
The kinetics of the polyurethane moisture curing reaction: a combined experimental and DFT mechanistic study†
Jie
Zhou
,
Zhen
Liu
 ,
Zhihua
Zhu
,
Zuoxiang
Zeng
and
Li
Sun
*
,
Zhihua
Zhu
,
Zuoxiang
Zeng
and
Li
Sun
*
East China University of Science and Technology, School of Chemical Engineering, Shanghai, 200237, P. R. China. E-mail: sunli@ecust.edu.cn
First published on 30th September 2024
Abstract
An examination of the temporal dynamics of the moisture curing process of polyurethane (PUR) hot melt adhesives under varied humidity (65–85% RH) and temperature (20–40 °C) was performed via in situ Fourier transform infrared (FTIR) spectroscopy. The commencement of the moisture curing for PUR was substantiated by a reduction in peak intensity at 2271 cm−1. The data revealed that the nascent phase of the moisture curing reaction aligns with the first-order reaction kinetics. Further, there was a noticeable acceleration in the rate of the PUR's moisture curing reaction with an increment in environmental humidity and temperature. Density functional theory (DFT) was harnessed to delve into the effects of additional water clusters on the PUR's moisture curing progression. The theoretical model proposed that these additional water molecules catalyzed the hydrogen transfer, thus bolstering the moisture curing reaction, a finding that corroborated with our empirical observations.
Introduction
Polyurethane (PUR) hot-melt adhesives are multifaceted adhesives that exhibit a range of favourable attributes, outperforming conventional adhesives in terms of high-temperature resistance, corrosion resistance, and early bonding.1–3 As a result, they play an essential role in a variety of fields, such as electronics, automotives, aerospace, and furniture.4–7 Efforts to develop systematic and reliable strategies for enhancing PUR performance are thus paramount for not only broadening its functionality but also for mitigating production expenditures.PUR consists of flexible soft chain segments derived from diols and rigid hard chain segments consisting of polyisocyanates linked to the diols by urethane bonds.8–12 There are two main curing methods for PUR, namely, moisture curing and light curing. Light curing employs ultraviolet (UV) or visible light to trigger a photoinitiator in PUR, which initiates the polymerization process and subsequent cross-linking of the polymer chains. In contrast, moisture curing is cost-effective and ensures a uniform cure thickness, and also has the unique ability to function in high humidity environments.13–17 The PUR curing process typically begins in the molten state, which cools and crystallizes at the substrate surface, leading to physical cross-linking of the molecules and the initial bond strength.18 Subsequently, the isocyanate groups in PUR interact with water on the surface of the substrate or in the surrounding atmosphere, ultimately forming cross-links and macromolecular network structures.19–22 This chemical curing process results in increased crystallization of the molecules in the colloid, resulting in the final bond strength.23–26Scheme 1 illustrates the diisocyanate cross-linking mechanism inherent in PUR.
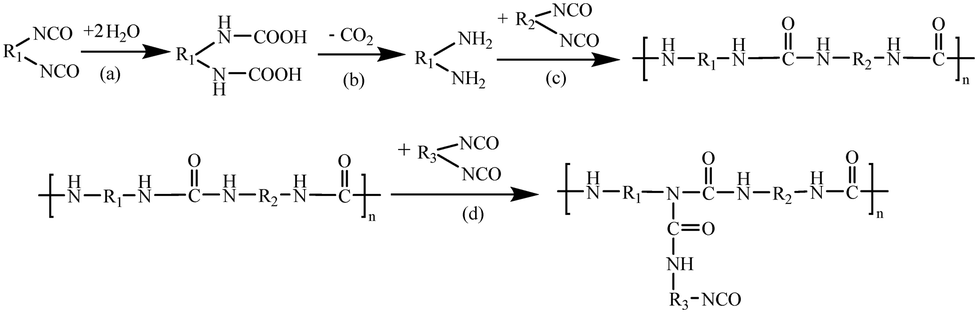 | ||
| Scheme 1 Diisocyanate crosslinking mechanism. (a) Electrophilic attack of water molecules on the –NCO group to generate carbamic acid. (b) Decomposition of carbamic acid into an amine and carbon dioxide. (c) Reaction of amine with isocyanate to form a substituted urea. (d) The active hydrogen on the secondary amine in the substituted urea attacks the –NCO group to form a biuret. | ||
Models proposed by scholars to understand the water molecule diffusion behavior inside polymers include the Langmuir-type model, the Drozdov model, and the mesoscopic model.27 External air temperature and humidity, as well as the rate of diffusion of moisture from the atmosphere or substrate, combine to affect the surface water content of polymer films.17,28 It has been shown that the moisture curing process as a mass transfer process conforms to Fick's second law. During the diffusion process, water molecules swiftly react with the terminal isocyanate reactive group NCO. Assuming a zero concentration of NCO groups within the diffusion path of water molecules, the reaction effect on mass transfer is overlooked, leading to a purely diffusive mass transfer process.29
Alterations in synthesis raw materials, additives, and curing conditions can manipulate PUR's properties and applications.30–33 Moreover, the curing speed of PUR can influence the colloid's structure and properties.34,35 Thus, it becomes imperative to explore and comprehend the curing kinetic mechanisms of different PUR reactions to achieve ideal curing conditions. A theoretical framework for regulating the curing process to acquire the most suitable and appealing PUR attributes can be provided by understanding PUR curing kinetics.25 The kinetics of PUR moisture curing have been examined via quantitative infrared spectroscopy, revealing that the curing reaction adheres to first-order reaction kinetics.31 Excess polyol was found to stimulate the curing kinetics of PUR, promoting the first-order reaction kinetics in one study.36 Another study probed the reaction kinetics between isocyanate groups (NCO) and hydroxyl groups (OH) at various sites, finding that the kinetics of each group followed the second-order equation.37 A study involving the crosslinking kinetics of polyol-cured polyurethanes with hyperbranched polyols showed a significant fit to the second-order equation.38 Dhevi et al. studied the curing process of hyperbranched polyurethanes, observing that the reaction kinetics followed second-order kinetics when the NCO and OH contents were similar, whereas third-order reaction kinetics were observed when NCO was in excess.39
The moisture curing mechanism of PUR is complex and ordinarily spans several hours.40 However, the initial phase of curing is often the most rapid and crucial.41 Previous studies have predominantly concentrated on the entire process without rigorous scrutiny of the initial stage. The objective of this study is to elucidate the initial moisture curing mechanism of PUR employing both experimental and computational techniques. In this context, in situ Fourier transform infrared spectroscopy was utilized to examine the kinetics of the initial moisture curing under diverse humidity and temperature conditions. Concurrently, density functional theory (DFT) was engaged to provide a theoretical basis for the moisture curing mechanism of PUR.
Experimental
Materials
The experimental raw materials, including polytetra-methylene glycol (PTMG, Mn = 2000, industrial grade) and polybutylene adipate diol (PBA, Mn = 2000, industrial grade), were purchased from BASF China Ltd. 4,4′-Methylene diphenyl diisocyanate (MDI) was supplied by Yantai Wanhua Polyurethanes Co., Ltd, China. The catalyst, 2,2′-dimorpholinediethylether (DMDEE, technical grade), was purchased from Shanghai Aladdin Biochemical Technology Co. The silane coupling agent, γ-(2,3-epoxypropoxy)-propytrimethoxysilane (KH-560, industrial grade), was purchased from Nanjing He Run Coupling Agent Co. Dilute monomer tetrahydrofuran acrylate (THFA), technical grade, was purchased from Shanghai Aladdin Biochemical Technology Co. Potassium bromide (KBr) for infrared compression, purity over 99.5%, was purchased from Shanghai Aladdin Biochemical Technology Co.Synthesis of polyurethane hot melt adhesive
PTMG-2000 (10.3 g) and PBA-2000 (10.3 g) were maintained under vacuum (100 Pa) at 120 °C for 6 hours in a 100 ml three-neck flask to remove water. After drying, MDI (5.15 g), DMDEE (0.5 g), KH-560 (0.5 g) and THFA (1 g) were added into the flask, and the mixture was further agitated for 2 h at 80–90 °C. The above reaction processes were carried out under vacuum (100 Pa).Preparation and testing method of PUR prepolymer samples
As the PUR prepolymer produced from MDI is extremely sensitive to water, the sample preparation process must be carried out in an anhydrous environment. In this work, dried KBr blanks were prepared in advance. In the early stages after the end of the polyurethane pre-polymerization reaction, when the product temperature and the residual content of the characteristic functional group –NCO have not yet been stabilized, in situ IR measurements can lead to experimental errors. The polyurethane prepolymer synthesized above was sealed and transferred to the glove box. The polyurethane film samples were prepared with a thickness of 1 mm by taking equal amounts of the prepolymer one at a time and applying them evenly and thinly to the prefabricated KBr blanks. The prepared samples were stored in an anhydrous environment for 2 hours for maturation.FTIR spectroscopy
The in situ FTIR spectroscopy was carried out using a Bruker VERTEX 80/80v FTIR spectrometer (Bruker, Germany), with spectral resolution of 4 cm−1, and the scanning range was from 4000 to 1000 cm−1 with scan interval of 30 seconds, using a Transmission Reflection (TR) accessory.In this study, an in situ infrared reactor was designed that can precisely control the humidity. Please refer to the ESI† for the corresponding experimental setup diagram and situation description. Equal amounts of polyurethane prepolymer were uniformly applied to KBr samples in a dry environment and moisture cured at 20–40 °C and 65–85% relative humidity (RH), and the data were recorded by the FTIR spectra.
Density functional theory (DFT)
All calculations were performed using the Gaussian 09 program package. Unconstrained optimization of the geometric structure of all intermediates was performed using the m062x functional in combination with the 6-31G(d,p) basis set.42 Truhlar's SMD solvation model with water as the model solvent and Grimme's dispersion were incorporated into the geometry optimizations.43 The electronic energies were further refined by using the 6-311G(d,p) basis set.44 Intrinsic reaction coordinate (IRC) calculations were used to verify each transition state property to indicate a direct relationship between the corresponding reactants and products.45 All transition states only have one imaginary frequency and have a vibration mode consistent with the reaction path.Results and discussion
Procedure of the FTIR spectra in urethane reaction kinetics
The real-time elucidation of the moisture-curing reaction process of polyurethane (PUR) largely employs in situ Fourier Transform Infrared (FTIR) spectroscopy due to its unparalleled accuracy and efficacy.19,24,46,47 Exhibited in Fig. 1a, the FTIR spectra of the PUR prepolymer shows salient features. The absorption peaks affiliated with the stretching vibrations of –CH3 and –CH2– are identified within the range of 2850 to 2990 cm−1. As these entities do not partake in the moisture curing reactions of PUR, their peaks serve as internal standards for conversion rate assessment.30,48 Prominent absorption peaks are attributed to –NCO groups at 2274 cm−1, carbamate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations at 1730 cm−1, and CO stretching vibrations in the urea bond at 1650 cm−1, acting as definitive markers of the PUR moisture curing reaction's inception. The N–H bending vibration absorption peaks and C–O–C stretching vibration absorption peaks, characteristic of amide II and amide III bands in carbamates, respectively, correspond to 1530 cm−1 and 1223 cm−1.9,49
O stretching vibrations at 1730 cm−1, and CO stretching vibrations in the urea bond at 1650 cm−1, acting as definitive markers of the PUR moisture curing reaction's inception. The N–H bending vibration absorption peaks and C–O–C stretching vibration absorption peaks, characteristic of amide II and amide III bands in carbamates, respectively, correspond to 1530 cm−1 and 1223 cm−1.9,49
 | ||
| Fig. 1 (a) Infrared spectrum of polyurethane hot-melt adhesives; (b) in situ infrared spectra of polyurethane moisture-curing. | ||
The evaluation of the PUR moisture curing progression is achievable through meticulous tracking of the hardener –NCO group spectral band's decline (proximate to 2273 cm−1) and the emergence of the carbonyl band (approximately at 1730 cm−1).24 The in situ FTIR spectra of PUR moisture curing, as represented in Fig. 1b, authenticate that the reaction was underway. The indicative functional group –NCO at 2273 cm−1 engages in persistent reactions with water, leading to its gradual consumption. The amplified CO vibrational peak at 1650 cm−1 in the urea group attests to the ongoing moisture curing reaction, thereby facilitating carbamate production.49,50 In addition, the presence of water in the sample can also be monitored by the hydroxyl group at 3000–3500 cm−1. It can be noticed from Fig. 1b that around 240 min the hydroxyl peak does not change much, at which point water can be considered to be close to equilibrium saturation.
Analysis of kinetic models
After contact with water, the PUR prepolymer undergoes continuous reactions involving the characteristic functional group –NCO, resulting in the formation of the final urea-based copolymer, with the by-product CO2 produced during the curing process. Hence, the degree of moisture curing of PUR can be assessed by monitoring the disappearance of the –NCO peak or the appearance of the CO peak. The conversion rate of –NCO groups for different temperature and humidity conditions can be estimated using eqn (1).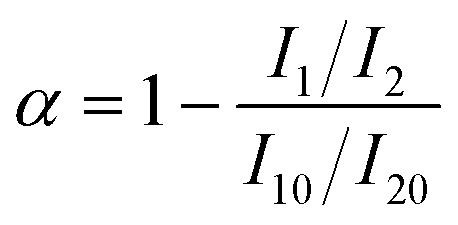 | (1) |
While it has been generally accepted that the PUR moisture cure kinetics can be described by the second-order reaction kinetics, Guo et al. demonstrated that the early and mid-late stages of PUR moisture curing follow distinct kinetic models.18 However, since the current study focuses on the reaction kinetics at the beginning of PUR moisture curing, the proposed first-order reaction model appears to be more suitable for this system.23 The kinetic model is shown in eqn (2):
| ln(1 − α) = − kt | (2) |
Thermodynamic parameters can be determined by plotting using the Arrhenius law (3) and Eyring formula (4):
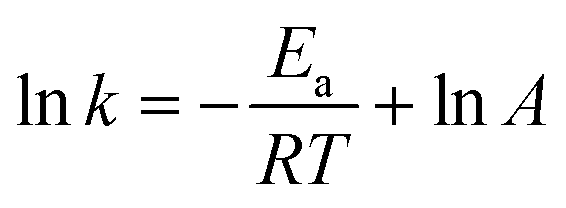 | (3) |
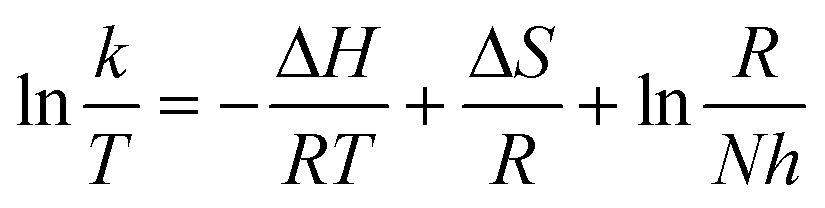 | (4) |
The curves fitting the kinetic model over a temporal course under a variety of humidity and temperature settings are exhibited in Fig. 2. The accompanying rate constants for each set of conditions are detailed in Table 1. The remarkable alignment of the fitted curves confirms the early phase of PUR moisture curing's adherence to first-order reaction kinetics, as suggested in Fig. 2. Moreover, we observe an amplified incline of the curve in line with elevated temperature and humidity, symbolizing an expedited reaction rate. The increase of temperature and humidity accelerated the moisture-curing rate of polyurethane. This is consistent with the research result of H. Xiang et al.51 Such a phenomenon is attributable to the heightened reactivity of the isocyanate group to water.17,52 Prior research studies have established that the moisture curing mechanism of PUR navigates through intricate reaction pathways, giving rise to by-products such as carbamates or biurets.30 The adhesive's curing phase comprises an intertwined process of physical diffusion and chemical reaction.29 During the initial stages of PUR moisture curing, the rate-determining step emerges as the chemical interaction between the reactive functional groups.13
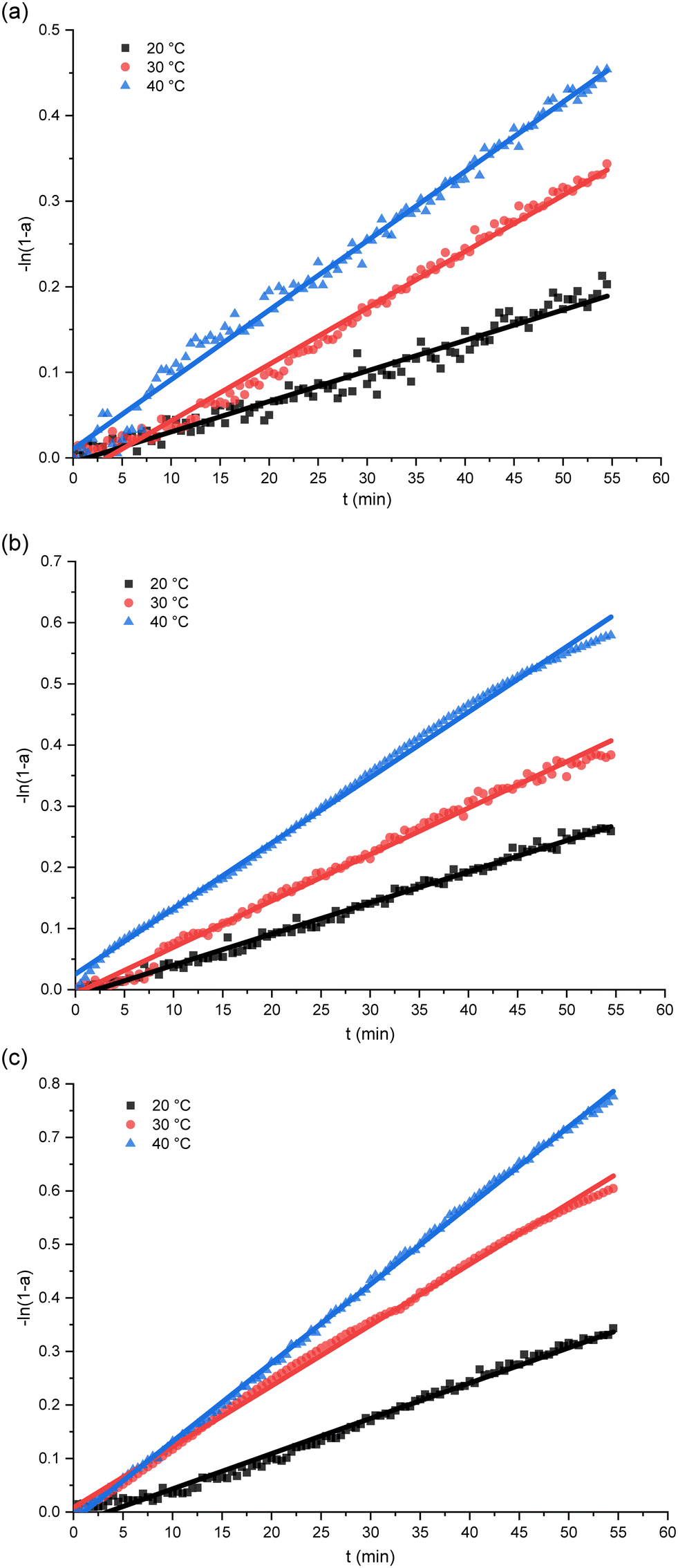 | ||
| Fig. 2 The relation plots of isocyanate group conversion versus time. (a) 65% RH; (b) 75% RH; (c) 85% RH. | ||
| k (×10−3 min−1) | 20 °C | 30 °C | 40 °C |
|---|---|---|---|
| 65% RH | 3.56 | 6.59 | 8.11 |
| 75% RH | 5.09 | 7.58 | 10.70 |
| 85% RH | 6.59 | 11.37 | 14.71 |
As the reaction unfolds, by-products are produced and an intricate cross-linked network structure evolves within the polymer, imposing a restriction on the water molecules' diffusion, which can modulate the future cure rate. Here, the cure rate appears to be dictated by the water's diffusion within the matrix.53 As corroborated by earlier investigations by He, Yan et al., the resin/wood mixture's moisture content drives distinct reactions during the curing process.17 An amplified moisture content in the substrate effectively diminishes the activation energy required for the moisture curing process, culminating in a rise in both the reaction enthalpy and rate.17 As delineated in Fig. 2, an escalated ambient humidity causes the curve's slope to steepen, signifying an accelerated PUR curing rate and suggesting an enhanced diffusion of water molecules within the polymer under conditions of higher ambient humidity. This observation aligns with the findings from Oliver Kläusler et al., who noted that the behaviour of PUR moisture curing undergoes significant alterations at varying relative humidity, ultimately influencing the final bond strength and mechanical attributes of the adhesive.28
Fig. 3 displays the thermodynamic equation curve, and the thermodynamic parameters obtained are presented in Table 2. The results show that an increase in the relative humidity of the environment significantly reduces the activation energy of the PUR moisture cure process, leading to an increase in the cure rate (Fig. 3a). This phenomenon is attributed to the fact that, in the early stage of moisture curing, the diffusion of water molecules is the rate-limiting step of the curing reaction.19 At this stage, the isocyanate groups are not heavily reacted, and the carbamates are not produced in large quantities, limiting the diffusive movement of water molecules.29 Additionally, a positive ΔH indicates that the PUR moisture curing process is an endothermic reaction. The negative value of ΔS may be due to the conjugation mechanism in the transition state (Table 2).31
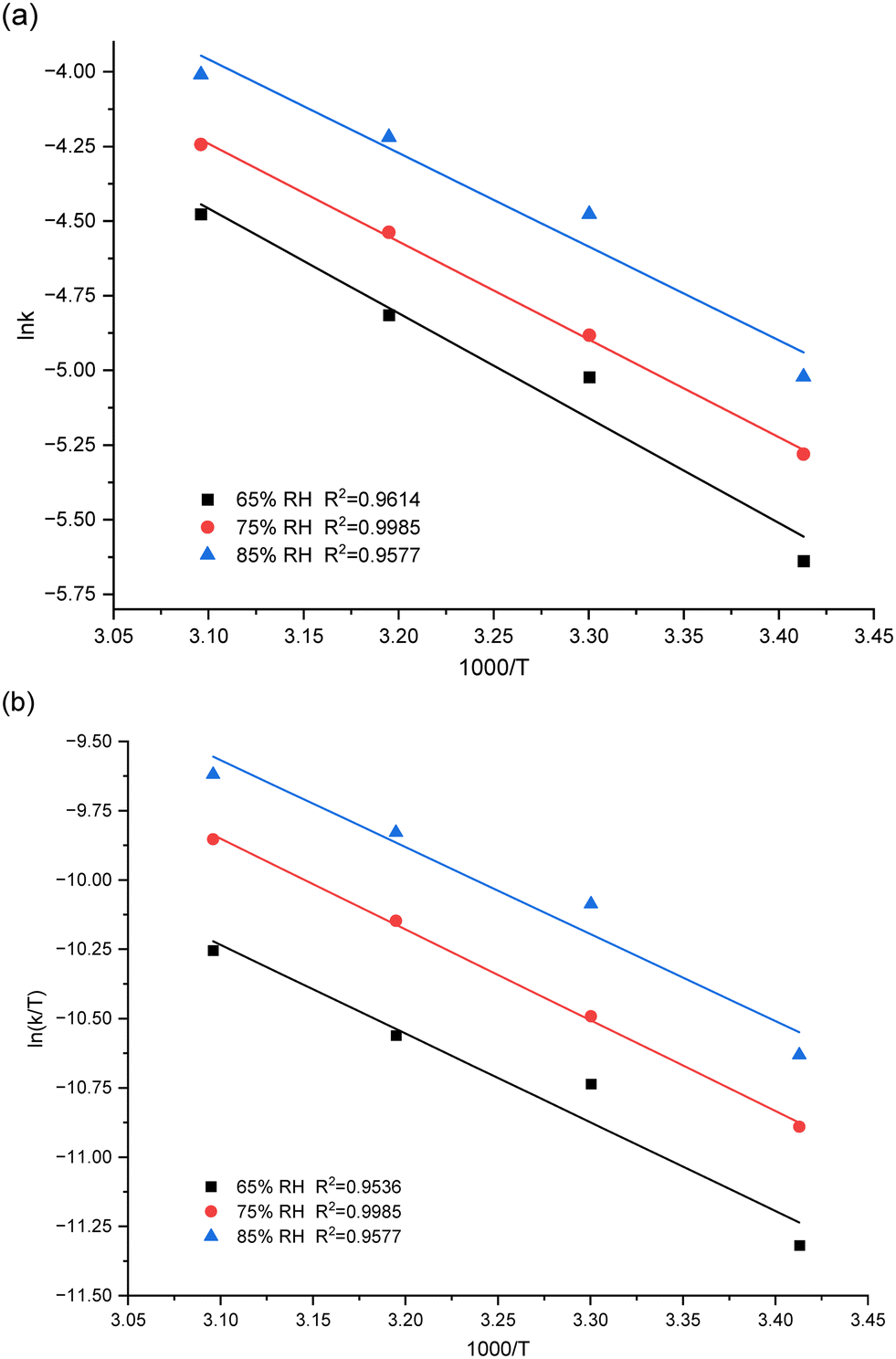 | ||
| Fig. 3 (a) Arrhenius and (b) Eyring plots for the curing reaction between MDI and H2O. | ||
| Humidity (% RH) | Arrhenius law | Eyring formula | ||
|---|---|---|---|---|
| E a (kJ mol−1) | A (min−1) | ΔH (kJ mol−1) | ΔS (J mol−1 K−1) | |
| 65 | 29.17 | 613.01 | 26.61 | −200.13 |
| 75 | 27.23 | 369.65 | 27.23 | −195.04 |
| 85 | 26.08 | 319.82 | 26.08 | −196.24 |
Moisture-curing mechanism of PUR
The moisture curing mechanism of PUR involves the nucleophilic attack of the oxygen atom in the water molecule on the carbon atom in the isocyanate group. This can occur via two possible reaction pathways. In one pathway, a single water molecule nucleophilically attacks the isocyanate group by breaking the CN bond, resulting in a rigid tetracyclic transition state TS2–4 and producing the carbamic acid 4 (path a, Scheme 2, Fig. 5a). During this reaction, the hydrogen transfer occurs, causing the O–H bond to break, forming a new N–H bond and a C–O bond. The energy barrier for this step of the reaction is 136.5 kJ mol−1. In the other pathway, a single water molecule breaks the CO bonds on the isocyanate to form CO bonds on the isocyanate to form a rigid tetrameric ring transition state TS2′–3, resulting in the unstable intermediate carbamide 3 (path b, Scheme 2, Fig. 5b). The energy barrier for this step is 165.3 kJ mol−1. Carbonimidic acid 3 further undergoes 1,3-hydrogen transfer to form carbamic acid (Fig. 5c). The energy barrier for the reaction broken via the CN bond is 28.8 kJ mol−1 lower than that for the reaction broken via the CO bond, indicating that the reaction tends to follow path a.
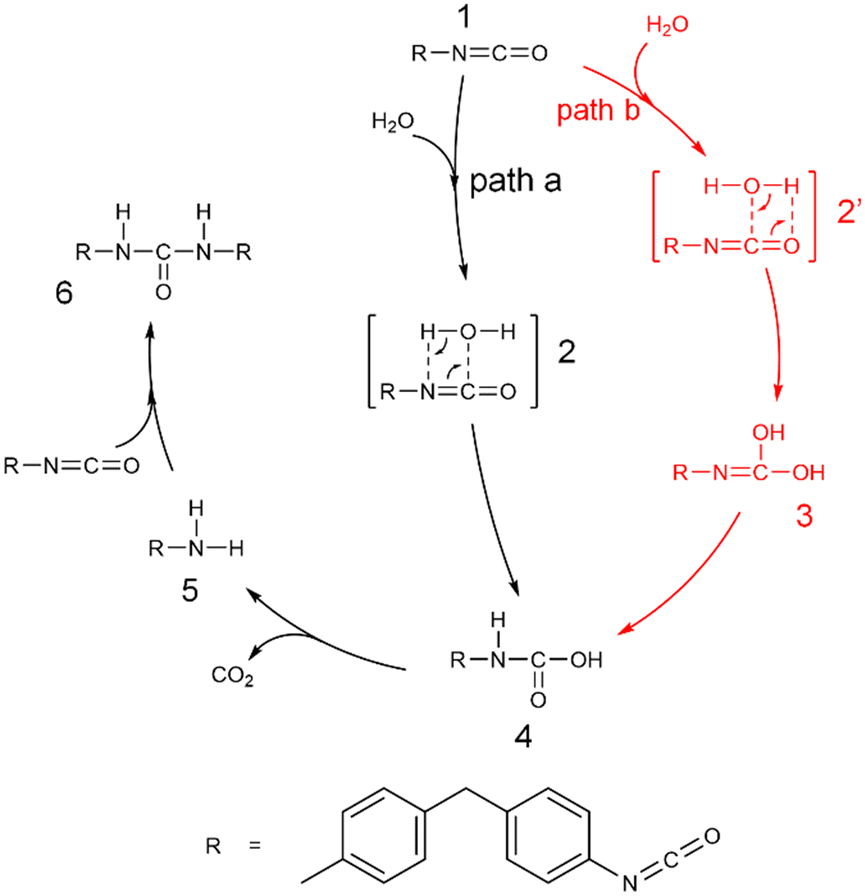 | ||
| Scheme 2 Moisture-curing mechanism of polyurethane. Path a: water molecule nucleophilically attacks the isocyanate group by breaking the CN bond. Path b: water molecule nucleophilically attacks the isocyanate group by breaking the CO bond. | ||
As shown in Scheme 2, after the formation of carbamic acid through path a or path b, it further decomposes to form a ring transition state TS4′′–5, which produces amine and carbon dioxide 5 (Fig. 5d). The energy barrier for this step is 155.8 kJ mol−1. The amine then nucleophilically attacks the remaining isocyanate groups, allowing for continuous reaction and resulting in the formation of a dense and complex cross-linked network of cured structures. This reaction leads to the formation of a rigid four-membered ring transition state TS5′–6 and ultimately to the formation of urea 6 (Fig. 5e). The energy barrier for this step is 86.7 kJ mol−1. The activation free energy of the reaction calculated from this two-step reaction is unrealistically high. It has been suggested that the additional water molecules may contribute to the moisture curing of PUR.
Effect of water on the moisture curing of PUR
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N bond breaking reaction.
The hydrolysis reaction of isocyanate was studied from a quantum chemical perspective by calculating reaction systems with 2 and 3 water molecules. Fig. 6 shows that when –NCO reacts with two water molecules, the two water molecules nucleophilically attack the NC bond in the isocyanate via a synergistic transition state mechanism, resulting in the formation of a six-membered ring synergistic transition state TS2–4-I. The hydrogen transfer results in the breakage of the H2–O2 bond (1.14 Å) and the formation of a new C–O2 bond and N–H1 bond (Fig. 6a). The energy barrier for this step is 50.9 kJ mol−1, which is much lower than the reaction with a single water molecule (Table 3). This is mainly because, in addition to a water molecule as a reactant, another water molecule acts as a H shuttling agent, facilitating the transfer of H from the hydroxyl group to the isocyanate group. The reaction of –NCO with two water molecules produces the carbamic acid. The product also contains one water molecule, confirming the theory that water molecules act as catalysts.
N bond breaking reaction.
The hydrolysis reaction of isocyanate was studied from a quantum chemical perspective by calculating reaction systems with 2 and 3 water molecules. Fig. 6 shows that when –NCO reacts with two water molecules, the two water molecules nucleophilically attack the NC bond in the isocyanate via a synergistic transition state mechanism, resulting in the formation of a six-membered ring synergistic transition state TS2–4-I. The hydrogen transfer results in the breakage of the H2–O2 bond (1.14 Å) and the formation of a new C–O2 bond and N–H1 bond (Fig. 6a). The energy barrier for this step is 50.9 kJ mol−1, which is much lower than the reaction with a single water molecule (Table 3). This is mainly because, in addition to a water molecule as a reactant, another water molecule acts as a H shuttling agent, facilitating the transfer of H from the hydroxyl group to the isocyanate group. The reaction of –NCO with two water molecules produces the carbamic acid. The product also contains one water molecule, confirming the theory that water molecules act as catalysts.
| Reaction phase | 0H2O | 1H2O | 2H2O | 3H2O |
|---|---|---|---|---|
| (a) CN |
— | 136.5 | 50.9 | 55.3 |
| (b) CO |
— | 165.3 | 56.9 | 47.6 |
| (c) H-transfer | 125.4 | 19.6 | 9.1 | 18.5 |
| (d) CO2↑ | 155.8 | 91.4 | 44.7 | — |
| (e) NH2 + NCO | 86.7 | 26.5 | 21.0 | — |
The three water molecules nucleophilically attack the NC bond in the isocyanate, forming a synergistic eight-membered ring transition state TS2–4-II, leading to carbamic acid (Fig. 6a). The energy barrier of this step is 55.3 kJ mol−1, which is significantly lower than that for the reaction with one water molecule, but higher than that for the reaction with two water molecules (Table 3). As the number of water molecules increases, the transition state structure becomes more stable and more favourable for hydrogen transfer, and the reaction energy barrier further decreases.
The above findings suggest that the nucleophilic addition reactions of –NCO with aqueous monomers, dimers, and trimers via opening of the NC bond at the isocyanate group form synergistic transition states characterized by tetra-, hexa- and octa-cycles. The energy barriers for the reactions of –NCO with one, two and three water molecules are 136.5, 50.9 and 55.3 kJ mol−1, respectively. This indicates that the transition state energy tends to stabilize as the number of water molecules increases compared to the reaction with the water monomer. The rate of hydrogen transfer is significantly faster when additional water molecules act as proton transport proteins. The hydrogen transfer rate is significantly faster with additional water molecules acting as a H shuttling agent. However, more water molecules do not lead to a further decrease in the energy barrier of the reaction, probably because the relative reactivity of this step of the reaction depends strongly on the formation of a stable synergistic transition state. The reaction energy barrier obtained from the DFT calculations is higher than the above experimental results. This may be due to the introduction of other catalysts during the production of the PUR prepolymer, which, together with water, promote the crosslinking reaction.
O bond breaking reaction.
When taking path b, two water molecules can nucleophilically attack the CO bond of the isocyanate to form a relatively relaxed six-membered ring transition state TS2′–3-I, forming an unstable carbamide (Fig. 6b). The transfer of hydrogen causes the H2–O3 bond (1.12 Å) to break and a new C1–O4 bond (1.61 Å) and H1–O1 bond (1.51 Å) are formed. This step creates two weak hydrogen bonds with a reaction energy barrier of 56.9 kJ mol−1. Compared to the reaction of a water molecule with isocyanate via CO bond breaking, the free energy barrier is reduced by approximately 108.4 kJ mol−1. Obviously, the additional water molecules have a promoting effect on the reaction.
Similarly, three water molecules can nucleophilically attack the CO bond of the isocyanate to form a relatively relaxed octa-cyclic transition state TS2′–3-II to form an unstable carbonimidic acid (Fig. 6b). This step leads to the formation of four weak hydrogen bonds with a reaction energy barrier of 47.6 kJ mol−1. Compared to the reaction of a single water molecule breaking the CO bond with isocyanate, the free energy barrier is reduced by about 117.7 kJ mol−1. Besides, compared to the reaction of two water molecules breaking the CO bond with isocyanate, the free energy barrier further decreases by about 9.3 kJ mol−1 (Table 3).
The additional water molecules are effective in reducing the reaction energy barrier, but further increase in the number of water molecules do not significantly reduce the reaction energy barrier, similar to the conclusion obtained for the CN bond breaking reaction. This may be due to the fact that the interaction between the reactant molecules increase as the number of reactants increase, although the additional water molecules have a facilitating effect on the cross-linking reaction. This is a coupling effect. On the other hand, although the reaction may tend to choose the CN bond breaking pathway when no additional water molecules are involved (Fig. 4), path b may be more advantageous with the introduction of additional water molecules (Table 3). This indicates that the two pathways are in competition.
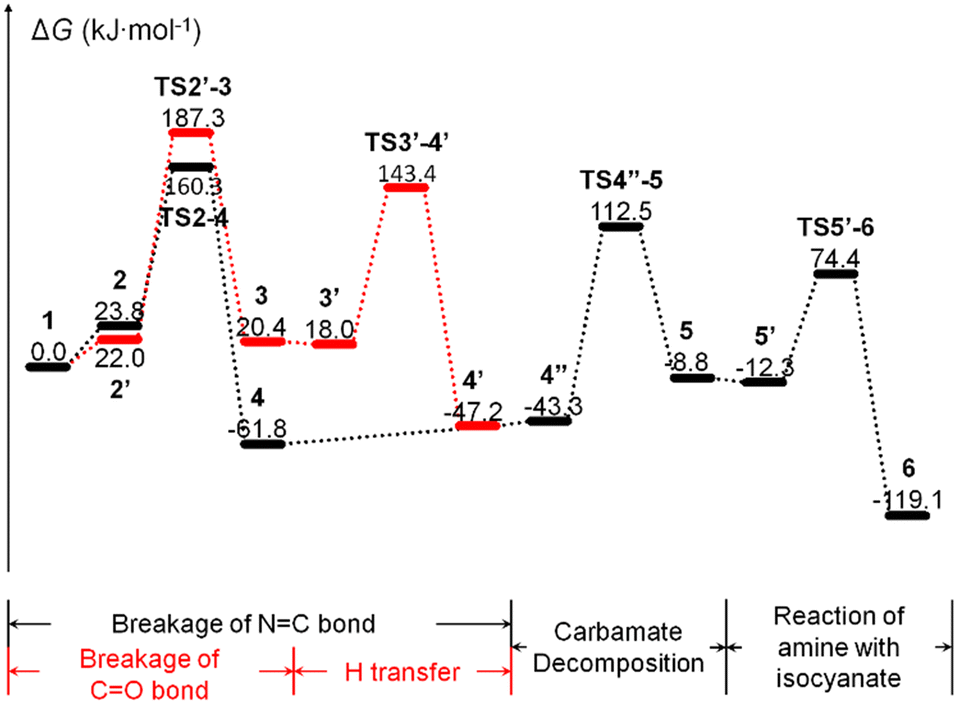 | ||
| Fig. 4 Gibbs free energy curve for moisture curing of PUR without the involvement of additional water molecules. | ||
Compared with the experimental results, the free energy barrier of the reaction obtained from DFT calculations when a single water molecule is used as a reactant is much higher than the experimental activation energy, which is an unrealistic value.
However, when additional water molecules are present, the calculated free energy barrier is closer to the experimental value. This phenomenon suggests that the number of water molecules has a significant effect on the PUR moisture curing process, but it is a more complex process. From an experimental point of view, this is reflected in the variation of ambient humidity. An increase in ambient humidity means that the PUR is in an environment with more water molecules, thus promoting the crosslinking reaction.
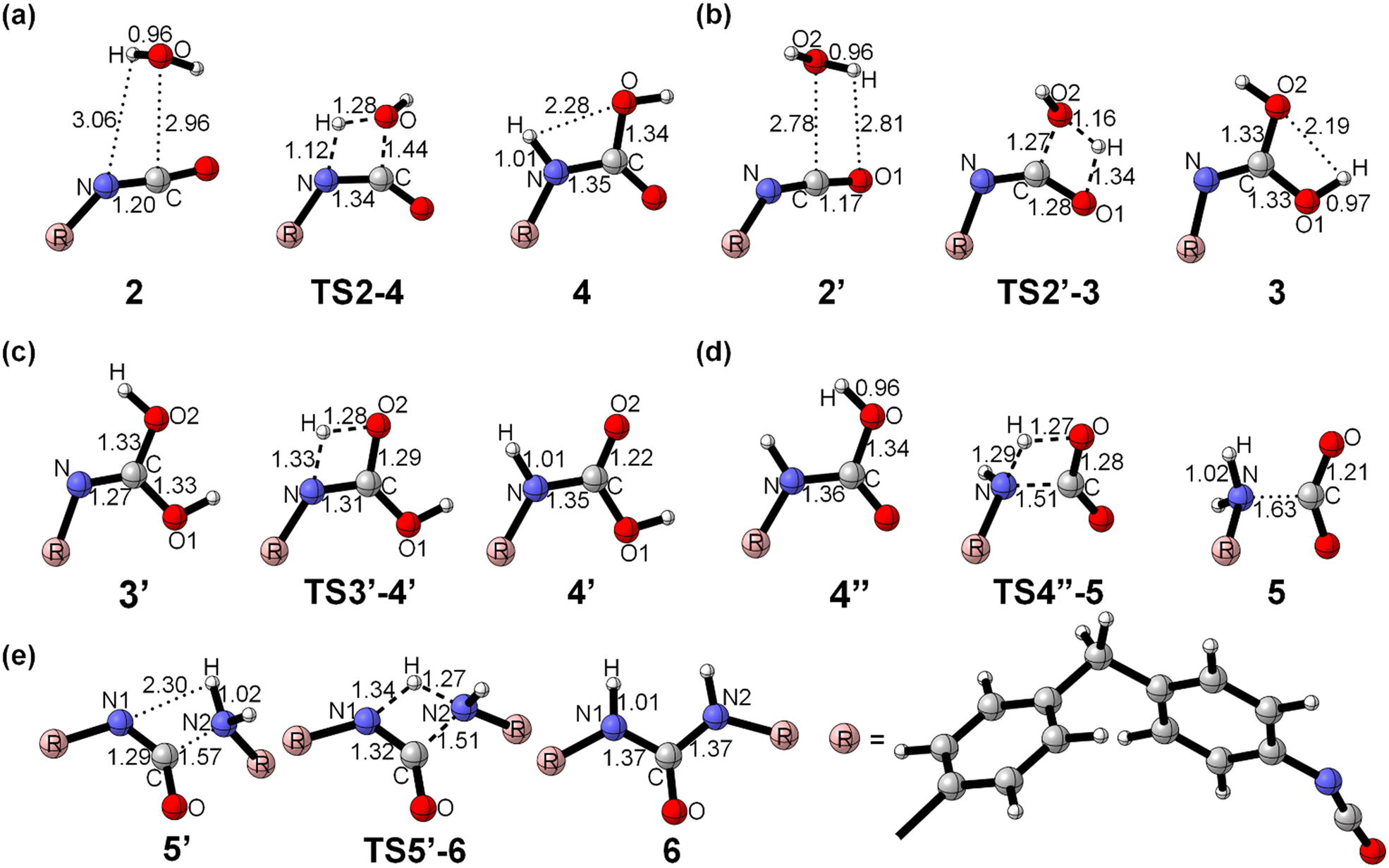 | ||
| Fig. 5 Optimized structure for PUR moisture curing reaction without the involvement of additional water molecules. (a) Breakage of CN bond; (b) breakage of CO bond; (c) H-transfer; (d) carbamic acid decomposition; (e) reaction of amine with isocyanate. *See the ESI† for the full conformation. | ||
To investigate whether the addition of water molecules would further affect the hydrogen transfer process, further DFT calculations were performed for the reactions with the addition of water dimers and trimers. The unstable carbonimidic acid undergoes hydrogen transfer catalyzed by the water dimer, forming a highly relaxed octa-ring transition state TS3′–4′-II to produce carbamic acid (Fig. 6c). Three hydrogen bonds are formed in this step, providing a more favourable hydrogen-bonded ring structure for the 1,3-hydrogen transfer. The reaction energy barrier in this step is 36.6 kJ mol−1, which is approximately 106.8 kJ mol−1 lower than the process without a water molecule and approximately 9.1 kJ mol−1 lower than the process with a water molecule. Similarly, carbamides undergo 1,3-hydrogen transfer catalyzed by aqueous trimers, forming a deca-ring transition state TS3′–4′-III to produce carbamic acids (Fig. 6c). The reaction energy barrier of this step is 18.5 kJ mol−1, even less advantageous than the first two cases (Table 3). This result, like the above, indicates a significant facilitation of the 1,3 hydrogen transfer by additional water molecules, but at the same time an increase in molecular interactions and a weak effect of subsequent water introduction. The research results of H. Moon et al.54 are consistent with our conclusions. As the relative humidity increased, the chemical reaction of polyurethane accelerated, however, even prolonged curing time and higher relative humidity did not lead to a continuous enhancement in the mechanical properties of polyurethane.
 | ||
| Fig. 6 Optimized structure for the PUR moisture curing reaction with the involvement of additional water molecules. (a) Breakage of CN bond; (b) breakage of CO bond; (c) H-transfer; (d) carbamic acid decomposition; (e) reaction of amine with isocyanate. *The Greek numbers indicate the number of additional water molecules involved in the reaction. See the ESI† for full conformation. | ||
The results show that the carbamic acid forms a six-membered ring transition state TS4′′–5-I in the presence of the water monomer, and the final composite products are all stable structures with a reaction energy barrier of 91.4 kJ mol−1. The old N–C bond (1.50 Å) and the H2–O2 bond (1.54 Å) break and a new N–H1 bond (1.40 Å) is formed (Fig. 6d). The water molecule acts as a carrier for hydrogen transfer, which significantly reduces the reaction energy barrier by 64.4 kJ mol−1. Water molecules also contribute to the decomposition of carboxylic acids.
In addition, carbamic acid forms a more relaxed octa-ring transition state TS4′′–5-II in the presence of a water dimer (Fig. 6d). This reaction step forms three strong hydrogen bonds with a reaction energy barrier of 44.7 kJ mol−1. Compared to the carboxylic acid decomposition reaction without the participation of water molecules, it has a significantly lower energy barrier of 111.1 kJ mol−1, and a further 46.7 kJ mol−1 compared to the reaction with the participation of a water molecule (Table 3). Contrary to the previous conclusion, the continued introduction of additional water molecules can effectively reduce the reaction energy barrier of carboxylic acid decomposition. The water molecule acts as a carrier for the transport of hydrogen, and the formation of a hydrogen bonding ring makes the reaction easier.
The results show that in the addition reaction of amines with MDI with the participation of additional water monomers, a relaxed six-membered ring transition state TS5′–6-I is formed with a reaction energy barrier of 26.5 kJ mol−1. Similarly, a more relaxed eight-membered ring transition state TS5′–6-II with a reaction energy barrier of 21.0 kJ mol−1 is formed in the reaction with additional water dimers (Fig. 6e). The additional water molecules have a catalytic effect on the reaction of amines with MDI, but the sustained introduction of water molecules does not further significantly lower the reaction energy barrier, which is probably mainly influenced by intermolecular interactions.
Conclusions
Utilizing in situ infrared spectroscopy, an investigation was conducted into the reaction kinetics characterizing the initial stages of polyurethane (PUR) materials' moisture curing, synthesized utilizing methylene diphenyl diisocyanate (MDI) as the curing agent. It was found that an escalation in ambient temperature and humidity notably spurred the moisture curing process of PUR. The incipient stage of PUR moisture curing appeared to adhere to first-order reaction kinetics, influenced by a melding of chemical reactions, and molecular diffusion consequences. Through the implementation of density functional theory (DFT), an in-depth examination into the mechanics of PUR moisture curing was conducted, as well as the influence of water on this process. The findings highlighted the indispensable role of supplementary water molecules within the broader framework of the moisture curing process. These molecules not only facilitated hydrogen transfer, but also considerably diminished the energy barrier associated with the PUR moisture curing reaction. Additionally, the research delineated two feasible reaction pathways in the context of competitive interaction. The presence of these additional water molecules was manifested in heightened humidity, which in turn expedited the PUR moisture curing process. This conclusion aligns coherently with the data derived from the experimental procedures.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant No. 22108072.References
- S. Camadanli, A. Hisir and S. Dural, J. Appl. Polym. Sci., 2022, 139, e51722 CrossRef.
- M. V. Pergal, J. V. Dzunuzovic, R. Poreba, M. Steinhart, M. M. Pergal, V. V. Vodnik and M. Spirkova, Ind. Eng. Chem. Res., 2013, 52, 6164–6176 CrossRef CAS.
- Q. Tang, J. He, R. Yang and Q. Ai, J. Appl. Polym. Sci., 2013, 128, 2152–2161 CrossRef CAS.
- X. Wei, D. Chen, L. Wang, Y. Ma and W. Yang, Appl. Surf. Sci., 2022, 579, 152153 CrossRef CAS.
- L. R. Bhagavathi, A. P. Deshpande, G. D. Janaki Ram and S. K. Panigrahi, Int. J. Adhes. Adhes., 2021, 105, 102771 CrossRef CAS.
- H. Gong, X. Guo, D. Cao, P. Gao, D. Feng, X. Zhang, Z. Shi, Y. Zhang, S. Zhu and Z. Cui, J. Mater. Chem. B, 2019, 7, 744–754 RSC.
- R. Prakash and P. Maiti, Energy Fuels, 2020, 34, 16847–16857 CrossRef CAS.
- D. Ren and C. E. Frazier, Int. J. Adhes. Adhes., 2013, 45, 118–124 CrossRef CAS.
- A. L. Daniel da Silva, J. M. Martin-Martinez and J. C. M. Bordado, Int. J. Adhes. Adhes., 2006, 26, 355–362 CrossRef CAS.
- M. Pramanik, S. K. Mendon and J. W. Rawlins, J. Appl. Polym. Sci., 2013, 130, 1530–1538 CrossRef CAS.
- T. Künniger, G. Clerc, S. Josset, P. Niemz, F. Pichelin and J.-W. G. van de Kuilen, Int. J. Adhes. Adhes., 2019, 92, 99–104 CrossRef.
- G. Clerc, M. Brülisauer, S. Affolter, T. Volkmer, F. Pichelin and P. Niemz, Int. J. Adhes. Adhes., 2017, 72, 130–138 CrossRef CAS.
- L. Sun, Z. Zong, W. Xue and Z. Zeng, Chem. Eng. J. Adv., 2020, 4, 100051 CrossRef CAS.
- E. N. Van Burns and B. J. Lear, J. Phys. Chem. C, 2019, 123, 14774–14780 CrossRef CAS.
- J. Xu, Y. Jiang, T. Zhang, Y. Dai, D. Yang, F. Qiu, Z. Yu and P. Yang, Prog. Org. Coat., 2018, 122, 10–18 CrossRef CAS.
- P. Feng, W. Li and Y. Q. Zou, J. Appl. Polym. Sci., 2014, 131, 40501 CrossRef.
- G. He and N. Yan, Int. J. Adhes. Adhes., 2005, 25, 450–455 CrossRef CAS.
- J. Guo, T. Chai, Y. Liu, J. Cui, H. Ma, S. Jing, L. Zhong, S. Qin, G. Wang and X. Ren, Coatings, 2018, 8, 175 CrossRef.
- N. Ducruet, L. Delmotte, G. Schrodj, F. Stankiewicz, N. Desgardin, M.-F. Vallat and B. Haidar, J. Appl. Polym. Sci., 2013, 128, 436–443 CrossRef CAS.
- B.-S. Chiou and P. E. Schoen, J. Appl. Polym. Sci., 2002, 83, 212–223 CrossRef CAS.
- J. Shi, T. Zheng, Y. Zhang, B. Guo and J. Xu, Polym. Chem., 2021, 12, 2421–2432 RSC.
- L. Sun, S. Cao, W. Xue, Z. Zeng and W. Zhu, J. Adhes. Sci. Technol., 2016, 30, 1212–1222 CrossRef CAS.
- P. Cong, A. Zhang, W. Ge and Y. Cheng, Constr. Build. Mater., 2022, 333, 127347 CrossRef CAS.
- C. Chai, J. Hou, X. Yang, Z. Ge, M. Huang and G. Li, Polym. Test., 2018, 69, 259–265 CrossRef CAS.
- S. Gogoi, S. Barua and N. Karak, Chem. Eng. Sci., 2015, 127, 230–238 CrossRef CAS.
- L. Sun, W. Zhang, W. Xue and Z. Zeng, J. Adhes. Sci. Technol., 2022, 37, 663–682 CrossRef.
- Q. Liu, D. De Kee and R. K. Gupta, AIChE J., 2008, 54, 364–371 CrossRef CAS.
- O. Kläusler, S. Clauß, L. Lübke, J. Trachsel and P. Niemz, Int. J. Adhes. Adhes., 2013, 44, 57–65 CrossRef.
- A. L. Daniel-da-Silva, J. C. Moura Bordado and J. M. Martín-Martínez, J. Appl. Polym. Sci., 2007, 104, 1049–1057 CrossRef CAS.
- A. L. Daniel-da-Silva, J. C. M. Bordado and J. M. Martín-Martínez, J. Appl. Polym. Sci., 2008, 107, 700–709 CrossRef CAS.
- W. P. Du, Y. Zhang, L. J. Tan and H. F. Chen, Polym. Sci., Ser. B, 2019, 61, 247–253 CrossRef CAS.
- N. A. Bratasyuk and V. V. Zuev, Thermochim. Acta, 2020, 687, 178598 CrossRef CAS.
- A. V. Palodkar, S. Mandal and A. K. Jana, Energy Fuels, 2016, 30, 7656–7665 CrossRef CAS.
- L. Xu and G. Shan, Ind. Eng. Chem. Res., 2013, 52, 8216–8222 CrossRef CAS.
- D. Ren and C. E. Frazier, Int. J. Adhes. Adhes., 2012, 34, 55–61 CrossRef CAS.
- V. de Lima, N. D. S. Pelissoli, J. Dullius, R. Ligabue and S. Einloft, J. Appl. Polym. Sci., 2010, 115, 1797–1802 CrossRef CAS.
- F. Burel, A. Feldman and C. Bunel, Polymer, 2005, 46, 15–25 CrossRef CAS.
- P. K. Maji and A. K. Bhowmick, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 731–745 CrossRef CAS.
- D. M. Dhevi, A. A. Prabu and K. J. Kim, Polym. Bull., 2016, 73, 2867–2888 CrossRef CAS.
- K. Hailu, G. Guthausen, W. Becker, A. Koenig, A. Bendfeld and E. Geissler, Polym. Test., 2010, 29, 513–519 CrossRef CAS.
- M. Stanko and M. Stommel, Polymers, 2018, 10, 698 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 2002, 14, 363–368 CrossRef.
- S. Monaghan and R. A. Pethrick, Macromolecules, 2012, 45, 3928–3938 CrossRef CAS.
- S. P. Wang, X. D. Yang, Q. Q. Bai and T. D. Li, Int. J. Polym. Anal. Charact., 2013, 18, 119–125 CrossRef CAS.
- M. K. Boulkadid, S. Touidjine, D. Trache and S. Belkhiri, Crit. Rev. Anal. Chem., 2022, 52, 1112–1121 CrossRef CAS PubMed.
- P. F. Yang, Y. D. Han, J. Y. Li and T. D. Li, Int. J. Polym. Anal. Charact., 2011, 16, 251–258 CrossRef CAS.
- D. M. Dhevi, A. Anand Prabu and K. J. Kim, Polym. Bull., 2016, 73, 2867–2888 CrossRef CAS.
- A. Tenorio-Alfonso, M. C. Sánchez and J. M. Franco, Pro. Org. Coat., 2021, 161, 106547 CrossRef CAS.
- H. Xiang, H. Xia, H. Chen and H. Mi, J. Mater. Sci., 2021, 56, 15069–15086 CrossRef CAS.
- S. Liu, X. Li, M. Ge, X. Du and M. Zou, Polymers, 2022, 14, 698 CrossRef.
- P. Cimavilla-Román, M. Santiago-Calvo and M. A. Rodríguez-Pérez, Polym. Test., 2021, 103, 107336 CrossRef.
- H. Moon, J. Park, W. Cho, J. Jeon and J. Wie, Eur. Polym. J., 2023, 201, 112579 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: http://doi.org/10.1039/d4re00385c |
| This journal is © The Royal Society of Chemistry 2025 |