
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature oxidation of ethanol to acetaldehyde over Mo-based catalysts
Chunhe Liuab,
Xiaqing Wangab,
Xiujuan Gaoa,
Yingquan Wu*a,
Xiaoxing Wanga,
Faen Songa,
Junfeng Zhang a,
Yizhuo Hana and
Qingde Zhang*a
a,
Yizhuo Hana and
Qingde Zhang*a
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, China. E-mail: qdzhang@sxicc.ac.cn; wuyq@sxicc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 6th January 2025
Abstract
The research and development of the green synthesis route of chemicals has become the focus of research in academia and industry. At present, the highly efficient oxidation of ethanol to acetaldehyde over non-precious metal catalysts under mild conditions is most promising, but remains a big challenge. Herein, the Mo–Sn oxide catalyst was designed to successfully realize low-temperature oxidation of ethanol to acetaldehyde, achieving an acetaldehyde selectivity of 89.3%, and ethanol conversion of 58.9% at 190 °C without COx formation. From the deep correlation of characterization and activity results, the weakened Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the enhanced mobility of lattice oxygen play crucial roles in the oxidation of Cα–H in the CH3CH2O* at lower temperatures. An optimal Mo/Sn ratio possessing multiple active centers can obviously promote the adsorption and dissociation of ethanol into CH3CH2O*. Furthermore, the reduced amount of medium-strong acid inhibited the formation of ethyl acetate as a byproduct.
O bond and the enhanced mobility of lattice oxygen play crucial roles in the oxidation of Cα–H in the CH3CH2O* at lower temperatures. An optimal Mo/Sn ratio possessing multiple active centers can obviously promote the adsorption and dissociation of ethanol into CH3CH2O*. Furthermore, the reduced amount of medium-strong acid inhibited the formation of ethyl acetate as a byproduct.
1 Introduction
Nowadays, fossil resources are gradually depleting and the environment is further deteriorating, which has accelerated the exploration of renewable chemicals.1–3 Wherein, biomass ethanol has become the most promising biomass resource due to its large annual production, wide distribution, renewability and environmental friendliness.4–6 Besides, ethanol can be obtained from coal-based DME, further expanding the sources of ethanol.7 In addition, bioethanol, as an important platform compound, can be dehydrated, dehydrogenated, and C–C coupled to produce ethylene, acetaldehyde, and aromatic compounds.8–10 Among them, acetaldehyde is an important raw material for many chemicals, which can be further condensed with other aldehydes or ketones to obtain oxygenated compounds with longer carbon chains. It is a very important organic chemical intermediate, which is widely used in the chemical, pharmaceutical, food, and other industries, and has a high industrial application value.11 Currently, in industry, acetaldehyde is mainly synthesized through ethylene oxidation and ethanol oxidative dehydrogenation. The catalyst used a solution composed of PdCl2–CuCl2–HCl–H2O in the ethylene oxidation method, which has high catalyst cost, acid corrosiveness, and high equipment investment.12,13 Therefore, a promising route appears to replace petroleum-based ethylene with renewable ethanol for more green and sustainable acetaldehyde production.The catalysts for the synthesis of acetaldehyde from ethanol can be classified as oxide catalysts,14,15 metal-based catalysts,10,16–19 and other catalysts.9,11,20,21 At present, Ag-based catalysts commonly used in industry often require high-temperature conditions (550–580 °C) and have the disadvantage of high energy consumption. Additionally, the catalyst is costly and the efficiency is relatively low. Compared to industrial Ag-based catalysts, the currently developed catalysts show a significant reduction in reaction temperature. However, a reaction temperature of 300 °C is still required to fully convert ethanol. Higher reaction temperatures will cause pyrolysis of acetaldehyde to COx, and will also cause secondary reactions of acetaldehyde to generate other by-products such as ethyl acetate and acetic acid.22
Due to the rich oxygen and variable valence coordination of molybdenum-based catalysts, they exhibit strong capabilities for C–H bond activation and cleavage, enabling low-temperature oxidation or dehydrogenation of reactants. Yang et al. disclosed that molybdenum species situated at unsaturated penta-coordinated Mo5c5+ sites significantly promoted the oxidation of the C–H bond in ethers at lower temperature (110 °C).23 Li found that the MoOx/TiO2 catalyst exhibited special activity to acetaldehyde for ethanol conversion to acetic acid (17% ethanol conversion and 94% selectivity at 200 °C).24 Subsequently, G. Pampararo discovered MoO3/γ-Al2O3 catalysts exhibited higher activity compared to MoO3/SiO2 catalysts, achieving optimal performance at lower temperatures (300–350 °C versus 400–450 °C). Consequently, the interaction between the metal and the support is crucial in the design and development of catalysts.25 Lucia G. Appel et al.26 investigated the ethanol oxidation reaction and found that isolated Mo tetrahedral species and catalyst's basicity promoted the rate of ethanol dehydrogenation. The catalyst exhibited highly oxidizing and more amount of acetic acid was produced in addition to acetaldehyde. Difficulties remain concerning how to control the oxidative properties of the catalyst to further increase the acetaldehyde selectivity. Therefore, it is of great significance to design a highly efficient catalyst for achieving low-temperature oxidation of ethanol to acetaldehyde under mild conditions.
In this work, Mo–Sn oxide catalyst was designed by adjusting the Mo/Sn ratio, and a variety of ethanol adsorption centers were formed, promoting the formation of ethoxy groups. The Mo1Sn12 catalyst exhibited remarkable low-temperature performance for ethanol oxidation at 190 °C, with a conversion of 58.9% and selectivity reaching up to 89.3%, about 40% higher than the one reported previously. The study found a gradual weakening of the MoO bond and an increase in the mobility of lattice oxygen on the catalyst surface as Mo content decreased, enhancing its reactivity and facilitating the oxidation of Cα–H bonds (the rate-determining step) at lower temperatures.10,27,28
2 Experimental
2.1 Catalyst preparation
The different Mo/Sn ratio catalysts were prepared as follows: add drops of ammonia solution to the SnCl4·5H2O solution at 60 °C using a peristaltic pump, adjusting the pH to 8. The obtained suspension was filtered and washed with deionized water until no chloride ions were detected with AgNO3 solution. Then the cake-like hydroxide was dried at 80 °C for 12 h to form tin hydroxide precursor. Subsequently added to the required (NH4)6Mo7O24·4H2O solution in molar ratios of Mo/Sn of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6, 1:9, 1:10, 1:12, 1:14, and 1:15. After evaporation of the solvent in a rotary evaporator at 60 °C, the solid obtained was dried at 80 °C for 12 h and annealed at 400 °C for 8 h in an oxygen atmosphere. The molar ratios of Mo/Sn catalysts were denoted as Mo1Sn6, Mo1Sn9, Mo1Sn10, Mo1Sn12, Mo1Sn14, and Mo1Sn15, successively.
:6, 1:9, 1:10, 1:12, 1:14, and 1:15. After evaporation of the solvent in a rotary evaporator at 60 °C, the solid obtained was dried at 80 °C for 12 h and annealed at 400 °C for 8 h in an oxygen atmosphere. The molar ratios of Mo/Sn catalysts were denoted as Mo1Sn6, Mo1Sn9, Mo1Sn10, Mo1Sn12, Mo1Sn14, and Mo1Sn15, successively.
2.2 Catalyst characterization
XRD measurements were conducted using a MiniFlex 600 X-ray diffractometer (Rigaku Corporation, Japan) with Cu Kα radiation. The scanning range was 5°–90°, with a scanning rate of 5° min−1, a current of 15 mA, and a voltage of 40 kV. Raman characterization was performed using a LABRAM-HR800 Raman spectrometer, with a laser wavelength of 532 nm and a scanning range of 100–1100 cm−1. The physical characterization of N2 adsorption was conducted using a Micromeritics ASAP 2460 surface area analyzer. The samples were pre-treated by heating and degassing under vacuum at 300 °C. Subsequently, low-temperature N2 adsorption–desorption experiments were performed using pure liquid nitrogen at −196 °C. The specific surface area of the samples was calculated using the BET method, and the pore distribution was analyzed using the BJH method. X-ray photoelectron spectroscopy (XPS) analysis was employed to obtain insights about the chemical state of metal oxide in the catalyst surface. For all elements, there is a characteristic of binding energy associated with each core atomic orbital. XPS was performed using a Thermo Fisher ESCALAB 250Xi X-ray photoelectron spectrometer, with an excitation source of Al Kα radiation (hv = 1486.6 eV, operating voltage 12.5 kV, filament current 16 mA). The binding energy of C 1s (284.8 eV) was used for calibration and comparison with the measured elements.CO2-TPD tests were conducted using a BELCAT-B chemisorption apparatus. A 100 mg catalyst sample (20–40 mesh) was placed in the sample tube and purged with an inert gas. The temperature was then programmed to rise to 300 °C for a 60 minutes pre-treatment, followed by cooling to 50 °C. At 50 °C, CO2 gas was introduced for 30 minutes (30 mL min−1). After purging with inert gas for 30 minutes, the temperature was increased at a rate of 10 °C min−1 to 650 °C for desorption, and the signal was recorded. The measurement process of NH3-TPD and O2-TPD are the same as that of CO2-TPD, except that NH3 is replaced. C2H5OH -TPSR-MS characterization was conducted using a micro fixed-bed reactor coupled with an OmniStar mass spectrometer. A 100 mg catalyst sample was weighed and heated in an argon atmosphere to 300 °C for 10 minutes, followed by cooling to 50 °C. After cooling, ethanol gas was introduced at a flow rate of 30 mL min−1. Once the baseline stabilized, the temperature was increased at a rate of 10 °C min−1 from 50 °C to 500 °C, with the exhaust gas connected to the mass spectrometer for detection.
In situ DRIFTS characterization was performed using a Bruker Tensor 27 spectrometer equipped with an MCT detector, with 64 scans and a resolution of 4 cm−1. During the in situ measurements, the sample was placed in the in situ cell and heated to 300 °C, followed by purging with argon for 20 minutes, after which the temperature was cooled to 190 °C. In an argon atmosphere, background signals were subtracted while ethanol/argon was introduced into the infrared cell as the adsorption gas for 20 minutes, during which the spectra were collected. Following the adsorption, argon was used to purge and desorb the sample, and the infrared spectra during the desorption process were recorded. Subsequently, ethanol/air was introduced into the infrared cell as the adsorption gas for another 20 minutes, with spectra collected throughout the adsorption process. Afterward, argon purging was employed for desorption, and the infrared spectra during this phase were also collected. Ethanol/argon was introduced into the infrared cell as the adsorption gas, and infrared spectra of ethanol adsorption were collected at different temperatures: 100 °C, 150 °C, 200 °C, 250 °C, 300 °C, and 350 °C. Subsequently, ethanol/air was introduced into the infrared cell, and infrared spectra of ethanol adsorption were similarly collected at the same temperature intervals: 100 °C, 150 °C, 200 °C, 250 °C, 300 °C, and 350 °C.
2.3 Catalytic test
The performance evaluation of ethanol oxidation to acetaldehyde over the Mo–Sn catalyst is conducted in a fixed-bed reactor. A catalyst volume of 1 mL (20–40 mesh) is diluted with an equal volume and particle size of ceramic rings to prevent rapid deactivation of the catalyst due to intense exothermic reactions. The gas (air) space velocity is set at 10000 h−1, while the liquid (ethanol) space velocity is maintained at 1.8 h−1.
The reaction products were analyzed through offline sampling using three chromatographic systems: organic oxygenated compounds were analyzed using a GC-2014CPF/SPL gas chromatograph (Shimadzu Corporation, Japan, equipped with an FID detector, 60 m × 0.25 mm DB-1 capillary column) and a GC-2014 (Shimadzu Corporation, Japan, equipped with a TCD detector and a 3 m Porapak T column). Permanent gases (H2, O2, CO, CO2, CH4, etc.) were analyzed using a GC-4000A chromatograph (Beijing East and West Analytical Instrument Co., Ltd). Quantitative analysis of the corresponding compounds was conducted by calibrating, correlating, and normalizing the peak areas from the FID and TCD spectra, with the conversion rate of ethanol and the selectivity of each product calculated using formulas (1) and (2), respectively.
| XEtOH/% = (1 − NEtOH,out/NEtOH,in) × 100 | (1) |
| Si/% = Niai/2(NEtOH,in − NEtOH,out) × 100 | (2) |
3 Results and discussion
3.1 Structure of the catalyst
The mesoporous properties of catalysts with different Mo/Sn ratios were analyzed using nitrogen adsorption–desorption characterization (see Table 1 and Fig. 1). They are mesoporous materials with well-developed pore structures. The specific surface area of all catalysts not much change and are all at 130–136 m2 g−1, and the average pore diameters fall between 2.67 and 3.08 nm, which provides favorable conditions for the extensive distribution of active sites.| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| Mo1Sn6 | 136 | 0.09 | 2.75 |
| Mo1Sn9 | 134 | 0.09 | 2.67 |
| Mo1Sn10 | 130 | 0.10 | 3.08 |
| Mo1Sn12 | 134 | 0.10 | 2.85 |
| Mo1Sn14 | 134 | 0.09 | 2.68 |
| Mo1Sn15 | 131 | 0.10 | 2.93 |
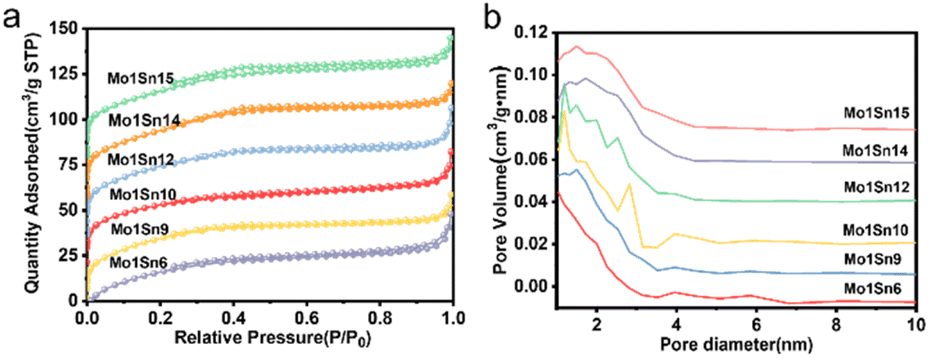 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms of catalysts with different Mo–Sn ratios, (b) pore size distribution of catalysts with different Mo–Sn ratios. | ||
X-ray diffraction (XRD) patterns (Fig. 2a) show the broadened characteristic diffraction peaks of tetragonal SnO2 crystalline (PDF# 41-1445) on catalysts with different Mo–Sn ratios. Whereas no diffraction peaks were assigned to MoO3 crystalline, indicating that the interaction between SnO2 and MoO3 disrupts the crystalline structure of MoO3, leading to the formation of an amorphous MoOx structure.29,30 The spectrum also reveals that as the molybdenum content decreases, the diffraction peaks of SnO2 shifts toward lower angles. According to Bragg's equation, this indicates an increase in the interplanar spacing of SnO2.31 Raman shifts (Fig. 2b) of 884 cm−1 and 945 cm−1 observed are significantly different from those of the MoO3 standard crystal (994 cm−1: terminal MoO stretching vibration; 819 cm−1: Mo–O–Mo bridged vibration).32–34 Such shifts further demonstrate the disruption of MoO3 crystallographic structure and the formation of amorphous MoOx species. As the molybdenum content decreases, the intensity of MoOx gradually diminishes. The characteristic peak for MoO in MoOx exhibits a red shift, indicating that the MoO bond energy gradually decreases, making it easier to cleavage and promoting the oxidation process.35–37
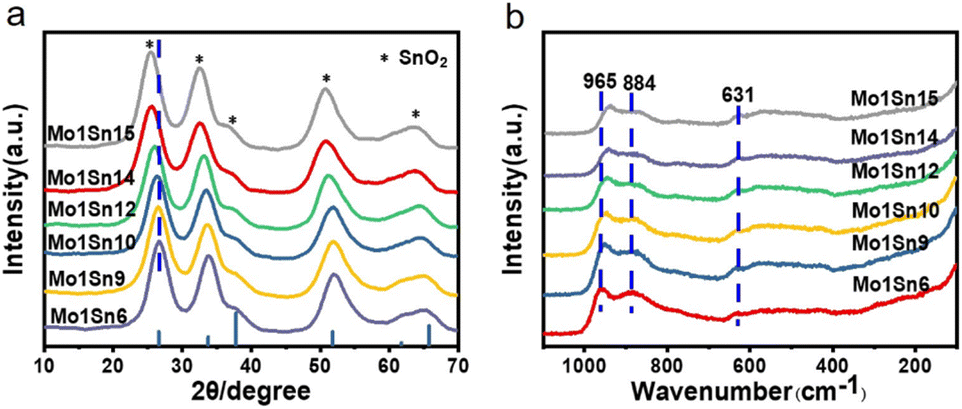 | ||
| Fig. 2 (a) XRD patterns of catalysts with different Mo–Sn ratios, (b) Raman spectra of catalysts with different Mo–Sn ratios. | ||
The electronic properties of Mo–Sn oxides with varying Mo/Sn ratios were characterized using XPS. Table 2 shows the quantitative analysis of the relative contents of Mo5+ and Mo6+ on the catalyst surface. As the molybdenum content decreases, the content of Mo5+ species first increases and then decreases (Fig. 3). The Mo1Sn12 catalyst has the highest Mo5+ content.38,39
| Catalyst | Mo3/26+ 3d (eV) | Mo5/26+ 3d (eV) | Mo3/25+ 3d (eV) | Mo5/25+ 3d (eV) | Mo6+ (%) | Mo5+ (%) |
|---|---|---|---|---|---|---|
| Mo1Sn6 | 235.96 | 232.84 | 237.71 | 231.67 | 82.44 | 17.56 |
| Mo1Sn9 | 236.26 | 233.09 | 235.20 | 231.97 | 74.88 | 25.12 |
| Mo1Sn10 | 236.36 | 233.21 | 235.58 | 232.29 | 75.45 | 24.55 |
| Mo1Sn12 | 236.33 | 233.20 | 235.43 | 232.52 | 62.04 | 37.96 |
| Mo1Sn14 | 236.27 | 233.18 | 235.27 | 232.35 | 72.75 | 27.25 |
| Mo1Sn15 | 236.18 | 233.08 | 234.97 | 231.90 | 76.92 | 23.08 |
 | ||
| Fig. 3 XPS Mo 3d spectra of catalysts with different Mo–Sn ratios. | ||
Oxygen species play a crucial role in facilitating O–H bond cleavage.40 Fig. 4 and Table 3 show that as the molybdenum content decreases, the relative content of lattice oxygen in the catalyst surface gradually increases. It can be inferred that lattice oxygen could facilitate the oxidation of ethanol, which can be further described and seen by O2-TPD.
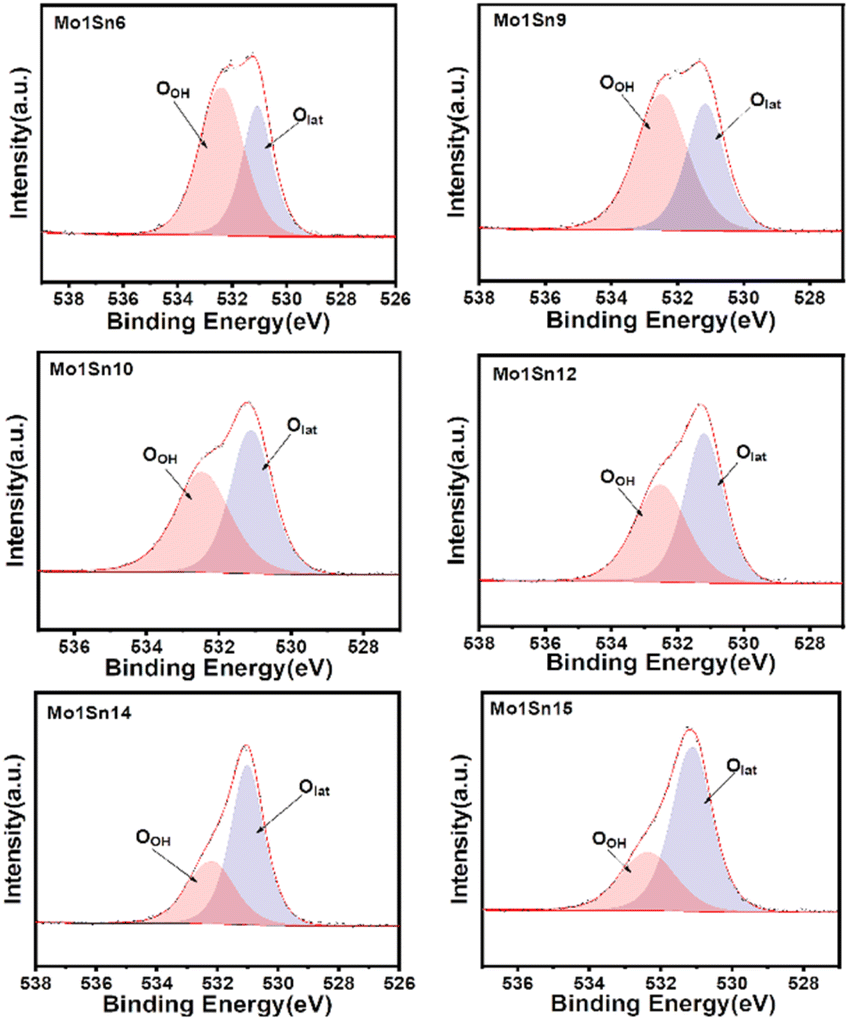 | ||
| Fig. 4 XPS O 1s spectra of catalysts with different Mo–Sn ratios. | ||
| Catalyst | OOH (eV) | Olat (eV) | OOH/OTotal | Olat/OTotal |
|---|---|---|---|---|
| Mo1Sn6 | 532.34 | 531.06 | 60.60 | 39.40 |
| Mo1Sn9 | 532.48 | 531.15 | 59.52 | 40.48 |
| Mo1Sn10 | 532.44 | 531.07 | 50.01 | 49.99 |
| Mo1Sn12 | 532.50 | 531.18 | 39.39 | 60.61 |
| Mo1Sn14 | 532.14 | 531.00 | 34.64 | 65.36 |
| Mo1Sn15 | 532.34 | 531.06 | 34.21 | 65.79 |
Subsequently, we further supplemented the ICP characterization to quantitatively analyze the Mo, Sn and O content in the Mo–Sn catalyst. As shown in Table 4, the Mo content consistently decreases from Mo1Sn6 to Mo1Sn15, from 6.36% to 3.99%, whereas the Sn content correspondingly increases from 53.88% to 63.23%. The remainder consists of oxygen.
| Catalyst | Mo (%) | Sn (%) | O (%) |
|---|---|---|---|
| Mo1Sn6 | 6.36 | 53.88 | 39.76 |
| Mo1Sn9 | 5.68 | 56.92 | 37.40 |
| Mo1Sn10 | 4.97 | 60.67 | 34.36 |
| Mo1Sn12 | 4.39 | 62.75 | 32.86 |
| Mo1Sn14 | 4.31 | 62.81 | 32.88 |
| Mo1Sn15 | 3.99 | 63.23 | 32.78 |
3.2 Surface properties of the catalyst
The formation and migration of oxygen species on the catalysts were identified using O2-TPD. The O2 desorption peaks (Fig. 5a) in the range of 50–200 °C are considered to represent weak chemical adsorption of oxygen. The O2 desorption peaks around 420 °C may be associated with the formation of O2− or O− from surface-adsorbed O2, while above 500 °C corresponding to the desorption of lattice oxygen.41 As the molybdenum content decreases, lattice oxygen desorption peaks begin to appear over the Mo1Sn12 catalyst. And the intensity of these peaks is increasing, suggesting the enhancement of lattice oxygen activity on the catalyst surface. Furthermore, XPS characterization confirms that the amount of lattice oxygen gradually increases with decreasing molybdenum content. Based on these findings, it can be inferred that when the Mo/Sn ratio exceeds 12, the mobility of surface lattice oxygen in the catalyst is enhanced. The oxygen vacancies formed upon desorption further activate O2, promoting the occurrence of redox cycles.42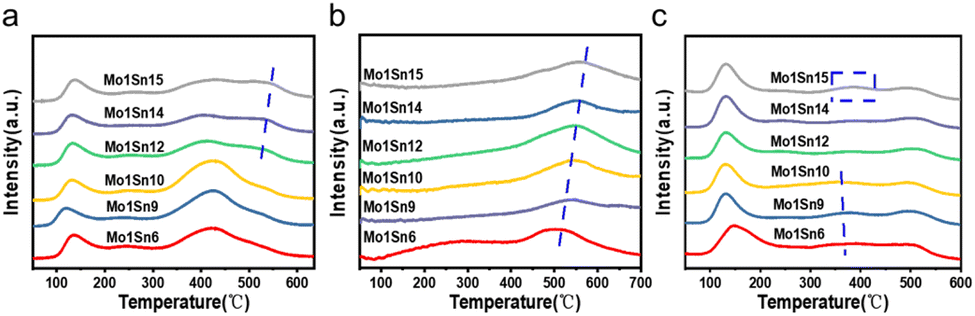 | ||
| Fig. 5 (a) O2-TPD spectra of catalysts with different Mo–Sn ratios, (b) CO2-TPD spectra of catalysts with different Mo–Sn ratios, and (c) NH3-TPD spectra of catalysts with different Mo–Sn ratios. | ||
Basic sites serve as active sites for the cleavage of O–H and Cα–H bonds in ethanol.43–45 The surface basicity was identified by CO2-TPD. Desorption peaks in the range of 300–500 °C (Fig. 5b) correspond to medium-strong basic sites on the catalysts. As the molybdenum content decreases, the CO2 desorption peaks shift overall toward higher temperatures, indicating a gradual increase in the basicity of medium-strong basic sites in the catalyst. Furthermore, a comparison of the CO2 desorption peak areas reveals that Mo1Sn12 has a greater amount of medium to strong basic sites, which facilitates the adsorption of more numbers of ethanol. Combining with the analysis of oxygen species from the XPS characterization, the increase in lattice oxygen may be a factor contributing to the enhanced basicity of catalysts. CO2 could react with basic sites on the catalyst surface to form carbonate species as Lewis acid, which exist in monodentate, bidentate, bridged carbonates, carboxylates, and also as polycarbonates forms. The thermal stability of these species varies with different coordination forms.46 From the CO2-TPD results, as the molybdenum content decreases, the CO2 desorption peak gradually shifts towards higher temperatures. It can be inferred that stronger basic sites have appeared on the catalyst surface, reacting with CO2 to form more stable carbonate species. This indicates that changes in the Mo/Sn ratio can alter the strength and quantity of basic centers on the catalyst surface. Thus, excessive strong basicity may form difficult-to-desorb species after CO2 adsorption, leading to a reduction in the CO2 desorption peak area exceeding that of Mo1Sn12. Mo–Sn catalysts are multifunctional catalysts exhibiting acidic, basic, and redox property. NH3-TPD characterization was used to investigate the effect of different Mo/Sn ratios on the acidity of catalysts surface. The desorption peaks in Fig. 5c, occurring at low (100–200 °C), medium (300–400 °C), and high (400–500 °C) temperatures, correspond to weak, medium, and strong acid sites, respectively. Based on the NH3-TPD results, it can be observed that the Mo1Sn6, Mo1Sn9 and Mo1Sn15 catalysts contain a higher amount of medium-strength acid sites. Since the formation of ethyl acetate and ethylene primarily stems from acid catalysis, these catalysts possibly produce more ethyl acetate. In contrast, Mo1Sn12 has the least medium-strength acid sites.47
3.3 Ethanol-TPSR-MS analysis
Fig. 6a shows the temperature-programmed surface reaction (TPSR) profiles of ethanol with air over different catalysts. It is evident that each catalyst exhibits a distinct acetaldehyde desorption peak around 190 °C, suggesting that this series of catalysts exhibits excellent performance in acetaldehyde synthesis at relatively low temperatures. Each catalyst shows two distinct water desorption regions, located around 190 °C and 350 °C (Fig. 6b). The low-temperature water desorption peak corresponds to the formation of acetaldehyde and is attributed to water produced during the conversion of ethanol to acetaldehyde. In contrast, the high-temperature desorption peak is associated with water generated during the formation of gaseous products, such as ethylene.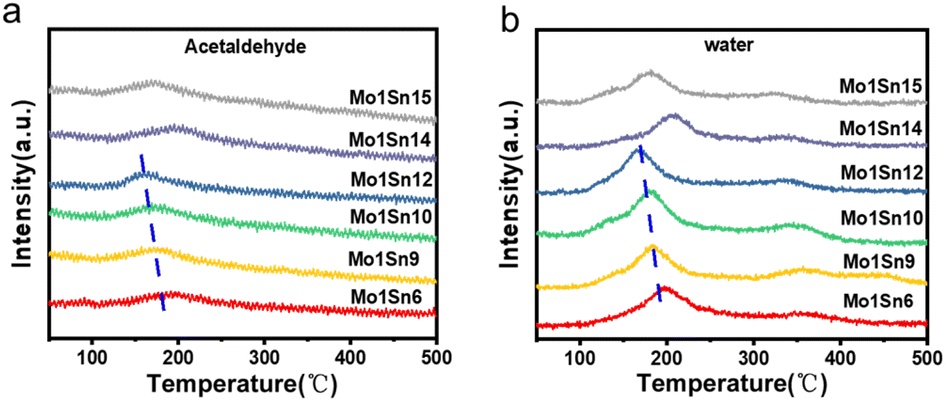 | ||
| Fig. 6 Ethanol-TPSR-MS spectra of catalysts with different Mo–Sn ratios (a) product acetaldehyde, (b) product water. | ||
3.4 In situ infrared analysis
The adsorption behavior of ethanol on the surface of Mo–Sn oxide catalysts was investigated by in situ DRIFTS, and the effect of different temperatures on the adsorption performance of ethanol was also examined. Fig. 7a shows the infrared spectra of ethanol adsorption on catalysts with different molybdenum contents under Ar atmosphere. As indicated, the Mo1Sn6 catalyst exhibits a characteristic peak at 3675 cm−1 corresponding to –OH, and a peak at 1050 cm−1 is related to ethoxy species.48 This suggests that the O–H bond cleavage occurs at the M–O ionic pairs on the catalyst surface, resulting in the formation of –OH and ethoxy species. The absorption peak at 1762 cm−1 corresponds to the carbonyl characteristic peak of acetaldehyde, while the peaks at 1457 cm−1 1333 cm−1 and 1363 cm−1 are associated with acetate species, and the peaks at 1245 cm−1 represent gas-phase ethanol.49 As the molybdenum content decreases, in addition to the characteristic peak at 3675 cm−1, a distinct negative peak appears at 3527 cm−1, indicating the consumption of –OH species on the catalyst surface.50 This suggests that ethanol reacts with the surface –OH groups, leading to the formation of water and ethoxy species, representing an alternative mode of ethanol adsorption. When Mo–Sn ratio changes to Mo1Sn15, only the –OH characteristic peak at 3675 cm−1 remains, and the negative peak disappears. This indicates that ethanol has only one mode of adsorption at this stage, with the M–O ionic pairs serving as the active adsorption sites, similar to the adsorption form observed in Mo1Sn6. Additionally, the weaker aldehyde carbonyl peak at 1762 cm−1 suggests a reduced oxidation activity of ethanol.
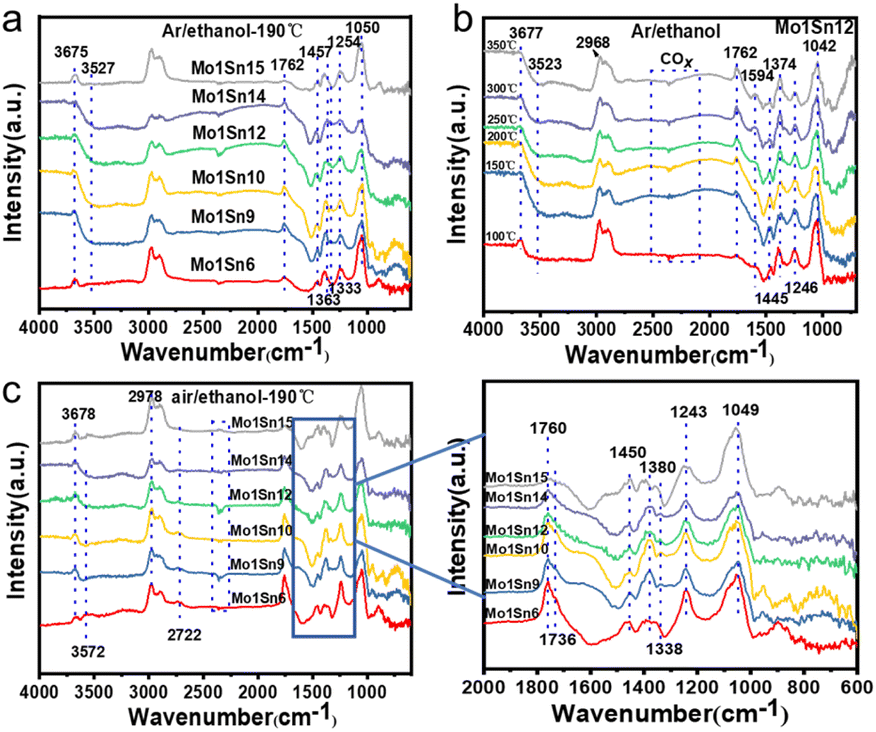 | ||
| Fig. 7 (a) Infrared spectra of ethanol adsorption on catalysts with different Mo–Sn ratios under Ar atmosphere, (b) the adsorption of ethanol on the Mo1Sn12 catalyst at different temperatures under Ar atmosphere. And (c) infrared spectra of ethanol adsorption on catalysts with different Mo–Sn ratios under air atmosphere. | ||
To further investigate the in situ adsorption and transformation of ethanol, we examined the infrared spectra of ethanol adsorption on catalysts with varying molybdenum contents under an air atmosphere (Fig. 7c). The forms of ethanol adsorption on different Mo–Sn ratio catalysts were consistent with the adsorption patterns observed under Ar atmosphere. As molybdenum content decreases, the carbonyl absorption peak at 1760 cm−1 (carbonyl (CO) vibration of acetaldehyde) diminishes, and the absorption peaks for acetate at 1450 cm−1 and 1380 cm−1 also decline.51 Notably, the lowest acetate content is found over the Mo1Sn10 and Mo1Sn12 catalysts.
According to the formation mechanism of ethyl acetate from acetate and ethoxy species, the formation of ethyl acetate over Mo1Sn10 and Mo1Sn12 catalysts would be inhibited. Further reduction of the molybdenum content resulted in another significant increase in acetate strength on the catalyst surface. Subsequently, we investigated the adsorption of ethanol on the Mo1Sn12 catalyst at different temperatures under Ar atmosphere (Fig. 7b). Ethoxylates (1042 cm−1) formed on the catalyst at low temperature (100 °C) suggest that the initial step of ethanol oxidation, the cleavage of the O–H bond, is relatively facile and the rate-determining step is the cleavage of the Cα–H bond. With rising temperature, the absorption peak at 1594 cm−1 becomes more pronounced, indicating an increase in acetate content and a decrease in ethoxy content. These findings indicate that with rising temperature, the ethanol conversion increases, whereas the selectivity for acetaldehyde decreases.
Table 5 displays the ethanol conversion and product selectivity at 190 °C for all catalysts. The primary product of ethanol oxidation is acetaldehyde, accompanied by byproducts including formaldehyde, acetone, methyl acetate, ethyl acetate, methanol, and ethylene. As the molybdenum content decreases, yield shows a trend of increasing and then decreasing. While the selectivity of ethyl acetate shows a counter-trend. The Mo1Sn12 catalyst yielded optimal results, achieving ethanol conversion of 58.9% and acetaldehyde selectivity of 89.3%.
| Mo/Sn | Con. (%) | Sel. (mol%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Formaldehyde | Acetaldehyde | Acetone | Methyl acetate | Ethyl acetate | Methanol | Ethylene | ||
| a Reaction conditions: temperature: 190 °C, 1 mL catalyst, LHSV (ethanol) = 1.8 h−1, at atmospheric pressure, GHSV = 10000 h−1, reaction time: 2 h. |
||||||||
| 1:6 |
29.7 | 1.2 | 84.9 | 1.0 | 0.0 | 12.6 | 0.1 | 0.2 |
| 1:9 |
36.4 | 1.1 | 89.0 | 1.0 | 0.0 | 8.7 | 0.0 | 0.2 |
| 1:10 |
46.9 | 2.0 | 90.0 | 3.0 | 0.2 | 4.5 | 0.2 | 0.1 |
| 1:12 |
58.9 | 2.2 | 89.3 | 2.7 | 0.0 | 5.8 | 0.0 | 0.0 |
| 1:14 |
37.3 | 0.8 | 88.4 | 1.7 | 0.0 | 9.1 | 0.0 | 0.0 |
| 1:15 |
30.3 | 1.2 | 83.6 | 1.6 | 0.0 | 13.6 | 0.0 | 0.0 |
The catalytic activity of pure molybdenum (Mo) or tin (Sn) in alcohol ether oxidation is significantly low, whereas Mo–Sn bimetallic mixed oxide catalysts exhibit excellent low-temperature performance in the ethanol oxidation reaction. This is due to the different interactions between molybdenum oxides and SnO2 within the Mo–Sn catalyst system.29 Variation in Mo content results in changes in the catalyst structure and surface properties. According to the BET results, the performance variations among different catalysts are not predominantly attributed to specific surface area differences. The catalyst structure has undergone distortion and deformation as Mo content decreases from XRD analysis, promoting the generation of defects and facilitating oxygen adsorption and activation. The MoO in MoOx gradually red shift as the Mo content decreases, as indicated by Raman spectroscopy. Consequently, the bond energy of MoO diminishes, facilitating bond dissociation and enhancing reactivity. This fosters the oxidation process, boosting catalytic activity and leading to the increase of ethanol conversion. However, as the Mo content further decreases, the concentration of MoO bonds on the catalyst surface also decreases, and the ethanol conversion begins to decline again. Furthermore, Yang observed that the presence of Mo5+ enhances the low-temperature oxidation properties of ethers. We have also identified the presence of Mo5+ in various catalysts, with the highest content observed in the Mo1Sn12 catalyst, which correlates with enhanced ethanol conversion efficiency. In situ DRIFTS analysis indicates that the active sites for the adsorption of activated ethanol over Mo1Sn6 comprise solely the M–O ion pair. Upon reducing the Mo content, the number of ethanol adsorption activation sites increases, and the surface active –OH groups can also activate ethanol. The presence of multiple active sites enhances ethanol conversion. However, when the molybdenum content is further reduced, only one type of ethanol adsorption site remains on the surface of the catalyst (Mo1Sn15), and the ethanol conversion begins to decrease. As shown in Fig. 7b, ethanol can dissociate and adsorb on the Mo1Sn12 catalyst at 100 °C to form ethoxy species, indicating that the first step (O–H bond cleavage) of the ethanol conversion process is facilitative. The weakening of the MoO bond significantly enhances ethanol conversion, as it is hypothesized to facilitate the second step (Cα–H bond cleavage) during the conversion of the ethanol active species. Thus, the MoO bond plays a crucial role in activating the decisive step.
It is well known that the formation of ethyl acetate occurs through an esterification reaction under acidic catalytic conditions. Typically, medium-strength acidic sites enhance the ester formation, whereas strong acidic sites favor ethylene production.52,53 In this work, an excess of ethanol is present, so only a small amount of acetic acid is needed to generate ethyl acetate at the acidic active centers. The number of medium-strength acidic centers on the surfaces of Mo1Sn6 and Mo1Sn15 catalysts is significantly higher than that on the Mo1Sn12 catalyst from NH3-TPD analysis. Combining this with the evaluation data, it is found that the content of acetic acid on the surfaces of Mo1Sn6 and Mo1Sn15 catalysts is higher, which reasonably explains Mo1Sn6 and Mo1Sn15 catalysts have more medium-strength acidic centers and thus produce a larger amount of ethyl acetate. A non-linear relationship between the medium-strength acid content and molybdenum content, characterized by an initial decrease followed by an increase. This trend is linearly correlated with the ethyl acetate selectivity. The reduced medium-strength acid content on the Mo1Sn12 catalyst likely contributes to its decreased ethyl acetate selectivity. Furthermore, Fig. 7c reveals that Mo1Sn6 and Mo1Sn15 catalysts exhibit higher contents of acetate and ethoxy species on their surfaces, whereas Mo1Sn12 catalysts have comparatively lower levels of these intermediates, accounting for the variations in ethyl acetate selectivity among the catalysts.
In combination with XPS and O2-TPD results, we indicate that the increased mobility of lattice oxygen on the catalyst surface also enhances the low-temperature oxidation performance of the catalyst. Meanwhile, the oxygen vacancies formed after the participation of active lattice oxygen in the reaction can further activate O2 and provide reactive oxygen species for the catalytic cycle.
Based on the above studies, we propose a possible reaction mechanism for the formation of acetaldehyde from ethanol over Mo–Sn catalysts. As shown in Fig. 8, initially, ethanol is adsorbed on two types of active sites on the catalyst surface to form CH3CH2O*. Subsequently, under the influence of the MoO active sites, the Cα–H bond is cleaved, resulting in the formation of acetaldehyde.
 | ||
| Fig. 8 A possible reaction mechanism for the oxidation of ethanol to acetaldehyde over the Mo–Sn catalyst. | ||
4 Conclusions
In summary, the highly efficient low-temperature oxidation of ethanol to acetaldehyde has been significantly realized and the acetaldehyde selectivity reaches 89.3% with ethanol conversion of 58.9% over the Mo1Sn12 catalyst at 190 °C. The combination of various characterizations (Raman, XPS, CO2-TPD, NH3-TPD and in situ DRIFTS, etc.) and activity results demonstrated that the weakened MoO bond and the enhanced mobility of lattice oxygen along with the presence of multiple adsorption and activation centers markedly promote the low-temperature oxidation of ethanol. Besides, adjusting the molybdenum content can decrease the amount of medium-strong acid, thereby inhibiting ethyl acetate production and enhancing the acetaldehyde selectivity. This work provides a sustainable strategy for designing highly efficient catalysts to synthesize acetaldehyde from ethanol oxidation under mild conditions.
Data availability
The data supporting this article have been included as part of the ESI.Author contributions
CRediT: Chunhe Liu data curation, investigation, methodology, writing-original draft, writing-review & editing; Xiaqing Wang formal analysis, methodology, writing-review & editing; Xiujuan Gao methodology, writing-review & editing; Yingquan Wu data curation, investigation, methodology, writing-review & editing; Xiaoxing Wang methodology, writing-review & editing; Faen Song methodology, writing-review; Junfeng Zhang methodology, writing-review & editing; Yizhuo Han writing-review & editing; Qingde Zhang conceptualization, funding acquisition, investigation, project administration, resources, supervision, validation, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22172187), the Central Guidance on Local Science and Technology Development Fund of Shanxi Province (YDZJSX2022A072), and the Youth Innovation Promotion Association CAS (2014155).Notes and references
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1494–1513 Search PubMed.
- F. Wang, X. Jiang, Y. Liu, G. Zhang, Y. Zhang, Y. Jin, S. Shi, X. Men, L. Liu, L. Wang, W. Liao, X. Chen, G. Chen, H. Liu, M. Ahmad, C. Fu, Q. Wang, H. Zhang and S. Y. Lee, Innov, 2024, 5, 100687 CAS.
- J. Sun and Y. Wang, ACS Catal., 2014, 4, 1078–1090 CrossRef CAS.
- R. A. Dagle, A. D. Winkelman, K. K. Ramasamy, V. Lebarbier Dagle and R. S. Weber, Ind. Eng. Chem. Res., 2020, 59, 4843–4853 CrossRef CAS.
- D. D. Petrolini, W. H. Cassinelli, C. A. Pereira, E. A. Urquieta-González, C. V. Santilli and L. Martins, RSC Adv., 2019, 9, 3294–3302 RSC.
- Y. Zhang, X. San, N. Tsubaki, Y. Tan and J. Chen, Ind. Eng. Chem. Res., 2010, 49, 5485–5488 CrossRef CAS.
- T. Takei, N. Iguchi and M. Haruta, Catal. Surv. Asia, 2011, 15, 80–88 CrossRef CAS.
- J. Pang, M. Yin, P. Wu, X. Li, H. Li, M. Zheng and T. Zhang, Green Chem., 2021, 23, 7902–7916 RSC.
- D. A. Patel, G. Giannakakis, G. Yan, H. T. Ngan, P. Yu, R. T. Hannagan, P. L. Kress, J. Shan, P. Deshlahra, P. Sautet and E. C. H. Sykes, ACS Catal., 2023, 13, 4290–4303 CrossRef CAS.
- X. Huang, S. Wu, Z. Xiao, D. Kong, T. Liang, X. Li, B. Luo, B. Wang and L. Zhi, Nano Today, 2022, 44, 101508 CrossRef CAS.
- R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034–9037 CrossRef CAS PubMed.
- J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038–9049 CrossRef CAS PubMed.
- D. Yun, Y. Wang and J. E. Herrera, ACS Catal., 2018, 8, 4681–4693 CrossRef CAS.
- V. V. Kaichev, Y. A. Chesalov, A. A. Saraev, A. Y. Klyushin, A. Knop-Gericke, T. V. Andrushkevich and V. I. Bukhtiyarov, J. Catal., 2016, 338, 82–93 CrossRef CAS.
- P. Liu and E. J. M. Hensen, J. Am. Chem. Soc., 2013, 135, 14032–14035 CrossRef CAS PubMed.
- E. Behravesh, M. M. Melander, J. Wärnå, T. Salmi, K. Honkala and D. Y. Murzin, J. Catal., 2021, 394, 193–205 CrossRef CAS.
- S. J. Raynes and R. A. Taylor, Sustainable Energy Fuels, 2021, 5, 2136–2148 RSC.
- Y. Injongkol, T. Maihom, S. Choomwattana, B. Boekfa and J. Limtrakul, RSC Adv., 2017, 7, 38052–38058 RSC.
- S. S. Dotsenko, V. A. Verkhov, V. A. Svetlichnyi, L. F. Liotta, V. La Parola, T. I. Izaak and O. V. Vodyankina, Catal. Today, 2020, 357, 503–510 CrossRef CAS.
- B. Jiang, R. Chang and Y. Lin, Ind. Eng. Chem. Res., 2012, 6, 37–42 Search PubMed.
- Y. Huang, B. Wang, H. Yuan, Y. Sun, D. Yang, X. Cui and F. Shi, Catal. Sci. Technol., 2021, 11, 1652–1664 RSC.
- Q. Yang, X. Gao, F. Song, X. Wang, T. Zhang, P. Xiong, Y. Bai, X. Liu, X. Liu, J. Zhang, G. Fu, Y. Tan, Y. Han and Q. Zhang, JACS Au, 2023, 3, 3141–3154 CrossRef CAS PubMed.
- X. Li and E. Iglesia, Chem.–Eur. J., 2007, 13, 9324–9330 CrossRef CAS PubMed.
- G. Pampararo, G. Garbarino, N. Ardoino, P. Riani and G. Busca, J. Chem. Technol. Biotechnol., 2021, 96, 3304–3315 CrossRef CAS.
- P. R. S. Medeiros, J. G. Eon and L. G. Appel, Catal. Lett., 2000, 69, 79–82 CrossRef CAS.
- S. T. Oyama and W. Zhang, J. Am. Chem. Soc., 1996, 118, 7173–7177 CrossRef CAS.
- L. He, B. Zhou, D. Sun, W. Li, W. Lv, J. Wang, Y. Liang and A. Lu, ACS Catal., 2023, 13, 11291–11304 CrossRef CAS.
- Q. Yang, X. Gao, R. Feng, M. Li, J. Zhang, Q. Zhang, Y. Han and Y. Tan, J. Fuel Chem. Technol., 2019, 47, 934–941 CrossRef CAS.
- Z. Zhang, Q. Zhang, L. Jia, W. Wang, S. P. Tian, P. Wang, H. Xiao, Y. Han, N. Tsubaki and Y. Tan, Catal. Sci. Technol., 2016, 6, 6109–6117 RSC.
- G. Liu, Q. Zhang, Y. Han, N. Tsubaki and Y. Tan, Green Chem., 2013, 15, 1501–1504 RSC.
- M. Dieterle, G. Weinberg and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 822–826 RSC.
- H. Liu, P. Cheung and E. Iglesia, J. Catal., 2003, 217, 222–232 CAS.
- L. Yan, J. Zhang, X. Gao, F. Song, X. Wang, T. Zhang, X. Liu, X. Meng, Q. Zhang, Y. Han and Y. Tan, Chem. Commun., 2021, 57, 13297–13300 RSC.
- M. Dieterle, G. Weinberg and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 812–821 RSC.
- G. Liu, Q. Zhang, Y. Han, N. Tsubaki and Y. Tan, Green Chem., 2013, 15, 1501–1504 RSC.
- Z. Zhang, Q. Zhang, L. Jia, W. Wang, T. Zhang, Y. Han, N. Tsubaki and Y. Tan, Catal. Sci. Technol., 2016, 6, 2975–2983 RSC.
- Y. Meng, T. Wang, S. Chen, Y. Zhao, X. Ma and J. Gong, Appl. Catal., B, 2014, 160–161, 161–172 CrossRef CAS.
- J. Światowska-Mrowiecka, S. de Diesbach, V. Maurice, S. Zanna, L. Klein, E. Briand, I. Vickridge and P. Marcus, J. Phys. Chem. C, 2008, 112, 11050–11058 CrossRef.
- F. Gonçalves, P. R. S. Medeiros, J. G. Eon and L. G. Appel, Appl. Catal., A, 2000, 193, 195–202 CrossRef.
- C. C. Díaz, M. Pilar Yeste, H. Vidal, J. M. Gatica, L. E. Cadús and M. R. Morales, Fuel, 2020, 262, 116564 CrossRef.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- G. V. Mamontov, M. V. Grabchenko, V. I. Sobolev, V. I. Zaikovskii and O. V. Vodyankina, Appl. Catal., A, 2016, 528, 161–167 CrossRef CAS.
- H. Liu, Y. Jiang, R. Zhou, Z. Chang and Z. Hou, Fuel, 2022, 321, 123980 CrossRef CAS.
- J. Velasquez Ochoa, E. Farci, F. Cavani, F. Sinisi, L. Artiglia, S. Agnoli, G. Granozzi, M. C. Paganini and L. Malfatti, ACS Appl. Nano Mater., 2019, 2, 3434–3443 CrossRef CAS.
- P. R. S. Medeiros and L. G. Appel, Appl. Catal., A, 2002, 231, 125–130 CrossRef CAS.
- W. Wang, X. Gao, P. Xiong, J. Zhang, F. Song, Q. Zhang, Y. Han and Y. Tan, J. Fuel Chem. Technol., 2021, 49, 1487–1494 CrossRef CAS.
- H. Song, X. Bao, C. M. Hadad and U. S. Ozkan, Catal. Lett., 2010, 141, 43–54 CrossRef.
- J. Raskó, A. Hancz and A. Erdőhelyi, Appl. Catal., A, 2004, 269, 13–25 CrossRef.
- X. Cao, W. Wang and X. Ma, Chem. Eng. Technol., 2009, 26, 44–49 CAS.
- J. M. Guil, N. Homs, J. Llorca and P. Ramírez de la Piscina, J. Phys. Chem. B, 2005, 109, 10813–10819 CrossRef CAS PubMed.
- S. Preedavijitkul, C. Autthanit, P. Praserthdam and B. Jongsomjit, J. Environ. Chem. Eng., 2022, 10, 107542 CrossRef CAS.
- C. Wang, G. Garbarino, L. F. Allard, F. Wilson, G. Busca and M. Flytzani-Stephanopoulos, ACS Catal., 2015, 6, 210–218 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |