
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemarkable utilization of quinazoline-based homosulfonamide for in vitro cytotoxic effects with triple kinase inhibition activities: cell cycle analysis and molecular docking profile †
Adel S. El-Azab *,
Alaa A.-M. Abdel-Aziz,
Ahmed H. Bakheit,
Hamad M. Alkahtani,
Ahmad J. Obaidullah,
Mohamed M. Hefnawy and
Ibrahim A. Al-Suwaidan
*,
Alaa A.-M. Abdel-Aziz,
Ahmed H. Bakheit,
Hamad M. Alkahtani,
Ahmad J. Obaidullah,
Mohamed M. Hefnawy and
Ibrahim A. Al-Suwaidan
Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, P.O. Box 2457, Riyadh 11451, Saudi Arabia. E-mail: adelazab@ksu.edu.sa
First published on 6th January 2025
Abstract
We tested newly synthesized compounds 1–13 on 59 cancer cell lines and found that acylhydrazones 5, 6, 7, 9, and 12 showed the best in vitro cytotoxic activity. They stopped the mean growth percentage (MG%) by an average of 23.5, 55.2, 89.4, 88.5, and 88.4%, respectively. Compound 5 was subjected to NCI tests at five-dose dilutions on 59 tumor cells. It is more effective in killing tumor cells than gefitinib (mean GI50: 7.7 μM) and erlotinib (mean GI50: 2.1 μM). Its mean GI50 value was 1.0 μM, and its LC50 value was over 100 μM, whereas gefitinib's was 95.6 μM and erlotinib's was 14.3 μM. Its TGI was 89.2 μM, while those drugs were 66.3 and 14.3 μM, respectively. We evaluated acylhydrazones 5, 6, 7, 9, and 12 for dose-dependent enzymatic inhibition of EGFR, HER2, and CDK9 kinases to study the mechanism of the in vitro cytotoxicity. With IC50 values of 84.4 and 51.5 nM, compounds 5 and 6 are the most potent EGFR inhibitor analogs, similar to Gefitinib (IC50 of 53.1 nM). Compounds 5, 6, and 12 blocked HER2 like Gefitinib did (IC50 = 38.8 nM); their IC50 values were 53.9, 44.1, and 110.6, respectively. Compounds 5, 6, and 7 had IC50 values of 146.9, 96.1, and 155.4 nM, which means they blocked CDK9 activity almost as well as Dinaciclib (IC50 53.1 nM). Flow cytometers count the amount of DNA in T-47D and MOLT4 cells treated with compounds 5 and 6. The IC50 value of compound 5 increases from 6.6% for the DMSO/T-47D control to 26.3% in the G2-M phase, while compound 6 goes from 61.4 for the DMSO/MOLT4 control to 89.0% in the G1 phase. The tested compounds cause early death, ranging from 0.4% and 0.6% (a DMSO control sample) to 9.3% and 19.2%, respectively. Derivatives 5 and 6 also increased late death from 0.1 to 14.8% and 12.6 to 0.3%, respectively, favoring the apoptotic route over the necrotic one for cell death to 50.5 μM. When tested for cell death against the standard WI-38 fibroblast cell line, imines 5 and 6 were less toxic than doxorubicin.
1 Introduction
A failure of certain enzymes and proteins that control cell division and transformation causes cancer, characterized by abnormal cell growth.1,2 Cancer is one of the most critical causes of death, with about 10 million deaths on average during the last few years.3 The quinazoline nucleus is one of the primary heterocyclic compounds with cytotoxic activity.4–7 Multikinase inhibitors with high selectivity and efficacy contain quinazoline moieties like erlotinib (I), afatinib (II), and gefitinib (III), which are effective chemotherapeutic agents (Fig. 1). Epidermal growth factor receptors (EGFRs) belong to the family of sizeable transmembrane growth factor receptors (PTKs), including homologous receptors such as HER1, HER2, HER3, and HER4.8 Many malignancies, such as colon, breast, ovarian, and NSC lung cancers, are characterized by overexpression of EGFRs.8 Designing a cytotoxic molecule that binds to the target enzyme's catalytic domain and treats different types of human cancers that compete with ATP, like an EGFR inhibitor, could be a primary method for a cytotoxic molecule.8 Chemotherapy has been one of the best methods for treating cancer. Hence, synthesizing new cytotoxic molecules with potential bioactivity and a high therapeutic index is the main goal for many pharmaceutical and medicinal researchers. Compounds containing a quinazoline core, on the other hand, exhibit inhibitor activities, such as COX-2,9–12 and carbonic anhydrase inhibitors,13–17 as well as tyrosine kinase inhibitor activities, such as EGFRs, CDK-9,18–27 anticonvulsant28–31 cytotoxic activity,32–39 and antimicrobial.40–43 Schiff base derivatives, such as aldimines or ketamine,44,45 can bind to and block EGFRs by interacting with their ATP-binding site, showing cytotoxic activities.45,46 Sulfonamides also showed many pharmacologic activities.47–60 Fig. 2 also shows several hydrazones that kill cancer cells, such as quinazolinylhydrazone (V) and PAC-1 (VI); arylhydrazone (VII and VIII) showed cytotoxic activity higher than erlotinib, gefitinib, and sorafenib against several human cancer cells.61–65 Some hydrazones stop cancer cells from growing by blocking EGFR, HER2, and COX-2 receptors.61–65 Here, aldimines and ketimines (4–13) are formed (Fig. 3) by combining various aldehydes or ketones with 4-((4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)methyl)benzenesulfonamide (1). We investigated the in vitro cytotoxic efficacy of target compounds in vitro in 59 human cancer cell lines, revealing the structure–activity relationship (SAR). We then used an enzymatic assay on effective cytotoxic acylhydrazones to determine EGFR, HER2, CDK9, and COX-2 inhibitions. We triggered apoptosis and examined cell cycles on the most active compounds to assess their in vitro cytotoxic potential. We performed molecular docking in the binding pockets of the EGFR, HER2, and CDK9 kinases to explore how the potential variants might line up. | ||
| Fig. 1 The EGFR and HER2 inhibitors antitumor agent (I–IV). | ||
 | ||
| Fig. 2 The reported hydrazones with antitumor activities. | ||
 | ||
| Fig. 3 The designed target quinazolinylhydrazone derivative (compounds 4–13) are based on the chemical structures of compounds I–XI. | ||
2 Result and discussion
4-((4-Oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)methyl)benzenesulfonamide (1) was obtained in excellent yield by the heating of 2-aminobenzoic acid with 4-isothiocyanatobenzenesulfonamide in ethanol (Scheme 1). Confirmation of the compound 1 structure was provided by the presence of the thioamide (![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) HCS) singlet peak at 13.15 ppm and doublet (NH2) peak of the sulfonamide moiety at 7.34 ppm, together with the singlet methylene peak of the phenylmethansulfonamide moiety at 5.37 ppm in 1H NMR. At the same time, 13C NMR showed a thioketone (NHCS) at 175.89 ppm and benzylic methylene peaks at 49.11 ppm, respectively. The reaction of compound 1 with ethyl 2-bromoacetate in acetone and potassium carbonate was a crucial step in our research. This reaction led to the formation of the ethyl 2-((4-oxo-3-(4-sulfamoylbenzyl)-3,4-dihydroquinazolin-2-yl)thio)acetate (2). The singlet thiomethylencarbonyl peak (–S
HCS) singlet peak at 13.15 ppm and doublet (NH2) peak of the sulfonamide moiety at 7.34 ppm, together with the singlet methylene peak of the phenylmethansulfonamide moiety at 5.37 ppm in 1H NMR. At the same time, 13C NMR showed a thioketone (NHCS) at 175.89 ppm and benzylic methylene peaks at 49.11 ppm, respectively. The reaction of compound 1 with ethyl 2-bromoacetate in acetone and potassium carbonate was a crucial step in our research. This reaction led to the formation of the ethyl 2-((4-oxo-3-(4-sulfamoylbenzyl)-3,4-dihydroquinazolin-2-yl)thio)acetate (2). The singlet thiomethylencarbonyl peak (–S![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CO–) at 4.11 ppm in 1H NMR, the quartet and triplet peaks of the ethoxide moiety (OCH2CH3) at 4.14 and 1.21 ppm, respectively, and fading of the thioamide (NH) singlet peak at 13.15 ppm confirming ester 2. 13C NMR of the ester 2 reveals the presence of thiomethylene peak (–SCH2) at 47.13 ppm and carbonyl (C
H2CO–) at 4.11 ppm in 1H NMR, the quartet and triplet peaks of the ethoxide moiety (OCH2CH3) at 4.14 and 1.21 ppm, respectively, and fading of the thioamide (NH) singlet peak at 13.15 ppm confirming ester 2. 13C NMR of the ester 2 reveals the presence of thiomethylene peak (–SCH2) at 47.13 ppm and carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) peaks at 168.58 ppm of the thiomethylenecarbonyl group (–SH2CO), the ethoxide peaks (OCH2CH3) at 61.58 and 14.61 ppm, respectively, and the disappearance of the thione group (CS) at 175.89 ppm, which supported the structure conformation. 4-((2-((2-Hydrazineyl-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (3) are obtained by stirring ester derivative 2 with hydrazine hydrate in ethanol. The acid hydrazide 3 was confirmed by the disappearance of ethoxide peaks (OCH2CH3) at (4.14 and 1.21 ppm) and (61.58 and 14.61 ppm) in 1H NMR and 13C NMR spectra, respectively. Additionally, it strongly supported the structure confirmation presence of the amidic proton (CONH–) peak at 9.40 ppm and (NH2) singlet peaks at 4.38 and 4.32 ppm due to an acid hydrazide moiety (CONHNH2) in 1H NMR and the carbonyl peak of an amide group (CONH) at 156.62 ppm in 13C NMR. The aldimines 4–9 and ketimines 10–13 were obtained by the heating of 4-((2-((2-hydrazineyl-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (3) in ethanol with various aldehydes and ketones. The formation of these imines 4–13 was confirmed by the disappearance of the singlet peaks of the amino group (NH2) at 4.38 and 4.32 ppm in the 1H NMR spectrum, which is attributed to the acid hydrazide moiety (CONHH2). Furthermore, the presence of the E and Z isomer peaks at 11.86–11.76 and 10.85–11.36 ppm of the imide group (COH–), together with the olefinic protons of the imine group (CONH–NC–H–) at 8.25–8.13 and 8.16–8.00 ppm in 1H NMR, strongly supported the confirmation of aldimines 4–9. Ketimines 10–13 were characterized by E and Z isomeric peaks at 10.97–10.93 and 10.80–10.77 ppm of the imide group (COH–) in 1H NMR, and the methyl peaks of the ethylidenehydrazineyl group (CONH–NC–CH3) at 2.34–2.30 and 2.30–2.27 ppm and 15.0–14.71 and 14.36–14.15 ppm in 1H NMR and 13C NMR respectively as isomeric mixtures.
O) peaks at 168.58 ppm of the thiomethylenecarbonyl group (–SH2CO), the ethoxide peaks (OCH2CH3) at 61.58 and 14.61 ppm, respectively, and the disappearance of the thione group (CS) at 175.89 ppm, which supported the structure conformation. 4-((2-((2-Hydrazineyl-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (3) are obtained by stirring ester derivative 2 with hydrazine hydrate in ethanol. The acid hydrazide 3 was confirmed by the disappearance of ethoxide peaks (OCH2CH3) at (4.14 and 1.21 ppm) and (61.58 and 14.61 ppm) in 1H NMR and 13C NMR spectra, respectively. Additionally, it strongly supported the structure confirmation presence of the amidic proton (CONH–) peak at 9.40 ppm and (NH2) singlet peaks at 4.38 and 4.32 ppm due to an acid hydrazide moiety (CONHNH2) in 1H NMR and the carbonyl peak of an amide group (CONH) at 156.62 ppm in 13C NMR. The aldimines 4–9 and ketimines 10–13 were obtained by the heating of 4-((2-((2-hydrazineyl-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (3) in ethanol with various aldehydes and ketones. The formation of these imines 4–13 was confirmed by the disappearance of the singlet peaks of the amino group (NH2) at 4.38 and 4.32 ppm in the 1H NMR spectrum, which is attributed to the acid hydrazide moiety (CONHH2). Furthermore, the presence of the E and Z isomer peaks at 11.86–11.76 and 10.85–11.36 ppm of the imide group (COH–), together with the olefinic protons of the imine group (CONH–NC–H–) at 8.25–8.13 and 8.16–8.00 ppm in 1H NMR, strongly supported the confirmation of aldimines 4–9. Ketimines 10–13 were characterized by E and Z isomeric peaks at 10.97–10.93 and 10.80–10.77 ppm of the imide group (COH–) in 1H NMR, and the methyl peaks of the ethylidenehydrazineyl group (CONH–NC–CH3) at 2.34–2.30 and 2.30–2.27 ppm and 15.0–14.71 and 14.36–14.15 ppm in 1H NMR and 13C NMR respectively as isomeric mixtures.
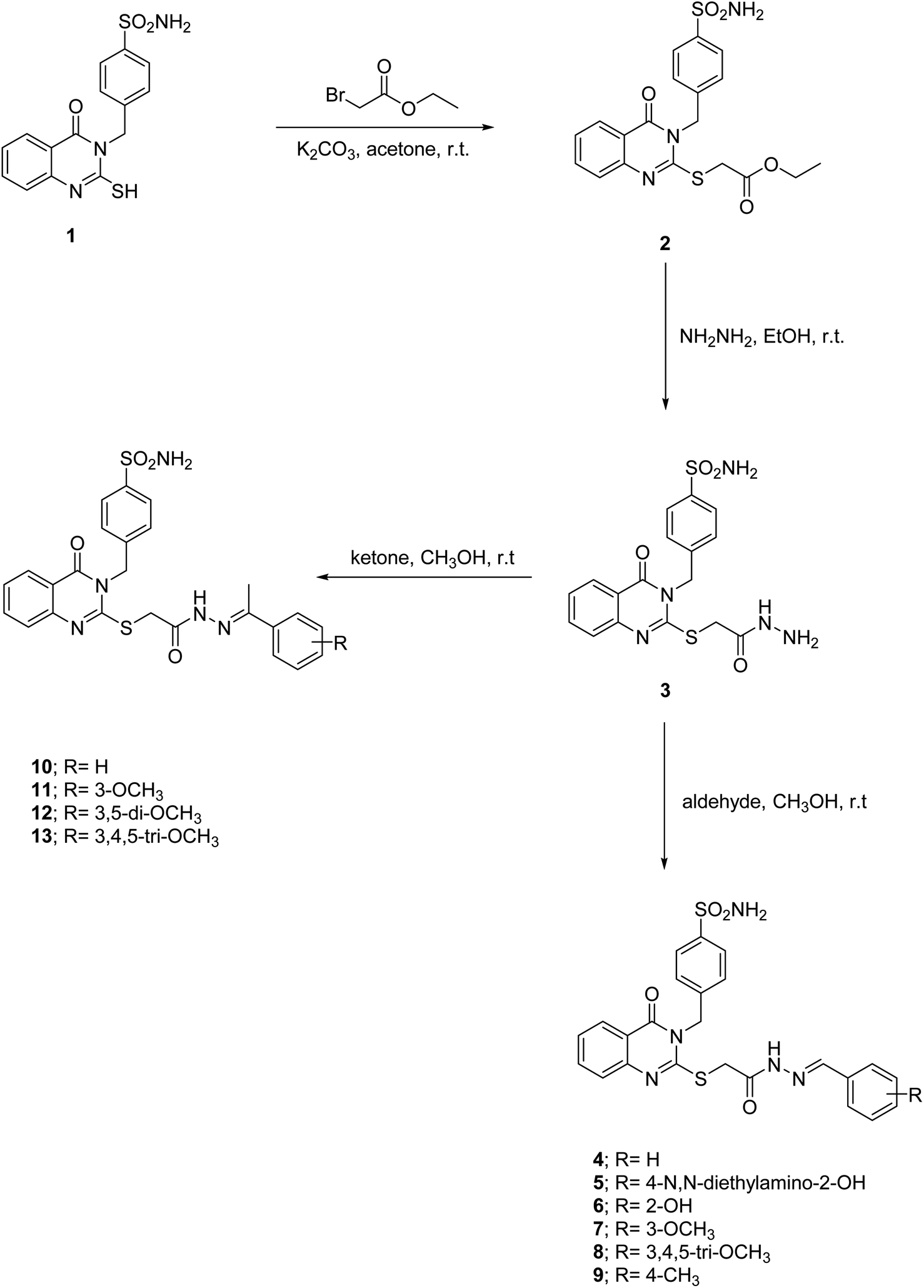 | ||
| Scheme 1 Synthesis of ester 2, acidhydrazide 3, acylhydrazones 4–13 based on quinazolines incorporating benzylulfonamide. | ||
2.1 Biological evaluation
| Compound no. | PCE* | Cancer cell line assays (10.0 μM in one dose, GI %) | **MG% |
|---|---|---|---|
| a *Most sensitive cell lines PCE: positive cytotoxic effect is defined as the ratio between the number of cell lines with percentage growth inhibition of >10% and the total number of cell lines **: mean growth percent. | |||
| 1 | 4/59 | Non-small cell Lung (HOP-92, 14%), C.N.S. (SNB-75, 23%), and Renal (CAKI-1, 11%; UO-31, 19%) | 97.13 |
| 2 | 2/59 | CNS (SNB-75, 15%), and Renal (UO-31, 16%) | 100.32 |
| 3 | 0/59 | — | 104.42 |
| 4 | 9/59 | Non-Small Cell Lung (EKVX, 14%; NCI–H522, 18%), CNS (SNB-75, 13%), Melanoma (UACC-62, 11%), Renal (CAKI-1, 16%; UO-31, 21%), Breast (MCF-7, 14%; MDA-MB-231/ATCC, 12%; HS-578T, 11%) | 99.39 |
| 5 | 59/59 | Leukemia (CCRF-CEM, 90%; HL-60(TB), 120%; K-562, 94%, MOLT-4, 89%; RPMI-8226, 83%; SR, 90%), Non-Small Cell Lung (A549/ATCC, 71%; EKVX, 65%; HOP-62, 79%; HOP-92, 42%; NCI-H226, 69%; NCI-H23, 61%; NCI-H322M, 67%; NCI-H460, 94%; NCI-H522, 126%), Colon (COLO-205, 102%; HCC-2998, 80%; HCT-116, 79%; HCT-15, 53%; HT-29, 68%; KM-12, 85%; SW-620, 79%), CNS (SF-268, 82%; SF-295, 80%; SF-539; 72%; SNB-19, 59%; SNB-75, 62%; U-251, 76%), Melanoma (LOX IMVI, 99%; MALME-3M, 69%; M14, 96%; MDA-MB-435, 87%; SK-MEL-2, 90%; SK-MEL-28, 67%; SK-MEL-5, 95%; UACC-257, 87%; UACC-62, 94%), Ovarian (IGROV1, 74%; IGROV-3, 85%; IGROV-4, 88%; IGROV-5, 45%; OVCAR-8, 80%; NCI/ADR-RES, 13%; SK-OV-3, 79%), Renal (786-0, 68%; ACHN, 67%; CAKI-1, 69%; RXF 393, 58%; SN-12C, 68%; TK-10, 85%; UO-31, 54%), Prostate (PC-3, 51%; DU-145, 62%), Breast (MCF-7, 87%; MDA-MB-231/ATCC, 75%; HS-578T, 37%; BT-549, 67%; T-47D, 94%; MDA-MB-468, 110%). | 23.46 |
| 6 | 58/59 | Leukemia (CCRF-CEM, 77%; HL-60(TB), 60%; K-562, 64%, MOLT-4, 80%; RPMI-8226, 51%; SR, 76%), Non-Small Cell Lung (A549/ATCC, 31%; EKVX, 40%; HOP-62, 65%; HOP-92, 46%; NCI-H226, 43%; NCI-H23, 37%; NCI-H322M, 53%; NCI-H460, 51%; NCI-H522, 101%), Colon (COLO-205, 71%; HCC-2998, 50%; HCT-116, 58%; HCT-15, 25%; HT-29, 14%; KM-12, 61%; SW-620, 40%), CNS (SF-268, 62%; SF-295, 35%; SF-539; 46%; SNB-19, 45%; SNB-75, 15%; U-251, 57%), Melanoma (LOX IMVI, 84%; MALME-3M, 41%; M14, 58%; MDA-MB-435, 29%; SK-MEL-2, 64%; SK-MEL-28, 43%; SK-MEL-5, 42%; UACC-257, 30%; UACC-62, 62%), Ovarian (IGROV1, 38%; IGROV-3, 87%; IGROV-4, 60%; IGROV-5, 32%; OVCAR-8, 56%; SK-OV-3, 37%), Renal (786-0, 29%; ACHN, 28%; CAKI-1, 46%; RXF 393, 28%; SN-12C, 44%; TK-10, 47%; UO-31, 39%), Prostate (PC-3, 33%; DU-145, 28%), Breast (MCF-7, 50%; MDA-MB-231/ATCC, 70%; HS-578T, 29%; BT-549, 44%; T-47D, 67%; MDA-MB-468, 48%). | 55.22 |
| 7 | 29/59 | Leukemia (CCRF-CEM, 46%; HL-60(TB), 16%; K-562, 40%, MOLT-4, 36%; RPMI-8226, 56%; SR, 17%), Non-Small Cell Lung (A549/ATCC, 14%; HOP-62, 14%; NCI-H23, 16%; NCI-H522, 30%), Colon (HCT-15, 26%; KM-12, 11%), CNS (SF-268, 15%; SF-295, 23%; SNB-75, 18%), Melanoma (LOX IMVI, 21%; SK-MEL-2, 19%; 67%; SK-MEL-5, 32%; UACC-62, 33%), Ovarian (IGROV1, 15%; IGROV-4, 10%; OVCAR-8, 23%), Renal (CAKI-1, 16%; SN-12C, 11%; UO-31, 29%), Prostate (PC-3, 18%), Breast (MCF-7, 17%; MDA-MB-231/ATCC, 13%; T-47D, 36%). | 89.35 |
| 8 | 15/59 | Leukemia (RPMI-8226, 13%), Non-Small Cell Lung (EKVX, 15%; NCI-H226, 11%; NCI-H322M, 15%; NCI-H522, 48%), CNS (SF-268, 15%; SF-539, 12%;), Renal (CAKI-1, 14%; RXF 393, 10%; SN-12C, 10%; UO-31, 25%), Breast (MCF-7, 11%; MDA-MB-231/ATCC, 15%; HS-578T, 15%; MDA-MB-468, 19%). | 96.77 |
| 9 | 28/59 | Leukemia (CCRF-CEM, 13%; K-562, 37%, MOLT-4, 14%; SR, 51%), Non-Small Cell Lung (A549/ATCC, 15%; EKVX, 10%; NCI-H226, 16%; NCI-H522, 58%), Colon (HCT-15, 11%; HT-29, 14%; KM-12, 14%), CNS (SF-268, 22%; SNB -19, 11%; SNB-75, 29%), Melanoma (LOX IMVI, 18%; MDA-MB-435, 54%; SK-MEL-5, 26%; UACC-62, 22%), Ovarian (OVCAR-3, 21%), Renal (CAKI-1, 36%; RXF 393, 14%; UO-31, 20%), Prostate (PC-3, 11%), Breast (MCF-7, 41%; MDA-MB-231/ATCC, 11%; HS-578T, 12%; T-47D, 19%; MDA-MB-468, 19%). | 88.50 |
| 10 | 6/59 | Non-small cell Lung (EKVX, 10%; NCI-H322M, 14%), CNS (SF-268, 14%; U-251, 15%), and Renal (CAKI-1, 16%; UO-31, 20%). | 100.92 |
| 11 | 17/59 | NSC Lung (NCI-H522, 52%), Colon (HCT-116, 20%), C.N.S. (SF-268, 25%; SF-539; 28%; SNB-19, 15%; SNB-75, 22%; U-251, 20%), Melanoma (SK-MEL-5, 10%), Ovarian (OVCAR-5, 12%), Renal (CAKI-1, 21%; RXF 393, 16%; UO-31, 21%), Breast (M.C.F.-7, 10%; MDA-MB-231/ATCC, 25%; HS-578T, 19%; T-47D, 11%; MDA-MB-468, 19%). | 94.54 |
| 12 | 25/59 | Leukemia (SR, 28%), N.S.C. Lung (NCI-H23, 17%; NCI-H522, 62%), Colon (HCT-116, 17%; KM-12, 30%), CNS (SF-268, 64%; SF-539; 64%; SNB-19, 49%; SNB-75, 39%; U-251, 26%), Melanoma (LOX IMVI, 20%; MALME-3M, 30%; M14, 11%; MDA-MB-435, 30%; SK-MEL-5, 32%; UACC-62, 17%), Ovarian (OVCAR-5, 14%), Renal (786-0, 15%; CAKI-1, 16%; RXF 393, 30%), Breast (MDA-MB-231/ATCC, 28%; HS-578T, 11%; BT-549, 57%; T-47D, 62%; MDA-MB-468, 27%). | 88.40 |
| 13 | 8/59 | Leukemia (S.R., 12%), C.N.S. (SNB-75, 13%), Renal (CAKI-1, 15%; UO-31, 24%), Breast (M.C.F.-7, 11%; MDA-MB-231/ATCC, 11%; T-47D, 15%; MDA-MB-468, 16%) | 97.44 |
| Imatinib | 20/59 | Leukemia (MOLT-4, 18%; PRMI-8226, 12.6%; SR, 14.6%), N.S.C. Lung (EKVX, 15.7%; NCI-H226, 10.6%; NCI-H23, 17.1%), Colon (HCT-116, 18.6%; HCT-15, 11.5%; HT-29, 47.1%), CNS (SF-295, 15.1%; SF-539, 24.5%; U251, 10.6%), Melanoma (LOX IMVI, 11.6%; SK-MEL-5, 22.3%), Renal (A-498, 13.7%), Prostate (PC-3, 10.6%; DU-145, 14.4%), Breast (MDA-MB-231/ATCC, 11.2%; T-47D, 18.6%; MDA-MB-468, 29.1%) | 92.62% |
Among ethylidene hydrazones, those with a methoxyphenyl group, like 11 and 12 (PCE = 17/59 and 25/59), were much better at killing tumors than those with an unsubstituted phenyl 10 (PCE = 6/59). The introduction of more than two methoxy groups to the phenyl ring, as in compound 13, slightly increased its effectiveness against tumors (PCE = 8/59) compared to unsubstituted phenyl 10 (PCE = 6/59); however, it was not as potent as methoxyphenyl 11 and 12, which had PCEs of 17/59 and 25/59, respectively. Hydrazones 4, 5, and 8 kill tumor cells more effectively (PCE = 9/59, 29/59, and 15/59, respectively) than the comparable ethylidene hydrazones 10, 11, and 13 (PCE = 6/59, 17/59, and 8/59, respectively).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 781) and gefitinib (759856) on in vitro subpanel tumor cell lines μma
781) and gefitinib (759856) on in vitro subpanel tumor cell lines μma
| Compound | Subpanel tumor cell lines | MG_MIDa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Activity | Leukemia | NSC lung cancer | Colon cancer | CNS cancer | Melanoma | Ovarian cancer | Renal cancer | Prostate cancer | Breast cancer | ||
| a aFull panel mean-graph midpoint (μM); c Compounds showed values > 100 μM; mean 50% cell growth inhibition (GI50); total cell growth inhibition (TGI); median lethal concentration (LC50); Mean GI50 graph midpoints (GI50 MG_MID). | |||||||||||
| 5 | GI50 | 0.384 | 1.059 | 1.557 | 1.494 | 1.501 | 15.11 | 1.917 | 0.886 | 1.23 | 1.096 |
| TGI | 68.371 | c | c | c | 59.09 | c | 91.54 | c | 84.17 | 89.24 | |
| LC50 | c | c | c | c | c | c | c | c | c | c | |
| Erlotinib | GI50 | 27.85 | 13.11 | 51.68 | 16.99 | 23.74 | 5.52 | 2.46 | 20.90 | 24.72 | 7.68 |
| TGI | 96.57 | 73.76 | c | 82.11 | 77.89 | 74.41 | 42.59 | c | 70.53 | 66.3 | |
| LC50 | c | 97.71 | c | c | 93.31 | 97.06 | 89.15 | c | 96.43 | 95.6 | |
| Gefitinib | GI50 | 2.56 | 2.05 | 5.23 | 5.64 | 3.68 | 3.05 | 1.41 | 3.29 | 4.67 | 2.10 |
| TGI | 12.07 | 13.86 | 18.47 | 19.62 | 12.49 | 33.29 | 12.50 | 31.62 | 18.62 | 14.3 | |
| LC50 | 93.85 | 94.68 | 51.74 | 50.56 | 36.40 | 83.58 | 52.82 | 89.72 | 52.47 | 51.9 | |
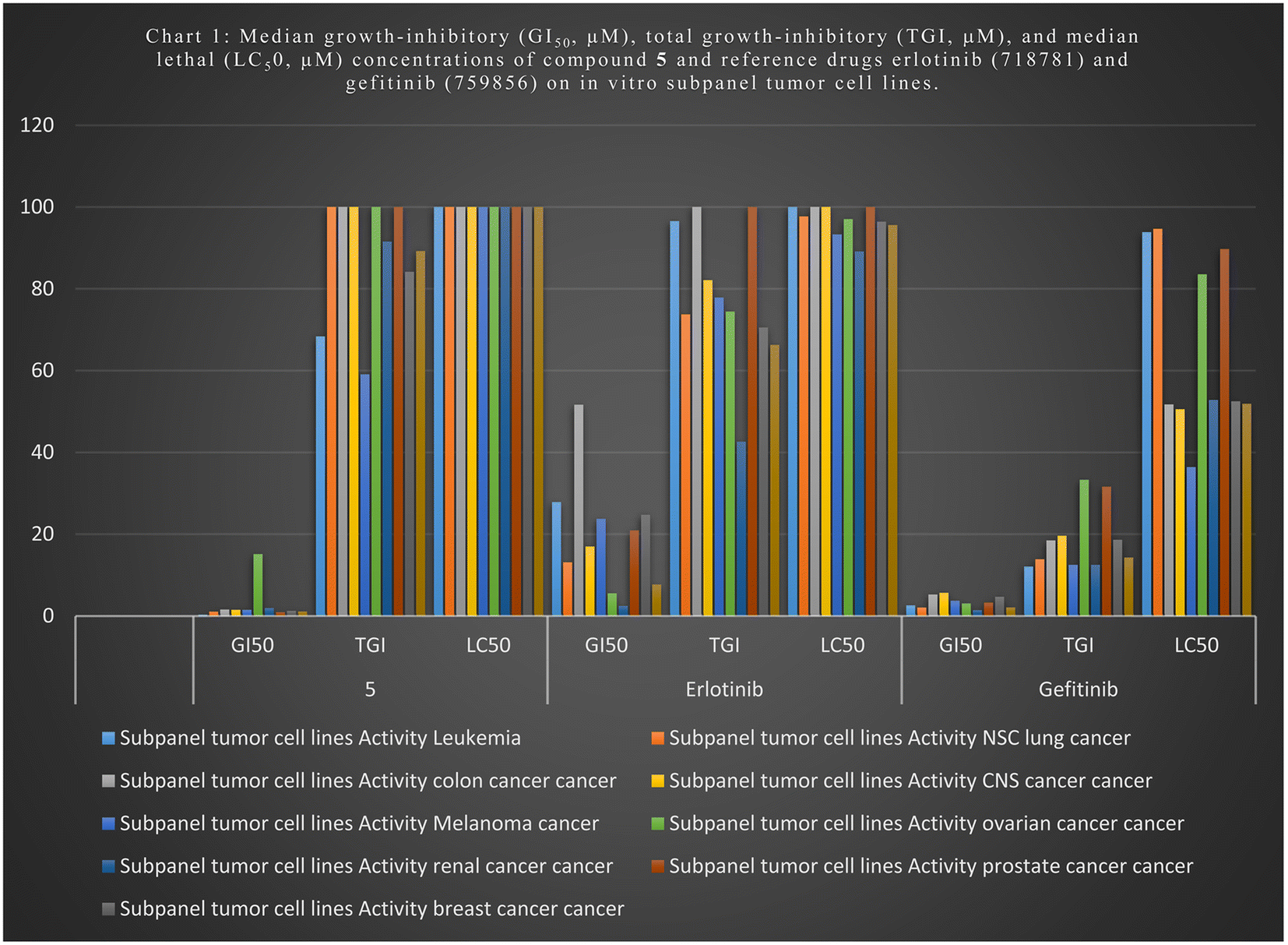 | ||
| Chart 1 Median growth-inhibitory (GI50, μM), total growth-inhibitory (TGI, μM), and median lethal (LC50, μM) concentrations of compound 5 and reference drugs erlotinib (718781) and gefitinib (759856) on in vitro subpanel tumor cell lines. | ||
| Subpanel tumor cell lines | GI50 (μM) | ||
|---|---|---|---|
| 5 | Erlotinib | Gefitinib | |
| a nt = not tested; c compounds showed values > 100 μM. | |||
| Leukemia | |||
| CCRF-CEM | 0.196 | 15.84 | 5.01 |
| HL-60(TB(K-562)) | 0.412 | 5.01 | 5.01 |
| 0.592 | 15.48 | 2.51 | |
| MOLT-4 | 0.372 | 5.01 | 3.98 |
| RPMI-8226 | 0.521 | 5.01 | 1.58 |
| SR | 0.212 | 6.30 | 3.16 |
|
|||
| Non-small cell lung cancer | |||
| A549/ATCC | 1.57 | 7.94 | 7.94 |
| EKVX | 0.650 | 0.005 | 0.005 |
| HOP-62 | 0.961 | 12.58 | 10.00 |
| HOP-92 | nt | 6.30 | 7.94 |
| NCI-H226 | 0.894 | 6.30 | 15.84 |
| NCI-H23 | 1.96 | 19.95 | 15.84 |
| NCI-H322M | 0.575 | 0.05 | 0.08 |
| NCI-H460 | 0.424 | 5.01 | 6.30 |
| NCI-H522 | 2.40 | 1.00 | 6.30 |
|
|||
| Colon cancer | |||
| COLO 205 | 1.51 | 31.62 | 6.30 |
| HCC-2998 | 0.882 | 79.34 | 10.00 |
| HCT-116 | 0.363 | 5.01 | 7.94 |
| HCT-15 | 3.62 | 3.16 | 5.01 |
| HT29 | 3.01 | 50.11 | 3.98 |
| KM12 | 0.569 | 63.09 | 7.94 |
| SW-620 | 0.949 | 5.01 | 7.94 |
|
|||
| CNS cancer | |||
| SF-268 | 0.766 | 19.95 | 7.94 |
| SF-295 | 1.47 | 15.84 | 1.99 |
| SF-539 | 1.26 | 12.58 | 10.00 |
| SNB-19 | 2.10 | 3.98 | 12.58 |
| SNB-75 | 2.86 | 12.58 | 6.30 |
| U251 | 0.510 | 19.95 | 10.00 |
|
|||
| Melanoma | |||
| LOX IMVI | 0.387 | 5.01 | 7.94 |
| MALME-3M | 1.09 | 5.01 | 3.16 |
| M14 | 0.452 | 6.30 | 5.01 |
| MDA-MB-435 | 2.07 | 15.84 | 3.16 |
| SK-MEL-2 | 2.41 | 12.58 | 12.58 |
| SK-MEL-28 | 1.62 | 31.62 | 0.31 |
| SK-MEL-5 | 1.57 | 15.84 | 3.98 |
| UACC-257 | 3.03 | 100 | 6.30 |
| UACC-62 | 0.886 | 1.25 | 5.01 |
|
|||
| Ovarian cancer | |||
| IGROV1 | 0.785 | 0.25 | 0.20 |
| OVCAR-3 | 0.287 | 3.16 | 5.01 |
| OVCAR-4 | 0.311 | 19.95 | 7.94 |
| OVCAR-5 | 2.09 | 19.95 | 10.00 |
| OVCAR-8 | 0.932 | 7.94 | 10.00 |
| NCI/ADR-RES | c | 6.30 | 12.58 |
| SK-OV-3 | 1.32 | 0.39 | 0.63 |
|
|||
| Renal cancer | |||
| 786-0 | 1.84 | 5.01 | 7.94 |
| A498 | nt | 1.58 | 0.40 |
| ACHN | 1.58 | 0.15 | 0.20 |
| CAKI-1 | 1.02 | 0.10 | 0.16 |
| RXF 393 | 2.50 | 6.30 | 5.01 |
| SN12C | 1.93 | 6.3 | 6.30 |
| TK-10 | 1.66 | 0.10 | 0.10 |
| UO-31 | 2.89 | 1.99 | 1.25 |
|
|||
| Prostate cancer | |||
| PC-3 | 0.829 | 50.11 | 0.79 |
| DU-145 | 0.944 | 1.58 | 2.51 |
|
|||
| Breast cancer | |||
| MCF7 | 0.457 | 100 | 10.00 |
| MDA-MB-231/ATCC | 0.643 | 1.99 | 12.58 |
| HS 578T | 2.11 | 6.30 | 10.00 |
| BT-549 | 2.68 | 39.81 | 7.94 |
| T-47D | 0.925 | 3.16 | 6.30 |
| MDA-MB-468 | 0.524 | 0.20 | 0.01 |
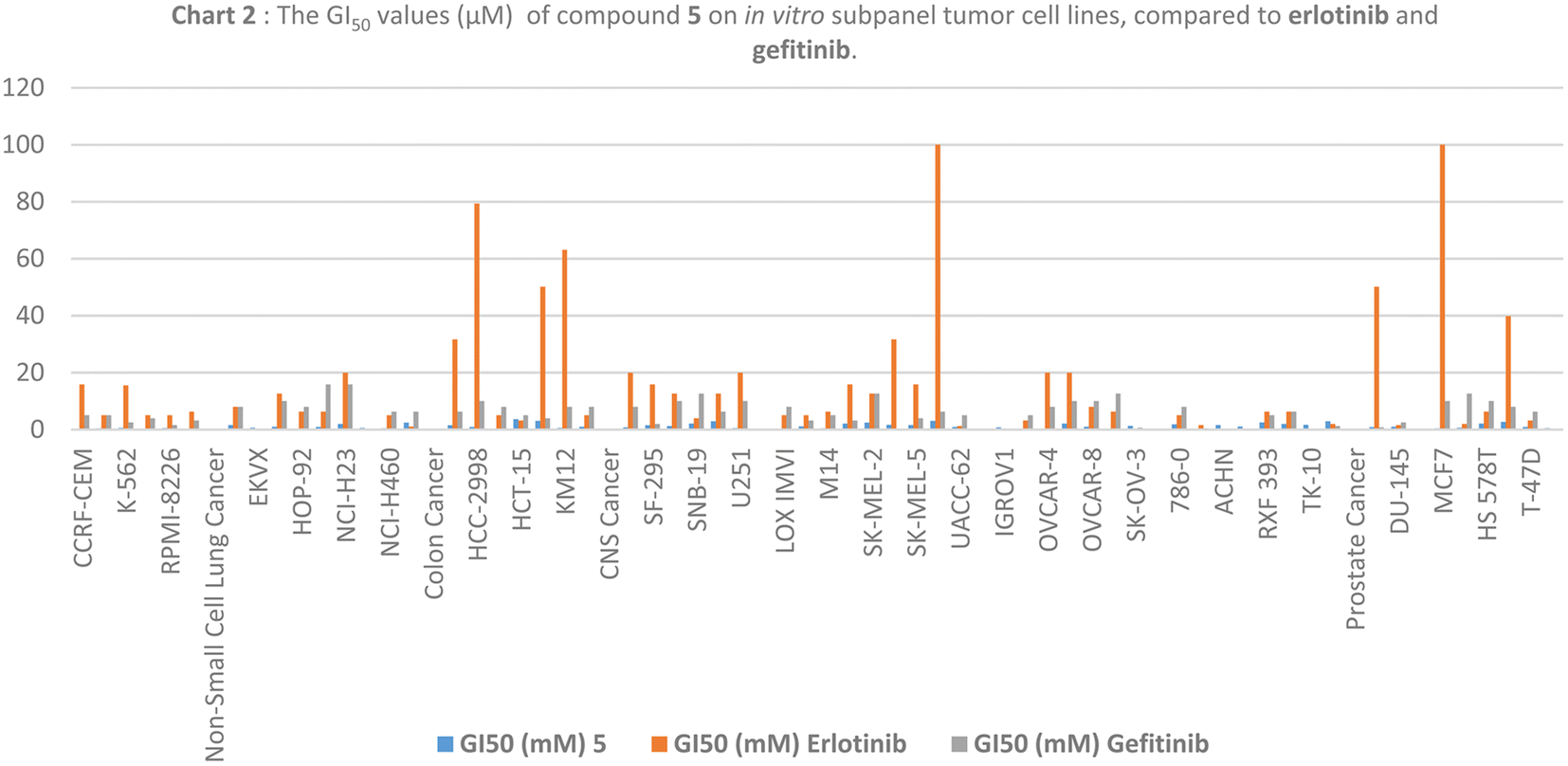 | ||
| Chart 2 The GI50 values (μM) of compound 5 on in vitro subpanel tumor cell lines, compared to erlotinib and gefitinib. | ||
| Compound no. | IC50a nM | IC50a (μM) | |||
|---|---|---|---|---|---|
| COX-2 inhibition | EGFR inhibition | HER2 inhibition | CDK9 inhibition | WI138 | |
| 5 | 3.32 ± 0.06 (μM) | 84.39 ± 2.07 | 53.91 ± 1.32 | 146.9 ± 3.60 | 45.326 ± 2.66 |
| 6 | 4.47 ± 0.09 (μM) | 51.52 ± 1.26 | 44.13 ± 1.08 | 96.07 ± 2.35 | 27.772 ± 1.63 |
| 7 | 50.48 ± 1.06 (μM) | 594.45 ± 14.5 | 387.01 ± 9.50 | 155.4 ± 3.81 | — |
| 9 | 15.41 ± 0.32 (μM) | 377.53 ± 9.26 | 228.53 ± 5.61 | 316.40 ± 7.76 | — |
| 12 | 21.27 ± 0.44 (μM) | 492.27 ± 12.1 | 110.64 ± 2.71 | 319.8 ± 7.85 | — |
| Celecoxib | 0.15 ± 0.003 (μM) | — | — | — | — |
| Gefitinib | 53.12 ± 1.30 | 38.81 ± 0.95 | — | ||
| Dinaciclib | — | — | — | 53.12 ± 1.30 | |
| Doxorubicin | — | — | — | — | 9.57 ± 0.59 |
2.2 Structure–activity relationship
Based on the data, we can conclude that the Schiff base series with a 2-hydroxyphenyl group on the acylhydrazone moiety is the most potent kinase inhibitor against EGFR (IC50; 84.4 and 51.5 nM), HER2 (IC50; 53.9 and 44.1 nM), and CDK9 (IC50; 146.9 and 96.1 nM), as compounds 5 and 6, respectively. Schiff base with a 3-methoxyphenyl group on the acylhydrazone moiety, such as compound 7, showed a potent CDK9 kinase inhibitor with an IC50 value of 155.4 nM, while compound 12 with the (3,5-dimethoxyphenyl)ethylidene)hydrazineyl) moiety showed vigorous HER2 kinase inhibitor activity with an IC50 value of 110.6 nM.2.3 In vitro cytotoxicity against WI-38 fibroblast cell line
The most active kinase inhibitors, acylhydrazones 5 and 6, were less toxic than doxorubicin when measured for their safety margin cytotoxicity against the standard WI-38 fibroblast cell line with IC50 values of 45.3 and 27.8 μM, respectively, compared to 9.6 μM for doxorubicin Table 4.2.4 Cell cycle arrest analysis and apoptosis detection
We are studying cell cycle arrest and apoptosis in T-47D and MOLT4 cells to determine the role of our promising derivatives 5 and 6 in the cell cycle (Tables 5 and 6). Flow cytometry assays quantify the DNA content. After being treated with compounds 5 and 6, the number of cells in the S phase dropped from 32.3 percent for DMSO-control cells to 20.9 percent for T-47D cells and 22.5 percent for MOLT4 cells. Also, compound 5 stopped the cells in the G2/M stage, raising the percentage of T-47D cells in the G2-M phase from 6.6% in the control cell to 26.3%. Acylhydrazone 6 stopped the cells in the G1 stage, lowering the percentage of MOLT4 cells in the G0-G1 phase from 89.0% in the control cell to 61.4%. Acylhydrazone 6 lowered the percentage of MOLT4 cells in the G0-G1 phase from 89.0% in the control cell to 61.4% at the G1 stage. Furthermore, compounds 5 and 6 boosted early apoptosis from 0.4 and 0.6 (DMSO control sample) to 9.3% and 19.2, respectively; also, derivatives 5 and 6 increased late apoptosis from 0.1 to 14.8% and 12.6 to 0.3%, respectively, when stained with annexin-5/PI in T-47D and MOLT4 cells compared to the control group treated with DMSO. These investigated that compounds 5 and 6 preferred the apoptotic pathway over the necrotic pathway for cell death (Fig. 4 and 5).| Compound no. | %G0-G1 | %S | %G2-M | Comment |
|---|---|---|---|---|
| Five/T-47D | 46.31 | 27.42 | 26.27 | Cell growth arrest at the G2/M phase |
| DMSO/T-47D | 61.11 | 32.29 | 6.6 | — |
| 6/MOLT4 | 89.04 | 9.56 | 1.40 | Cell growth arrest at the G1 phase |
| DMSO/MOLT4 | 61.37 | 22.54 | 16.09 | — |
| Compound no. | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 5/T-47D | 31.07 | 9.28 | 14.75 | 7.04 |
| DMSO/T-47D | 2.37 | 0.37 | 0.14 | 1.86 |
| 6/MOLT4 | 35.06 | 19.22 | 12.61 | 3.23 |
| DMSO/MOLT4 | 1.71 | 0.55 | 0.31 | 0.85 |
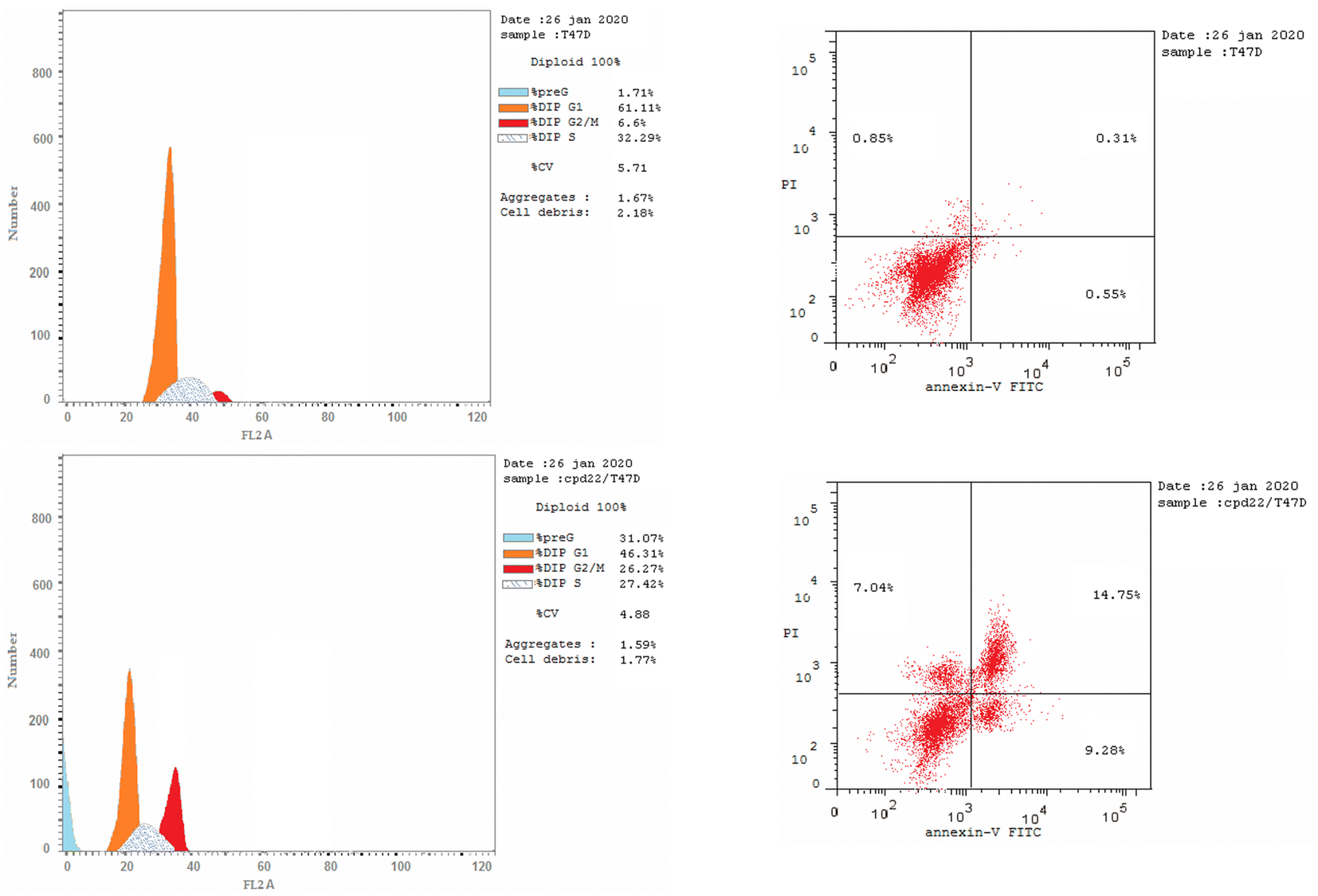 | ||
| Fig. 4 Effect of DMSO (upper right panel) and 5 (lower right panel) on the percentage of annexin V-FITC-positive staining in MCF-7 cell. Cell cycle analysis of T47D cells treated with DMSO (upper left panel) and cell cycle analysis of T47D cells treated with compound 5 (lower left panel). | ||
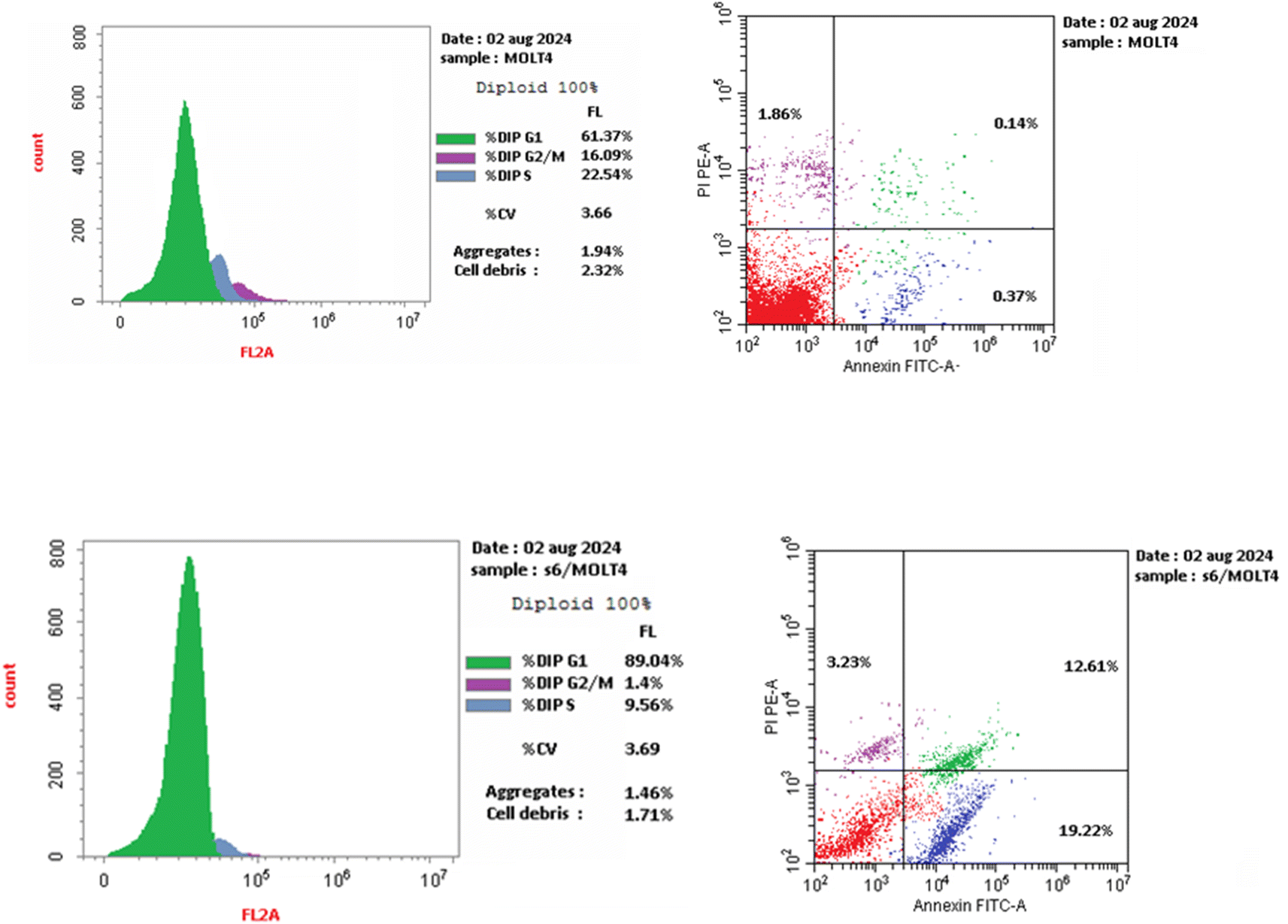 | ||
| Fig. 5 Effect of DMSO (upper right panel) and 6 (lower right panel) on the percentage of annexin V-FITC-positive staining in MCF-7 cells. Cell cycle analysis of MOLT4 cells treated with DMSO (upper left panel) and MOLT4 cells treated with compound 6 (lower left panel). | ||
2.5 Molecular docking
Molecular docking simulations of compound 5 and compound 6 with the HER2 receptor (PDB: 7PCD), EGFR kinase domain (PDB: 1XKK), and CDK9 (PDB: 3RCD) reveal significant insights into their potential as inhibitors, highlighting key hydrogen bond and hydrophobic interactions contributing to their binding affinities.2.6 HER2 receptor interactions
Compound 5 exhibits a strong binding affinity of −9.2 kcal mol−1 with the HER2 receptor, attributed to a network of interactions within the active site (Table 7). It forms hydrogen bonds with PHE864, LYS753, THR862, and MET801 as an acceptor and LEU796 as a donor. These hydrogen bonds, particularly those with THR862 and MET801 within the critical ATP-binding pocket, likely underpin its strong affinity. Additionally, compound 5 engages in hydrophobic interactions with LEU726, VAL734, ALA751, LYS753, GLU770, ALA771, THR798, LEU800, and PHE864, further stabilizing its binding within the hydrophobic environment of the active site (Fig. 6A). Compound 6 shows a weaker binding affinity of −7.9 kcal mol−1, forming fewer hydrogen bonds within the active site. It acts as a hydrogen bond acceptor with MET801, LYS753, LYS860, and ARG784. While the interaction with MET801 in the ATP-binding site is promising, the fewer hydrogen bonds suggest a weaker overall interaction than compound 5. Derivative 6 also benefits from hydrophobic interactions with LEU726, ILE767, THR798, LEU800, THR862, and PHE864, contributing to its binding stability (Fig. 6B).| Compound | Ligand | Receptor | Interaction | Distance | Binding affinities (kcal mol−1) |
|---|---|---|---|---|---|
| EGFR (PDB: 1XKK) | |||||
| 5 | C 15 | OD2 ASP 855 (A) | H-donor | 3.42 | −8.513 |
| O 40 | O SER 720 (A) | H-donor | 3.01 | ||
| O 26 | N MET 793 (A) | H-acceptor | 3.54 | ||
| O 36 | HA ASP 855 (A) | H-acceptor | 3.33 | ||
| 6 | N 1 | O LEU 788 (A) | H-donor | 3.22 | −8.109 |
| O 20 | N MET 793 (A) | H-acceptor | 3.67 | ||
|
|||||
| HER2 (PDB: 7PCD) | |||||
| 5 | O 38 | O PHE 864 (A) | H-acceptor | 3.83 | −9.159 |
| N 21 | HZ1 LYS 753 (A) | H-acceptor | 3.84 | ||
| O 14 | OG1 THR 862 (A) | H-acceptor | 3.35 | ||
| O 26 | N MET 801 (A) | H-acceptor | 3.38 | ||
| N 38 | O LEU 796 (A) | H-donor | 3.1 | ||
| 6 | O 20 | N MET 801 (A) | H-acceptor | 3.23 | −7.854 |
| O 24 | NZ LYS 753 (A) | H-acceptor | 3.56 | ||
| N 27 | O LYS 860 (A) | H-acceptor | 3.51 | ||
| O 35 | N ARG 784 (A) | H-acceptor | 4.09 | ||
|
|||||
| CDK9 (PDB: 3BLR) | |||||
| 5 | O 26 | N CYS 106 (A) | H-acceptor | 3.47 | −7.425 |
| N 38 | OD1 ASP 109 (A) | H-donor | 2.96 | ||
| 6 | N 1 | OD2 ASP 109 (A) | H-donor | 2.92 | −7.656 |
| O 20 | N CYS 106 (A) | H-acceptor | 3.12 | ||
| O 33 | N LYS 151 (A) | H-acceptor | 4.09 | ||
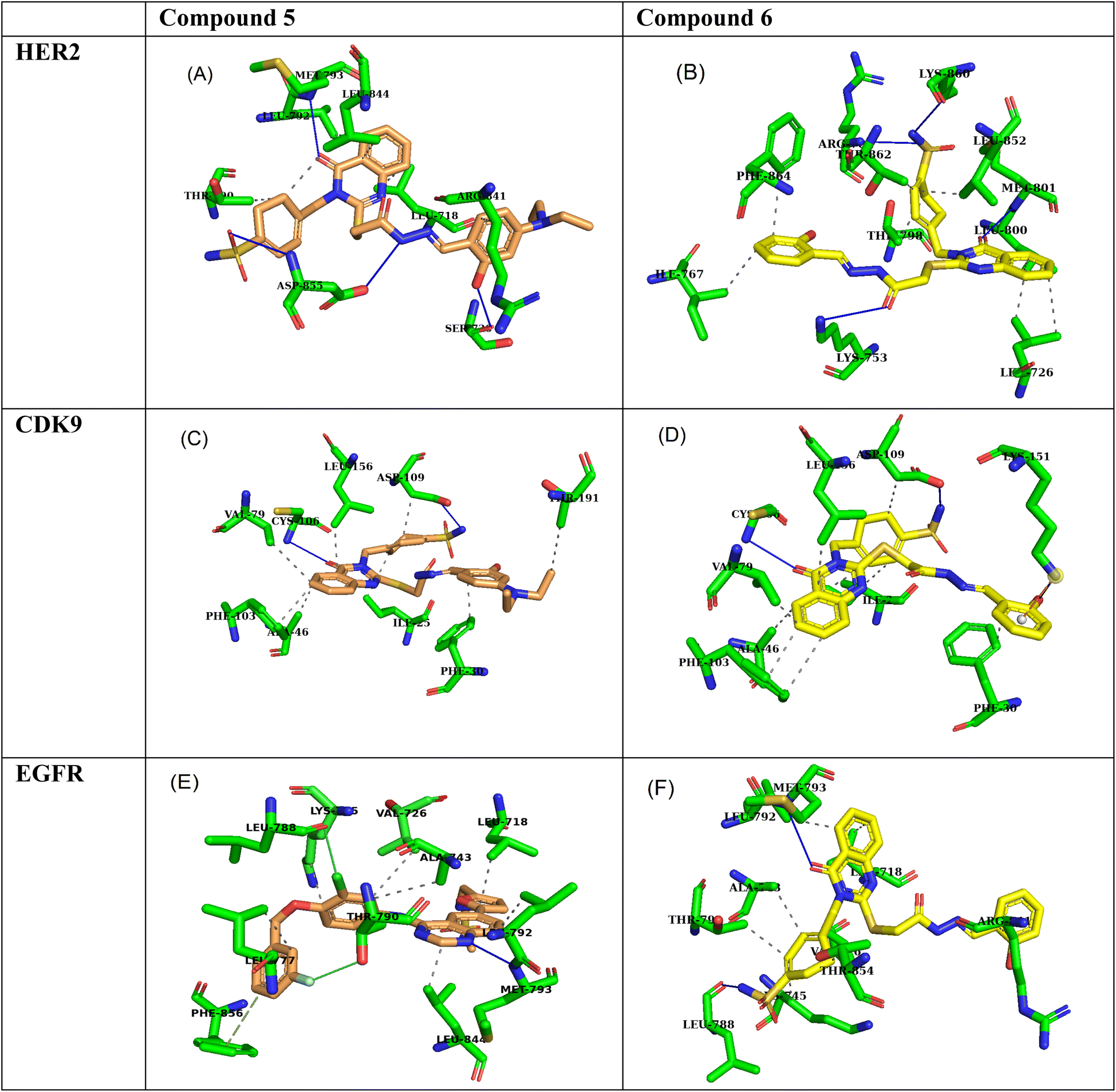 | ||
| Fig. 6 3D interaction poses of compound 5 and compound 6 within the active sites of (A and B) HER2, (C and D) EGFR, and (E and F) CDK9. Compound 5 is depicted in orange, and compound 6 is in yellow. | ||
2.7 EGFR kinase domain interactions
Compound 5 demonstrates a strong binding affinity of −8.5 kcal mol−1 with the EGFR kinase domain, engaging in a network of hydrogen bonds and hydrophobic interactions within the active site (Table 7). It forms hydrogen bonds with ASP855 as both a donor and acceptor, SER720 as a donor, and MET793 as an acceptor. The interaction with MET793, a key residue within the hinge region of the ATP-binding pocket, is particularly noteworthy. Moreover, compound 5 engages in hydrophobic interactions with LEU718, THR790, LEU792, ARG841, and LEU844, further enhancing its binding stability within the hydrophobic environment of the active site (Fig. 6C).Quinazoline 6 exhibits a slightly weaker binding affinity of −8.1 kcal mol−1, forming fewer hydrogen bonds within the active site. It acts as a hydrogen bond donor with LEU788 and an acceptor with MET793. The interaction with MET793 in the ATP-binding pocket is favorable, but the lower number of hydrogen bonds suggests a slightly weaker interaction than compound 5. Schiff base 6 also benefits from hydrophobic interactions with a broader range of residues, including LEU718, VAL726, ALA743, LYS745, THR790, LEU792, ARG841, and THR854, contributing to its overall binding stability (Fig. 6D).
2.8 CDK9 kinase interactions
Derivative 5 exhibits a binding affinity of −7.4 kcal mol−1 with CDK9, primarily driven by two key hydrogen bonds. It acts as a hydrogen bond acceptor with CYS106 and a donor with ASP109. These interactions likely position compound 5 within the active site to effectively interfere with ATP binding or catalytic activity. Furthermore, compound 5 benefits from hydrophobic interactions with ILE25, PHE30, ALA46, VAL79, PHE103, ASP109, LEU156, and THR191, contributing to its overall binding stability within the hydrophobic environment of the active site (Table 7, Fig. 6E). Compound 6 demonstrates a slightly stronger binding affinity of −7.7 kcal mol−1, attributed to more hydrogen bonds. It forms a hydrogen bond as a donor with ASP109 and an acceptor with CYS106 and LYS151. The interaction with LYS151, while at a longer distance (4.09 Å), could provide additional binding energy. Compound 6 also engages in hydrophobic interactions with ILE25, PHE30, ALA46, VAL79, PHE103, ASP109, and LEU156, further stabilizing its binding within the active site (Fig. 6F).3 Conclusion
The NCI, Bethesda, MD, USA, evaluated the newly synthesized quinazolines 1–13 for their in vitro cytotoxic activity against 59 cell lines. Acylhydrazones 5, 6, 7, 9, and 12 were the most effective at killing cancer cells, lowering the mean growth percentage (MG%) by an average of 23.5, 55.2, 89.4, 88.5, and 88.4%, in that order. Compound 5 was selected for other investigation by NCI, Bethesda, MD, USA, at five-dose dilutions on 59 tumor cells. Regarding tumor cell death, compound 5 compared to gefitinib (mean GI50: 7.7 μM) and erlotinib (mean GI50: 2.1 μM), with a mean GI50 value of 1.1 μM. It had an LC50 value above 100 μM, whereas the values for gefitinib and erlotinib were 95.6 μM and 14.3 μM, respectively. Those medications' TGIs were 14.3 μM and 66.3 μM, respectively, compared to compound 5, with a TGI value of 89.2 μM. acylhydrazones 5, 6, 7, 9, and 12 exhibited the highest cytotoxic activity of the newly synthesized compounds 1–13 evaluated against the cell lines by reducing the mean growth percentage (MG%) by an average of 23.5, 55.2, 89.4, 88.5, and 88.4%, respectively. The acylhydrazones 5, 6, 7, 9, and 12 exhibit superior cytotoxic efficacy against the cell lines under investigation. The dose-dependent enzymatic inhibition experiments were conducted on EGFR, HER2, and CDK9 kinases at different dosages to determine their IC50 magnitudes in the nanomolar range. The Schiff base series with 2-hydroxyphenyl moiety, represented by compounds 5 and 6, is the most effective kinase inhibitor against EGFR (IC50; 84.4 and 51.5 nM), HER2 (IC50; 53.9 and 44.1 nM), and CDK9 (IC50; 146.9 and 96.1 nM). Compound 12 with the (3,5-dimethoxyphenyl)ethylidene)hydrazineyl) moiety demonstrated intense HER2 kinase inhibitor activity with an IC50 value of 110.6 nM. Schiff bases 7, with a 3-methoxyphenyl group on the acylhydrazone moiety, demonstrated a potent CDK9 kinase inhibitor with an IC50 value of 155.4 nM. Throughout the investigation of cell cycle analysis for acylhydrazones 5 and 6, the S phase cell count decreased from 32.3% and 9.56 in the DMSO-control cells to 20.9% in T-47D cells and 22.5% in MOLT4 cells. Additionally, compound 5 increased the proportion of T-47D cells from 6.6% in the control cell to 26.3% by halting the G2/M stage cells. After adding substance 6, the percentage of MOLT4 cells in the G0-G1 phase decreased from 89.0% in the control cell to 61.4%. Compounds 5 and 6 enhanced early death from 0.4 and 0.6% (DMSO control sample) to 9.3% and 19.2%, respectively, as measured by flow cytometers using annexin-5/PI staining of T-47D and MOLT4 cells, respectively. The late death rate increased from 0.1 to 14.8% with derivative 5 and 12.6 to 0.3% with derivative 6. These results demonstrate that acylhydrazones 5 and 6 preferred the apoptotic pathway than the necrotic pathway. Acylhydrazones 5 and 6 were less toxic than doxorubicin, with IC50 values of 45.3 and 27.8 μM, respectively, compared to 9.6 μM for doxorubicin when testing the cell death against the standard WI-38 fibroblast cell line. 2-Hydroxyacylhydrazones 5 and 6 showed potential inhibition of HER2, EGFR, and CDK9, due to their potential interaction. Due to the greater number of hydrogen bonds, 2-hydroxylamine 5 shows stronger binding affinities within the ATP-binding pockets, and the hydrophobic interactions contribute to their stability within the active sites.4 Experimental section
4.1 Chemistry
Melting points were recorded on a Barnstead 9100 Electrothermal melting apparatus. IR spectra (KBr) were recorded on an FT-IR PerkinElmer spectrometer (n cm−1). 1H and 13C NMR were recorded on Bruker 700 MHz spectrometers using DMSO-d6 as the solvent. We obtained microanalytical data (C, H, and N) using a PerkinElmer 240 analyzer, and the proposed structures were within approximately 0.4% of the theoretical values. Acquity UPLC machine (the UPH model with serial number H10UPH) and the Acquity TQD MS instrument (the TQD model with serial number QBB1203) are used for mass spectra recording. Compound 1 was prepared according to the reported method,66 with a melting point of 301–303 °C compared to 286–287 °C.4.2 4-((4-Oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)methyl)benzenesulfonamide (1)
A mixture of 2-aminobenzoic acid (411 mg, 3.0 mmol) with 4-isothiocyanatobenzenesulfonamide (642.00 mg, 3.00 mmol) in ethanol (19.0 ml) and trimethylamine (404.00 mg, 4.00 mmol) was heated for 5 hours. The quinazoline-2-thione 3 was obtained by filtration, washing with iced ethanol (70%), and drying. M.p 301–303°, 90% yield; ν: 3203, 3134 (NH), 1777 (CO), 1320, 1155 (OSO); 1H NMR (700 MHz, DMSO) δ 13.15 (s, 1H), 7.97 (dd, J = 7.8, 1.6 Hz, 1H), 7.87–7.65 (m, 3H), 7.47 (dd, J = 14.4, 8.3 Hz, 3H), 7.34 (d, J = 17.8 Hz, 3H), 5.73 (s, 2H). 13C NMR (176 MHz, DMSO) δ 175.89, 160.07, 143.09, 141.21, 140.08, 136.10, 127.81, 127.68, 126.15, 125.01, 116.57, 116.00, 49.11; Ms; [M+1, 348].
O), 1340, 1154 (OSO); 1H NMR (700 MHz, DMSO) δ 8.12 (dd, J = 8.0, 1.5 Hz, 1H), 7.87–7.79 (m, 3H), 7.52–7.33 (m, 6H), 5.42 (s, 2H), 4.14 (q, J = 7.1 Hz, 2H), 4.11 (s, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (176 MHz, DMSO) δ 168.63, 161.24, 156.43, 147.14, 143.78, 139.91, 135.60, 127.57, 127.19, 126.81, 126.51, 126.34, 119.11, 61.61, 47.15, 34.67, 14.62; Ms; [M+1, 434].O), 1329, 1159 (OSO); 1H NMR (700 MHz, DMSO) δ 9.40 (s, 1H), 8.13 (d, J = 7.9 Hz, 1H), 7.86–7.84 (m, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 8.2 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.37 (s, 2H), 5.42 (s, 2H), 4.38 & 4.32 (ss, 2H), 3.97 (s, 2H), 13C NMR (176 MHz, DMSO) δ 166.48, 161.38, 156.62, 147.26, 143.65, 140.06, 135.47, 127.57, 127.11, 126.70, 126.62, 126.53, 119.14, 47.08, 34.68; Ms; [M+1, 420].4.2.3.1 4-((2-((2-(2-Benzylidenehydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (4). M.p 267–268°, 93% yield; ν: 3363, 3249 (NH), 1667, 1602 (C
O), 1333, 1152 (OSO); 1H NMR (500 MHz, DMSO) δ 11.85 & 11.68 (s, 1H), 8.28 & 8.08 (s, 1H), 8.12 (dd, J = 7.9, 1.7 Hz, 1H), 7.83–7.78 (m, 3H), 7.71 (td, J = 6.2, 2.0 Hz, 2H), 7.57–7.41 (m, 6H), 7.37 (d, J = 2.2 Hz, 2H), 5.45 (d, J = 3.0 Hz, 2H), 4.60 & 4.12 (s, 2H). 13C NMR (176 MHz, DMSO) δ 169.10, 163.89, 161.33, 161.31, 156.85, 156.70, 147.22, 147.15, 143.91, 143.70, 143.65, 140.08, 140.03, 135.52, 134.57, 134.54, 130.61, 130.42, 129.31, 127.63, 127.60, 127.58, 127.33, 127.17, 126.74, 126.67, 126.53, 126.49, 126.47, 126.43, 119.17, 119.12, 47.18, 35.61, 34.49; Ms; [M+1, 508].
4.2.3.2 4-((2-((2-(2-(4-(Diethylamino)-2-hydroxybenzylidene)hydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (5). M.p 270–271°, 92% yield; ν: 3429 (OH), 3303, 3201 (NH), 1780, 1636 (C
O), 1331, 1153 (OSO); 1H NMR (700 MHz, DMSO) δ 11.76 & 11.36 (s, 1H), 11.14 & 9.98 (s, 1H), 8.25 & 8.16 (s, 1H), 8.12 (ddd, J = 8.3, 4.4, 1.6 Hz, 1H), 7.85–7.76 (m, 3H), 7.59–7.53 (m, 1H), 7.51–7.44 (m, 3H), 7.40–7.32 (m, 2H), 7.21 (d, J = 8.8 Hz, 1H), 6.25 & 6.18 (dd, J = 8.8, 2.5 Hz, 1H), 6.12 & 6.08 (d, J = 2.4 Hz, 1H), 5.43 (d, J = 5.8 Hz, 2H), 4.50 (s, 1H), 4.08 (s, 1H), 3.35–3.31 (m, 4H), 1.10–1.07 (m, 6H), 13C NMR (176 MHz, DMSO) δ 167.18, 162.34, 160.79, 159.42, 158.26, 156.35, 156.14, 150.09, 149.97, 148.74, 146.71, 146.69, 143.14, 143.07, 139.55, 139.48, 134.99, 134.96, 131.38, 127.10, 127.04, 126.64, 126.60, 126.20, 126.11, 125.99, 125.95, 125.93, 125.91, 118.62, 118.58, 107.04, 106.11, 103.86, 103.59, 97.29, 97.16, 46.61, 43.74, 43.71, 34.80, 34.29, 12.44; Ms; [M+1, 595].
4.2.3.3 4-((2-((2-(2-(2-Hydroxybenzylidene)hydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (6). M.p 288–290°, 94% yield; ν: 3372, 3265, 3183 (NH), 1671, 1611 (C
O), 1338, 1156 (OSO); 1H NMR (500 MHz, DMSO) δ 11.80 & 11.62 (s, 1H), 9.66 (s, 1H), 8.17 & 8.00 (s, 1H), 8.13–8.10 (m, 1H), 7.85–7.76 (m, 3H), 7.59–7.43 (m, 4H), 7.38 (d, J = 2.7 Hz, 2H), 7.28–7.06 (m, 3H), 6.84–6.82 (m, 1H), 5.45 (d, J = 5.8 Hz, 2H), 4.58 & 4.10 (s, 2H); 13C NMR (176 MHz, DMSO) δ 168.74, 163.87, 161.35, 161.33, 157.70, 156.89, 156.84, 156.62, 147.37, 147.21, 143.67, 143.62, 141.63, 140.10, 140.02, 135.55, 135.52, 131.97, 131.72, 129.63, 127.65, 127.59, 127.18, 127.16, 126.84, 126.78, 126.69, 126.54, 126.50, 126.46, 126.42, 120.51, 119.91, 119.88, 119.15, 119.10, 116.82, 116.64, 47.19, 35.34, 34.61; Ms; [M+1, 524].
4.2.3.4 4-((2-((2-(2-(3-Methoxybenzylidene)hydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (7). M.p 279–280°, 91% yield; ν: 3362, 3246 (NH), 1669, 1590 (C
O), 1351, 1151 (OSO); 1H NMR (500 MHz, DMSO) δ 11.86 & 11.70 (s, 1H), 8.24 & 8.05 (s, 1H), 8.12 (dt, J = 8.0, 2.0 Hz, 1H), 7.80 (qd, J = 7.9, 1.6 Hz, 3H), 7.56–7.43 (m, 4H), 7.37 (q, J = 2.4 Hz, 2H), 7.35–7.25 (m, 3H), 7.03–6.98 (m, 1H), 5.45 (d, J = 3.9 Hz, 2H), 4.61& 4.12 (s, 2H), 3.79 (d, J = 2.6 Hz, 3H), 13C NMR (176 MHz, DMSO) δ 169.10, 163.94, 161.33, 161.30, 160.01, 159.98, 156.86, 156.69, 147.21, 147.03, 143.76, 143.69, 143.65, 140.08, 140.03, 136.00, 135.96, 135.53, 135.50, 130.43, 127.63, 127.61, 127.17, 126.74, 126.66, 126.53, 126.50, 126.47, 126.39, 120.47, 119.93, 119.16, 119.12, 116.78, 116.33, 112.01, 111.70, 55.64, 55.60, 49.08, 47.18, 35.59, 34.57; Ms; [M+1, 538].
4.2.3.5 4-((4-Oxo-2-((2-oxo-2-(2-(3,4,5-trimethoxybenzylidene)hydrazineyl)ethyl)thio)quinazolin-3(4H)-yl)methyl)benzenesulfonamide (8). M.p 210–212°, 91% yield; ν: 3300, 3249 (NH), 1655, 1611 (C
O), 1331, 1149 (OSO); 1H NMR (700 MHz, DMSO) δ 11.82 & 11.71 (ss, 1H), 9.39 (s, 1H), 8.13 (d, J = 7.7 Hz, 1H), 7.89–7.78 (m, 3H), 7.65–7.59 (m, 1H), 7.52–7.45 (m, 3H), 7.36 (s, 2H), 7.07–6.98 (m, 1H), 6.79 (s, 1H), 5.47–5.39 (m, 2H), 4.50–4.11 (m, 2H), 4.69–3.66 (m, 9H); 13C NMR (176 MHz, DMSO) δ 166.47, 161.38, 156.64, 153.64, 153.48, 147.27, 143.66, 140.06, 138.67, 137.51, 135.47, 132.58, 127.57, 127.12, 126.70, 126.62, 126.52, 119.15, 106.06, 104.78, 104.58, 102.73, 60.50, 56.42, 56.35, 56.18, 47.07, 34.69; \Ms; [M+1, 598].
4.2.3.6 4-((2-((2-(2-(4-Methylbenzylidene)hydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (9). M.p 273–274°, 95% yield; ν: 3302, 3236 (NH), 1665, 1610 (C
O), 1334, 1153 (OSO); 1H NMR (700 MHz, DMSO) δ 11.77 & 11.61 (s, 1H), 8.23 & 8.04 (s, 1H), 8.13–8.12 (m, 1H), 7.84–7.79 (m, 3H), 7.61–7.46 (m, 6H), 7.36 (s, 2H), 7.27–7.23 (m, 2H), 5.45 (d, J = 4.5 Hz, 2H), 4.59 & 4.11 (s, 2H), 2.34 (s, 3H); 13C NMR (176 MHz, DMSO) δ 168.95, 163.73, 161.34, 161.31, 156.88, 156.71, 147.23, 143.99, 143.66, 140.21, 140.08, 135.51, 131.85, 129.91, 127.63, 127.59, 127.56, 127.31, 127.17, 126.66, 126.53, 126.49, 126.47, 119.13, 47.18, 35.61, 34.52, 21.50; Ms; [M+1, 522].
4.2.4.1 4-((4-Oxo-2-((2-oxo-2-(2-(1-(p-tolyl)ethylidene)hydrazineyl)ethyl)thio)quinazolin-3(4H)-yl)methyl)benzenesulfonamide (10). M.p 279–280°, 90% yield; ν: 3353, 3201 (NH), 1668, 1610 (C
O), 1338, 1156 (OSO); 1H NMR (700 MHz, DMSO) δ 10.93 & 10.80 (s, 1H), 8.13 (t, J = 9.3 Hz, 1H), 7.84–7.78 (m, 5H), 7.61–7.38 (m, 9H), 5.45 & 5.42 (s, 2H), 4.65 (d, J = 3.1 Hz, 1.3H), 4.25 (d, J = 3.2 Hz, 0.7H), 2.33 (s, 1H), 2.30 (d, J = 2.7 Hz, 2H), 13C NMR (176 MHz, DMSO) δ 169.96, 161.35, 156.92, 152.64, 148.52, 147.26, 147.23, 143.68, 143.63, 140.09, 140.05, 138.45, 135.55, 129.84, 129.66, 128.87, 128.81, 127.62, 127.57, 127.17, 126.81, 126.66, 126.56, 126.49, 126.40, 119.12, 47.15, 35.52, 35.20, 14.71, 14.15; Ms; [M+1, 522].
4.2.4.2 4-((2-((2-(2-(1-(3-Methoxyphenyl)ethylidene)hydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (11). M.p 263–265°, 88% yield; ν 3356, 3255, 3200 (NH), 17
674, 1619 (CO), 133, 1157 (OSO); 1H NMR (700 MHz, DMSO) δ 10.95 & 10.80 (s, 1H), 8.13 (t, J = 9.5 Hz, 1H), 7.83 (td, J = 17.3, 7.5 Hz, 3H), 7.59–7.29 (m, 9H), 7.00 (d, J = 7.9 Hz, 1H), 5.46 (s, 2H), 4.67 & 4.25 (s, 2H), 3.78 (s, 3H), 2.32 & 2.29 (s, 3H); 13C NMR (176 MHz, DMSO) δ 169.97, 161.35, 159.74, 159.65, 156.94, 156.90, 148.31, 147.25, 143.70, 143.64, 140.10, 140.05, 139.93, 135.52, 129.96, 129.88, 127.59, 126.51, 126.40, 119.35, 119.13, 119.07, 115.42, 115.01, 112.20, 112.14, 55.59, 47.18, 35.44, 14.88, 14.27; Ms; [M+1, 552].
4.2.4.3 4-((2-((2-(2-(1-(3,5-Dimethoxyphenyl)ethylidene)hydrazineyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)methyl)benzenesulfonamide (12). M.p 295–296°, 89% yield; ν: 3331, 3261 (NH), 1674, 1591 (C
O), 1336, 1158 (OSO); 1H NMR (700 MHz, DMSO) δ 10.95 & 10.80 (s, 1H), 8.13 (dd, J = 12.3, 7.4 Hz, 1H), 7.84–7.79 (m, 3H), 7.48 (td, J = 15.1, 7.1 Hz, 4H), 7.38 (d, J = 4.0 Hz, 2H), 6.99 & 6.92 (s, 2H), 6.62–6.51 (m, 1H), 5.45 (s, 2H), 4.66 & 4.24 (s, 2H), 3.77 (d, J = 2.9 Hz, 6H), 2.30 (s, 1H), 2.27 (d, J = 2.7 Hz, 2H), 13C NMR (176 MHz, DMSO) δ 169.97, 160.88, 160.77, 156.94, 148.27, 147.24, 143.62, 140.53, 140.10, 135.48, 127.63, 127.58, 127.15, 126.66, 126.51, 126.38, 119.11, 105.02, 104.97, 101.63, 101.17, 55.73, 47.18, 35.56, 35.48, 14.95, 14.31; Ms; [M+1, 582].
4.2.4.4 4-((4-Oxo-2-((2-oxo-2-(2-(1-(3,4,5-trimethoxyphenyl)ethylidene)hydrazineyl)ethyl)thio)quinazolin-3(4H)-yl)methyl)benzenesulfonamide (13). M.p 283–284°, 90% yield; ν: 3322, 3243, 3168 (NH), 1659, 1612 (C
O), 1333, 1162 (OSO); 1H NMR (700 MHz, DMSO) δ 10.91 & 10.77 (t, J = 4.2 Hz, 1H), 8.13 (p, J = 6.7 Hz, 1H), 7.90–7.79 (m, 3H), 7.59–7.42 (m, 4H), 7.39–7.38 (m, 2H), 7.13–7.06 (m, 2H), 5.46 (s, 2H), 4.66 & 4.24 (t, J = 4.1 Hz, 2H), 3.81 (s, 6H), 3.70 (s, 3H), 2.34–2.30 (m, 3H); 13C NMR (176 MHz, DMSO) δ 169.81, 164.22, 161.34, 156.99, 156.91, 153.16, 153.09, 148.60, 147.25, 143.64, 140.10, 139.17, 135.52, 134.08, 133.99, 127.65, 127.55, 127.19, 126.67, 126.52, 126.35, 119.11, 104.41, 104.31, 60.60, 56.38, 47.20, 35.56, 15.00, 14.36; Ms; [M+1, 612].
4.3 Biological evaluation
4.4 Apoptosis assay
The apoptosis of cancer cell lines T-47D and MOLT4 cells was measured by Annexin 5-FITC/PI apoptosis detection kit using FACSCalibur flow cytometer for analysis74 ESI.†4.5 Cell cycle analysis
Cancer cell lines T-47D and MOLT4 were stained with the DNA fluorochrome PI and analyzed by FACSCalibur flow cytometer.754.6 Docking methodology
We performed molecular docking simulations using AutoDock 4.2,76 which employed the Lamarckian Genetic Algorithm (LGA) with defined parameters. We used Accelrys Discovery Studio Visualizer (version 4.0) to visualize binding poses and interactions and analyze the binding dynamics. We further characterized intermolecular interactions using the PLIP web server.77–82Data availability
Data supporting this study are included in the article and ESI.†Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R1049), King Saud University, Riyadh, Saudi Arabia, for funding this research.References
- M. Mareel and A. J. P. r. Leroy, Clinical, cellular, and molecular aspects of cancer invasion, Physiol. Rev., 2003, 83, 337–376 CrossRef CAS PubMed.
- J. Wesche, K. Haglund and E. M. J. B. J. Haugsten, Fibroblast growth factors and their receptors in cancer, Biochem. J., 2011, 437, 199–213 CrossRef CAS PubMed.
- J. Ferlay, M. Ervik, F. Lam, M. Colombet, L. Mery, M. Piñeros, A. Znaor, I. Soerjomataram and F. J. L. I. a. f. r. o. c. Bray, Global Cancer Observatory: Cancer Today, 2020, 20182020 Search PubMed.
- A. M. Alanazi, I. A. Al-Suwaidan, A.-M. Alaa, M. A. Mohamed, A. M. El_Morsy and A. S. El-Azab, Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents, Med. Chem. Res., 2013, 22, 5566–5577 CrossRef CAS.
- A. S. El-Azab, A. A. Abdel-Aziz, H. A. Ghabbour and M. A. Al-Gendy, Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues, J. Enzyme Inhib. Med. Chem., 2017, 32, 1229–1239 CrossRef CAS PubMed.
- A. M. Alanazi, A. A.-M. Abdel-Aziz, T. Z. Shawer, R. R. Ayyad, A. M. Al-Obaid, M. H. Al-Agamy, A. R. Maarouf and A. S. El-Azab, Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4 (3 H)-quinazolinone analogues: in silico studies, J. Enzyme Inhib. Med. Chem., 2016, 31, 721–735 CrossRef CAS PubMed.
- A. M. Alanazi, A.-M. Alaa, I. A. Al-Suwaidan, S. G. Abdel-Hamide, T. Z. Shawer and A. S. El-Azab, Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents, Eur. J. Med. Chem., 2014, 79, 446–454 CrossRef CAS PubMed.
- J.-H. Cheng, C.-F. Hung, S.-C. Yang, J.-P. Wang, S.-J. Won and C.-N. Lin, Synthesis and cytotoxic, anti-inflammatory, and anti-oxidant activities of 2′, 5′-dialkoxylchalcones as cancer chemopreventive agents, Bioorg. Med. Chem., 2008, 16, 7270–7276 CrossRef CAS PubMed.
- A.-M. Alaa, L. A. Abou-Zeid, K. E. H. ElTahir, M. A. Mohamed, M. A. A. El-Enin and A. S. El-Azab, Design, synthesis of 2, 3-disubstitued 4 (3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies, Bioorg. Med. Chem., 2016, 24, 3818–3828 CrossRef PubMed.
- A. A. Abdel-Aziz, L. A. Abou-Zeid, K. E. H. ElTahir, R. R. Ayyad, M. A. El-Sayed and A. S. El-Azab, Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4(3H)-quinazolinones, Eur. J. Med. Chem., 2016, 121, 410–421 CrossRef CAS PubMed.
- A.-M. Alaa, L. A. Abou-Zeid, K. E. H. ElTahir, R. R. Ayyad, A.-A. Magda and A. S. El-Azab, Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4 (3H)-quinazolinones, Eur. J. Med. Chem., 2016, 121, 410–421 CrossRef PubMed.
- A.-M. Alaa, L. A. Abou-Zeid, K. E. H. ElTahir, M. A. Mohamed, M. A. A. El-Enin and A. S. El-Azab, Design, synthesis of 2, 3-disubstitued 4 (3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies, Bioorg. Med. Chem., 2016, 24, 3818–3828 CrossRef PubMed.
- A. S. El-Azab, A. A. Abdel-Aziz, S. Bua, A. Nocentini, M. A. El-Gendy, M. A. Mohamed, T. Z. Shawer, N. A. AlSaif and C. T. Supuran, Synthesis of benzensulfonamides linked to quinazoline scaffolds as novel carbonic anhydrase inhibitors, Bioorg. Chem., 2019, 87, 78–90 CrossRef CAS PubMed.
- A. S. El-Azab, A. A. Abdel-Aziz, H. E. A. Ahmed, S. Bua, A. Nocentini, N. A. AlSaif, A. J. Obaidullah, M. M. Hefnawy and C. T. Supuran, Exploring structure-activity relationship of S-substituted 2-mercaptoquinazolin-4(3H)-one including 4-ethylbenzenesulfonamides as human carbonic anhydrase inhibitors, J. Enzyme Inhib. Med. Chem., 2020, 35, 598–609 CrossRef CAS PubMed.
- A. S. El-Azab, A. A. Abdel-Aziz, S. Bua, A. Nocentini, N. A. AlSaif, M. M. Alanazi, M. A. El-Gendy, H. E. A. Ahmed and C. T. Supuran, S-substituted 2-mercaptoquinazolin-4(3H)-one and 4-ethylbenzensulfonamides act as potent and selective human carbonic anhydrase IX and XII inhibitors, J. Enzyme Inhib. Med. Chem., 2020, 35, 733–743 CrossRef CAS PubMed.
- A. S. El-Azab, A. A.-M. Abdel-Aziz, S. Bua, A. Nocentini, N. A. AlSaif, M. M. Alanazi, M. A. El-Gendy, H. E. Ahmed and C. T. Supuran, S-substituted 2-mercaptoquinazolin-4 (3H)-one and 4-ethylbenzensulfonamides act as potent and selective human carbonic anhydrase IX and XII inhibitors, J. Enzyme Inhib. Med. Chem., 2020, 35, 733–743 CrossRef CAS PubMed.
- A. S. El-Azab, A.-M. Alaa, S. Bua, A. Nocentini, M. A. El-Gendy, M. A. Mohamed, T. Z. Shawer, N. A. AlSaif and C. T. Supuran, Synthesis of benzensulfonamides linked to quinazoline scaffolds as novel carbonic anhydrase inhibitors, Bioorg. Chem., 2019, 87, 78–90 CrossRef CAS PubMed.
- A. S. El-Azab, A. Al-Dhfyan, A. A. Abdel-Aziz, L. A. Abou-Zeid, H. M. Alkahtani, A. M. Al-Obaid and M. A. Al-Gendy, Synthesis, anticancer and apoptosis-inducing activities of quinazoline-isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies, J. Enzyme Inhib. Med. Chem., 2017, 32, 935–944 CrossRef CAS PubMed.
- H. M. Alkahtani, A. N. Abdalla, A. J. Obaidullah, M. M. Alanazi, A. A. Almehizia, M. G. Alanazi, A. Y. Ahmed, O. I. Alwassil, H. W. Darwish, A. A. Abdel-Aziz and A. S. El-Azab, Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2, Bioorg. Chem., 2020, 95, 103461 CrossRef CAS PubMed.
- A. J. Barker, K. H. Gibson, W. Grundy, A. A. Godfrey, J. J. Barlow, M. P. Healy, J. R. Woodburn, S. E. Ashton, B. J. Curry, L. Scarlett, L. Henthorn and L. Richards, Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer, Bioorg. Med. Chem. Lett., 2001, 11, 1911–1914 CrossRef CAS PubMed.
- R. T. Dungo and G. M. Keating, Afatinib: first global approval, Drugs, 2013, 73, 1503–1515 CrossRef CAS PubMed.
- J. E. Frampton, Lapatinib: a review of its use in the treatment of HER2-overexpressing, trastuzumab-refractory, advanced or metastatic breast cancer, Drugs, 2009, 69, 2125–2148 CrossRef CAS PubMed.
- A. S. El-Azab, A. A. Abdel-Aziz, N. A. AlSaif, H. M. Alkahtani, M. M. Alanazi, A. J. Obaidullah, R. O. Eskandrani and A. Alharbi, Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: Cell cycle analysis, Bioorg. Chem., 2020, 104, 104345 CrossRef CAS PubMed.
- A. Hamdi, H. W. El-Shafey, D. I. Othman, A. S. El-Azab, N. A. AlSaif and A.-M. J. B. C. Alaa, Design, synthesis, antitumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: Molecular modeling studies, 122 ( 2022) Search PubMed.
- A. S. Altamimi, A. S. El-Azab, S. G. Abdelhamid, M. A. Alamri, A. H. Bayoumi, S. M. Alqahtani, A. B. Alabbas, A. I. Altharawi, M. A. Alossaimi and M. A. J. M. Mohamed, Synthesis, anticancer screening of some novel trimethoxy quinazolines and VEGFR2, EGFR tyrosine kinase inhibitors assay; molecular docking studies, 26 ( 2021) 2992 Search PubMed.
- A. S. El-Azab, A.-M. Alaa, N. A. AlSaif, H. M. Alkahtani, M. M. Alanazi, A. J. Obaidullah, R. O. Eskandrani, A. J. B. C. Alharbi, Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: Cell cycle analysis, 104 ( 2020) Search PubMed.
- M. A.-A. El-Sayed, W. M. El-Husseiny, N. I. Abdel-Aziz, A. S. El-Azab, H. A. Abuelizz and A. A.-M. Abdel-Aziz, Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: molecular docking study, J. Enzyme Inhib. Med. Chem., 2018, 33, 199–209 CrossRef CAS PubMed.
- A. S. El-Azab, S. G. Abdel-Hamide, M. M. Sayed-Ahmed, G. S. Hassan, T. M. El-Hadiyah, O. A. Al-Shabanah, O. A. Al-Deeb and H. I. El-Subbagh, Novel 4 (3 H)-quinazolinone analogs: synthesis and anticonvulsant activity, Med. Chem. Res., 2013, 22, 2815–2827 CrossRef CAS.
- A. S. El-Azab, K. E. J. B. ElTahir and M. C. Letters, Design and synthesis of novel 7-aminoquinazoline derivatives: Antitumor and anticonvulsant activities, 22 ( 2012) 1879–1885 Search PubMed.
- A. S. El-Azab and K. E. ElTahir, Synthesis and anticonvulsant evaluation of some new 2, 3, 8-trisubstituted-4 (3H)-quinazoline derivatives, Bioorg. Med. Chem. Lett., 2012, 22, 327–333 CrossRef CAS PubMed.
- A. S. El-Azab, K. E. ElTahir and S. M. Attia, Synthesis and anticonvulsant evaluation of some novel 4 (3H)-quinazolinones, Monatsh. fur Chem., 2011, 142, 837–848 CrossRef CAS.
- A. S. El-Azab, A. A.-M. Abdel-Aziz, H. A. Ghabbour and M. A. Al-Gendy, Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3, 4, 5-trimethoxybenzyl)-4 (3H)-quinazolinone analogues, J. Enzyme Inhib. Med. Chem., 2017, 32, 1229–1239 CrossRef CAS PubMed.
- A. S. El-Azab, A. Al-Dhfyan, A. A.-M. Abdel-Aziz, L. A. Abou-Zeid, H. M. Alkahtani, A. M. Al-Obaid, M. A. J. J. o. E. I. Al-Gendy, M. Chemistry, Synthesis, anticancer and apoptosis-inducing activities of quinazoline–isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies, 32 ( 2017) 935–944 Search PubMed.
- I. A. Al-Suwaidan, A. A.-M. Abdel-Aziz, T. Z. Shawer, R. R. Ayyad, A. M. Alanazi, A. M. El-Morsy, M. A. Mohamed, N. I. Abdel-Aziz, M. A.-A. El-Sayed, A. S. J. J. o. e. i. El-Azab, m. chemistry, Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4 (3H) quinazolinone analogues, 31 ( 2016) 78–89 Search PubMed.
- M. A. Mohamed, R. R. Ayyad, T. Z. Shawer, A.-M. Alaa, A. S. J. E. J. O. M. C. El-Azab, Synthesis and antitumor evaluation of trimethoxyanilides based on 4 (3H)-quinazolinone scaffolds, 112 ( 2016) 106–113 Search PubMed.
- A. M. Alanazi, A.-M. Alaa, I. A. Al-Suwaidan, S. G. Abdel-Hamide, T. Z. Shawer, A. S. J. E. J. o. M. C. El-Azab, Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents, 79 ( 2014) 446–454 Search PubMed.
- A. M. Alanazi, I. A. Al-Suwaidan, A. A.-M. Abdel-Aziz, M. A. Mohamed, A. M. El_Morsy, A. S. J. M. C. R. El-Azab, Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents, 22 ( 2013) 5566–5577 Search PubMed.
- I. A. Al-Suwaidan, A. M. Alanazi, A.-M. Alaa, M. A. Mohamed, A. S. J. B. El-Azab and M. C. Letters, Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study, 23 ( 2013) 3935–3941 Search PubMed.
- A. S. El-Azab, M. A. Al-Omar, A.-M. Alaa, N. I. Abdel-Aziz, A.-A. Magda, A. M. Aleisa, M. M. Sayed-Ahmed and S. G. Abdel-Hamide, Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study, Eur. J. Med. Chem., 2010, 45, 4188–4198 CrossRef CAS PubMed.
- A. M. Alanazi, A. A.-M. Abdel-Aziz, T. Z. Shawer, R. R. Ayyad, A. M. Al-Obaid, M. H. Al-Agamy, A. R. Maarouf and A. S. El-Azab, Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4 (3 H)-quinazolinone analogues: in silico studies, J. Enzyme Inhib. Med. Chem., 2016, 31, 721–735 CrossRef CAS PubMed.
- A. M. Alafeefy, A. S. El-Azab, M. A. Mohamed, M. A. Bakhat and S. Abdel-Hamid, Synthesis of some new substituted iodoquinazoline derivatives and their antimicrobial screening, J. Saudi Chem. Soc., 2011, 15, 319–325 CrossRef CAS.
- A. S. El-Azab, Synthesis of some new substituted 2-mercaptoquinazoline analogs as potential antimicrobial agents, Phosphorus, Sulfur Relat. Elem., 2007, 182, 333–348 CrossRef CAS.
- M. Aziza, M. Nassar, S. AbdelHamide, A. ElHakim and A. El-Azab, Synthesis and antimicrobial activities of some new 3-heteroaryl-quinazolin-4-ones, Indian J. Heterocycl. Chem., 1996, 6, 25–30 CAS.
- A. S. El-Azab, H. M. Alkahtani, N. A. AlSaif, I. A. Al-Suwaidan, A. J. Obaidullah, M. M. Alanazi, A. M. Al-Obaid, M. H. Al-Agamy, A.-M. J. J. o. M. S. Alaa, Synthesis, antiproliferative and enzymatic inhibition activities of quinazolines incorporating benzenesulfonamide: Cell cycle analysis and molecular modeling study, 1278 ( 2023) Search PubMed.
- A. A.-M. Abdel-Aziz, A. S. El-Azab, N. A. AlSaif, A. J. Obaidullah, A. M. Al-Obaid and I. A. Al-Suwaidan, Synthesis, potential antitumor activity, cell cycle analysis, and multitarget mechanisms of novel hydrazones incorporating a 4-methylsulfonylbenzene scaffold: a molecular docking study, J. Enzyme Inhib. Med. Chem., 2021, 36, 1520–1538 CrossRef CAS PubMed.
- G. Xu, M. C. Abad, P. J. Connolly, M. P. Neeper, G. T. Struble, B. A. Springer, S. L. Emanuel, N. Pandey, R. H. Gruninger, M. J. B. Adams, m. c. letters, 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors, 18 ( 2008) 4615–4619 Search PubMed.
- A. A.-M. Abdel-Aziz, A. S. El-Azab, N. A. AlSaif, M. M. Alanazi, M. A. El-Gendy, A. J. Obaidullah, H. M. Alkahtani, A. A. Almehizia, I. A. J. J. o. E. I. Al-Suwaidan, M. Chemistry, Synthesis, anti-inflammatory, cytotoxic, and COX-1/2 inhibitory activities of cyclic imides bearing 3-benzenesulfonamide, oxime, and β-phenylalanine scaffolds: a molecular docking study, 35 ( 2020) 610–621 Search PubMed.
- A. S. El-Azab, A. A.-M. Abdel-Aziz, H. E. Ahmed, S. Bua, A. Nocentini, N. A. AlSaif, A. J. Obaidullah, M. M. Hefnawy and C. T. Supuran, Exploring structure-activity relationship of S-substituted 2-mercaptoquinazolin-4 (3H)-one including 4-ethylbenzenesulfonamides as human carbonic anhydrase inhibitors, J. Enzyme Inhib. Med. Chem., 2020, 35, 598–609 CrossRef CAS PubMed.
- H. M. Alkahtani, A. N. Abdalla, A. J. Obaidullah, M. M. Alanazi, A. A. Almehizia, M. G. Alanazi, A. Y. Ahmed, O. I. Alwassil, H. W. Darwish, A.-M. J. B. C. Alaa, Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2, 95 ( 2020) Search PubMed.
- A. S. El-Azab, A.-M. Alaa, S. Bua, A. Nocentini, M. M. Alanazi, N. A. AlSaif, I. A. Al-Suwaidan, M. M. Hefnawy, C. T. J. B. C. Supuran, Synthesis and comparative carbonic anhydrase inhibition of new Schiff's bases incorporating benzenesulfonamide, methanesulfonamide, and methylsulfonylbenzene scaffolds, 92 ( 2019) Search PubMed.
- A. S. El-Azab, A.-M. Alaa, S. Bua, A. Nocentini, N. A. AlSaif, A. A. Almehizia, M. M. Alanazi, M. M. Hefnawy and C. T. Supuran, New anthranilic acid-incorporating N-benzenesulfonamidophthalimides as potent inhibitors of carbonic anhydrases I, II, IX, and XII: Synthesis, in vitro testing, and in silico assessment, J. Enzyme Inhib. Med. Chem., 2019, 181, 111573 CAS.
- A.-M. Alaa, A. S. El-Azab, S. Bua, A. Nocentini, M. A. A. El-Enin, M. M. Alanazi, N. A. AlSaif, M. M. Hefnawy, C. T. J. B. c. Supuran, Design, synthesis, and carbonic anhydrase inhibition activity of benzenesulfonamide-linked novel pyrazoline derivatives, 87 ( 2019) 425–431 Search PubMed.
- A. Abdel-Aziz, A. Angeli, A. El-Azab, M. Hammouda, M. El-sherbeny, C. J. B. C. Supuran, Synthesis and auti-inflammatory activity of sulphonamides and carboxygenase/carbonic anhydrase inhibitory actions, 84 ( 2019) 260–268 Search PubMed.
- A.-M. Alaa, A. S. El-Azab, A. H. Ghiaty, P. Gratteri, C. T. Supuran, A. J. B. C. Nocentini, 4-Substituted benzenesulfonamides featuring cyclic imides moieties exhibit potent and isoform-selective carbonic anhydrase II/IX inhibition, 83 ( 2019) 198–204 Search PubMed.
- A.-M. Alaa, A. S. El-Azab, M. A. A. El-Enin, A. A. Almehizia, C. T. Supuran and A. J. Nocentini, Synthesis of novel isoindoline-1, 3-dione-based oximes and benzenesulfonamide hydrazones as selective inhibitors of the tumor-associated carbonic anhydrase IX, Bioorg. Chem., 2018, 80, 706–713 CrossRef PubMed.
- A. Angeli, A.-M. Alaa, A. Nocentini, A. S. El-Azab, P. Gratteri, C. T. J. B. Supuran, M. Chemistry, Synthesis and carbonic anhydrase inhibition of polycyclic imides incorporating N-benzenesulfonamide moieties, 25 ( 2017) 5373–5379 Search PubMed.
- M. A. Mohamed, A.-M. Alaa, H. M. Sakr, A. S. El-Azab, S. Bua, C. T. J. B. Supuran, m. chemistry, Synthesis and human/bacterial carbonic anhydrase inhibition with a series of sulfonamides incorporating phthalimido moieties, 25 ( 2017) 2524–2529 Search PubMed.
- A.-M. Alaa, A. Angeli, A. S. El-Azab, M. A. A. El-Enin, C. T. J. B. Supuran, M. Chemistry, Synthesis and biological evaluation of cyclic imides incorporating benzenesulfonamide moieties as carbonic anhydrase I, II, IV and IX inhibitors, 25 ( 2017) 1666–1671 Search PubMed.
- A. S. El-Azab, A.-M. Alaa, R. R. Ayyad, M. Ceruso, C. T. J. B. Supuran and M. Chemistry, Inhibition of carbonic anhydrase isoforms I, II, IVVII and XII with carboxylates and sulfonamides incorporating phthalimide/phthalic anhydride scaffolds, 24 ( 2016) 20–25 Search PubMed.
- A.-M. Alaa, A. S. El-Azab, M. Ceruso, C. T. J. B. Supuran and M. C. Letters, Carbonic anhydrase inhibitory activity of sulfonamides and carboxylic acids incorporating cyclic imide scaffolds, 24 ( 2014) 5185–5189 Search PubMed.
- A. A. Abdel-Aziz, A. S. El-Azab, N. A. AlSaif, A. J. Obaidullah, A. M. Al-Obaid and I. A. Al-Suwaidan, Synthesis, potential antitumor activity, cell cycle analysis, and multitarget mechanisms of novel hydrazones incorporating a 4-methylsulfonylbenzene scaffold: a molecular docking study, J. Enzyme Inhib. Med. Chem., 2021, 36, 1521–1539 Search PubMed.
- G. Xu, M. C. Abad, P. J. Connolly, M. P. Neeper, G. T. Struble, B. A. Springer, S. L. Emanuel, N. Pandey, R. H. Gruninger, M. Adams, S. Moreno-Mazza, A. R. Fuentes-Pesquera and S. A. Middleton, 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors, Bioorg. Med. Chem. Lett., 2008, 18, 4615–4619 CrossRef CAS PubMed.
- S. Şenkardeş, M. Han, N. Kulabaş, M. Abbak, Ö. Çevik, İ. Küçükgüzel and G. Küçükgüzel Ş, Synthesis, molecular docking and evaluation of novel sulfonyl hydrazones as anticancer agents and COX-2 inhibitors, Mol. Diversity, 2020, 24, 673–689 CrossRef PubMed.
- S. Vogel, D. Kaufmann, M. Pojarová, C. Müller, T. Pfaller, S. Kühne, P. J. Bednarski and E. von Angerer, Aroyl hydrazones of 2-phenylindole-3-carbaldehydes as novel antimitotic agents, Bioorg. Med. Chem., 2008, 16, 6436–6447 CrossRef CAS PubMed.
- R. F. George, Facile synthesis of simple 2-oxindole-based compounds with promising antiproliferative activity, Future Med. Chem., 2018, 10, 269–282 CrossRef CAS PubMed.
- R. H. Shoemaker, The NCI60 human tumour cell line anticancer drug screen, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- M. R. Grever, S. A. Schepartz and B. A. Chabner, The National Cancer Institute: cancer drug discovery and development program, Semin. Oncol., 1992, 19, 622–638 CAS.
- M. A. El-Sayed, N. I. Abdel-Aziz, A. A. Abdel-Aziz, A. S. El-Azab, Y. A. Asiri and K. E. Eltahir, Design, synthesis, and biological evaluation of substituted hydrazone and pyrazole derivatives as selective COX-2 inhibitors: Molecular docking study, Bioorg. Med. Chem., 2011, 19, 3416–3424 CrossRef CAS PubMed.
- A. M. Alanazi, A. S. El-Azab, I. A. Al-Suwaidan, K. E. ElTahir, Y. A. Asiri, N. I. Abdel-Aziz and A. A. Abdel-Aziz, Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities, Eur. J. Med. Chem., 2015, 92, 115–123 CrossRef CAS PubMed.
- J. L. Nakamura, The epidermal growth factor receptor in malignant gliomas: pathogenesis and therapeutic implications, Expert Opin. Ther. Targets, 2007, 11, 463–472 CrossRef CAS PubMed.
- R. M. Hudziak, J. Schlessinger and A. Ullrich, Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7159–7163 CrossRef CAS PubMed.
- A. Badache and A. Gonçalves, The ErbB2 signaling network as a target for breast cancer therapy, J. Mammary Gland Biol. Neoplasia, 2006, 11, 13–25 CrossRef PubMed.
- U. Asghar, A. K. Witkiewicz, N. C. Turner and E. S. Knudsen, The history and future of targeting cyclin-dependent kinases in cancer therapy, Nat. Rev. Drug Discovery, 2015, 14, 130–146 CrossRef CAS PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS PubMed.
- P. K. Parida, B. Mahata, A. Santra, S. Chakraborty, Z. Ghosh, S. Raha, A. K. Misra, K. Biswas and K. Jana, Inhibition of cancer progression by a novel trans-stilbene derivative through disruption of microtubule dynamics, driving G2/M arrest, and p53-dependent apoptosis, Cell Death Dis., 2018, 9, 448 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell, A. J. J. J. O. C. C. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, 30 ( 2009) 2785–2791 Search PubMed.
- N. K. K. Ikram, J. D. Durrant, M. Muchtaridi, A. S. Zalaludin, N. Purwitasari, N. Mohamed, A. S. A. Rahim, C. K. Lam, Y. M. Normi, N. A. J. J. o. c. i. Rahman, Modeling, A virtual screening approach for identifying plants with anti H5N1 neuraminidase activity, 55 ( 2015) 308–316 Search PubMed.
- A. Hamdi, W. M. Elhusseiny, D. I. Othman, A. Haikal, A. H. Bakheit, A. S. El-Azab, M. H. Al-Agamy, A.-M. J. E. J. O. M. C. Alaa, Synthesis, antitumor, and apoptosis-inducing activities of novel 5-arylidenethiazolidine-2, 4-dione derivatives: histone deacetylases inhibitory activity and molecular docking study, 244 ( 2022) Search PubMed.
- A. S. El-Azab, Y. S. Mary, C. Y. Panicker, A.-M. Alaa, M. A. El-Sherbeny and C. Van Alsenoy, DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3, 4-dihydroquinazolin-2-ylthio)-N-(3, 4, 5-trimethoxyphenyl) acetamide, J. Mol. Struct., 2016, 1113, 133–145 CrossRef CAS.
- A. S. El-Azab, A. M. Alanazi, N. I. Abdel-Aziz, I. A. Al-Suwaidan, M. A. El-Sayed, M. A. El-Sherbeny and A.-M. Alaa, Synthesis, molecular modeling study, preliminary antibacterial, and antitumor evaluation of N-substituted naphthalimides and their structural analogues, Med. Chem. Res., 2013, 22, 2360–2375 CrossRef CAS.
- A. A. Abdel-Aziz, A. S. El-Azab, A. M. Alanazi, Y. A. Asiri, I. A. Al-Suwaidan, A. R. Maarouf, R. R. Ayyad and T. Z. Shawer, Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies, J. Enzyme Inhib. Med. Chem., 2016, 31, 796–809 CrossRef CAS PubMed.
- H. A. Abuelizz, M. Marzouk, A. Bakhiet, M. M. Abdel-Aziz, E. Ezzeldin, H. Rashid and R. J. M. P. Al-Salahi, In silico study and biological screening of benzoquinazolines as potential antimicrobial agents against methicillin-resistant Staphylococcus aureus, carbapenem-resistant Klebsiella pneumoniae, and fluconazole-resistant Candida albicans, 160 ( 2021) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07174c |
| This journal is © The Royal Society of Chemistry 2025 |