
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro and in silico hybrid approach to unveil triterpenoids from Helicteres hirsuta leaves as potential compounds for inhibiting Nrf2†
Minh Hien Nguyen *ab,
Nguyen Thien Han Leab,
Bui Quoc Huy Nguyenc,
Mai Thanh Thi Nguyenbde,
Truong Nhat Van Dobde,
Tho Huu Lebde,
Vu Thanh Nguyenf and
Chia-Hung Yengh
*ab,
Nguyen Thien Han Leab,
Bui Quoc Huy Nguyenc,
Mai Thanh Thi Nguyenbde,
Truong Nhat Van Dobde,
Tho Huu Lebde,
Vu Thanh Nguyenf and
Chia-Hung Yengh
aUniversity of Health Sciences, Vietnam National University Ho Chi Minh City, YA1 Administrative Building, Hai Thuong Lan Ong Street, Dong Hoa Ward, Di An City, Binh Duong Province 75308, Vietnam. E-mail: nmhien@uhsvnu.edu.vn; Tel: (+84) 373 696 894
bVietnam National University Ho Chi Minh City, Quarter 6, Linh Trung Ward, Thu Duc District, Ho Chi Minh City 70000, Vietnam
cThe University of Danang – VN-UK Institute for Research and Executive Education, 41 Le Duan Street, Hai Chau 1 Ward, Hai Chau District, Danang City 50000, Vietnam
dFaculty of Chemistry, University of Science, Ho Chi Minh City, Vietnam
eResearch Lab for Drug Discovery and Development, University of Science, Ho Chi Minh City, Vietnam
fDivision of Aquacultural Biotechnology, Biotechnology Center of Ho Chi Minh City, 2374, Highway 1, Quarter 2, Trung My Tay Ward, District 12, Ho Chi Minh City 70000, Vietnam
gGraduate Institute of Natural Products, College of Pharmacy, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Road, Sanmin District, Kaohsiung City 80708, Taiwan
hNational Natural Product Libraries and High-Throughput Screening Core Facility, Kaohsiung Medical University, No. 100, Shih-Chuan 1st Road, Sanmin District, Kaohsiung City 80708, Taiwan
First published on 21st January 2025
Abstract
Cancer is a leading global health concern, with over 20 million new cases and 9.7 million deaths reported in 2022. Chemotherapy remains a widely used treatment, but drug resistance, which affects up to 90% of treatment outcomes, significantly hampers its effectiveness. The transcription factor Nrf2, which is crucial for cellular defense against oxidative stress, plays a dual role in cancer treatment. Although Nrf2 activation can suppress early carcinogenesis, its overexpression in cancer cells contributes to drug resistance, resulting in poor patient outcomes. Thus, inhibiting Nrf2 has emerged as a promising strategy for overcoming cancer drug resistance. Natural compounds such as luteolin and brusatol have shown potential in inhibiting Nrf2, although with limitations. This study isolates and characterizes seven triterpenoids from the n-hexane sub-fraction of Helicteres hirsuta, a plant traditionally used for medicinal purposes, to evaluate their ability to modulate Nrf2 activity in Huh7 cancer and HaCaT normal cells. Additionally, molecular docking and dynamic simulations were utilized to assess the binding potential of these compounds to the PI3Kα receptor, which regulates downstream signaling pathways, thereby suppressing Nrf2 activity in cancer cells. Our findings provide insights into new strategies seeking triterpenoids as promising structures to reverse chemoresistance by regulating Nrf2. The results also reveal the potential of 3β-O-trans-caffeoylbetulinic acid from H. hirsuta leaves as the unprecedented compound inhibiting Nrf2 activity, with an IC50 of 74.5 μg mL−1 in Huh7 cancer cells.
Introduction
Cancer, with more than 20 million new cases and nearly 9.7 million cancer deaths according to GLOBOCAN statistics in 2022, remains a major public health concern worldwide.1 Cancer is also the leading cause of mortality in adults in most countries, along with noncommunicable diseases and cardiovascular diseases.2 Among the various methods applied in cancer treatment, chemotherapy still stands out as a popular method.3 It is predicted that by 2030, the number of patients needing first-course chemotherapy annually will increase by more than 50% to 15 million patients.4Cancer drug resistance is a condition in which cancer cells develop the ability to resist the effects of chemotherapy drugs. This results in drug treatment partially or completely losing its expected effectiveness.5 Ineffective drug treatment resulting from drug resistance is the cause of up to 90% of treatment failures and deaths.6 Drug resistance can arise through different mechanisms, which can be classified into intrinsic and acquired resistance, depending on the time of occurrence.7 In general, the goal of overcoming drug resistance in cancer cells is to optimize the sensitivity of the treatment method.
Nuclear factor erythroid-2 p45-related factor 2 (Nrf2) is a transcription factor that regulates the cellular defense against toxic agents and oxidative stress by expressing genes involved in oxidative stress response and detoxification.8 Nrf2 maintains cellular redox homeostasis and exerts anti-inflammatory and anti-cancer activities, thereby supporting cell survival. Some evidence suggests that Nrf2 activation can suppress carcinogenesis, especially at early stages.9 However, Nrf2 expression can be considered a double-edged sword for cancer patients. Indeed, overexpression of Nrf2 in cancer cells is closely related to the development of drug resistance in cancer cells.10 Nrf2 protects cells against toxic agents, which are currently understood as chemotherapy drugs, enhancing their resistance to chemotherapeutic agents.11 Clinically, overexpression of Nrf2 is consistently observed with poor prognosis in cancer patients.12 Therefore, direct or indirect inhibition of Nrf2 expression may increase the sensitivity of cancer cells to chemotherapy drugs and reverse drug resistance.13 The inhibition of Nrf2 expression is now considered a promising target for strategies to combat cancer drug resistance. The dual functions of Nrf2 pose a complex challenge when considering Nrf2 inhibition as a therapeutic strategy. However, this approach carries potential risks, such as increased vulnerability of normal cells to oxidative stress and toxic agents, potentially leading to adverse effects.14,15 Thus, a thorough risk-benefit strategies should prioritize selective inhibition of Nrf2 in cancer cells, while preserving Nrf2 activity and sparing normal tissues to minimize collateral toxicity.
Luteolin is a flavone that has been reported to be a potent inhibitor of Nrf2 activity. Luteolin reduced the effective inhibitory concentration of Nrf2 mRNA by 34% when co-administered with actinomycin D for 30 min, and by 43% after 1.5 h in A549 human lung carcinoma cells.16 In vivo studies in mice showed that single treatment with increasing doses of luteolin, and co-treatment with luteolin and cisplatin significantly reduced tumor sizes compared to solely treating with cisplatin.17 Brusatol was also shown to enhance the therapeutic effect of gemcitabine by inhibiting cell growth and inducing apoptosis in human pancreatic cancer cells compared to others through inhibition of the Nrf2 pathway when combined with 1 μM brusatol and 20 μM gemcitabine for 48 h of treatment.18 However, the Nrf2 inhibition by brusatol was reported to be nonspecific, and it also reduced the viability of healthy human colon cells.19 Other natural compounds, such as ochratoxin A and trigonelline in coffee beans, were also reported to block the nuclear accumulation of Nrf2 in leukemic cells.20,21 Malabaricone A, a plant-derived antioxidant, effectively inhibited Nrf2 transcriptional activity as reflected by a decrease in HO-1 protein levels, and led to ROS accumulation and subsequent cell apoptosis.22 Overall, the number of compounds demonstrated to selectively inhibit Nrf2 in cancer cells without affecting healthy cells is currently very limited, and concentrated in the flavonoid group.
Nrf2 inhibitors exhibit regulatory effects through a variety of complex pathways, both upstream and downstream mechanisms, and influence the expression and activity of Nrf2.23 For instance, the phosphoinositide 3-kinase (PI3K) pathway has been identified as one of the modulators of Nrf2.24 Thus, inhibiting the PI3K/AKT pathway can effectively restore cancer cells' sensitivity to chemotherapy, thereby overcoming drug resistance.25 This approach targets a key survival pathway that is often hyperactivated in tumors, leading to overexpression of the Nrf2 activity. However, the exact mechanisms regulating Nrf2 expression and activity remain unclear and complicated.26 The use of in silico approaches for predicting the Nrf2 inhibitory signalling pathway offers a valuable tool for reassessing the potential of Nrf2 inhibition, specifically through the PI3K regulatory receptor.
Helicteres hirsuta, a plant traditionally used for various medicinal purposes in Southeast Asia, has been reported to possess anti-inflammatory and antioxidant activities.27 Previous phytochemical studies have identified various compounds from this plant, including flavonoids, terpenoids, and phenolics.28 However, the potential of H. hirsuta and its constituents in targeting Nrf2 and overcoming drug resistance in cancer remains largely unexplored. Based on our previous in vitro screening results, the n-hexane subfraction of H. hirsuta leaves demonstrated a significant decrease in Nrf2 activity on cancer cells and no cytotoxicity on HaCaT normal cells.28 Furthermore, in an in vivo study using a zebrafish model, the n-hexane subfraction from H. hirsuta leaves showed antioxidant activity by protecting wild-type larvae against oxidative stress, while significantly reducing both Nrf2 expression and the expression of its target genes on an overexpressed Nrf2 zebrafish model.29 These findings underscore the potential of this subfraction as a selective Nrf2 modulator with minimal adverse events to normal tissues. Therefore, our study aims to isolate and elucidate the structure of triterpenoids from the n-hexane sub-fraction of H. hirsuta. We then evaluated their ability to regulate Nrf2 activity on Huh7 cancer cells and HaCaT normal cells. Compounds demonstrating Nrf2 inhibition in Huh7 cancer cells, while simultaneously activating Nrf2 in HaCaT normal cells, were identified as promising candidates for further investigation. Furthermore, we employed docking methods and dynamic simulations to evaluate the binding ability of these triterpenoids to the Nrf2 activity-regulating receptor PI3Kα.
Materials and methods
Chemicals and instrument
NMR spectra were taken on a Bruker Avance III 500 spectrometer (Brucker BioSpin AG) with deuterated solvents as an internal standard, and chemical shifts are expressed in δ values. Analytical and preparative TLCs were performed on precoated Kieselgel 60F254 or RP-18F254 plates (Merck KGaA). Dulbecco's modified Eagle's medium (DMEM), Gibco fetal bovine serum, penicillin–streptomycin, non-essential amino acids, L-glutamine, and alamarblue were sourced from Thermo Fisher Scientific, USA. Luciferase buffer, luciferin and cell culture lysis 5× were obtained from Promega, USA, while dimethyl sulfoxide (DMSO) was sourced from Nacalai Tesque Inc., Japan.Sample collection
The mature leaves of H. hirsuta were collected from Tinh Bien district, An Giang province, Vietnam, in December 2023. The plant was taxonomically verified by Dr Tuan Anh Le Dang of the Faculty of Biology and Biotechnology, VNUHCM-University of Science. A voucher specimen (No. MDC-9008) has been catalogued and securely stored in the herbarium of the Department of Medicinal Chemistry, Faculty of Chemistry, VNUHCM-University of Science.Plant extraction and isolation
A total of 4 kg of fresh plant material was cleaned, naturally air-dried, and stored in a dry place. The moisture content of the dried material was measured to be 10 ± 1.9%. The dried material was then chopped into a fine powder. This powdered material was subjected to Soxhlet extraction with methanol (500 g of sample × 2 L of solvent × 8 hours), yielding a methanolic-soluble extract. This extract was concentrated under reduced pressure to afford a crude methanol extract (100.2 g). This crude extract was fully dispersed in water and then subjected to liquid–liquid partitioning using solvents of increasing polarity, including n-hexane and EtOAc, to yield three distinct sub-fractions: n-hexane (6.2 g), EtOAc (3.4 g), and H2O (150.9 g).A part of the n-hexane sub-fraction (AxH; 4.1 g) was fractionated by normal-phase silica gel column chromatography (NP-CC) using a gradient of n-hexane–acetone (0%, 5%, 10%, 20%, and 50% acetone), yielding six major fractions: AxH1 (626.3 mg), AxH2 (425.0 mg), AxH3 (335.0 mg), AxH4 (1.2 g), AxH5 (771.6 mg), and AxH6 (327.4 mg). Fraction AxH2 underwent further separation via NP-CC with a gradient of n-hexane–isopropanol (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 97:3, 95:5, 90:10, 85:15, 80:20, and 70:30), producing two subfractions: AxH2.1 and AxH2.2. Subfraction AxH2.1 (64.7 mg) was recrystallized in MeOH, affording 1 (5.0 mg). Subfraction AxH2.2 (290.2 mg) was subjected to NP-CC using an n-hexane–EtOAc gradient (0–80% EtOAc), followed by preparative normal-phase thin-layer chromatography (NP-PTLC) with n-hexane–isopropanol (95:5), which led to the isolation of 7 (5.2 mg). Fraction AxH4 (1.2 g) underwent further separation by NP-CC using a gradient of CHCl3–MeOH (100:0, 97:3, 95:5, 90:10, 85:15, and 70:30), resulting in six subfractions: AxH4.1 − AxH4.6. Subfraction AxH4.2 (166.9 mg) was further purified by NP-CC using an n-hexane–CH2Cl2 gradient (0–100% CH2Cl2), yielding 2 (5.3 mg). Subfraction AxH4.6 (224.8 mg) was divided by NP-CC utilizing a gradient of CH2Cl2–acetone (0–30% acetone), followed by RP-PTLC using H2O–ACN–MeOH system (50:40:10), resulting in the isolation of 6 (5.3 mg). Subfraction AxH4.4 (325.4 mg) was processed via RP-CC with H2O–acetone gradient (10–100% acetone), yielding 4 (4.1 mg) and 5 (3.7 mg). Fraction AxH5 (771.6 mg) was subjected to RP-CC using a gradient of H2O–MeOH (10–100% MeOH), resulting in three subfractions: AxH5.1 − AxH5.3. Subfraction AxH5.2 (324.7 mg) was further fractionated by RP-CC using an H2O–ACN gradient (80:20, 50:50, and 30:70), followed by NP-PTLC with n-hexane–CH2Cl2–isopropanol (50:45:5), yielding 3 (2.0 mg).
:0, 97:3, 95:5, 90:10, 85:15, 80:20, and 70:30), producing two subfractions: AxH2.1 and AxH2.2. Subfraction AxH2.1 (64.7 mg) was recrystallized in MeOH, affording 1 (5.0 mg). Subfraction AxH2.2 (290.2 mg) was subjected to NP-CC using an n-hexane–EtOAc gradient (0–80% EtOAc), followed by preparative normal-phase thin-layer chromatography (NP-PTLC) with n-hexane–isopropanol (95:5), which led to the isolation of 7 (5.2 mg). Fraction AxH4 (1.2 g) underwent further separation by NP-CC using a gradient of CHCl3–MeOH (100:0, 97:3, 95:5, 90:10, 85:15, and 70:30), resulting in six subfractions: AxH4.1 − AxH4.6. Subfraction AxH4.2 (166.9 mg) was further purified by NP-CC using an n-hexane–CH2Cl2 gradient (0–100% CH2Cl2), yielding 2 (5.3 mg). Subfraction AxH4.6 (224.8 mg) was divided by NP-CC utilizing a gradient of CH2Cl2–acetone (0–30% acetone), followed by RP-PTLC using H2O–ACN–MeOH system (50:40:10), resulting in the isolation of 6 (5.3 mg). Subfraction AxH4.4 (325.4 mg) was processed via RP-CC with H2O–acetone gradient (10–100% acetone), yielding 4 (4.1 mg) and 5 (3.7 mg). Fraction AxH5 (771.6 mg) was subjected to RP-CC using a gradient of H2O–MeOH (10–100% MeOH), resulting in three subfractions: AxH5.1 − AxH5.3. Subfraction AxH5.2 (324.7 mg) was further fractionated by RP-CC using an H2O–ACN gradient (80:20, 50:50, and 30:70), followed by NP-PTLC with n-hexane–CH2Cl2–isopropanol (50:45:5), yielding 3 (2.0 mg).
ARE-luciferase reporter assay and cell viability assay
HaCaT/ARE and Huh7/ARE cell lines were cultured as described in our previous publication. The luciferase reporter assay and cell viability assay were performed based on methods described in the same paper.30 The relative luciferase activity was calculated by normalizing the luciferase activity to the cell viability. The average relative luciferase activity of DMSO wells was defined as the control, and assigned to a relative Nrf2 activity of 100%.Molecular docking
Molecular docking was performed to obtain the binding energies and the interactions between the PI3Kα receptor and the filtered ligands. The 3D crystal structure of the PI3Kα complex was obtained from the RCSB Protein Data Bank database (PDB ID: 5XGJ), and SMILES formats of natural compounds were converted to 3D structures using the OpenBabel tool. All protein and compound structures were prepared prior to docking by removing water molecules and other non-standard residues, adding missing hydrogen atoms, and assigning charges using the Dock Prep tool of UCSF Chimera program version 1.17.3. Molecular docking on PI3Kα was conducted on five allosteric binding sites.31 The docking box dimensions for sites 1 and 2 are 43.21 Å × 40.10 Å × 36.10 Å, with center coordinates at x = 5.25, y = 38.6, and z = 62.80. For site 3, the docking box size was 34.07 Å × 34.40 Å × 29.31 Å, at x = −0.70, y = 64.14, and z = 26.90. The dimensions of site 4 are 35.24 Å × 25.30 Å × 35.00 Å, centered at x = −0.80, y = 29.90, and z = 15.40. Lastly, the docking box for site 5 has dimensions of 35.62 Å × 48.00 Å × 39.00 Å, with the center at x = 25.80, y = 15.00, and z = 12.80. Next, the docking process was conducted using AutoDock Vina 1.1.2. Finally, PyMOL 3.0 software was used to visualize the docking poses and the interaction types between the PI3K and ligands.Molecular dynamics
MD simulations were performed on PI3Kα (PDB ID: 5XGJ) and the docked complexes of PI3Kα with 3β-O-trans-caffeoylbetulinic acid, betulinic acid, and β-sitosterol ligands using Gromacs 2024.1.32. The topology of PI3Kα was prepared using CHARMS-36 force field and TIP3P GROMACS recommended water model. The ligand topologies were prepared using the CGENFF web server tool. After that, the GROMACS compatible topology files of the ligands were converted using a Python script downloaded from the MacKerell-lab website. Then, the topology files of PI3Kα and the ligands were manually merged together. Next, the complex was placed in a dodecahedron box with a minimum distance of 1 nm between the solute and the box wall. All systems were solvated using the simple point charge-216 explicit water model (spc216.gro), and then neutralized using Na+ ions. Energy minimization was run using the steepest descent algorithm until all atomic forces in the systems were below 100 kJ mol−1 nm−1. Under position restraints, all systems were equilibrated in two stages with a time step of 2 fs and a duration of 1 ns. In the first stage, all systems were equilibrated in an NVT ensemble using the V-rescale thermostat at the temperature of 300 K. The second equilibration stage was conducted in an NPT ensemble using the C-rescale barostat at the pressure of 1 bar. To account for electrostatic forces, the Ewald Particle Mesh (PME) method was used. A 1 nm cutoff was applied to treat short-range electrostatics and van der Waals interactions. Hydrogen bonds were constrained in both equilibration and production steps using the LINCS algorithm. Finally, the production run was conducted for 100 ns with a trajectory snapshot saved every 10 ps.In silico ADME and toxicology profile
Compounds underwent in silico ADME predictions using the SwissADME web tool (https://www.swissadme.ch/).32 The SMILES format of each compound was input to calculate physicochemical properties, including molecular weight, hydrogen bond acceptor/donor counts, topological polar surface area (TPSA), lipophilicity, and water solubility. Additional assessments included pharmacokinetic behavior and drug-lead likeness. The in silico toxicity profiles of the compounds were analyzed using the ProTox 3.0 web tool (https://tox.charite.de/protox3/).33 Predicted toxicity was evaluated based on toxicity classification and median lethal dose (LD50), providing valuable insights into the safety and potential risks associated with these compounds.Result and discussion
Isolation and structure identification of triterpenoids from H. hirsuta
Phytochemical investigation of the n-hexane sub-fraction of H. hirsuta leaves resulted in the isolation of seven known compounds. The structure elucidation of these compounds was accomplished through extensive-NMR spectroscopy analysis and supported by comparison with reference literature (Fig. S1–S14†). The identified compounds include betulinic acid (1),34 lupeol (2),35 3β-O-trans-caffeoylbetulinic acid (3),36 oleanolic acid (4),37 maslinic acid (5),38 ursolic acid (6)39 and β-sitosterol (7)40 (Fig. 1).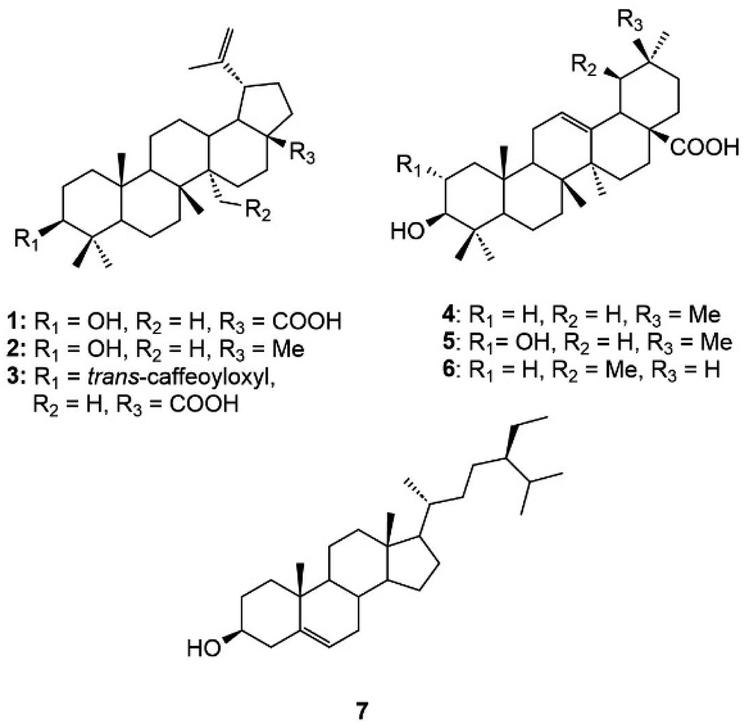 | ||
| Fig. 1 Compounds isolated from the n-hexane sub-fraction of H. hirsuta leaves. | ||
Screening Nrf2 activity on Huh7 and HaCaT cells
The Nrf2 activity screening results of the triterpenoid compounds isolated from the n-hexane fraction of H. hirsuta on Huh7 liver cancer cells and HaCaT normal cells are summarized in Fig. 2 and Table 1. Out of the seven compounds tested, three compounds, including betulinic acid (1), 3β-O-trans-caffeoylbetulinic acid (3), and β-sitosterol (7), demonstrated dual activity, inhibiting Nrf2 activity in Huh7 cells, while activating it in HaCaT cells. Specifically, compound 3 reduced Nrf2 activity by 16% in Huh7 cells, while tripling Nrf2 activation in HaCaT cells. Compounds 1 and 7 inhibited Nrf2 by more than 50% and 70%, respectively, in Huh7 cells, while boosting Nrf2 activity by nearly 400% in normal cells. Neither compound showed cytotoxicity in either cell line, whereas compound 3 exhibited mild cytotoxicity (less than 30%) in both Huh7 and HaCaT cells.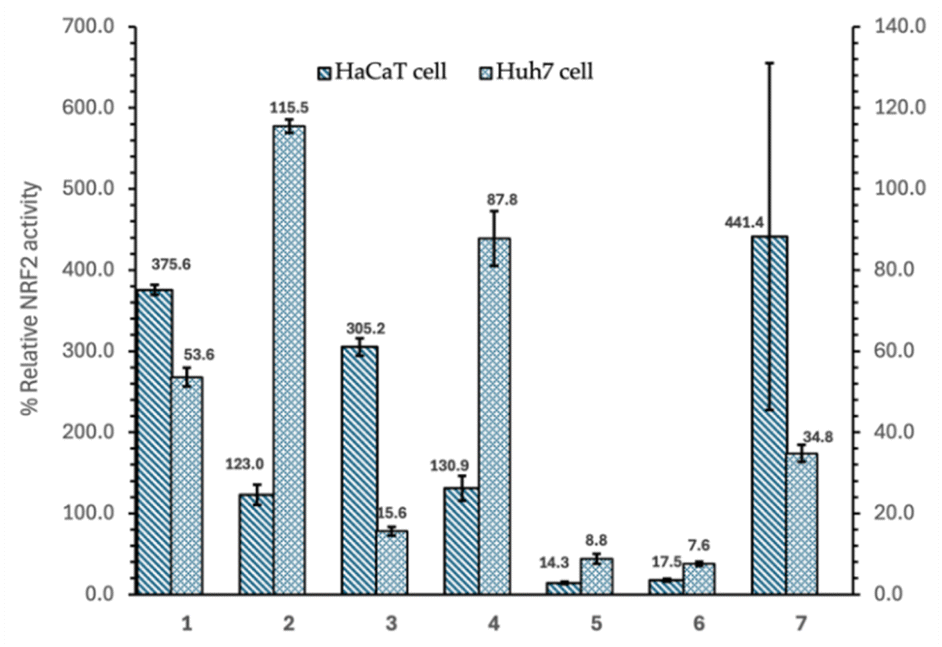 | ||
| Fig. 2 Relative Nrf2 activity on HaCaT and Huh7 cells of the following seven triterpenoids obtained from the n-hexane sub-fraction of Helicteres hirsuta leaves: betulinic acid (1), lupeol (2), 3β-O-trans-caffeoylbetulinic acid (3), oleanolic acid (4), maslinic acid (5), ursolic acid (6), and β-sitosterol (7). | ||
| Compound | Relative Nrf2 activity in HaCaT cell (%) | Relative Nrf2 activity in Huh7 cell (%) | HaCaT cell viability (%) | Huh7 cell viability (%) |
|---|---|---|---|---|
| a Betulinic acid (1), lupeol (2), 3β-O-trans-caffeoylbetulinic acid (3), oleanolic acid (4), maslinic acid (5), ursolic acid (6), and β-sitosterol (7). DMSO*: negative control. tBHQ** (tert-butylhydroquinone 10 μM): positive control for HaCaT. Luteolin*** (50 μM): positive control for Huh7. | ||||
| DMSO* | 100 | 100 | 100 | 100 |
| tBHQ** | 1144.7 ± 135.2 | — | 101.0 ± 0.4 | — |
| Luteolin*** | — | 7.6 ± 0.1 | — | 88.1 ± 1.8 |
| 1 | 375.6 ± 6.2 | 53.6 ± 2.3 | 104.8 ± 0.6 | 116.4 ± 2.9 |
| 2 | 123.0 ± 12.5 | 115.5 ± 1.6 | 99.2 ± 0.3 | 109.9 ± 0.4 |
| 3 | 305.2 ± 10.7 | 15.6 ± 1.1 | 88.8 ± 2.1 | 72.9 ± 1.7 |
| 4 | 130.9 ± 15.3 | 87.8 ± 6.7 | 101.1 ± 1.0 | 120.7 ± 0.6 |
| 5 | 14.3 ± 1.5 | 8.8 ± 1.2 | 49.5 ± 1.8 | 31.2 ± 0.6 |
| 6 | 17.5 ± 1.8 | 7.6 ± 0.5 | 36.6 ± 2.0 | 36.7 ± 0.7 |
| 7 | 441.4 ± 213.7 | 34.8 ± 2.1 | 110.4 ± 1.2 | 175.4 ± 4.6 |
These preliminary results underscore the potential application of H. hirsuta triterpenoids as promising compounds selectively inhibiting Nrf2 in Huh7 cancer cells. Compounds 5 and 6, while not selective, were able to reduce Nrf2 activity by over 80% in both cell types, showing further potential for anti-cancer strategies targeting Nrf2.
Molecular docking and molecular dynamics simulation
Based on in vitro results, the study selected compounds that demonstrated selective inhibition of Nrf2 activity in Huh7 cancer cells, while preserving Nrf2 activity in HaCaT normal cells for in silico studies, including 3β-O-trans-caffeoylbetulinic acid (3), betulinic acid (1), and β-sitosterol (7). The docking results of the three compounds isolated from H. hirsuta leaves, showing promising inhibition of Nrf2 and PI3Kα, are summarized in Table 2 and Fig. 3. The molecular docking results indicated that compound 3 exhibited the strongest binding affinity to the PI3Kα receptor. Among the five binding sites, compound 3 had the highest affinity to binding site 5 of PI3Kα with a docking score of −10.7 kcal mol−1. Similar patterns were also observed in compounds 1 and 7. This binding site is surrounded by several key residues capable of forming hydrogen bonds and van der Waals interaction, including Glu365, Leu540, Glu542, Ileu543, Leu570, Cys604, Asn605 and Phe1016 of p110α and residues Glu341, Asn344 and Asn377 on the p85α subunit, which is similar to the previous study – compound 3 with an aromatic ring and a hydroxyl group interacted with the Glu542 residue.31 The binding site 5 is located at the interface of the helical kinase, C2, and nSH2 domains, commonly referred to as the phosphopeptide-binding site.41 Upon upstream activation, the nSH2 domain of PI3K binds to auto-phosphorylated tyrosine residues, subsequently triggering the activation of the kinase activity. The docking pose of compound 3 observed in binding sites 1 and 2 also demonstrated significant potential in inhibiting PI3K. The aromatic ring of the caffeoyl group was inverted deep within the binding pocket, establishing interactions with residues, including Pro168, Asn170, Val166, Arg575, Arg662, Val851, and Ala758. These residues are similar to those PI3K inhibitors currently under investigation, targeting the region situated between the adaptor-binding domain (ABD) and the kinase domain of PI3Kα.42,43 Targeting this specific pocket with an inhibitor is expected to enhance the interaction between the ABD and kinase domain, thereby stabilizing their interface and potentially inhibiting PI3Kα activity.44 Current research on PI3Kα inhibitors focuses on the competitors on ATP-binding site. However, this approach often results in undesirable side effects. Additionally, PI3K plays a crucial role in activating the Nrf2 signaling pathway and regulating other signaling pathways in normal cells.45 To reduce side effects and avoid inhibiting PI3K in healthy cells, research studies increasingly focus on developing inhibitors that target allosteric sites, distinct from the active site.44 This strategy offers the potential to achieve selective inhibition of the PI3K activity, reducing adverse effects while maintaining therapeutic efficacy. Overall, the docking results suggest that PI3Kα can serve as a promising target protein for triterpenoid 3 derived from H. hirsuta. Triterpenoid 3 effectively inhibits the kinase activity of PI3K, regulating the downstream signaling pathways; thereby suppressing Nrf2 activity in cancer cells.| Compounds | Docking score (kcal mol−1) | Residue | |
|---|---|---|---|
| Sites 1&2 | 3β-O-trans-caffeoylbetulinic acid (3) | −9.8 | H-bond: Pro168 (2.5 Å), Asn170 (2.7 Å), Val166 (2.9 Å) |
| Alkyl: Arg575 (3.7 Å), Arg662 (3.7 Å), Val851 (4.2 Å) | |||
| π-sigma: Ala758 (3.8 Å) | |||
| Betulinic acid (1) | −8.2 | H-bond: Glu259 (2.3 Å), Lys924 (2.7 Å) | |
| β-Sitosterol (7) | −8.0 | H-bond: Arg852 (2.0 Å) | |
| Unfavorable donor–donor: Arg852 (2.1 Å) | |||
| Site 3 | 3β-O-trans-caffeoylbetulinic acid (3) | −8.9 | H-bond: Glu707 (2.4 Å) |
| π-sigma: Thr86 (3.8 Å) | |||
| Betulinic acid (1) | −8.8 | H-bond: Arg88 (2.8 Å), Asp746 (2.9 Å) | |
| π-sigma: Phe119 (3.6 Å) | |||
| Unfavorable donor–donor: Asn703 (2.2 Å) | |||
| β-Sitosterol (7) | −7.6 | Alkyl: Arg87 (4 Å), Arg115 (3.9 Å), Phe119 (4.2 Å) | |
| Site 4 | 3β-O-trans-caffeoylbetulinic acid (3) | −9.6 | H-bond: Ser464 (3.0 Å), Thr679 (2.2 Å), Ser681 (2.5 Å), Gln682 (2.0 Å) |
| Betulinic acid (1) | −8.2 | H-bond: Asn428 (1.9 Å) | |
| Alkyl: Val437 (5.7 Å) | |||
| Unfavorable donor–donor: Lys132 (2.2 Å) | |||
| β-Sitosterol (7) | −7.8 | H-bond: Gln682 (2.2 Å), Ser681 (2.7 Å) | |
| Alkyl: Pro466 (4.9 Å) | |||
| Site 5 | 3β-O-trans-caffeoylbetulinic acid (3) | −10.7 | H-bond: Glu365 (2.9 Å), Asn605 (4.2 Å), Glu348-chain B (2.0 Å and 2.2 Å) |
| π-anion: Asp1018 (4.2 Å) | |||
| Betulinic acid (1) | −10.1 | H-bond: Ile453 (2.5 Å), Arg574-chainB (1.5 Å) | |
| β-Sitosterol (7) | −9.1 | H-bond: Arg574-chainB (2.3 Å) | |
| Alkyl: His419 (4.8 Å); Ile571-chainB (4.1 Å) |
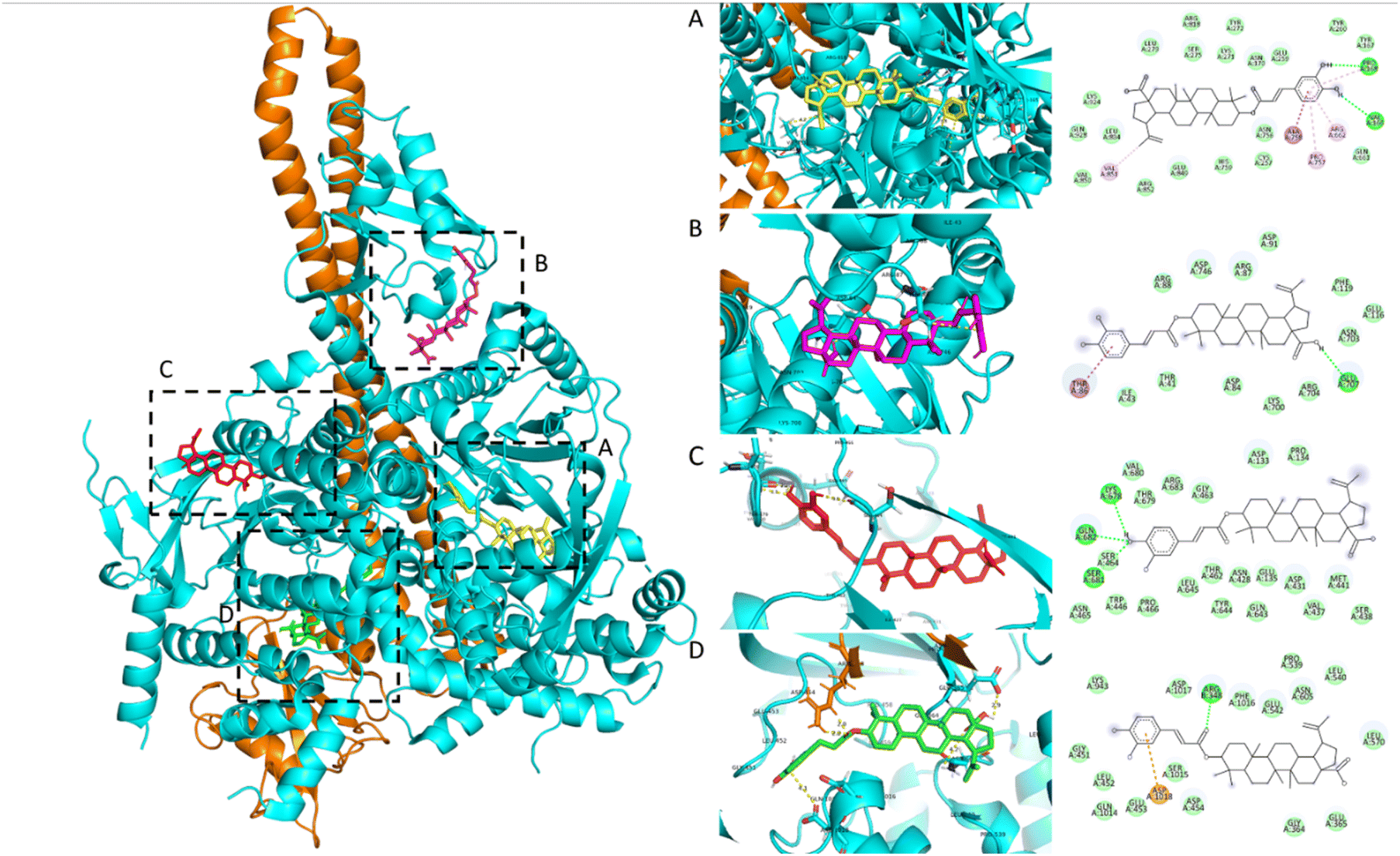 | ||
| Fig. 3 2D and 3D molecular interaction between 3β-O-trans-caffeoylbetulinic acid (3) and five active sites on PI3Kα: (A) site 1 and 2, (B) site 3, (C) site 4, and (D) site 5. | ||
Based on the results of the in vitro study, PI3Kα and the complex of PI3Kα with compounds 1, 3, and 7 were subjected to MD simulation in 100 ns. The root mean square deviation (RMSD) analysis indicated that the PI3Kα-ligand complexes exhibited greater stability compared to free PI3Kα (Fig. 4A). For the entire MD simulation, the free PI3Kα and its complexes displayed fluctuations within the 3–4.1 Å range. Notably, the PI3Kα-3 complex reached a stable state during MD simulation and exhibited RMSD values below 2.5 Å, much less than that of the free PI3Kα and other complexes, indicating their significantly enhanced stability. The radii of gyration (Rg) were consistent with the RMSD values when they maintained the lowest stable values within 3.45–3.50 nm throughout the simulation time, as shown in Fig. 4B. To determine the effect of fluctuations on protein residues, the root mean square fluctuations (RMSF) were calculated for chain A and chain B of PI3Kα, as demonstrated by Fig. 4C and D, respectively. The peaks observed in the RMSF curves of chain A (p110α) around residues 500–560 and 1000 are consistent with the residues mentioned in Table 2 and Fig. 3D (binding site 5). From residues 500–530 and around residue 600 on chain B (p85α), there were also RMSF peaks that corresponded well with the surrounding residues in binding site 5. The peaks observed in the RMSF curves of chain A, 200–300 residues, were closely aligned with the residues identified in Fig. 3A, corresponding to binding sites 1 and 2. In particular, the residues that formed hydrogen bonds contributed to peaks in RMSF. When in combination with compounds, the PI3Kα backbone atoms showed a distinguished decrease in the fluctuation of residues; specifically, around the region covering the amino acids mentioned. Further analysis of the hydrogen bond interactions revealed that compound 3 formed the greatest number of hydrogen bonds with PI3Kα (up to 6), demonstrating higher binding affinity compared to other compounds. In contrast, compound 1 established only a single hydrogen bond, leading to lower stability within the complex (Fig. 4E). Furthermore, solvent accessible surface area (SASA) analysis was carried out to evaluate the surface area of all systems that are available to water molecules (Fig. 4F). It is noted that the SASA values follow a similar trend demonstrated by Rg values, with compound 3 almost having the lowest SASA values. Regions with a decrease in the SASA value of the complexes indicate a relative contraction of their structure, suggesting a more compact conformation compared to free PI3Kα. These results align with the in vitro findings, where compound 3 exhibited great Nrf2 inhibitory activity on Huh7 cells, followed by compounds 1 and 7. These findings also support the hypothesis that PI3Kα is a regulator of the Nrf2 signaling pathway, suggesting that inhibiting the PI3Kα activity may contribute to the suppression of the Nrf2 activity in cancer cells.
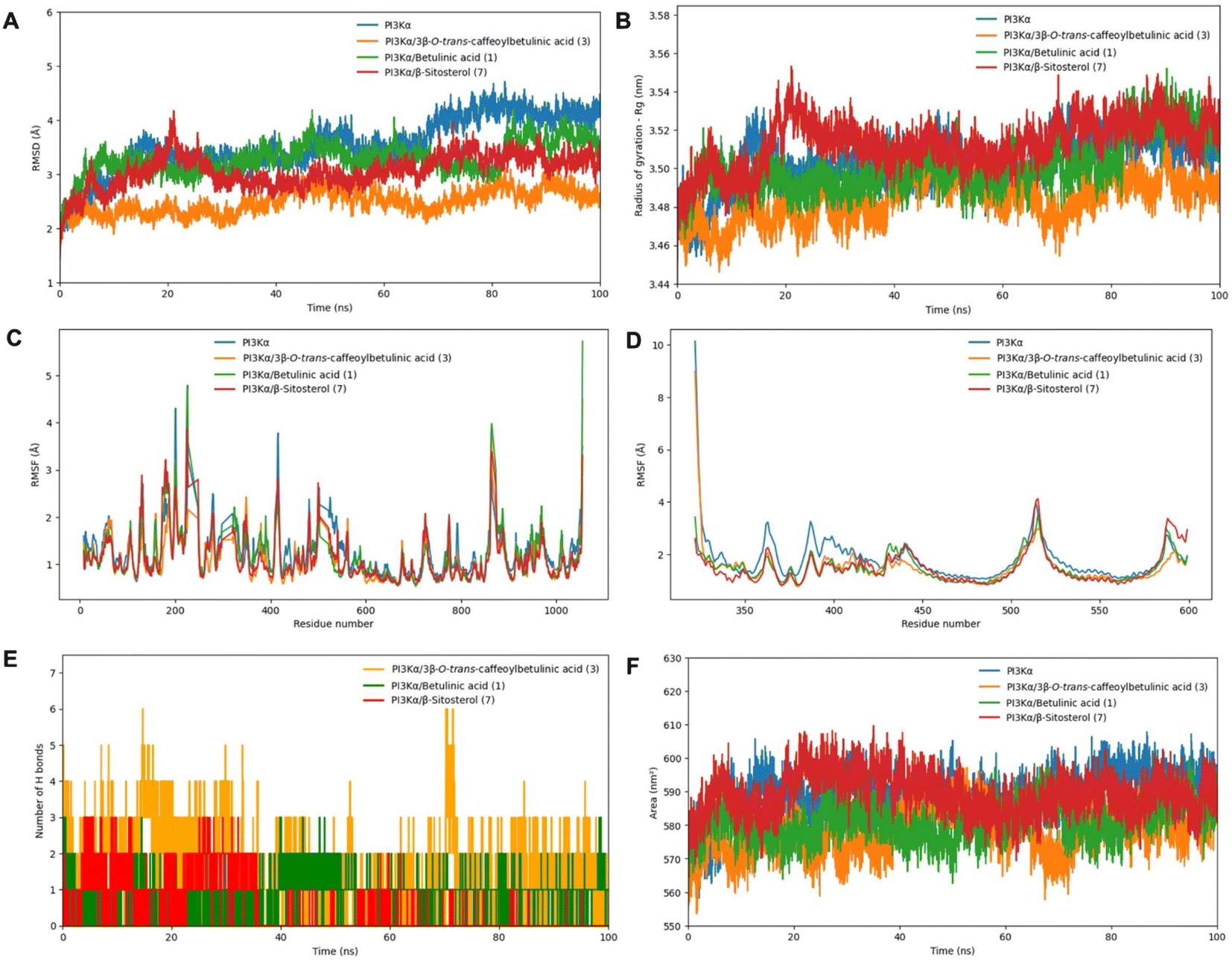 | ||
| Fig. 4 MD simulation analysis of PI3Kα (blue), PI3Kα/3β-O-trans-caffeoylbetulinic acid (orange), PI3Kα/betulinic acid (green), PI3Kα/β-sitosterol (red), (A) RMSD values, (B) Rg values, (C) RMSF values of PI3Kα chain A (p110α), (D) RMSF values of PI3Kα chain B (p110α), (E) hydrogen bond numbers, and (F) solvent-accessible surface area (SASA) values. | ||
In silico ADME and toxicology profile
In silico computational tools were used to assess the predicted ADME properties of the compounds, which are presented in Table 3. The physicochemical properties analysis revealed that betulinic acid and β-sitosterol exhibited favorable drug-likeness profiles, according to Lipinski's Rule of Five. While both compounds showed a single violation due to the logP values exceeding 5, other parameters, including molecular weight and the number of hydrogen bond donors and acceptors, complied with the established criteria. Meanwhile, 3β-O-trans-caffeoylbetulinic acid violated Lipinski's Rule of Five as the molecular weights exceeded 500 g mol−1 and the logP values were higher than 5. All three compounds were predicted to have poor solubility in water, poor gastrointestinal absorption, and negative logKp values, indicating limited skin permeability. The ADME predictions also revealed that none of the compounds are expected to bind to P-glycoprotein, potentially reducing the likelihood of drug efflux.
| Parameter | Betulinic acid | 3β-O-trans-caffeoylbetulinic acid | β-Sitosterol |
|---|---|---|---|
| a Probability that a compound may have >10% bioavailability on rat model. | |||
| Molecular weight | 456.70 | 618.84 | 414.71 |
| Num. H-bond acceptors | 3 | 6 | 1 |
| Num. H-bond donors | 2 | 3 | 1 |
| Num. Rotatable bonds | 2 | 6 | 6 |
| Molar refractivity | 136.91 | 180.31 | 133.23 |
| TPSA (Å2) | 57.53 | 104.06 | 20.23 |
| Lipophilicity (logPo/w) |
5.82 | 5.87 | 6.73 |
| Water solubility (logS) |
−7.71 | −9.77 | −7.90 |
| GI absorption | Low | Low | Low |
| BBB permeant | No | No | No |
| P-gp substrate | No | No | No |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | No | No | No |
| CYP2C9 inhibitor | Yes | No | No |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | No | Yes | No |
| Skin permeation (logKp cm s−1) |
−3.26 | −2.87 | −2.20 |
| Lipinski violation | 1 Violation | 2 Violations | 1 Violation |
| Bioavailability scorea | 0.85 | 0.56 | 0.55 |
The oral toxicity predictions from ProTox 3.0 revealed varying safety profiles among the compounds. 3β-O-trans-caffeoylbetulinic acid is predicted to be non-toxic, with a LD50 of 9600 mg kg−1, an accuracy level of 69.26%, and a confidence level of 72.94%. Betulinic acid has an LD50 of 2610 mg kg−1 and an accuracy level of 69.26%. β-Sitosterol exhibited higher toxicity, with an LD50 of 890 mg kg−1 and an accuracy level of 70.97%, similar to other compounds in databases up to 89.38%. The pharmacophore models also indicated that none of the compounds interact with toxicity targets, including adenosine receptor A2a, β-2 adrenergic receptor, or androgen receptor, avoiding the neurotoxic potentials of these compounds. These findings underscore the toxicity levels of the compounds, emphasizing the need for careful safety assessment in future studies.
Based on the results from both in vitro and in silico screening the relative Nrf2 activity, compound 3 was selected to determine the concentration that inhibits 50% of Nrf2 activity in Huh7 cells. The result indicated that the IC50 of compound 3 was 74.2 μg mL−1 or 119 nM (Fig. 5). This result reveals that the triterpenoid derived from H. hirsuta, 3β-O-trans-caffeoylbetulinic acid, is a compound with promising inhibition of Nrf2 activity in cancer cells without affecting healthy cells.
 | ||
| Fig. 5 Concentration response of 3β-O-trans-caffeoylbetulinic acid toward Nrf2 activity on the Huh7 cell line. | ||
Conclusions
Among the seven triterpenoids isolated and identified from the n-hexane sub-fraction of H. hirsuta leaves, 3β-O-trans-caffeoylbetulinic acid was revealed as a potential inhibitor of Nrf2 activity in Huh7 cells, with an IC50 value of 74.2 μg mL−1. Additionally, molecular docking and simulations showed that these compounds have potential binding affinity for PI3Kα, modulating downstream pathways and reducing Nrf2 activity in cancer cells. These findings underscore the potential of triterpenoids as Nrf2 inhibitors, especially 3β-O-trans-caffeoylbetulinic acid, emerging as a promising candidate. However, the regulatory mechanisms of Nrf2 remain unclear, and further in vitro studies are needed to confirm the relationship between Nrf2 regulation and PI3Kα.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: M. H. N.; isolation and identification: M. T. T. N., T. N. V. D. and T. H. L.; bioassay: C. H. Y and V. T. N.; computational studies: B. Q. H. N., M. H. N. and T. H. N. L.; writing – original draft preparation: M. H. N., B. Q. H. N., T. H. N. L., T. N. V. D. and T. H. L.; writing – review and editing: M. H. N., B. Q. H. N. and T. H. N. L.; supervision: M. H. N.; project administration: M. H. N. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is funded by Vietnam National University HoChiMinh City (VNU-HCM) under grant number B2023-44-01.Notes and references
- International Agency for Research on Cancer, Cancer Today, https://gco.iarc.who.int/today/en, accessed 21 Oct, 2024.
- F. Bray, M. Laversanne, E. Weiderpass and I. Soerjomataram, Cancer, 2021, 127, 3029–3030 CrossRef PubMed.
- B. Liu, H. Zhou, L. Tan, K. T. H. Siu and X.-Y. Guan, Signal Transduction Targeted Ther., 2024, 9, 175 CrossRef.
- B. E. Wilson, S. Jacob, M. L. Yap, J. Ferlay, F. Bray and M. B. Barton, Lancet Oncol., 2019, 20, 769–780 CrossRef PubMed.
- K. Bukowski, M. Kciuk and R. Kontek, Int. J. Mol. Sci., 2020, 21, 3233 CrossRef CAS.
- J. Rueff and A. S. Rodrigues, Cancer Drug Resistance: A Brief Overview from a Genetic Viewpoint, Springer New York, New York, NY, 2016 Search PubMed.
- Z. N. Lei, Q. Tian, Q. X. Teng, J. N. D. Wurpel, L. Zeng, Y. Pan and Z. S. Chen, MedComm, 2023, 4, e265 CrossRef CAS PubMed.
- Q. Ma, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 401–426 CrossRef CAS PubMed.
- S. Wu, H. Lu and Y. Bai, Cancer Med., 2019, 8, 2252–2267 CrossRef.
- X.-J. Wang, Z. Sun, N. F. Villeneuve, S. Zhang, F. Zhao, Y. Li, W. Chen, X. Yi, W. Zheng, G. T. Wondrak, P. K. Wong and D. D. Zhang, Carcinogenesis, 2008, 29, 1235–1243 CrossRef CAS.
- H. S. Khalil, A. Goltsov, S. P. Langdon, D. J. Harrison, J. Bown and Y. Deeni, J. Biotechnol., 2015, 202, 12–30 CrossRef CAS PubMed.
- L. M. Solis, C. Behrens, W. Dong, M. Suraokar, N. C. Ozburn, C. A. Moran, A. H. Corvalan, S. Biswal, S. G. Swisher, B. N. Bekele, J. D. Minna, D. J. Stewart and I. I. Wistuba, Clin. Cancer Res., 2010, 16, 3743–3753 CrossRef CAS.
- D. Li, X. Hong, F. Zhao, X. Ci and S. Zhang, Cancer Cell Int., 2021, 21, 116–125 CrossRef CAS PubMed.
- M. Negrette-Guzmán, Eur. J. Pharmacol., 2019, 859, 172513 CrossRef PubMed.
- D. D. Zhang and E. Chapman, Nat. Prod. Rep., 2020, 37, 797–826 RSC.
- Z. Hong, X. Cao, N. Li, Y. Zhang, L. Lan, Y. Zhou, X. Pan, L. Shen, Z. Yin and L. Luo, Br. J. Pharmacol., 2014, 171, 2842–2853 CrossRef CAS.
- H. Wang, Y. Luo, T. Qiao, Z. Wu and Z. Huang, J. Ovarian Res., 2018, 11, 93–104 CrossRef CAS.
- Y. Xiang, W. Ye, C. Huang, D. Yu, H. Chen, T. Deng, F. Zhang, B. Lou, J. Zhang, K. Shi, B. Chen and M. Zhou, Oxid. Med. Cell. Longevity, 2018, 2018, 2360427 CrossRef PubMed.
- J. P. Evans, B. K. Winiarski, P. A. Sutton, R. P. Jones, L. Ressel, C. A. Duckworth, M. D. Pritchard, Z.-X. Lin, V. L. Fretwell, E. M. Tweedle, E. Costello, C. E. Goldring, I. M. Copple, K. B. Park, D. H. Palmer and N. R. Kitteringham, Oncotarget, 2018, 9, 27104 CrossRef.
- A. Limonciel and P. Jennings, Toxins, 2014, 6, 371–379 CrossRef CAS PubMed.
- A. Arlt, S. Sebens, S. Krebs, C. Geismann, M. Grossmann, M. L. Kruse, S. Schreiber and H. Schäfer, Oncogene, 2013, 32, 4825–4835 CrossRef CAS PubMed.
- A. Manna, S. De Sarkar, S. De, A. K. Bauri, S. Chattopadhyay and M. Chatterjee, Phytomedicine, 2015, 22, 713–723 CrossRef CAS PubMed.
- J. Zhang, H. X. Xu, J. Q. Zhu, Y. X. Dou, Y. F. Xian and Z. X. Lin, Int. J. Biol. Sci., 2023, 19, 3029 CrossRef CAS PubMed.
- M. Hammad, M. Raftari, R. Cesário, R. Salma, P. Godoy, S. N. Emami and S. Haghdoost, Antioxidants, 2023, 12, 1371 CrossRef CAS PubMed.
- C. Dong, J. Wu, Y. Chen, J. Nie and C. Chen, Front. Pharmacol., 2021, 12, 628690 CrossRef CAS.
- S. Adinolfi, T. Patinen, A. Jawahar Deen, S. Pitkänen, J. Härkönen, E. Kansanen, J. Küblbeck and A.-L. Levonen, Redox Biol., 2023, 63, 102726–102735 CrossRef CAS PubMed.
- V. V. Chi, Dictionary of Vietnamese Medicinal Plants, Publishing House Medicine: Hanoi, Vietnam, 2012 Search PubMed.
- M. H. Nguyen, N. Y. T. Nguyen, Y.-S. Chen, H. T. Nguyen Le, H. T. Vo and C.-H. Yen, Heliyon, 2024, 10, e38411 CrossRef CAS.
- N. T. Vu, L. T. N. Tam, L. N. T. Han, N. K. Anh, T. K. Minh, L. M. Tri and N. M. Hien, J. Community Med., 2024, 65, 26–31 Search PubMed.
- Y.-S. Chen, H.-S. Chang, H.-H. Hsiao, Y.-F. Chen, Y.-P. Kuo, F.-L. Yen and C.-H. Yen, Antioxidants, 2021, 10, 544–559 CrossRef CAS PubMed.
- M. S. Miller, S. Maheshwari, F. M. McRobb, K. W. Kinzler, L. M. Amzel, B. Vogelstein and S. B. Gabelli, Bioorg. Med. Chem., 2017, 25, 1481–1486 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef.
- P. Banerjee, E. Kemmler, M. Dunkel and R. Preissner, Nucleic Acids Res., 2024, 52, W513–W520 CrossRef.
- A. Yili, Mutalipu, H. A. Aisa and M. I. Isaev, Chem. Nat. Compd., 2009, 45, 592–594 CrossRef CAS.
- A. H. Laghari, S. Memon, A. Nelofar and K. M. Khan, Ind. Crops Prod., 2011, 34, 1141–1145 CrossRef CAS.
- H. Pan, L. N. Lundgren and R. Andersson, Phytochemistry, 1994, 37, 795–799 CrossRef CAS.
- H. Jae-Kwan, S. Jae-Seok, P. Kyung-Min and C. Jae-Youn, Prev. Nutr. Food Sci., 2002, 7, 215–218 CrossRef.
- L. Y. Mooi, N. A. Wahab, N. H. Lajis and A. M. Ali, Chem. Biodiversity, 2010, 7, 1267–1275 CrossRef CAS PubMed.
- D. D. Tshilanda, D. N. Onyamboko, P. Babady-Bila, K.-t.-N. Ngbolua, D. S. Tshibangu, E. dia Fita Dibwe and P. T. Mpiana, Nat. Prod. Bioprospect., 2015, 5, 215–221 CrossRef CAS.
- T. T. Le Nguyen, T. T. Pham, P. E. Hansen and P. K. P. Nguyen, STDJ, 2019, 22, 106–113 Search PubMed.
- D. Mandelker, S. B. Gabelli, O. Schmidt-Kittler, J. Zhu, I. Cheong, C. H. Huang, K. W. Kinzler, B. Vogelstein and L. M. Amzel, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 16996–17001 CrossRef CAS.
- H. Cheng, S. T. M. Orr, S. Bailey, A. Brooun, P. Chen, J. G. Deal, Y. L. Deng, M. P. Edwards, G. M. Gallego, N. Grodsky, B. Huang, M. Jalaie, S. Kaiser, R. S. Kania, S. E. Kephart, J. Lafontaine, M. A. Ornelas, M. Pairish, S. Planken, H. Shen, S. Sutton, L. Zehnder, C. D. Almaden, S. Bagrodia, M. D. Falk, H. J. Gukasyan, C. Ho, X. Kang, R. E. Kosa, L. Liu, M. E. Spilker, S. Timofeevski, R. Visswanathan, Z. Wang, F. Meng, S. Ren, L. Shao, F. Xu and J. C. Kath, J. Med. Chem., 2021, 64, 644–661 CrossRef CAS.
- J. Huang, L. Chen, J. Wu, D. Ai, J.-Q. Zhang, T.-G. Chen and L. Wang, J. Med. Chem., 2022, 65, 16033–16061 CrossRef CAS.
- M. Zhang, H. Jang and R. Nussinov, Chem. Sci., 2020, 11, 5855–5865 RSC.
- K. Sotolongo, J. Ghiso and A. Rostagno, Alzheimer's Res. Ther., 2020, 12, 13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07646j |
| This journal is © The Royal Society of Chemistry 2025 |