. Xue, S.-K. Ji, Y. Zhou, Y. Gao, Y.-C. Zheng, H.-M. Liu and H.-M. Liu, Eur. J. Med. Chem., 2023, 249, 115101 CrossRef CAS PubMed.
- L. Chen, R. Huang, X. H. Yun, T. H. Hao and S. J. Yan, Chem. Commun., 2021, 57, 7657–7660 RSC.
- D. N. Lyapustin, E. N. Ulomsky, T. O. Zanakhov and V. L. Rusinov, J. Org. Chem., 2019, 84, 15267–15275 CrossRef CAS PubMed.
- B. Zhang, H. Yang, T. Wang, X. Han, H. Sun, L. Fang, H. Zhai and B. Cheng, J. Org. Chem., 2021, 86, 18304–18311 CrossRef CAS PubMed.
- H. Tan and Y. Wang, ACS Comb. Sci., 2020, 22, 468–474 CrossRef CAS PubMed.
- F. P. L. Lim and A. V. Dolzhenko, Eur. J. Med. Chem., 2014, 85, 371–390 CrossRef CAS PubMed.
- P. Zhao, Y. Zhou, X.-X. Yu, C. Huang, Y.-D. Wu, G. Yin and A.-X. Wu, Org. Lett., 2020, 22, 8528–8532 CrossRef CAS PubMed.
- G. F. S. Fernandes, J. R. Lopes, J. L. Dos Santos and C. B. Scarim, Drug Dev. Res., 2023, 84, 1346–1375 CrossRef CAS PubMed.
- Ó. Vázquez-Vera, J. S. Sánchez-Badillo, A. Islas-Jácome, M. A. Rentería-Gómez, S. G. Pharande, C. J. Cortes-García, M. A. Rincón-Guevara, I. A. Ibarra, R. Gámez-Montaño and E. González-Zamora, Org. Biomol. Chem., 2017, 15, 2363–2369 RSC.
- N. Kise, S. Isemoto and T. Sakurai, J. Org. Chem., 2011, 76, 9856–9860 CrossRef CAS PubMed.
- H. Yoda, A. Nakahama, T. Koketsu and K. Takabe, Tetrahedron Lett., 2002, 43, 4667–4669 CrossRef CAS.
- D. A. Nalawansha and C. M. Crews, Cell Chem. Biol., 2020, 27, 998–1014 CrossRef CAS PubMed.
- Z. Liu, M. Hu, Y. Yang, C. Du, H. Zhou, C. Liu, Y. Chen, L. Fan, H. Ma, Y. Gong and Y. Xie, Mol. Biomed., 2022, 3, 46 CrossRef CAS PubMed.
- J. Yang, B. Zhao, Y. Xi, S. Sun, Z. Yang, Y. Ye, K. Jiang and Y. Wei, Org. Lett., 2018, 20, 1216–1219 CrossRef CAS PubMed.
- B. Martin, H. Sekljic and C. Chassaing, Org. Lett., 2003, 5, 1851–1853 CrossRef CAS PubMed.
- X. Wang, W. Xiong, Y. Huang, J. Zhu, Q. Hu, W. Wu and H. Jiang, Org. Lett., 2017, 19, 5818–5821 CrossRef CAS PubMed.
- A. Alizadeh, B. Farajpour, T. O. Knedel and C. Janiak, J. Org. Chem., 2021, 86, 574–580 CrossRef CAS PubMed.
- N. Jeedimalla, M. Flint, L. Smith, A. Haces, D. Minond and S. P. Roche, Eur. J. Med. Chem., 2015, 106, 167–179 CrossRef CAS PubMed.
- Q. G. Nguyen Thi, G. Le-Nhat-Thuy, T. A. Dang Thi, P. Hoang Thi, A. Nguyen Tuan, T. H. Nguyen Thi, T. T. Nguyen, T. Nguyen Ha, H. Hoang Mai and T. Van Nguyen, Bioorg. Med. Chem. Lett., 2021, 37, 127841 CrossRef CAS PubMed.
- M. Lusardi, A. Spallarossa and C. Brullo, Int. J. Mol. Sci., 2023, 24, 7834 CrossRef CAS PubMed.
- T. Parvin, Top. Curr. Chem., 2023, 381, 19 CrossRef CAS PubMed.
- S. B. Annes, R. Saritha, K. Chandru, P. K. Mandali and S. Ramesh, J. Org. Chem., 2021, 86, 16473–16484 CrossRef CAS PubMed.
- M. A. Halcrow, Coord. Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS.
- J. Kim, D. Lee, C. Park, W. So, M. Jo, T. Ok, J. Kwon, S. Kong, S. Jo, Y. Kim, J. Choi, H. C. Kim, Y. Ko, I. Choi, Y. Park, J. Yoon, M. K. Ju, J. Kim, S. J. Han, T. H. Kim, J. Cechetto, J. Nam, P. Sommer, M. Liuzzi, J. Lee and Z. No, ACS Med. Chem. Lett., 2012, 3, 678–682 CrossRef CAS PubMed.
- H. Y. Li, W. T. McMillen, C. R. Heap, D. J. McCann, L. Yan, R. M. Campbell, S. R. Mundla, C. H. R. King, E. A. Dierks, B. D. Anderson, K. S. Britt, K. L. Huss, M. D. Voss, Y. Wang, D. K. Clawson, J. M. Yingling and J. S. Sawyer, J. Med. Chem., 2008, 51, 2302–2306 CrossRef CAS PubMed.
- M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2013, 52, 7219–7223 CrossRef PubMed.
- R. Barroso, M. P. Cabal, A. Jiménez and C. Valdés, Org. Biomol. Chem., 2020, 18, 1629–1636 RSC.
- Z. Zheng and P. J. Walsh, Adv. Synth. Catal., 2021, 363, 800–807 CrossRef CAS.
- H. Jiang, J. Yang, X. Tang, J. Li and W. Wu, J. Org. Chem., 2015, 80, 8763–8771 CrossRef CAS PubMed.
- H. Andersson, F. Almqvist and R. Olsson, Org. Lett., 2007, 9, 1335–1337 CrossRef CAS PubMed.
- X. He, Y. Shang, Z. Yu, M. Fang, Y. Zhou, G. Han and F. Wu, J. Org. Chem., 2014, 79, 8882–8888 CrossRef CAS PubMed.
- B. Zhu, J. He, K. Zou, A. Li, C. Zhang, J. Zhao and H. Cao, J. Org. Chem., 2023, 88, 11450–11459 CrossRef CAS PubMed.
- V. V. Kumbhar, B. B. Khairnar, M. G. Chaskar, R. A. Pawar and G. S. Gugale, Synth. Commun., 2022, 52, 1209–1244 CrossRef CAS.
- M. Sharma and P. Prasher, Synth. Commun., 2022, 52, 1337–1356 CrossRef CAS.
- S. Samanta, S. Kumar, E. K. Aratikatla, S. R. Ghorpade and V. Singh, RSC Med. Chem., 2023, 14, 644–657 RSC.
- S. Mohana Roopan, S. M. Patil and J. Palaniraja, Res. Chem. Intermed., 2016, 42, 2749–2790 CrossRef CAS.
- C. Enguehard-Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888–899 CrossRef CAS PubMed.
- G. Brahmachari, N. Nayek, I. Karmakar, K. Nurjamal, S. K. Chandra and A. Bhowmick, J. Org. Chem., 2020, 85, 8405–8414 CrossRef CAS PubMed.
- M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardarson, J. Med. Chem., 2018, 61, 10996–11020 CrossRef CAS PubMed.
- I. E. Soria-Mercado, A. Prieto-Davo, P. R. Jensen and W. Fenical, J. Nat. Prod., 2005, 68, 904–910 CrossRef CAS PubMed.
- D. Kumar, P. Sharma, H. Singh, K. Nepali, G. K. Gupta, S. K. Jain and F. Ntie-Kang, RSC Adv., 2017, 7, 36977–36999 RSC.
- G. Le-Nhat-Thuy, T. A. Dang Thi, P. Hoang Thi, Q. G. Nguyen Thi, H.-T. Nguyen, D. Vu Ngoc, T.-A. Nguyen and T. Van Nguyen, Tetrahedron Lett., 2021, 75, 153215 CrossRef CAS.
- X. H. Wang, Y. W. Xue, C. Y. Bai, Y. Bin Wang, X. H. Wei and Q. Su, J. Org. Chem., 2023, 88, 16216–16228 CrossRef CAS PubMed.
- X.-H. Wang, X. Liu, Y.-W. Xue, X.-H. Wei, Y.-B. Wang and Q. Su, J. Org. Chem., 2024, 89, 8531–8536 CrossRef CAS PubMed.
- J. Huang, J. F. Chen, X. Cui, J. Z. Zhao, Z. Tang and G. X. Li, J. Org. Chem., 2022, 87, 3311–3318 CrossRef CAS PubMed.
- S. Funayama, M. Ishibashi, Y. Anraku, K. Komiyama and S. Omura, Tetrahedron Lett., 1989, 30, 7427–7430 CrossRef CAS.
- K. H. Narasimhamurthy, A. M. Sajith, M. N. Joy and K. S. Rangappa, ChemistrySelect, 2020, 5, 5629–5656 CrossRef CAS.
- A. Petrou, M. Fesatidou and A. Geronikaki, Molecules, 2021, 26, 3166 CrossRef CAS PubMed.
- S. Nayak and S. L. Gaonkar, Mini-Rev. Med. Chem., 2018, 19, 215–238 CrossRef PubMed.
- M. A. M. Alhamami, J. S. Algethami and S. Khan, Crit. Rev. Anal. Chem., 2023, 1–25 Search PubMed.
- A. Dessì, M. Calamante, A. Sinicropi, M. L. Parisi, L. Vesce, P. Mariani, B. Taheri, M. Ciocca, A. Di Carlo, L. Zani, A. Mordini and G. Reginato, Sustainable Energy Fuels, 2020, 4, 2309–2321 RSC.
- S. Kumar and R. Aggarwal, Mini-Rev. Org. Chem., 2019, 16, 26–34 CrossRef CAS.
- Z. Jin, Nat. Prod. Rep., 2006, 23, 464–496 RSC.
- A. Nitelet, P. Gérard, J. Bouche and G. Evano, Org. Lett., 2019, 21, 4318–4321 CrossRef CAS PubMed.
- R. Banerjee, D. Ali, N. Mondal and L. H. Choudhury, J. Org. Chem., 2024, 89, 4423–4437 CrossRef CAS PubMed.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- Z. Zhao, X. Dai, C. Li, X. Wang, J. Tian, Y. Feng, J. Xie, C. Ma, Z. Nie, P. Fan, M. Qian, X. He, S. Wu, Y. Zhang and X. Zheng, Eur. J. Med. Chem., 2020, 186, 111893 CrossRef CAS PubMed.
- J. C. J. M. D. S. Menezes, RSC Adv., 2017, 7, 9357–9372 RSC.
- S. A. Patil, R. Patil and S. A. Patil, Eur. J. Med. Chem., 2017, 138, 182–198 CrossRef CAS PubMed.
- P. Sharma, N. Taneja, S. Singh and C. K. Hazra, Chem.–Eur. J., 2023, 29, e202202956 CrossRef CAS PubMed.
- M. A. Tallarida, F. Olivito, C. D. Navo, V. Algieri, A. Jiritano, P. Costanzo, A. Poveda, M. J. Moure, J. Jiménez-Barbero, L. Maiuolo, G. Jiménez-Osés and A. De Nino, Org. Lett., 2023, 25, 3001–3006 CrossRef CAS PubMed.
- A. Abdullahi and K. Y. Yeong, Med. Chem. Res., 2024, 33, 406–438 CrossRef CAS.
- T. Horch, E. M. Molloy, F. Bredy, V. G. Haensch, K. Scherlach, K. L. Dunbar, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2022, 61, e202205409 CrossRef CAS PubMed.
- Y. Xu, L. Xiao, S. Sun, Z. Pei, Y. Pei and P. Yi, Chem. Commun., 2014, 50, 7514–7516 RSC.
- D. Wee, S. Yoo, Y. H. Kang, Y. H. Kim, J.-W. Ka, S. Y. Cho, C. Lee, J. Ryu, M. H. Yi and K.-S. Jang, J. Mater. Chem. C, 2014, 2, 6395–6401 RSC.
- S. Soni, N. Sahiba, S. Teli, P. Teli, L. K. Agarwal and S. Agarwal, RSC Adv., 2023, 13, 24093–24111 RSC.
- H. Sharghi, J. Aboonajmi and M. Aberi, J. Org. Chem., 2020, 85, 6567–6577 CrossRef CAS PubMed.
- M. T. M. Sayed, R. A. Hassan, P. A. Halim and A. K. El-Ansary, Med. Chem. Res., 2023, 32, 659–681 CrossRef CAS.
- P. Lagardère, C. Fersing, N. Masurier and V. Lisowski, Pharmaceuticals, 2021, 15, 35 CrossRef PubMed.
- S. Sundriyal, S. Moniot, Z. Mahmud, S. Yao, P. Di Fruscia, C. R. Reynolds, D. T. Dexter, M. J. E. Sternberg, E. W. F. Lam, C. Steegborn and M. J. Fuchter, J. Med. Chem., 2017, 60, 1928–1945 CrossRef CAS PubMed.
- J. S. Disch, G. Evindar, C. H. Chiu, C. A. Blum, H. Dai, L. Jin, E. Schuman, K. E. Lind, S. L. Belyanskaya, J. Deng, F. Coppo, L. Aquilani, T. L. Graybill, J. W. Cuozzo, S. Lavu, C. Mao, G. P. Vlasuk and R. B. Perni, J. Med. Chem., 2013, 56, 3666–3679 CrossRef CAS PubMed.
- T. Shi, L. Kaneko, M. Sandino, R. Busse, M. Zhang, D. Mason, J. Machulis, A. J. Ambrose, D. D. Zhang and E. Chapman, ACS Sustain. Chem. Eng., 2019, 7, 1524–1528 CrossRef CAS PubMed.
- O. Méndez-Lucio and J. L. Medina-Franco, Drug Discovery Today, 2017, 22, 120–126 CrossRef PubMed.
- J. Cao, F. Yang, J. Sun, Y. Huang and C. G. Yan, J. Org. Chem., 2019, 84, 622–635 CrossRef CAS PubMed.
- P. Zamani, J. Phipps, J. Hu, F. Cheema, H. Amiri Rudbari, A. K. Bordbar, A. R. Khosropour and M. H. Beyzavi, ACS Comb. Sci., 2019, 21, 557–561 CrossRef CAS PubMed.
- M. Cao, Y. L. Fang, Y. C. Wang, X. J. Xu, Z. W. Xi and S. Tang, ACS Comb. Sci., 2020, 22, 268–273 CrossRef CAS PubMed.
- H. Z. Zhang, Z. L. Zhao and C. H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS PubMed.
- S. Kakkar and B. Narasimhan, BMC Chem., 2019, 13, 16 CrossRef PubMed.
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discovery Today, 2017, 22, 1572–1581 CrossRef CAS PubMed.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- J. K. Aronson and A. R. Green, Br. J. Clin. Pharmacol., 2020, 86, 2114–2122 CrossRef CAS PubMed.
- J. C. F. Fang, A. E. Friedman, W. E. Macdonald, R. J. Lewis and R. L. Tatken, Drug Chem. Toxicol., 1980, 3, 35–45 CrossRef CAS PubMed.
- D. V. Sweet, Approaches to Safe Nanotechnology, 1997, p. 186 Search PubMed.
- R. Nudelman, G. Kocks, B. Mouton, D. J. Ponting, J. Schlingemann, S. Simon, G. F. Smith, A. Teasdale and A. L. Werner, Org. Process Res. Dev., 2023, 27, 1719–1735 CrossRef CAS.
- K. M. Manchuri, M. A. Shaik, V. S. R. Gopireddy and S. Gogineni, Chem. Res. Toxicol., 2024, 37, 1456–1483 Search PubMed.
- J. C. Beard and T. M. Swager, J. Org. Chem., 2021, 86, 2037–2057 CrossRef CAS PubMed.
- N. S. Murphy, D. C. O'Connor, G. C. Gavins, L. James, J. P. Lockett, J. A. McManus, G. Packer, R. Lopez-Rodríguez, S. J. Webb and M. J. Burns, Org. Process Res. Dev., 2023, 27, 1812–1819 CrossRef CAS.
- M. J. Burns, A. Teasdale, E. Elliott and C. G. Barber, Org. Process Res. Dev., 2020, 24, 1531–1535 CrossRef CAS.
- M. J. Burns, M. A. Ott, A. Teasdale, S. A. Stalford, V. Antonucci, J. C. Baumann, R. Brown, E. M. Covey-Crump, D. Elder, E. Elliott, J. W. Fennell, F. Gallou, N. D. Ide, T. Itoh, G. Jordine, J. M. Kallemeyn, D. Lauwers, A. R. Looker, L. E. Lovelle, R. Molzahn, D. Schils, R. Schulte Oestrich, G. W. Sluggett, N. Stevenson, P. Talavera, M. W. Urquhart, D. L. Varie and D. S. Welch, Org. Process Res. Dev., 2019, 23, 2470–2481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06681b |
| This journal is © The Royal Society of Chemistry 2025 |