
Upcycling polymethyl methacrylate to methacrylic acid†
Yanfa
Zhuang
a,
Nooshin
Saadatkhah
a,
Tien-Dat
Nguyen
a,
Jacopo
De Tommaso
a,
Clive Yi Jie
Ng
a,
Chunyu
Wang
ab,
Abdellah
Ajji
a and
Gregory S.
Patience
 *a
*a
aDepartment of Chemical Engineering, Polytechnique Montreal, C.P. 6079, Succ. “CV”, Montreal, H3C 3A7, Quebec, Canada. E-mail: gregory-s.patience@polymtl.ca; Fax: +1 (514) 340 4159; Tel: +1 (514) 340 4711 Ext. 3439
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, China
First published on 13th November 2024
Abstract
Waste polymethyl methacrylate (PMMA) has become a more prominent contributor to global plastic waste in the aftermath of the COVID-19 pandemic. Recycling PMMA relies either on mechanical recycling or thermal depolymerization. Mechanical properties deteriorate after several mechanical recycling cycles. Depolymerization technologies operate in an inert atmosphere and require costly monomer purification downstream. Therefore, neither chemical nor mechanical recycling of PMMA is economically viable. Here, we demonstrate a sustainable recycling method through catalytic hydrolysis to upcycle PMMA while reaching higher product purity. PMMA reacts over zeolites and produces methacrylic acid instead of methyl methacrylate offering technical, economical, and market benefits. Direct hydrolysis of PMMA over an H-type zeolite with an SiO2/Al2O3 ratio of 80 produced methacrylic acid with a yield of 56% and a selectivity of 58%. Coke formed within the framework of large-pore zeolites, causing reversible deactivation of medium–strong acid sites and Brønsted acid sites. The catalytic decarboxylation of methacrylic acid primarily produces acetone and CO, and six-membered glutaric anhydride forms in solid residues.
1 Introduction
Plastic waste is a continuing challenge that spans generations. The Organization for Economic Cooperation and Development (OECD) reported that annual plastic production more than doubled from 2000 to 2019, reaching 400 million tonnes,1 yet this number surged to 530 million tonnes within just the first seven months of the COVID-19 pandemic.2 The pandemic exacerbated the already existing plastic waste crisis.3 The primary short-term impact of the pandemic was an unprecedented demand for personal protective equipment (PPE) in the healthcare and public sectors to combat the spread of the virus.4 Simultaneously, single-use plastic ban legislation faced setbacks in many countries, including the USA, Canada, UK, France, India, and South Korea.5Fundamentally, the pandemic triggered a shift in behavior towards remote consumption modes, such as e-commerce and food delivery, which heavily rely on disposable plastics.2 Consequently, the ongoing global economic slowdown, coupled with supply chain disruptions, has led to a reduction in post-industrial plastic waste and an increase in post-consumer waste, which is more challenging to collect and recycle. The disruption of recycling initiatives at both local and international levels has hindered the ability to manage this transition,1,6 particularly concerning single-use healthcare waste streams, where low manufacturing cost outweighs high collection and treatment expenses.7
In the current scenario, to achieve absolute sustainability in human activities by 2030, defined as adhering to the safe operating space (SOS) to prevent destabilizing the Earth's planetary system, hinges on a global mass-to-mass plastic recycling rate exceeding 71%.8 This target is unreachable, even with the current most advanced recycling technologies. By 2050, 94% of plastics are projected to be recycled or incinerated, while 6% are expected to be discarded.9 In fact, even assuming the recycling of the maximum theoretically possible plastic amount (94%), the material losses in the recycling chain impede reaching complete sustainability. Achieving this target requires higher plastic-to-plastic yielding technologies (e.g. plastics to monomers or chemicals).8
Polymethyl methacrylate (PMMA), known as Plexiglas, Lucite, and Perspex, stands out as one of the prime candidates for achieving high yield plastic-to-plastic recycling.10 PMMA attracted considerable attention during the pandemic, serving as protective barriers. This thermoplastic is distinguished by its tensile and flexural strength, UV resistance, and transparency, making it a versatile and multifunctional material. In 2021, the global PMMA market exceeded 4.5 billion USD,10,11 and it is projected to reach an annual production of 3 million tonnes by 2028, with its primary applications spanning the automotive, construction, electronics, signage, and coating industries.
Europe, Asia Pacific, and America each account for roughly one-third of the global PMMA market, collectively representing 93% of the market.12 Despite Europe's above-average ranking in overall macro-regional plastic recycling,1 Europe recycled only 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of PMMA in 2019, which is approximately 10% of the total PMMA waste.13 This results in an annual volume of 800000 t to 1000000 t of unaddressed waste PMMA worldwide.14
000 tonnes of PMMA in 2019, which is approximately 10% of the total PMMA waste.13 This results in an annual volume of 800000 t to 1000000 t of unaddressed waste PMMA worldwide.14
PMMA has a ceiling temperature of 192 °C15,16 and pyrolyzes to MMA with a yield greater than 90% between 350 °C and 450 °C17–19 (Fig. 1a). The European MMAtwo project successfully demonstrated a pilot-scale, extrusion-based pyrolysis process to depolymerize selected cast, injection, and extrusion grade scraps, achieving a plastic-to-monomer yield of 80% to 90% and a MMA purity of 99.8%.20 Pyrolysis of PMMA to MMA holds promise for maintaining carbon within the economy. However, it is not a universal solution for all types of PMMA-based waste.
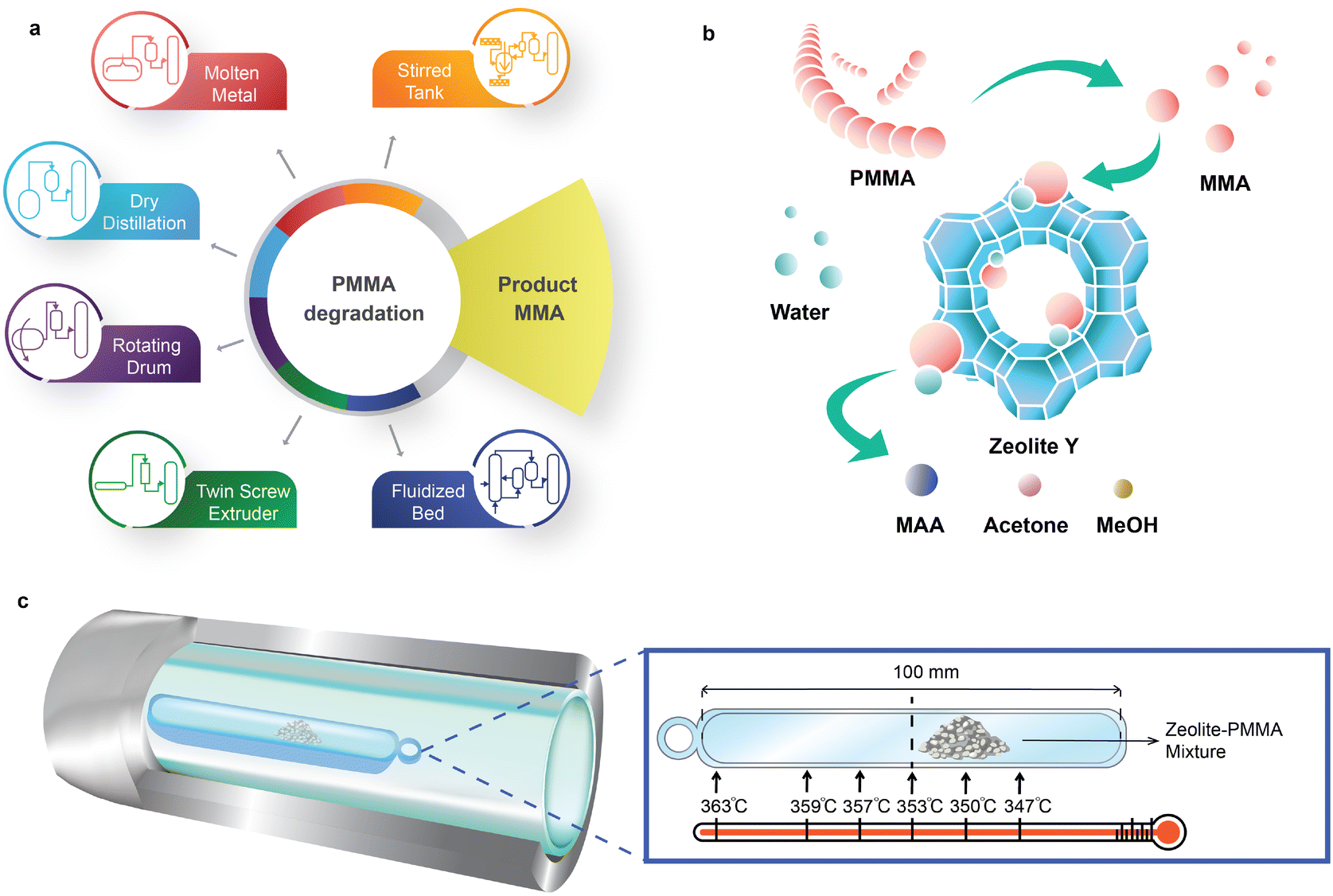 | ||
| Fig. 1 PMMA chemical recycling schemes. a. Conventional PMMA chemical recycling routes rely on PMMA thermal depolymerization to produce the MMA monomer. b. In contrast to conventional PMMA chemical recycling, direct upcycling of PMMA to the MAA monomer over zeolite catalysts yields a more versatile chemical block over MMA and so circumvents the issues related to MMA repolymerization. c. A double-layer 316L SS-quartz liner reaction tube and fused quartz boat with axial temperature difference for catalyst screening. Reaction temperature tolerance was ±3 °C. | ||
Depending on the nature of the scraps or the depolymerization process, the resulting crude regenerated MMA (r-MMA) may contain impurities, such as methyl pyruvate, methyl or ethyl propionate, methyl isobutyrate, methyl acrylate, or ethyl acrylate, which have close boiling points and chemical structures to MMA.21,22 For example, ethyl acrylate shares the same boiling point as MMA (100 °C) and has an odour threshold 200 times higher,23 posing challenges for the pyrolysis process and the quality of the resulting product. PMMA polymerization follows a radical mechanism,18,24 and acrylates generally create radicals more effectively than MMA in most situations, so even a small fraction triggers repolymerization at 25 °C to 180 °C,25–28 resulting in pipe blockages (Fig. S1†). Low quality scraps produce lower quality r-MMA with more impurities,10,13 which hinders its commercialization in strictly regulated markets, or those requiring high-quality monomers.29 Similarly, some batch depolymerization technologies like dry distillation or rotating drums maximize the contact between depolymerizing PMMA and the char formed from the reaction. This facilitates hydrogen transfer reactions of MMA, such as MMA to methyl isobutyrate, a volatile compound with the same boiling point as MMA.13
PMMA upcycling to methacrylic acid (MAA) instead of MMA can enhance the polymer recycling capacity and represents an alternative solution that offers significant technical, economic, and market benefits.30,31 MAA, as a functional monomer, is widely used in synthesis of copolymers with other monomers, its esters (e.g. MMA), and MAA salts. In addition, MAA serves as a valuable specialty chemical in applications such as paints, leather and textile treatment agents, adhesives, and ion-exchange resins.32,33 In contrast to MMA, the market volumes of MAA (in terms of import and export) are approximately one-tenth the size, with roughly 150000 t traded globally, compared to the 1500000 t of MMA.34 Due to its smaller volumes and its niche applications, MAA has fewer suppliers, and there is consumer interest in having new producers to enhance market elasticity and decouple MAA dynamics from those of MMA.
In a recent study, Chub et al.30 synthesized MAA by integrating the depolymerization of virgin PMMA to MAA with its subsequent catalytic hydrolysis of MMA in a fluidized bed reactor. They evaluated the effect of various catalysts Al2O3, FCC, Cs-HPA, zeolite Y, ZrO2MoO3/SiO2 and sand, and found that zeolite Y achieved the highest yield of MAA at 33%. However, their study outlined a simplified mechanism for MAA production and did not fully explore side reactions that affect the overall yield. Additionally, it lacked detailed analysis of zeolite properties such as micropore shape-selectivity and the influence of the SiO2/Al2O3 molar ratio. In follow-up research, the team developed a tandem reactor system, initially producing MMA in a fluidized sand bed, followed by MMA hydrolysis to MAA using zeolite Y in a fixed bed reactor.35 Despite its innovative design, this system only achieved a maximum PMMA conversion of 53% and an MAA yield of up to 48%.
Here, we introduce a sustainable catalytic hydrolysis method to directly upcycle PMMA to the MAA monomer over zeolite catalysts from 330 °C to 370 °C in a fixed bed reactor. Upcycling PMMA to MAA integrates PMMA thermal depolymerization and in situ MMA hydrolysis (Fig. 1b). We investigated the activity of zeolite Y, ZSM-5, zeolite beta, mordenite (MOR) and desilicated zeolite Y in a double layer reaction tube (Fig. 1c). Direct hydrolysis of PMMA over zeolite Y-80 (SiO2/Al2O3 = 80) exhibited the highest MAA yield (56%) and MAA selectivity (58%) at 350 °C. MAA can be purified and converted to MMA for polymerization purposes, or sold as is.
2 Experimental
2.1 Materials
We purchased zeolites, including HY(80), HY(60), HY(30), Hβ(360), NH4-MOR(20), NH4-ZSM-5(200–400), and NH4-ZSM-5(80), as well as virgin PMMA (Mw = 550000 g mol−1), from Thermo Scientific Chemicals. All ammonium type zeolites were calcined at 550 °C for 2 hours (heating up by a 2 °C min−1 ramp) to form proton type HMOR(20), HZSM-5(200–400) and HZSM-5(80). Prior to each experiment, all samples were vacuum dried at 70 °C for 24 h. For all zeolites, the number in parenthesis indicates the SiO2/Al2O3 molar ratio.
2.2 Preparation of desilicated zeolite Y-80
Commercial HY(80) was desilicated in 0.05 M, 0.1 M, 0.2 M and 0.3 M NaOH solutions at 60 °C for 30 min, followed by filtration. The filtered samples were washed with deionized water until reaching neutral pH and dried overnight at 110 °C. The dried samples were further exchanged with 0.5 M NH4NO3 three times and then dried overnight at 110 °C. The NH4+ exchanged samples were finally calcined at 550 °C for 6 h in air. The desilicated HY(80) catalysts were labelled M0.5, M1, M2 and M3 corresponding to the NaOH solution concentration.2.3 Regeneration of coked zeolite Y-80
Reacted HY(80) at 350 °C for 35 min was calcined at 550 °C for 6 h (2 °C min−1 ramp) to remove coke. R-HY(80) represents regenerated HY(80).2.4 Characterization
000 g mol−1) was used to measure the molecular weight (Mw) of unreacted PMMA. The GPC system, calibrated with a Shodex PMMA calibration kit STANDARD M-75 (Mw range: 2.79 × 103 to 1.01 × 106 g mol−1), employed a refractive index (RI) detector and tetrahydrofuran (THF) as the eluent. The flow rate of the THF eluent was 1 mm min−1. The temperature of the RI detector and column remained at 35 °C. THF dissolved unreacted PMMA in the solid residues and was left to stand at room temperature for 12 h. The supernatant was injected into the GPC system.
2.5 Gas, liquid and solid product analysis
An Agilent 7890A GC system equipped with a thermal conductivity detector (TCD) was used to analyze the gas effluent. The GC comprised a two-column system. An Agilent Hayesep Q 80/100 UltiMetal column (0.5 m × 1/8′′ × 2 mm) separated CH4 and CO2, while an Agilent J&W MolSieve 13× 80/100 UltiMetal column (1.5 m × 1/8′′ × 2 mm) separated CO and H2. An extra Agilent Hayesep Q 80/100 UltiMetal column (1 m × 1/8′′ × 2 mm) as a precolumn was used in a backflush to the detector.An Agilent 7890A GC system equipped with an Agilent 7693 Autosampler and Agilent 5975C mass selective detector (MSD) mass spectrometry qualitatively and quantitatively analyzed the liquid products. An ultra inert (UI) capillary column, Agilent J&W DB-Wax UI (polyethylene glycol stationary phases, length 30 m, inner diameter 0.25 mm, film thickness 0.25 μm), separated the components. The MSD scanned m/z (mass-to-charge ratio) from 29 to 500 in the qualitative analysis. The selected ion monitoring (SIM) mode was used to quantitatively analyze acetone (selected m/z 43 & 58), methanol (selected m/z 31), MMA (selected m/z 41, 69 & 100) and MAA (selected m/z 41 & 86). The concentration of the four components was determined by external calibration. External calibration standards were prepared by mixing acetone (HPLC grade, ≥99.9%, Sigma-Aldrich), methanol (HPLC grade, ≥99.9%, Sigma-Aldrich), MMA (≥99.6%, stabilized with 6-tert-butyl-2,4-xylenol, TCI America™) and MAA (99%, containing 250 ppm MEHQ as an inhibitor, Sigma-Aldrich) in a 4:1 (volume ratio) solvent mixture of ultrapure water (HPLC grade, Thermo Scientific™) and anhydrous ethanol (HPLC grade, Commercial Alcohols).
The PMMA conversion (XPMMA), product selectivity (Si) and yield (Yi) follow equations:
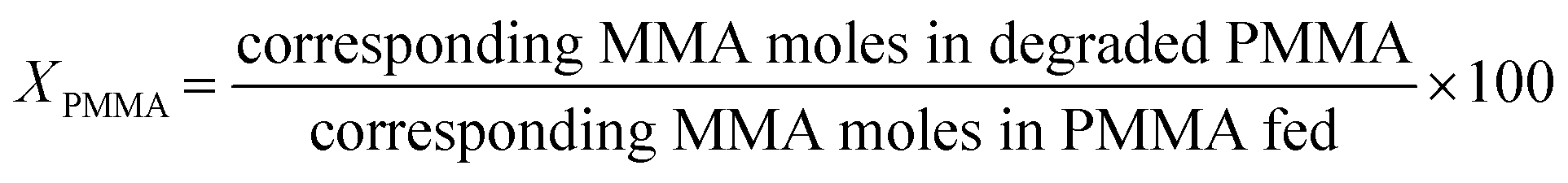 | (1) |
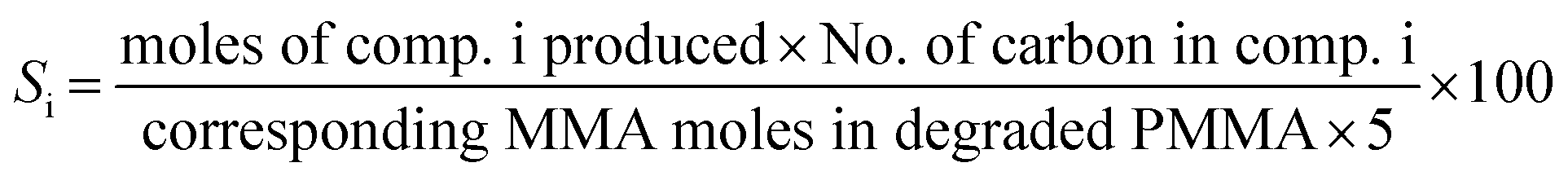 | (2) |
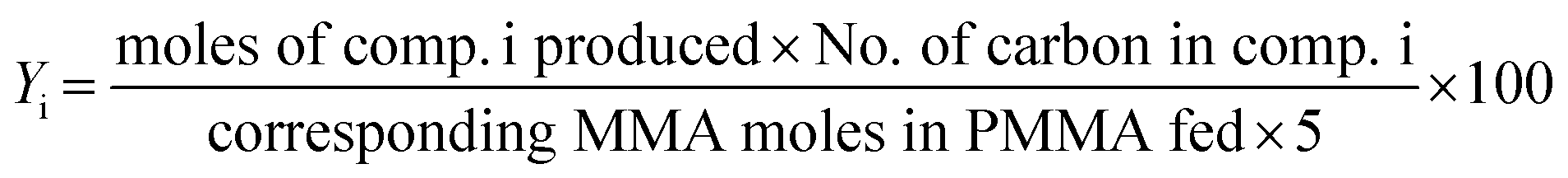 | (3) |
A TA Instruments Q500 thermogravimetric analyzer was used to determine the mass of unreacted PMMA and the coke formed in the solid residues. The residue samples were heated from 25 °C to 700 °C with a ramp of 5 °C min−1 under 60 mL min−1 of air flow. A LECO CS744 analyzer equipped with an IR detector was applied to measure the mass fraction of total carbon remaining in the reacted solid residue by combusting samples at 1300 °C to 1400 °C with tungsten and iron accelerators.
2.6 Experimental set-up and procedure
PMMA was hydrolyzed in a 450 mm of ultra-high-polished (interior smoothness Ra = 10) 316L stainless steel (SS) tubing (OD = 25.4 mm, ID = 22 mm) with 450 mm of quartz liner tubing (OD = 21.6 mm, ID = 18 mm). A Carbolite Gero horizontal split tube furnace was used to heat a 316L SS-quartz double-walled reaction tube. A mixture of 0.2 g PMMA and 0.2 g zeolite was placed in a fused quartz boat (100 mm × 15 mm × 7.5 mm) in the middle of the reaction tube. A thermocouple above the fused quartz boat recorded the temperature in real time. A Swagelok 316L SS sample cylinder (150 mm) wrapped with fibreglass heater tape evaporated 2 mL min−1 of water and generated steam at 450 °C. Helium (170 mL min−1) carried the steam into the reaction tube. To avoid interference with the H2 peak during gas analysis, we opted for helium over nitrogen as the carrier gas. The gaseous products from the reaction tube condensed in a Synthware Graham condenser, followed by a Synthware liquid nitrogen cold trap. A Supel inert multi-layer foil gas bag collected all produced gases. Each reaction lasted for 35 min.2.7 Kinetic diameter estimation
Kinetic diameter estimation referred to a universal electron density isosurface method proposed by Mehio et al.36 Gaussian 16W optimized and generated the wavefunction of MMA and MAA molecules at the PBE0/def2-TZVP level. Based on the wavefunction, the Multiwfn program37 quantitatively analyzed the molecular surface using 0.0015 a.u. as an electron density isovalue, and yielded all surface vertices. VMD software visualized molecular surface vertices and measured the distance between two surface vertices reflecting kinetic diameter characteristics.2.8 In situ liquid and gas product analysis
A Synthware Graham condenser cooled down condensable gaseous products that were generated from 160 °C to 350 °C. Dozens of vials with 2 mm of ethanol were used to collect liquid products from the Graham condenser every minute. An Agilent 7890A-6975C GC-MSD was used to analyze each sample corresponding to a specific reaction temperature. A HIDEN HPR-20 QIC R&D quadrupole mass spectrometer was used to online monitor the gas composite after condensation.3 Results and discussion
3.1 PMMA hydrolysis activities over zeolites
The temperature gradient along the axial direction of the fused quartz boat exceeded 16 °C (Fig. 1c). Consequently, we positioned the samples in a zone where the temperature ranged from 347 °C to 353 °C with a temperature setpoint at 350 °C. The evaporation of products during sample collection inevitably results in carbon loss (Fig. 2a and Table S1†). Also, a small fraction of liquid products evaporates and enters the gas bag, further restricting quantitative detection of liquid products. The carbon recovery over fresh zeolite Y with SiO2/Al2O3 ratios of 80, 60 and 30, and Hβ(360) was >85% at 350 °C. The best carbon balance was 95% over fresh HY(80) at 330 °C due to higher unreacted PMMA residues. The carbon content in solid residues is easier to quantify than liquid and gaseous products. In reactions over ZSM-5, MOR, and HY(80), the carbon recovery fell below 85%. The decrease in the SiO2/Al2O3 ratio in zeolite Y, from 80 to 30, and ZSM-5, from 200 to 80, led to a lower MAA yield, which corresponded to lower carbon recovery. The highest MAA yield was over HY(80) followed by Hβ(360). HZSM-5(80) and desilicated zeolite Y (M0.5, M1 and M2) had the lowest carbon balance (≤60%) and MAA yield (≤24%).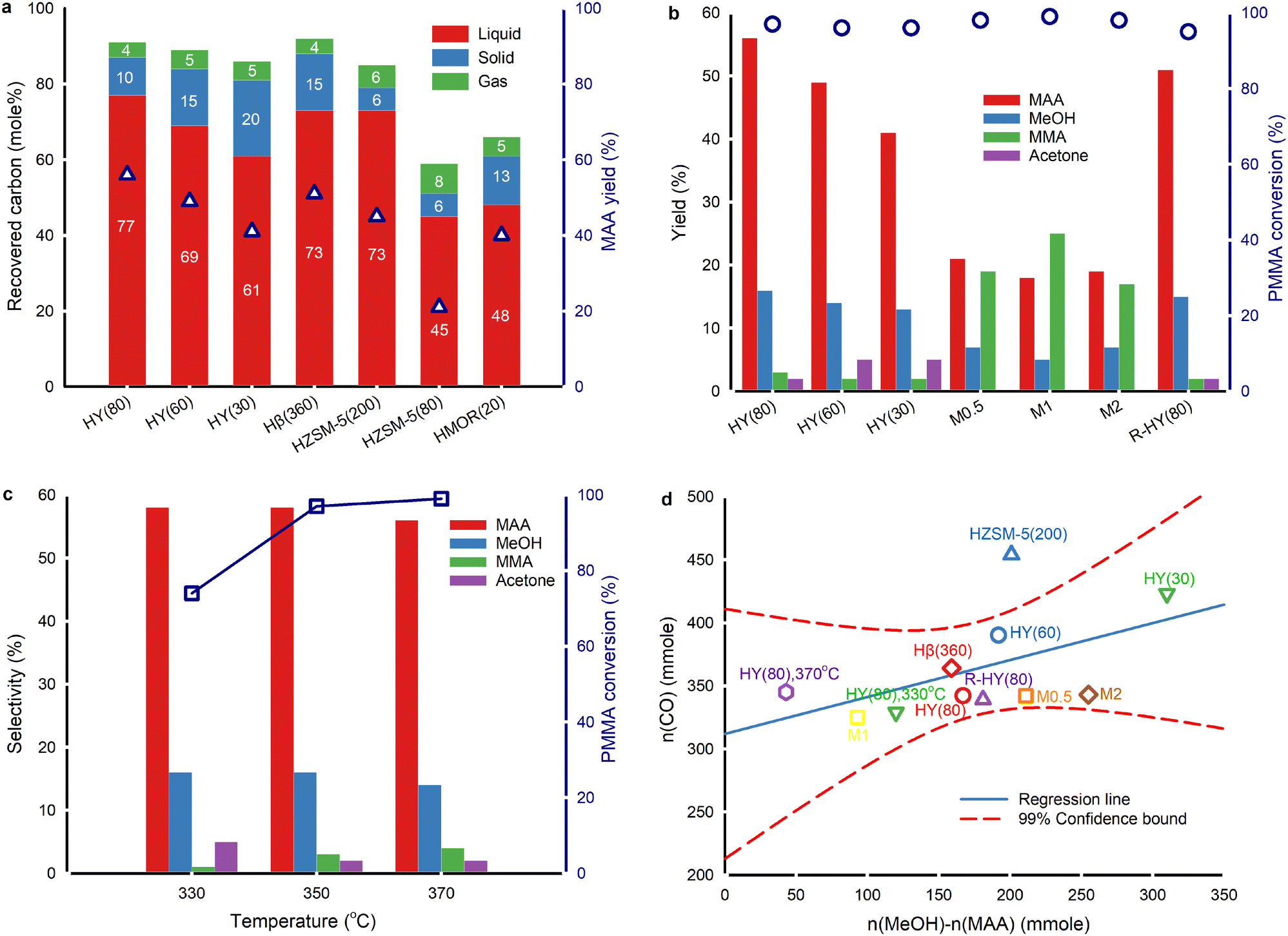 | ||
| Fig. 2 PMMA hydrolysis over fresh, activated, regenerated and desilicated zeolite catalysts. Each experiment contained 0.2 g virgin PMMA and 0.2 g zeolite. a. Recovered carbon and MAA yield (right axis, hollow triangles) at 350 °C for 35 min with 2 mL min−1 liquid water injection. b. MAA, MeOH, MMA and acetone yields, and PMMA conversion (right axis, hollow circles) over fresh, desilicated and regenerated zeolite Y at 350 °C for 35 min with 2 mL min−1 liquid water injection. c. The effect of reaction temperature on the selectivity to MAA, MeOH, MMA and acetone, and PMMA conversion (right axis, hollow squares) over fresh HY(80) with 35 min reaction time and 2 mL min−1 liquid water injection. d. The correlation between the moles of CO and the difference of the moles of MeOH and MAA over various catalysts at 350 °C for 35 min with 2 mL min−1 water injection. HMOR(20) and HZSM-5(80) are excluded due to a huge deviation between n(CO) and n(MeOH)–n(MAA). All fresh, regenerated and desilicated zeolite Y, and Hβ(360) lie within 99% confidence interval. | ||
Based on the catalyst screening, the Y type zeolite produced MAA with the highest yield. Consequently, we focused on the Y type zeolite in this work. All fresh, desilicated and regenerated zeolite Y converted ≥95% of PMMA at 350 °C (Fig. 2b). The acidity of zeolite Y influences the yields of MAA and methanol. A lower SiO2/Al2O3 ratio, which corresponds to higher acidity (Table S3†), led to a decreased yield of MAA and methanol but an increased yield of acetone. From HY(80) to HY(30) (SiO2/Al2O3 = 80 to SiO2/Al2O3 = 30), the MAA yield decreased by 37% (from 56% to 41%), and the methanol yield decreased by 23% (from 16% to 13%), while the acetone yield increased 2.5-fold (from 2% to 5%). Specifically, the acetone yield was 4.8% over HY(60) and 5.1% over HY(30) based on the original experimental results. Due to rounding off, both are presented to be 5% in Fig. 2b. The more pronounced reduction in the MAA yield compared to methanol, coupled with the increase in acetone production, suggests potential over-cracking of MAA and/or MMA into acetone. Desilicated zeolite Y, treated with varying concentrations of NaOH, produced more MMA but less MAA and methanol compared to fresh zeolite Y. Among the desilicated samples, M1 showed the highest yield of MMA but the lowest yields of MAA and methanol. Interestingly, none of the desilicated zeolites generated acetone. This suggests that specific active sites within the zeolitic framework are responsible for acetone formation. The desilication treatment may have compromised the zeolitic framework, leading to the loss of some active sites. R-HY(80) displayed an equivalent MMA yield to fresh HY(80) but slightly lower yields of MAA, methanol and acetone.
The PMMA conversion increased by 31% from 330 °C to 350 °C and slightly rose from 350 °C to 370 °C over fresh HY(80) (Fig. 2c). The selectivity to MAA and methanol remained constant at 330 °C and 350 °C, but decreased at 370 °C. At 330 °C, there was lower selectivity to MMA but higher selectivity to acetone compared to 350 °C and 370 °C. Higher temperature favoured faster PMMA depolymerization. Nevertheless, excessive temperature resulted in lower selectivity to MAA and methanol due to further undesired reactions of both MMA and MAA. The optimal PMMA hydrolysis performance was on HY(80) at 350 °C.
PMMA depolymerizes to a CH3O(CO)· free radical fragment, converting to either methanol and CO or CO2 and CH4via combining with one H· proton abstracted from other H-containing components.18 Among all the catalysts, only HY(80) at 370 °C and HMOR(20) at 350 °C formed CH4 (≤0.1 mol%) (Table S1†). This confirmed that the CH3O(CO)· fragment primarily formed CO and methanol.
PMMA hydrolysis without any catalyst at 350 °C with 2 mL min−1 H2O injection resulted in approximately the same CO2 yield as PMMA hydrolysis over zeolites except M2 and unblended HY(80) on PMMA (Table S1†). We attributed the primary detected CO2 to external sources, i.e. CO2 entering during H2O injection and gas collection. Small amounts of CO2 might result from thermal degradation of MMA/MAA. If PMMA depolymerizes to MMA and partially to the CH3O(CO)· fragment, followed by MMA hydrolysis to MAA and methanol along with CH3O(CO)· cracking to CO and methanol, the CO amount would be theoretically equal to the mole difference between the collected methanol and MAA. The CO yield, however, did not correlate to the difference between methanol and MAA by the 1:1 linear relationship over all zeolites (Fig. 2d). This discrepancy was attributed to the dehydration of methanol to dimethyl ether (DME).30 The formation of acetone is also likely to affect this relationship. Fresh, regenerated and desilicated zeolite Y and Hβ(360) followed a linear distribution and fell within 99% confidence intervals, indicating a similar reaction mechanism to generate CO and methanol among zeolites. HZSM-5(200), HZSM-5(80) and HMOR(20) significantly deviated from the linear regression confidence intervals. More aggressive cracking reactions occurred on ZSM-5 and MOR, resulting in more CO (Table S3†). As proton-type ZSM-5 is a common solid acid catalyst for methanol dehydration to DME,38 we attributed part of this deviation to more aggressive methanol dehydration.
As the acidity of fresh zeolite Y increased from HY(80) to HY(30), the CO yield gradually rose (Fig. 2d). At 350 °C, PMMA hydrolysis produced 3.4 mol% CO over HY(80), 3.9 mol% CO over HY(60), and 3.9 mol% CO over HY(30) (Table S1†). Along with this increase in acidity, the acetone yield also increased, while the yield of MAA decreased. The similar upward trends of CO and acetone, as the acidity increased, suggest that MAA cracking may co-generate both CO and acetone.
3.2 Effect of the zeolite structure and acidity on PMMA hydrolysis
Commercial H-type zeolite Y and β, activated ZSM-5, and activated MOR displayed typical diffraction peaks compared with simulated XRD patterns for zeolites collected in ref. 39 (Fig. 3a and S2†). R-HY(80) retained the same crystallographic structure as fresh zeolite Y, indicating no framework destruction after regeneration at 550 °C. However, diffraction peaks nearly disappeared in M0.5, M1 and M2, suggesting a damaged crystal structure post-desilication. Only two small and broad crystal faces of (220) and (531) were detected. Broad peaks revealed smaller or zero crystallites on desilicated samples.40 Even though desilication created meso-pores on HY(80), NaOH treatment corrupted the crystal structure.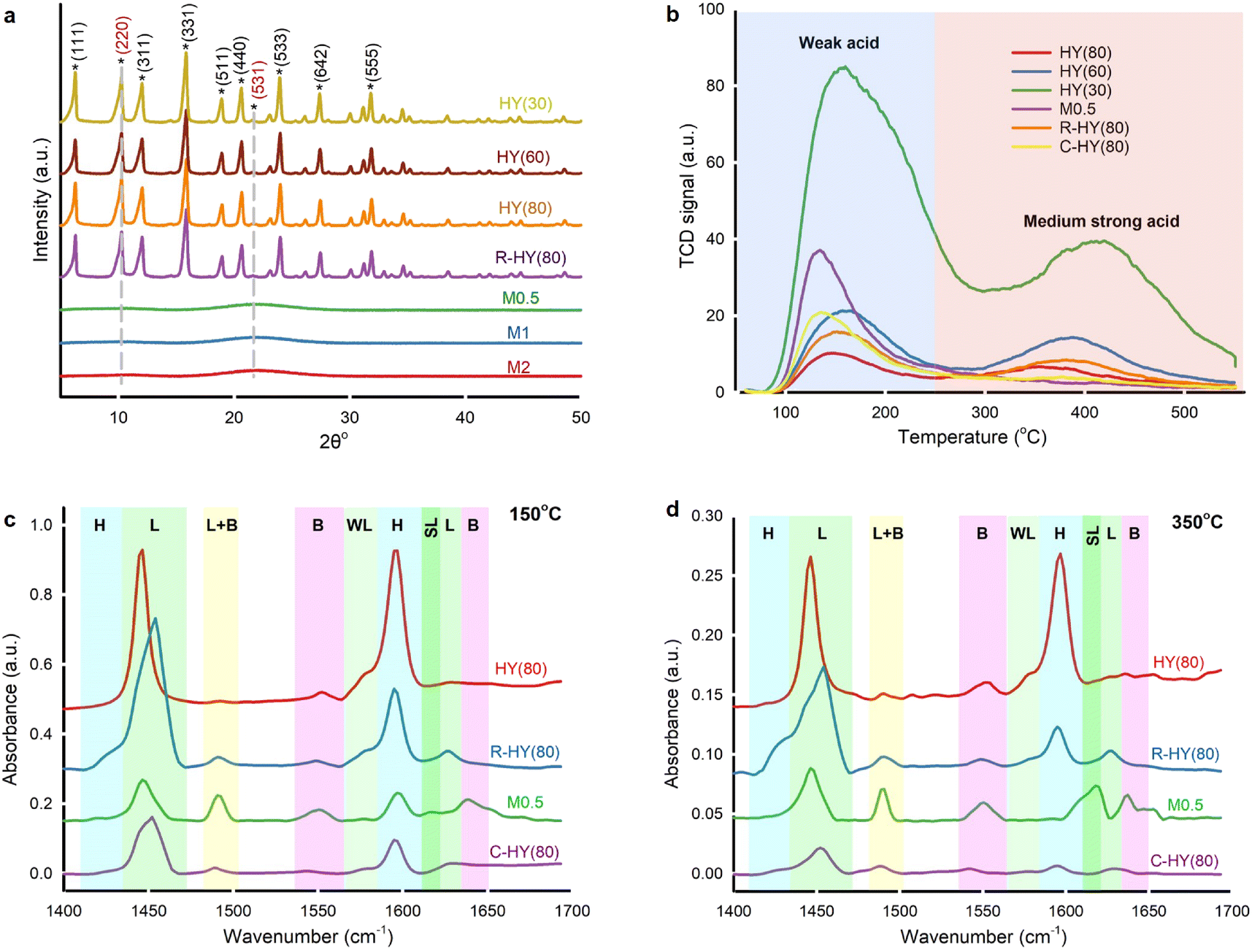 | ||
| Fig. 3 Characterization of fresh, activated, regenerated, desilicated and coked zeolite catalysts. a. X-ray powder diffraction over fresh, regenerated and desilicated zeolite Y. b. NH3-TPD curves over fresh, desilicated, regenerated and coked zeolite Y starting from 50 °C to 550 °C. c and d. FTIR spectra of adsorbed pyridine on fresh, regenerated, desilicated and coked HY(80) with a pyridine desorption temperature of 150 °C (c) and 350 °C (d). Characteristic bands identified hydrogen-bonded pyridine (H), Lewis acid sites (L), Brønsted acid sites (B), weak Lewis acid sites (WL), strong Lewis acid sites (SL) and the combination of Lewis and Brønsted acid sites (L + B). | ||
Desilicated zeolite Y (M0.5, M1 and M2) exhibited low specific surface areas from 43 m2 g−1 to 128 m2 g−1 and insignificant micropore volumes, but higher average pore diameters (39 to 87) compared to fresh HY(80) (Table S2†). In sample M3, the Si/Al ratio indicated that too concentrated NaOH solution caused excessive desilication. Even though desilication generated more mesoporous channels which facilitated molecular diffusion, the framework collapse weakened the microporous networks' shape-selectivity41 and caused amorphous aluminosilicate with substantial mesoporosity.42
NH3-TPD identified and quantified weak acid sites at 100 °C to 250 °C and medium strong acid sites at 250 °C to 550 °C on fresh, regenerated, desilicated and coked zeolite Y (Fig. 3b and Table S3†).43 With the increase in the SiO2/Al2O3 ratio, fresh H-type zeolite Y displayed lower acidity on weak and medium strong acid sites simultaneously. The amount of weak acid sites and medium strong acid sites on R-HY(80) increased by 55% and rose fourfold, respectively, compared with fresh HY(80). The Si/Al ratio of R-HY(80) was slightly higher than that of fresh HY(80) (Table S2†). Slight dealumination happened during catalyst regeneration at 550 °C. Weak acid sites are mainly derived from NH3 desorption from Si–OH groups and the extra-framework Al species, while medium strong acid sites principally come from NH3 desorption from bridging Si–OH–Al acidic hydroxyls in the framework.44 We attributed the increase of weak acid strength on R-HY(80) to the extra-framework Al species resulting from framework Al extraction. Meanwhile, partial extra-framework Al interacted with Si–OH–Al, which enhances medium strong acidity.45 Surprisingly, medium strong acid sites on M0.5 and C-HY(80) disappeared entirely. NaOH treatment at 60 °C destroyed the Si–O–Al bridge-linking framework of zeolite Y and produced amorphous aluminosilicate that only exhibited weak acid sites. Zero medium strong acid sites on C-HY(80) confirmed that coke mainly formed on the surface of Si–OH–Al groups in the zeolite framework and wholly deactivated these active sites and blocked zeolitic channels. A higher amount of weak acid strength on C-HY(80), compared to fresh HY(80), implied that carbon-based solid residues may contain acid components interacting with the NH3 probe molecule.
Pyridine-adsorbed FT-IR (Py-IR) analysis further distinguished Lewis and Brønsted acid sites on fresh, regenerated, desilicated and coked HY(80) at pyridine desorption temperatures of 150 °C and 350 °C (Fig. 3c and d and Table S3†). We attributed spectral bands at 1450, 1577, 1615 and 1628 cm−1 to C–C bond stretching vibrations of pyridine adsorbed on Lewis acid sites (LAS),46,47 and spectral bands at 1425 and 1596 cm−1 to hydrogen-bonded pyridine combined with the zeolite surface's silanol groups.48,49 Pyridine ions protonated by Brønsted acid sites (BAS) generated two spectral bands at 1550 and 1640 cm−1.46 The spectral band at 1490 cm−1 corresponded to pyridine adsorbed on both Lewis and Brønsted acid sites.50 We further clarified weak LAS at 1577 cm−1 and strong LAS at 1615 cm−1.47,51 The four-coordinate framework Al in zeolite Y contributes to BAS, yet extra-framework Al species generated by dealumination induced LAS.52,53
Compared with fresh HY(80), R-HY(80) had more LAS centered at 1450 cm−1 but less SiOH groups located at 1596 cm−1 and less BAS at 1550 cm−1. Meanwhile, a new spectral band at 1425 cm−1 related to SiOH groups appeared for R-HY(80). Steam treatment reversibly transforms strong LAS into BAS, whereas thermal treatment causes an irreversible conversion of some BAS into LAS.54 We attributed the reduction of BAS and the increase of LAS on R-HY(80) to zeolite dealumination during calcination at 550 °C. Desilication enhanced BAS located at 1550 cm−1 and acid sites corresponding to the combination of LAS and BAS at the 1490 cm−1 band, but reduced both LAS centered at 1450 cm−1 and SiOH groups located at 1596 cm−1. Presumably desilication caused some extra-framework Al species due to framework collapse and these Al species interacted with Si–OH–Al groups strengthening the peak at 1490 cm−1. C-HY(80) showed only one predominant peak at 1450 cm−1 corresponding to LAS at a pyridine desorption temperature of 350 °C. The marked decline of BAS on C-HY(80) indicated that coke primarily formed on the surface of BAS generated by Si–OH–Al in the zeolitic framework, which is consistent with the disappearance of medium strong acid sites on C-HY(80) measured by NH3-TPD. Both HY(80) and R-HY(80) presented weak LAS centered at 1577 cm−1 and lacked strong LAS located at 1615 cm−1. Nevertheless, M0.5 displayed the exact opposite result. PMMA blended well with zeolite Y catalysts produced acetone during hydrolysis, but desilicated zeolites (M0.5, M1 and M1) entirely eliminated the production of acetone. This LAS type's transformation was likely to be responsible for the production of acetone.
3.3 Solid residue analysis
PMMA powder (Mw = 550000 g mol−1) used in this work has a midpoint glass transition temperature Tg of 123 °C (Fig. S6†), which indicates atactic tacticity.55 Thermal depolymerization of this virgin PMMA powder underwent three weight loss stages (Fig. 4a) under either N2 or steam. Steam caused less weight loss compared with an inert atmosphere at stage I because the steam thermogravimetric test kept the sample at 130 °C for 30 min prior to the temperature increase. We inferred that PMMA degradation started since 130 °C during steam equilibrium, whereas the PMMA weight loss began at 140 °C in N2. The weight loss at stages II and III remained consistent under both steam and inert gas atmospheres. PMMA non-catalytic depolymerization under inert gas and steam follows almost identical degradation behaviour. The thermogravimetric degradation of PMMA, synthesized via free radical polymerization, follows three distinct weight loss stages.18 Stage I involved depolymerization initiated by the least stable head-to-head (H–H) linkages.56 In stage II, the weight loss was attributed to depolymerization initiated at the unsaturated vinyl ends, while stage III was characterized by random scission within the polymer chains.
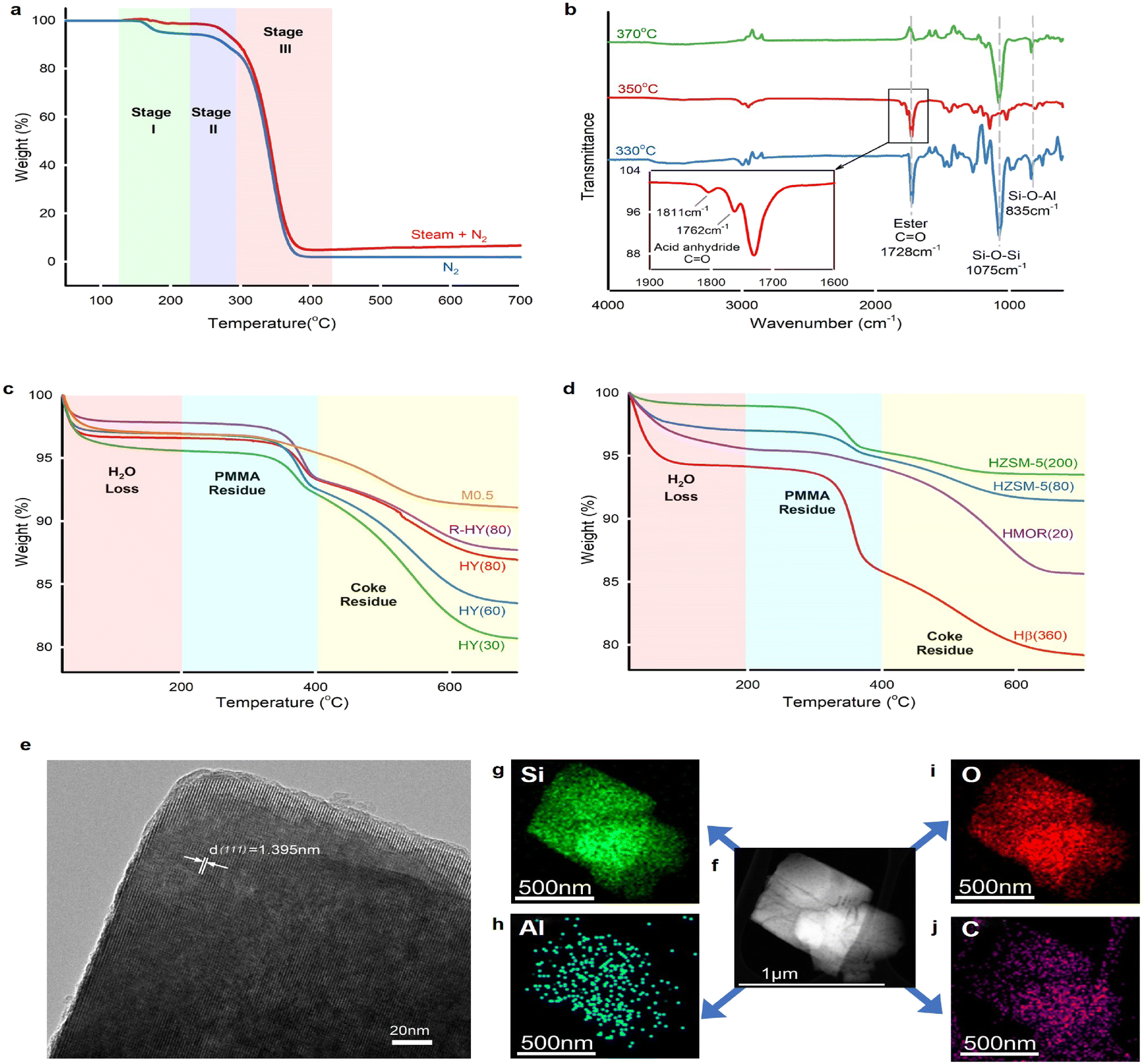 | ||
| Fig. 4 Solid residue analysis. a. Thermal gravimetric analysis of pure PMMA under a N2 atmosphere (60 mL min−1, blue curve) and steam (mixture atmosphere of 70 mL min−1 of steam and 30 mL min−1 of N2, red curve) with a heating ramp of 5 °C min−1. b. FTIR spectra of acetone-dissolved residues on coked HY(80) reacted at 330 °C, 350 °C and 370 °C. Groups Si–O–Al and Si–O–Si were from HY(80) particle residues in acetone. c and d. Thermal gravimetric analysis of solid residues for fresh, regenerated and desilicated zeolite Y reacted at 350 °C (c), and activated HZSM-5(200), HZSM-5(80), HMOR(20) and fresh Hβ(360) reacted at 350 °C (d) under an air atmosphere (60 mL min−1) with a heating ramp of 5 °C min−1. e–j. TEM images of coked HY(80) reacted at 350 °C. The interplanar spacing of 1.395 nm corresponded to the (111) plane in the zeolite Y crystal (e). Dark-field TEM image of C-HY(80) (f) and element mapping images of Si (g), Al (h), O (i) and C (j) using an X-ray energy dispersive spectrometer (EDS). | ||
FTIR identified the functional groups of unreacted PMMA remaining in HY(80) (Fig. 4b). The spectral band at 1728 cm−1 corresponded to carbonyl group C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O on the PMMA chain. Solid residues obtained at 330 °C and 350 °C left unreacted PMMA, while the band at 1728 cm−1 completely disappeared over residues obtained at 370 °C. Carbon balance showed 99% PMMA conversion over fresh HY(80) at a reaction temperature of 370 °C (Fig. 2c and Table S1†). Notwithstanding analytical and statistical uncertainties, the data suggests PMMA completely degraded at 370 °C. PMMA residues reacting over HY(60) reached a weight averaged molecular weight (Mw) of 18000 g mol−1 measured by GPC (Fig. S5†). However, FTIR detected two small peaks at 1811 and 1762 cm−1 for HY(80) solid residues reacted at 350 °C. We attributed these two peaks to anhydride carbonyl twin peaks. Double peaks at 1811 and 1762 cm−1 were more likely to be intramolecular six-membered glutaric anhydride formed between adjacent carboxyl groups.57,58 Polymethacrylic acid (PMAA)–PMMA copolymers were formed by direct hydrolysis of partial carbomethoxy groups on the PMMA chain to carboxyl groups during PMMA depolymerization, followed by dehydration of two adjacent carboxyl groups to form six-membered glutaric anhydride.
O on the PMMA chain. Solid residues obtained at 330 °C and 350 °C left unreacted PMMA, while the band at 1728 cm−1 completely disappeared over residues obtained at 370 °C. Carbon balance showed 99% PMMA conversion over fresh HY(80) at a reaction temperature of 370 °C (Fig. 2c and Table S1†). Notwithstanding analytical and statistical uncertainties, the data suggests PMMA completely degraded at 370 °C. PMMA residues reacting over HY(60) reached a weight averaged molecular weight (Mw) of 18000 g mol−1 measured by GPC (Fig. S5†). However, FTIR detected two small peaks at 1811 and 1762 cm−1 for HY(80) solid residues reacted at 350 °C. We attributed these two peaks to anhydride carbonyl twin peaks. Double peaks at 1811 and 1762 cm−1 were more likely to be intramolecular six-membered glutaric anhydride formed between adjacent carboxyl groups.57,58 Polymethacrylic acid (PMAA)–PMMA copolymers were formed by direct hydrolysis of partial carbomethoxy groups on the PMMA chain to carboxyl groups during PMMA depolymerization, followed by dehydration of two adjacent carboxyl groups to form six-membered glutaric anhydride.
Thermogravimetric analysis (TGA) of solid residues in air recognized three weight loss stages (Fig. 4c and d). The weight loss below 200 °C belonged to H2O adsorbed on solid residues, while weight losses between 200 and 400 °C, and above 400 °C corresponded to unreacted PMMA and coke deposits, respectively (Fig. S7†). Solid residues remaining in the fresh and regenerated zeolite Y displayed two distinct weight loss stages corresponding to PMMA and coke residues. With the decrease of the SiO2/Al2O3 ratio from 80 to 30 (thus stronger acidity and a higher amount of Al species), reacted zeolite Y presented more coke deposits. The increased coke deposits at the lower SiO2/Al2O3 ratio of zeolite Y confirmed that coke formed on the surface of Al sites and blocked Al-containing LAS and BAS as well as zeolite pore channels. Desilicated zeolite Y, M0.5, demonstrated strong cracking ability, which almost completely degraded PMMA and yielded a low amount of coke. We attributed the low coke deposits on M0.5 to the increased zeolite pore size. Further TGA investigation on reacted HZSM-5, HMOR(20) and Hβ(360) found that the zeolite pore size plays a decisive role in coke formation (Fig. 4d). HZSM-5(80) and HZSM-5(200) exhibited ultra-low coke deposits, even though HZSM(80) had higher coke formation than HZSM(200) owing to the strengthened acidity. HMOR(20) almost completely degraded PMMA, like M0.5, whereas it accumulated more coke species than M0.5. Hβ(360) exhibited not only high PMMA residues, but also high coke deposits.
We further calculated the kinetic diameters of MMA and MAA molecules based on an electron density isosurface method.36 The shape selectivity effect of zeolite limits molecules with a kinetic diameter higher than the pore channel size to enter size-defined pores and access internal acid sites of the zeolitic framework.59 The kinetic diameter of MMA, 6.6 Å, is marginally higher than the MAA kinetic diameter, 6.5 Å (Fig. 5b and d). The four types of zeolites have specific pore dimensions (Fig. 5e–h and Table S4†). Owing to large-pore channels (7.4 × 7.4 Å), zeolite Y allows MMA and MAA to readily access its pores (Fig. 5e). Zeolite β possesses 6.6 × 6.7 Å and 5.6 × 5.6 Å interconnected channels, which restricts the diffusion and transfer of MMA and MAA in the pores (Fig. 5f). Mordenite has 12-ring 6.5 × 7.0 Å large-pore channels as well as an 8-ring 2.6 × 5.7 Å micro-pore channel, which not only allow MMA and MMA to enter the pores but also confine the diffusion and transfer of MMA and MAA in the pores (Fig. 5g). ZSM-5 possesses 5.3 × 5.6 Å and 5.1 × 5.5 Å channels that only allow MMA and MAA to access its external surface (Fig. 5h).
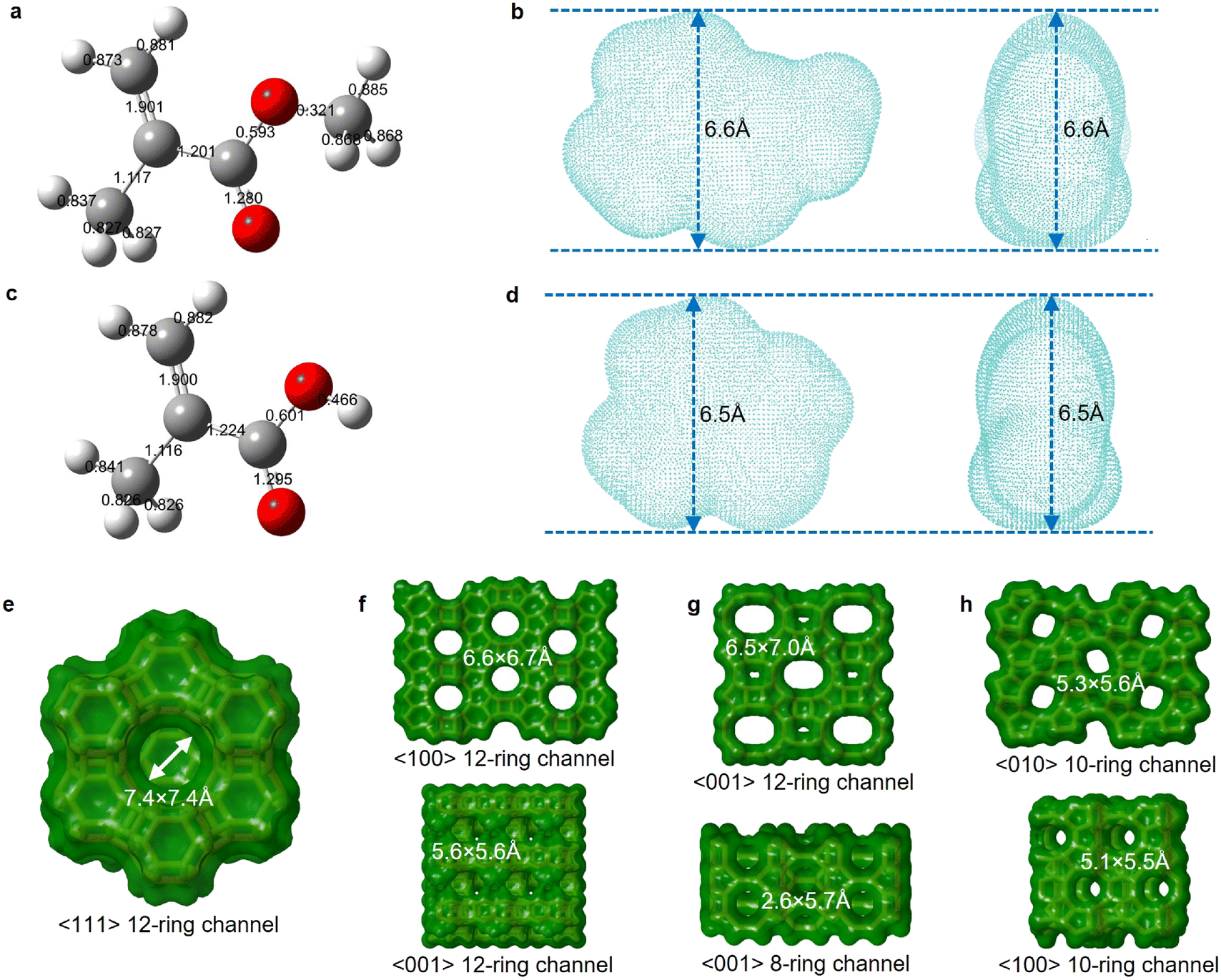 | ||
| Fig. 5 Relationship between the molecular kinetic diameter and the zeolite structure size. a–d. 3D molecular ball-and-stick structures labelled with bond orders of MMA (a) and MAA (c) via Gaussview 6, and electron density isosurfaces of MMA (b) and MAA (d) visualized using VMD. The estimated kinetic diameter is 6.6 Å for MMA (b) and 6.5 Å for MAA (d) based on the electron density isovalue of 0.0015 a.u. e–h. Channel diameter, ring size and crystal plane of zeolite Y (e), zeolite β (f), mordenite (g) and ZSM-5 (h). All structure information referred to the database of zeolite structures.60 | ||
The microporous confinement effect directly influences coke formation. Due to the entire channel constraint, the reacted ZSM-5 exhibited mild coke deposits on the external surface (Fig. 4d). Primary coke deposits formed on the internal surface of the pores over zeolite Y, β and MOR. The transmission electron microscopy (TEM) image of the reacted HY(80) at 350 °C demonstrated an interplanar spacing of 1.395 nm, which corresponds to the zeolite Y (111) crystal plane (Fig. 4e). Furthermore, the element mapping scan found that the carbon distribution remains consistent with Al, which confirmed that coke mainly deposits on the surface of Al-containing acid sites (Fig. 4f–j).
Through kinetic diameter calculations of MMA and MAA, we determined that their diffusion within the zeolitic framework is a key factor in the deactivation of the zeolites. Both MMA and MAA can diffuse into the large-pore channels of zeolite Y, zeolite β, and MOR, leading to coke formation within the zeolitic framework. In contrast, the microporous channels of ZSM-5 prevent MMA and MAA from entering its internal pores, thereby avoiding coke formation. These kinetic diameter calculations provided us with a quantitative understanding of the relationship between zeolite deactivation and the diffusion behaviour of MMA and MAA within the zeolitic framework.
3.4 PMMA hydrolysis mechanism over HY(80)
PMMA thermolysis follows a free radical depolymerization mechanism.17 TGA of PMMA degradation under steam without any catalysts confirms that PMMA thermal depolymerization in steam also follows a free radical depolymerization mechanism (Fig. 4a). To gain further insights into PMMA hydrolysis and its by-product, we conducted in situ liquid and gas analysis using GC-MS, detecting MAA, methanol, MMA and acetone every minute (Fig. 6a). Two peaks appeared in product analysis at time zero and 15 min, corresponding to weight loss stage I and stages II & III together (Fig. 4a). PMMA hydrolysis over solid acid catalysts began with PMMA thermolysis to MMA, followed by catalytic hydrolysis of MMA to MAA and methanol.30 We observed that PMMA depolymerizes and MMA hydrolyzes immediately at the beginning of the reaction. In a lower temperature range (175 °C to 250 °C), we detected equivalent amounts of MAA, methanol and MMA. MMA hydrolysis over HY(80) produces MAA and methanol.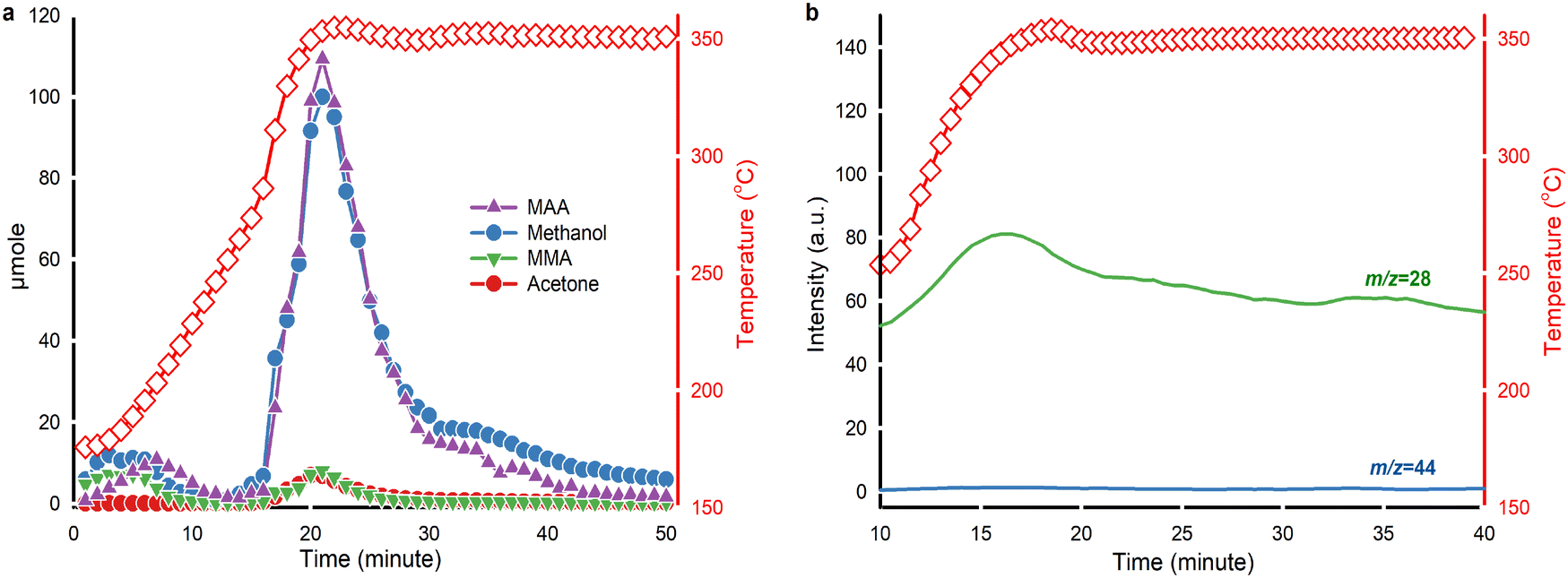 | ||
| Fig. 6 In situ product monitoring. a. Moles of the products measured every minute by GC-MS (left axis, solid symbols) with reaction temperature (right axis, red hollow squares). DME was excluded. b. Continuous mass-to-charge ratio (m/z) recorded using an on-line mass spectrometer (left axis, green and blue curves) evolved with reaction temperature (right axis, red hollow squares) and reaction time. m/z = 44 corresponds to CO2, while m/z = 28 represents CO. | ||
We observed the evolution of PMMA powder during melting to prepare the highest contact between PMMA and the catalyst. When feeding the reactor with mechanically blended PMMA and HY(80), the catalyst powder partially covered melted PMMA and so some of the produced MMA exited the reactor without passing through HY(80).
In contrast, blanketing PMMA with HY(80) powder kept PMMA covered even after melting (Table S1†). PMMA spheres slightly melted and became like a “Ferrero” chocolate with HY(80) particles adhered on the surface (Fig. S3c and d†).
PMMA underwent end-chain scission to produce MMA in the low-temperature zone (175 °C to 250 °C),61 but did not form acetone. Above 250 °C, the yield of MAA and methanol increased because random chain scission started and accelerated PMMA depolymerization.61 By-product acetone formed above 250 °C and maintained a concentration change pattern similar to MMA. At 350 °C from t = 30 min, the quantity of methanol surpassed that of MAA. Extra methanol may be a result of the direct hydrolysis of PMMA to Poly(MMA-co-MAA) copolymer chains, which subsequently forms acid anhydride groups on the chain via dehydration (Fig. 7).
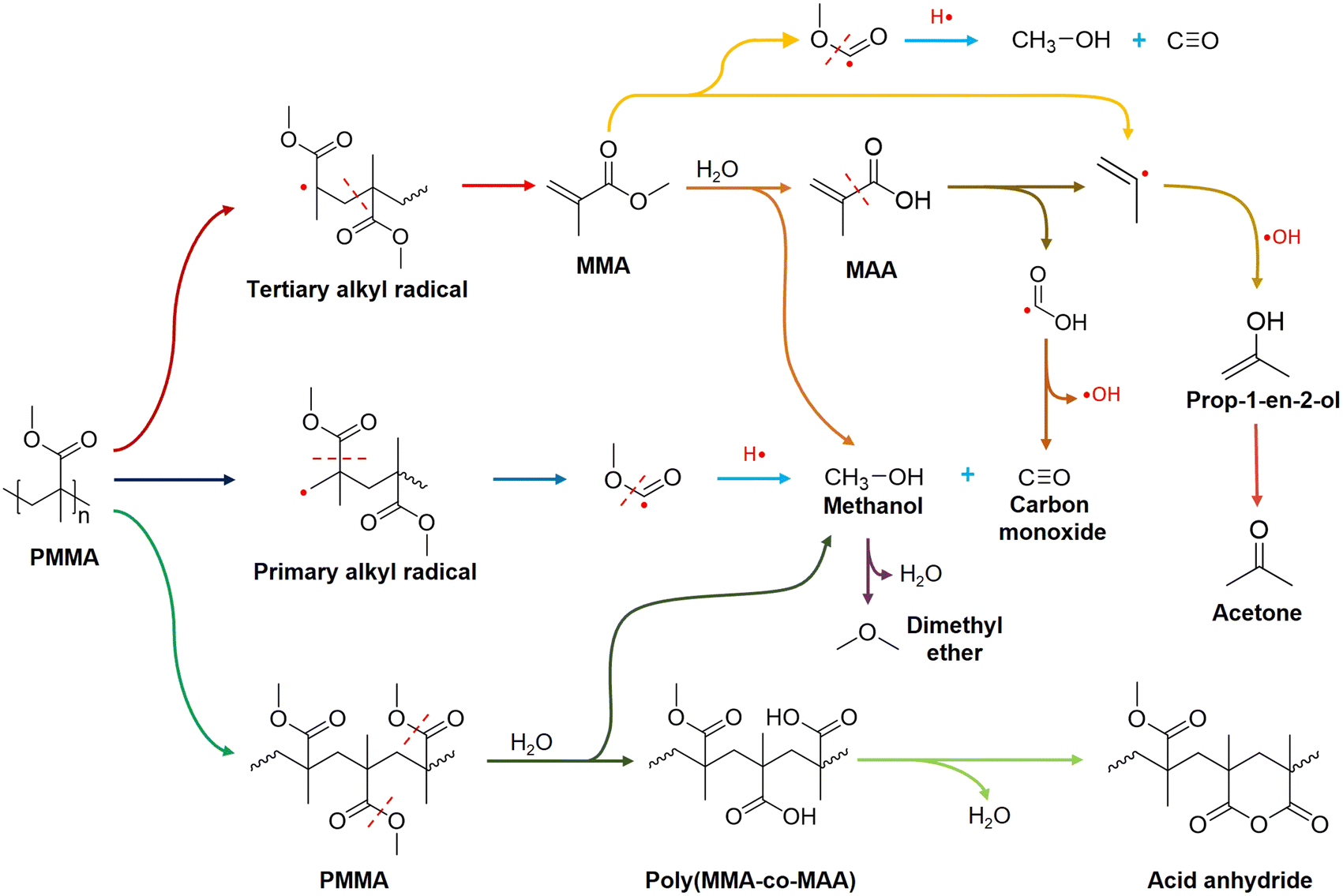 | ||
| Fig. 7 PMMA hydrolysis mechanism over zeolite Y. Coloured arrows represent different reactions. FTIR spectra of PMMA residues in C-HY(80) reacted at 350 °C validated the formation of Poly(MMA-co-MAA) acid anhydride. Primary by-products, acetone and CO, were derived from MAA cracking over acid sites on zeolite Y. | ||
On-line MS monitored the evolution of CO and CO2 during the reaction (Fig. 6b). A weak signal of m/z = 44 suggested that PMMA hydrolysis yielded minor production of CO2. The signal of CO increased starting from 250 °C and peaked at around 350 °C. Acetone and CO both appeared at the same temperature.
To further elucidate the relationship between CO and acetone, we performed blank tests by feeding only MMA or MAA into the reactor (Fig. S4†). Non-catalytic hydrolysis of MMA at 350 °C did not produce any liquid product. However, catalytic hydrolysis of MMA over HY(80) at 350 °C produced a small amount of MAA, methanol and a trace of acetone (Fig. S4a†). While non-catalytic hydrolysis of MAA also did not generate any decomposition products at 350 °C, catalytic hydrolysis of MAA over HY(80) at 350 °C yielded a considerable amount of acetone (Fig. S4b†). Catalytic hydrolysis of MAA produced CO under the same conditions (Fig. S4d†).
Higher bond orders indicate stronger molecular stability. Even though the C–C bond (bond order = 1.224) between the carboxyl group and CC double bond in MAA has a higher bond order than the C–C bond (bond order = 1.201) between the ester group and CC double bond in MMA (Fig. 5a and c), the dominant formation route of acetone and CO is the scission of the C–C bond between the carboxyl group and CC double bond in MAA since MAA predominated over HY(80) during the reaction.
Here, we proposed a PMMA hydrolysis mechanism over HY(80) at 350 °C based on experimental data and the literature (Fig. 7). Random chain scission of PMMA at 350 °C generated tertiary alkyl radicals, followed by unzipping to produce the MMA monomer. Subsequently, HY(80) hydrolyzed MMA to MAA and methanol on acid sites. MAA further cracked into formic acid radicals and 2-propenyl radicals at 350 °C. Dehydroxylation of formic acid radicals resulted in CO, while 2-propenyl radicals combined with one hydroxyl group to form prop-1-en-2-ol that was further isomerized to acetone. Cracking of MMA may produce 2-propenyl radicals and CH3O(CO)· free radicals directly. Random chain scission of PMMA generated a small amount of primary alkyl radicals which readily resulted in the CH3O(CO)· free radical fragment. Protonation of the CH3O(CO)· fragment produced methanol and CO. Further dehydration of methanol yielded a trace of dimethyl ether (DME). A small amount of PMMA was partially hydrolyzed to the poly(MMA-co-MAA) copolymer, followed by dehydration to form six-membered glutaric anhydride (Fig. 7).
Conclusions
Upcycling PMMA to MAA instead of MMA is a viable alternative to recycle both post-processed industrial scraps and cuttings and end-of-life PMMA, offering prominent technical, economic, and market benefits. In this study, we introduced a heterogeneous catalytic hydrolysis method to directly convert PMMA to MAA over zeolite -fresh, activated, regenerated and desilicated zeolites. H-type zeolite Y with a SiO2/Al2O3 ratio of 80 exhibited the highest MAA yield and selectivity. PMMA thermolysis to MMA followed by MMA catalytic hydrolysis over zeolite Y to MAA produced a high concentration of the MAA monomer. Coke primarily formed on the surface of medium strong acid sites and so temporarily deactivated Brønsted acid sites. Kinetic diameter estimation of MMA and MAA suggested that coke predominantly formed in the channels of zeolites with large pores. While small-pore ZSM-5 had stronger cracking capacity, while large-pore zeolite Y demonstrated superior hydrolysis performance. Acetone appeared with CO at higher hydrolysis temperature (>250 °C). Acetone was produced during PMMA hydrolysis primarily by decarboxylation of the MAA monomer. The existence of the acid anhydride group in the reacted residues proved partial hydrolysis of PMMA chains to the poly(MMA-co-MAA) copolymer, and subsequent dehydration to six-membered glutaric anhydride. For subsequent research, we suggest to extend the feed from virgin PMMA to post-processed industrial scraps and cuttings and end-of-life PMMA plastic, and design specific pilot and industrial scale reactors for direct conversion of PMMA to MAA, along with a techno-economic assessment.Data availability
The data supporting the findings of this study are available in the ESI† or can be obtained from the corresponding author, Prof. Gregory Patience, upon request.The database and software used in this manuscript have been appropriately cited.
For clarity, the key databases and software are listed below:
1. Zeolite structure information:
All structural information of zeolites presented in Fig. 5 is referenced from the Database of Zeolite Structures. This database is cited in citation 54:
• C. Baerlocher and L. McCusker, Database of Zeolite Structures, 2023, http://www.iza-structure.org/databases/ (accessed: January 2023).
2. Kinetic diameter estimation (section 2.7):
• Gaussian 16W software: the license for Gaussian 16W is provided by Shanghai Jiao Tong University as stated in the Acknowledgements.
• VMD (Visual Molecular Dynamics): VMD is a free molecular visualization program for academic research purposes. The software can be downloaded from https://www.ks.uiuc.edu/Research/vmd/.
• Multiwfn program: Multiwfn is a free wavefunction analysis program. The software can be downloaded from http://sobereva.com/multiwfn/. This program is cited in citation 36: T. Lu and F. Chen, Journal of Computational Chemistry, 2012, 33, 580–592.
Author contributions
Yanfa Zhuang: conceptualization, investigation, methodology, formal analysis, visualization, validation, writing – original draft preparation. Nooshin Saadatkhah: conceptualization, validation, project administration, writing – review & editing. Tien-Dat Nguyen: investigation, visualization, writing – review & editing. Jacopo De Tommaso: validation, writing – review & editing. Clive Yi Jie Ng: investigation, visualization, formal analysis. Chunyu Wang: software, resources, validation. Abdellah Ajji: resources, supervision, writing – review & editing. Gregory S. Patience: funding acquisition, resources, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr. Jean-Luc Dubois at Trinseo France SAS for discussion and revision, Prof. Liang Wu at Shanghai Jiao Tong University for the Gaussian 16 license, Tian Lu at the Beijing Kein Research Centre for Natural Sciences for the Multiwfn program, and Jean-Philippe Masse at Polytechnique Montreal for the TEM-EDS and XRD analysis. This work was supported by the Natural Sciences and Engineering Research Council of Canada (ALLRP-573784-22 CRSNG) and Arkema & Lavergne (CDT 051087).References
- OECD, Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, OECD Publishing, Paris, 2022 Search PubMed.
- X. Yuan, X. Wang, B. Sarkar and Y. S. Ok, Nat. Rev. Earth Environ., 2021, 2, 659–660 CrossRef CAS PubMed.
- K. S. Khoo, L. Y. Ho, H. R. Lim, H. Y. Leong and K. W. Chew, J. Hazard. Mater., 2021, 417, 126108 CrossRef CAS PubMed.
- T. M. Adyel, Science, 2020, 369, 1314–1315 CrossRef.
- N. Ebner and E. Iacovidou, Sustain. Prod. Consumption, 2021, 28, 726–735 CrossRef.
- A. L. P. Silva, J. C. Prata, T. R. Walker, D. Campos, A. C. Duarte, A. M. Soares, D. Barcelo and T. Rocha-Santos, Sci. Total Environ., 2020, 742, 140565 CrossRef.
- B. Joseph, J. James, N. Kalarikkal and S. Thomas, Adv. Ind. Eng. Polym. Res., 2021, 4, 199–208 CAS.
- M. Bachmann, C. Zibunas, J. Hartmann, V. Tulus, S. Suh, G. Guillen-Gosalbez and A. Bardow, Nat. Sustain., 2023, 6, 599–610 CrossRef.
- R. Geyer, R. J. Jenna and L. L. Kara, Sci. Adv., 2017, 3, e1700782 CrossRef.
- J. De Tommaso and J. Dubois, Polymers, 2021, 13, 2724 CrossRef CAS.
- Markets and Markets, Polymethyl Methacrylate (PMMA) Market, 2022, https://www.marketsandmarkets.com/Market-Reports/polymethyl-methacrylate-pmma-market-715.html, (accessed: August 2022) Search PubMed.
- P. Research, Polymethyl Methacrylate (PMMA) Market Size, Report, 2021, https://www.precedenceresearch.com/polymethylmethacrylate-market, (accessed: April 2023) Search PubMed.
- D. D'Hooge, Y. W. Marien and J. L. Dubois, Polymer Circularity Roadmap: Recycling of Poly(methyl Methacrylate) as a Case Study, Walter de Gruyter, Berlin, 2022 Search PubMed.
- T. C. Research and D. I. S. (CORDIS), Innovative Acrylic(PMMA-Polymethyl methacrylate) Recycling Technology Complying with Regulations, 2022, https://cordis.europa.eu/project/id/856103/reporting, (accessed: August 2022) Search PubMed.
- M. Sponchioni and S. Altinok, Advances in Chemical Engineering, Elsevier, New York, 2022, vol. 60, pp. 269–287 Search PubMed.
- T. Kashiwagi, T. Hirata and J. E. Brown, Macromolecules, 1985, 18, 1410–1418 CrossRef.
- E. Esmizadeh, S. Khalili, A. Vahidifar, G. Naderi and C. Dubois, Handbook of Ecomaterials, Springer International Publishing, Cham, 2018, pp. 1–33 Search PubMed.
- E. K. Moens, K. De Smit, Y. W. Marien, A. D. Trigilio, P. H. Van Steenberge, K. M. Van Geem, J. Dubois and D. R. D'hooge, Polymers, 2020, 12, 1667 CrossRef CAS PubMed.
- J. Dubois, Guidelines for PMMA depolymerization at pilot and industrial scale, 2022, https://www.researchgate.net/, (accessed: April 2022) Search PubMed.
- MMAtwo, MMAtwo in position to benchmark its results with state of the art virgin and regenerated MMA, 2022, https://www.mmatwo.eu, (accessed: March 2023) Search PubMed.
- C. B. Godiya, S. Gabrielli, S. Materazzi, M. S. Pianesi, N. Stefanini and E. Marcantoni, J. Environ. Manage., 2019, 231, 1012–1020 CrossRef CAS.
- W. Kaminsky and J. Franck, J. Anal. Appl. Pyrolysis, 1991, 19, 311–318 CrossRef CAS.
- USEPA, Ethyl Acrylate, 2016, https://www.epa.gov/sites/default/files/2016-09/documents/ethyl-acrylate.pdf, (accessed: March 2023) Search PubMed.
- E. K. Moens, Y. W. Marien, F. L. Figueira, A. D. Trigilio, K. De Smit, K. M. Van Geem, P. H. Van Steenberge and D. R. D'hooge, Chem. Eng. J., 2023, 475, 146105 CrossRef CAS.
- H. Narita, S. Okamoto and S. Machida, Macromol. Chem. Phys., 1969, 125, 15–23 CrossRef CAS.
- R. Buter, Y. Y. Tan and G. Challa, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 2975–2989 CrossRef CAS.
- F. Fenouillot, J. Terrisse and T. Rimlinger, J. Appl. Polym. Sci., 1999, 72, 1589–1599 CrossRef CAS.
- IARC, IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Geneva, 1994, pp. 1–560 Search PubMed.
- BMT, Methyl methacrylate (MMA), 2023, https://cargohandbook.com, (accessed: March 2023) Search PubMed.
- O. V. Chub, N. Saadatkhah, J. Dubois and G. S. Patience, Appl. Catal., A, 2022, 638, 118637 CrossRef CAS.
- O. V. Chub, J. Dubois and G. S. Patience, Chem. Eng. J., 2023, 459, 141479 CrossRef CAS.
- K. Sunitha, R. Sadhana, D. Mathew and C. P. Reghunadhan Nair, Des. Monomers Polym., 2015, 18, 512–523 CrossRef CAS.
- G. Swift, Encyclopedia of Polymer Science and Technology, 2002, vol. 1 Search PubMed.
- T. Map, 291614 Esters of methacrylic acid, 2023, https://www.trademap.org/, (accessed: March 2023) Search PubMed.
- O. V. Chub, J. Dubois and G. S. Patience, Appl. Catal., A, 2022, 647, 118887 CrossRef CAS.
- N. Mehio, S. Dai and D. Jiang, J. Phys. Chem. A, 2014, 118, 1150–1154 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- D. Masih, S. Rohani, J. N. Kondo and T. Tatsumi, Appl. Catal., B, 2017, 217, 247–255 CrossRef CAS.
- M. M. J. Treacy and J. B. Higgins, Collection of simulated XRD powder patterns for zeolites, Elsevier, Amsterdam, 5th revised edn, 2007 Search PubMed.
- P. B. Raja, K. R. Munusamy, V. Perumal and M. N. M. Ibrahim, Nano-Bioremediation: Fundamentals and Applications, Elsevier, Amsterdam, 2022, pp. 57–83 Search PubMed.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- M. Gackowski and J. Datka, Molecules, 2020, 25, 1044 CrossRef CAS PubMed.
- J. Zuo, W. Chen, J. Liu, X. Duan, L. Ye and Y. Yuan, Sci. Adv., 2020, 6, eaba5433 CrossRef CAS.
- J. Lu, S. Zhou, K. Ma, M. Meng and Y. Tian, Chin. J. Catal., 2015, 36, 1295–1303 CrossRef CAS.
- M. A. Makarova and J. Dwyer, J. Phys. Chem., 1993, 97, 6337–6338 CrossRef CAS.
- M. Xie, Y. Li, U. J. Etim, H. Lou, W. Xing, P. Wu, X. Liu, P. Bai and Z. Yan, Ind. Eng. Chem. Res., 2019, 58, 5455–5463 CrossRef CAS.
- L. Wang, A. Wang, X. Li, F. Zhou and Y. Hu, J. Mater. Chem., 2010, 20, 2232–2239 RSC.
- W. Dong, Z. Shen, B. Peng, M. Gu, X. Zhou, B. Xiang and Y. Zhang, Sci. Rep., 2016, 6, 26713 CrossRef CAS.
- Z. Shen, L. Kong, W. Zhang, M. Gu, M. Xia, X. Zhou and Y. Zhang, RSC Adv., 2019, 9, 18989–18995 RSC.
- N. V. Vlasenko, Y. N. Kochkin, G. M. Telbiz, O. V. Shvets and P. E. Strizhak, RSC Adv., 2019, 9, 35957–35968 RSC.
- W. Zhang, P. Wang, C. Yang and C. Li, Catal. Lett., 2019, 149, 1017–1025 CrossRef CAS.
- S. Li, A. Zheng, Y. Su, H. Zhang, L. Chen, J. Yang, C. Ye and F. Deng, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS.
- D. S. Zasukhin, I. A. Kasyanov, Y. G. Kolyagin, A. I. Bulygina, K. C. Kharas and I. I. Ivanova, ACS Omega, 2022, 7, 12318–12328 CrossRef CAS PubMed.
- J. N. Kondo, R. Nishitani, E. Yoda, T. Yokoi, T. Tatsumi and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 11576–11586 RSC.
- L. Chang and E. Woo, Polym. Chem., 2010, 1, 198–202 RSC.
- T. Kashiwagi, A. Inaba, J. E. Brown, K. Hatada, T. Kitayama and E. Masuda, Macromolecules, 1986, 19, 2160–2168 CrossRef CAS.
- B. Ho, Y. Lee and W. Chin, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2389–2397 CrossRef CAS.
- J. Semen and J. Lando, Macromolecules, 1969, 2, 570–575 CrossRef CAS.
- D. Breck, Zeolite Molecular Sieves: Structure, Chemistry, and Use, Wiley, Hoboken, 1973 Search PubMed.
- C. Baerlocher and L. McCusker, Database of Zeolite Structures, 2023, http://www.iza-structure.org/databases/, (accessed: January 2023) Search PubMed.
- Y.-H. Hu and C.-Y. Chen, Polym. Degrad. Stab., 2003, 82, 81–88 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00341a |
| This journal is © The Royal Society of Chemistry 2025 |