DOI:
10.1039/D4RA08643K
(Paper)
RSC Adv., 2025,
15, 1896-1914
1,2,3-Triazole-tethered fluoroquinolone analogues with antibacterial potential: synthesis and in vitro cytotoxicity investigations†
Received
8th December 2024
, Accepted 27th December 2024
First published on 22nd January 2025
Abstract
The antibacterial efficacy of some newly developed bis- and C3-carboxylic moieties of fluoroquinolone-linked triazole conjugates was studied. Twenty compounds from two different series of triazoles were synthesized using click chemistry and evaluated for their antibacterial activity against a Gram-positive strain, i.e. Enterococcus faecalis (ATCC29212), and its clinical isolate and a Gram-negative bacterial strain, i.e. Escherichia coli (ATCC25922), and its clinical isolate. Among the compounds, 7, 9a, 9d, 9i, 10(a–d), and 10i showed excellent activity with MIC values of up to 6.25 μg mL−1, whereas the control ciprofloxacin showed MIC values of up to 12.5 μg mL−1 towards the various strains. Cytotoxicity was evaluated against Vero cells (kidney epithelial cells of an African green monkey), and results revealed that compounds 9a, 9c, 10g, 10h, and 10 are toxic. Molecular docking and MD analysis were performed using the protein structure of E. coli DNA gyrase B and further corroborated with an in vitro assay to evaluate the inhibition of DNA gyrase. The analysis revealed that compound 10d was a more potent inhibitor of DNA gyrase compared to ciprofloxacin, which was employed as the positive control.
Introduction
Antimicrobial resistance (AMR) poses a severe threat to public health as current antibiotics are becoming less effective in preventing and treating infectious diseases worldwide.1 Increasing reports of multidrug-resistant (MDR) bacteria pose a significant burden on the healthcare system since MDR infections are associated with increased mortality rates, higher risk of complications, and more extended hospital stays, resulting in substantial financial strain.2 Enterococcus faecalis (E. faecalis) is a commensal bacterium found in the human intestines and a major opportunistic pathogen common in immunocompromised and elderly patients.3 The pathogenesis of E. faecalis infection relies in part on its capacity to colonize the gut. Following the disruption of intestinal homeostasis, E. faecalis can overgrow and enter the lymphatic system and bloodstream, crossing the intestinal barrier.4 Over 30 years, E. faecalis has become a leading cause of healthcare-associated infections worldwide.5 It is still a significant health concern and has developed multiple drug resistance, thus causing severe problems in the medical treatment of nosocomial infections,6 while Escherichia coli (E. coli) is one of the most clinically significant Gram-negative bacteria, which is responsible for a wide range of nosocomial infections including urinary tract infections and ventilator-associated pneumonia.7 According to the World Health Organization (WHO) classification, E. coli is a critical bacterial species resistant to most commercially available drugs.8 Escherichia coli (E. coli) bacteria inhabit the human GI tract and are versatile pathogens. At the same time, Enterococcus faecalis (E. faecalis) is a facultative, anaerobic, Gram-positive coccus that inhabits the human GI tract.9,10 Both E. coli and E. faecalis are commensal pathogens, and both colonize immediately after birth, thus, they are good representatives of their corresponding phyla.11 From a technical perspective, both species can be easily isolated, grown, and maintained in a laboratory and are frequently used as potential human fecal indicators. Hence, it is urgent to find alternative antimicrobial agents that can reduce the problems of drug resistance in bacteria.
Fluoroquinolones (FQs) are some of the most prominent, widely used classes of synthetic antibacterial agents in treating several infectious diseases. This is because they offer many of the attributes of an ideal antibacterial, combining high potency, broad-spectrum activity, good bioavailability, oral and intravenous formulations, high serum levels, and large distribution volume, indicating good concentrations in different tissues.12,13 They play a key role in the treatment of urinary tract infections (UTIs), upper and lower respiratory tract infections (RTI), and sexually transmitted diseases (STDs). Besides their effects in curing skin, soft tissue, gastrointestinal, bones, and joint infections,14,15 they exert their bactericidal activity by targeting bacterial DNA gyrase and topoisomerase IV (Topo IV), where they bind to complexes that are formed between DNA and DNA gyrase or Topo IV, forming a ternary complex of DNA enzyme-quinolone. The produced complexes inhibit DNA replication and cell growth and are responsible for the antibacterial activity of fluoroquinolones.16 However, alone fluoroquinolones cannot control the problems of drug resistance in bacteria, and thus the development of new drugs with effective antibacterial action in a cost-effective manner is necessary. Accordingly, the development of hybrid molecules that inhibit the emergence of drug resistance in bacteria through affinity and efficacy compared to the parent drugs can address these issues.
1,2,3-Triazole is a heterocyclic five-membered ring system possessing three nitrogen and two carbon atoms. Furthermore, 1,2,3-triazole has been a fruitful source of inspiration for medicinal chemists for many years and has attracted our attention due to its synthetic accessibility and diverse inhibitory activities.17–19 It can be synthesized via the Huisgen 1,3-dipolar cycloaddition of azides and alkynes through copper-catalyzed click reaction.20–23 Our literature search showed that the pharmacological effects of 1,2,3-triazole are very alluring and promising for the design of antibacterial agents. Specifically, 1,2,3-triazole conjugated with a wide range of moieties was reported to exhibit potent antibacterial properties.24–26
Previously, we reported a pool of novel fluoroquinolone (ciprofloxacin)-tethered 1,2,3-triazole hybrids with improved antibacterial activity, as shown in Fig. 1. Most of the synthesized conjugates showed enhanced antibacterial activity compared to the parent drug (ciprofloxacin).25–27 The study of new hybrid systems (bis and C3-carboxylic group of fluoroquinolone linked triazole hybrids) in which 1,2,3-triazole and fluoroquinolone are combined is an unexplored field of research to date. Thus, the above-mentioned findings encouraged us to investigate the potential synergistic effects of substituted 1,2,3-tiazoles and fluoroquinolone scaffolds. Herein, the synthesized hybrids tested against Gram-positive (E. faecalis) and Gram-negative (E. coli). At the same time, the toxicity of the compounds was evaluated in Vero cells using the MTT assay, where compounds 9a, 9c, 10g, 10h, and 10i were found to be toxic. Also, they were further tested using the DNA gyrase assay as a possible target, and molecular docking studies were conducted to investigate their binding mechanisms.
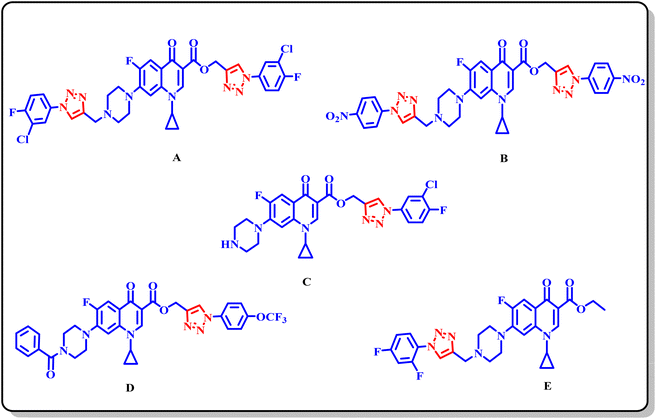 |
| | Fig. 1 Structure of some previously reported fluoroquinolone 1,2,3-triazole analogues. | |
Results and discussion
Chemistry
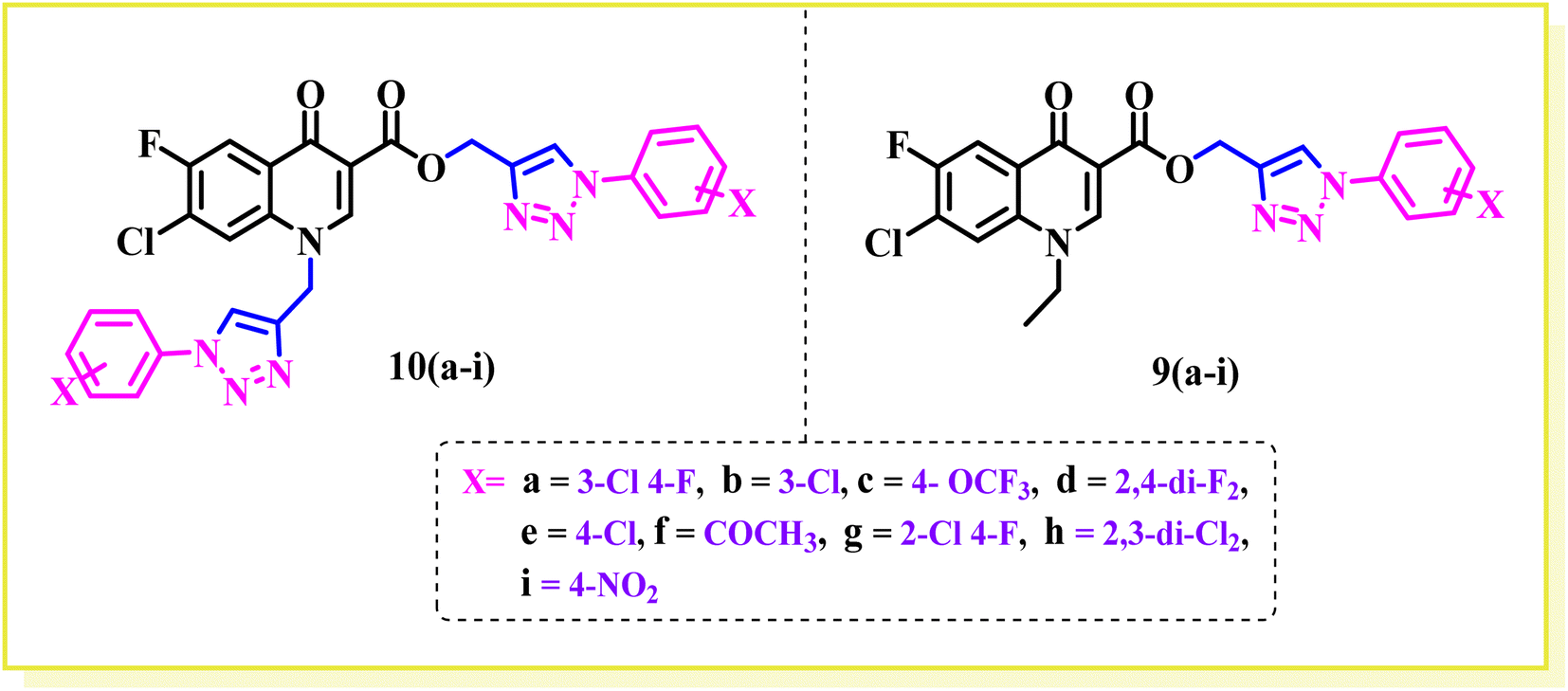
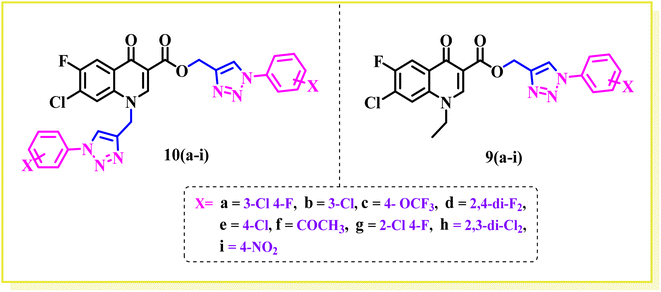
Bis-fluoroquinolone triazole and fluoroquinolone carboxylic group-linked triazole analogues were synthesized in various steps, as outlined in Scheme 1. In brief, the condensation of 3-chloro-4-fluoroaniline with diethyl(ethoxymethylene) malonate ester at 100 °C yielded the product (1), which underwent a cyclization reaction in diphenyl ether at 250 °C to produce 7-chloro-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (2) in good yield. Consequently, compound (2) was alkylated with ethyl bromide using K2CO3 in DMF to the corresponding N-ethylated product (3). The compounds (ethyl 7-chloro-6-fluoro)/(ethyl 7-chloro-1-ethyl-6-fluoro)-4-oxo-1,4-dihydroquinoline-3-carboxylate(2)/(3) were hydrolyzed into the corresponding acid (4)/(5) (ethyl 7-chloro-6-fluoro)/(ethyl-7-chloro-1-ethyl-6-fluoro)-4-oxo-1,4-dihydroquinoline-3-carboxylicacid using 2N NaOH solution, respectively. Further, compounds (4) and (5) were propargylated using propargyl bromide and NaHCO3 in DMF to the corresponding N-/C-propargylated products (6) and (7), respectively. Again, the above-propargylated products (6) and (7) were used to synthesize 1,2,3 compounds 9 and 10(a–i) via click chemistry. All the synthesized compounds were characterized by 1H, 13C NMR, and HRMS.
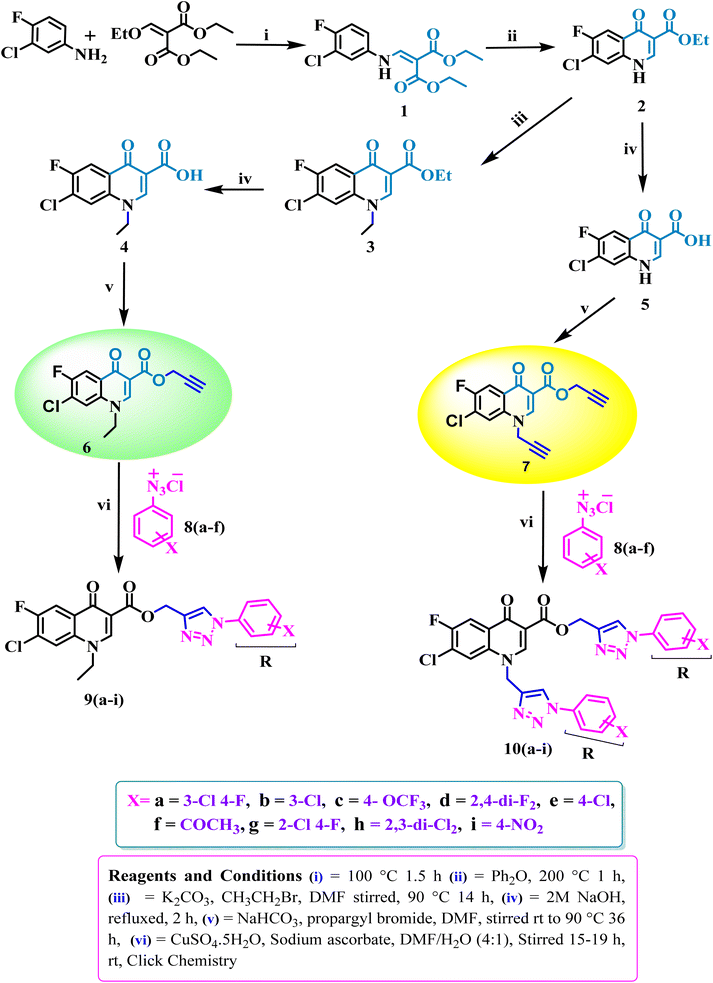 |
| | Scheme 1 Schematic representation of the synthesis of fluoroquinolone-triazole analogues. | |
Biological activity
Antibacterial activity
The antibacterial assay was performed for twenty compounds (fluoroquinolone-triazole hybrids), i.e., 6, 7, 9(a–i), and 10(a–i), which were synthesized from propargylated fluoroquinolones (intermediates) using click chemistry and copper-catalyzed azide-alkyne [3 + 2] cycloaddition. These hybrid compounds, i.e., 6, 7, 9(a–i), and 10(a–i), were tested for their antibacterial property against two-Gram + ve strains, i.e. Enterococcus faecalis (ATCC 29212) and Enterococcus faecalis (clinical isolate) and two-Gram −ve strains, i.e., Escherichia coli (ATCC 25922) and Escherichia coli (clinical isolate). The MIC results showed that the nature, position, and isomeric effect of the substituents on the phenyl ring significantly impact the antibacterial activity of the prepared compounds (Table 1). According to Table 1, most of the compounds showed greater or equal antibacterial activity in terms of MIC compared to the standard drugs against the investigated strains, i.e., E. faecalis (ATCC 29212), E. faecalis (clinical isolate), E. coli (ATCC 25922) and E. coli (clinical isolate) except 9b, 9c, 9e, 9g, 9h, 10g, and 10h.
Table 1 Antibacterial activity (MIC μg mL−1) and cytotoxicity (μg mL−1) of compounds 6, 7 & 9–10(a–i)a
| Compound no. |
R |
Gram + ve strains |
Gram −ve strains |
CC50 value (μg mL−1) |
| E. faecalis (ATCC 29212) |
E. faecalis (clinical isolate) |
E. coli (ATCC25922) |
E. coli (clinical isolate) |
| nd = determined. |
| 6 |
— |
1.56 |
25 |
≥0.195 |
1.56 |
>100 |
| 7 |
— |
≥0.195 |
12.5 |
0.195 |
≥0.195 |
>100 |
| 9a |
 |
0.781 |
50 |
12.5 |
0.195 |
<100 |
| 9b |
 |
3.12 |
100 |
25 |
12.5 |
>100 |
| 9c |
 |
1.56 |
50 |
6.25 |
3.12 |
<100 |
| 9d |
 |
≥0.195 |
12.5 |
≥0.195 |
0.781 |
>100 |
| 9e |
 |
50 |
100 |
0.781 |
12.5 |
>100 |
| 9f |
 |
25 |
50 |
0.391 |
0.781 |
>100 |
| 9g |
 |
50 |
50 |
0.781 |
6.25 |
>100 |
| 9h |
 |
50 |
100 |
12.5 |
25 |
>100 |
| 9i |
 |
≥0.195 |
50 |
0.195 |
0.781 |
>100 |
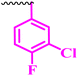
| 10a |
 |
≥0.195 |
6.25 |
0.195 |
0.195 |
>100 |
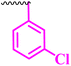
| 10b |
 |
≥0.195 |
12.5 |
0.195 |
≥0.195 |
>100 |
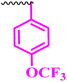
| 10c |
 |
0.391 |
12.5 |
0.195 |
≥0.195 |
>100 |
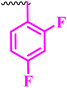
| 10d |
 |
0.195 |
6.25 |
0.195 |
≥0.195 |
>100 |
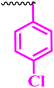
| 10e |
 |
1.56 |
50 |
6.25 |
0.781 |
>100 |
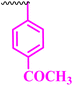
| 10f |
 |
0.781 |
50 |
6.25 |
0.781 |
>100 |
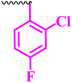
| 10g |
 |
25 |
50 |
3.12 |
6.25 |
<100 |
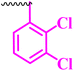
| 10h |
 |
3.12 |
50 |
6.25 |
12.5 |
<100 |
| 10i |
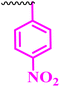 |
1.56 |
12.5 |
0.195 |
0.781 |
<100 |
| Standard |
Ciprofloxacin |
0.781 |
12.5 |
≥0.195 |
0.781 |
nd |
| Standard |
Chloroquine |
nd |
nd |
nd |
nd |
>100 |
Structure–activity relationship
In this part, we summarize the effect of the substituents on the antibacterial activity of the synthesized compounds, as shown in Fig. 2. Compound 6 has an ethyl group on its N-atom and propargyl moiety on the carboxylic oxygen of fluoroquinolone, showing good activity against E. coli (ATCC 25922) with MIC of ≥0.195 μg mL−1, which is equal to the standard drug. In the other strains, compound 6 was found to be less active than the control with MIC of 1.56 μg mL−1 against E. coli (clinical isolate), E. faecalis (ATCC 29212), and E. faecalis (clinical isolate) with MIC of 25 μg mL−1.
 |
| | Fig. 2 Structure of fluoroquinolone mono and bis-1,2,3-triazole analogues. | |
Compound 7 is a bis propargylated fluoroquinolone, which showed excellent activity against E. coli (clinical isolate) and E. faecalis (ATCC 29212) with MIC of ≥0.195 μg mL−1, which is four times greater than that of the standard drug against E. coli (ATCC 25922) and found to be slightly more active (MIC 0.195 μg mL−1) in comparison to the control drug, while in E. faecalis (clinical isolate), it showed equal activity with a MIC of 12.5 μg mL−1, the same as ciprofloxacin showed.
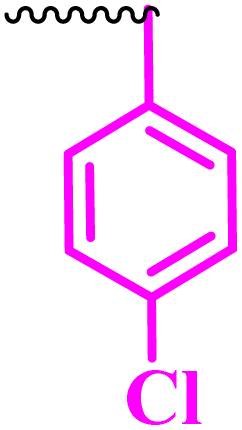
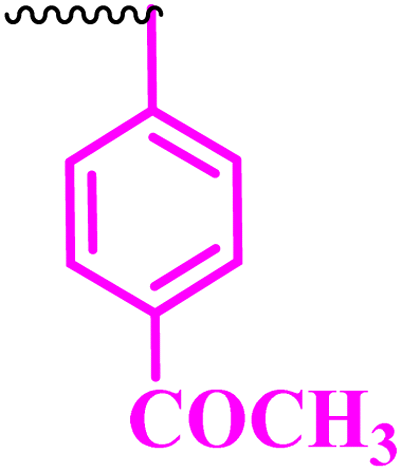
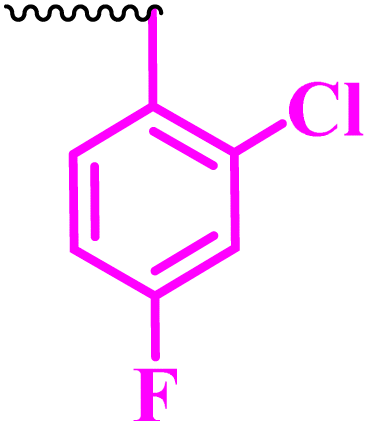
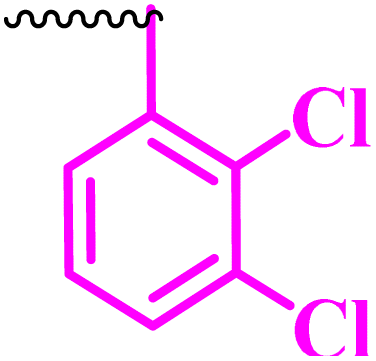
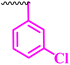
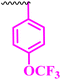
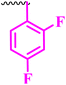
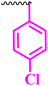
Compound 9a, a C-3 linked triazole hybrid having 3-Cl and 4-F groups, was found to be four times more active with MIC of 0.195 μg mL−1 in comparison to the standard (0.781 μg mL−1) against E. coli (clinical isolate). This compound was also found to be equally active as the standard with MIC of 0.781 μg mL−1 against E. faecalis (ATCC 29212), while against the other strains, it was found to be active. Compounds 9b and 9c, having 3-Cl and 4-OCF3 groups substituted on the phenyl triazole ring, were found to be less active and showed lower MIC values compared to ciprofloxacin against the respective strains. Compound 9d, which is a 2,4-difluoro-substituted phenyl triazole ring, was found to be very active (MIC ≥0.195 μg mL−1) against E. faecalis (ATCC 29212) and active against the remaining three strains i.e., E. coli (ATCC 25922), E. coli (clinical isolate), and E. faecalis (clinical isolate) with MIC ≥ 0.195 μg mL−1, MIC ≥ 0.781 μg mL−1 and MIC ≥ 12.5 μg mL−1, which is similar to the standard drug. Compounds 9e, 9f, 9g, and 9h having 4-Cl, 4-COCH3, 2-Cl 4-F, and 2,3-dichloro group-substituted phenyl ring, respectively, were all found to have equal activity and were less active than the ciprofloxacin in the tested strains. Compound 9i, a 4-Cl substituted-phenyl triazole ring, showed potent activity against E. coli (ATCC 25922) and E. coli (clinical isolate), while it was four times more active (MIC ≥ 0.195 μg mL−1) against E. faecalis (ATCC 29212) and less active against E. faecalis (clinical isolate) in comparison to the standard drug with MIC ≥ 0.195 μg mL−1 and 50 μg mL−1, respectively.
Compounds 10(a–i) are bis-triazole fluoroquinolone hybrids, as shown in Fig. 2, among which compound 10a, having 3-Cl, 4-F-substituted benzene triazole, showed the best antibacterial activity against the tested strains. It showed MIC of 0.195 μg mL−1, which was slightly higher against E. coli (ATCC 25922) and four times greater against E. coli (clinical isolate) in comparison to ciprofloxacin. Again, the same compound was found to be nearly four times more active (MIC ≥ 0.195 μg mL−1) against E. faecalis (ATCC 29212), which is similar to compound 9i, and two times more active with MIC of 6.25 μg mL−1 against E. faecalis (clinical isolate) in comparison to ciprofloxacin. Compound 10b with 3-Cl substituted phenyl triazole showed similar activity as compound 10a except against E. coli (clinical isolate) and E. faecalis (clinical isolate), showing MIC of ≥ 0.195 μg mL−1, which is nearly four times greater than the control drug, and MIC of 12.5 μg mL−1, a similar value to the control drug. Compound 10c, a 4-OCF3-substituted phenyl triazole, was found to be more active against E. coli (ATCC 25922) and four times more active against E. coli (clinical isolate) in comparison to the standard drug ciprofloxacin with MIC of 0.195 μg mL−1 and ≥0.195 μg mL−1, respectively. The same compound was found to be less active with MIC of 0.391 μg mL−1 against E. faecalis (ATCC 29212), while it showed similar activity as the standard drug (MIC 12.5 μg mL−1) against E. faecalis (clinical isolate). Compound 10d, a 2,4-difluoro-containing phenyl triazole hybrid, showed the second most potent activity after 10a compound, showing similar activity as compound 10a against E. coli (ATCC 25922) and E. faecalis (clinical isolate), while it was found to be four times more potent (MIC 0.195 μg mL−1) than the standard drug against E. faecalis (ATCC 29212) and E. coli (clinical isolate). Compounds 10(e–h), having 4-Cl, 4-COCH3, 2-Cl 4-F, and 2,3-dichloro group-substituted phenyl triazoles, respectively, were found to be equal or less potent than ciprofloxacin against the tested strains. At the same time, compound 10i was also found to be equal or less active than the standard drug except against E. coli (ATCC 25922), which was found to be slightly more active with MIC of 0.195 μg mL−1. Thus, according to their activity, most of the compounds were found to be active against the investigated strains, while a certain number of compounds were found to be more potent than the standard drugs. Further, this work can be tuned to obtain useful results, whereas second-generation synthesis, depending on the activity data, is required, and lead compounds should be used in SAR experiments.
Molecular docking studies
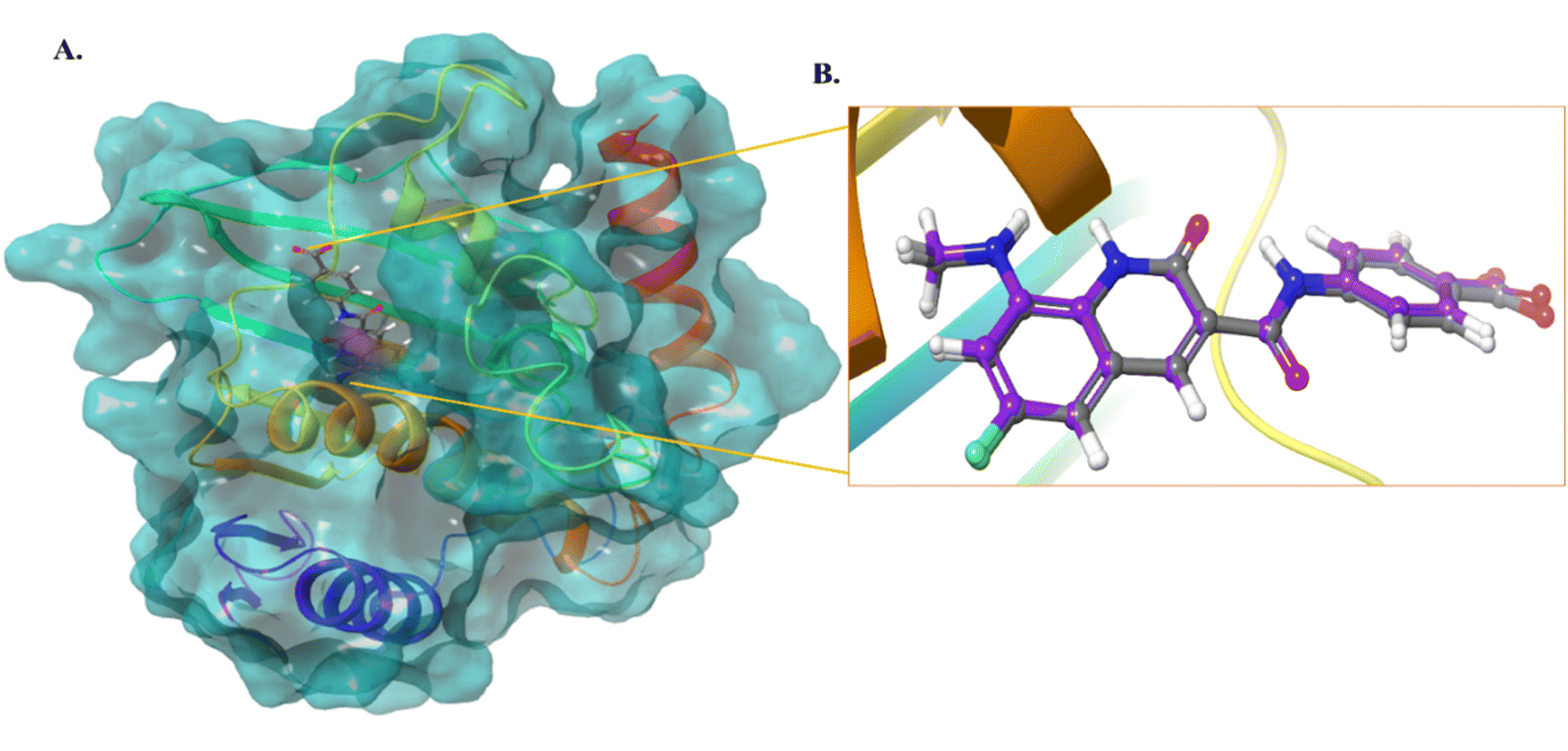
We selected the most active compound from the in vitro studies, 10d, and the least active compound, 9a, for the docking studies to understand the fundamental interactions between the target protein and selected compounds. Both compounds were subjected to molecular docking studies using the Maestro Schrodinger suite. The protein and ligands were prepared at a pH setting of 7 ± 2 using the Epik module of the Schrodinger software to generate the possible conformers states. The protein used for the docking studies was E. coli DNA gyrase B crystal structure in complex with inhibitor 6-fluoro-8-(methylamino)-2-oxo-1,2-dihydroquinoline (PDB ID: 7C7N).28 Initially, the docking protocol was validated by calculating the root mean square deviation (RMSD), where the co-crystallized ligand was again redocked to the same cavity (Fig. 3). The docking score of compound 10d was 7.0 kcal mol−1, which is comparable to that of the standard ciprofloxacin with a value of 7.7 kcal mol−1, and crystallized, as shown in Fig. 4. Compound 10d interacted with the Asn46, Asp73, and Lys103 amino acid residues. Compound 9a exhibited a docking score of 4 kcal mol−1.
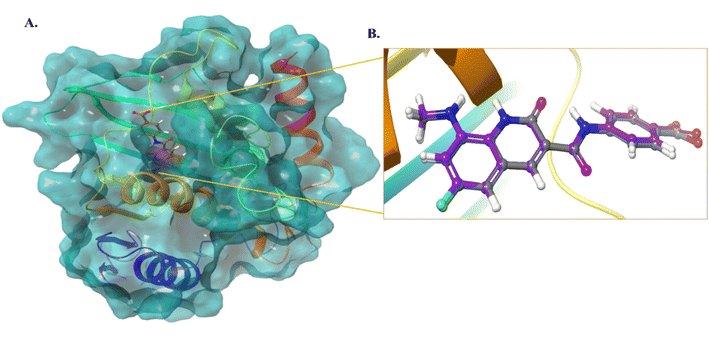 |
| | Fig. 3 (A) Protein structure with the co-crystallized ligand. (B) RMSD calculation of the co-crystallized ligand and redocked pose of the co-crystallized ligand (0.1 Å). | |
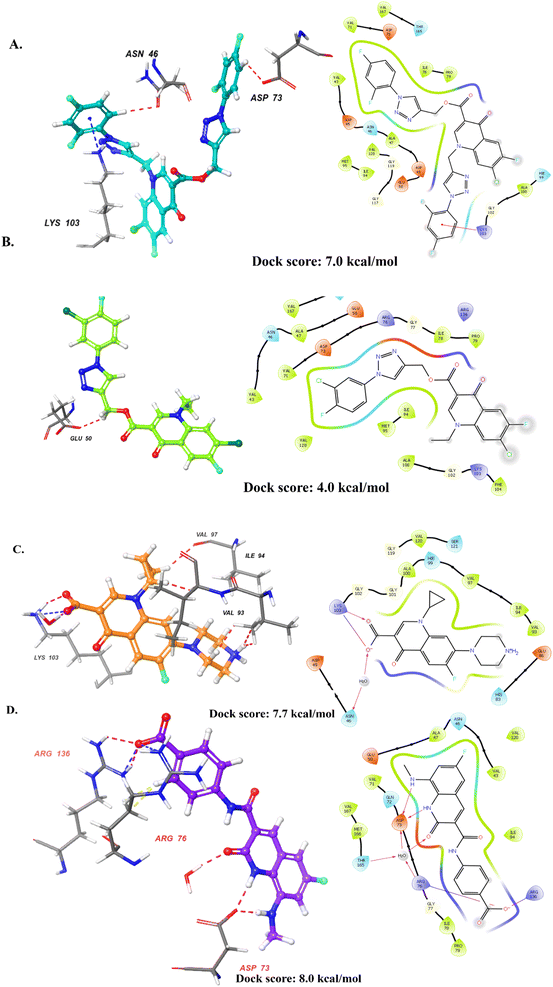 |
| | Fig. 4 (A) 3D and 2D interaction of compound 10d. (B) 3D and 2D interaction of compound 9a. (C) 3D and 2D interaction of the compound standard ciprofloxacin. (D) 3D and 2D interaction of the compound co-crystallized ligand. | |
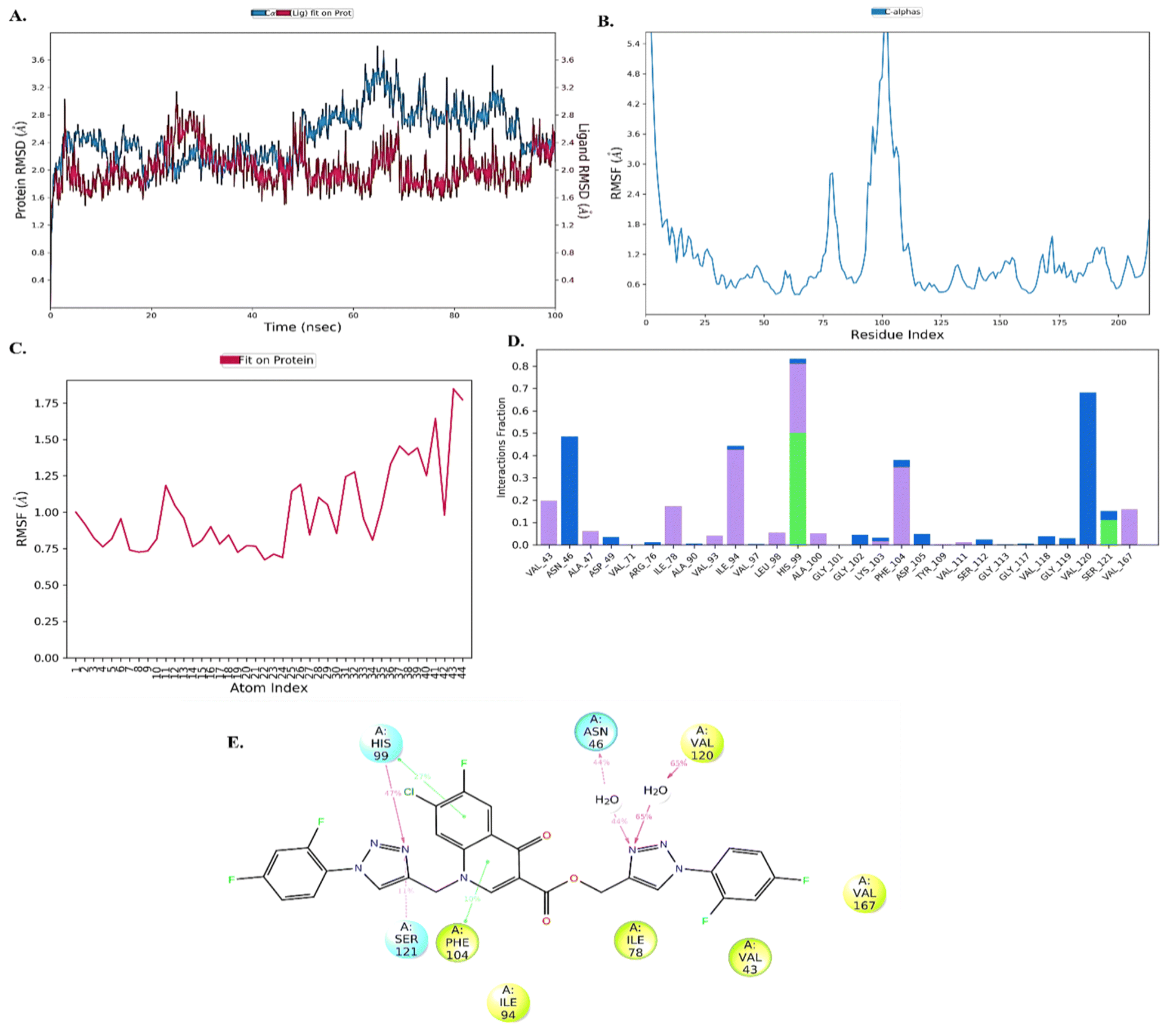
Molecular dynamic (MD) analysis
Further, to understand the stability of protein–ligand (PL), complex molecular dynamics (MD) studies were performed. Compound 10d in complex with DNA gyrase B protein (PDB ID: 7C7N) was selected for the MD simulation study. The root-mean-square deviation (RMSD) analysis revealed that the protein showed little fluctuations of around 1.6–2.8 Å throughout the 100 ns simulation time. The ligand was stable with minimal fluctuation within the range of 1.6–2.4 Å, indicating the ligand remains in the active site of the protein, as shown in Fig. 5(A). Next, the root mean square fluctuation (RMSF) plot was employed to understand the change occurring with time throughout the simulation. The protein and ligand RMSF showed that amino acid residues 100–125 (5.4 Å) [Fig. 5(B)] and 44 atoms (1.75) of the ligand fluctuated, as shown in Fig. 5(C). The PL histogram diagram showed H-bonding interactions with amino acid residues His99 (47%), Ser121 (11%), Asn46 (44%), and Val120 (65%) and π–π interaction with amino acid residues His99 (27%) and Phe104 (10%), as shown in Fig. 5(D and E).
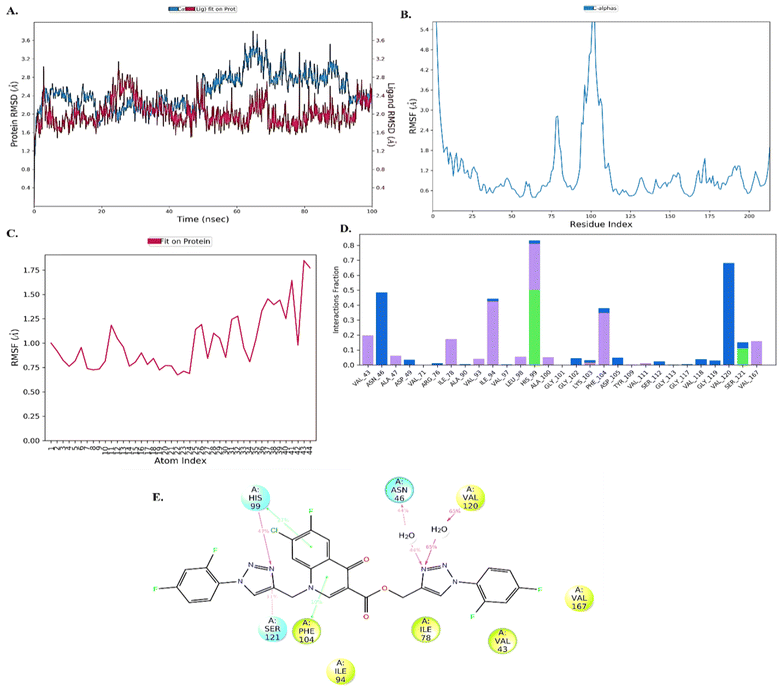 |
| | Fig. 5 Molecular dynamic analysis. (A) RMSD analysis of the protein–ligand (10d) complex, (B) RMSF analysis of protein, (C) RMSF analysis of the ligand, (D) and protein–ligand histogram analysis and (E) 2D diagram of the ligand with interaction with amino acids. | |
DNA gyrase inhibition assay
We performed the DNA gyrase-based inhibition assay to validate the in silico finding of 10d. The assay was performed in the presence of ciprofloxacin as a positive control. DNA gyrase is reported to relieve the negative supercoils in DNA during replication. Usually, upon running agarose gel, the supercoiled form of the DNA molecules tends to migrate faster than the linear form of DNA. Thereby, any decrease in the concentration of the supercoiled form of DNA and an increase in the relaxed form of the DNA product is linked with DNA gyrase inhibition. The present investigation (Fig. 6) with 9a and 10d at 10 μM concentration treatment on DNA gyrase inhibition revealed that compound 10d (as revealed by docking investigations previously) was more potent than 9a, thus further corroborating the in silico findings. The gel image and densitometric plot revealing the decrease in supercoiled DNA are shown in Fig. 6(A and B), respectively.
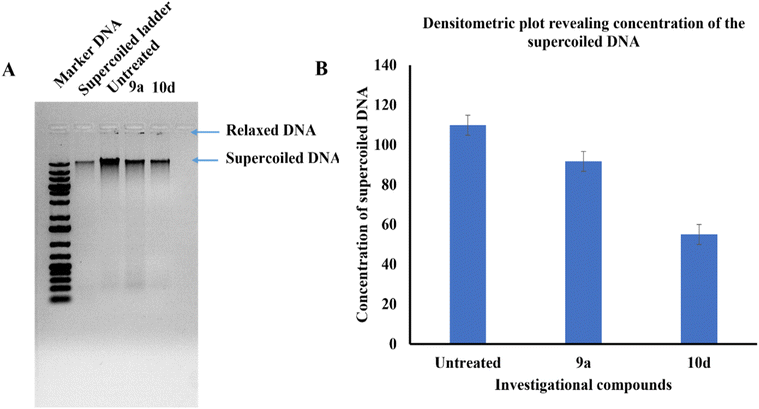 |
| | Fig. 6 (A) Agarose gel (inverted image) showing the supercoiled DNA concentration of the treated samples with investigational compounds with respect to untreated DNA. (B) A densitometric plot revealing the concentration of supercoiled DNA which indicates DNA gyrase inhibition. | |
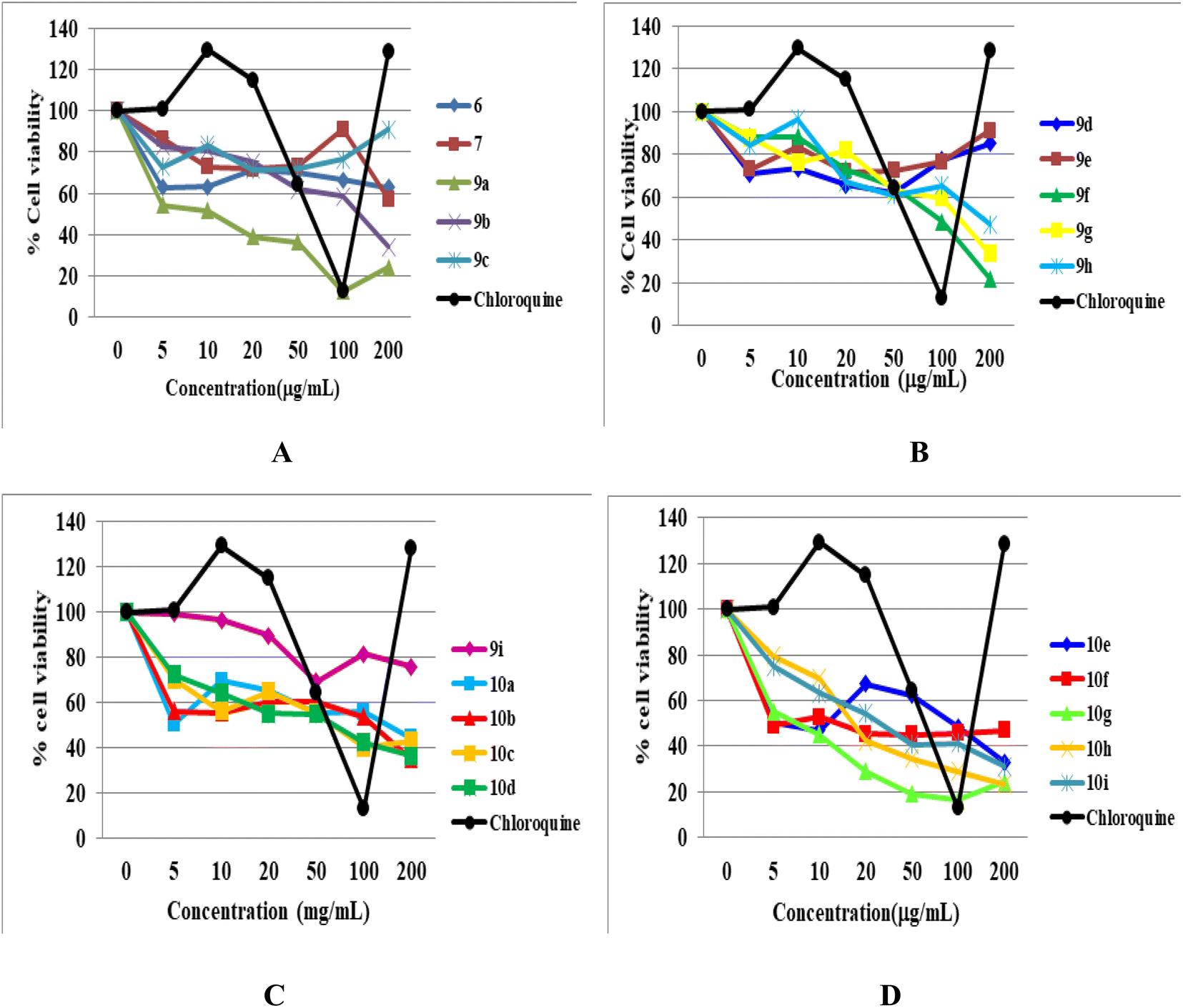
Cytotoxicity
The cytotoxicity of all the newly synthesized compounds, 6, 7, 9(a–i), and 10(a–i), were evaluated in Vero cells in vitro using the published protocol.29,30 The results showed that fifteen compounds are free of any considerable cytotoxicity at higher levels of test concentration (100 μg mL−1), as shown in Table 1. In contrast, five newly synthesized analogues (9a, 9c, 10g, 10h and 10i) showed toxicity. It is evident from the cytotoxicity results that the functional group position, linkage, nature, and number affect the cytotoxicity of the compounds. For example, compound 9a, having C-linked monosubstituted triazole with 3-Cl and 4-F phenyl ring and N-ethyl moiety, showed cytotoxicity against Vero cells. Compound 9c has the same connectivity with para-substituted trifluoromethoxy (-OCF3) phenyltriazole ring also showed cytotoxicity, while the bis triazoles (10a and 10c) of the same compounds did not show any toxicity. In conclusion, we deduced that the N-ethyl moiety with monosubstituted triazole having 3-Cl, 4-F, and -OCF3-substituted phenyl ring is responsible for the cytotoxicity. Similarly, compound 10g having bis triazole(C/N-linked triazole) with 2-Cl, 4-F-substituted phenyl ring exhibited cytotoxicity compared to the standard drug at a higher concentration (100 μg mL−1). Compounds 10h and 10i having 2,3-dichloro and 4-NO2 groups, respectively, showed toxicity, while monosubstituted triazoles (9g, 9h, and 9i) of the same compounds were free from cytotoxicity. Thus, we hypothesized that the compounds (10g, 10h, and 10i) with more functional groups (-Cl, –F, di-Cl, and –NO2) showed cytotoxicity (Fig. 7).
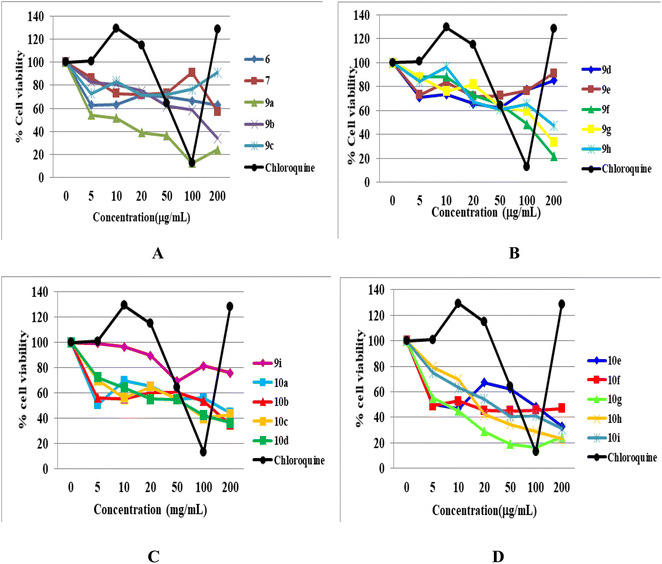 |
| | Fig. 7 MTT cytotoxicity. (A) Cell viability of Vero cells treated with compounds 6, 7, and 9(a–c) and chloroquine. (B) Cell viability of Vero cells treated with compounds 9(d–h) and chloroquine. (C) Cell viability of Vero cells treated with compounds 9i and 10(a–d), and chloroquine. (D) Cell viability of Vero cells treated with compounds 10(e–i) and chloroquine. | |
Experimental
All the chemicals and solvents used in the current study were purchased from E. Merck (India) and Sigma-Aldrich. On precoated silica gel 60 F254 (mesh), the reactions during synthesis were monitored via thin layer chromatography (TLC), and spots were visualized using UV light. Silica gel (60–120 mesh) was employed for column chromatography. The melting points of all the synthesized compounds were determined using the open capillary method and may be uncorrected. The structural assignments of the synthesized products were based on 1H NMR, 13C NMR, HRMS, FT-IR, and single-crystal XRD. NMR data were collected using 600 Bruker Avance Neo 600MHZ and 400 MHz, JEOL JNM-ECS spectrometer in DMSO-d6 and CDCl3 using TMS as an internal standard and Delta software to process the data. In reporting spectra, abbreviations such as s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, t = triplet, and m = multiple are used. Mass data was produced using a Bruker Compass spectrometer. X-ray analysis was performed using Rigaku XtaLAB Synergy-i Single Crystal X-ray Diffractometer with a CCD detector (HyPix-Bantam) using graphite mono-chromatized Cu-Kα radiation (λ = 1.54184 Å).
Method for the synthesis of diethyl-2-(((3-chloro-4-fluorophenyl)amino)methylene)malonate (1)
The synthesis of diethyl 2-(((3-chloro-4-fluorophenyl)amino)methylene)malonate was achieved according to the published procedure.31 Briefly, 3-chloro-4-fluoroaniline (24.73 mmol, 3.6 g) and diethyl ethoxymethylenemalonate (27.20 mmol, 5.5 mL) were heated at 100 °C for 1.5 h. After this period, the reaction mixture was cooled at room temperature, and ethanol formed during the reaction was removed under vacuum to yield the crude product. It was purified by recrystallization with n-hexane to give the corresponding malonate ester (7.2 g). White solid; yield 7.2 g (92.3%); NMR & mp: reported 1.31
Method for the synthesis of ethyl-7-chloro-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (2)
The synthesis of ethyl-7-chloro-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate was achieved via the cyclization of malonate ester (1). In short, heating diphenyl ether (50 mL) in an oil bath at 250 °C and the above-mentioned malonate ester (1) (20 mmol, 6.3 g) was added slowly. The reaction mixture was refluxed for 1 h with stirring, and a white solid was formed on cooling. The solid was filtered, washed with hexane, and purified through recrystallization from DMF to give 2. White solid; yield 2.15 g (70%); NMR & mp reported 2.31
Procedure for the synthesis of ethyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (3)
A mixture of compound 2 (4.0 mmol, 1.08 g), K2CO3 (20.0 mmol, 2.76 g), and ethyl bromide (20.0 mmol, 2.17 g) in 50 mL DMF was heated at 90 °C. After 14 h, the reaction mixture was in ice-cold water to get a white solid precipitate, which was collected by vacuum filtration. The crude products were purified by column chromatography using CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (99:1) as the eluent to afford the corresponding N-1 ethyl-substituted quinolones (3).32
:MeOH (99:1) as the eluent to afford the corresponding N-1 ethyl-substituted quinolones (3).32
White solid; yield 2.1 g (70.5%); mp: 133–135 °C; 1H NMR (600 MHz, DMSO-d6) δ: 8.47 (s, 1H, Ar–H), 8.23 (d, J = 9.0 Hz, 1H, Ar–H), 7.54 (d, J = 5.6 Hz, 1H, Ar–H), 4.39 (q, J = 7.1 Hz, 2H, –OCH2 of –OCH2CH3), 4.24 (q, J = 7.2 Hz, 2H, –NCH2, of –NCH2CH3), 1.57 (t, J = 7.2 Hz, 3H, –CH3 of –OCH2CH3), 1.41 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (151 MHz, DMSO-d6) δ: 172.86 (–CO), 165.45 (–CO), 156.39, 154.73, 148.86, 135.46, 129.60 (d, J = 5.6 Hz), 127.33 (d, J = 20.3 Hz), 118.22, 114.45 (d, J = 22.5 Hz), 111.09, 61.20, 49.39, 14.58.
General procedure for the synthesis of 7-chloro-1-ethyl/7-chloro-6-fluoro-4-oxo-1,4 dihydroquinoline-3-carboxylic acid (4)/(5)
Compounds 2 and 3 (10.0 mmol, 2.7 g) were refluxed with 2N NaOH (25.0 mL) for 2 h. The mixture was allowed to cool at room temperature and acidified with acetic acid. The solid was filtered, washed with water, and dried under a vacuum. The solid was recrystallized with DMF to give 4 and 5 (2.30 g).32
7-Chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (4)
White solid; yield 630 mg (97.83%); mp: 245–247 °C; 1H NMR (600 MHz, DMSO-d6) δ: 14.76 (s, 1H, –COOH), 9.07 (s, 1H, Ar–H), 8.44 (s), 8.20 (d, J = 7.5 Hz, 1H, Ar–H), 4.61 (s, 2H, –NCH2, of –NCH2CH3), 1.39 (s, 3H, –CH3 of –NCH2CH3); 13C NMR (151 MHz, DMSO-d6) δ: 176.39 (–CO of –COOH), 166.69 (–CO), 155.37, 153.73, 147.43, 146.13, 138.82, δ 126.05 (d, J = 20.5 Hz), 125.98, 124.71 (d, J = 6.6 Hz), 123.58, 121.91, 110.85 (d, J = 22.4 Hz), 51.65, 16.39.
7-Chloro-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (5)
White solid; yield 1.10 g (91.28%); mp: 287–287 °C; 1H NMR (600 MHz, DMSO-d6) δ: 15.26 (s, 1H, –COOH), 8.91 (s, 1H, Ar–H), 8.07 (d, J = 9.3 Hz, 1H, Ar–H), 7.99 (d, J = 6.2 Hz, 1H, Ar–H); 13C NMR (151 MHz, DMSO-d6) δ 176.90 (–CO of –COOH), 166.66 (–CO), 155.73, 154.09, 147.10, 138.16, 126.62 (d, J = 20.7 Hz), 124.94, 123.20, 111.24 (d, J = 22.4 Hz), 107.78.
Procedure for the synthesis of prop-2-yn-1-yl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (6)
Propargylation was performed using a method reported in the literature.33 To a solution of compound 4 (4.81 mmol, 1.3 g) in dry N,N-dimethylformamide (40 mL), in separate round-bottom flasks, 25 mL of NaHCO3 solution (7.2 mmol, 0.60 g) and (7.2 mmol, 0.86 g) of propargyl bromide were added under vigorous stirring at room temperature. The mixture was allowed to react at 90 °C for 36 h. The progress of the reaction was checked by TLC. After evaporating the solvent, the residue was purified by column chromatography using a CHCl3:MeOH (97:3) mixture as the eluent to obtain the desired propargylated product 6.
Prop-2-yn-1-yl 7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (6)
White solid; yield 590 mg (79.65%); mp: 165–167 °C; 1H NMR (600 MHz, DMSO-d6) δ: 8.51 (s, 1H, Ar–H), 8.22 (d, J = 9.0 Hz, 1H, Ar–H), 7.55 (d, J = 5.6 Hz, 1H, Ar–H), 4.91 (s, 2H, –OCH2), 4.25 (q, J = 7.3 Hz, 2H, –NCH2 of –NCH2CH3), 2.51 (s, 1H, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 1.58 (t, J = 7.3 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (151 MHz, DMSO-d6) δ: 172.35 (–CO), 164.06 (–CO), 156.22, 154.56, 148.88, 135.08, 129.23 (d, J = 5.7 Hz), 127.21 (d, J = 20.2 Hz), 118.01, 114.24 (d, J = 23.0 Hz), 109.70, 77.86, 77.74, 52.03, 49.22, 14.27; IR (KBr) cm−1: 3283, 3062, 1697, 1610, 1486, 1311, 1218, 1172, 1033, 807, 734; HRMS (ESI): anal. calcd. For C15H11ClFNO3, 307.7054 [M]+; found 308.0491 [M + H]+.
CH), 1.58 (t, J = 7.3 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (151 MHz, DMSO-d6) δ: 172.35 (–CO), 164.06 (–CO), 156.22, 154.56, 148.88, 135.08, 129.23 (d, J = 5.7 Hz), 127.21 (d, J = 20.2 Hz), 118.01, 114.24 (d, J = 23.0 Hz), 109.70, 77.86, 77.74, 52.03, 49.22, 14.27; IR (KBr) cm−1: 3283, 3062, 1697, 1610, 1486, 1311, 1218, 1172, 1033, 807, 734; HRMS (ESI): anal. calcd. For C15H11ClFNO3, 307.7054 [M]+; found 308.0491 [M + H]+.
Procedure for the synthesis of prop-2-yn-1-yl 7-chloro-6-fluoro-4-oxo-1-(prop-2-yn-1-yl)-1,4-dihydroquinoline-3-carboxylate (7)
Propargylation was done using a method reported in the literature.33 To a solution of compound 5 (4.1 mmol, 1 g) in dry N,N-dimethylformamide (40 mL), in separate round-bottom flasks, 25 mL of NaHCO3 solution (16.4 mmol, 1.4 g) and (1.2 mmol, 1.95 g) of propargyl bromide were added under vigorous stirring at room temperature. The mixture was allowed to react at 100 °C for 48 h. The progress of the reaction was checked by TLC. After evaporating the solvent, the residue was purified by column chromatography using a CHCl3:MeOH (97:3) mixture as the eluent to obtain the desired propargylated products 7.
Prop-2-yn-1-yl-7-chloro-6-fluoro-4-oxo-1-(prop-2-yn-1-yl)-1,4-dihydroquinoline-3-carboxylate (7)
White solid; yield 460 mg (70%); mp: 193–195 °C; 1H NMR (600 MHz, DMSO-d6) δ: 8.89 (s, 1H, Ar–H), 8.16 (s, 1H, Ar–H), 8.05 (d, J = 8.8 Hz, Ar–H), 5.36 (s, 2H, –OCH2), 4.87 (s, 2H, –NCH2), 3.70 (s, 1H, –CCH), 3.57 (s, 1H, –CCH); 13C NMR (151 MHz, DMSO-d6) δ: 171.40 (–CO), 163.17 (–CO), 155.54, 153.89, 149.89, 148.64, 135.53, 128.55 (d, J = 5.2 Hz), 125.53 (d, J = 20.0 Hz), 120.48, 112.73 (d, J = 22.6 Hz), 109.25, 78.79 (d, J = 6.6 Hz), 77.67, 77.20, 51.72, 42.90; IR (KBr) cm−1: 3200, 2125, 1682, 1615, 1481, 1213, 1157, 1028, 755, 549; HRMS (ESI): anal. calcd. For C16H9ClFNO3, 317.7004 [M]+; found 318.0347 [M + H]+.
General procedure for the synthesis of azides (8a–i)
The azides were synthesized according to the established method.34 Briefly, aniline (1 eq.) was dissolved in 6 N HCl solution (10 mL mmol−1 of aniline) at room temperature and cooled to 0 °C and further supplemented with NaNO2 (1.2 eq.) solution under stirring. After 10 min, sodium azide (1.2 eq.) was added to the reaction mixture at the same temperature under stirring. Again, this mixture was stirred at room temperature for 2–3 h. The reaction was worked up by extraction with ethyl acetate. The organic layer was washed with brine solution and dried over Na2SO4. After evaporation of the solvent, the crude product (8a–i) was pure enough for further reactions.
General procedure for the synthesis of 1,2,3-triazole scaffolds 9–10(a–i)
The triazoles were synthesized according to the literature-reported method.35 In brief, compounds 6 and 7 (1 eq.) and substituted aromatic azide (9–10(a–i), 1.2 eq.) were suspended in N,N-dimethylformamide (25 mL mmol−1 of alkyne). A solution of sodium ascorbate (0.4 eq. in minimum water) was added, followed by copper(II) sulfate pentahydrate solution (0.2 eq. in minimum water). At room temperature, the heterogeneous mixture was rapidly stirred until alkyne was consumed, and the progress of the reaction was checked by TLC. After completion of the reaction the reaction, the mixture was poured into ice water to get the precipitate and the precipitate was collected by filtration. The required products were purified by column chromatography using CHCl3:MeOH (96:4) mixture as the eluent.
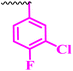
(1-(3-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9a)
Off white solid; yield 67 mg (86%); mp: 180–182 °C; 1H NMR (400 MHz, DMSO-d6) δ: 9.02 (s, 1H, Ar–H), 8.95 (s, 1H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH of triazole), 8.30 (d, J = 6.0 Hz, 1H, Ar–H), 8.23 (dd, J = 6.4, 2.7 Hz, 1H, Ar–H), 8.12 (d, J = 9.3 Hz, 1H, Ar–H), 7.99 (ddd, J = 9.0, 4.1, 2.7 Hz, 1H, Ar–H), 7.72 (t, J = 9.0 Hz, 1H, Ar–H), 5.93 (s, 2H, –OCH2), 4.32 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.36 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 172.10 (–CO), 164.75 (–CO), 156.23, 150.92, 143.50, 136.53, 133.86, 129.20, 123.03, 121.58 (d, J = 8.2 Hz), 120.98, 118.62 (d, J = 22.7 Hz), 113.06 (d, J = 22.7 Hz), 110.84, 60.57, 48.50.14.81; IR (KBr) cm−1: 3093, 2923, 1718, 1610, 1512, 1302, 1223, 1028, 796, 716; HRMS (ESI): anal. calcd. For C21H14Cl2F2N4O3, 479.2648 [M]+; found 479.0510 [M + H]+.
CH of triazole), 8.30 (d, J = 6.0 Hz, 1H, Ar–H), 8.23 (dd, J = 6.4, 2.7 Hz, 1H, Ar–H), 8.12 (d, J = 9.3 Hz, 1H, Ar–H), 7.99 (ddd, J = 9.0, 4.1, 2.7 Hz, 1H, Ar–H), 7.72 (t, J = 9.0 Hz, 1H, Ar–H), 5.93 (s, 2H, –OCH2), 4.32 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.36 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 172.10 (–CO), 164.75 (–CO), 156.23, 150.92, 143.50, 136.53, 133.86, 129.20, 123.03, 121.58 (d, J = 8.2 Hz), 120.98, 118.62 (d, J = 22.7 Hz), 113.06 (d, J = 22.7 Hz), 110.84, 60.57, 48.50.14.81; IR (KBr) cm−1: 3093, 2923, 1718, 1610, 1512, 1302, 1223, 1028, 796, 716; HRMS (ESI): anal. calcd. For C21H14Cl2F2N4O3, 479.2648 [M]+; found 479.0510 [M + H]+.
(1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9b)
Creamy white solid; yield 71 mg (95%); mp: 218–220 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.96 (d, J = 6.9 Hz, 2H, Ar–H, –CCH of triazole), 8.23 (d, J = 6.0 Hz, 1H, Ar–H), 8.02 (dd, J = 17.1, 5.6 Hz, 2H, Ar–H), 7.89 (d, J = 6.9 Hz, 1H, Ar–H), 7.66–7.49 (m, 2H, Ar–H), 5.88 (s, 1H, 2H, –OCH2), 4.27 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.31 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 172.10 (–CO), 164.75 (–CO), 150.92, 143.51, 137.93, 136.56, 134.68, 132.12, 129.20, 122.84, 120.73 (d, J = 49.3 Hz), 119.30, 113.07 (d, J = 22.8 Hz), 110.87, 60.56, 48.55, 14.82; IR (KBr) cm−1: 3095, 2929, 1730, 1612, 1485, 1315, 1215, 1162, 1043, 782, 672; HRMS (ESI): anal. calcd. For C21H15Cl2FN4O3, 461.2744 [M]+; found 461.0607 [M + H]+.
(1-(4-(Trifluoromethoxy)phenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9c)
White solid; yield 65 mg (78%); mp: 228–230 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.96 (s, 1H, Ar–H), 8.91 (s, 1H, –CCH of triazole), 8.25 (d, J = 5.8 Hz, 1H, Ar–H), 8.01 (dd, J = 13.4, 9.1 Hz, 4H, Ar–H), 7.59 (d, J = 8.8 Hz, 1H, Ar–H), 5.86 (s, 1H, 2H, –OCH2), 4.25 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.29 (t, J = 7.0 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 172.10 (–CO), 164.75 (–CO), 153.79 (d, J = 5 Hz), 150.92, 143.51, 137.93, 136.56, 134.68, 132.12, 129.14 (d, J = 11.5 Hz), 125.84 (d, J = 20.3 Hz), 122.84, 120.73 (d, J = 49.3 Hz), 119.30, 113.07 (d, J = 22.8 Hz), 110.87, 60.56, 48.55, 14.82; IR (KBr) cm−1: 3103, 1728, 1610, 1486, 1270, 1218, 1157, 1038, 801; HRMS (ESI): anal. calcd. For C22H15ClF4N4O4, 510.8296 [M]+; found 511.0821 [M + H]+.
(1-(2,4-Difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9d)
Off-white solid; yield 59 mg (78%); mp: 218–220 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.95 (s, 1H, Ar–H), 8.71 (s, 1H, –CCH of triazole), 8.31 (d, J = 5.8 Hz, 1H, Ar–H), 7.99 (d, J = 9.1 Hz, 1H, Ar–H), 7.84 (dd, J = 14.7, 8.7 Hz, 1H, Ar–H), 7.64 (d, J = 20.0 Hz, 1H, Ar–H), 7.29 (t, J = 8.0 Hz, 1H, Ar–H), 5.82 (s, 2H, –OCH2), 4.21 (q, J = 7.0 Hz, 2H, –NCH2 of –NCH2CH3), 1.26 (t, J = 7.0 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz DMSO-d6) δ: 172.04 (–CO), 164.76 (–CO), 153.77, 150.92, 142.68 (d, J = 20.5 Hz), 136.54, 128.12 (d, J = 10.4 Hz), 126.24, 121.06, 113.16 (dd, J = 22.7, 15.8 Hz), 110.79, 60.59, 48.13, 14.82; IR (KBr) cm−1: 3079, 1722, 1613, 1520, 1216, 1142, 1040, 840, 762, 603; HRMS (ESI): anal. calcd. For C21H14ClF3N4O3, 462.8132 [M]+; found 463.0793 [M + H]+.
(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9e)
Light-pink solid; yield 65 mg (87%); mp: 248–250 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.95 (s, 1H, Ar–H), 8.89 (s, 1H, –CCH of triazole), 8.25 (d, J = 6.0 Hz, 1H, Ar–H), 8.04 (d, J = 9.3 Hz, 1H, Ar–H), 7.90 (d, J = 6.9 Hz, 2H, Ar–H), 7.65 (d, J = 7.0 Hz, 2H, Ar–H), 5.85 (s, 2H, –OCH2), 4.25 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.29 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C-NMR (101 MHz, DMSO-d6) δ: 172.10 (–CO), 164.75 (–CO), 150.92, 143.51, 137.93, 136.56, 134.68, 132.12, 129.20, 122.84, 120.73 (d, J = 49.3 Hz), 119.30, 113.07 (d, J = 22.8 Hz), 110.87, 60.56, 48.55, 14.82; IR (KBr) cm−1: 3093, 1733, 1617, 1488, 1317, 1161, 1042, 781, 674; HRMS (ESI): anal. calcd. ForC21H15Cl2FN4O3, 461.2744 [M]+; found 461.0609 [M + H]+.
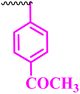
(1-(4-Acetylphenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9f)
Pale-white solid; yield 49 mg (64%); mp: 190–192 °C; 1H NMR (600 MHz, DMSO-d6) δ: 9.02 (s, 1H, Ar–H), 8.74 (s, 1H, –CCH of triazole), 8.23 (d, J = 5.8 Hz, 1H, Ar–H), 8.13–8.05 (m, 3H, Ar–H), 7.99 (dd, J = 8.8, 4.5 Hz, 2H, Ar–H), 5.41 (s, 2H, –OCH2), 4.41 (q, J = 7.2 Hz, 2H, –NCH2 of –NCH2CH3), 2.62 (s, 3H, –COCH3), 1.34 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (151 MHz, DMSO-d6) δ: 197.18 (–COCH3), 171.65 (–CO), 164.03 (–CO), 155.48, 153.84, 150.07, 143.62 (d, J = 120.6 Hz), 139.66, 136.27 (d, J = 117.0 Hz), 130.26 (d, J = 11.5 Hz), 128.87, 125.89 (d, J = 19.8 Hz), 123.39, 122.55, 120.55–118.35 (m) 112.79 (d, J = 22.5 Hz), 109.38, 57.16, 48.70.27.04, 14.58; IR (KBr) cm−1: 3132, 2916, 1617, 1596, 1484, 1227, 1157, 1047, 794, 604; HRMS (ESI): anal. calcd. For C23H18ClFN4O4, 468.8694 [M]+; found 469.1105 [M + H]+.
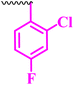
(1-(2-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9g)
Pale-yellow solid; yield 50 mg (64%); mp: 224–226 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.99 (s, 1H, Ar–H), 8.74 (s, 1H, –CCH of triazole), 8.31 (d, J = 5.7 Hz, 1H, Ar–H), 8.01 (dd, J = 20.2, 5.6 Hz, 2H, Ar–H), 7.70 (dd, J = 30.3, 8.5 Hz, 1H, Ar–H), 5.87 (s, 2H, –OCH2), 4.25 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.30 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 172.10 (–CO), 164.75 (–CO), 156.23, 154.00, 150.92, 143.50, 136.53, 133.86, 129.20, 125.86 (d, J = 20.2 Hz), 123.03, 121.58 (d, J = 8.2 Hz), 120.98, 118.62 (d, J = 22.7 Hz), 113.06 (d, J = 22.7 Hz), 110.84, 60.57, 48.50, 14.81; IR (KBr) cm−1: 3090, 2920, 1720, 1612, 1510, 1221, 1026, 794, 714.
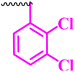
(1-(2,3-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9h)
Pale-yellow solid; yield 55 mg (68%); mp: 199–201 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.99 (s, 1H, Ar–H), 8.76 (s, 1H, –CCH of triazole), 8.31 (d, J = 6.0 Hz, 1H, Ar–H), 8.04 (d, J = 9.3 Hz, 1H, Ar–H), 7.91 (dd, J = 8.1, 1.4 Hz, 2H, Ar–H), 7.70 (dd, J = 8.0, 1.4 Hz, 2H, Ar–H), 7.59 (t, J = 8.1 Hz), 5.89 (s, 2H, –OCH2), 4.26 (q, J = 7.1 Hz, 2H, –NCH2 of –NCH2CH3), 1.30 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 171.97 (–CO), 164.70 (–CO), 156.23, 150.89, 141.84, 132.74, 129.58, 127.82, 127.01, 121.13, 118.92, 113.64, 112.96 (d, J = 4.8 Hz), 111.36, 60.58, 48.13, 14.83; IR (KBr) cm−1: 3147, 1685, 1608, 1480, 1229, 1160, 1026, 789.
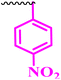
(1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-ethyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (9i)
Yellow solid; yield 61 mg (80%); mp: 230–232 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.95 (s, 1H, Ar–H), 8.89 (s, 1H, –CCH of triazole), 8.25 (d, J = 6.0 Hz, 1H, Ar–H), 8.04 (d, J = 9.3 Hz, 1H, Ar–H), 7.90 (d, J = 6.9 Hz, 2H, Ar–H), 7.65 (d, J = 7.0 Hz, 2H, Ar–H), 5.85 (s, 2H, –OCH2), 4.25 (q, J = 7.1 Hz, 2H, 2H, –NCH2 of –NCH2CH3), 1.29 (t, J = 7.1 Hz, 3H, –CH3 of –NCH2CH3); 13C NMR (101 MHz, DMSO-d6) δ: 171.98 (–CO), 164.70 (–CO), 156.35, 150.93, 143.88, 137.31, 136.58, 135.70, 133.68, 130.37, 122.57 (d, J = 34.3 Hz), 121.01, 113.19, 112.99 (d, J = 5.9 Hz), 110.85, 60.57, 48.49, 14.83; IR (KBr) cm−1: 3091, 2119, 1720, 1612, 1524, 1338, 1225, 1030, 860, 744.
(1-(3-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-1-((1-(3-chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10a)
Light-pink solid; yield 103 mg (99%); mp: 228–230 °C; 1H NMR (600 MHz, DMSO-d6) δ: 9.03–8.83 (m, 3H, Ar–H, 2× –CCH of triazole), 8.22 (d, J = 0.5 Hz, 2H, Ar–H), 8.13 (d, J = 1.0 Hz, 1H, Ar–H), 8.06–7.93 (m, 2H, Ar–H), 7.89 (d, J = 1.6 Hz, 1H, Ar–H), 7.72–7.55 (m, 2H, Ar–H), 5.87 (s, 2H, –OCH2), 5.42 (s, 2H, –NCH2); 13C NMR (151 MHz, DMSO-d6) δ: 165.87 (–CO), 158.00, 152.06, 150.41, 149.69, 148.05, 145.03, 137.85, 137.19, 130.29, 127.71 (d, J = 21.6 Hz), 122.94 (d, J = 5.1 Hz), 119.76 (d, J = 20.1 Hz), 116.79 (d, J = 12.4 Hz), 115.02 (d, J = 11.9 Hz), 115.29, 115.24, 115.06, 114.92 (d, J = 11.1 Hz), 112.51 (d, J = 9.7 Hz), 112.36 (d, J = 9.8 Hz), 106.87 (d, J = 22.5 Hz), 103.86, 51.32, 42.36; IR (KBr) cm−1: 3088, 1734, 1615, 1512, 1223, 1157, 1028, 801, 719.
(1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-((1-(3-chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10b)
Off-white solid; yield 50 mg (51%); mp: 225–227 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.13–8.82 (m, 3H, Ar–H, 2× –CCH of triazole), 8.22 (s, 1H, Ar–H), 8.08–7.82 (m, 5H, Ar–H), 7.64–7.50 (m, 4H, Ar–H), 5.87 (s, 2H, –OCH2), 5.44 (s, 2H, –NCH2); 13C NMR (151 MHz, DMSO-d6) δ: 171.60 (–CO), 163.73 (–CO), 155.40, 153.76, 150.33, 143.60, 142.92, 137.47 (d, J = 16.2 Hz), 136.01, 134.21 (d, J = 10.8 Hz), 131.63 (d, J = 7.4 Hz), 128.66 (d, J = 11.6 Hz), 125.47 (d, J = 20.0 Hz), 123.32, 122.33, 120.58, 119.94, 118.75 (d, J = 6.3 Hz), 112.58 (d, J = 22.3 Hz), 109.60, 57.07, 48.12; IR (KBr) cm−1: 3098, 2928, 1734, 1615, 1492, 1218, 1157, 1028, 776, 673.
(1-(4-(Trifluoromethoxy)phenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-6-fluoro-4-oxo-1-((1-(4-(trifluoromethoxy)phenyl)-1H-1,2,3-triazol-4-yl)methyl)-1,4-dihydroquinoline-3-carboxylate (10c)
Off-white solid; yield 55 mg (48%); mp: 210–212 °C; 1H NMR (600 MHz, DMSO-d6) δ: 8.96–8.92 (m, 3H, Ar–H, 2× –CCH of triazole), 8.22 (s, 1H, ArH), 8.04–7.85 (m, 5H, Ar–H), 7.62–7.52 (m, 4H, Ar–H), 5.87 (s, 2H, –OCH2), 5.44 (s, 2H, –NCH2); 13C NMR (151 MHz, DMSO-d6) δ: 172.11 (–CO), 164.33 (–CO), 163.97, 157.25, 156.20, 155.00, 154.22, 153.99, 151.43 (d, J = 25.8 Hz), 144.14, 143.47, 136.61, 135.84 (d, J = 20.8 Hz), 133.72 (d, J = 13.0 Hz), 130.47 (d, J = 7.8 Hz), 129.26, 125.55 (d, J = 18.2 Hz), 123.80, 122.86, 122.47 (d, J = 36.4 Hz), 121.12 (d, J = 25.0 Hz), 79.42, 78.25, 57.67, 52.28, 48.67, 43.49; IR (KBr) cm−1: 3103, 2928, 1728, 1610, 1486, 1259, 1213, 1162, 1043, 791.
(1-(2,4-Difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-1-((1-(2,4-difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10d)
White solid; yield 88 mg (89%); mp: 227–229 °C; 1H NMR (600 MHz, DMSO-d6) δ: 9.04 (s, 1H, Ar–H), 8.71 (m, 2H, 2× –CCH of triazole), 8.34 (d, J = 5.2 Hz, 1H, Ar–H), 8.02 (d, J = 9.0 Hz, 1H, Ar–H), 7.97–7.82 (m, 2H, Ar–H), 7.67 (dd, J = 21.8, 10.5 Hz, 2H, Ar–H), 7.41–7.28 (m, 2H, Ar–H), 5.87 (s, 2H, –OCH2), 5.45 (s, 2H, –NCH2); 13C NMR (151 MHz, DMSO-d6) δ: 171.83 (–CO), 164.15 (–CO), 163.36 (d, J = 11.9 Hz), 161.71 (d, J = 11.9 Hz), 155.55 (d, J = 44.5 Hz), 154.13–153.56 (m), 151.06, 143.22, 142.32, 136.32, 128.94 (d, J = 6.4 Hz), 127.93 (dd, J = 16.7, 10.2 Hz), 126.75 (d, J = 5.0 Hz), 126.05 (d, J = 5.5 Hz), 125.78 (d, J = 20.0 Hz), 121.91 (d, J = 20.9 Hz), 120.94, 113.15 (d, J = 5.0 Hz), 113.00 (d, J = 5.6 Hz), 112.83, 109.86, 106.19 (d, J = 6.6 Hz), 106.11–105.97 (m), 105.85 (d, J = 6.7 Hz), 57.36, 48.04; IR (KBr) cm−1: 3078, 1728, 1615, 1522, 1213, 1146, 1043, 842, 606.
(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10e)
Off white solid; yield 64 mg (65%); mp: 248–250 °C; 1H NMR (600 MHz, DMSO-d6) δ; 9.05–8.97 (m, 3H, Ar–H, 2× –CCH of triazole), 8.28 (s, 1H, Ar–H), 8.10–7.91 (m, 5H, Ar–H), 7.69–7.57 (m, 4H, Ar–H), 5.92 (s, 2H, –OCH2), 5.50 (s, 2H, –NCH2); 13C NMR (151 MHz, DMSO-d6) δ: 171.51 (–CO), 163.38 (–CO), 155.42 (d, J = 8.0 Hz), 153.78 (d, J = 6.5 Hz), 150.85 (d, J = 25.8 Hz), 149.85 (d, J = 15.0 Hz), 143.57, 142.90, 136.04, 135.26 (d, J = 19.1 Hz), 133.15 (d, J = 12.8 Hz), 133.15 (d, J = 12.8 Hz), 129.90 (d, J = 8.1 Hz), 128.69, 125.49 (d, J = 19.9 Hz), 123.22 (d, J = 4.5 Hz), 122.29 (d, J = 4.8 Hz), 122.12–121.62 (m), 120.62 (d, J = 6.5 Hz), 112.68 (d, J = 5.9 Hz), 112.55, 109.43 (d, J = 33.3 Hz), 51.70, 48.10; IR (KBr) cm−1: 3097, 2931, 1731, 1611, 1483, 1313, 1219, 1165, 1045, 780, 671.
(1-(4-Acetylphenyl)-1H-1,2,3-triazol-4-yl)methyl 1-((1-(4-acetylphenyl)-1H-1,2,3-triazol-4-yl)methyl)-7-chloro-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10f)
Light pink solid; yield 97 mg (96%); mp: 258–260 °C; 1H NMR (600 MHz, DMSO-d6) δ: 9.04–8.98 (m, 3H, Ar–H, 2× –CCH of triazole), 8.25 (d, J = 5.7 Hz, 1H, Ar–H), 8.19–7.94 (m, 6H, Ar–H), 8.03–8.00 (m, 3H, Ar–H), 5.87 (s, 2H, –OCH2), 5.45 (s, 2H, –NCH2), 2.62 (d, J = 11.9 Hz, 6H, 2× –COCH3); 13C NMR (151 MHz, DMSO-d6) δ: 197.19 (–COCH3), 171.68 (–CO), 164.03 (–CO), 155.26, 153.61, 150.98, 143.97, 143.24, 139.59 (d, J = 24.5 Hz), 136.72 (d, J = 9.7 Hz), 130.28 (d, J = 10.1 Hz), 123.46, 122.55, 120.78, 120.10 (d, J = 6.9 Hz), 112.80 (d, J = 21.8 Hz), 109.62, 57.25, 48.16, 27.05; IR (KBr) cm−1: 3134, 2918, 1672, 1599, 1486, 1229, 1049, 796, 606; HRMS (ESI): anal. calcd. ForC32H23ClFN7O5, 640.0284 [M]+; found 640.1549 [M + H]+.
(1-(2-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl-7-chloro-1-((1-(2-chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10g)
Off-white solid; yield 102 mg (98%); mp: 225–227 °C; 1H NMR (600 MHz, DMSO-d6) δ: 9.03 (s, 1H, Ar–H), 8.70 (d, J = 29.4 Hz, 2× –CCH of triazole), 8.29 (d, J = 5.4 Hz, 1H, Ar–H), 8.03 (d, J = 9.1 Hz, 1H, Ar–H), 7.89 (t, J = 8.7 Hz, 2H, Ar–H), 7.67 (dd, J = 17.7, 7.8 Hz, 2H, Ar–H), 7.58 (dt, J = 16.0, 8.0 Hz, 2H, Ar–H), 5.87 (s, –OCH2), 5.45 (s, –NCH2); 13C NMR (151 MHz, DMSO-d6) δ: 171.57 (–CO), 163.73 (–CO), 163.02 (d, J = 6.7 Hz), 161.35 (d, J = 6.3 Hz), 155.31, 153.67, 150.62, 142.42, 141.37, 135.85, 131.09 (d, J = 5.0 Hz), 130.91 (d, J = 4.5 Hz), 130.01 (dd, J = 12.5, 7.6 Hz), 128.53, 126.97, 126.52, 125.43 (d, J = 19.8 Hz), 120.57, 117.94 (d, J = 5.1 Hz), 117.76 (d, J = 5.3 Hz), 115.62 (d, J = 22.9 Hz), 112.52 (d, J = 22.6 Hz), 109.45, 57.01, 47.81; IR (KBr) cm−1: 3079, 2921, 1730, 1610, 1510, 1381, 1314, 1220, 1024, 800, 763.
(1-(2,3-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-1-((1-(2,3-dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (10h)
White solid; yield 101 mg (92%); mp: 198–200 °C; 1H NMR (600 MHz, DMSO-d6) δ: 9.03 (s, 1H, Ar–H), 8.70 (d, J = 29.4 Hz, 2× –CCH of triazole), 8.29 (s, 1H, Ar–H), 8.03 (d, J = 9.1 Hz, 1H, Ar–H), 7.91 (t, J = 8.7 Hz, 2H, Ar–H), 7.67 (dd, J = 17.7, 7.8 Hz, 2H, Ar–H), 7.58 (dt, J = 16.0, 8.0 Hz, 2H, Ar–H), 5.87 (s, –OCH2), 5.45 (s, –NCH2): 13C NMR (151 MHz, DMSO-d6) δ: 171.58 (–CO), 163.67 (–CO), 155.32, 153.68, 155.32, 153.68, 150.61, 142.46, 141.44, 136.05–135.68 (m), 132.87 (d, J = 8.0 Hz), 132.13 (d, J = 12.0 Hz), 128.99, 128.49, 127.54 (d, J = 14.5 Hz), 127.16 (d, J = 6.2 Hz), 126.88, 126.42, 125.44 (d, J = 19.7 Hz), 120.58, 112.52 (d, J = 22.5 Hz), 109.45, 56.98, 47.78; IR (KBr) cm−1: 3149, 1687, 1610, 1481, 1231, 1162, 1028, 791.
(1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl 7-chloro-6-fluoro-1-((1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (10i)
Pale-yellow solid; yield 100 mg (98%); mp: 278–280 °C; 1H NMR (600 MHz, DMSO-d6) δ: 8.99–8.92 (m, 2H, Ar–H, –CCH of triazole), 8.22 (s, 1H, –CCH of triazole), 8.06–7.99 (m, 2H, ArH), 7.94 (d, J = 19.8 Hz, 2H, ArH), 7.85 (s, 2H, ArH), 7.65–7.50 (m, 5H, ArH), 5.87 (s, –OCH2), 5.44 (s, –NCH2); 13C-NMR (151 MHz, DMSO-d6) δ: 171.60 (–CO), 163.73 (–CO), 155.40, 153.76, 150.77, 143.26 (d, J = 103.2 Hz), 137.47 (d, J = 23.4 Hz), 136.01, 134.21 (d, J = 10.8 Hz), 131.63 (d, J = 7.4 Hz), 128.66 (d, J = 11.6 Hz), 125.40, 123.32, 123.32, 122.33, 120.58, 119.94, 118.75 (d, J = 6.3 Hz), 112.58 (d, J = 22.3 Hz), 109.60, 57.07, 48.12; IR (KBr) cm−1: 3093, 2120, 1723, 1610, 1522, 1337, 1223, 1028, 858, 744.
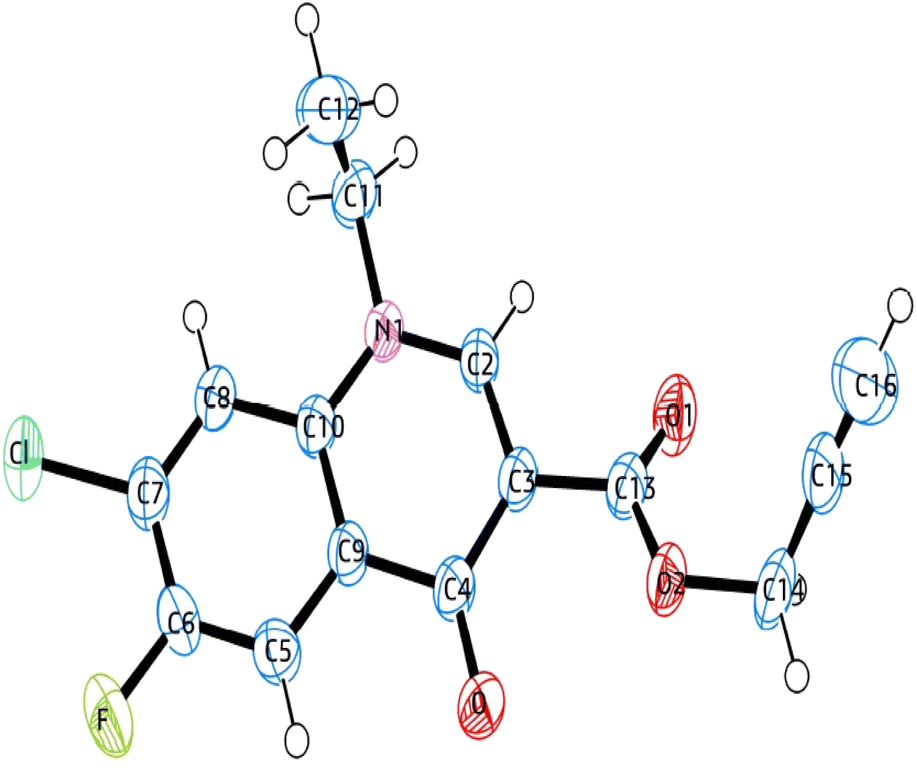
X-ray crystallographic analysis
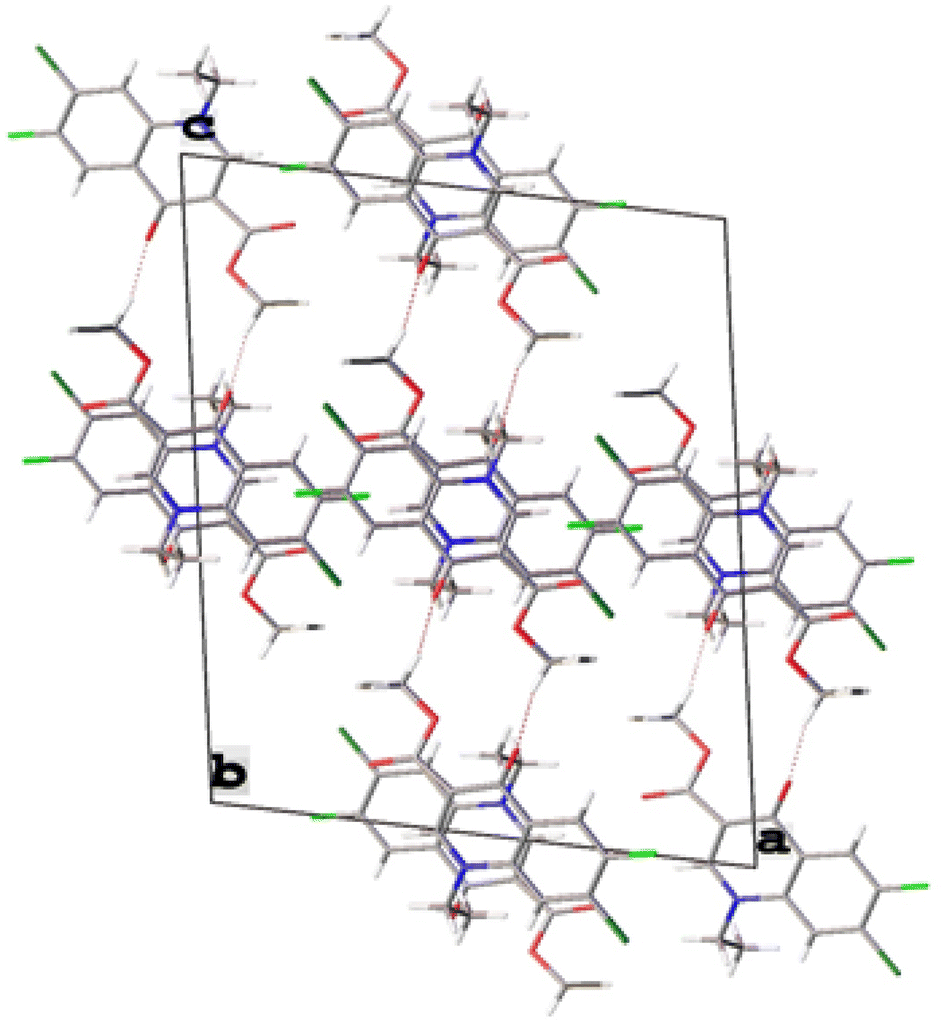
The single-crystal X-ray analysis further verified the structure of synthesized intermediate 6. Briefly, the crystals were formed via the slow evaporation solution technique with ethyl acetate. Graphite monochromatized Cu Kα radiation (λ = 1.54184 Å) was used to measure the X-ray diffraction intensity data at 293 K using the X-ray scan method on a Rigaku XtaLAB Synergy-i single-crystal X-ray diffractometer with a CCD-detector (HyPix-Bantam). The structure of compound 6 was established by the direct method using the Olex2-1.5 software.36 Then, it was refined using the full-matrix least-squares method on F2 by SHELXL.37 Fig. 8 show the thermal ellipsoid plot prepared using ORTEP III38 of compound 6, which was crystallized in monoclinic system with the C2/c space group. Table 2 provides information about the single-crystal X-ray crystallographic structure of compound 6 (Fig. 9).
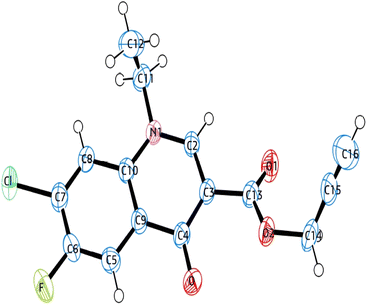 |
| | Fig. 8 ORTEP diagram of compound 6 based on single X-ray crystallographic analysis. | |
Table 2 Crystal data, data collection and structure refinement details for compound 6
| CCDC no. |
2345449 |
| Empirical formula |
C15H11ClFNO3 |
| Formula weight |
307.70 |
| Temperature |
293(2) K |
| Wavelength |
1.54184 Å |
| Crystal system |
Monoclinic |
| Space group |
C2/c |
| Hall group |
–C 2yc |
| Unit cell dimensions |
a = 14.9992(2) Å α = 90 |
| b = 9.55020(10) Å β = 100.19 |
| c = 20.3352(2) Å ϒ = 90 |
| Volume |
2866.96(6) Å3 |
| Z |
8 |
| Density |
1.426 g cm−3 |
| Absorption coefficient |
2.561 mm−1 |
| F(000) |
1264.0 |
| Theta range for data collection |
8.836 to 144.096 |
| Index ranges |
−17 ≤ h ≤ 18 |
| −11 ≤ k ≤ 11 |
| −25 ≤ l ≤ 24 |
| Reflections collected |
15237 |
| Completeness to theta |
100% |
| Absorption correction |
Multi-scan |
| Refinement method |
Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 |
1.115 |
| Final R indices [I > 2 sigma(I)] |
R1 = 0.0559, wR2 = 0.1621 |
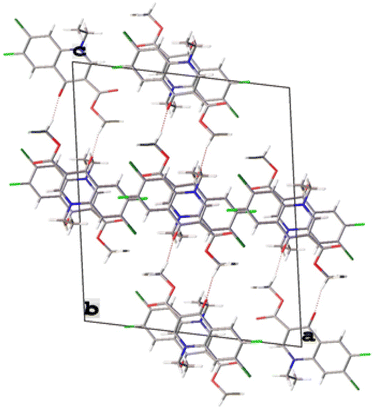 |
| | Fig. 9 Crystal packing of compound 6. | |
Biological assays
Determination of antibacterial activity
The bacterial strains used in the in vitro antibacterial investigations included E. faecalis (ATCC 29212), E. faecalis (clinical isolate), E. coli (ATCC 25922), and E. coli (clinical isolate). The cultures used in this study were preserved at the Department of Microbiology, Institute of Medical Sciences, Banaras Hindu University, Varanasi, India. All cultures and clinical strains were obtained from the American Type Culture Collection (ATCC). Prior to the screening process, the new microbial broth cultures were prepared in a regular saline solution. The reference medication for evaluating antibacterial effectiveness was ciprofloxacin. The micro-dilution approach was used to calculate the minimum inhibitory concentration (MIC) using a series of dilutions (10-fold) of each chemical.39 In a microtiter plate, the various chemical concentrations were serially diluted. In each tube of the microtiter plate, 10 μL of standardized inoculum (1–2 × 107 cfu per mL) was introduced. Subsequently, the plates were incubated for 24 h aerobically at 37 °C. Compared to the wells containing the control, the lowest concentration of the compounds at which they illustrated no sign of bacterial growth and no turbidity in the solution was regarded as the MIC.
Molecular docking
The top selected compounds (10d and 9a) were subjected to molecular docking to understand the fundamental interactions of the compound with the target protein. For the docking studies, the Maestro Suite was used in the Schrodinger software. Docking studies were performed in four steps, as follows: (1) protein preparation, (2) ligand preparation, (3) grid generation, and (4) molecular docking. After molecular docking, the top potent compound (10d) complex was subjected to a molecular dynamics (MD) study.
Molecular dynamics (MD) study
Further, the molecular dynamics (MD) study of the selected compound (10d) was carried out by using the Desmond Module of the Schrodinger to find the protein–ligand (PL) complex stability.40 The PL complex was initially solvated for MD simulation studies with the single point charge solvent model, and the OPLS2005 force field was used. According to the system builder, the orthorhombic box was selected, and ions and a salt concentration were added to reach the physiological conditions. Next, the energy minimization of the PL complex was done. Then, the MD study was performed for 100 ns with 1.01 bar atmospheric pressure and a temperature of 310 K at the NPT ensemble.41
DNA gyrase assay
Escherichia coli was grown in 5 mL Luria broth (Himedia) at 37 °C and incubated for 24 h to determine gyrase activity. After incubation of 10 mg mL−1 ciprofloxacin, samples 9a and 10d 1 mg mL−1 were treated with bacterial culture, respectively. Bacterial cells were harvested after 24 h of incubation. A DNA easy (Blood and Tissue) kit from Qiagen (Cat. No. 69504) was used to isolate genomic DNA, and the manufacturer's protocol was followed to isolate the DNA from the control and treated strain. Isolated DNA was checked in 0.8% agarose gel (Ameresco, USA) against a 1 kb marker from Fermentas, USA. The DNA was quantified, and its purity was calculated at 260 nm and 280 nm absorbance using a NanoDrop Spectrometer (Thermo Fisher).
Method of cytotoxicity
The cytotoxicity of the compounds was evaluated in Vero cells (kidney epithelial cells of an African green monkey), using the MTT assay and their CC50 values were calculated. Assays were performed in 96-well microtiter plates, each well containing 100 μL of DMEM medium supplemented with 1% antibiotic solution and 10% fetal bovine serum, and 2 × 104 Vero cells. The cells were treated with varying concentrations (5, 10, 20, 50, 100, and 200 μg mL−1) of each derivative and incubated for 72 h at 37 °C in a CO2 incubator. After 72 h of incubation, the plates were inspected under an inverted microscope to ensure the growth of the controls and sterile conditions. Then, 10 μL of MTT reagent (5 mg MTT dissolved in 1 mL PBS) was added to each well, and the plates were incubated for 4 h in a cell culture incubator. The formazan crystals formed in each well were dissolved in 100 μL of MTT solubilizing agent (250 μL of 2 M HCl in methanol to 12.5 mL isopropanol) and the absorbance was measured at a wavelength of 590 nm using a SpectraMax iD3 multi-mode microplate reader. The cellular viability in terms of percentage cell survival was calculated as follows:
| % Cell survival = 100 × (average OD of treated cells)/(average OD of control cells). |
Conclusions
In total, 20 new synthetic fluoroquinolone 1,2,3-triazole analogues from two series were tested against four different antibacterial stains. Seven compounds from a small library pool, i.e., 7, 9i, 10(a–d), and 10i, showed excellent activity against E. coli (ATCC 25922) strains with MIC of 0.195 μg mL−1, while compounds 7, 9a, and 10(a–d) showed excellent activity against E. coli (clinical isolate), with a MIC of ≥0.195 μg mL−1 and 0.195 μg mL−1. Moreover, six compounds, i.e., 7, 9d, 9i, 10a, 10b, and 10d, showed excellent activity with MIC in the range of ≥0.195–0.195 μg mL−1 against E. faecalis (ATCC 29212), whereas the control drug, ciprofloxacin, showed an MIC of 0.781 μg mL−1. Two compounds, 10a and 10d, were found to be more active with an MIC of 6.25 μg mL−1 compared to the control drug, which showed an MIC of 12.5 μg mL−1. Further, it was found that electron-withdrawing groups such as OCF3, F, Cl, and Br in the benzene ring at the para-position played a significant role in defining the antibacterial activity. The cytotoxicity testing results showed that compounds 9a, 9c, 10g, 10h and 10i possess toxicity. It is necessary to analyze the structure–activity relationship (SAR) of potential lead molecules and lower their further effective doses while increasing their antibacterial action. Subsequently, the lead molecules from the synthesized compounds should be coupled with current clinical medications to create multifunctional hybrids. This approach will undoubtedly help to reduce the problem of bacterial drug resistance to a certain extent. Moreover, in silico studies were performed on the most potent compounds, and it was found that compound 10d showed the highest docking score of 7.0 kcal mol−1. Thus, due to its highest docking score, it was further subjected to MD analysis with E. coli DNA gyrase B complex protein for 100 ns. Also, the in vitro assay indicated that 10d was a more potent inhibitor of DNA gyrase compared to ciprofloxacin employed as a positive control. The single-crystal X-ray analysis of the compounds further confirmed their structure and design criteria. Extensive studies involving a large number of compounds are required to reach any meaningful conclusion in the future.
Data availability
All relevant data are included in the manuscript and its additional files. The data are available from the corresponding author on reasonable request.
Author contributions
Upendra Kumar Patel did the conceptualization, methodology, software, visualization, experimental work, and writing the main text. Alka did the formal analysis and IR data validation. Punit Tiwari did the antibacterial and hemolytic activity under the supervision of Ragini Tilak. Gaurav Joshi and Roshan Kumar did molecular docking studies, molecular dynamic (MD) analysis, DNA gyrase expression assay, software, and visualization. Alka Agarwal supervised, conceptualization, reviewed, and edited the original draft. All authors reviewed the manuscript.
Conflicts of interest
The authors have no conflict of interest regarding manuscript publication.
Acknowledgements
Upendra Kumar Patel is thankful to the Council of Scientific & Industrial Research (CSIR) HRDG New Delhi, India (Grant No. 09/013(0933)/2020-EMR-I) for the Junior Research Fellowship and Senior Research Fellowship. Alka is thankful to CSIR-UGC, New Delhi, India, for providing the Junior Research Fellowship and Senior Research Fellowship (Award Ref. No. 105/(CSIR-UGC NET JUNE 2019)). Alka Agarwal is thankful to Banaras Hindu University and the Institute of Eminence (IoE No. Dev Scheme No 6031), Varanasi, India, for the financial support. The authors are also thankful to CDC-Banaras Hindu University, India, for providing instrumentation facilities.
References
- P. Dadgostar, Antimicrobial Resistance: Implications and Costs, Infect. Drug Resist., 2019, 12, 3903–3910 CrossRef CAS PubMed.
- C. Llor and L. Bjerrum, Antimicrobial resistance: risk associated with antibiotic overuse and initiatives to reduce the problem, Ther. Adv. Drug Saf., 2014, 5, 229–241 CrossRef PubMed.
- L. Rigottier-Gois, C. Madec, A. Navickas, R. C. Matos, E. Akary-Lepage, M. Y. Mistou and P. Serror, The Surface RhamnopolysaccharideEpa of Enterococcus faecalis Is a Key Determinant of Intestinal Colonization, J. Infect. Dis., 2015, 211, 62–71 CrossRef CAS PubMed.
-
(a) Z. Chen, K. Song, Y. P. Shang, Y. P. Xiong, Z. H. Lyu, J. W. Chen, J. X. Zheng, P. Y. Li, Y. Wu, C. J. Gu, Y. H. Xie, Q. W. Deng, Z. J. Yu, J. Zhang and D. Qu, Selection and Identification of Novel Antibacterial Agents against Planktonic Growth and Biofilm Formation of Enterococcus faecalis, J. Med. Chem., 2021, 64, 15037–15052 CrossRef CAS PubMed;
(b) S. Manoharadas, M. Altaf, N. Ahmad, A. F. Alrefaei and B. F. Al-Rayes, Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis, Antibiotics, 2023, 12, 388 CrossRef CAS PubMed.
-
(a) X. M. Zhou, Y. Y. Hu, B. Fang and C. H. Zhou, Benzenesulfonylthiazoloimines as unique multitargeting antibacterial agents towards Enterococcus faecalis, Eur. J. Med. Chem., 2023, 248, 115088 CrossRef CAS PubMed;
(b) A. A. Iannetta, N. E. Minton, A. A. Uitenbroek, J. L. Little, C. R. Stanton, C. J. Kristich and L. M. Hicks, IreK-Mediated, Cell Wall-Protective Phosphorylation in Enterococcus faecalis, J. Proteome Res., 2021, 20, 5131–5144 CrossRef CAS PubMed.
- C. Muller, S. Massier, Y. L. Breton and A. Rince, The role of the CroR response regulator in resistance of Enterococcus faecalis to D-cycloserine is defined using an inducible receiver domain, Mol. Microbiol., 2018, 107, 416–427 CrossRef CAS PubMed.
- E. Ruppe, P.-L. Woerther and F. Barbier, Mechanisms of antimicrobial resistance in Gram-negative bacilli, Ann. Intensive Care, 2015, 5, 21–36 CrossRef PubMed.
- World Health Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, 2017(WHO/EMP/IAU/2017.12), WHO, Geneva, Switzerland, 2017 Search PubMed.
- J. A. Mohamed and D. B. Huang, Biofilm formation by enterococci, J. Med. Microbiol., 2007, 56, 1581–1588 CrossRef CAS PubMed.
- O. Tenaillon, D. Skurnik, B. Picard and E. Denamur, The population genetics of commensal Escherichia coli, Nat. Rev. Microbiol., 2010, 8, 207–217 CrossRef CAS PubMed.
- E. Jimenez, M. L. Marin, R. Martin, J. M. Odriozola, M. Olivares, J. Xaus, L. Fernandez and J. M. Rodriguez, Is meconium from healthy newborns actually sterile?, Res. Microbiol., 2008, 159, 187–193 CrossRef CAS PubMed.
- M. I. Andersson and A. P. MacGowan, Development of the quinolones, J. Antimicrob. Chemother., 2003, 51, 1–11 CrossRef CAS PubMed.
- A. L. Aguirre, P. R. Chheda, S. R. C. Lentz, H. A. Held, N. P. Groves, H. Hiasa and R. J. Kerns, Identification of an ethyl 5,6-dihydropyrazolo[1,5-c]quinazoline-1-carboxylate as a catalytic inhibitor of DNA gyrase, Bioorg. Med. Chem., 2020, 28, 115439 CrossRef CAS PubMed.
- E. N. Esfahani, M. Mohammadi-Khanaposhtani, Z. Rezaei, Y. Valizadeh, R. Rajabnia, M. Hassankalhori, F. Bandarian, M. A. Faramarzi, N. Samadi, M. R. Amini, M. Mahdavi and B. Larijani, New ciprofloxacin–dithiocarbamate–benzyl hybrids: design, synthesis, antibacterial evaluation, and molecular modeling studies, Res. Chem. Intermed., 2019, 45, 223–236 CrossRef CAS.
- Z. Xu, S. J. Zhao, Z. S. Lv, F. Gao, Y. Wang, F. Zhang, L. Bai and J. L. Deng, Fluoroquinolone-isatin hybrids and their biological activities, Eur. J. Med. Chem., 2019, 162, 396–406 CrossRef CAS PubMed.
- R. J. Reece and A. Maxwell, DNA gyrase: structure and function, Crit. Rev. Biochem. Mol. Biol., 1991, 26, 335–375 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, The growing impact of click chemistry on Drug Discovery, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Click Chemistry: 1,2,3-Triazoles as Pharmacophores, Chem.–Asian J., 2011, 6, 2696 CrossRef CAS PubMed.
- R. Huisgen, G. Szeimies and L. Mobius, 1,3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionenorganischer Azide an CC-Mehrfachbindungen, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- A. Massarotti, S. Aprile, V. Mercalli, E. D. Grosso, G. Grosa, G. Sorba and G. C. Tron, Are 1,4- and 1,5-disubstituted 1,2,3-triazoles good pharmacophoric groups?, ChemMedChem, 2014, 9, 2497–2508 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Click Chemistry: 1,2,3-Triazoles as Pharmacophores, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- D. Gonzalez-Calderon, M. G. Mejía-Dionicio, M. A. Morales-Reza, A. Ramírez-Villalva, M. Morales-Rodríguez, B. Jauregui-Rodríguez, E. Díaz-Torres, C. Gonzalez-Romero and A. Fuentes-Benítes, Azide-enolate 1,3-dipolar cycloaddition in the synthesis of novel triazole-based miconazole analogues as promising antifungal agents, Eur. J. Med. Chem., 2016, 112, 60–65 CrossRef CAS PubMed.
- R. Kant, V. Singh, G. Nath, S. K. Awasthi and A. Agarwal, Design, synthesis and biological evaluation of ciprofloxacin tethered bis-1,2,3-triazole conjugates as potent antibacterial agents, Eur. J. Med. Chem., 2016, 124, 218–228 CrossRef CAS PubMed.
- M. K. Singh, R. Tilak, G. Nath, S. K. Awasthi and A. Agarwal, Design, synthesis and antimicrobial activity of novel benzothiazole analogs, Eur. J. Med. Chem., 2013, 63, 635–644 CrossRef CAS PubMed.
- A. Agarwal, P. Singh, A. Maurya, U. K. Patel, A. Singh and G. Nath, Ciprofloxacin-Tethered 1,2,3-Triazole Conjugates: New Quinolone Family Compounds to Upgrade Our Antiquated Approach against Bacterial Infections, ACS Omega, 2022, 7, 2725–2736 CrossRef CAS PubMed.
- U. K. Patel, P. Tiwari, R. Tilak, G. Joshi, R. Kumar and A. Agarwal, Synthesis of ciprofloxacin-linked 1,2,3-triazole conjugates as potent antibacterial agents using click chemistry: exploring their function as DNA gyrase inhibitors via in silico- and in vitro-based studies, RSC Adv., 2024, 14, 17051 RSC.
- F. Ushiyama, H. Amada, Y. Mihara, T. Takeuchi, N. Tanaka-Yamamoto, M. Mima, M. Kamitani, R. Wada, Y. Tamura, M. Endo, A. Masuko, I. Takata, K. Hitaka, H. Sugiyama and N. Ohtake, Lead optimization of 8-(methylamino)-2-oxo-1,2-dihydroquinolines as bacterial type II topoisomerase inhibitors, Bioorg. Med. Chem., 2020, 28, 115776 CrossRef CAS PubMed.
- P. Price and T. J. McMillan, Use of the tetrazolium assay in measuring the response of human tumor cells to ionizing radiation, Cancer Res., 1990, 50, 1392–1396 CAS.
- P. Singh, C. Sharma, B. Sharma, A. Mishra, D. Agarwal, D. Kannan, J. Held, S. Singh and S. K. Awasthi, N-sulfonylpiperidinedispiro-1,2,4,5-tetraoxanes exhibit potent in vitro antiplasmodial activity and in vivo efficacy in mice infected with P. berghei ANKA, Eur. J. Med. Chem., 2022, 15, 114774 CrossRef PubMed.
- H. Koga, A. Itoh, S. Murayama, S. Suzue and T. Irikura, Structure-activity relationships of antibacterial 6,7- and 7,8-disubstituted 1-alkyl-1,4-dihydro-4-oxoquinoline-3-carboxylic acids, J. Med. Chem., 1980, 23, 1358–1363 CrossRef CAS PubMed.
- S. K. Dixit, N. Mishra, M. Sharma, S. Singh, A. Agarwal, S. K. Awasthi and V. K. Bhasin, Synthesis and in vitro antiplasmodial activities of fluoroquinolone analogs, Eur. J. Med. Chem., 2012, 51, 52–59 CrossRef CAS PubMed.
- J. C. Mc Pherson III, R. Runner, T. B. Buxton, J. F. Hartmann, D. Farcasiu, I. Bereczki, E. Roth, S. Tollas, E. Ostorházi, F. Rozgonyi and P. Herczegh, Synthesis of osteotropichydroxybisphosphonate derivatives of fluoroquinolone antibacterials, Eur. J. Med. Chem., 2012, 47, 615–618 CrossRef PubMed.
- M. Hu, J. Li and S.-Q. Yao, In situ “click” assembly of small molecule matrix metalloprotease inhibitors containing zinc-chelating groups, Org. Lett., 2008, 10, 5529–5531 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, iotbx.cif: a comprehensive CIF toolbox, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT-Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A, 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, WinGX and ORTEP for Windows: An Update, J. Appl. Crystallogr., 2012, 4, 5849–5854 Search PubMed.
- S. F. Nielsen, M. Larsen, T. Boesen, K. Schønning and H. Kromann, Cationic Chalcone Antibiotics. Design, Synthesis, and Mechanism of Action, J. Med. Chem., 2005, 48, 2667–2677 CrossRef CAS PubMed.
- P. Vishwakarma, N. F. Siddiqui, S. Thakur and H. Jadhav, FDA approved fused-pyrimidines as potential PI3K inhibitors: a computational repurposing approach, J. Biomol. Struct. Dyn., 2023, 1–18, DOI:10.1080/07391102.2023.2276315.
- S. A. Hollingsworth and R. O. Dror, Molecular Dynamics Simulation for All, Neuron., 2018, 99, 1129–1143 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Alka
a,
Alka