
Mechanistic insights into C–O bond cleavage in erythritol during hydrodeoxygenation on an Ir–ReOx catalyst†
Ajin
Rajan
and
Jithin John
Varghese
 *
*
Department of Chemical Engineering, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: jithinjv@iitm.ac.in
First published on 26th July 2024
Abstract
1,4-Butanediol (1,4-BDO) is a key ingredient in the polymer industry. When derived from renewable erythritol, it can pave the way for sustainable poly(butylene terephthalate), polyurethane and polyester manufacturing. Hydrodeoxygenation (HDO) of erythritol on Brønsted acidic metal–metal oxide catalysts can result in 1,4-BDO, among other alcohols. Selective synthesis of 1,4-BDO requires deep insights into the preference for the cleavage of the different C–O bonds and the energy landscape for the formation of other polyol intermediates. In this work, we used density functional theory (DFT) simulations to investigate HDO of erythritol and other polyol intermediates on an inverse Ir–ReOx catalyst, where rhenium oxide is dispersed on iridium. While Ir nanoparticles can drive HDO through dehydroxylation, a protonation and dehydration mechanism happening at the Ir–ReOx interface has greater kinetic relevance. We show the kinetic preference for secondary C–O cleavage in erythritol to explain the predominant formation of 1,2,4-butanetriol (1,2,4-BTO) during erythritol HDO. The kinetic preference for 1,4-BDO formation from 1,2,4-BTO makes it the most prominent butanediol during erythritol HDO. C–O bond cleavage in 1,4-BDO has a high barrier making 1,4-BDO less reactive in a polyol mixture. This indicates potential selective formation of 1,4-BDO, with a possibility of tuning reaction conditions and reaction time to maximise its yield. Our analyses reveal that C–O cleavage is not always the kinetically relevant step and it can be the hydrogenation that follows the C–O cleavage. Hence, reactions at high hydrogen pressure and lower temperatures might suit higher selectivity towards desired alcohols such as 1,4-BDO.
Introduction
1,4-Butanediol (1,4-BDO) is a C4 platform chemical and finds applications in the production of tetrahydrofuran,1 poly(butylene terephthalate),2 polyurethane,3 and polyester.4 It is typically produced through various fossil-based processes such as ethynylation of formaldehyde,5 epoxidation of butadiene,6 hydroformylation of allyl alcohol,7 and hydroformylation of allyl acetate,8 followed by subsequent conversion to 1,4-BDO. Biomass-derived polyols are attracting significant attention as alternatives to non-renewable fossil resource-based polyols for both fuel and chemical applications.9 Erythritol, a C4 sugar alcohol derived from the fermentation of glycerol and glucose,10 is one such example.11 Erythritol could serve as a renewable alternative source for 1,4-BDO synthesis. However, erythritol possesses four hydroxyl groups, potentially leading to the production of various products as shown in Fig. 1 such as 1,2,3-butanetriol (1,2,3-BTO), 1,2,4-butanetriol (1,2,4-BTO), 1,2-butanediol (1,2-BDO), 1,3-butanediol (1,3-BDO), 1,4-butanediol (1,4-BDO), 2,3-butanediol (2,3-BDO), 1-butanol (1-BO), and 2-butanol (2-BO). Hence, selectively converting erythritol to 1,4-butanediol is highly challenging and often requires specialized catalysts and processes.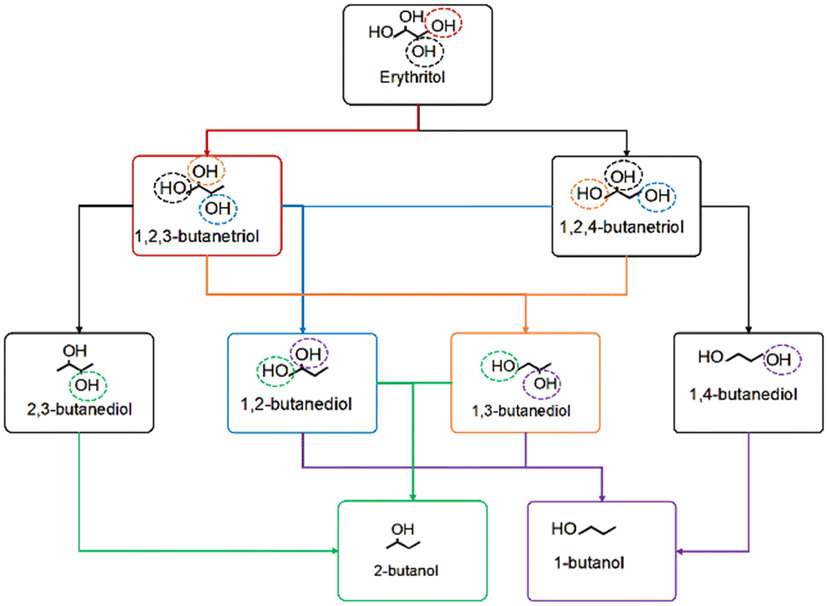 | ||
| Fig. 1 Possible products that can form during hydrodeoxygenation of erythritol showing the OH group that needs to be eliminated to obtain the product. Colored dotted circle represents the hydroxyls to be cleaved to reach the product connected by the same-coloured line. | ||
Hydrodeoxygenation (HDO) stands out as one of the most widely used processes for reducing the oxygen content of polyols.12–14 It involves treating polyols with hydrogen at high pressures to remove the OH groups in the form of water. Selecting appropriate catalysts which can facilitate targeted C–O cleavage for HDO is crucial. For instance, metallic catalysts based on Cu mostly lead to the product 1,2-BDO.15 Selective formation of 1,4-BDO from erythritol requires cleaving only the interior hydroxyls while preserving the terminal hydroxyls.
In this context, Brønsted acid (BA) site-containing metal-supported oxophilic metal oxide-based inverse catalysts, such as Pt–WOx and Ir–ReOx,16–20 have emerged as promising candidates for selective secondary C–O bond cleavage during HDO. Virgilio et al.21 reported 51% conversion, with just 4.5% selectivity to 1,4-BDO for erythritol hydrogenolysis with an Ir/ReOx/TiO2 catalyst at 2.5 MPa hydrogen pressure and 150 °C. By conducting the HDO reaction at 190 °C and a relatively higher hydrogen pressure of 5 MPa over a Pt/WOx/TiOx–SBA-15 catalyst, Bhowmik et al.17 reported a 1,4-BDO selectivity of 35% at 94% conversion of erythritol. While the above results may indicate Ir–ReOx catalysts to be ineffective for erythritol HDO, it was found to be selective for 1,3-propanediol (terminal diol) production from glycerol. Lujie et al. reported a 1,3-PDO selectivity of 47% at a glycerol conversion of 69% using an Ir–ReOx/SiO2 catalyst during a batch reaction of 24 h at 8 MPa PH2 and 120 °C.
Density functional theory (DFT) simulation-based mechanistic investigations of glycerol HDO on Pt–WOx and Ir–ReOx catalysts have revealed that the protonation dehydration mechanism is highly relevant for C–O bond cleavage in these catalysts.16,18 Here, the BA from the catalyst is proposed to combine with the hydroxyl group of the glycerol and becomes eliminated as water. These investigations have given mechanistic insights into the key reaction step involving C–O cleavage, and clarity on factors influencing the product distribution noticed during glycerol HDO. However strategies to improve 1,3-PDO yield are still elusive.
Glycerol is a simpler polyol molecule compared to erythritol, with very few possible HDO by-products, whereas erythritol is a reasonably complex polyol with numerous possible HDO products. Therefore, a detailed mechanistic understanding of the thermodynamic and kinetic preference for formation of different products and their sequences is crucial in designing catalysts and operating reactors for achieving high 1,4-BDO yields during erythritol HDO. Through detailed DFT simulations, this work aims to provide a deep understanding of the mechanistic complexity involved in C–O bond cleavages in erythritol on Ir–ReOx catalysts for the targeted production of 1,4-BDO.
Methods
Computational methods
DFT simulations were performed using the Vienna ab initio simulation package (VASP version 5.4.4)22 with the Perdew–Burke–Ernzerhof (PBE)23 exchange–correlation functional. The projector augmented wave (PAW)24 method was employed to handle the core–valence electron interactions. A plane wave expansion cutoff energy of 400 eV was used in all the simulations. The van der Waals corrections were incorporated using the semi-empirical DFT-D3 method of Grimme,25 as implemented in the VASP. The solvation effect of the aqueous environment in which the reactions are typically carried out was accounted by carrying out the reactions using the implicit solvation scheme VASPsol26 as implemented in the VASP with water solvent. The convergence criteria for all the simulations were set at 10−4 eV for the electronic self-consistent steps, and were set at 0.1 eV Å−1 for atomic residual forces during geometry optimization. Various orientations of each of the polyols on the catalysts were investigated. Only certain configurations of the polyols on the catalyst facilitate protonation and dehydration. The most favorable among such a constrained set of configurations were chosen to be the reactant/initial state for each elementary step and the reaction energies and activation barriers were reported with reference to that specific configuration. As an example, the erythritol configuration that enables removal of the primary and secondary hydroxyls on different catalyst models and via different mechanisms is shown in Fig. S6.† The nudged elastic band (NEB) method implemented in VASP was used to locate the transition state (TS). The TS nature of the identified stationary point was verified by the existence of a single imaginary frequency in the normal mode vibrational analysis within the harmonic oscillator approximation. Each identified TS was further refined with a quasi-Newton–Raphson optimizer.Catalyst models
The catalyst model for the computational investigation was developed based on detailed characterization of Ir–ReOx as reported by Liu et al.27 Their X-ray diffraction (XRD) analyses revealed a prominent crystalline peak for Ir with a dominant [111] facet. High-resolution transmission electron microscopy (HRTEM) also detected a d-spacing of 2.22 Å, corresponding to the Ir[111] planes. Hence, the Ir[111] surface was considered in the catalyst model to represent the metallic Ir identified during the characterization. Despite the absence of detectable ReOx crystals in the XRD analysis, possibly due to ReOx being finely dispersed to a non-detectable range of XRD, energy-dispersive X-ray (EDX) spectroscopy confirmed the presence of ReOx and suggested an even distribution of ReOx.27 Their extended X-ray absorption fine structure (EXAFS) analysis identified the presence of a Re–Ir bond with a bond length of 0.27 nm, suggesting a direct contact of the ReOx cluster on the Ir surface and possibly the formation of the inverse metal oxide (ReOx) on the metal (Ir) catalyst structure. The Re3O6 moiety is a representative partially reduced ReOx cluster, with three Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and three Re–O–Re species, and has Re in the +4 oxidation state which is in agreement with the +4 oxidation state of Re identified in XPS analysis in the literature.18 Hence, a model featuring an Re3O6H unit directly interacting with a 4-layer (bottom layer frozen) p5 × 5 Ir[111] surface to represent a BA site (Re–OH) containing inverse Ir–ReOx catalyst was considered. This Re3O6H/Ir[111] surface was incorporated with 4H* on the Ir surface to represent dissociated hydrogen that is expected under the typical reaction conditions. The catalyst model incorporates a 17 Å vacuum thickness to separate the periodic images in the z-direction. This Re3O6H/Ir[111] catalyst model with 4H* is shown in Fig. S1a.† There is potential variability in the numbers and density of the BA sites on the ReOx clusters and the surface hydrogen coverage during the reaction. To study the effect of Brønsted acid concentration on Ir–ReOx catalysts, Re3O6H2/Ir(111) and Re3O6H3/Ir(111) surfaces with 4H*coverage were modelled, as shown in Fig. S1b and c.† To study the effect of H* coverage on the Ir surface, a Re3O6H/Ir[111] surface with 10H* (representing 50% coverage on the surface) was modeled as shown in Fig. S1d.† After testing, a 3 × 3 × 1 Monkhorst–Pack k-point grid was employed for Brillouin zone sampling in the simulations involving the slab models. To study the potential role of undercoordinated Ir sites in the HDO reaction, an Ir225 catalyst model comprising a 225 atom Ir nanoparticle in a 25.5 Å cubic cell as shown in Fig. S1f† was considered. Only the Γ point was used for the Brillouin zone sampling in these simulations.
O and three Re–O–Re species, and has Re in the +4 oxidation state which is in agreement with the +4 oxidation state of Re identified in XPS analysis in the literature.18 Hence, a model featuring an Re3O6H unit directly interacting with a 4-layer (bottom layer frozen) p5 × 5 Ir[111] surface to represent a BA site (Re–OH) containing inverse Ir–ReOx catalyst was considered. This Re3O6H/Ir[111] surface was incorporated with 4H* on the Ir surface to represent dissociated hydrogen that is expected under the typical reaction conditions. The catalyst model incorporates a 17 Å vacuum thickness to separate the periodic images in the z-direction. This Re3O6H/Ir[111] catalyst model with 4H* is shown in Fig. S1a.† There is potential variability in the numbers and density of the BA sites on the ReOx clusters and the surface hydrogen coverage during the reaction. To study the effect of Brønsted acid concentration on Ir–ReOx catalysts, Re3O6H2/Ir(111) and Re3O6H3/Ir(111) surfaces with 4H*coverage were modelled, as shown in Fig. S1b and c.† To study the effect of H* coverage on the Ir surface, a Re3O6H/Ir[111] surface with 10H* (representing 50% coverage on the surface) was modeled as shown in Fig. S1d.† After testing, a 3 × 3 × 1 Monkhorst–Pack k-point grid was employed for Brillouin zone sampling in the simulations involving the slab models. To study the potential role of undercoordinated Ir sites in the HDO reaction, an Ir225 catalyst model comprising a 225 atom Ir nanoparticle in a 25.5 Å cubic cell as shown in Fig. S1f† was considered. Only the Γ point was used for the Brillouin zone sampling in these simulations.
Results and discussion
HDO of erythritol: butanediol formation via sequential C–O bond cleavages versus via butanetriol intermediates
Erythritol conversion to products like 1,2-BDO (requires primary and secondary hydroxyl removal) and 1,4-BDO (requires two secondary hydroxyl removal) may proceed via (1) sequential elimination of adjacent hydroxyl groups or (2) through triol intermediates. In the case of (1), a protonation dehydration mechanism eliminating one of the hydroxyls is likely to be followed immediately by an adjacent C–O bond cleavage by the metal, without involving a BA site. In the case of (2), the HDO via the protonation dehydration forms a triol product. This product subsequently undergoes another protonation dehydration-based HDO to form the diol. Both these possibilities were investigated for 1,2-BDO and 1,4-BDO formation on Ir–ReOx catalysts, and the energy profiles are depicted in Fig. 2.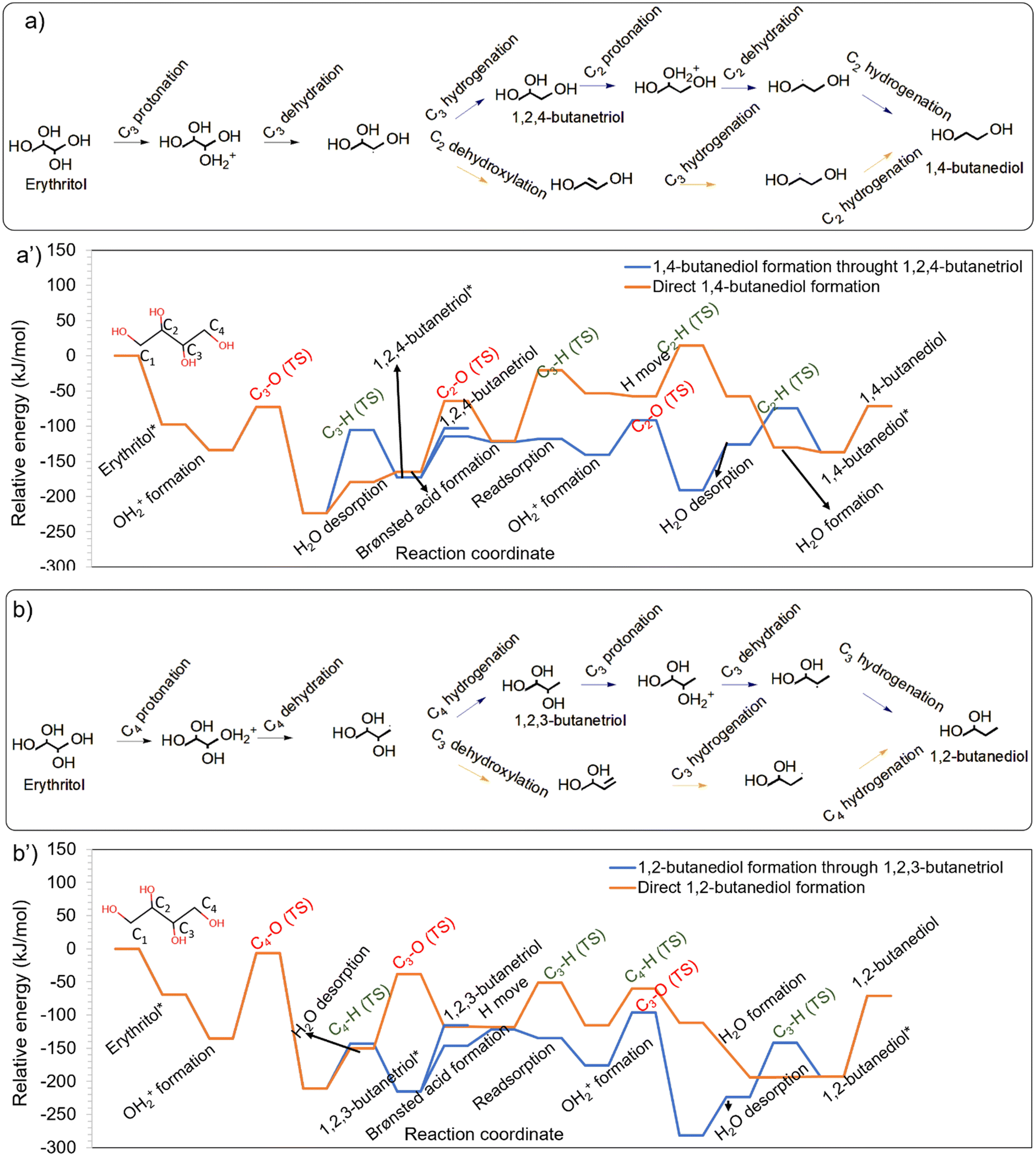 | ||
| Fig. 2 HDO of erythritol on Ir–ReOx catalysts showing formation of a) 1,4-butanediol and b) 1,2-butanediol. Parts a′) and b′) represent the energy profile corresponding to the reaction scheme shown in a) and b), respectively. Images of transition states in these energy profiles are available in Fig. S2 and S3 of the ESI.† | ||
Considering the kinetic preference for the elimination of the secondary hydroxyl that we reported for glycerol HDO,16 the HDO pathway of erythritol, initiated by the protonation dehydration of one of the secondary hydroxyls, was investigated. One difference here is the formation of the stable protonated state in an energetically favourable reaction step and the subsequent elimination of the water molecule. In our previous work on glycerol HDO, we observed and reported concerted protonation and dehydration.16 Here, the first C–O cleavage by protonation and dehydration (OH at C3 removed) had an activation barrier of 61 kJ mol−1 as shown in Fig. 2a and was highly favourable. The hydrogenation at C3 and 1,2,4-BTO formation had an activation barrier of 119 kJ mol−1, making it the kinetically relevant step. Moreover, this was an energetically unfavourable step. Alternatively, after desorption of the formed water during dehydration, another secondary C–O cleavage (OH at C2 removed) by direct dehydroxylation by Ir, leading to elimination of the adjacent hydroxyl, can follow. The activation barrier for the direct dehydroxylation was 115 kJ mol−1 and was energetically unfavourable. Sequential hydrogenation at C3 and C2, with activation barriers of 101 kJ mol−1 and 72 kJ mol−1 respectively, resulted in 1,4-BDO. Among these, the first hydrogenation step was highly unfavourable while the second one was nearly thermoneutral. In this sequence, the C–O cleavage by Ir is the kinetically relevant step as the hydrogenation barriers are much lower.
If the BA site that was consumed during the protonation dehydrogenation step leading to 1,2,4-BTO formation was regenerated immediately, 1,2,4-BTO can undergo another protonation and dehydration step to eliminate the other secondary hydroxyl (OH at C2 removed). This step had an activation barrier of 49 kJ mol−1 and was an energetically favourable step. Hydrogenation of this intermediate, with an activation barrier of 52 kJ mol−1 (following desorption of the formed water), resulted in 1,4-BDO and was energetically favourable. It is to be noted here that the C–O cleavage at C2 has a substantially lower barrier via the protonation and dehydration mechanism involving the BA site than the direct C–O cleavage by Ir. The low barriers for both C–O cleavage and the hydrogenation steps suggest that 1,2,4-BTO is likely to undergo quick transformation to 1,4-BDO. Based on the reaction profile shown in Fig. 2a, 1,4-BDO is likely formed via the 1,2,4-BTO intermediate, especially at a high hydrogen pressure. High hydrogen pressure will enhance the likelihood of BA site regeneration and also make the hydrogenation steps more likely to occur, both of which will favour 1,2,4-BTO formation and its subsequent HDO. Formation of other possible products from 1,2,4-BTO is described in later sections of the manuscript.
Certain orientations of erythritol over the catalyst may facilitate primary C–O bond cleavage by protonation and dehydration as the first step (Fig. S6b†). The primary C–O cleavage barrier (OH at C4 removed) was 129 kJ mol−1 as shown in Fig. 2b. Despite the reaction being favourable, this has a substantially higher barrier than the barrier for the secondary C–O cleavage described earlier. Hydrogenation of the formed intermediate with a comparatively lower barrier of 68 kJ mol−1 resulted in 1,2,3-BTO. Here, C–O cleavage is the kinetically relevant step unlike in the case of 1,2,4-BTO formation. Alternatively, after desorption of the formed water, another secondary C–O cleavage by Ir followed, leading to elimination of the adjacent hydroxyl at C3 with an activation barrier of 112 kJ mol−1. This step is not energetically favourable though. Sequential hydrogenation steps at C3 and C4 with activation barriers of 67 kJ mol−1 and 56 kJ mol−1, respectively, resulted in 1,2-BDO formation.
If the BA site that was consumed during the protonation dehydration to form 1,2,3-BTO is immediately regenerated, the formed 1,2,3-BTO may undergo secondary C–O cleavage. This is by protonation and dehydration and leads to elimination of the adjacent hydroxyl at C3 with an activation barrier of 80 kJ mol−1 in a highly favourable step. Following the desorption of the formed water, hydrogenation of the previous intermediate with an activation barrier of 82 kJ mol−1 formed 1,2-BDO in an energetically unfavourable step. Analysis of the profiles in Fig. 2b suggests that 1,2-BDO formation is kinetically and energetically preferred via the 1,2,3-BTO intermediate than via sequential C–O bond cleavages in erythritol, especially in reactions carried out at high hydrogen pressure as discussed earlier.
In summary, consecutive C–O cleavages, eliminating two adjacent hydroxyls in erythritol and eventually leading to butanediol formation seems to be energetically and kinetically unfavourable and formation of butanediols is expected to be via butanetriol intermediates.
The prediction of 1,2,4-BTO and 1,2,3-BTO being intermediates in the HDO of erythritol is supported by experiments of erythritol HDO on an Ir–ReOx catalyst reported by Gu et al.28 Table S1† shows 1,2,4-BTO and 1,2,3-BTO to be the prominent products during erythritol HDO with 37% and 27% selectivity, respectively. Within the 4 hours of batch reaction, the cracking products were negligible (around 1%), indicating that the HDO reactions are more prominent than cracking of the polyols on this catalyst and under these conditions. Hence, we restricted our analysis to the HDO reaction network and have not considered cracking reactions of polyols. Our prediction of 1,2,4-BTO being the precursor for 1,4-BDO is also supported by findings of Gu et al.28 who showed (Table S1,† entry 3) 1,4-BDO to be the primary butanediol with 80% selectivity during HDO of 1,2,4-BTO on the Ir–ReOx catalyst. They did not observe 1,2-BDO as a product of erythritol HDO but 1,3-BDO was observed instead. Its formation is discussed in the next section.
HDO of butanetriols to butanediols
In addition to 1,4-BDO, erythritol HDO also resulted in 1,3-BDO as a prominent product (Table S1†).28 1,3-BDO was obtained during HDO of 1,2,4-BTO and 1,2,3-BTO. Moreover, HDO of 1,2,3-BTO also formed 2,3-BDO. However, 2,3-BDO was not observed as a product during erythritol HDO on the Ir–ReOx catalyst. To understand the formation of butanediols, all the potential C–O cleavages from 1,2,3-BTO and 1,2,4-BTO were systematically studied on Ir–ReOx catalysts to understand the experimentally observed trends (Table S1†). The energy profiles for the protonation and dehydration based HDO of butanetriols and transformation to diols are shown in Fig. 3.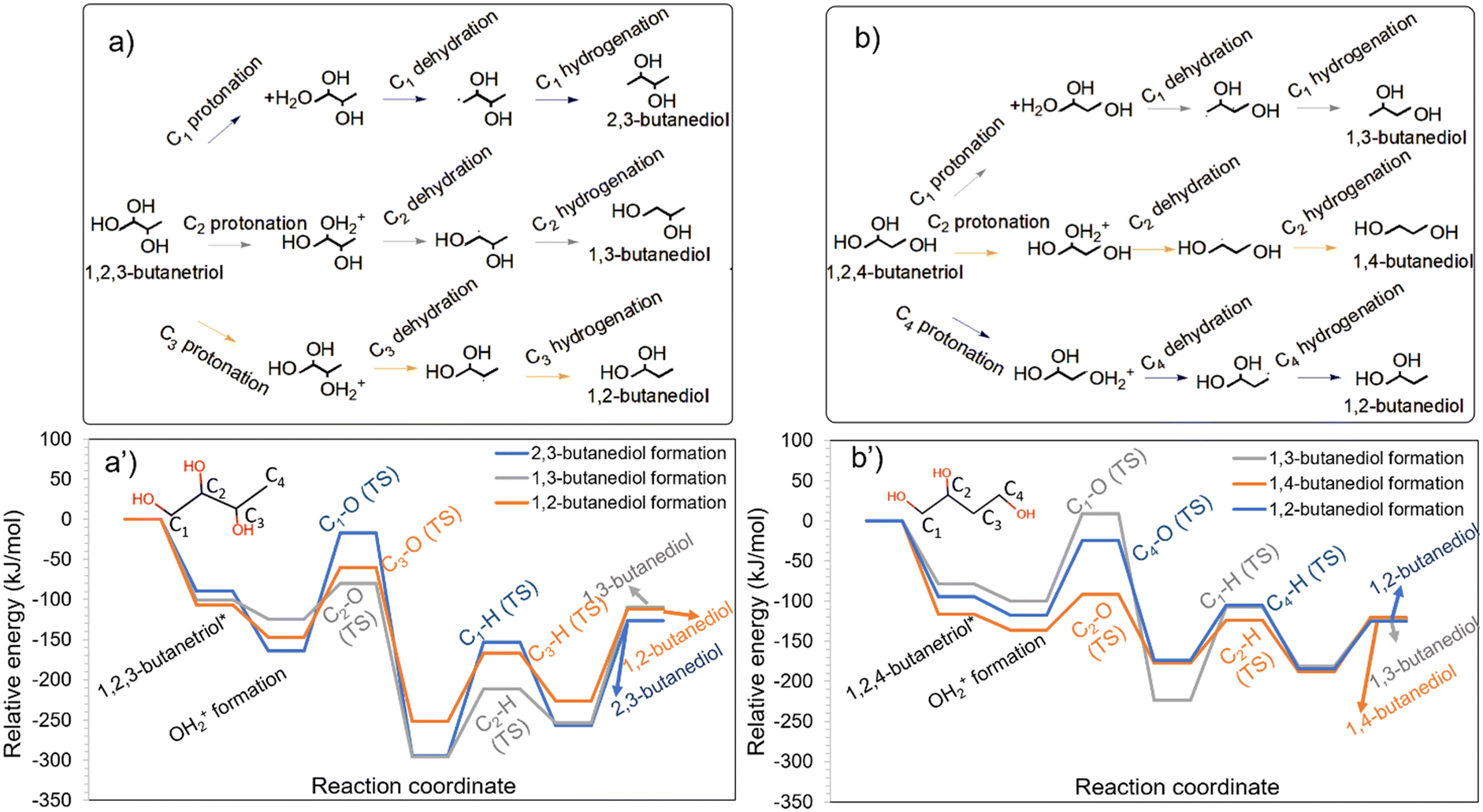 | ||
| Fig. 3 Formation of different butanediols during HDO of a) 1,2,3-butanetriol and b) 1,2,4-butanetriol on the Ir–ReOx catalyst. The energy profiles corresponding to HDO of 1,2,3-BTO and 1,2,4-BTO are provided in a′) and b′), respectively. Images of transition states in these energy profiles are available in Fig. S4 and S5 of the ESI.† | ||
Protonation of the hydroxyls at different positions in 1,2,3-BTO showed primary hydroxyl protonation at C1 to be the most energetically favourable, followed by secondary hydroxyl at C3 and secondary hydroxyl at C2 as can be seen in Fig. 3a. Given the relatively unstable protonated state in 1,2,3-BTO with protonation at C2, secondary C–O cleavage (OH at C2 removed) leading to the formation of 1,3-BDO had a very low barrier of only 45 kJ mol−1 and was highly favourable. However, the hydrogenation at C2 had a much higher activation barrier of 85 kJ mol−1, making it the kinetically relevant step. Moreover, this step was energetically not favourable. The secondary C–O cleavage (OH at C3 removed) leading to the formation of 1,2-BDO had a barrier of 80 kJ mol−1 and was not as favourable as C2–OH removal. Following this, the hydrogenation at C3 had a barrier of 85 kJ mol−1 and was also not energetically favourable. In contrast to the secondary C–O cleavage, the primary C–O cleavage (OH at C1 removed) had a high barrier of 146 kJ mol−1, despite the favourable energetics. Following this, the hydrogenation at C1 also had a high barrier of 142 kJ mol−1 and was energetically unfavourable, leading to the formation of 2,3-BDO. Analysis of trends in Fig. 3a shows that the protonation of C1–OH is the most favourable, but the reaction steps following this have comparatively high barriers. In contrast, protonation of C2–OH is the least favourable, but the reaction steps following this have comparatively low barriers. The protonated state at C3–OH has intermediate stability and the reaction steps have barriers exceeding 80 kJ mol−1. These trends indicate 1,3-BDO to be the most likely product during HDO of 1,2,3-BTO, especially when the reaction is carried out at low temperatures. This was confirmed by the 1,3-BDO selectivity of 61% during 1,2,3-BTO HDO reported by Gu et al.28 (Table S1,† entry 2). The high barrier for the primary C–O cleavage and the subsequent hydrogenation in 1,2,3-BTO on the other hand resulted in the low selectivity of 13% towards 2,3-BDO during HDO of 1,2,3-BTO. 1,2-BDO is also likely to form as per our computations but was not observed in the experiments of HDO of 1,2,3-BTO by Gu et al. (Table S1†).28 A possible explanation for this is provided in the last section.
It is to be noted here that the adsorption energy of 1,2,4-BTO configurations that facilitate protonation and dehydration of its different hydroxyls is different as can be seen in Fig. 3b. The configuration leading to the cleavage of C2–OH was the most stable and the configuration leading to C1–OH was the least stable. With reference to these adsorbed states, the protonation of different hydroxyls of 1,2,4-BTO showed a nearly similar preference. The primary C–O cleavage in 1,2,4-BTO (OH at C4 removed) had an activation barrier of 93 kJ mol−1 and was energetically favourable. Following this, the hydrogenation at C4 had an activation barrier of 68 kJ mol−1, resulting in the formation of 1,2-BDO. The primary C–O cleavage (OH at C1 removed) had a higher activation barrier of 109 kJ mol−1 but the reaction was extremely favourable leading to a highly stable state. Following this, the hydrogenation barrier at C1 was also high at 116 kJ mol−1, leading to the formation of 1,3-BDO. Moreover, this reaction was unfavourable, given the extremely stable precursor state. In contrast to these primary C–O cleavages, the secondary C–O bond cleavage barrier in 1,2,4-BTO for the formation of 1,4-BDO (OH at C2 removed) was extremely low at 45 kJ mol−1 and the reaction was energetically favourable. The subsequent hydrogenation at C2 had a barrier of 53 kJ mol−1 and was energetically favourable. These trends in C–O cleavage barriers and the observed thermodynamic preferences in Fig. 3b suggest that 1,4-BDO is the most likely product from 1,2,4-BTO. This is confirmed by the high selectivity of 80% to 1,4-BDO (Table S1†). The relatively lower stability of the 1,2,4-BTO configuration leading to 1,3-BDO formation and the comparatively high barriers for the reaction steps leading to the formation of 1,3-BDO make it a less preferred product. This is evident from the 8% selectivity to 1,3-BDO during HDO of 1,2,4-BTO (Table S1† entry 3).28 Our simulations indicate that 1,2-BDO formation is possible under the conditions at which 1,3-BDO might form. However, experiments of HDO of 1,2,4-BTO by Gu et al.28 did not report the formation of 1,2-BDO. A possible explanation for this is provided in the last section.
HDO of butanediols to butanols
Although HDO of erythritol did not yield appreciable quantities of butanols, HDO of butanetriols (Table S1†) and butanediols (Table S2†) showed 1-BO and 2-BO as products.28 The formation of butanols from butanetriols is likely to occur via the butanediol intermediates.28 1-BO may form from 1,2-BDO (OH at C2 removed), 1,3-BDO (OH at C3 removed) and 1,4-BDO (any of the OHs removed), while 2-BO can form from 1,2-BDO (OH at C1 removed), 1,3-BDO (OH at C1 removed) and 2,3-BDO (any of the OHs removed). It is worthwhile to note that 1,2-BDO and 1,3-BDO can form both 1-BO and 2-BO, while 1,4-BDO can form only 1-BO, and 2,3-BDO can form only 2-BO.Among the configurations of 1,2-BDO, 1,3-BDO and 1,4-BDO that facilitate 1-BO formation, 1,2-BDO had the strongest adsorption on the catalyst and led to the most stable protonated state as can be seen from Fig. 4a. The interaction of 1,3-BDO and 1,4-BDO was substantially weaker and led to slightly less stable protonated states. Among the configurations of 1,2-BDO, 1,3-BDO and 2,3-BDO that facilitate the formation of 2-BO, 2,3-BDO had the strongest interaction with the catalyst and led to the most stable protonated state as can be seen in Fig. 4b. The interaction of 1,2-BDO and 1,3-BDO was much weaker.
 | ||
| Fig. 4 HDO of different butanediols to form a) 1-butanol and b) 2-butanol on the Ir–ReOx catalyst. The energy profiles for the HDO of butanediols to form 1-butanol and 2-butanol are shown in a′) and b′), respectively. Images of transition states in these energy profiles are available in Fig. S7 and S8 of the ESI.† | ||
The primary C–O cleavage in 1,4-BDO towards the formation of 1-BO had an activation barrier of 103 kJ mol−1 and was favourable as shown in Fig. 4a. Following this, the hydrogenation had an activation barrier of 61 kJ mol−1 and was energetically favourable. The experiments of HDO of 1,4-BDO on Ir–ReOx (ref. 28) showed only 1-BO as the alcohol product, as reported in Table S2.† The comparatively high C–O cleavage barrier explains the low 1,4-BDO conversion of 16% in a four-hour reaction (Table S2†). In contrast to the primary C–O cleavage in 1,4-BDO, the secondary C–O cleavage in 2,3-BDO towards 2-BO formation had a lower activation barrier of 85 kJ mol−1 and the reaction was highly favourable as shown in Fig. 4b. Following this, the hydrogenation barrier was 80 kJ mol−1. The high stability of the protonated state and the comparatively low barrier for the HDO steps are likely to make 2,3-BDO much more reactive than 1,4-BDO.
The secondary C–O cleavage in 1,2-BDO towards forming 1-BO had a low activation barrier of 65 kJ mol−1 and the reaction was extremely favourable as shown in Fig. 4a. Following this, the hydrogenation barrier was 105 kJ mol−1 and the reaction was slightly unfavourable. In contrast, the primary C–O cleavage in 1,2-BDO towards 2-BO formation had a high barrier of 167 kJ mol−1 as shown in Fig. 4b, although the reaction was highly favourable. Following this, the hydrogenation barrier was 74 kJ mol−1. The high barrier for the primary C–O cleavage (Fig. 4a and b) explains the predominant formation of 1-BO with a selectivity of 86% during HDO of 1,2-BDO while 2-BO was observed at a lower selectivity (10%) on this catalyst as reported in Table S2.†
The secondary C–O cleavage in 1,3-BDO towards 1-BO formation had an activation barrier of 62 kJ mol−1 and the reaction was highly favourable as can be seen in Fig. 4a. Following this, the hydrogenation barrier was 74 kJ mol−1. Surprisingly, the primary C–O cleavage in 1,3-BDO towards 2-BO formation had a relatively low activation barrier of 77 kJ mol−1 and the reaction was energetically favourable. Following this, the hydrogenation barrier was only 56 kJ mol−1 and this step was highly favourable. The minimal difference in the activation barriers for the kinetically relevant step for 1-BO and 2-BO formation from 1,3-BDO explains the nearly similar selectivity of 1-BO (47%) and 2-BO (45%) obtained on this catalyst28 as reported in Table S2.†
Effect of H* coverage and Brønsted acid concentration on the ReOx cluster and undercoordinated Ir sites
The mechanistic analyses presented so far are based on the Re3O6H/Ir[111] catalyst model containing 4H*. A single model may not be able to capture insights into some variabilities associated with realistic catalysts. In this section, we showcase the impact of a few catalytic features on the reaction mechanisms and energy profiles.Lin et al.29 in their study on the ring-opening of tetrahydrofurfuryl alcohol over a WOx-modified Pt catalyst revealed that H* coverage added a 0.5 eV repulsive interaction weakening the tetrahydrofurfuryl alcohol adsorption, making its binding on the Pt surface unfavourable in the presence of H* for further activation. Similarly, Wang et al.30 studied the effect of H* coverage on the selectivity towards hydrogenation and decarbonylation of furfural on Pd(111) and reported that increasing the hydrogen coverage made decarbonylation unfavourable, while hydrogenation was favoured. To understand if similar impacts are relevant in the HDO of erythritol, 1,2,4-BTO and 1,2,3-BTO formation via the secondary and primary C–O cleavage, respectively, was compared at 0.2 ML and 0.5 ML H* coverage. The energy profiles are shown in Fig. 5a and b, respectively. Our analysis shows that the protonation and dehydration steps facilitated by the BA site on the ReOx cluster are only marginally affected by the H* coverage on Ir. However, the subsequent hydrogenation step is impacted more significantly, with slightly different trends for hydrogenation of the secondary and primary carbon. During 1,2,4-BTO formation at 0.2 ML H* coverage, the secondary C–O cleavage barrier was 61 kJ mol−1 (ΔHr = −90 kJ mol−1), and the subsequent hydrogenation step, which was the kinetically relevant step, had a barrier of 119 kJ mol−1 (ΔHr = +50). At 0.5 ML H* coverage, the secondary C–O cleavage barrier was only marginally impacted with a barrier of 65 kJ mol−1 (ΔHr = −81). Interestingly, the hydrogenation barrier dropped to 93 kJ mol−1 (ΔHr = +45) which is a substantial drop in the activation barrier. During 1,2,3-BTO formation, the primary C–O cleavage had a barrier of 129 kJ mol−1 (ΔHr = −75 kJ mol−1) at 0.2ML H* coverage which became 133 kJ mol−1 (ΔHr = −84 kJ mol−1) at 0.5 ML H* coverage. However, the hydrogenation barrier increased from 68 kJ mol−1 (ΔHr = −4) at 0.2ML H* coverage to 76 kJ mol−1 (ΔHr = −4) at 0.5ML H*. Hence, a higher hydrogen pressure which is likely to increase the H* coverage on the Ir surface is expected to promote HDO via secondary C–O cleavage, thereby enhancing the selectivity towards 1,2,4-BTO.
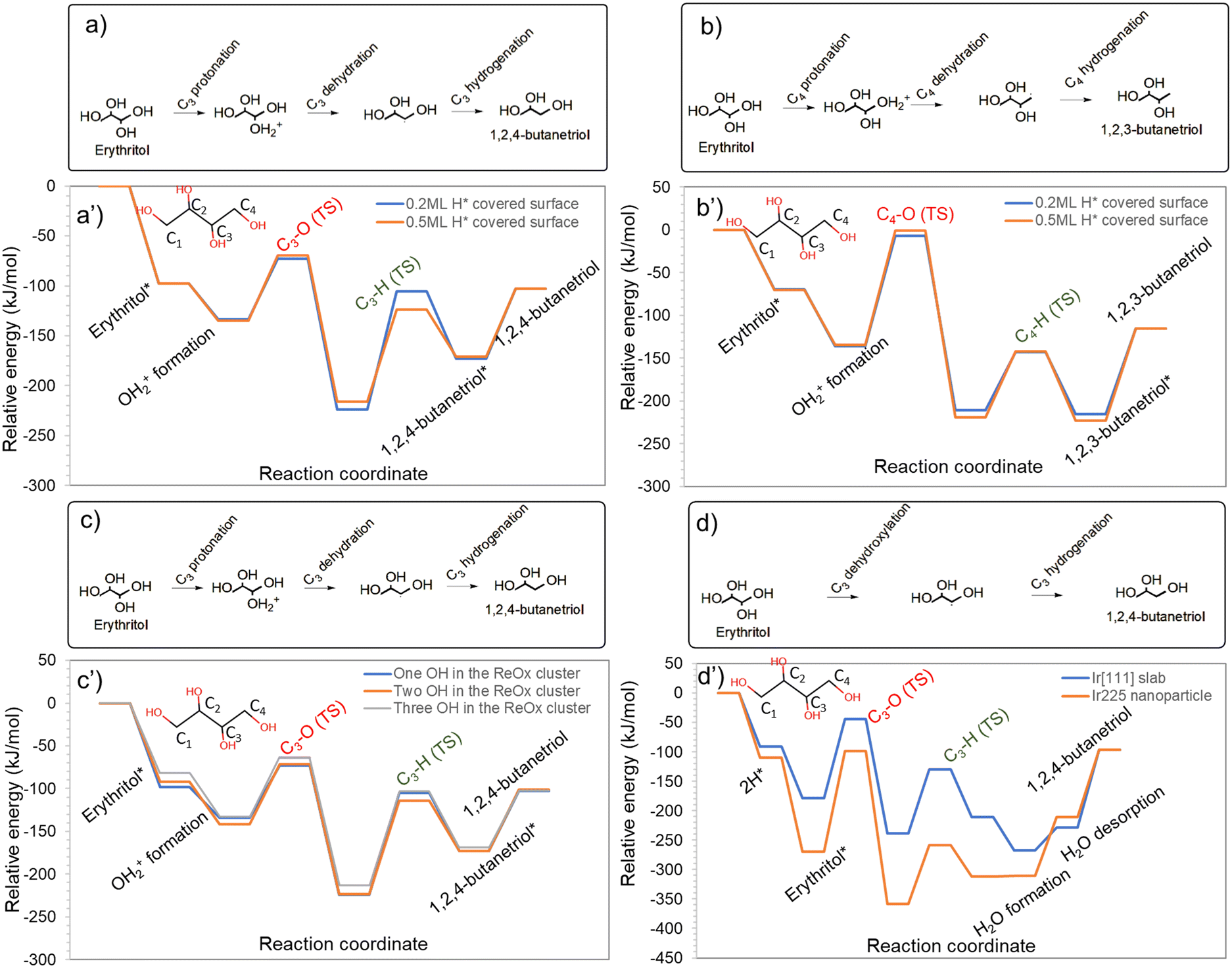 | ||
| Fig. 5 HDO of erythritol to 1,2,4-butanetriol (a) and energy profiles comparing the HDO of erythritol to 1,2,4-BTO at surface hydrogen coverage of 0.2 monolayer and 0.5 monolayer (a′). HDO of erythritol to 1,2,3-butanetriol (b) and energy profiles comparing the HDO of erythritol to 1,2,3-BTO at surface hydrogen coverage of 0.2 monolayer and 0.5 monolayer (b′). HDO of erythritol to 1,2,4-BTO (c) and energy profiles showing the effect of Brønsted acid density on the ReOx cluster on 1,2,4 butanediol formation from erythritol (c′). HDO of erythritol to 1,2,4-BTO in a mechanism involving C–O cleavage by direct dehydroxylation by the Ir atoms (d) and the energy profiles comparing the direct dehydroxylation mechanism at the undercoordinated sites on an Ir225 nanoparticle model and at the terrace sites on an Ir[111] slab model (d′). | ||
The Brønsted acid density on the partially reduced WOx on Pt in a Pt–WOx/SiO2 inverse catalyst which exhibits similar characteristics to the ReOx–Ir inverse catalyst was reported to be dynamic during the reaction.31 Hence, a single model may not be representative of the operando catalyst. To understand the extent of influence of the Brønsted acid density on the ReOx cluster in the ReOx–Ir inverse catalyst on polyol adsorption, we considered two additional catalyst models: Re3O6H2/Ir(111) and Re3O6H3/Ir(111) with systematically increasing BA density. The adsorption energy of erythritol was computed to be −98 kJ mol−1, −92 kJ mol−1, and −82 kJ mol−1 on Re3O6H/Ir(111), Re3O6H2/Ir(111), and Re3O6H3/Ir(111), respectively. In each of these cases, it should be noted that erythritol adsorption was considered in a configuration that facilitates secondary C–O bond cleavage and formation of 1,2,4-BTO. Since we observed a non-negligible variation in erythritol adsorption strength with BA density on the ReOx cluster, we investigated the entire reaction sequence of 1,2,4-BTO formation on Re3O6H/Ir(111), Re3O6H2/Ir(111), and Re3O6H3/Ir(111) catalyst models and the energy profiles are shown in Fig. 5c. The protonation of erythritol became more favourable with increasing BA density, with the protonation energy changing from −36 kJ mol−1 on Re3O6H/Ir(111) to −49 kJ mol−1 and −51 kJ mol−1 on Re3O6H2/Ir(111) and Re3O6H3/Ir(111) catalyst models, respectively. The stabilization of the protonated erythritol with an increase in BA density resulted in a marginal increase in the secondary C–O cleavage barrier from 61 kJ mol−1 (ΔHr = −90) on Re3O6H/Ir(111) to 70 kJ mol−1 (ΔHr = −79) and 69 kJ mol−1 (ΔHr = −80) on Re3O6H2/Ir(111) and Re3O6H3/Ir(111) catalyst models, respectively. With the slight destabilization of the dehydrated state with the increase in BA density, the hydrogenation barriers decreased from 119 kJ mol−1 (ΔHr = +51) on Re3O6H/Ir(111) to 109 kJ mol−1 (ΔHr = +50) and 110 kJ mol−1 (ΔHr = +45) on Re3O6H2/Ir(111) and Re3O6H3/Ir(111) catalyst models, respectively. Our results here indicate that the increase in BA density on the ReOx cluster may favour the HDO reaction by lowering the barrier for the kinetically relevant hydrogenation step at moderate BA density, but the trend is unlikely to be monotonic with BA density. Thus, increasing the hydrogen pressure in the reactor which is likely to increase the BA density on the ReOx cluster due to hydrogen spillover from the Ir is likely to favor HDO for a certain range of hydrogen pressure.
Marlowe et al.32,33 showed a clear role of undercoordinated metal sites in determining catalytic performance and product selectivity using the HDO of dihydroeugenol as a model reaction. To understand potential contributions of undercoordinated Ir sites during HDO of erythritol, we compared the erythritol HDO mechanism at the undercoordinated sites on an Ir225 nanoparticle model and at the terrace sites on the Ir(111) catalyst. In this HDO mechanism, the C–O bond cleavage proceeded by direct dehydroxylation by the Ir atoms, unlike the protonation and dehydration described for the Re3O6H/Ir(111) catalyst. The energy profiles are compared in Fig. 5d. Compared to terrace sites on the Ir(111) surface (erythritol adsorption energy of −89 kJ mol−1), erythritol adsorbs much more strongly at the undercoordinated sites of the Ir225 nanoparticle with an adsorption energy of −159 kJ mol−1. The secondary C–O bond cleavage by direct dehydroxylation had an activation barrier of 135 kJ mol−1 (ΔHr = −59 kJ mol−1) at the terrace site on Ir(111) while it was much higher at 170 kJ mol−1 (ΔHr = −89 kJ mol−1) at the undercoordinated sites on the Ir225 nanoparticle. This might be due to the much more stable adsorbed state of erythritol at the undercoordinated sites of the Ir225 nanoparticle. It is to be noted that these secondary C–O cleavage barriers are substantially higher than the barrier by the protonation and dehydration mechanism which was 61 kJ mol−1 on the Re3O6H/Ir(111) catalyst. After the dehydroxylation, the hydrogenation barrier was 109 kJ mol−1 (ΔHr = +30) on the terrace site of Ir(111) which was slightly lower at 100 kJ mol−1 (ΔHr = +47) at the undercoordinated site on the Ir225 nanoparticle. Based on these results and the comparison with the mechanistic analysis on the Re3O6H/Ir(111) catalyst, it can be inferred that, for straight chain polyols such as erythritol, the HDO on the Ir–ReOx catalysts is most likely to occur at the Ir–ReOx interface involving active sites across the Ir nanoparticle and the ReOx cluster via the protonation and dehydration mechanism.
One of the observations from this work is that the BA site is consumed during the protonation step of the reaction. BA site formation is possible by heterolytic dissociation of H2 on the ReOx cluster or H* diffusion/spillover from Ir sites to the ReOx cluster. Analyses of these BA site regeneration mechanisms showed that the heterolytic dissociation of H2 on ReOx has a high barrier of 166 kJ mol−1 while H2 activation on Ir and subsequent H* diffusion/spillover have a much lower barrier of just 41 kJ mol−1. With the latter likely being the prominent BA site formation and regeneration mechanism, high hydrogen pressures will facilitate the seamless formation and regeneration of the BA sites for sustained and selective HDO activity. Such a mechanism will also benefit from the inverse nature of the catalyst with finely dispersed ReOx on the Ir surface which maximises the ReO–Ir interface.
Rationalizing product selectivity trends during HDO of erythritol
Fig. 6 summarises the various routes for the formation of the different products starting from erythritol, together with activation energy barriers and reaction energies for the key steps along these routes. The data pertaining to the protonation step are provided in blue font, the C–O cleavage is in black font while those pertaining to the hydrogenation are in purple font. During HDO of erythritol on the Ir–ReOx catalyst, the substantially lower barrier (61 kJ mol−1vs. 129 kJ mol−1, Fig. 2 and 6) for the secondary C–O cleavage compared to the primary led to the formation of 1,2,4-BTO (selectivity = 37%) as the primary butanetriol, along with smaller quantities of 1,2,3-BTO (selectivity = 27%). 1,2,4-BTO is highly reactive (Table S1†). Among the HDO products from 1,2,4-BTO, 1,4-BDO is the most preferred butanediol based on analysis of the reaction profiles in Fig. 3b and 6 and data in Table S1.† Hence, 1,4-BDO is the prominent butanediol during erythritol HDO (Table S1†). Our simulations predicted the likelihood of formation of both 1,2-BDO and 1,3-BDO from 1,2,4-BTO. However, 1,2-BDO is not observed as a product during erythritol or 1,2,4-BTO HDO (Tables S1 and S2†) while 1,3-BDO is observed in both cases. Based on these, we believe that 1,3-BDO and 1,4-BDO originate from 1,2,4-BTO, with the latter being the predominant product. 1,2,3-BTO is substantially less reactive than 1,2,4-BTO (Table S1†). Among the HDO products of 1,2,3-BTO, 1,3-BDO is the most likely to form, based on analysis of the reaction profiles in Fig. 3a and 6 and data in Table S1.† Our simulations predicted the likelihood of formation of 1,2-BDO from 1,2,3-BTO as well. However, it is not observed during HDO of erythritol or 1,2,3-BTO (Table S1†). The stability of the protonated state of 1,2,3-BTO with protonation at C1–OH may lead to 2,3-BDO formation although it is not a kinetically preferred product (Fig. 3a). The HDO of erythritol did not however result in 2,3-BDO (Table S1†) although it was a minor product during HDO of 1,2,3-BTO.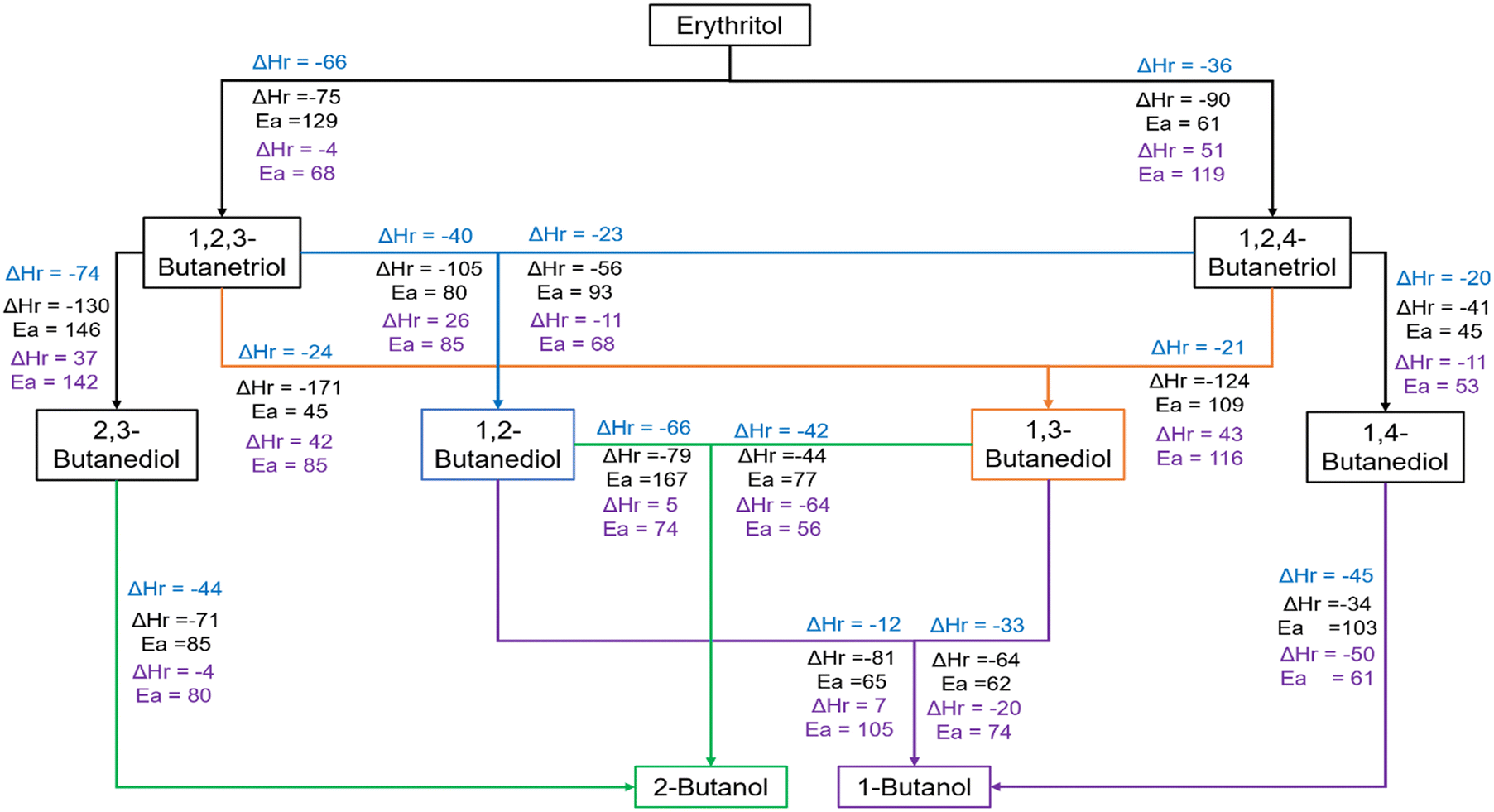 | ||
| Fig. 6 Summary of reaction routes to various alcohols during HDO of erythritol (data from Fig. 2–4) showing the reaction energy (ΔHr in kJ mol−1) for the protonation step (blue font), activation barrier (Ea in kJ mol−1) and reaction energy for the C–O bond cleavage step (black font), and the activation barrier and reaction energy for the hydrogenation step (purple font) on the Ir–ReOx catalyst. | ||
Although HDO of 1,3-BDO and 1,4-BDO gives butanols (Table S2†), these butanediols do not seem to react much in the presence of butanetriols and erythritol in the reaction mixture (Table S1†) in the short duration of 4 h. Upon analysis of the energy profiles in Fig. 4b and 6, it appears that the formation of 2-BO is most likely from 1,3-BDO. Hence, most of the 2-BO observed during HDO of 1,2,3-BTO (Table S1†) is likely to have originated from 1,3-BDO which was the dominant diol observed. However, we do not preclude its formation from 2,3-BDO which is a minor product of the HDO of 1,2,3-BTO. Based on these trends, 1,3-BDO is also likely to be the source of the trace amount of 2-BO observed during erythritol HDO (Table S1†).
Analysis of the energy profiles in Fig. 4a and 6 suggests that 1,2-BDO and 1,3-BDO are the most likely sources for 1-BO formation. Considering the preferential formation of 1-BO from 1,2-BDO (86% selectivity, Table S2†) and its substantially higher reactivity (conversion of 48%, Table S2†), we believe that 1,2-BDO is the primary source of 1-BO during HDO of 1,2,3-BTO and 1,2,4-BTO. This might be a possible explanation for the absence of 1,2-BDO reported as a product during HDO of erythritol, 1,2,3-BTO and 1,2,4-BTO (Table S1†). Based on analysis of trends in Fig. 4a and the low conversion of 1,4-BDO (Table S2†), 1,4-BDO formed during the HDO of erythritol and 1,2,4-BTO are less likely to react further to give 1-BO in a reaction mixture containing 1,2-BDO, 1,3-BDO or larger polyols.
Summary and conclusion
A thorough analysis of the reaction pathways involved in the hydrodeoxygenation (HDO) of erythritol and all possible intermediates from erythritol was done on inverse Ir–ReOx catalysts using DFT simulations. The kinetic preference for eliminating the secondary hydroxyl groups in polyols is a unique feature of Brønsted acid (BA) site-containing inverse catalysts such as Ir–ReOx. While the direct dehydroxylation mechanism is possible at the undercoordinated Ir sites, the protonation followed by dehydration initiated at the BA sites on the ReOx cluster is likely to be the kinetically preferred HDO mechanism. Hence, erythritol HDO reactions proceed by protonation of the polyol followed by elimination of the water molecule and subsequent hydrogenation from the Ir.We show that conversion of erythritol to butanediols is via formation of an intermediate butanetriol such as 1,2,3-butanetriol (1,2,3-BTO) or 1,2,4-butanetriol (1,2,4-BTO). We show that among the butanetriols, 1,2,4-BTO is the kinetically preferred product. However, an alternative initial orientation of erythritol over the catalyst can result in the formation of 1,2,3-BTO. In these reactions, we show that the C–O cleavage is not always the kinetically relevant step and it is in many cases the hydrogenation following the C–O cleavage. Hence, reactions carried out at high hydrogen pressure will benefit from the higher concentration of hydrogen for both generation of the BA sites on the ReOx cluster and also reduction of the barrier for the hydrogenation steps on the Ir surface. Considering the hydrogen spillover to be the primary mechanism for BA site formation and regeneration, maximising the Ir–ReOx interface by having finely dispersed ReOx would be beneficial for efficient BA generation/regeneration.
We show that 1,4-BDO is the most probable butanediol during HDO of erythritol. However, 1,3-BDO formation is unavoidable as it can form from both 1,2,3-BTO and 1,2,4-BTO. Our simulations indicated the possibility of formation of 1,2-BDO during erythritol HDO, although experiments do not report this product. Based on analysis of our simulations and experimental data, we posit that 1,2-BDO, if formed, will quickly react further to form 1-butanol. 1,3-BDO is the likely source for 2-butanol formation. 1,4-BDO is likely to be less reactive and remain as a product due to the comparatively high barrier for the primary C–O cleavage. This is useful as it might be possible to tune reaction conditions and time to maximize the production of this desired product.
While we can explain most of the experimental trends based on our extensive reaction network analyses, more detailed configurational sampling of the polyols over the catalyst and investigation of competitive interactions of various components in the reaction mixture with the catalyst would be necessary to be more conclusive and predictive. Additionally, explicit sampling of the solvent species might be necessary to make quantitative predictions of reaction routes and rates. We also demonstrate the potential variability in computational predictions arising from the differences in the catalytic states that are likely to occur during actual experiments.
Data availability
The DFT data generated as part of this investigation are reported in the manuscript and images of structures are provided in the ESI.†Author contributions
Ajin R.: conceptualization, data curation, formal analysis, investigation, writing – original draft. Jithin J. V.: conceptualization, resources, software, validation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support for this work from the Indian Institute of Technology Madras. The authors acknowledge the High-Performance Computing (HPC) facility provided by the Indian Institute of Technology Madras (IITM) for computing resources.Notes and references
- Z. Wang, C. Luo, B. Wang, T. Lai, Y. Hong and G. Gao, Fuel, 2024, 363, 130806 CrossRef CAS
.
- P.-H. Tsai, C.-H. Wang, L.-S. Kan and C. W. Chen, J. Polym. Res., 2020, 27, 121 CrossRef CAS
- C.-F. Lee, C.-W. Chen, S.-P. Rwei and F.-S. Chuang, Appl. Sci., 2021, 11, 698 CrossRef CAS
- B. M. Stadler, A. Brandt, A. Kux, H. Beck and J. G. de Vries, ChemSusChem, 2020, 13, 556–563 CrossRef CAS PubMed
- G. Yang, L. Yang and J. Chen, Ind. Eng. Chem. Res., 2023, 62, 21067–21077 CrossRef CAS
- T. Haas, B. Jaeger, R. Weber, S. F. Mitchell and C. F. King, Appl. Catal., A, 2005, 280, 83–88 CrossRef CAS
-
W. E. Smith, G. R. Chambers, R. C. Lindberg, J. N. Cawse, A. J. Dennis, G. E. Harrison and D. R. Bryante, in Catalysis of Organic Reactions, CRC Press, 2020, pp. 151–167 Search PubMed
- G.-H. Shi, X.-C. Chen, T. Jing, Y.-Y. Zhuang, Y. Lu and Y. Liu, J. Mol. Catal., 2023, 545, 113209 CrossRef CAS
- S. M. Prasanth, P. S. Kumar, S. Harish, M. Rishikesh, S. Nanda and D.-V. N. Vo, Chemosphere, 2021, 280, 130723 CrossRef CAS PubMed
- M. Jeya, K.-M. Lee, M. K. Tiwari, J.-S. Kim, P. Gunasekaran, S.-Y. Kim, I.-W. Kim and J.-K. Lee, Appl. Microbiol. Biotechnol., 2009, 83, 225–231 CrossRef CAS PubMed
- Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520–2530 CrossRef CAS
- L. Zhu, M. Sun, X. Zhao, Y. Zhang, H. Luo, W. Liu, G. Miao and L. Kong, Sustainable Energy Fuels, 2021, 1747–1755 RSC
- H. Li, Y. Wang, C. Zhang, Z. Huang, J. Han, X. Nie and F. Wang, Appl. Catal., A, 2023, 325, 122342 CrossRef CAS
- E. M. Virgilio, C. L. Padró and M. E. Sad, ChemCatChem, 2021, 13, 3889–3906 CrossRef CAS
- W. Wang, K. Nakagawa, T. Yoshikawa, T. Masuda, E. Fumoto, Y. Koyama, T. Tago and H. Fujitsuka, Appl. Catal., A, 2021, 619, 118152 CrossRef
- A. Rajan and J. J. Varghese, J. Catal., 2023, 423, 94–104 CrossRef CAS
- S. Bhowmik, N. Enjamuri, B. Marimuthu and S. Darbha, Catalysts, 2022, 12, 1070 CrossRef CAS
- J. J. Varghese, L. Cao, C. Robertson, Y. Yang, L. F. Gladden, A. A. Lapkin and S. H. Mushrif, ACS Catal., 2018, 9, 485–503 CrossRef
- S. Bhowmik, V. Akula, G. Sethia, B. Marimuthu and S. Darbha, Appl. Catal., A, 2023, 666, 119425 CrossRef CAS
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 2375–2380 RSC
- E. M. Virgilio, M. E. Sad and C. L. Padró, Appl. Catal., A, 2022, 643, 118691 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS
- J. A. White and D. M. Bird, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 4954–4957 CrossRef CAS
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef
- L. Liu, S. Kawakami, Y. Nakagawa, M. Tamura and K. Tomishige, Appl. Catal., A, 2019, 256, 117775 CrossRef
- M. Gu, L. Liu, Y. Nakagawa, C. Li, M. Tamura, Z. Shen, X. Zhou, Y. Zhang and K. Tomishige, ChemSusChem, 2021, 14, 642–654 CrossRef CAS PubMed
- Z. Lin, S. Deshpande, S. R. Denny, W. N. Porter, C. Wang, J. Marlowe, P. Christopher, W. Zheng, S. Caratzoulas, D. G. Vlachos and J. G. Chen, ACS Catal., 2023, 13, 8014–8024 CrossRef CAS
- S. Wang, V. Vorotnikov and D. G. Vlachos, ACS Catal., 2015, 5, 104–112 CrossRef CAS
- Y. Wu, S. Sourav, A. Worrad, J. Zhou, S. Caratzoulas, G. Tsilomelekis, W. Zheng and D. G. Vlachos, ACS Catal., 2023, 13, 7371–7382 CrossRef CAS
- J. Marlowe, S. Deshpande, D. G. Vlachos, M. M. Abu-Omar and P. Christopher, J. Am. Chem. Soc., 2024, 146, 13862–13874 CrossRef CAS PubMed
- J. Marlowe, P. C. Ford, M. M. Abu-Omar and P. Christopher, Catal. Sci. Technol., 2023, 13, 5662–5678 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00245h |
| This journal is © The Royal Society of Chemistry 2025 |