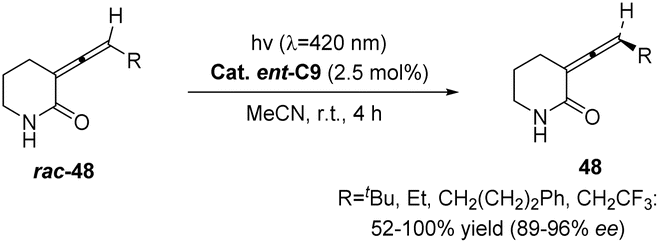DOI:
10.1039/D4RA08581G
(Review Article)
RSC Adv., 2025,
15, 2874-2889
Asymmetric photoreactions catalyzed by chiral ketones
Received
5th December 2024
, Accepted 9th January 2025
First published on 30th January 2025
Abstract
Asymmetric catalytic reactions are essential for synthesizing chiral drugs and fine chemicals, with their stereoselectivity influenced significantly by interactions between catalysts and substrates. Ketone catalysts have garnered considerable attention in the realm of asymmetric photoreactions because of their highly controllable structures, ease of availability, and environmental friendliness. This review highlights the application of various reported ketone catalysts in a range of asymmetric photoreactions, including [2 + 2] photocycloaddition, photoderacemization, photochemical rearrangement, asymmetric electrophilic amination, and asymmetric alkylation of aldehydes. This review discusses the design concepts, catalytic mechanisms, as well as the advantages and limitations of these catalysts within each reaction. The aim is to provide valuable insights for developing more effective asymmetric catalytic systems.
1. Introduction
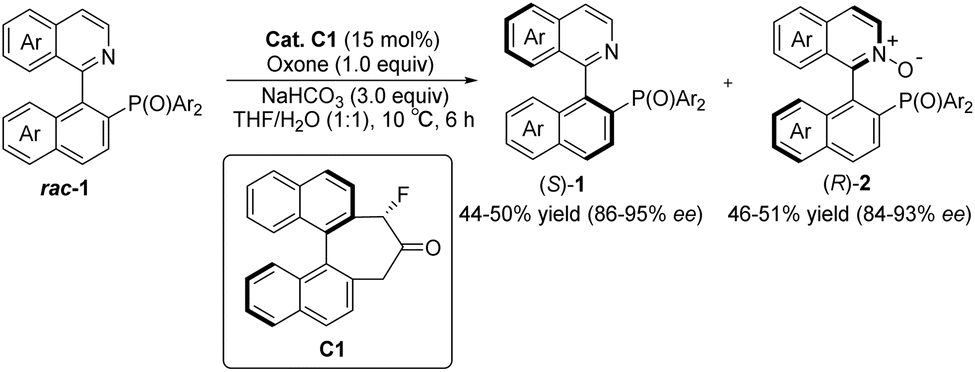
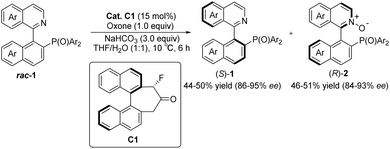
Asymmetric catalytic reactions play an important role in modern synthetic organic chemistry. To achieve asymmetric catalytic reactions, designing chiral catalysts with high enantioselectivity and universality is crucial. Before the introduction of the concept of organic catalysis, ketone-based catalysts emerged as one of the earliest investigated types among small molecule catalysts. They garnered extensive attention because of their cost-effectiveness, ready availability, eco-friendliness, and remarkable structural controllability.1–7 The carbonyl group of ketone catalysts can undergo oxidation to form dioxiranes that serve as mild and selective oxidants. Leveraging this characteristic, in conjunction with precise spatial structure regulation, ketone catalysts find extensive applications in the asymmetric epoxidation of olefins. Dioxiranes in these catalysts have two directions for oxygen transfer when approaching olefins, both of which can be approached by olefins. Rationally designing the structure of a catalyst to control the reaction's direction with regard to the substrate is a challenge in studying this type of reaction. The design of catalysts in asymmetric epoxidation using the oxidizability of ketone catalysts has been covered in detail in numerous high-quality reviews.8–12 These studies primarily focused on the period prior to 2015; therefore, this review does not delve into a detailed discussion on research pertaining to ketone catalysts in asymmetric epoxidation. Additionally, among the ketone catalysts used in asymmetric epoxidation reactions, the sugar-derived ketones developed by Shi's research group are the most widely used.13 This type of catalyst has been successfully used in the synthesis of more than 20 key drug intermediates, such as KRAS G12C inhibitors, pradilolide A, and aplysiasecosterols A. As of August 2024, the relevant literature continues to increase annually and has a wide range of application values.14–38 In addition, Tan et al. recently reported a ketone-catalyzed reaction for the enantioselective oxidation of N atoms that can be applied effectively to the enantioselective kinetic resolution of QUINAPO and related compounds.39 The two highly enantiomeric QUINAPO N-oxide products can be converted easily to QUINAP without losing chiral integrity (Scheme 1). This is the first report of chiral ketone catalysts for the enantioselective oxidation of N atoms, and it is anticipated that there will be significant prospects for future research.
 |
| | Scheme 1 Enantioselective oxidation of N atoms catalyzed by ketone C1. | |
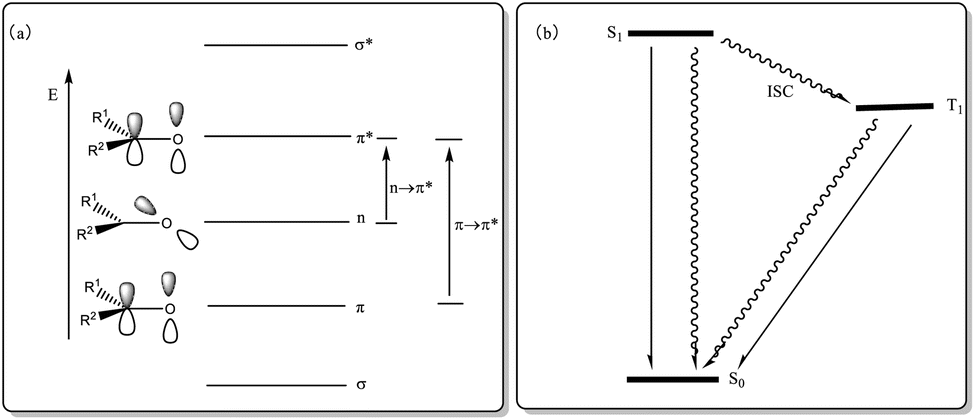
In addition to utilizing the oxidation characteristics of the ketone structure for designing ketone catalysts in catalytic reactions, ketone compounds can undergo np–π* and π–π* transitions because their oxygen atoms have two pairs of lone electrons in nonbonding orbitals (occupying two nondegenerate orbitals, np and nsp, respectively). The intersystem crossing (ISC) process can occur in S1 (np, π*) and T1 (π, π*), making it possible to utilize their optical properties for designing photosensitizers (PSs) that catalyze photochemical reactions (Fig. 1). The direct excitation of photoreactions with low absorption coefficients via light absorption is challenging, as it may lead to competitive processes and unnecessary side reactions; however, using a sensitizer allows for absorbing light and subsequently transferring excitation energy to the substrate at a lower level, thus enhancing chemical selectivity. Hence, the sensitization method offers distinct advantages. Sensitization is a classic mode of photocatalysis, with triplet sensitization being the most commonly used in organic synthesis. Triplet PSs are compounds that can be effectively excited to the triplet excited state and act as catalysts in photochemical reactions. A triplet PS meeting these conditions should exhibit the following characteristics: (1) strong absorption of excitation light; (2) higher energy than the receptor; (3) high-yield ISC for efficiently producing triplet states; (4) long triplet lifetimes to complete energy transfer.40,41 The organic ketone molecule acts as a photocatalyst, absorbing energy and transitioning to S1. Through the ISC process, S1 is converted to T1. At this point, the PS in the T1 state interacts with the substrate, providing energy for the substrate to undergo a π–π* transition while the ketone PS returns to its original state (S0) to complete the catalytic cycle (Fig. 1). Compared with aliphatic ketones,42 aromatic ketones have picosecond ISC, while alkyl ketones have nanosecond ISC, which means that most ketone PSs are aromatic.
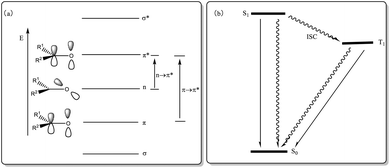 |
| | Fig. 1 (a) Electronic energy level and electronic transition of carbonyl group; (b) ISC process diagram of S1 and T1. | |
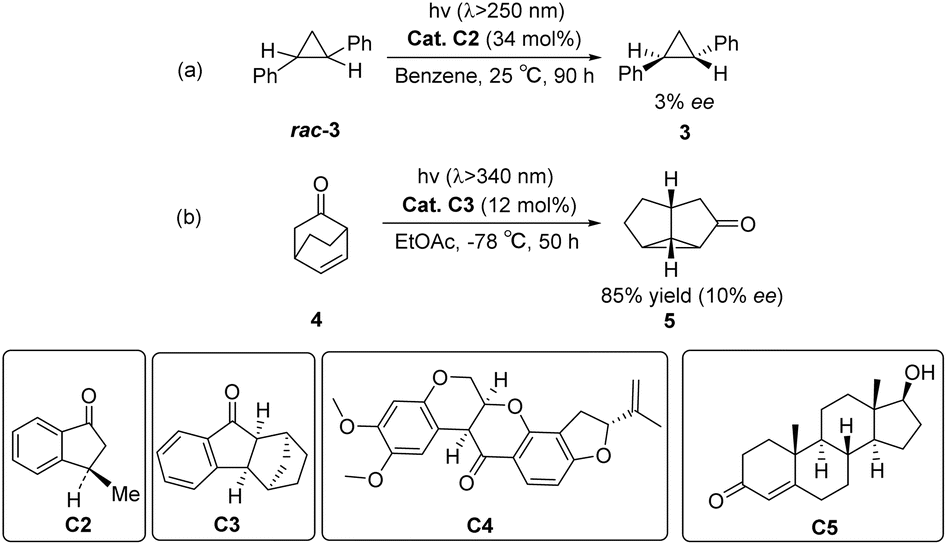
In 1973, Ouannès et al. pioneered the utilization of the triplet sensitizer (R)-(−)-3-methyl-1-indanone C2 to catalyze the enantioselective isomerization of a substrate. The earliest example involves the conversion of racemic trans-diphenylcyclopropane rac-3 (DPC) into cis-DPC 3, albeit with a modest ee value of only 3% (Scheme 2(a)).43 The reason for the enantioselectivity may be due to the kinetic difference between the two enantiomers of racemic DPC during the energy transfer process from the triplet state. In 1980, Demuth et al. carried out photorearrangement of β,γ-unsaturated ketone 4 by chiral indanone sensitizer C3, and the yield of the isolated product 5 was 80–90%, and the ee value was 10% (Scheme 2(b)).44 Subsequently, Rau et al. subjected the racemic mixture of optically active trans-3,5-diphenylpyrazoline to photolysis using (−)-rotenone C4 and (+)-testosterone C5 as sensitizers (Scheme 2). In comparison with the direct photolysis of diphenylpyrazoline under circularly polarized light, rotenone sensitization resulted in a 150% increase in reaction asymmetry; however, it still failed to provide effective enantioselectivity.45 The reported PSs in the early stage exhibit sufficient ISC rates for substrate sensitization and reaction initiation; however, the resulting enantioselectivity remains significantly low. This could potentially be attributable to the rapid dissociation of the sensitizer–substrate complex upon excitation, thus hindering the well-defined chiral environment necessary for subsequent reactions. At the beginning of the 21st century, Bach designed aromatic ketone-type photocatalysts for asymmetric [2 + 2] photocycloaddition reactions46 and photoderacemization reactions.47 The application of ketones as PSs in the field of asymmetric photocatalysis has ushered in new developments. Currently, ketone-type asymmetric photocatalysts are mainly classified into three categories: diphenyl ketone-type photocatalysts, xanthone-type photocatalysts, and thioxanthone-type photocatalysts. These ketone catalysts have been widely applied in various asymmetric reactions, including asymmetric [2 + 2] photodimerization, photodecarboxylation, asymmetric electrophilic amination, asymmetric hydroxylation, and asymmetric alkylation of aldehydes. Although there are several reviews on asymmetric photocatalytic reactions in the literature,48,49 this article specifically focuses on the design strategies for ketone-type catalysts used in these reactions. The synergistic effects between the PS unit and the chiral control unit are explored, providing insights into how the optimization of the design of these two components can significantly enhance the efficiency and selectivity of asymmetric photocatalytic reactions. Notably, this specific aspect of design strategies for ketone catalysts in asymmetric photochemical reactions has not been discussed in depth in the existing literature. Therefore, we contend that our review offers a valuable contribution to the current body of knowledge, providing new perspectives for developing catalysts in asymmetric light reactions. Additionally, Table 1 provides a summary of the types of reactions and substrates that are compatible with various catalysts, along with their corresponding enantiomeric excess (ee) values and yields.
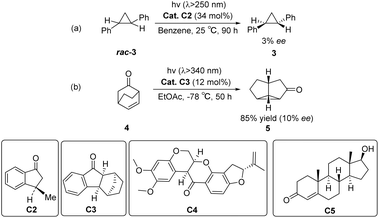 |
| | Scheme 2 Early exploration of chiral aromatic ketones as sensitizers in photocatalytic reactions. | |
Table 1 Types and effects of photoreactions catalyzed by chiral ketones
| Cat. |
Reaction type |
λ (nm) |
Substrate characteristics |
Example |
ee (%) |
Yield (%) |
Ref. |
| C1 |
Kinetic resolution |
— |
QUINAPO |
18 |
44–50 |
86–95 |
13 |
| C2 |
Asymmetric isomerization reaction |
>250 |
Indanones |
1 |
3 |
— |
43 |
| C3 |
Asymmetric photorearrangement reaction |
>340 |
Unsaturated ketones |
2 |
10–13 |
80–90 |
44 |
| C4–C5 |
Asymmetric photolysis reaction |
315 |
Trans-3,5-diphenylpyrazoline |
2 |
<5 |
— |
45 |
| C6 |
Intramolecular [2 + 2] photocycloaddition reaction |
>310 |
Olefins and enones |
1 |
19 |
100 |
50 |
| C7 |
>300 |
α-Aminoalkyl and ketene |
1 |
70 |
64 |
46 |
| ent-C7 |
Photoderacemization reaction |
366 |
5-Substituted 3-phenylimidazolidine-2,4-dione |
27 |
81–99 |
69–99 |
51 |
| 3-Substitution indolone |
13 |
79–99 |
83–96 |
52 |
| C8 |
Intramolecular [2 + 2] photocycloaddition reaction |
366 |
2-Quinolone and intramolecular double bond |
6 |
6–90 |
21–75 |
53 |
| Asymmetric photorearrangement reaction |
366 |
Spirooxindole epoxide |
8 |
16–33 |
85–98 |
54 |
| Photoderacemization reaction |
366 |
Benzothiazinone-1-oxide |
5 |
20–55 |
52–95 |
55 |
| ent-C8 |
Intermolecular [2 + 2] photocycloaddition reaction |
>366 |
2-Pyridone and acetylene dicarboxylate |
12 |
59–92 |
40–88 |
56 |
| C9 |
Intramolecular [2 + 2] photocycloaddition reaction |
420 |
Quinolones and double bonds |
18 |
87–99 |
79–99 |
57,58 |
| Intermolecular [2 + 2] photocycloaddition reaction |
419 |
Quinolones and electron – deficient olefins |
12 |
80–95 |
44–94 |
59 |
| Photoderacemization reaction |
420 |
Racemic alkylidenecyclobutane and alkylidenecyclopentane |
16 |
81–96 |
51–99 |
60 |
| Di-π-methane rearrangement reaction |
420 |
Substituted quinolone |
9 |
32–55 |
88–96 |
61 |
| ent-C9 |
Intermolecular Paternò–Büchi reaction |
420 |
Quinoxalinones and olefins |
13 |
86–98 |
50–99 |
62 |
| Photoderacemization reaction |
420 |
Racemic 3-(1′-vinylidene)piperidin-2-one |
17 |
89–97 |
52–99 |
63 |
| 420 |
3-(1′-Alkenylmethylene)pyrrolidin-2-one |
13 |
86–98 |
60–96 |
64 |
| 420 |
Acrylamide |
19 |
70–93 |
74–99 |
65 |
| C10 |
Photocyclization |
419 |
2-(4-Bromophenoxy)-3,5,5-trimethylcyclohex-2-enone |
1 |
12 |
26 |
66 |
| C11 |
Intermolecular [2 + 2] photocycloaddition reaction |
437 |
β-Carboxyl substituted cycloenones and alkenes |
5 |
46–85 |
26–61 |
67 |
| 459 |
N,O-Acetals and olefins |
10 |
83–95 |
54–93 |
68 |
| C12 |
Intramolecular [2 + 2] photocycloaddition reaction |
405 |
Quinolones and olefins |
3 |
91–97 |
88–95 |
69 |
| Intermolecular [2 + 2] photocycloaddition reaction |
4 |
73–98 |
24–99 |
| C13 |
Intermolecular [2 + 2] photocycloaddition reaction |
393 |
Quinoxalinone and styrene |
8 |
75–95 |
46–90 |
70 |
| C14 |
Asymmetric electrophilic amination reaction |
420–510 |
Diazene and enamine |
15 |
91–99 |
30–95 |
71 |
| C15 |
405 |
Alkenylcarbamate |
5 |
95–99 |
53–66 |
72 |
| C16 |
Asymmetric alkylation reaction |
>400 |
Aldehyde |
13 |
84–99 |
55–99 |
73 |
| C17 |
Asymmetric α-hydroxylation reaction |
>400 |
β-Ketoesters and β-ketoamides |
21 |
85–95 |
78–98 |
74 |
2. Benzophenone-type photocatalysts
Early exploration of photocatalytic reactions using chiral aromatic ketones as sensitizers has shown promising yields;44 however, the ee values remain relatively low. This could be attributable to the rapid dissociation of the sensitizer from the excited substrate after sensitization, leading to inadequate control over stereoselectivity and resulting in near-zero or very low enantioselectivity; therefore, controlling the reaction direction of the substrate to increase the ee value is crucial for achieving efficient asymmetric photocatalytic reactions. This goal can be achieved by introducing a chiral control unit into the structure of the PS, thus effectively regulating the selectivity of the PS regarding the substrate reaction direction when transmitting energy. Benzophenone was initially applied to this concept because of its good light absorption ability and potential to decompose into free radicals under light conditions, allowing it to exhibit a variety of roles in different light reactions.
2.1 Asymmetric [2 + 2] photocycloaddition reactions
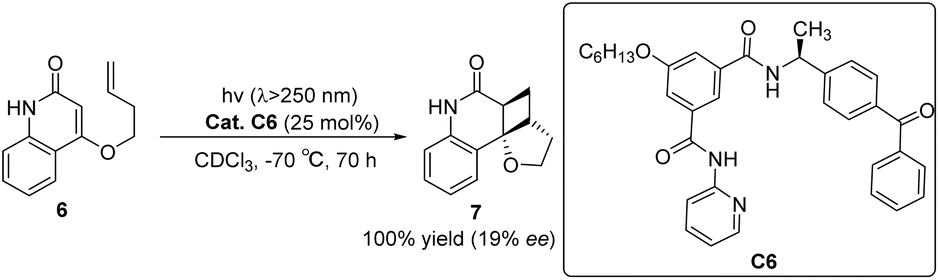
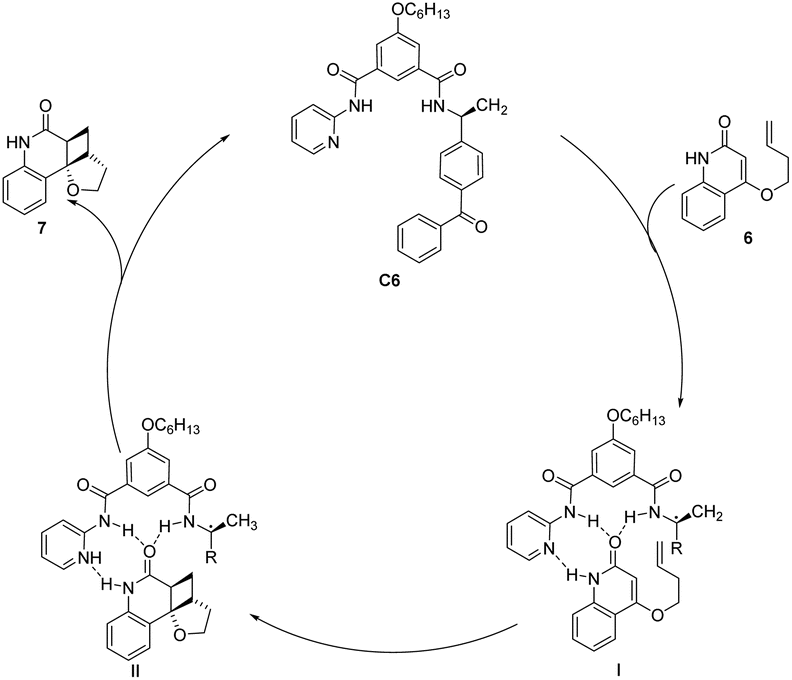
The asymmetric [2 + 2] photocycloaddition reaction is a crucial strategy for constructing optically active cyclobutane fragments found in natural products. In 2003, Krische developed a [2 + 2] asymmetric cycloaddition reaction catalyzed by ketone C6; however, the ee value was only 19% (Scheme 3).50 The reaction mechanism is shown in Fig. 2. Catalyst C6 and quinolone 6 form complex I. Due to the distance dependence of energy transfer, the cycloaddition reaction only occurs on the I complex to control the formation of chirality and produce the II complex. Finally, the cycloadduct 7 dissociates, and ketone C6 is regenerated to complete the catalytic cycle. The catalyst consists of two parts. The diphenyl ketone part is used as a sensitizing unit to transfer energy to the substrate to promote the reaction, and the other part has many N atoms that drive the substrate to form hydrogen bonds and control the spatial position of the substrate; however, the ee value obtained by the authors is not high, which may be due to the fact that the interaction between the single enantiomers of the catalyst and the substrate does not have the advantage of photoreaction kinetics. Benzophenone has a low triplet state energy and a short absorption wavelength that can lead to low sensitization efficiency. Furthermore, before the cyclization step of the sensitized substrate is completed, the catalyst separates from the reactant, resulting in most of the substrate not being in a chiral environment, thus reducing the enantiomeric selectivity of the products.
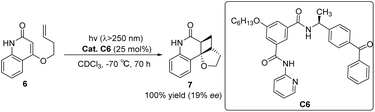 |
| | Scheme 3 Ketone C6 catalyzed asymmetric [2 + 2] photocycloaddition of ketenes. | |
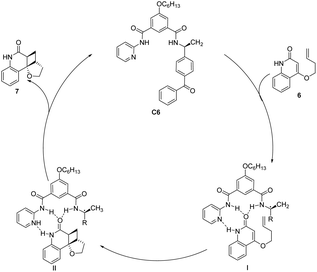 |
| | Fig. 2 Mechanism of asymmetric photo [2 + 2] cycloaddition of ketone C6 to quinolone. | |
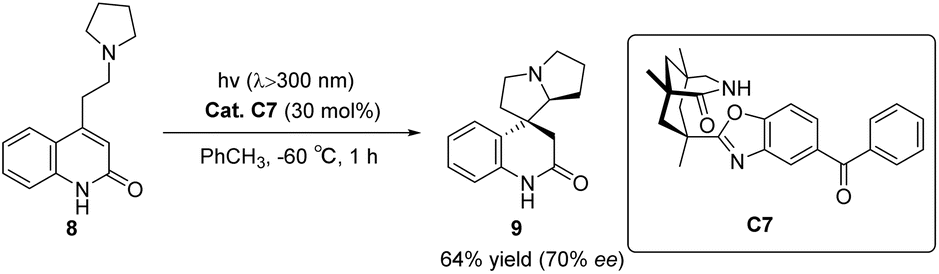
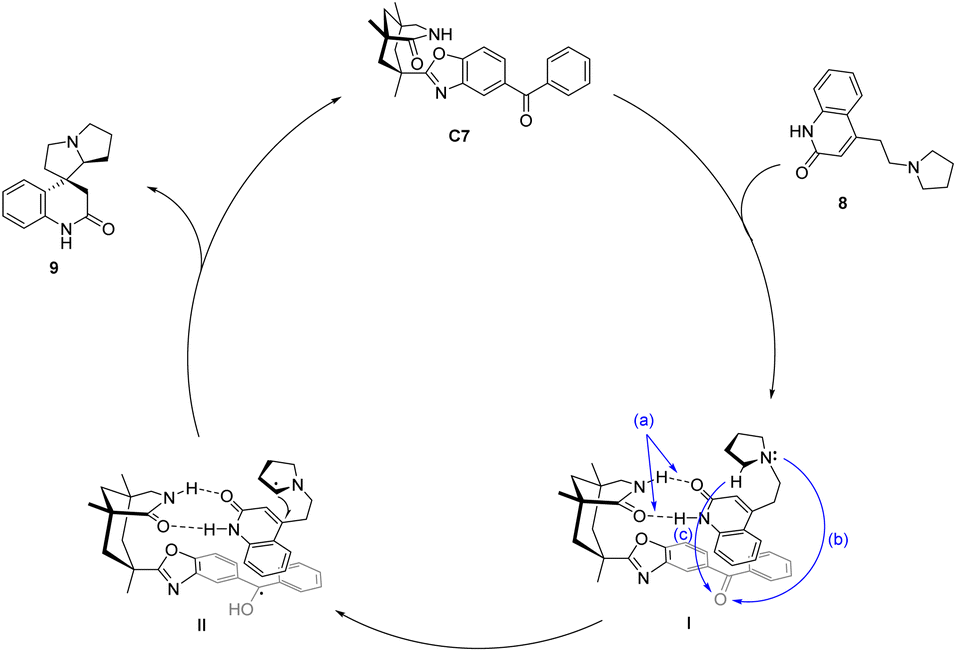
In 2005, Bach designed ketone C7 for the asymmetric catalytic conjugate addition of α-aminoalkyl and ketene and achieved a significant breakthrough in the field of asymmetric photocatalysis.46 A chiral spirocyclic pyrrolizidine compound was synthesized through the utilization of a 30 mol% catalyst C7 for substrate catalysis. The resulting yield was determined to be 64%, accompanied by an ee value of 70% (Scheme 4). The catalyst's design concept bears a resemblance to that proposed by Krische.50 By utilizing hydrogen bonding, the substrate's spatial position is immobilized, thus establishing a chiral environment. Subsequently, introducing an aromatic ketone sensitizer endows the catalyst with the capability to facilitate energy or electron transfer. This mechanism is visually depicted in Fig. 3.34 The catalyst controls the spatial position through hydrogen bonds (shown as “a” in Fig. 3), and when activated by light, the benzophenone group accepts an electron from the N atom in a pyrrolidine (shown as “b” path in Fig. 3). The transfer of electron-deficient nitrogen ortho-hydrogen protons to benzophenone radical anions (shown as the “c” path in Fig. 3) and the remaining free radicals undergo intramolecular cyclization. The hydrogen initially transferred to benzophenone is returned to the carbon radical of the product to complete the reaction cycle. Ketone C7 not only promotes the required enantiomeric differentiation but also acts as a photooxidant.
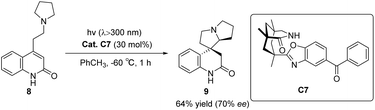 |
| | Scheme 4 Ketone C7 catalyzed asymmetric conjugate addition of α-aminoalkyl to ketenes. | |
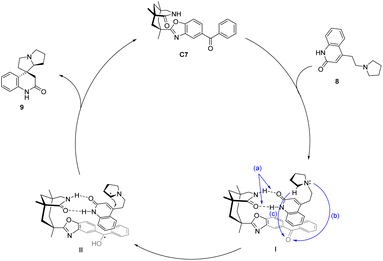 |
| | Fig. 3 Mechanism of asymmetric conjugate addition of α-aminoalkyl and ketene catalyzed by xanthone C7. | |
2.2 Photoderacemization reactions
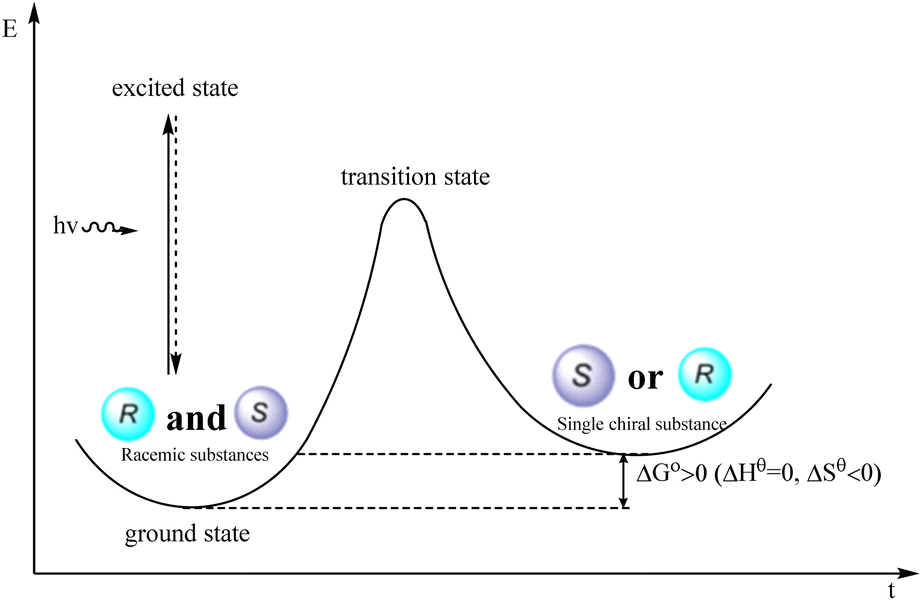
The deracemization reaction pertains to the transformation in which the quantity of one enantiomer is augmented while the quantity of the other enantiomer is diminished, resulting in the conversion of a racemic mixture into a nonracemic form.75 This process involves reducing the entropy, necessitating energy expenditure. Thermodynamic approaches are incapable of transforming racemates into enantiomers under equilibrium conditions; however, the photochemical deracemization reaction effectively surmounts the kinetic obstacles and thermodynamic difficulties encountered in chemical conversion. Through this reaction, the racemic compound is converted directly into its enantiomer, resulting in the acquisition of a singular chiral compound while compensating for the entropy loss incurred during the energy compensation process. Additionally, optical racemization exhibits a 100% atomic efficiency, surpassing the traditional resolution process, which can only achieve a maximum enantiomeric yield of 50%, resulting in a wastage of 50% of the enantiomer. Hence, it possesses distinct advantages compared with conventional kinetic resolution (Fig. 4).
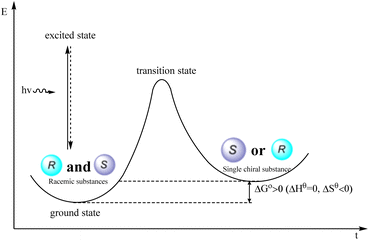 |
| | Fig. 4 Thermodynamic process of photocatalytic deracemization reaction. | |
In 2021, Bach proposed a new photochemical deracemization method based on the selective hydrogen atom transfer (HAT) of photocatalysts. Using the chiral diarylketone photocatalyst ent-C7 to catalyze the photoderacemization of rac-10 (Scheme 5),51 it should be noted that the reaction occurs through selective HAT rather than ent. From this principle, it is not difficult to find that other compounds with two hydrogen bonding sites may also be deracemized by the catalyst ent-C7, and its application range needs to be further explored.
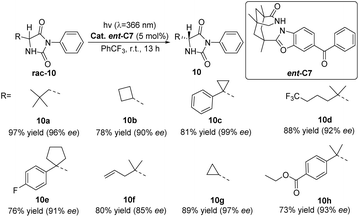 |
| | Scheme 5 The photochemical deracemization of 5-substituted 3-phenylimidazolidine-2,4-dione (hydantoin) catalyzed by ent-C7. | |
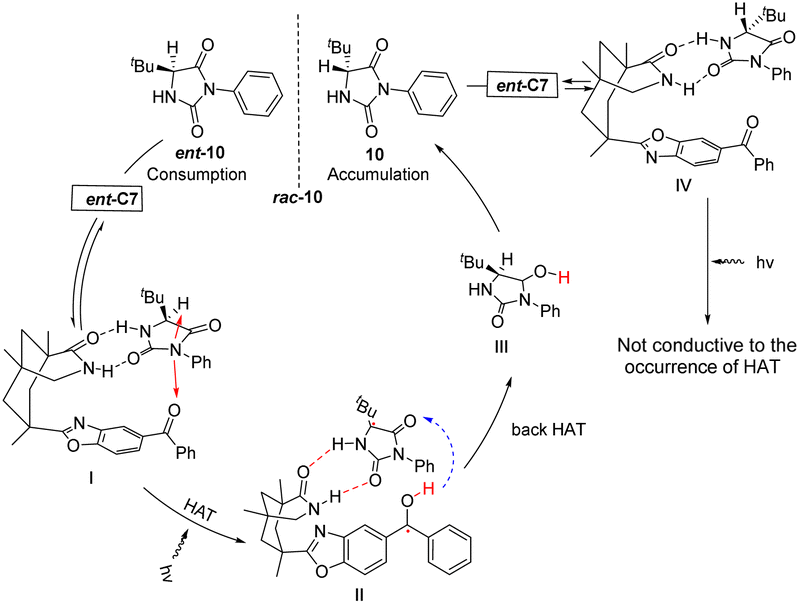
Bach provided a more detailed elucidation in 2023 on the racemization mechanism of chiral imidazolidine-2,4-dione (hydantoin) catalyzed by chiral benzophenone ent-C7.76 Furthermore, they once again confirmed that the catalytic cycle is achieved through reversible HAT. The catalyst was confirmed to bind two enantiomers through two hydrogen bonds using nuclear magnetic resonance titration. Specifically, I forms a ground state conformation through these hydrogen bonds. The distance between the carbonyl oxygen atom and the hydrogen atom in the stereocenter is very close, leading to HAT upon excitation. Subsequently, under photoexcitation, benzophenone extracts the hydrogen atom from the sp3 hybrid carbon center, resulting in the breaking of the C–H bond via a nonlinear transition state to form an achiral radical known as II. Enol intermediate III is generated by back HAT, followed by a tautomerization reaction that produces two enantiomers; however, only 10 returns to the catalytic cycle while ent-10 is depleted continuously. As a result, enantiomer 10 becomes increasingly enriched. In complex IV, the hydrogen atom is positioned on the opposite side of the 3-phenylimidazolidine-2,4-dione ring, an inaccessible location (Fig. 5). The keto radical intermediate was identified through transient absorption spectroscopy, and it was further confirmed that HAT exclusively occurred on a single enantiomer, thus precluding the aforementioned process. Consequently, this led to the accumulation of 10 chiral compounds.
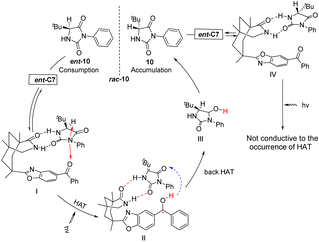 |
| | Fig. 5 Mechanism of deracemization of rac-10 catalyzed by ketone ent-C7. | |
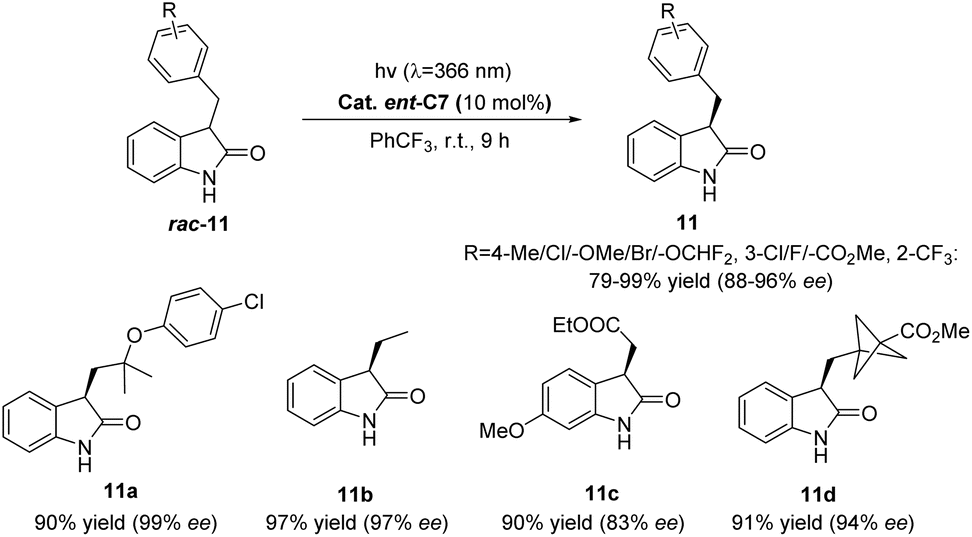
Subsequently, Bach reported that catalyst ent-C7 (10 mol%) could catalyze the deracemization of racemic 3-substituted oxindoles (Scheme 6),52 which occurred through reversible HAT in the substrate–catalyst complex. In addition, this method is characterized by its ability to predictably edit the chiral center at the C3 position of racemic 3-substituted oxindoles (rac-11), and the light energy compensates for the related entropy loss, which has important theoretical and application value for drug synthesis that requires controllable editing of a chiral center.
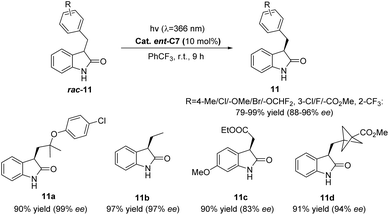 |
| | Scheme 6 Deracemization of racemic 3-substituted oxindoles catalyzed by ent-C7. | |
3. Xanthone-type photocatalysts
Although benzophenone possesses the basic characteristics of a PS, its relatively low triplet state energy limits its catalytic performance as a photocatalyst, resulting in low catalytic efficiency and selectivity; therefore, optimizing its structure to design photocatalysts with better photochemical reactivity and enantioselectivity has become a key area of focus.
3.1 Asymmetric [2 + 2] photocycloaddition reactions
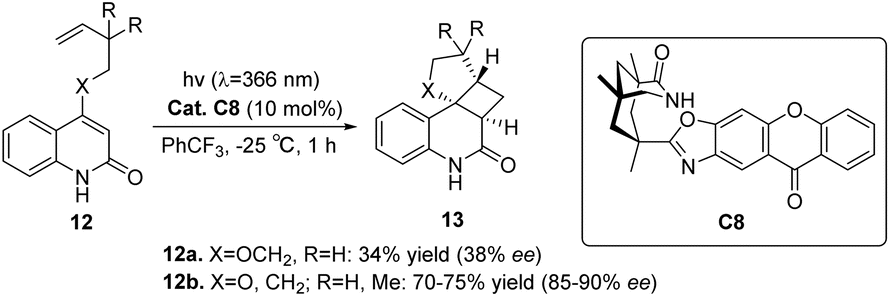
Considering that both ketone C7 and ketone C6 are diphenyl ketone derivatives, is it plausible to assume that ketone C7 can also catalyze the intramolecular asymmetric [2 + 2] photocycloaddition reaction, similar to ketone C6. A study conducted by Bach revealed that the reaction rate facilitated by ketone C7 is superior to that facilitated by ketone C6, albeit the enantioselectivity remains considerably low. This can be attributed to the relatively low triplet energy and short wavelength absorption of benzophenone C7, resulting in poor enantioselectivity (39% ee). Consequently, the catalyst structure was optimized to design a xanthone-type catalyst C8. The bicyclic lactam unit structure in ketone C8 can be pre-combined with the substrate to ensure that the sensitizing part is close to the substrate chromophore. The xanthone catalyst C8 exhibits a significantly elevated absorption coefficient at a wavelength exceeding 350 nm (ε350 = 9200 in PhCF3). Moreover, a substantial degree of enantioselectivity can be achieved with a mere 5–10 mol% of catalyst C8 (Scheme 7).53,77
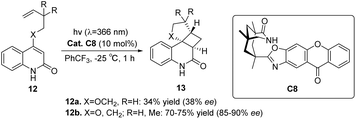 |
| | Scheme 7 Asymmetric intramolecular [2 + 2] photocycloaddition of 2-quinolone substrates catalyzed by 2-quinolone C8. | |
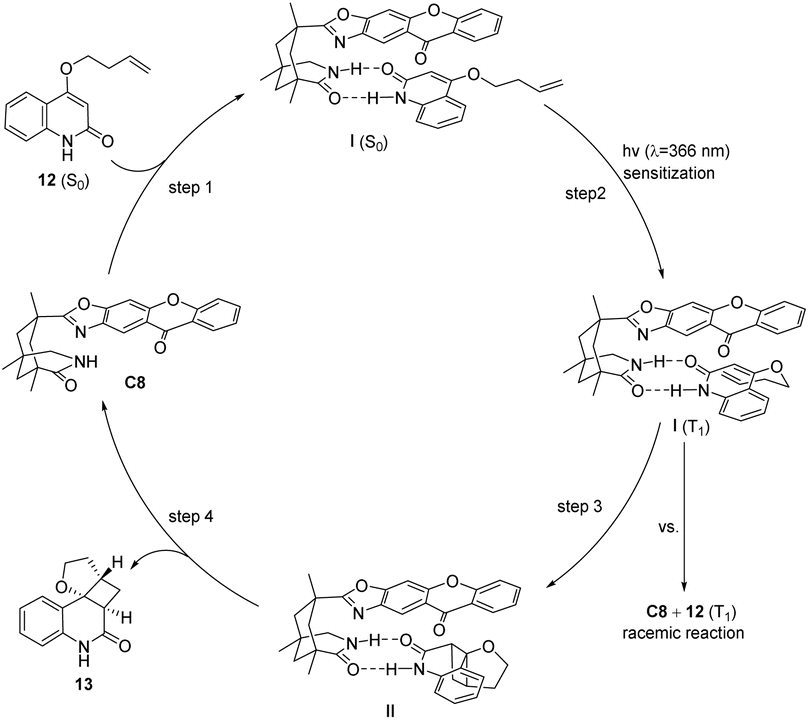
The enantioselectivity observed in the intramolecular [2 + 2] photocycloaddition reaction is contingent upon the extent of substrate–catalyst association mediated by hydrogen bonding (step 1, Fig. 6), the partial effective sensitization of xanthones (step 2, Fig. 6), and the subsequent rate of cyclization (step 3, Fig. 6). The association behavior of the substrate quinolone compounds with the catalyst was studied using NMR titration experiments.78 It was discovered that the catalyst was fully associated with the substrate prior to the initiation of the reaction. The increase in enantioselectivity for C7 and C8 is determined by the efficiency of sensitization and the subsequent cyclization rate. The disparity in enantioselectivity between substrates quinolone 12a and quinolone 12b (X = O, R = H) in Scheme 7 arises from the dissociation of the substrate from the catalyst prior to photocycloaddition, resulting in a reduction in enantioselectivity.
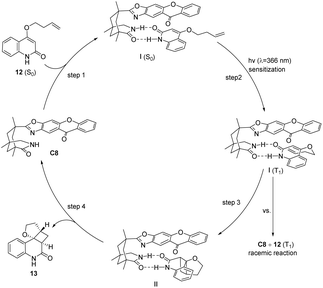 |
| | Fig. 6 Mechanism of asymmetric [2 + 2] photocycloaddition of quinolones catalyzed by ketone C8. | |
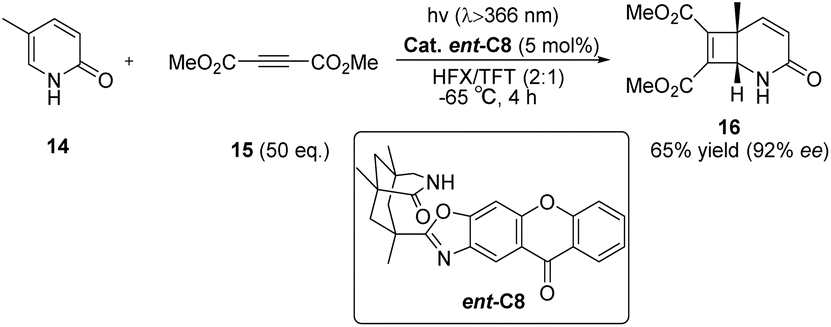
Subsequently, Bach also reported the intermolecular [2 + 2] photocycloaddition reaction between 2-pyridone and acetylene dicarboxylate catalyzed by ent-C8 that can achieve high enantioselectivity (Scheme 8).56 These excellent outcomes were achieved as a result of the following factors: (1) the rapid addition of alkynes to the excited substrate, (2) the effective binding of the substrate to the catalyst through hydrogen bonds, and (3) the high enantiomeric differentiation ensured by the 1,5,7-trimethyl-3-azabicyclo[3.3.1]nonane-2-one scaffold.79
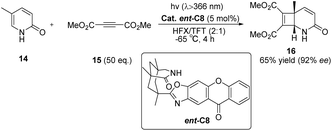 |
| | Scheme 8 Intermolecular [2 + 2] asymmetric photocycloaddition between 2-pyridone and acetylene dicarboxylate catalyzed by ent-C8. | |
Comparing the structures of the two PS components, xanthone C8 and benzophenone C7, it can be observed that the triplet energy of xanthone C8 is higher than that of benzophenone C7. Additionally, since the shape of the chromophore is approximately planar in the excited state, it facilitates closer proximity for energy transfer with the substrate. In contrast, the photochemically excited diphenyl ketone is not planar, resulting in a faster dissociation rate of the substrate during the reaction, leading to a decrease in the ee value. It can be seen that for the [2 + 2] photocycloaddition reaction, the photocatalyst of xanthone type C8 has more advantages than the photocatalyst of benzophenone type C7.
3.2 Other asymmetric photocatalytic reactions
Xanthone-type photocatalysts, in addition to their application in asymmetric [2 + 2] photocycloaddition reactions, have also been utilized in various other reactions, such as asymmetric photorearrangement reactions and photoderacemization reactions.
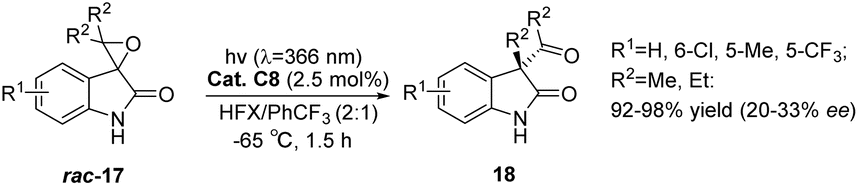
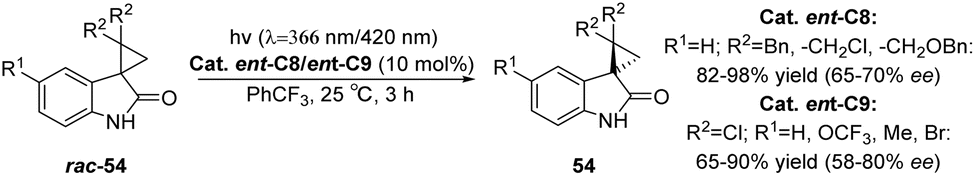
Inspired by the application of benzophenone in the rearrangement reaction of spirophenol indole epoxides reported by Zhang et al.,80 Bach used xanthone C8 to study the photochemical rearrangement of spirooxindole epoxides (Scheme 9).54 At λ = 366 nm, the yield of spirooxindole rac-17 rearranged to 3-acyl-3-alkylindole-2-one 18 can reach 92–98%, and the ee value is 20–33%. The low enantioselectivity may be due to the relatively slow rearrangement steps after sensitization that generate intermediates. Additionally, most intermediates undergo rearrangement after dissociation from the chiral catalyst, resulting in low ee values. This provides a new example for applying bifunctional ketone catalysts in other asymmetric photocatalytic reactions, further proving the universality of this type of catalyst.
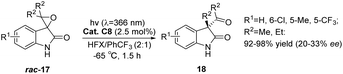 |
| | Scheme 9 Xanthone C8 catalyzes the photochemical rearrangement of spirooxindole epoxides (HFX = hexafluoro-m-xylene). | |
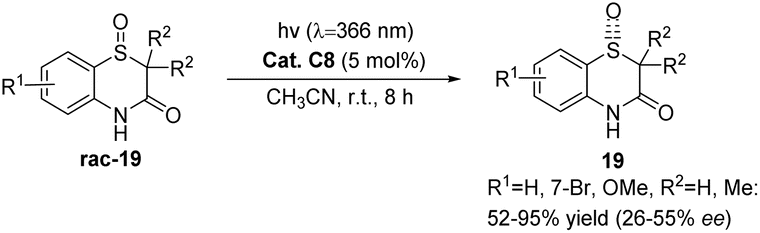
Ketone C8 also exhibits catalytic activity in the deracemization reaction of racemic benzothiazinone-1-oxide. Despite a high substrate conversion rate, the resulting product's ee value is significantly low (Scheme 10).55 This could potentially be attributable to the instability of the catalyst xanthone employed in this reaction.
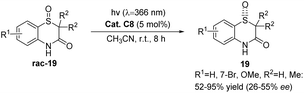 |
| | Scheme 10 Deracemization reaction of benzothiazinone-1-oxide catalyzed by ketone C8. | |
From the above discussion, it is clear that the appropriate choice of excitation wavelength, along with the absorption characteristics of the substrate and the corresponding substrate–photocatalyst complex, influences the reaction significantly. The triplet energy of the PS must be higher than the triplet energy of the substrate so rapid energy transfer can occur even at low temperatures. Additionally, if excitation can occur in the absence of a photocatalyst, the enantioselectivity will be compromised because of background reactions. The oxoxanthone photocatalyst ent-C8 possesses a significantly higher extinction coefficient compared with the substrate and effectively absorbs nearly all photons. Consequently, the racemic background reaction is attenuated, leading to enhanced enantioselectivity. Conversely, benzophenone derivative C7 exhibits a much lower extinction coefficient at 366 nm that inadequately weakens the competing racemic background reaction, resulting in reduced selectivity. Henceforth, it is crucial to design photosensitive units based on reaction characteristics rationally.
4. Thioxanthone photocatalysts
Common chromophores, such as carbonyl compounds, ketenes, and aromatic hydrocarbons, require a wavelength region of λ = 250–350 nm for direct excitation. Medium-pressure mercury lamps are commonly used as light sources for this purpose. Nevertheless, advancing scientific research often relies on employing uncomplicated techniques to achieve intended outcomes. Is employing visible light as an illuminating source to successfully catalyze the reaction feasible? The energy associated with a photon of wavelength λ = 400 nm is approximately 300 kJ mol−1, surpassing the triplet energy of numerous photochemical substrates. This observation suggests the feasibility of the proposed concept. Furthermore, the irradiation of xanthones in toluene at a wavelength of 366 nm results in swift decomposition, with a duration of less than 10 minutes. Xanthones are stimulated by ultraviolet light wavelengths, and the higher energy of these shorter wavelengths may directly excite the substrate, leading to undesired side reactions. The utilization of a suitable sensitizer and the substitution of ultraviolet light with visible light for exciting photoreactions are evident strategies that significantly enhance experimental convenience and reaction selectivity.
4.1 Asymmetric [2 + 2] photocycloaddition reactions
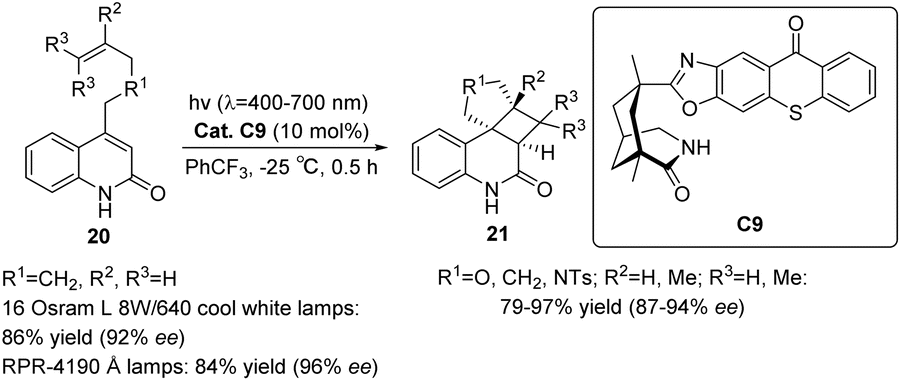
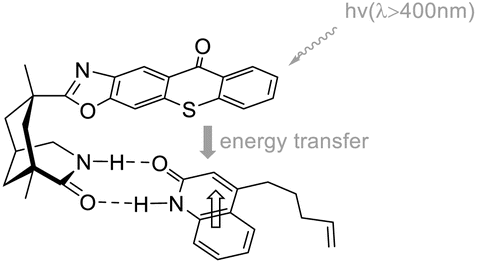
In 2014, Bach devised a novel chiral thioxanthone C9 (Scheme 11) capable of facilitating the intramolecular [2 + 2] asymmetric cycloaddition reaction through absorbing visible light, employing a mere 10 mol% of ketone C9.57 The resulting ee reaches an impressive 92%, while the attained yield is 86%. This reaction maintains a high degree of enantioselectivity even under ambient conditions. Furthermore, the author employed an alternative set of lamps (RPR-4190 Å) that do not emit ultraviolet light in order to address the small emission of ultraviolet light from the visible light source utilized. By conducting the experiment under identical conditions as previously mentioned, the substrate was obtained with a yield of 84% and an ee of 96%.
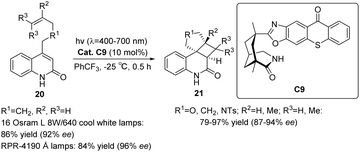 |
| | Scheme 11 Ketone C9 catalyzed intramolecular [2 + 2] asymmetric photocycloaddition reaction. | |
The intramolecular [2 + 2] photocycloaddition transition state of the thioxanthone-type catalyst is similar to that of the xanthone-type catalyst (Fig. 7). The differentiation of enantiomers is provided by the planar xanthone through hydrogen bonds, and the energy from absorbed photons is transferred to quinolone by triple energy transfer; therefore, the attack on the quinolone double bond occurs with high selectivity.
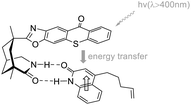 |
| | Fig. 7 Transition state model for sensitization and enantioface differentiation in the coordination complex of catalyst C9 and quinolone. | |
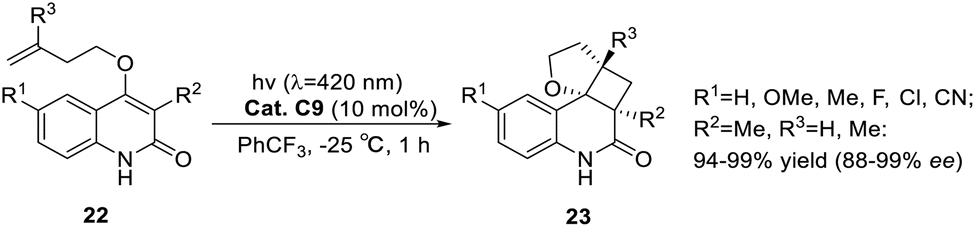
The known asymmetric [2 + 2] photocycloaddition reactions are all applied to substrates without alkyl substituents at the α-position because the substituents at this position may damage the coordination of the carbonyl group from a structural point of view. In 2020, Bach posed a challenge to the synthesis of cyclobutanes with a high degree of substitution.58 Surprisingly, compared with unsubstituted quinolones, the alkyl substituents at the α-position of the carbonyl group not only did not block the binding to the catalyst but also resulted in a significant decrease in triplet energy (ΔET ≈ 25 kJ mol−1), which produced a beneficial effect. The intramolecular asymmetric [2 + 2] photocycloaddition of 3-alkylquinolone catalyzed by ketone C9 can be obtained in good yields and ee values (Scheme 12).
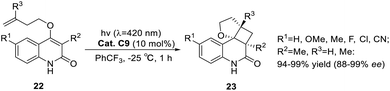 |
| | Scheme 12 Ketone C9 catalyzed intramolecular [2 + 2] asymmetric photocycloaddition of 3-alkyl substituted quinolones. | |
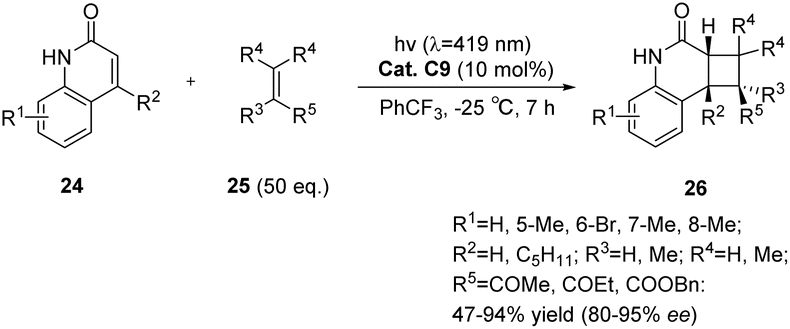
In comparison with xanthone compounds, thioxanthone compounds exhibit a red shift in their maximum absorption wavelength (λmax = 390 nm).81 Furthermore, thioxanthone C9 demonstrates a detectable lifetime at λ = 400–700 nm, suggesting its suitability for facilitating photochemical reactions when exposed to visible light sources. The intermolecular [2 + 2] asymmetric photocycloaddition reaction of quinolone compounds with electron-deficient olefins was catalyzed by chiral thioxanthone C9 (10 mol%).59 The resulting cyclobutane product exhibited an ee value of up to 95% (Scheme 13). An increase in the amount of coupling agent olefins in this reaction is associated with an increase in both the reaction rate and enantioselectivity. This phenomenon can be attributed to a higher proportion of cyclization occurring prior to catalyst dissociation, thus leading to enhanced enantioselectivity. Additionally, kinetic data indicate that the enantioselectivity is influenced by the sluggish reaction rate. It is postulated that other substrates featuring lactam binding motifs and exhibiting rapid intermolecular reaction rates would similarly be suitable for enantioselectively catalyzed intermolecular [2 + 2] photocycloaddition reactions.
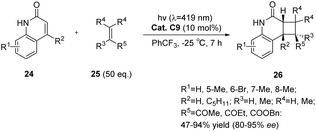 |
| | Scheme 13 Ketone C9 catalyzed intermolecular asymmetric [2 + 2] photocycloaddition reaction. | |
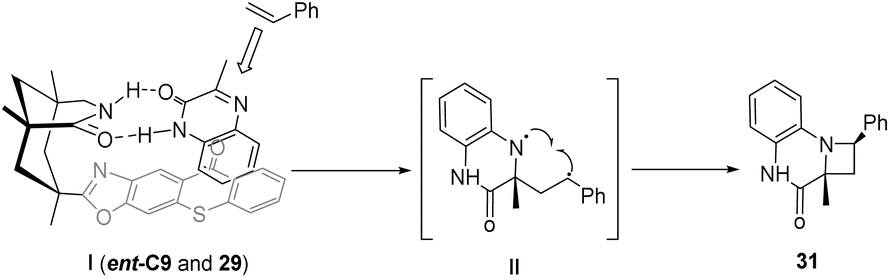
In the presence of a chiral sensitizer ent-C9 (10 mol%), 3-substituted quinoxalinones can engage in intramolecular or intermolecular asymmetric aza-Paternò–Büchi reactions with diverse aryl-substituted or chain olefins (Scheme 14), resulting in the formation of chiral azetidines.62 When the C3 position of the quinolone (27 or 29) is not replaced, this reaction does not produce azamine (28 or 31), indicating that the reaction mode of quinoxalinone is largely dependent on the 3-substituent, and the 3-substituent plays an important role in a certain intermediate process.
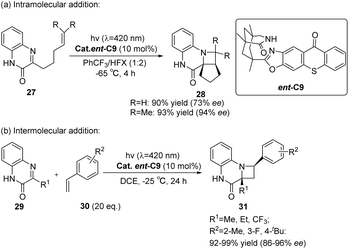 |
| | Scheme 14 Asymmetric [2 + 2] photocycloaddition of carbonyl-olefins catalyzed by ent-C9 (HFX = hexafluoro-m-xylene). | |
For the intermolecular [2 + 2] cycloaddition transition state, as shown in Fig. 8, the Si face of 29 is shielded by thioxanthone so that styrene only attacks the Re face. The intermediate 1,4-diradical II collapses after the ISC process to form the azetidine product 31. Although the triplet energy transfer from ent-C9 to styrene is energetically feasible, this pathway is less likely because the enantioselectivity is so high when the catalyst loading is only 10 mol%, proving that ent-C9 does not undergo triplet energy transfer to styrene.62
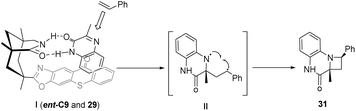 |
| | Fig. 8 Transition state analysis of intermolecular [2 + 2] photocycloaddition catalyzed by thioxanthone ent-C9. | |
Catalysts primarily employ hydrogen bonding interactions with the substrate in their structure to regulate the direction of the reaction, utilizing the chiral control elements of the aforementioned PSs; however, the current hydrogen bond skeleton exhibits relatively limited diversity, and designing different hydrogen bond skeleton units is of great significance for expanding more substrates.
In 2017, Bach conducted a novel endeavor to advance the development of a hydrogen bond skeleton,66 incorporating a thiourea group as a chiral control unit and devised a thiourea–thioxanthone compound C10. The findings indicated that ketone C10 effectively facilitated the photocyclization of 2-(4-bromophenoxy)-3,5,5-trimethylcyclohex-2-enone 32 (Scheme 15). Although the yield and enantioselectivity of the product are relatively low, the catalyst's success lies in introducing a novel chiral hydrogen bond scaffold into the chiral control unit. The inclusion of thiourea on this scaffold enables the formation of two hydrogen bond sites, thus expanding the scope of existing bifunctional catalysts and providing valuable insights for the future design of new chiral control units.
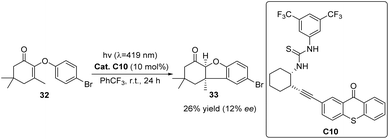 |
| | Scheme 15 Ketone C10 catalyzed photocyclization reaction. | |
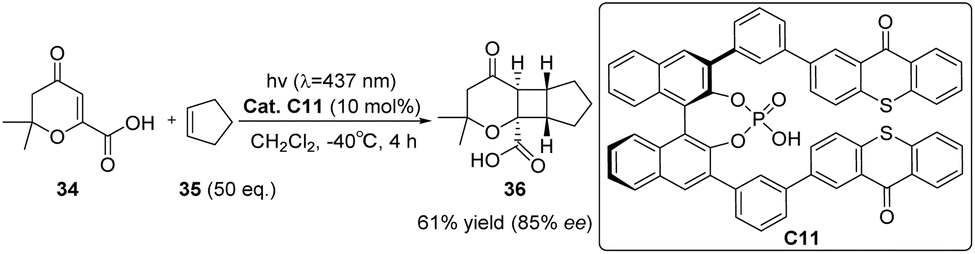
In 2020, Bach introduced chiral phosphoric acid (CPA) into asymmetric photochemical conversion for the first time and introduced two thioxanthone groups as PS units at the 3,3′-position of the BINOL skeleton to obtain a new CPA binding site catalyst C11.67 CPA is a representative Brønsted acid in thermochemical reactions.82 Here, its role is to generate a chiral environment and activate the substrate through hydrogen bonds during the conversion process. The catalyst effectively decreases the triplet energy of the substrate through hydrogen bonding, resulting in notable enantiomeric discrimination. It demonstrates high efficacy as a catalyst for the intermolecular [2 + 2] photocycloaddition reaction of β-carboxyl substituted cycloenones, with a 61% product yield with an 85% ee (Scheme 16).
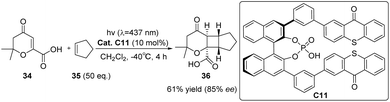 |
| | Scheme 16 Ketone C11 catalyzed intermolecular [2 + 2] asymmetric photocycloaddition of β-carboxyl substituted cyclic ketenes. | |
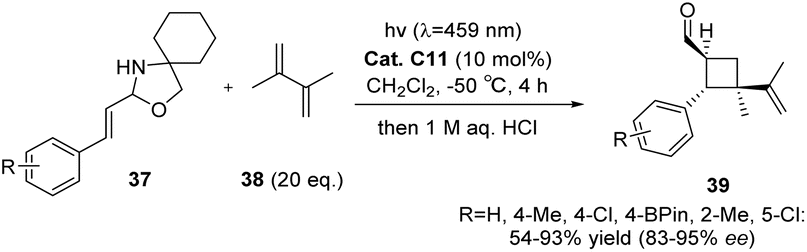
The asymmetric [2 + 2] photocycloaddition of N,O-acetals with olefins can also be catalyzed by thioxanthone C11 (10 mol%), resulting in yields ranging from 54% to 96% and ee values ranging from 84% to 98% (Scheme 17). The successful reaction relies on the triplet energy transfer of thioxanthone and the efficient enantiomeric differentiation of CPA.68
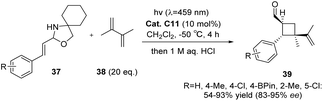 |
| | Scheme 17 The [2 + 2] asymmetric cycloaddition reaction of N,O-acetals with olefins catalyzed by C11 was studied. | |
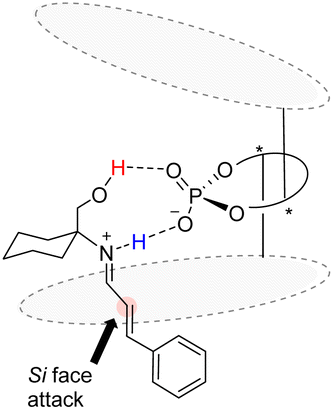
The Bach reaction is based on the prechiral imine ion addition model proposed by Akiyama et al.83 An analogous transition state model for catalyst C11 and substrate enantiomer differentiation is proposed, wherein the aryl group of the acid undergoes attack from the Si face (Fig. 9).
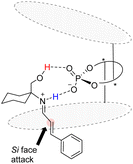 |
| | Fig. 9 Transition state model of asymmetric [2 + 2] cycloaddition catalyzed by catalyst C11. | |
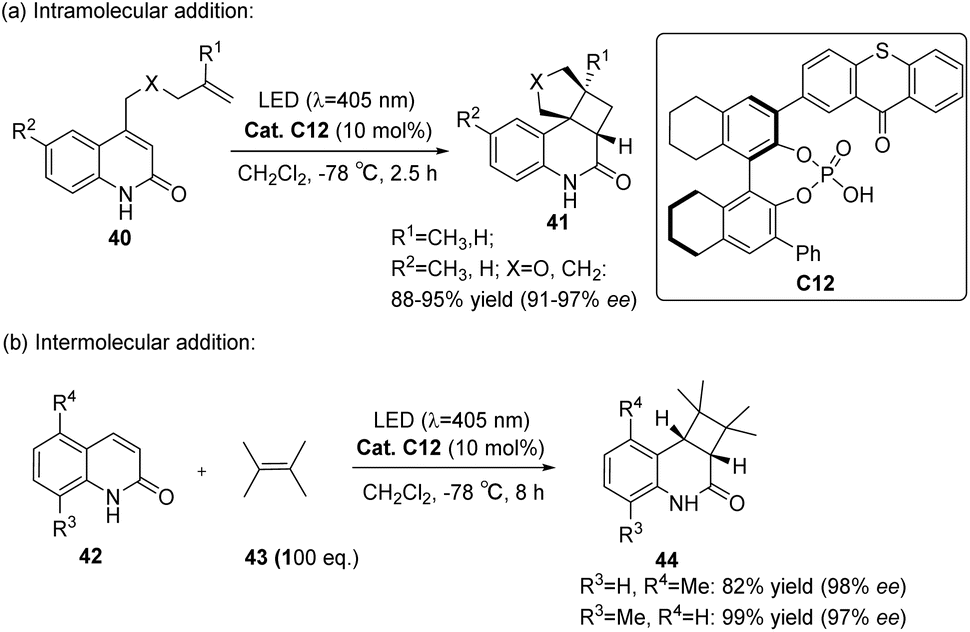
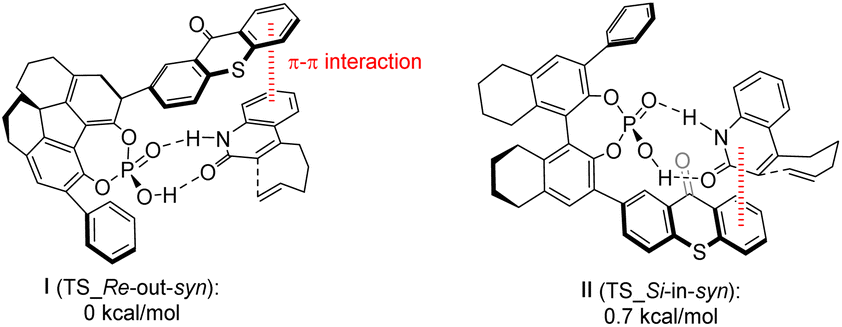
Takagi et al.69 employed a C1 symmetric CPA as the main chiral chain. They introduced thioxanthone as a photosensitive component to synthesize catalyst C12. Subsequently, they conducted intramolecular and intermolecular [2 + 2] photocycloaddition reactions of quinolones, resulting in the attainment of higher ee values (91–98%) (Scheme 18). In photoconversion, the chiral environment is established through the complexation of phosphoric acid and quinolone. The enantioselectivity is influenced significantly by the π–π interaction between the thioxanthone moiety of phosphoric acid and the substrate. The transition state with the lowest energy for each enantiomer in the reaction is depicted in Fig. 10. In both instances, the quinolones are oriented towards the thioxanthone ring on CPA. Notably, the main product obtained corresponds to the I transition state, thus confirming the experimental findings.
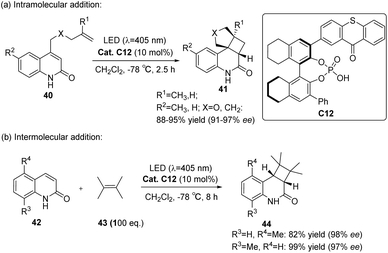 |
| | Scheme 18 Intramolecular and intermolecular photocycloaddition of quinolones catalyzed by ketone C12. | |
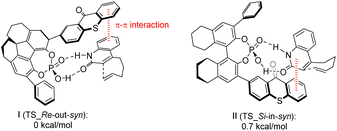 |
| | Fig. 10 Transition state energy calculation of ketone C12 with the substrate (the schematic structure and relative Gibbs energy of each enantiomer at 195.15 K in the intramolecular [2 + 2] photocycloaddition reaction). | |
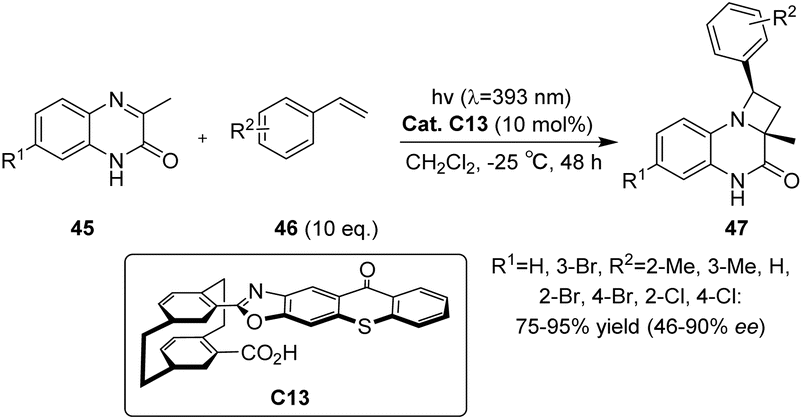
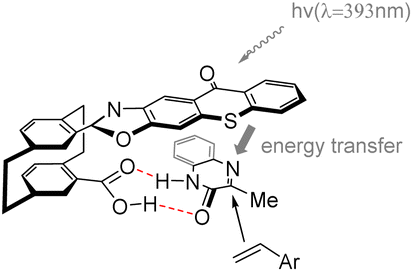
In 2023, Huo et al.70 developed a novel photocatalyst C13 based on thioxanthone, featuring a hamburger-shaped structure and planar chirality (Scheme 19). This catalyst was successfully employed in the enantioselective Paternò–Büchi reaction of quinoxalinone 45 and styrene 46 under visible light irradiation to afford azetidines. The catalyst C13 and the substrate amide 45 establish a stabilized spatial position through hydrogen bonding interactions. Upon irradiation, the thiazolone group of the intermediate undergoes excitation, leading to intramolecular energy transfer to the substrate amide. Due to the spatial shielding provided by the thioxanthone group, styrene is unable to approach the Si face of the transition state model (Fig. 11).
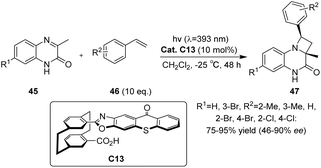 |
| | Scheme 19 Enantioselective Paternò–Büchi reaction catalyzed by ketone C13. | |
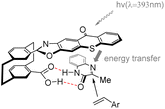 |
| | Fig. 11 Transition state model is proposed for the enantioselective Paternò–Büchi reaction catalyzed by ketone C13. | |
Through the above content, it can be concluded that the successful use of ketone catalysts in asymmetric reactions depends on two basic factors: (a) including a sensitizing ketone unit for energy or electron transfer and (b) incorporating chiral control units that interact with the substrate to ensure effective enantiomer or group differentiation. The rate of substrate dissociation is slower than the enantioselectivity determination step of the reaction. Consequently, even after sensitization, the excited substrate still encounters the spatial bias imposed by the control element to ensure a distinct chiral environment.
4.2 Photoderacemization reactions
In 2018, Bach first reported the pioneering achievement of a successful photoderacemization reaction. By employing a catalytic amount (2.5 mol%) of the triple sensitizer ent-C9, various racemic 3-(1′-methylene)piperidin-2-ones were effectively transformed into their corresponding single enantiomers under visible light irradiation at a specific wavelength of 420 nm. The resulting yield exceeded 56%, while the ee value ranged from 89% to 97% (Scheme 20).63 The sensitizer is transferred to allene via triplet energy transfer and subsequently separated based on the distinct energy transfer efficiencies of the two enantiomers.
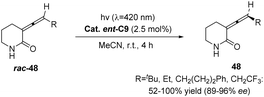 |
| | Scheme 20 Deracemization of 3-(1′-alkenyl)piperidin-2-ones catalyzed by ketone ent-C9. | |
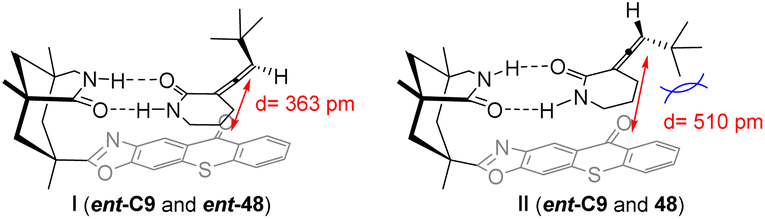
The proximity between the sensitizer ent-C9 chromophore and the allene double bond of substrate ent-48 is closer compared with that of substrate 48, resulting in a faster triplet energy transfer between ent-48 and the sensitizer. The disparity in association constants (ground state thermodynamics) and sensitization efficiency (excited state kinetics) between 48 and ent-48 with the catalyst leads to distinct total sensitization rates. Consequently, the intramolecular sensitization mechanism of 48 is unfavorable, enabling the accumulation of 48 (Fig. 12).63
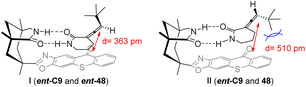 |
| | Fig. 12 The deracemization transition state model of 3-(1′-alkenyl)piperidin-2-one catalyzed by ent-C9. | |
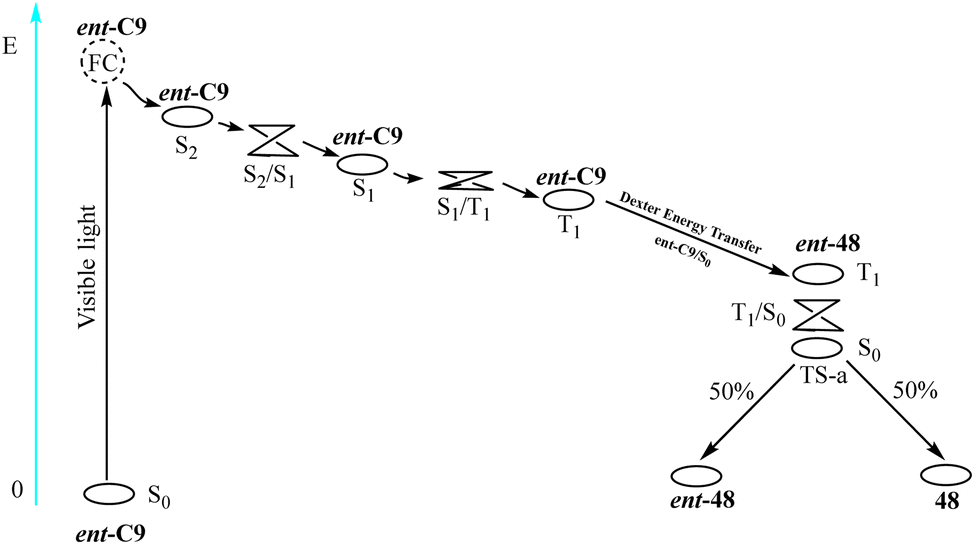
Yuan et al. conducted a theoretical investigation using density functional theory (DFT).84 Using the enantiomeric conversion of 3-(3,3-dimethyl-1-butyl-1-subunit)-2-piperidone from Scheme 20 as a case study (R = tBu), they elucidated the mechanism underlying the deracemization reaction catalyzed by ent-C9 (see Fig. 13). The PS ent-C9 is excited under visible light (λ = 420 nm) irradiation (S0 → S2) because of the potential surface crossing between the S1 and S2 states; therefore, a fast internal conversion (S2 → S1) occurs near the intersection. At this time, the energy of the T1 state is slightly lower than that of the S1 state, and the PS undergoes ISC (S1 → T1). The T1 state of the PS ent-C9 undergoes Dexter-type energy transfer to the S0 state of ent-48, forming the S0 state of the PS ent-C9 and the T1 state of ent-48. Finally, the T1 state of ent-48 can pass through the potential energy surface of the triplet state and transform into the transition state between ent-48 and 48 of the ground state S0. This transition state then forms a 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ent-48 ratio of 1:1. The newly generated ent-48 can be resensitized to form TS-a by catalyzing thioxanthone ent-C9, but 48 cannot. By repeating this process, the amount of ent-48 is halved in each photocatalytic cycle, while the amount of 48 increases accordingly. The mechanism reveals that complete conversion of ent-48 to 48 is unattainable, thus limiting the ee value infinitely close but not equal to 100%. From the theoretical calculation, deracemization occurs because limited occurrence of energy transfer within ent-C9 and 48, consistent with the spatial effects analysis results (Fig. 12). It can be observed that designing a new deracemization photocatalyst based on the disparity between the ground state association constant and excited state sensitization efficiency of two diastereomeric complexes proves to be an effective strategy.
:ent-48 ratio of 1:1. The newly generated ent-48 can be resensitized to form TS-a by catalyzing thioxanthone ent-C9, but 48 cannot. By repeating this process, the amount of ent-48 is halved in each photocatalytic cycle, while the amount of 48 increases accordingly. The mechanism reveals that complete conversion of ent-48 to 48 is unattainable, thus limiting the ee value infinitely close but not equal to 100%. From the theoretical calculation, deracemization occurs because limited occurrence of energy transfer within ent-C9 and 48, consistent with the spatial effects analysis results (Fig. 12). It can be observed that designing a new deracemization photocatalyst based on the disparity between the ground state association constant and excited state sensitization efficiency of two diastereomeric complexes proves to be an effective strategy.
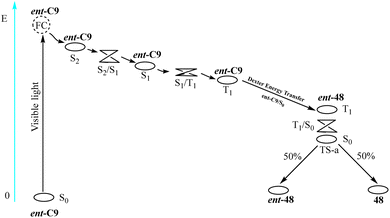 |
| | Fig. 13 Photosensitive deracemization mechanism of racemates. | |
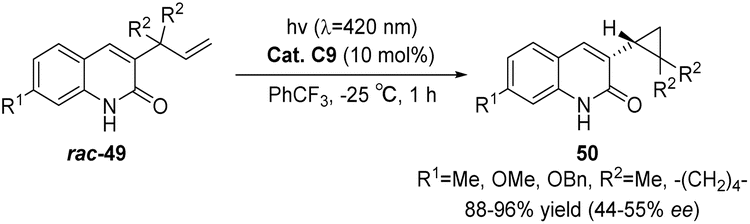
In 2019, Bach reported that under irradiation with visible light (λ = 420 nm), rac-49, a 3-allyl-substituted quinolone, underwent a triple-sensitized di-π-methane rearrangement reaction to yield the corresponding rac-50, a 3-cyclopropyl quinolone. By employing chiral thioxanthone sensitizer C9, cyclopropane underwent a triple-sensitized deracemization reaction, forming enantiomerically pure 3-cyclopropylquinolone 50. The yields ranged from 88% to 96%, while the ee values were between 44% and 55% (Scheme 21).61
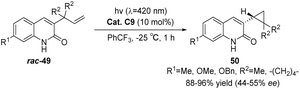 |
| | Scheme 21 Deracemization of 3-cyclopropylquinolone catalyzed by ketone C9. | |
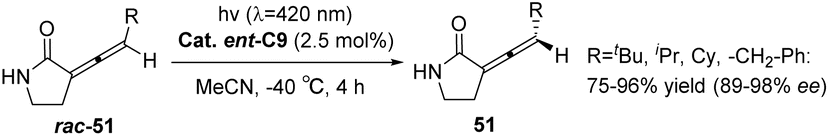
In the subsequent year, Bach employed the chiral triplet sensitizer ent-C9 (2.5 mol%) to facilitate the racemization reaction of rac-51, resulting in an enhanced ee value of 86–98% for product 51 (Scheme 22). The photocatalyst also operates through selective recognition of an olefin enantiomer and a hydrogen bond-mediated triplet sensitization process.64
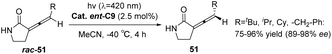 |
| | Scheme 22 Deracemization of rac-49 catalyzed by ketone ent-C9. | |
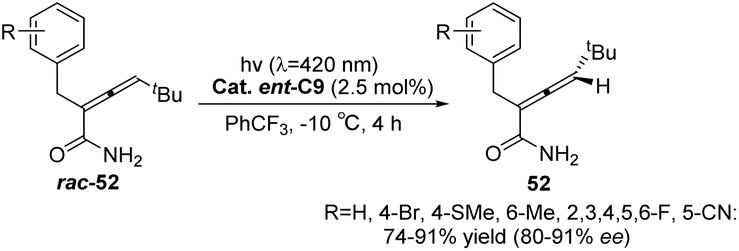
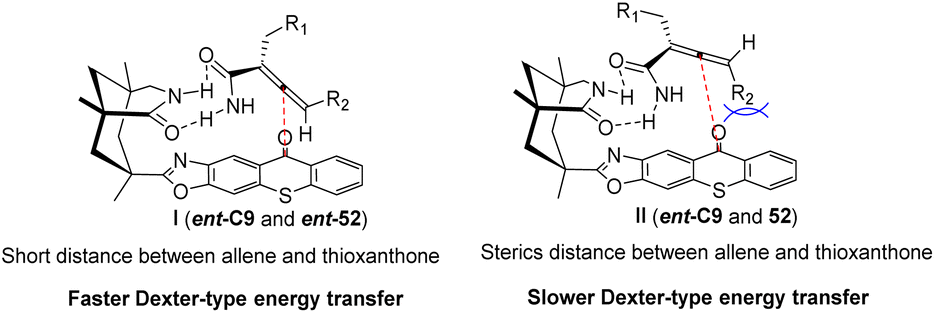
Similarly, the chiral sensitizer thioxanthone ent-C9 (2.5 mol%) is capable of catalyzing the photoderacemization of enamide rac-52 at 420 nm. The resulting acrylamide 52 exhibits an ee value ranging from 80% to 91% (Scheme 23).65 In addition to the difference in the degree of association between the enantiomer and the catalyst caused by the hydrogen bonding between the catalyst and the substrate, it was also found via DFT calculations that this result was affected by the attractive dispersion interaction between the π system of the propylene and ketone catalyst. One of the enantiomers, the C–H or C–halogen bond, is perpendicular to the π system, forcing propylene away from the thioxanthone chromophore. In the case of another propylene enantiomer, the interaction between the C–H bond at the terminal carbon atom of propylene and the outer benzene ring of thioxanthone, known as the CH–π interaction, leads to propylene being in close proximity to the chromophore. Enantiomers in close proximity to the chromophore exhibit a higher reaction rate, ultimately resulting in the observed outcome (Fig. 14). This discovery represents the first instance in which a dispersion interaction has been identified as a valuable means to enhance the enantioselectivity of photochemical reactions, thus offering a novel approach for the future design of enantioselective photoinduced conversions.
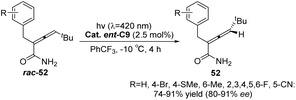 |
| | Scheme 23 Ketone ent-C9 catalyzed photochemical deracemization of enamides. | |
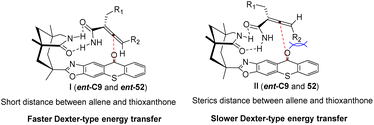 |
| | Fig. 14 Transition state model of deracemization of acrylamide by ent-C9. | |
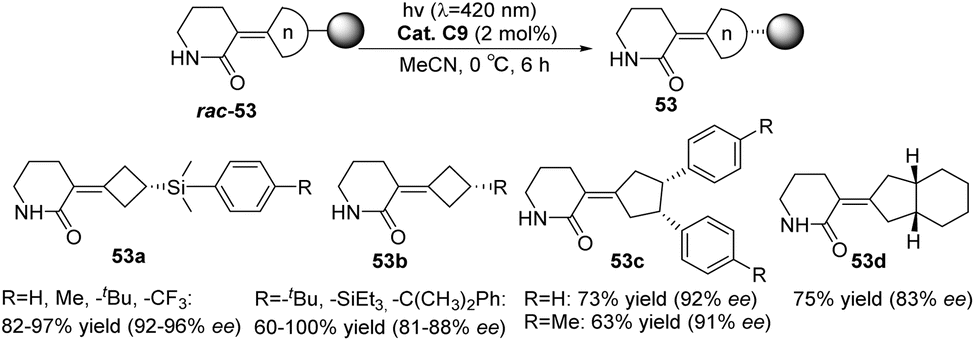
In 2022, Bach reported a visible-light-mediated photocatalytic deracemization method for synthesizing axially chiral olefins. A variety of racemic alkylidenecyclobutanes (n = 4) and alkylidenecyclopentanes (n = 5) can be deracemized by triplet energy transfer with only 2 mol% chiral thioxanthone C9, achieving up to 81–96% ee (Scheme 24).60
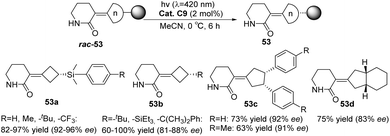 |
| | Scheme 24 Substrate range of photoderacemization of axially chiral olefins catalyzed by ketone C9. | |
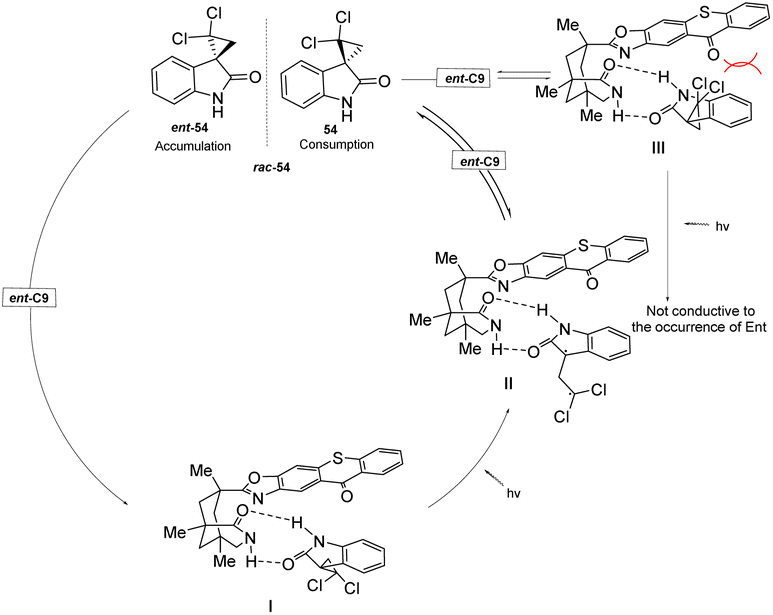
In the presence of the catalysts ent-C8 or ent-C9, the photoderacemization reaction of 3-spirocyclopropyl 2-oxindoles yields a high ee value (Scheme 25) and exhibits a broad substrate scope. Notably, this method enables the construction of a full-carbon stereocenter on cyclopropane, which holds significant potential for synthesizing chiral drugs analogous to cyclopropane derivatives.85 Its good enantioselectivity is obtained through the synergistic effect of three mechanisms: (1) the difference in the binding constant with the chiral thiazolone, (2) the molecular distance of the enantiomer in the complex, and (3) the lifetime of the intermediate 1,3-diradical. According to NMR titration experiments, transient absorption spectroscopy measurements, and DFT calculations, the reaction mechanism was elucidated (Fig. 15). Due to steric hindrance, the interaction between 54 and the PS ent-C9 to form III is thermodynamically unfavorable; however, the formation of I is more favorable and ultimately leads to the enrichment of enantiomer 54.
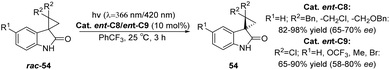 |
| | Scheme 25 Deracemization of 3-spirocyclopropyl 2-oxindoles. | |
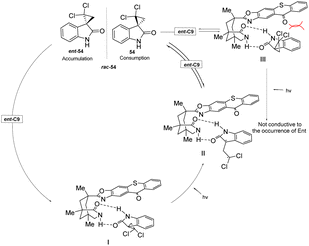 |
| | Fig. 15 Deracemization mechanism of 3-spirocyclopropyl 2-oxindoles catalyzed by ketone ent-C9. | |
In conclusion, the successful completion of catalytic deracemization in terms of PS units depends on the synergistic effect between chiral catalytic cycles and the photoenergy input cycle that overcomes thermodynamic and kinetic challenges. In addition, from the perspective of the chiral control unit, whether a product with high enantioselectivity can be obtained depends on the difference between the spatial effect (such as steric hindrance) and the electronic effect (such as the degree of hydrogen bond binding and the influence of dispersion force) when the two enantiomers in the racemic substance are close to the catalyst. The spatial effect and the electronic effect lead to different degrees of association between the substrate and the photocatalyst, and the sensitization efficiency differ, ultimately affecting the enantioselectivity; therefore, the key points to consider when designing and selecting the catalyst for the photoderacemization reaction are the matching of the photosensitive unit with the reaction and the rational design of the chiral control unit.
4.3 Other asymmetric photocatalytic reactions
In recent years, thioxanthone-type catalysts have been utilized in various reactions, including the asymmetric [2 + 2] photocycloaddition reaction and racemization reaction mentioned earlier. Additionally, these catalysts have found application in other reactions, such as asymmetric electrophilic amination, asymmetric alkylation of aldehydes, and asymmetric α-hydroxylation reactions.
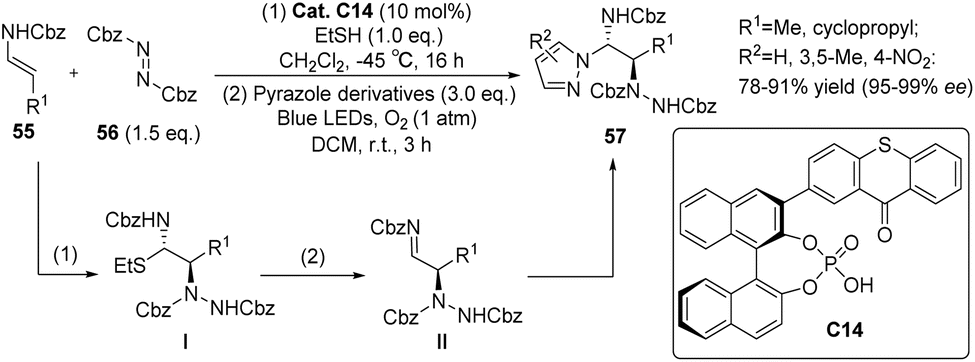
Masson et al. successfully synthesized a mono-substituted C1-symmetric photocatalyst C14 by combining phosphoric acid and thioxanthone (Scheme 26).71 The mono-substituted C1-symmetric photocatalyst exhibited superior catalytic efficiency compared with the corresponding C2-symmetric photocatalyst. Using catalyst C14, enecarbamate and azodicarboxylate were employed as starting materials for the reaction, which was conducted in two distinct steps. In the initial step, the nucleophilic enecarbamate reacted with the electrophilic azodicarboxylate to yield an imine, subsequently reacting with ethanethiol to form α-carbamoyl sulfide I. In the subsequent step, pyrazole was introduced, and the reaction was subjected to irradiation with blue light (λ = 420–510 nm). This led to the formation of imine II, which underwent a diastereothermal reaction with pyrazole to produce the ultimate product. The ee value achieved ranged from 95% to 99%.
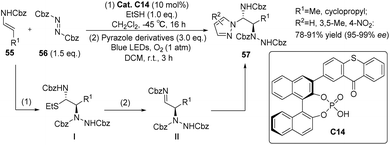 |
| | Scheme 26 Catalyst C14 as photocatalyst enantioselective synthesis of 1,2-diamine. | |
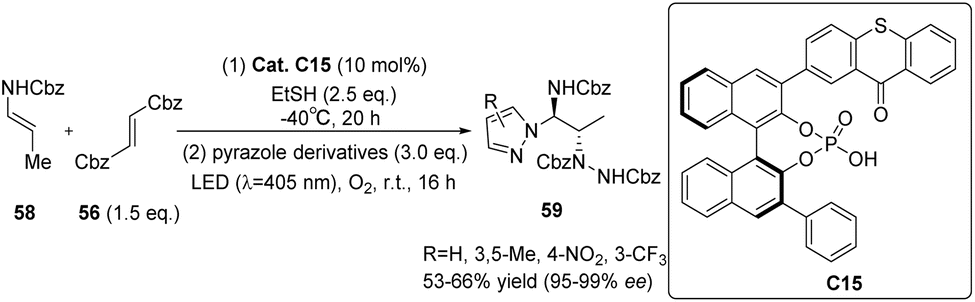
The asymmetric electrophilic amination of α-unsubstituted enecarbamates was observed under 10 mol% catalyst C15 upon irradiation with 405 nm. This reaction yielded chiral 1,2-diamides. The performance of catalyst C15 was found to be satisfactory,72 with only a minor disparity compared with catalyst C14.71 This suggests that modifying the 3′-position substituents of the C1 symmetrical thioxanthone catalyst does not play a crucial role in determining the ee value of the substrate. In order to verify the universality of the results obtained by catalyst C15, several 1-(hetero)aryl-1,2-diamines were synthesized by changing the pyrazole ligand (Scheme 27). Good yield and enantioselectivity were obtained.
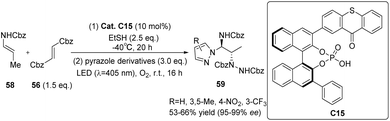 |
| | Scheme 27 Asymmetric electrophilic amination of enecarbamates catalyzed by ketone C15. | |
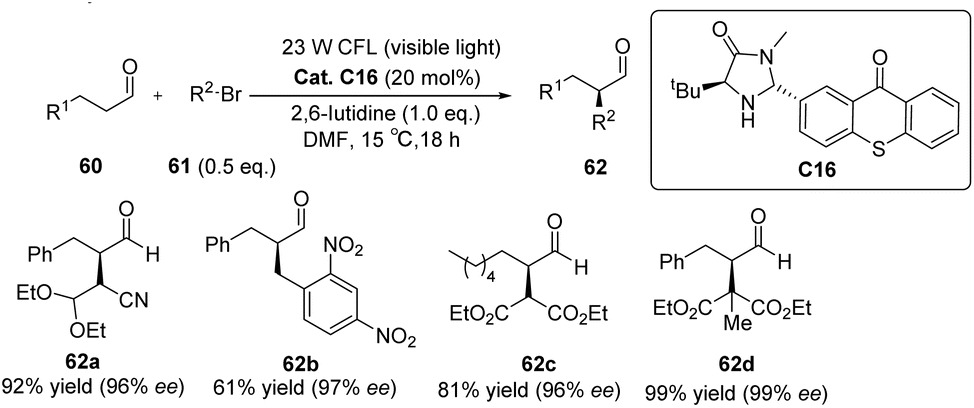
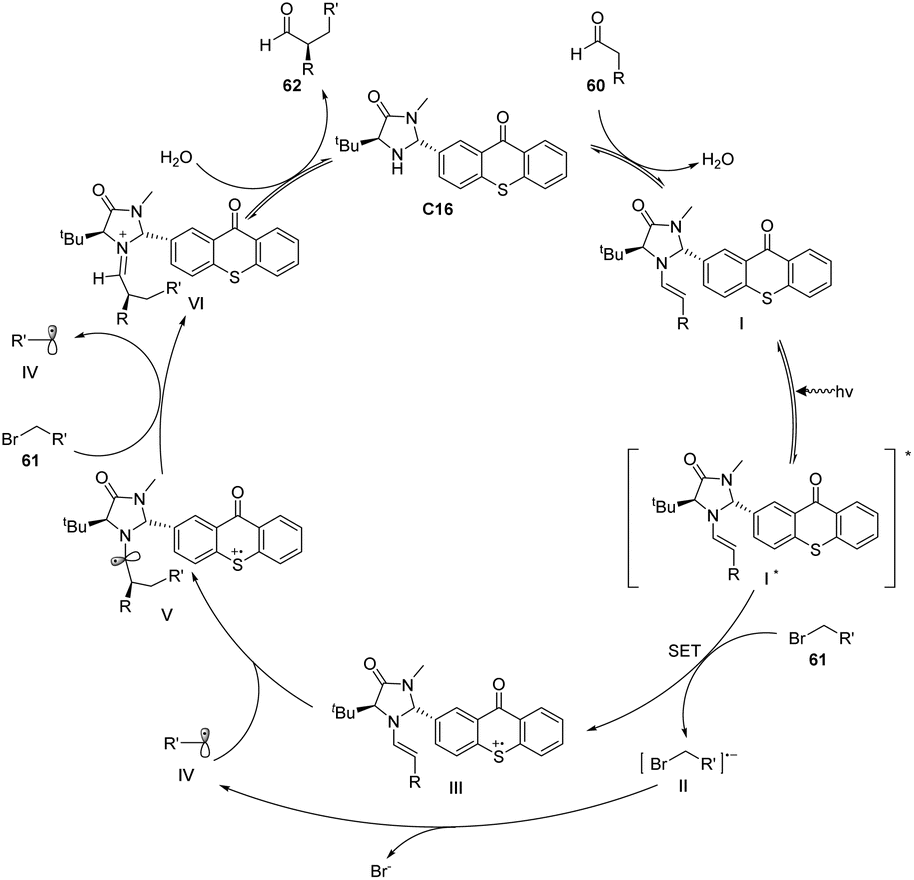
Alemán et al. successfully synthesized a novel catalyst, designated as catalyst C16, derived from thioxanthone through a two-step synthesis involving imidazolidinone and thioxanthone. This particular structure exhibits a convenient spatial property that can be readily adjusted. Notably, the catalyst demonstrates remarkable enantioselectivity and yield in the alkylation reaction of aldehydes, as depicted in Scheme 28.73 The reaction mechanism is elucidated in Fig. 16. The thioxanthone of intermediate I is excited by light to produce the excited ketone I*. Thioxanthone can promote the single electron transfer process (SET). The excited intermediate I* is used as a free radical chain initiator to reduce the bromoalkane derivative 61 through one SET, generating a free radical anion II and an enamine cation III. The bromine anion is left behind to produce free radical IV that reacts with the enamino cation intermediate III to produce α-amino radical V. Further, VI is obtained, and product 62 is obtained by hydrolysis, along with the chiral imidazolidinone–thioxanthone catalyst C16. The highlight of this work is the combination of photoredox catalysis and organic catalysis to achieve the asymmetric alkylation of aldehydes.
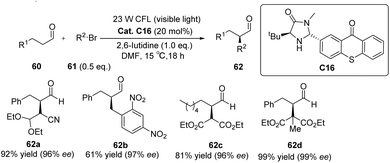 |
| | Scheme 28 Asymmetric alkylation of aldehydes catalyzed by ketone C16. | |
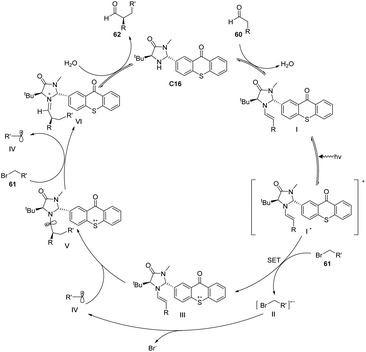 |
| | Fig. 16 Mechanism of asymmetric alkylation of aldehydes catalyzed by ketone C16. | |
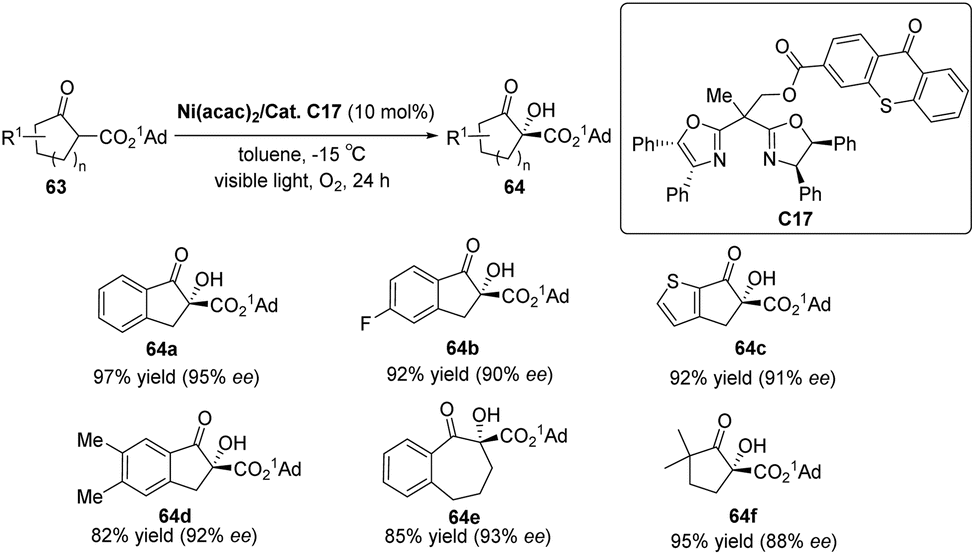
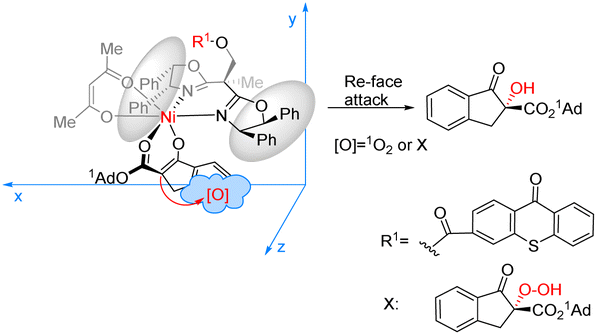
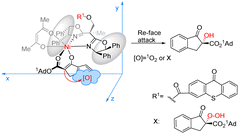
Xiao et al. designed a bifunctional catalyst that differs from Bach and developed a new photocatalyst, C17, by grafting the thioxanthone motif to the chiral BOX ligand.74 The ground state of the thioxanthone component absorbs visible light and triplet state and then sensitizes the nonreactive triplet state of molecular oxygen (3O2) to produce the reactive singlet state of molecular oxygen (1O2). The chiral metal complexes derived from the BOX component activate the β-keto carbonyl group, thus controlling the selective formation of C–O bonds in the reaction. The ligand complexed with Ni (acac)2 to catalyze the asymmetric α-hydroxylation of β-ketoesters to obtain products with high yields and ee values (Scheme 29). It should be noted that the catalyst does not directly act on the substrate but rather links the metal complex to the PS. The metal complex then activates the substrate while also controlling the chirality. In this context, the PS does not engage in direct activation of the substrate through energy or electron transfer. Instead, it indirectly sensitizes oxygen, inducing its transition from a nonreactive triplet state to a reactive singlet state, thus enabling oxygen's involvement in the reaction. Consequently, the successful achievement of the asymmetric aerobic oxidation of β-ketoesters and β-ketoamides was realized through visible light-induced metal photocatalysis, resulting in the production of α-hydroxy-β-dicarbonyl products. A possible stereoinduction model (Fig. 17) is proposed that assumes that the adamantyl group (1Ad–) is positioned far away from the chiral ligand to avoid steric hindrance. The si-face of the β-ketoester formed by the enol is obstructed by the phenyl group behind it; therefore, favorably, the oxidant attack from the Re-face activates 1O2 or peroxide X, resulting in an R configuration for the hydroxylated product.
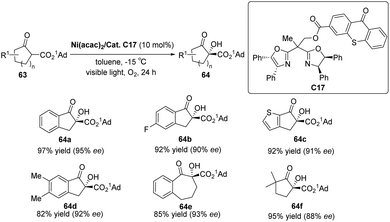 |
| | Scheme 29 Asymmetric α-hydroxylation of β-ketoesters catalyzed by the complexation of ketone C17 with Ni (acac)2 (1Ad = 1-adamantyl). | |
 |
| | Fig. 17 Stereoinduction model of asymmetric α-hydroxylation catalyzed by complexation of ketone C17 with Ni (acac)2. | |
The aforementioned diverse reaction studies demonstrate the continued efficacy of bifunctional ketone catalysts incorporating sensitization and chiral control units in other asymmetric photoreactions, with ample potential for further exploration across various reaction types. Consequently, their future utilization in additional photochemical reactions is anticipated; however, the prerequisite for applying this type of catalyst to other reactions is the presence of a chiral control site in the substrate and a suitable reaction that can be initiated by the ketone PS unit, so that the ketone PS unit and chiral control unit can be reasonably designed according to the structure of the substrate to synthesize the required new ketone catalyst.
5. Summary and prospect
In the past decade, chiral ketone catalysts have shown great potential in the field of asymmetric catalysis. Chiral aromatic ketone catalysts exhibit high triplet energies and long triplet lifetimes, making them excellent PS units. These catalysts have become a powerful tool for asymmetric photocatalytic reactions; however, there is still room for future development in this field: (1) the modular formation of all-carbon quaternary stereocenters and multiple stereocenters remains a daunting challenge. (2) From the perspective of the PS unit of the catalyst, introducing various chiral skeletons to modify the chiral aromatic ketone photosensitive unit has shown limited effects. This limitation arises because the chiral aromatic ketone photosensitive unit itself may encounter issues such as mismatched redox potential or insufficient triplet energy. Furthermore, from the perspective of the chiral control unit of the catalyst, the chiral skeleton form is relatively simple. As a result, it can only exhibit an asymmetric catalytic role in specific substrates capable of forming hydrogen bonds, thus restricting its applicability to certain types of reactions.
In order to address the aforementioned issues, the future focus of development may lie in the design of new ketone catalysts. First, when designing the PS unit of the ketone catalyst, considering its match with the required light energy for the reaction is necessary, and this structure should be able to complete the catalytic cycle through processes such as ent, HAT, or SET, which are decisive factors for initiating photosensitive reactions. Furthermore, obtaining a single chiral compound relies on an appropriate chiral skeleton. The design of a new chiral control unit can be achieved through various forces such as spatial effects, conjugation effects, induction effects, π–π stacking interactions, van der Waals forces, and hydrogen bonding. By combining different ketone PS units and chiral control units in diverse ways, it is possible to expand their application to more types of asymmetric photochemical reactions in the future. In summary, the application of ketone catalysts in [2 + 2] photocycloaddition reactions, deracemization reactions, photochemical rearrangement reactions, asymmetric electrophilic amination reactions, and the asymmetric alkylation of aldehydes were reviewed. The structural characteristics of these catalysts, the mechanism of catalytic reaction, and their advantages and disadvantages are discussed. It is hoped that it can provide the references for developing more applicable asymmetric catalytic methodologies in the future.
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this review. The findings presented in this review are based on previously published works cited in the reference list.
Author contributions
B. J. B. collected the literature and wrote the manuscripts L. Y. and L. X. Q. revised the manuscript. Q. Z. and W. H. supervised the work and revised the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research is supported by the Xijing Innovation Institute Joint Fund (No. LHJJ2023-YX04), the Qin Chuangyuan's Team Building Project (No. 2022KXJ-101), the Science and Technology Program of Gansu Province (No. 23ZDFA003), the National Natural Science Foundation of China (No. 21272272), the Project of Research and Development of Key Technologies (No. 2023-YBGY-486), and the Natural Science Basic Research Program of Shaanxi (No. 2023-JC-QN-0149, 2023-JC-QN-0868).
References
- I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521–2535 CrossRef CAS.
- L. D'Accolti, C. Annese and C. Fusco, Chemistry, 2019, 25, 12003–12017 CrossRef PubMed.
- N. Hoffmann, Photochem. Photobiol. Sci., 2021, 20, 1657–1674 CrossRef CAS PubMed.
- A. Antenucci, S. Dughera and P. Renzi, ChemSusChem, 2021, 14, 2785–2853 CrossRef CAS PubMed.
- T. H. El-Assaad, J. Zhu, A. Sebastian, D. V. McGrath, I. Neogi and K. N. Parida, Org. Chem. Front., 2022, 9, 5675–5725 RSC.
- M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed.
- J. Wang, X. Lv and Z. Jiang, Chem.–Eur. J., 2023, 29, e202204029 CrossRef CAS PubMed.
- Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS PubMed.
- D. Yang, Acc. Chem. Res., 2004, 37, 497–505 CrossRef CAS PubMed.
- O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef CAS PubMed.
- R. L. Davis, J. Stiller, T. Naicker, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2014, 53, 7406–7426 CrossRef CAS PubMed.
- Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199–8256 CrossRef CAS PubMed.
- Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806–9807 CrossRef CAS.
- Z. Xiong and E. J. Corey, J. Am. Chem. Soc., 2000, 122, 4831–4832 CrossRef CAS.
- D. W. Hoard, E. D. Moher, M. J. Martinelli and B. H. Norman, Org. Lett., 2002, 4, 1813–1815 CrossRef CAS PubMed.
- C. M. Adams, I. Ghosh and Y. Kishi, Org. Lett., 2004, 6, 4723–4726 CrossRef CAS PubMed.
- Y. Morimoto, Y. Nishikawa and M. Takaishi, J. Am. Chem. Soc., 2005, 127, 5806–5807 CrossRef CAS PubMed.
- A. B. Smith, S. P. Walsh, M. Frohn and M. O. Duffey, Org. Lett., 2005, 7, 139–142 CrossRef CAS PubMed.
- F. Lecornué, R. Paugam and J. Ollivier, Eur. J. Org Chem., 2005, 2005, 2589–2598 CrossRef.
- D. F. Taber and Y. He, J. Org. Chem., 2005, 70, 7711–7714 CrossRef CAS PubMed.
- J. Bian, M. Van Wingerden and J. M. Ready, J. Am. Chem. Soc., 2006, 128, 7428–7429 CrossRef CAS PubMed.
- R. M. Kanada, D. Itoh, M. Nagai, J. Niijima, N. Asai, Y. Mizui, S. Abe and Y. Kotake, Angew. Chem., Int. Ed., 2007, 46, 4350–4355 CrossRef CAS PubMed.
- A. R. Van Dyke and T. F. Jamison, Angew Chem. Int. Ed. Engl., 2009, 48, 4430–4432 CrossRef CAS PubMed.
- M. Yu and B. B. Snider, Org. Lett., 2009, 11, 1031–1032 CrossRef CAS PubMed.
- T. J. Harrison, S. Ho and J. L. Leighton, J. Am. Chem. Soc., 2011, 133, 7308–7311 CrossRef CAS PubMed.
- D. J. Clausen, S. Wan and P. E. Floreancig, Angew Chem. Int. Ed. Engl., 2011, 50, 5178–5181 CrossRef CAS PubMed.
- P. Yang, P.-F. Li, J. Qu and L.-F. Tang, Org. Lett., 2012, 14, 3932–3935 CrossRef CAS PubMed.
- A. K. Ghosh and D. D. Anderson, Org. Lett., 2012, 14, 4730–4733 CrossRef CAS PubMed.
- V. P. Kumar and S. Chandrasekhar, Org. Lett., 2013, 15, 3610–3613 CrossRef CAS PubMed.
- C. An, J. A. Jurica, S. P. Walsh, A. T. Hoye and A. B. Smith, J. Org. Chem., 2013, 78, 4278–4296 CrossRef CAS PubMed.
- T. Kodama, S. Aoki, S. Kikuchi, T. Matsuo, Y. Tachi, K. Nishikawa and Y. Morimoto, Tetrahedron Lett., 2013, 54, 5647–5649 CrossRef CAS.
- D. J. Mack and J. T. Njardarson, Angew. Chem., Int. Ed., 2013, 52, 1543–1547 CrossRef CAS PubMed.
- Y. Morimoto, E. Takeuchi, H. Kambara, T. Kodama, Y. Tachi and K. Nishikawa, Org. Lett., 2013, 15, 2966–2969 CrossRef CAS PubMed.
- J. Rodríguez-López, F. Pinacho Crisóstomo, N. Ortega, M. López-Rodríguez, V. S. Martín and T. Martín, Angew Chem. Int. Ed. Engl., 2013, 52, 3659–3662 CrossRef PubMed.
- S. Sittihan and T. F. Jamison, J. Am. Chem. Soc., 2019, 141, 11239–11244 CrossRef CAS PubMed.
- D. Rhoades, A. L. Rheingold, B. W. O'Malley and J. Wang, J. Am. Chem. Soc., 2021, 143, 4915–4920 CrossRef CAS PubMed.
- T. Ohyoshi, H. Iizumi, S. Hosono, H. Tano and H. Kigoshi, Org. Lett., 2023, 25, 4725–4729 CrossRef CAS PubMed.
- Z. Tan, X. Ju, H. Wu, W. Dong, J. C. Leung, X. Hou, H. Lee, A. Granger, J. M. Paolillo, S. V. DiMeo, C. Chen, L. Wu, J. C. Lorenz, M. Sarvestani, F. Buono, R. Frutos, T. G. Tampone, X. Huang, G. Zhang, Y. Wang, E. Spinelli, Z. Lei and J. J. Song, Org. Process Res. Dev., 2024, 28, 78–91 CrossRef CAS.
- P. Jiang, S. Wu, G. Wang, S. Xiang and B. Tan, Angew. Chem., Int. Ed., 2023, 40, e202309272 Search PubMed.
- C. Michelin and N. Hoffmann, ACS Catal., 2018, 8, 12046–12055 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- D. E. Damschen, C. D. Merritt, D. L. Perry, G. W. Scott and L. D. Talley, J. Phys. Chem., 1978, 82, 2268–2272 CrossRef CAS.
- C. Ouannès, R. Beugelmans and G. Roussi, J. Am. Chem. Soc., 1973, 95, 8472–8474 CrossRef.
- M. Demuth, P. R. Raghavan, C. Carter, K. Nakano and K. Schaffner, Helv. Chim. Acta, 1980, 63, 2434–2439 CrossRef CAS.
- H. Rau and M. Hörmann, J. Photochem., 1981, 16, 231–247 CrossRef CAS.
- A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139–1140 CrossRef CAS PubMed.
- R. Sebesta, Beilstein J. Org. Chem., 2022, 18, 240–242 CrossRef CAS PubMed.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed.
- D. F. Cauble, V. Lynch and M. J. Krische, J. Org. Chem., 2003, 68, 15–21 CrossRef CAS PubMed.
- J. Großkopf, M. Plaza, A. Seitz, S. Breitenlechner, G. Storch and T. Bach, J. Am. Chem. Soc., 2021, 143, 21241–21245 CrossRef PubMed.
- J. Großkopf, A. A. Heidecker and T. Bach, Angew. Chem., Int. Ed., 2023, 62, e202305274 CrossRef PubMed.
- C. Müller, A. Bauer, M. M. Maturi, M. C. Cuquerella, M. A. Miranda and T. Bach, J. Am. Chem. Soc., 2011, 133, 16689–16697 CrossRef PubMed.
- M. M. Maturi, A. Pöthig and T. Bach, Aust. J. Chem., 2015, 68, 1682–1692 CrossRef CAS.
- L. Wimberger, T. Kratz and T. Bach, Synthesis, 2019, 51, 4417–4424 CrossRef CAS.
- M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 7661–7664 CrossRef CAS PubMed.
- R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
- X. Li, C. Jandl and T. Bach, Org. Lett., 2020, 22, 3618–3622 CrossRef CAS PubMed.
- A. Tröster, R. Alonso, A. Bauer and T. Bach, J. Am. Chem. Soc., 2016, 138, 7808–7811 CrossRef PubMed.
- T. Kratz, P. Steinbach, S. Breitenlechner, G. Storch, C. Bannwarth and T. Bach, J. Am. Chem. Soc., 2022, 144, 10133–10138 CrossRef CAS PubMed.
- A. Tröster, A. Bauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 3538–3541 CrossRef PubMed.
- X. Li, J. Großkopf, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2021, 60, 2684–2688 CrossRef CAS PubMed.
- A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed.
- M. Plaza, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 12785–12788 CrossRef CAS PubMed.
- M. Plaza, J. Großkopf, S. Breitenlechner, C. Bannwarth and T. Bach, J. Am. Chem. Soc., 2021, 143, 11209–11217 CrossRef CAS PubMed.
- F. Mayr, L.-M. Mohr, E. Rodriguez and T. Bach, Synthesis, 2017, 49, 5238–5250 CrossRef CAS.
- F. Pecho, Y. Zou, J. Gramüller, T. Mori, S. M. Huber, A. Bauer, R. M. Gschwind and T. Bach, Chem.–Eur. J., 2020, 26, 5190–5194 CrossRef CAS PubMed.
- F. Pecho, Y. Sempere, J. Gramüller, F. M. Hörmann, R. M. Gschwind and T. Bach, J. Am. Chem. Soc., 2021, 143, 9350–9354 CrossRef CAS PubMed.
- R. Takagi and T. Tanimoto, Org. Biomol. Chem., 2022, 20, 3940–3947 RSC.
- S. C. Huo, R. R. Indurmuddam, B.-C. Hong, C.-F. Lu and S.-Y. Chien, Org. Biomol. Chem., 2023, 21, 9330–9336 RSC.
- J. Lyu, A. Claraz, M. R. Vitale, C. Allain and G. Masson, J. Org. Chem., 2020, 85, 12843–12855 CrossRef CAS PubMed.
- J. Lyu, M. Leone, A. Claraz, C. Allain, L. Neuville and G. Masson, RSC Adv., 2021, 11, 36663–36669 RSC.
- T. Rigotti, A. Casado-Sánchez, S. Cabrera, R. Pérez-Ruiz, M. Liras, V. A. de la Peña O'Shea and J. Alemán, ACS Catal., 2018, 8, 5928–5940 CrossRef CAS.
- W. Ding, L.-Q. Lu, Q.-Q. Zhou, Y. Wei, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 63–66 CrossRef CAS PubMed.
- W. H. Pirkle and D. S. Reno, J. Am. Chem. Soc., 1987, 109, 7189–7190 CrossRef CAS.
- R. J. Kutta, J. Großkopf, N. Van Staalduinen, A. Seitz, P. Pracht, S. Breitenlechner, C. Bannwarth, P. Nuernberger and T. Bach, J. Am. Chem. Soc., 2023, 145, 2354–2363 CrossRef CAS PubMed.
- C. Müller, A. Bauer and T. Bach, Angew. Chem., Int. Ed., 2009, 48, 6640–6642 CrossRef PubMed.
- A. Bakowski, M. Dressel, A. Bauer and T. Bach, Org. Biomol. Chem., 2011, 9, 3516–3529 RSC.
- M. M. Maturi, M. Wenninger, R. Alonso, A. Bauer, A. Pöthig, E. Riedle and T. Bach, Chem.–Eur. J., 2013, 19, 7461–7472 CrossRef CAS PubMed.
- L. Wang, Z. Li, L. Lu and W. Zhang, Tetrahedron, 2012, 68, 1483–1491 CrossRef CAS.
- E. C. Lathioor and W. J. Leigh, Photochem. Photobiol., 2006, 82, 291–300 CrossRef CAS PubMed.
- Z.-L. Xia, Q.-F. Xu-Xu, C. Zheng and S.-L. You, Chem. Soc. Rev., 2020, 49, 286–300 RSC.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed.
- K. Yuan, P. Wang, H.-X. Li, Y.-Z. Liu and L.-L. Lv, Org. Chem. Front., 2020, 7, 3656–3663 RSC.
- X. Li, R. J. Kutta, C. Jandl, A. Bauer, P. Nuernberger and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 21640–21647 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Wei He
*a and
Wei He