
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C12 aromatic triol-furoin and diol-furil from bio-based 5-(hydroxymethyl)furfural: enhanced selective synthesis, scale-up and mechanistic insight into cyclic catalysis†
Thi Tuyet Thuy
Vu
a,
Shentan
Liu
*ab,
Mantas
Jonušis
 c,
Simona
Jonušienė
c,
Jinsik
Choi
d,
Mohamed
Ismail
a,
Nicola
Rehnberg
e,
Rajni
Hatti-Kaul
a and
Sang-Hyun
Pyo
*a
c,
Simona
Jonušienė
c,
Jinsik
Choi
d,
Mohamed
Ismail
a,
Nicola
Rehnberg
e,
Rajni
Hatti-Kaul
a and
Sang-Hyun
Pyo
*a
aBiotechnology, Department of Chemistry, Faculty of Engineering, Lund University, SE-22100 Lund, Sweden. E-mail: sang-hyun.pyo@biotek.lu.se
bCollege of Geology and Environment, Xi'an University of Science and Technology, Xi'an, 710054, Shaanxi, China. E-mail: liushentan@xust.edu.cn
cVU LSC Institute of Biochemistry, Mokslininku st 12A, LT-08412 Vilnius, Lithuania
dChemical R&D Center, Samyang Corporation, 730 Daeduck-daero, Daejeon, 34055, South Korea
eR&D, Bona Sweden AB, Box 210 74, 200 21 Malmö, Sweden
First published on 7th October 2024
Abstract
In this study, we investigate the valorization of 5-(hydroxymethyl)furfural (5-HMF), a versatile and pivotal renewable C6 platform chemical, into a C12 heteroaromatic triol, 5,5′-bis(hydroxymethyl)furoin (DHMF), and a C12 heteroaromatic diol, 5,5′-bis(hydroxymethyl)furil (BHMF). The carboligation of 5-HMF to DHMF is catalyzed by an N-heterocyclic carbene, 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene (TPT), generated in situ from its stable methoxy adduct, 5-methoxy-1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazoline (TPA-OMe). This reaction achieves quantitative yield in dimethyl carbonate, a more environmentally friendly solvent. The resulting DHMF precipitate was readily purified via simple filtration and washing. Moreover, an enhanced selective oxidation was conducted at the secondary hydroxyl group of DHMF to generate the ketone group of BHMF in quantitative yield by using organo-catalysts, anionic exchanger, and NaOH. We proposed and subsequently validated a cyclic catalysis mechanism for the oxidation through the colorimetric detection of the by-product, H2O2, in the reaction. All synthetic processes to produce these C12 triol-furoin and diol-furil compounds were successfully demonstrated on a scale ranging from 20 to 400 grams. The feasibility of these processes was established with high yields achieved under moderate reaction conditions and ambient pressure, making them suitable for large-scale production. Consequently, these C12 multi-functional chemicals can find applications in the production of bio-based aromatic polymers such as polyesters, polyurethanes, and polycarbonates.
Introduction
Fossil-derived resources currently serve as the primary sources of carbon building blocks for fuels, chemicals, and polymers.1,2 The pursuit of sustainable production for these materials from bio-based renewable resources, utilizing environmentally friendly processes, has prompted both academia and industry to explore bio-based alternatives with green processes.3–95-(Hydroxymethyl)furfural (5-HMF) stands out as a widely studied bio-platform chemical, thanks to its versatile chemical reactivity and functionality.10,11 Recent efforts have focused on investigating new synthetic conditions, catalyst development (both homocatalysts and heterocatalysts), raw materials (such as glucose, fructose, and lignocellulosic hydrolysates), and process optimization (both batch and continuous modes) to achieve high-yield production of 5-HMF in a sustainable manner, aligning with commercialization interest.12,13 In our recent work, we reported a high yield of HMF obtained from a high concentration (20–30 wt%) of fructose by acid catalysis in a water–dimethyl carbonate two-phase system8 and by H+ exchange heterogeneous catalysis, respectively.9,14 Moreover, various valuable chemicals, including dimethylfuran (DMF),15 levulinic acid,16 alkyl levulinate,17 2,5-furandicarboxylic acid (FDCA),7,18–20 5-(hydroxymethyl)furan-2-carboxylic acid (HMFCA),21 2,5-diformylfuran (DFF),22 5-hydroxymethyl-2-furfurylamine,23,24 2,5-bis(hydroxymethylfuran),25 5-hydroxy-4-keto-2-pentenoic acid (HKPA),26 γ-valerolactone (gVL),27 1,6-hexanediol (1,6-HD),28 and adipic acid (AA),29–31 can be derived through the oxidation, reduction, esterification, amination, and hydrogenation of HMF (Scheme 1). These compounds can serve as crucial building blocks in the polymer industry.
 | ||
| Scheme 1 Production and valorization of HMF from biomass. Diformylfuran (DFF), 5-(hydroxymethyl)furan-2-carboxylic acid (HMFCA), 2,5-furan dicarboxylic acid (FDCA), 2,5-bis(hydroxymethyl)furan (BHMF), 4-hydroxy-2-keto-pentanoic acid (HKPA), γ-valerolactone (gVL), 2,5-bis(hydroxymethyl)tetrahydrofuran (BHMTF), 2,5-dimethylfuran (DMF), 5-hydroxymethyl-2-furfurylamine (HMFA), levulinic acid (LA), alkyl levulinate (AL), 1,6-hexanediol (1,6-HD) and adipic acid (AA). The bold arrows indicate the synthesis route of C12 triol 5,5′-bis(hydroxymethyl)furoin (DHMF) and C12 diol 5,5′-bis(hydroxymethyl)furil (BHMF) through 5-HMF from biomass-derived fructose. | ||
As a means of further valorization to synthesize larger multi-functional chemicals, the C–C bond formation through benzoin-type self-condensation of 5-HMF by various chemicals and biocatalysts, such as N-heterocyclic carbenes (NHCs), thiazolium ionic liquids, and enzymes, has been reported to yield C12 aromatic triol, 5,5′-bis(hydroxymethyl)furoin (DHMF).32–36 The process involves carboligation through an aldehyde of 5-HMF, leading to its self-coupling to form DHMF. DHMF, a novel bio-based difuranic polyol scaffold featuring 12 carbons, 3 hydroxyl groups, 2 substituted furan rings, and 1 carbonyl group (Scheme 1), is proposed as a precursor for oxygenated diesel fuels with high energy density or high-quality C12 linear alkane fuels.37,38 It has recently found application as a monomer for the preparation of cross-linked polyesters and polyurethane.36,39,40
Furthermore, DHMF can undergo further oxidation to yield the diol, 5,5′-bis(hydroxymethyl)furil (BHMF),36,40 featuring 12 carbons, 2 hydroxyl groups, 2 substituted furan rings, and 2 carbonyl groups (Scheme 1), with an organo-catalyst, active manganese dioxide and biocatalyst.36,40,41 BHMF stands out as a particularly attractive bio-based diol monomer due to the intriguing physical and mechanical properties conferred by its rigid heteroaromatic structure to the derived polymers.36,40,42 Given the growing interest in these C12 platform chemicals, there is a need for further development in the production processes, specifically focusing on refining reaction conditions, catalysts, and mechanisms to facilitate efficiency and scaling-up for industrial applications, all while addressing environmental and safety concerns.
In this study, we explored a straightforward and environmentally friendly synthetic route for the production of C12 aromatic multi-functional platform chemicals from bio-based 5-HMF (Scheme 1), taking into account both green chemistry and economic considerations. The carboligation of 5-HMF, involving C–C bond formation, was further developed to achieve quantitative yields of DHMF in various solvents by employing an NHC generated in situ within the reaction medium. Subsequently, the obtained DHMF was transformed into C12 BHMF through the selective oxidation of secondary alcohols. This oxidation process was examined using various base organocatalysts, heterogeneous anion exchangers, and NaOH. Moreover, we proposed and subsequently confirmed a cyclic catalysis mechanism for the oxidation, initially introducing the concept and later validating it through colorimetric detection of the by-product, H2O2, in the mechanism. Consequently, these processes offer facile and resource-efficient approaches, positioning them as valuable tools for industrial-scale applications.
Experimental
Materials
5-HMF (98% purity, Angene International Limited, China), the anionic exchanger Ambersep 900 (Hydroxy form, Fluka), and sodium hydroxide (VWR) were employed in this study. The cationic exchanger, SPC 260H (Hydrogen form, dry and wet), was generously provided by Samyang Corporation (Republic of Korea). L-Proline (99%), 4-(dimethylamino)pyridine (DMAP), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), and titanium(IV) butoxide (Ti(OBu)4), along with tetrahydrofuran (THF), ethyl acetate, dichloromethane (DCM), dimethyl carbonate (DMC), acetonitrile, diethyl ether, and dimethylformamide (DMF), were procured from Sigma-Aldrich. All chemicals were used without further treatment.Carboligation of pure and partially purified 5-HMF to produce DHMF through HNC catalysis
An NHC catalyst (TPA-OMe), 5-methoxy-1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazoline, was prepared as almost colorless to cream-colored crystals, following a modified procedure outlined in a previous report.43 The resulting product was characterized using 1H-NMR and 13C-NMR, and the crystals were stored at room temperature for subsequent use (Fig. S1†).The carboligation of 5-HMF was conducted using a method reported earlier with modifications.35,40 For a small-scale experiment, 0.5 g of purified 5-HMF and 1 mL of various solvents such as diethyl ether, dichloromethane (DCM), THF, MIBK, ethyl acetate, and DMC, respectively, were placed in a 4 mL glass vial. Subsequently, 1.1 mol% of the TPA-OMe catalyst was added to the vial. The solution was heated and shaken using a thermomixer (HTMR 131, HLC BioTech, Germany) at 40–60 °C and 600 rpm for 1 hour. During the reaction, the product (DHMF) precipitated. After cooling to room temperature, the solid was isolated by filtration and washed with a solvent. The isolated DHMF and the residual 5-HMF in solution were analyzed to calculate the product yield using GC–MS and 1H-NMR.
For the carboligation of 5-HMF on a larger scale, 400 g of 5-HMF and 800 mL of DMC were introduced into a 2 L glass vessel, followed by the addition of the TPA-OMe catalyst (1.1 mol% relative to 5-HMF). The reaction was conducted in a water bath at 60 °C with magnetic stirring for 1 hour. After cooling to room temperature, the solid product was isolated by filtration and washed with DMC. In the end, 380 g of purified DHMF was obtained, demonstrating a 95% yield, as confirmed by 1H-NMR.
Selective oxidation of DHMF to BHMF by various catalysts
The selective oxidation of DHMF to BHMF was conducted under various solvent conditions using different catalysts. The reaction was explored with various types of catalysts, including base organocatalysts (4-(dimethylamino)pyridine (DMAP, pKa 9.6), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKa 13.5 ± 1.5), and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, pKa 15.2 ± 1.0)), anion exchange resins (Ambersep 900, hydroxy form, wet), cation exchange resins (SPC 260H, hydrogen form, dry), L-proline, Ti(OBu)4, and NaOH with or without molecular sieves in THF, respectively. Additionally, the solvent effect on DBU was investigated in various solvents such as methyltetrahydrofuran (M-THF), ethanol, or dimethylformamide (DMF), with or without molecular sieves, respectively.For screening and optimization under aerobic conditions with open access to O2, 0.5 g of DHMF and 15 mL of solvent were placed in a 50 mL flask equipped with a condensing cylinder. Subsequently, the catalyst was added at a specific ratio. The reaction was carried out with slow air bubbling at 65 °C and magnetic stirring, both with and without molecular sieves.
Under closed conditions, 50 mg of DHMF and 1.5 mL of solvent were placed in a 4 mL vial, followed by the addition of the catalyst. The reaction was conducted in a closed system at 60 °C with shaking at 600 rpm using a thermomixer (HTMR 131, HLC BioTech, Germany).
For the large-scale reaction, 20 g of DHMF in 500 mL of THF was placed in a 1 L flask equipped with a condensing cylinder, followed by the addition of 10 g of molecular sieves and 0.6 g of DBU (5 mol% to DHMF). The reaction was carried out with slow air bubbling at 65 °C and magnetic stirring, both with molecular sieves. Samples (500 μL each) were collected at different reaction times and analyzed to assess the conversion of DHMF and the production of BHMF using HPLC or silica-TLC in ethyl acetate. After the reaction was complete, the reaction mixture was cooled to room temperature, followed by evaporation to remove THF. The resulting solid product was washed with diethyl ether and analyzed by 1H-NMR.
Additionally, 10 g (0.5 ratio to DHMF) of NaOH and 10 g (0.5 ratio to DHMF) of anionic exchanger were employed in separate reactions for 20 g DHMF, replacing DBU, under the same conditions.
To determine the presence of hydrogen peroxide, a by-product, potassium iodide (KI) in glacial acetic acid was employed as the colorimetric reagent.44 As previously reported with further optimization, 0.75 mg mL−1 of DBU in DI water and 25 mg mL−1 of DHMF in DI water were prepared. Following the oxidation reaction of DHMF with and without molecular sieves using DBU in THF at 9 hours, 200 μL reaction samples were extracted and dried. The resulting dried samples were treated with 200 μL of 100% acetic acid, 200 μL of deionized water, and 16 mg of KI, respectively. The color changes before and after the addition of these agents were observed after 1 minute when the triiodide was produced from the oxidation of KI with H2O2.45
Analytical procedures
The conversion of the substrates and the concentrations of the products formed were calculated from the standard curves on the GC and HPLC chromatograms or by 1H-NMR.Quantitative analysis of 5-HMF was conducted using gas chromatography–mass spectrometry (GC–MS, 431-GC and 210-MS, Varian, USA) equipped with a FactorFour Capillary column, VF-1ms (Varian, 15 M × 0.25 mm). The initial column oven temperature was raised from 50 to 250 °C at a rate of 20 °C min−1. The samples, diluted with acetonitrile to a concentration of 0.1–0.5 mg mL−1, were injected in the split injection mode of 10% at 275 °C. The conversion and concentration of the substrates were calculated from the calibration curves of the standard materials on the chromatograms.
The concentrations of 5-HMF, DHMF, and BHMF were determined using an HPLC system (HP Agilent 1100, Palo Alto, USA) equipped with a diode-array detector and a reversed-phase chromatography column (C18, Kromasil, Sweden) connected to a C18 guard column (Kromasil, Sweden). Chromatography was conducted at a column temperature of 30 °C, employing a 20–80% (v/v) acetonitrile gradient at 0.6 mL min−1, and monitored by UV detection at 254 and 280 nm. Prior to injection, the samples were diluted with acetonitrile and filtered. A 10 μL aliquot was then injected. The peaks of different compounds were confirmed and quantified using external standards. The calculated reaction parameters included the percent substrate conversion (mol mol−1) and percent product yield (mol mol−1).
The chemical structures of HMF, DHMF, and BHMF were confirmed using external standard materials in HPLC and GC chromatograms or elucidated by 1H-NMR and 13C-NMR (DMSO-d6 or CDCl3) using a 400 MHz NMR instrument (Bruker, UltraShield Plus 400, Germany).
All data were obtained from two independent experiments and are presented as the average of replicates ± standard deviation.
Results and discussion
After extensive investigations and reporting on the production of 5-HMF from C6 sugars and hemicellulose hydrolysates over the past decade,11 interest and focus have shifted towards upgrading it to more value-added and useful fine chemicals for polymer production and liquid fuel applications.38 These fine chemicals primarily include C5–C6 or their derivatives, such as aromatic single-furan compounds (HMFCA and FDCA) and aliphatic compounds (adipic acid, hexanediol, and levulinic derivatives), derived through the oxidation, reduction, esterification, and dehydration of HMF (Scheme 1). Simultaneously, carbon–carbon (C–C) bond formation stands out as a fundamental process in chemistry, where two carbon atoms unite to create a covalent bond.46,47 The carboligation of the aldehyde group in 5-HMF is crucial as a new C–C bond formation pathway, providing a means to produce larger molecules, specifically C12 aromatic multi-functional substances.48 This is achieved through organocatalytic self-condensation coupling of HMF to DHMF, followed by its oxidation to BHMF.49,50Solvent selection for carboligation of 5-HMF to DHMF through in situ NHC catalysis
In previous reports,34–37,39–42 C–C bond formation through a benzoin-type self-condensation of 5-HMF was employed to produce C12 DHMF using various chemicals and biocatalysts such as NHCs, thiazolium ionic liquids, whole cells, and enzymes. A methanol adduct of 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene (TPT), 5-methoxy-1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazoline (TPT-OMe), was directly employed for the self-condensation coupling of HMF in dichloromethane (DCM) at 40 °C, resulting in 95% isolated yield of DHMF.40 In our recent investigation, the carboligation reaction was studied using whole cells of Escherichia coli with recombinant P. fluorescens benzaldehyde lyase in a fed-batch reaction with a feed of 20 g L−1 5-HMF in 10% dimethyl carbonate (DMC).36In this study, the carboligation of 5-HMF and the resulting DHMF purification were further developed in various solvents, including DMC, THF, diethyl ether, ethyl acetate, DCM, and MIBK (Fig. 1). Except for diethyl ether, which underwent phase separation with 5-HMF, all the solvents dissolved 5-HMF before the reaction, and the resulting product precipitated during the reaction. For purification purposes, DMC, DCM, and EA were preferred due to the low solubility of DHMF and high solubility of 5-HMF. These solvents could be used for simple washing to remove any remaining 5-HMF and the organocatalyst. Consequently, high-purity DHMF was obtained with a high recovery yield using a straightforward separation process without the need for chromatography or recrystallization. DMC is the preferred solvent and is considered one of the green solvents.51 It is a nonpolar aprotic solvent with good miscibility with water and is non-toxic. DMC readily biodegrades in the atmosphere and is classified in the greenest “recommended” bracket according to the solvent selection guide.52 New alternative production processes of DMC from CO and CO2 are being developed.53
 | ||
| Fig. 1 Effects of solvents on the carboligation of 5-HMF (0.5 g) in 2.5 mL (a) dimethylcarbonate, (b) THF, (c) diethylether, (d) ethyl acetate, (e) dichloromethane, and (f) MIBK using TPT-Ome (1.1 mol%) as the catalyst at 40–60 °C. (A) Conversion of 5-HMF and yield of DHMF in the precipitate and solution. (B) Picture of the reaction solutions at 1 h. (C) Reaction at 400 g HMF scale in dimethyl carbonate. (D) DHMF purified from the reaction (C). | ||
Meanwhile, the carboligation was performed using TPT-OMe, which can be converted to its carbene form under the various reaction conditions. This can eliminate the need for an additional step to convert TPT-OMe to TPT in the synthesis and enable the upgrading of 5-HMF to DHMF. Thus, the modified mechanism of the carboligation of 5-HMF to DHMF in DMC, involving in situ NHC generation from TPA-OMe in cyclic catalysis, is proposed in Fig. S2.† TPA-OMe in solid form is exceptionally stable under ambient conditions for up to a year, even when exposed to air and moisture during storage and handling. However, it was slightly decomposed in DCM at 40 °C for 1 h (reaction conditions), which was performed without a substrate. Additionally, the recovered catalyst solution from the first batch reaction (conversion of 5-HMF to DHMF) exhibited very low activity in the second batch, with almost no reaction observed. Therefore, the catalyst could not be reused in this study.
The process was scaled up to a 400 g scale of 5-HMF in DMC, quantitatively producing DHMF, with a 95% isolated yield after simple separation (Fig. 1C and D). Thus, it is noteworthy that the upgrading of 5-HMF to C12 triol DHMF could be performed by in situ NHC generation from the stable precursor NHC-OMe in cyclic catalysis in a more environmentally friendly solvent, DMC, instead of dichloromethane. These factors are very important for scaling up and industrialization.
Enhanced selective oxidation of DHMF to BHMF by various catalysts
The selective oxidation of secondary alcohols over primary alcohols in a molecule holds significance due to the distinct chemical reactivity and functional groups present in the compound. The triol DHMF can undergo further conversion to the diol BHMF through the selective oxidation of the secondary alcohol in DHMF using basic catalysts. This specifically targets the secondary alcohol groups, converting them into carbonyl (ketone) functional groups. BHMF, being a more stable chemical, is formed through the slow auto-oxidation of DHMF. It is also employed as a functional monomer in the production of polymers, including (co)-polyurethane and (co)-polyesters.Consequently, both DHMF and BHMF can serve as valuable monomers for the formation of cross-linked or linearly linked structures during polymerization.40
In a prior study by Mou et al., DBU was found to catalyze the oxidation of DHMF to BHMF with an 86% yield (after chromatography) over 24 hours, while a 95% BHMF yield was achieved after separation using a 200% (mole) ratio of active manganese dioxide (Table 1). In order to enhance the conversion rate of DHMF and the yield of BHMF while avoiding the use of excess manganese dioxide, further investigation into the oxidation was carried out using various types of catalysts, both with and without scavengers, such as molecular sieves. Additionally, the oxidation mechanism of DHMF to BHMF was initially explored to gain insight into and improve the organo-base catalysis.
| Run | DHMF (g, scale) | Catalyst (weight % to DHMF) | Solvent | Conditions (with air) | BHMF yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a Isolation yield. b Hexadecyltrimethylammonium bromide (CTAB). c Tetramethylammonium bromide (TMAB). d 1,8-Diazabicyclo(5.4.0)undec-7-ene (DBU). e Bioconversion using benzaldehyde lyase (BAL, 1 mg mL−1), 40 mM thiamine-diphosphate in 40 mL potassium phosphate buffer solution (PBS) and 20% vol DMSO. f Bioconversion using recombinant E. coli cells expressing P. fluorescens benzaldehyde lyase (0.2–10 gcdw L−1) in 4 mL potassium phosphate buffer solution (PBS) and 10% vol dimethyl carbonate. g Anionic exchanger, Ambersep 900 (Hydroxy form) (A900). | ||||||
| 1 | 0.25 | [CTAB]4Mo8O26 (10)b | Acetic acid | 110 °C, 2 h | 91.5a | 2018, ref. 54 |
| 2 | 0.25 | [TMAB]4Mo8O26 (10)c | Acetic acid | 110 °C, 2 h | 61.5 | 2018, ref. 54 |
| 3 | 2.52 | Active MnO2 (200 mol%) | THF | RT, 20 h | 95a | 2016, ref. 40 |
| 4 | 0.756 | DBU (5 mol%)d | THF | 70 °C, 24 h | 86a | 2016, ref. 40 |
| 5e | 250 mM | BAL (1 mg mL−1) | 40 mL PBS/20% DMSO | pH 8, RT, 18 h | 10 | 2015, ref. 42 |
| 6f | 5–10 g L−1 | 0.2–10 gcdw L−1 per cell (BAL) | 4 mL PBS/10% DMC | pH 8, 30 °C, 72 h | 96.7 | 2023, ref. 36 |
| 7 | 20 | DBU (5 mol%)/MS | THF | 70 °C, 11 h | 98(94)a | This study |
| 8 | 10 | A900g (50)/MS | THF | 70 °C, 11 h | 99(93)a | This study |
| 9 | 10 | NaOH (50) | THF | 70 °C, 11 h | 95(89)a | This study |
The catalytic performances of typical catalysts, including organo-bases, metal bases, cation and anion exchangers, and inorganic bases, such as 4-(dimethylamino)pyridine (DMAP, pKa 9.6), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKa 13.5 ± 1), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, pKa 15.2 ± 1.0), titanium butoxide (Ti(OBu)4), L-proline, NaOH, and cation and anion exchangers, were compared based on DHMF conversion to BHMF in the oxidation process.
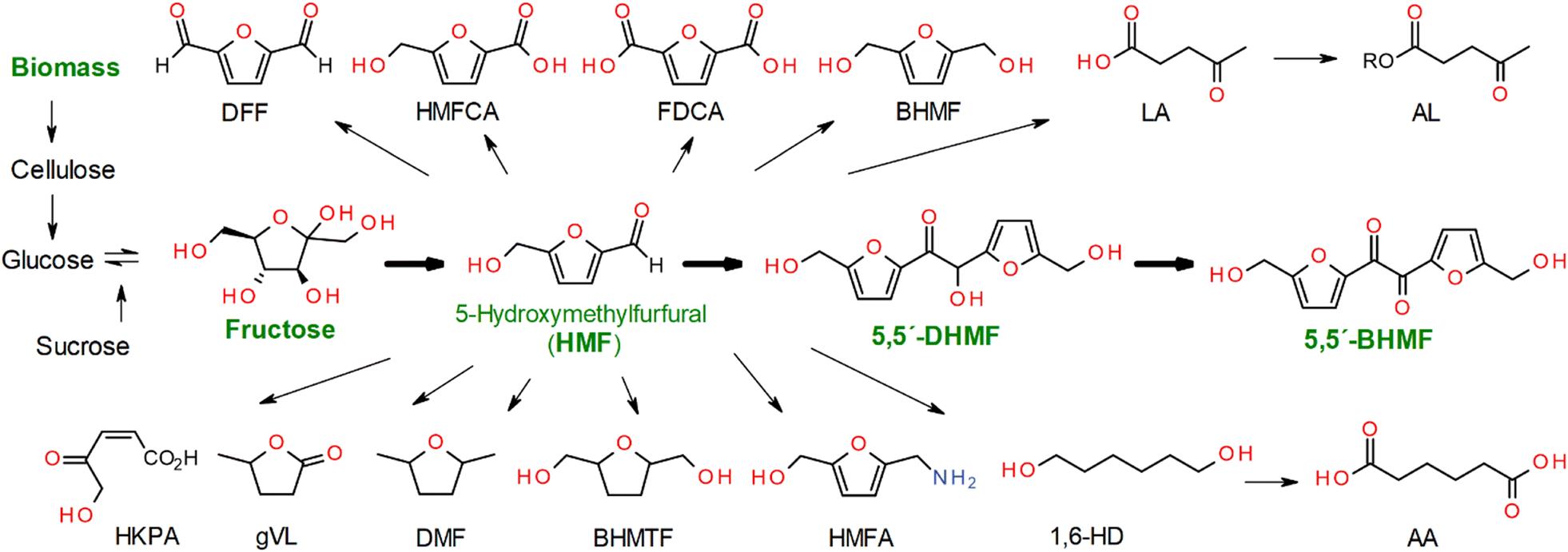
Under non-aerated conditions using an organo-catalyst (5 mol% relative to DHMF), BHMF was obtained in a 34.8% yield at 12 hours using DBU, while yields of 14.1%, 34.3%, and 7.8% were achieved at 12 hours using 5 mol% DMAP, TBD, and L-proline (10 wt%), respectively (Fig. 2A).
 | ||
| Fig. 2 Selective oxidation of DHMF to BHMF. (A) BHMF yield (%) using various 5% (mol mol−1) organo- and metal-based catalysts, respectively, after 12 h of the reaction under closed conditions at 65 °C. (B) BHMF yield (%) after 12 h of the reaction using DBU and TBD under open and closed conditions in air with or without MS in THF. | ||
Additionally, 23.2% and 8.5% of BHMF were obtained using inorganic bases, titanium butoxide (5 mol%) and NaOH (10 wt%), respectively. In the case of ion exchangers (20 wt%), the anionic exchanger showed a higher yield (25.1%) compared to the cationic exchanger (2.5%) under the same conditions. These results suggest that the oxidation process is more effective in the presence of a basic organocatalyst than with a metal-base or acidic catalyst.
The greater the basicity, the greater the catalytic power of the organo-base catalyst. While this observation is consistent with the behavior of DMAP, DBU exhibited similar catalytic ability to TBD, a catalyst known for its bifunctional activity and higher basicity.55 We hypothesize that TBD loses its bifunctional activity after protonation due to the presence of hydroxyl groups in DHMF and water molecules generated from the decomposition of H2O2 in the cyclic catalysis. A similar phenomenon is responsible for the increase in DBU activity, where protonation leads to the formation of a base–conjugate acid pair [DBU/DBUH]+, creating a bifunctional mode of action.55,56
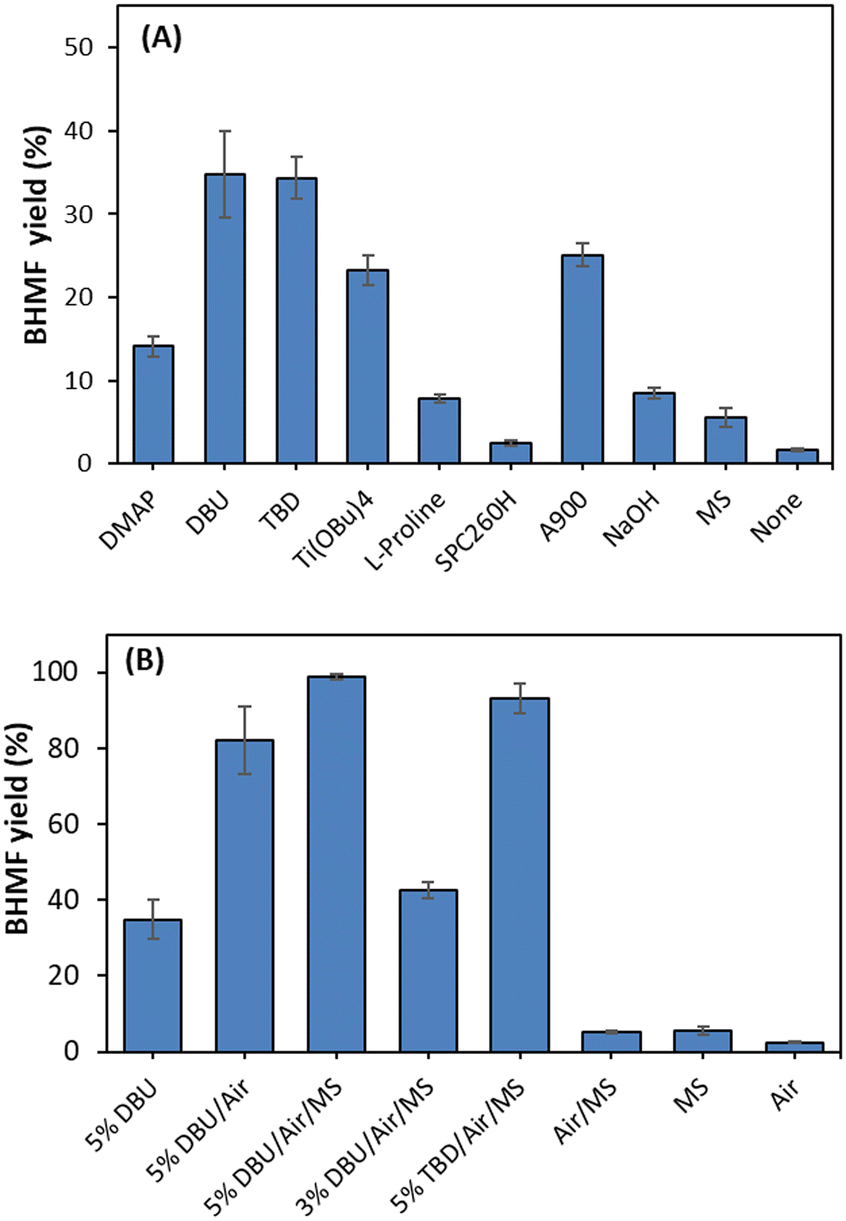
Auto-oxidation was observed, but with a low conversion (1.7%) of DHMF in the absence of a catalyst. The catalytic effect of DBU was further explored with molecular sieves and aeration (O2), where the role of molecular sieves and aeration was also monitored in the absence of DBU (Fig. 2). A quantitative yield of BHMF was achieved at 11 hours using 5 mol% DBU with molecular sieves in THF (Fig. 2B and 3). This was compared to the oxidation of DHMF without molecular sieves under slow aeration conditions within the same reaction time (Fig. 2B and 3). The oxidation of DHMF was significantly enhanced with molecular sieves under slow aeration conditions, resulting in a quantitative yield of BHMF at 11 hours, while an 82.1% yield of BHMF was obtained without the use of molecular sieves. Compared to the result without molecular sieves and aeration (34.8%), aeration played an important role in the oxidation, making it necessary.
 | ||
| Fig. 3 Solvent effects on the selective oxidation of DHMF to BHMF using 5% (mol mol−1) DBU in THF with and without molecular sieves (MSs), and methyl-THF (M-THF) and ethanol. | ||
The solvent effect was investigated using ethanol and methyltetrahydrofuran, which are renewable solvents. However, lower yields (79% and 82%) of BHMF were obtained under the same conditions, respectively (Fig. 3).
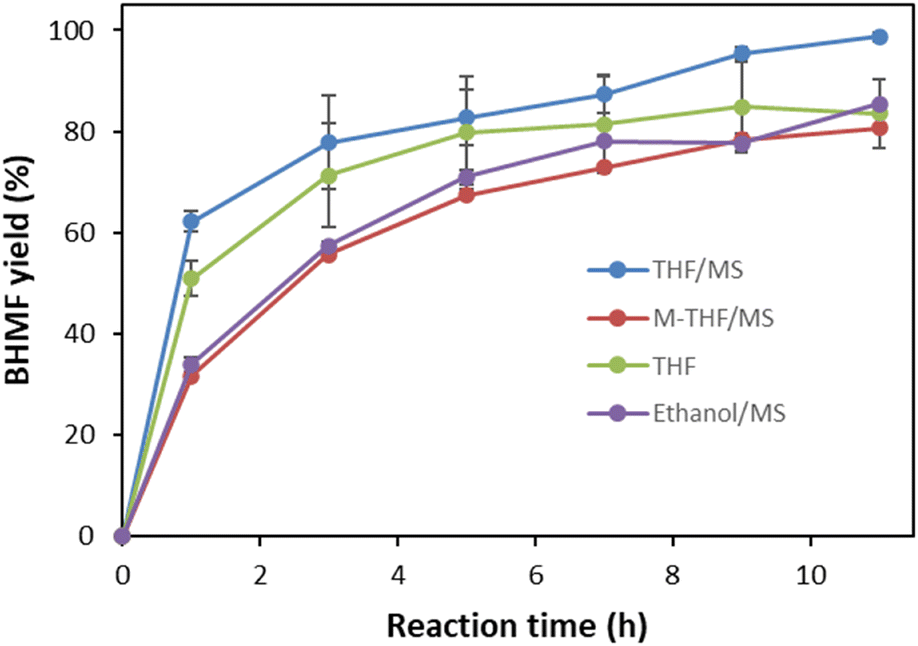
The process was scaled up to a 20 g scale of DHMF in THF, producing BHMF with a 92% isolated yield after simple separation (Fig. 4; Table 1, run 7). Thus, it is noteworthy that the oxidation of C12 triol DHMF to C12 diol BHMF could be performed quantitatively with organo-catalysts. The resulting BHMF was purified by washing the catalyst with diethyl ether after THF removal. Therefore, this process provides an enhanced reaction and simple purification method for the production of BHMF, and thus can be applied in large scale.
 | ||
| Fig. 4 Scaling up of the selective oxidation of 20 g DHMF to BHMF using 5% (mol mol−1) DBU in THF with molecular sieves (MSs) at 65 °C. HPLC chromatograms of (A) the reactant at 8 h, (B) the reactant at 12 h, and (C) purified BHMF. (D) Picture of the reaction setup equipped with a condenser and air bubbling for the 20 g DHMF reaction using 5% (mol mol−1) DBU in THF with molecular sieves (MSs). (E) Picture of purified BHMF. | ||
Heterogeneous catalysis using NaOH and anion exchanger A900 was employed for the oxidation of DHMF to BHMF in THF (Fig. 5). Although high ratios (0.5 and 1 (w/w) to DHMF) of NaOH and A900 were required, they could oxidize DHMF to BHMF with high yield under optimum conditions at 11 hours and 7 hours, respectively.
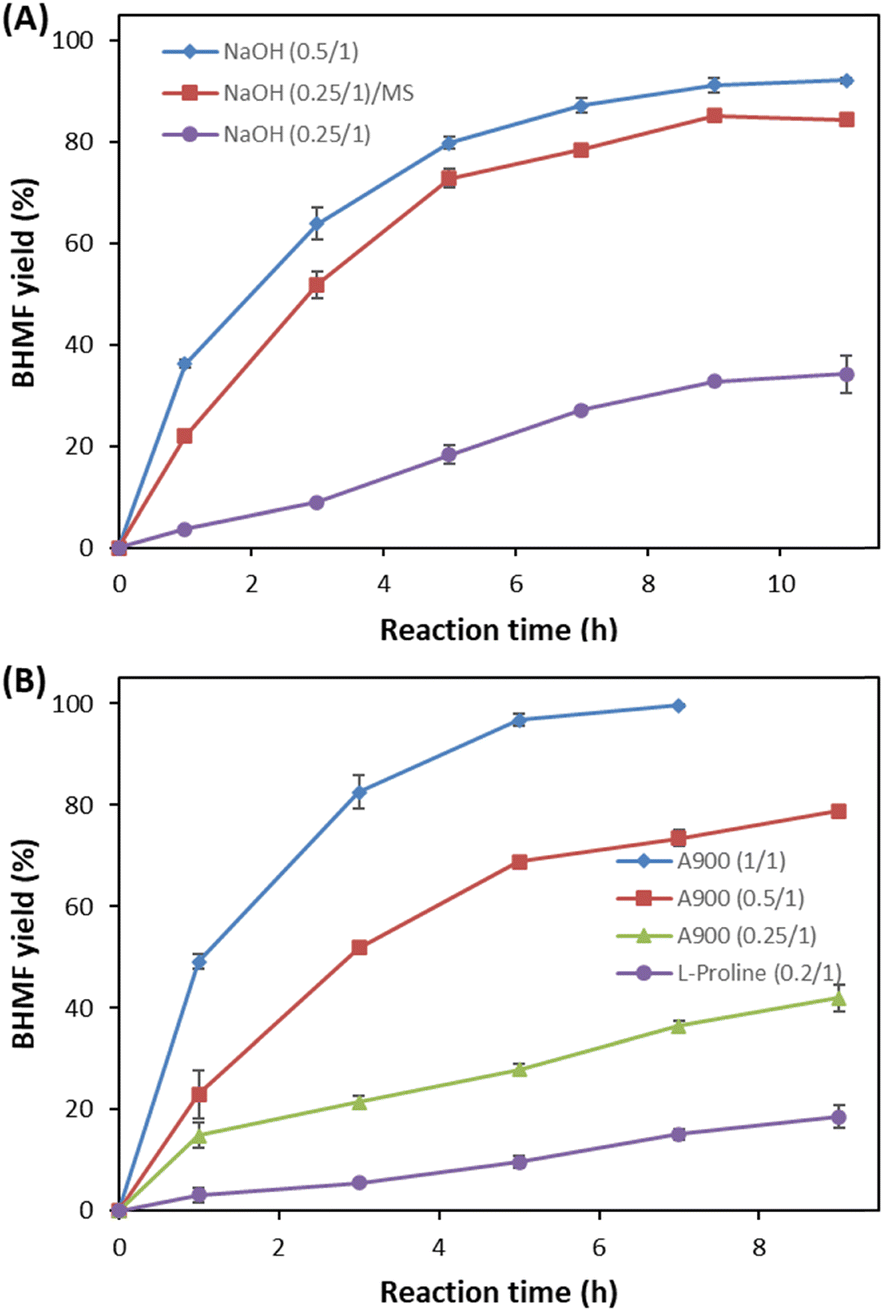 | ||
| Fig. 5 Reaction time course on the selective oxidation of 20 g DHMF to BHMF in THF at 65 °C using (A) NaOH at 0.25 and 0.5 ratios to DHMF with and without molecular sieves (MSs) and (B) the anion exchanger (A900) at 0.25, 0.5 and 1 ratios (w/w) to DHMF, and L-proline at a 0.2 ratio (w/w) to DHMF with molecular sieves. | ||
L-Proline could perform the oxidation but showed slow and low conversion at a 0.2 (w/w) ratio to DHMF under the same conditions (Fig. 5B). It served as a heterogeneous catalyst since it was not dissolved in THF. Therefore, we confirmed that most basic catalysts can be employed for the oxidation, providing high conversion and yield, although different ratios of catalysts were required. DBU, NaOH, and A900 were preferable catalysts to obtain a quantitative yield, and the results were obtained at a 20 g DHMF scale, respectively (Fig. 5).
Moreover, based on the results obtained from the oxidation, yielding quantitative BHMF (Fig. 2 and 3), the enhancement of the reaction rate and yield using molecular sieves can be explained by the in situ removal of H2O2, a by-product, which might be eliminated by molecular sieves in the reaction system, as illustrated in the proposed mechanisms, leading to further improvement in the yield. The proposed mechanism for the oxidation of DHMF to BHMF using DBU as the accelerator is depicted in Fig. 6A. Initially, deprotonation of DHMF into the enolate anion occurs, followed by a single-electron transfer between DHMF and triplet oxygen, forming a superoxide anion radical and a radical intermediate. The radical intermediate then reacts with the superoxide anion radical and undergoes an elimination of the hydroperoxide anion to produce BHMF. Furthermore, the hydroperoxide anion quickly reacts with the protonated DBU to form H2O2 as a by-product. The increased reaction rate and improved yield by the removal of a possible by-product in the mechanism support the proposed mechanism, which was further confirmed by the detection of H2O2 in the reaction solution (Fig. 6B and C). In the colorimetric test with potassium iodide, the sample collected from the oxidation of DHMF without molecular sieves exhibited a strong color change from red to brown after adding KI and acetic acid, while the sample from the reaction with molecular sieves exhibited no significant color change. These observations indicate that H2O2 was produced during the reaction and can be removed through molecular sieves, thereby supporting the proposed reaction mechanism.
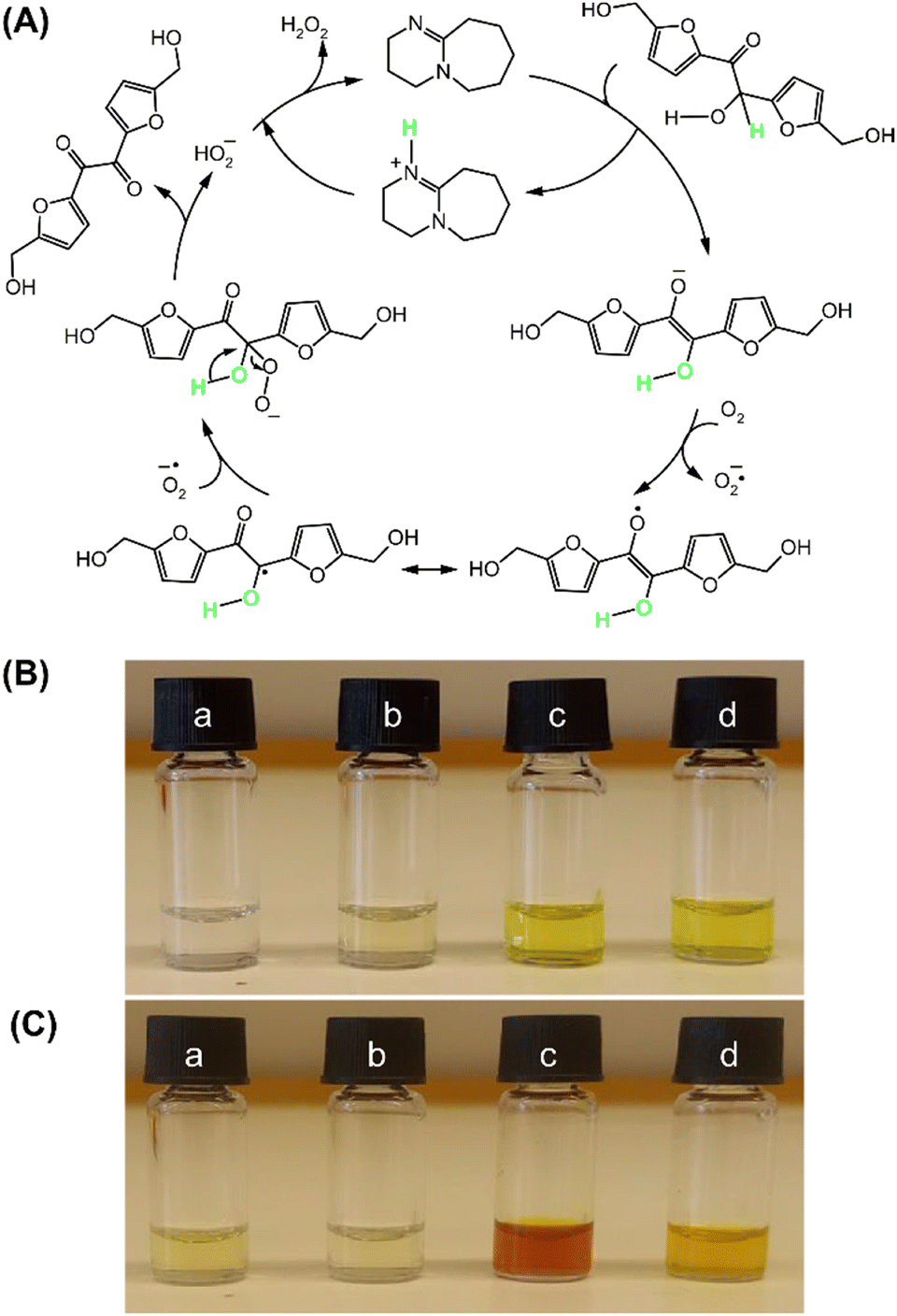 | ||
| Fig. 6 (A) Proposed mechanism for the selective oxidation of the secondary alcohol of DHMF to the ketone of BHMF using DBU as a base catalyst. Colorimetric test to detect the by-product in the oxidative mechanism: (B) before and (C) after addition of colorimetric agents in a. DBU in deionized water, b. DHMF in deionized water, c. samples from the reaction using 5 mol% DBU without molecular sieves, and d. samples from the reaction using 5 mol% DBU with molecular sieves. | ||
Therefore, the significance of this oxidation process lies in its ability to selectively convert DHMF to BHMF based on the proposed mechanism. The optimized reaction conditions, catalyst selection, and the use of molecular sieves contribute to the efficiency and scalability of the process, enhancing its potential for industrial applications in the synthesis of bio-based chemicals and polymers.
Conclusions
5-HMF has garnered significant attention as a bio-based platform chemical due to its potential in production, conversion, and application. The comprehensive process outlined in this study establishes a sustainable value chain that connects renewable C6 sugars to C12 aromatic polyols, serving as versatile chemicals for polymer production through the intermediary 5-HMF.The initial step involves the carboligation of 5-HMF through self-coupling to yield DHMF, an aromatic triol containing two primary alcohol moieties and one secondary alcohol moiety. As a more environmentally friendly option, alternative solvents to DCM (a halogenated solvent) were proposed. The scalability of the reaction was demonstrated at the 400 g scale, yielding a quantitative amount of product.
Subsequently, DHMF undergoes quantitative oxidation in the secondary alcohol group, resulting in the production of BHMF, an aromatic diol, utilizing a basic organocatalyst. The oxidation was further characterized by the proposed mechanism, which was confirmed through colorimetric detection of the by-product, H2O2. These processes have been successfully demonstrated on a larger scale, highlighting the efficient reaction and separation, to achieve a quantitative yield using only a catalytic amount (5%) of DBU along with molecular sieves.
In adherence to the principles of green chemistry, the overall process and value chain exhibit sustainability by utilizing renewable resources, employing recyclable materials, ensuring high catalytic efficiency, and maintaining cost-effectiveness through the elimination of high-cost and challenging procedures. Consequently, the comprehensive processes presented in this study underscore the economic feasibility of upgrading 5-HMF to C12 DHMF and BHMF, offering efficient catalysis in a more environmentally friendly solvent for scalable and industrial applications.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
SHP, JC, NR and RHK conceived and designed the study. TTTV, SL, MJ, SJ, MI, and SHP performed the experiments and analyses. TTTV, SL, MI and SHP wrote the initial draft of the manuscript, and all authors revised the manuscript. All authors have read and approved the final manuscript for submission.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the Technology Innovation Program (20014436, Development of biodegradable polycarbonate material and mass production process technology) funded by the Ministry of Trade, Industry & Energy (MOTIE, Republic of Korea), Mistra (The Swedish Foundation for Strategic Environmental Research) for the project Sustainable Plastics & Transition Pathways (STEPS, project no. 2016/1489) and Energimyndigheten within the call: RE:Source 2023 (Cirkulär ekonomi och resursanvändning inom planetens gränser).References
- Petrochemical Industry Overview – Chemical Economics Handbook (CEH) | S&P Global, https://www.spglobal.com/commodityinsights/en/ci/products/petrochemical-industry-chemical-economics-handbook.html, (accessed 23 February 2024).
- G. Lopez, D. Keiner, M. Fasihi, T. Koiranen and C. Breyer, Energy Environ. Sci., 2023, 16, 2879–2909 RSC.
- New Publication – Bio-Based Chemicals – a 2020 Update | Bioenergy, https://www.ieabioenergy.com/blog/publications/new-publication-bio-based-chemicals-a-2020-update/, (accessed 23 February 2024).
- L. Goswami, R. Kayalvizhi, P. K. Dikshit, K. C. Sherpa, S. Roy, A. Kushwaha, B. S. Kim, R. Banerjee, S. Jacob and R. C. Rajak, Chem. Eng. J., 2022, 448, 137677 CrossRef CAS.
- Z. Chen, L. Chen, K. S. Khoo, V. K. Gupta, M. Sharma, P. L. Show and P. S. Yap, Biotechnol. Adv., 2023, 69, 108265 CrossRef CAS PubMed.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- A. He, L. Dong, N. Xu, S. I. El-Hout, J. Xia, Z. Qiu, J. He, Y. Deng, X. Liu, L. Hu and J. Xu, Chem. Eng. J., 2022, 449, 137797 CrossRef CAS.
- M. Sayed, N. Warlin, C. Hulteberg, I. Munslow, S. Lundmark, O. Pajalic, P. Tunå, B. Zhang, S. H. Pyo and R. Hatti-Kaul, Green Chem., 2020, 22, 5402–5413 RSC.
- T. T. T. Vu, S. Liu, M. Jonušis, S. Jonušienė, J. Choi, R. Kawano, N. Rehnberg, R. Hatti-Kaul and S. H. Pyo, ACS Sustainable Chem. Eng., 2023, 11, 17130–17141 CrossRef.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- N. Li and M. H. Zong, ACS Catal., 2022, 12, 10080–10114 CrossRef CAS.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- S. H. Pyo, M. Sayed and R. Hatti-Kaul, Org. Process Res. Dev., 2019, 23, 952–960 CrossRef CAS.
- B. Saha and M. M. Abu-Omar, ChemSusChem, 2015, 8, 1133–1142 CrossRef CAS PubMed.
- S. H. Pyo, S. J. Glaser, N. Rehnberg and R. Hatti-Kaul, ACS Omega, 2020, 5, 14275–14282 CrossRef CAS PubMed.
- Q. Ding, P. Yan, H. Li, Z. Chen, Y. Guan, Y. Jiao, J. Zou and X. Gao, Chem. Eng. Sci., 2023, 265, 118258 CrossRef CAS.
- W. Yang, X. Tang, W. Li, X. Luo, C. Zhang and C. Shen, Chem. Eng. J., 2022, 442, 136110 CrossRef CAS.
- N. Wierckx, T. D. Elink Schuurman, L. M. Blank and H. J. Ruijssenaars, Whole-Cell Biocatalytic Production of 2,5-Furandicarboxylic Acid, in Microorganisms in Biorefineries, ed. B. Kamm, Microbiology Monographs, Springer, Berlin, Heidelberg, 2015, vol. 26, DOI:10.1007/978-3-662-45209-7_8.
- Z. Y. Yang, M. Wen, M. H. Zong and N. Li, Catal. Commun., 2020, 139, 105979 CrossRef CAS.
- M. Sayed, S. H. Pyo, N. Rehnberg and R. Hatti-Kaul, ACS Sustainable Chem. Eng., 2019, 7, 4406–4413 CrossRef CAS.
- M. Zhang, Z. Li, X. Xin, J. Zhang, Y. Feng and H. Lv, ACS Catal., 2020, 10, 14793–14800 CrossRef CAS.
- C. Wu, Q. Li, J. Di, Y. C. He and C. Ma, Fuel, 2023, 343, 127830 CrossRef CAS.
- R. Gao, Q. Li, J. Di, Q. Li, Y. C. He and C. Ma, Ind. Crops Prod., 2023, 193, 116199 CrossRef CAS.
- L. Wang, J. Zuo, Q. Zhang, F. Peng, S. Chen and Z. Liu, Catal. Lett., 2022, 152, 361–371 CrossRef CAS.
- C. Marisa, D. S. Ilaria, R. Marotta, A. Roberto and C. Vincenzo, J. Photochem. Photobiol., A, 2010, 210, 69–76 CrossRef.
- T. Raj, K. Chandrasekhar, R. Banu, J. J. Yoon, G. Kumar and S. H. Kim, Fuel, 2021, 303, 121333 CrossRef CAS.
- H. Kim, S. Lee and W. Won, Energy, 2021, 214, 118974 CrossRef CAS.
- S. H. Pyo, M. Sayed, O. E. Örn, J. A. Gallo, N. F. Ros and R. Hatti-Kaul, Microb. Cell Fact., 2022, 21, 1–10 CrossRef PubMed.
- S. H. Pyo, J. H. Park, V. Srebny and R. Hatti-Kaul, Green Chem., 2020, 22, 4450–4455 RSC.
- M. Lang and H. Li, ChemSusChem, 2022, 15, e202101531 CrossRef CAS PubMed.
- D. Liu, Y. Zhang and E. Y. X. Chen, Green Chem., 2012, 14, 2738–2746 RSC.
- B. Yan, H. Zang, Y. Jiang, S. Yu and E. Y. X. Chen, RSC Adv., 2016, 6, 76707–76715 RSC.
- H. Zang and E. Y. X. Chen, Int. J. Mol. Sci., 2015, 16, 7143–7158 CrossRef CAS PubMed.
- D. Liu and E. Y. X. Chen, ACS Catal., 2014, 4, 1302–1310 CrossRef CAS.
- S. J. Glaser, S. H. Pyo, N. Rehnberg, D. Rother and R. Hatti-Kaul, Microb. Cell Fact., 2023, 22, 1–13 CrossRef PubMed.
- H. Zang, K. Wang, M. Zhang, R. Xie, L. Wang and E. Y. X. Chen, Catal. Sci. Technol., 2018, 8, 1777–1798 RSC.
- F. Deng and A. S. Amarasekara, Ind. Crops Prod., 2021, 159, 113055 CrossRef CAS.
- S. Baraldi, G. Fantin, G. Di Carmine, D. Ragno, A. Brandolese, A. Massi, O. Bortolini, N. Marchetti and P. P. Giovannini, RSC Adv., 2019, 9, 29044–29050 RSC.
- Z. Mou, S. Feng and E. Y. X. Chen, Polym. Chem., 2016, 7, 1593–1602 RSC.
- D. Liu, E. Y.-X. Chen, D. J. Liu and E. Y.-X. Chen, ChemSusChem, 2013, 6, 2236–2239 CrossRef CAS PubMed.
- J. Donnelly, C. R. Müller, L. Wiermans, C. J. Chuck and P. D. De María, Green Chem., 2015, 17, 2714–2718 RSC.
- D. Enders, K. Breuer, U. Kallfass and T. Balensiefer, Synthesis, 2003, 34, 1292–1295 Search PubMed.
- S. Junglee, L. Urban, H. Sallanon, F. Lopez-Lauri, S. Junglee, L. Urban, H. Sallanon and F. Lopez-Lauri, Am. J. Anal. Chem., 2014, 5, 730–736 CrossRef CAS.
- V. Alexieva, I. Sergiev, S. Mapelli and E. Karanov, Plant, Cell Environ., 2001, 24, 1337–1344 CrossRef CAS.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed.
- A. Erkkilä and P. M. Pihko, Eur. J. Org. Chem., 2007, 2007, 4205–4216 CrossRef.
- B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2008, 47, 4632–4634 CrossRef CAS PubMed.
- S. H. Pyo, J. H. Park, T. S. Chang and R. Hatti-Kaul, Curr. Opin. Green Sustainable Chem., 2017, 5, 61–66 CrossRef.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- E. Leino, P. Mäki-Arvela, V. Eta, D. Y. Murzin, T. Salmi and J. P. Mikkola, Appl. Catal., A, 2010, 383, 1–13 CrossRef CAS.
- R. Xie, H. Zang, H. Ding, B. Yan and B. Cheng, J. Mol. Liq., 2018, 249, 486–493 CrossRef CAS.
- A. Dzienia, P. Maksym, B. Hachuła, M. Tarnacka, T. Biela, S. Golba, A. Ziȩba, M. Chorażewski, K. Kaminski and M. Paluch, Polym. Chem., 2019, 10, 6047–6061 RSC.
- L. Li, Q. Wang, P. Liu, H. Meng, X. L. Kan, Q. Liu and Y. L. Zhao, Org. Biomol. Chem., 2015, 14, 165–171 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental methods, preparation of the N-heterocyclic carbene (NHC) compound, carboligation mechanism of 5-HMF and the NMR spectra for 5-methoxy-1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazoline, DHMF, and BHMF. See DOI: https://doi.org/10.1039/d4re00212a |
| This journal is © The Royal Society of Chemistry 2025 |