
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg(I) emitters with ultrafast spin-flip dynamics for high-efficiency electroluminescence†
Ao
Ying‡
a,
Nengquan
Li‡
 b,
Xingyu
Chen
c,
Jianlong
Xia
c,
Chuluo
Yang
*b and
Shaolong
Gong
*a
b,
Xingyu
Chen
c,
Jianlong
Xia
c,
Chuluo
Yang
*b and
Shaolong
Gong
*a
aCollege of Chemistry and Molecular Sciences, Hubei Key Laboratory on Organic and Polymeric Optoelectronic Materials, Wuhan University, Wuhan 430072, China. E-mail: slgong@whu.edu.cn
bShenzhen Key Laboratory of New Display and Storage Materials, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: clyang@szu.edu.cn
cState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Center of Smart Materials and Devices, International School of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China
First published on 22nd November 2024
Abstract
Carbene-metal-amide (CMA) complexes are appealing emitters for organic light-emitting diodes (OLEDs). However, little is known about silver(I)-CMA complexes, particularly electroluminescent ones. Here we report a series of Ag(I)-CMA complexes prepared using benzothiophene-fused carbazole derivatives as amide ligands. These complexes emit via thermally activated delayed fluorescence (TADF), together with high photoluminescence quantum yields of up to 72% in thin films. By strengthening the π-donating ability of the amide ligands, ultrashort emission lifetimes of down to 144 ns in thin films and 11 ns in solution (with a radiative rate constant of ∼107 s−1) are realized, among the shortest lifetimes for TADF emitters. Key to this unique feature is the ultrafast spin-flip dynamics consisting of forward and reverse intersystem crossing rates of up to ∼109 s−1 and ∼108 s−1, respectively, verified by the transient absorption spectroscopic study. The resulting solution-processed OLEDs based on the optimal complex afford record external quantum efficiencies of 16.2% at maximum and 13.4% at 1000 nits, representing the state-of-the-art performance for Ag(I) emitters. This work presents an effective approach for the development of short-lived TADF materials for high-efficiency OLEDs.
Introduction
In comparison with well-established iridium(III) and platinum(II) phosphors, luminescent coinage metal(I) complexes, particularly copper(I) and silver(I) complexes, have been studied for decades as promising cost-effective alternatives for applications in photocatalysis and optoelectronics.1–3 Phosphorescence from metal-to-ligand charge transfer (MLCT) excited states is the main focus for coinage metal(I) emitters at the early stage.4,5 However, with respect to Ir(III) and Pt(II) complexes, Cu(I) and Ag(I) complexes usually show inefficient phosphorescence because of their slower intersystem crossing (ISC) rates from S1 to T1 and from T1 to S0, originating from moderate spin–orbital coupling (SOC) parameters of the Cu (857 cm−1) and Ag (1830 cm−1) nuclei.6,7 In the past decade, thermally activated delayed fluorescence (TADF) has been demonstrated in numerous Cu(I) complexes by introducing MLCT, halogen-to-ligand charge transfer (XLCT) and/or ligand-to-ligand charge transfer (LLCT) into excited states.8–14 Compared with Cu(I) complexes, Ag(I) complexes are more appealing candidates for short-lived TADF emitters due to the larger atomic number of the silver atom according to semi-classical Marcus theory.15 Despite the fact that some Ag(I) complexes have been developed to exhibit temperature-dependent photophysical behaviors,16 the first TADF Ag(I) complex was reported by Osawa and co-workers only in 2013 (Fig. 1a).17 The TADF Ag(I) complexes are still scarce. The main limitations are the considerably high oxidation potential and low-lying d-orbitals of the Ag(I) center, preventing the participation of d electrons of the Ag atom in frontier occupied orbitals.18–23 As a result, the excited states of the Ag(I) complexes often feature a ligand-centered nature, leading to long-lived phosphorescence together with low radiative rates (kr).13,14,24–26 To access the TADF channel of Ag(I) emitters, one of the mainstream approaches is to use strongly electron-donating ligands such as phosphines, which can destabilize the d-orbital of the central Ag atom and thus allow for metal-involved charge transfer in excited states.18–24,27–29 For instance, Yersin and co-workers developed a series of four-coordinate Ag(I) complexes using phosphine and phenanthroline derivatives as ligands (Fig. 1a).19,21,23–25,27,29 With the introduction of carborane subunits, the phosphine ligands have strong donor strength and thus render efficient TADF from excited states featuring MLCT and LLCT.19,29 Similarly, Lu et al. constructed a series of TADF Ag(I) complexes (Fig. 1a) bearing P^P and N^N ligands in a donor–acceptor motif.30 The resulting complexes feature LLCT nature with less metal contribution (<20%) in excited states. Despite the above promising results, the emission lifetimes of these Ag(I) complexes are still on the μs-scale (>1 μs) and are comparable with conventional TADF or phosphorescent emitters.31–34 How to further accelerate the spin-flip process of Ag(I) complexes is still a formidable challenge. In addition, only two electroluminescent mononuclear Ag(I) emitters have been reported so far, yet with external quantum efficiencies (EQEs) of <15%.35,36 This device performance is far away from the theoretical limit of 20–30% EQEs for organic light-emitting diodes (OLEDs).37 In this regard, the search for high-performance Ag(I) complexes is of vital importance for both fundamental investigation and practical applications.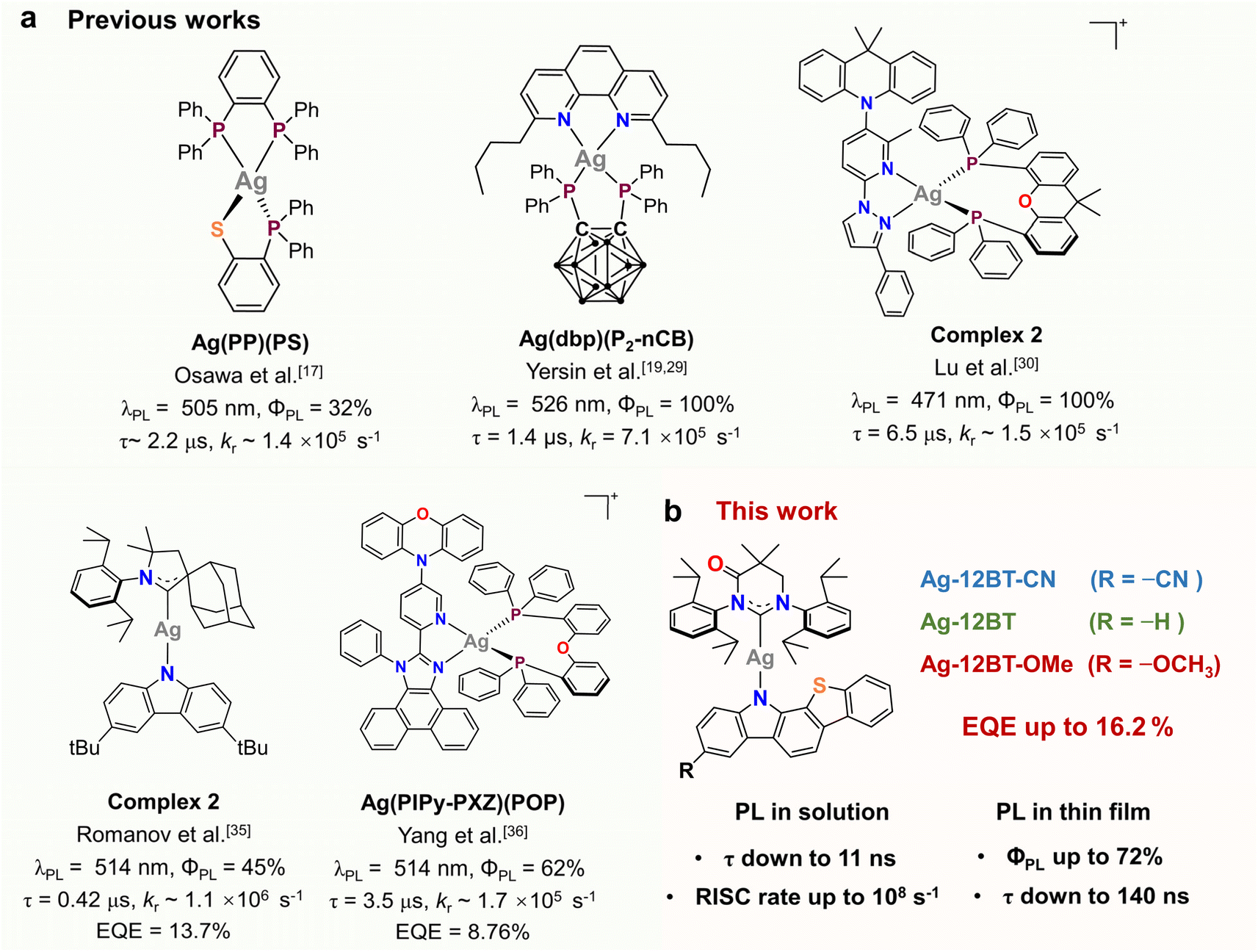 | ||
| Fig. 1 (a) Selected examples of TADF Ag(I) complexes reported in the literature. (b) Chemical structures of the Ag(I)-CMA complexes in this work. | ||
Recently, paramount attention has been paid to a new category of coinage metal(I) complexes bearing a carbene-metal-amide (CMA) motif, due to their distinct TADF properties and high emission performance.7,38 In these complexes, the central metal serves as an electronic bridge to facilitate the long-range p-orbital coupling between the carbene and amide ligands, with limited metal contribution (<15%) to this CT process.39 As a result, CMA complexes have a unique metal-perturbed LLCT nature in excited states, giving rise to distinct TADF properties and fast radiative rates. Despite the small metal contribution to excited states of CMA complexes, the introduction of the metal linkage can still enhance the SOC, accelerate the spin-flip dynamics and thus shorten the exciton lifetimes.38h–j,39,40 Therefore, CMA complexes are ideal candidates for the development of short-lived TADF emitters. In the past few years, the development of CMA complexes has mainly focused on Cu(I) and Au(I) complexes, and some of them have achieved comparable emission performance to Ir(III) and Pt(II) complexes.39 In contrast to MLCT-featured phosphorescent complexes, the emission lifetimes of CMA complexes are mainly governed by the singlet–triplet energy difference (ΔEST), rather than the SOC of the central metals.38l,n In general, Ag(I)-CMA complexes exhibit shortened emission lifetimes compared with Cu(I) and Au(I) analogues, due to reduced ΔEST originating from the prolonged metal–ligand bond lengths.38e,i This feature makes Ag(I)-CMA complexes appealing emitters for OLEDs with low efficiency roll-off. However, there are only a few reports on Ag(I)-CMA complexes. Only one example of OLEDs based on Ag(I)-CMA complexes was demonstrated by Romanov et al. (Fig. 1)35 but with moderate EQEs of 13.7% and 11.0% in vacuum- and solution-processed devices, respectively. This proof-of-concept application in OLEDs pinpoints the large potential of Ag(I)-CMA emitters.
Ligand engineering is the most popular way of modulating the optoelectronic properties of CMA complexes. We recently demonstrated that chalcogen-heterocyclic engineering on the amide ligand can optimize the conformation and the related optical properties of Cu(I)-CMA complexes, giving rise to a record EQE of 28.6% for Cu(I)-OLEDs.41 Following this revelation, we herein developed a series of Ag(I)-CMA complexes, Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe, using benzothiophene-fused carbazole derivatives, 12H-benzo[4,5]thieno[2,3-a]carbazole-3-carbonitrile (12BT-CN), 12H-benzo[4,5]thieno[2,3-a]carbazole (12BT), and 3-methoxy-12H-benzo[4,5]thieno[2,3-a]carbazole (12BT-OMe), as amide ligands to combine with a widely used carbene ligand of MAC* (Fig. 1b). With the fusion of a benzothiophene subunit, Ag-12BT-OMe demonstrated a distinct secondary metal–ligand interaction between the Ag center and amide ligand, as verified by theoretical simulation and single-crystal structure analysis. These complexes exhibited distinct TADF nature with high photoluminescence quantum yields (ΦPL) of up to 72% and kr of up to 2.2 × 106 s−1 in thin films. Intriguingly, Ag-12BT and Ag-12BT-OMe delivered ultrashort emission lifetimes of 70 and 11 ns in solution, respectively, representing the shortest lifetime for TADF emitters. This unique feature is strongly associated with ultrafast spin-flip dynamics consisting of ultrahigh rate constants of ∼109 s−1 and ∼108 s−1 for the forward and reverse intersystem crossing (ISC) between T1 and S1, respectively, as verified by the transient absorption spectroscopic study. The solution-processed OLEDs based on the optimal emitter Ag(I)-12BT realized outstanding EL performance with record EQEs of 16.2% and 13.4% at maximum and 1000 cd m−2, respectively.
Results and discussion
Before synthesis, theoretical calculations were conducted on these Ag(I) complexes to study their electronic structures and excited states. As shown in Fig. S1,† all the Ag(I) complexes exhibited nearly coplanar conformations with small ligand–ligand dihedral angles of 10° to 12°, along with nearly linear ∠C–Ag–N angles of 173° to 175°. Meanwhile, these complexes possessed C–Ag and Ag–N bond lengths of ∼2.1 Å, similar to the values reported in the literature.38l In comparison with their Cu(I) and Au(I) analogues, the Ag(I) complexes possess prolonged Ccarbene⋯Ncarbazole distances, resulting in more separated hole–electron distributions and longer-range LLCT (Fig. 2). For Ag-12BT-CN and Ag-12BT, the center of the hole in their S1 excited states mainly lied near the Ncarbazole atom, whereas the –OCH3 group of Ag-12BT-OMe dispersed the positive charge and thus shifted the hole center away from the Ncarbazole atom. Therefore, Ag-12BT-OMe had the longest hole–electron distance of 6.45 Å in its S1 excited state. Moreover, all the complexes had dominant LLCT nature in both S1 and T1 excited states together with the ca. 2–4% contributions from the central silver (Fig. 2a and Table S1†), leading to a metal-perturbed intraligand charge-transfer feature. Meanwhile, the small transition contributions from the metal center also prevented the Renner–Teller distortion and direct phosphorescence, supported by the CT-featured emission profiles at 300 and 77 K (Fig. S2†). Furthermore, the introduction of the –CN/–OCH3 group can finely modulate the π-donating ability of the amide ligands and thus render HOMO levels of the complexes tunable (Fig. S3†). Therefore, the S1 levels of the Ag(I) complexes follow the order of 2.47 eV (Ag-12BT-CN, 502 nm) > 2.22 eV (Ag-12BT, 558 nm) > 2.02 eV (Ag-12BT-OMe, 614 nm), depending on the π-donating strength of the amide ligands. Meanwhile, with respect to Ag-12BT-CN and Ag-12BT, Ag-12BT-OMe delivered the smallest ΔEST of 0.05 eV (Fig. S3†), which stemmed from the prolonged hole–electron distance. Thanks to the large steric hindrance between the carbene and benzothiophene subunits, these complexes have slightly twisted S1 conformations with lower energies compared with the orthogonal conformers (Fig. S4 and S5†), different from the orthogonal S1 conformation in most CMA complexes. Furthermore, the root mean square deviation (RMSD) between S0 and S1 in these complexes follows the trend of 0.29 Å (Ag-12BT-CN) ≈ 0.22 Å (Ag-12BT-CN) < 0.78 Å (Ag-12BT-OMe), suggesting that Ag-12BT-CN and Ag-12BT have limited structure reorganization and could be better emitters (Fig. S6†).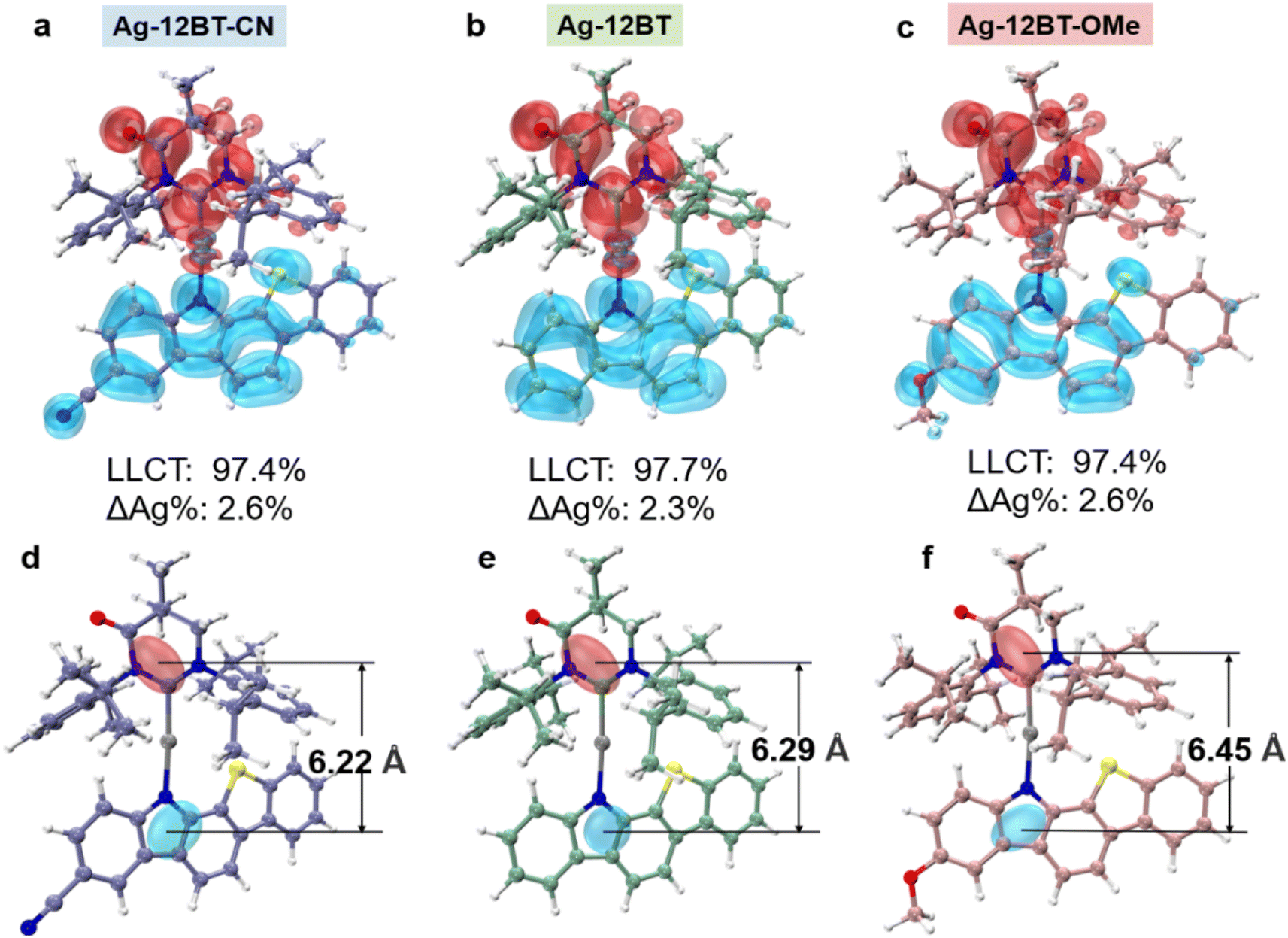 | ||
| Fig. 2 The hole–electron distribution of S1 excited states for (a) Ag-12BT-CN, (b) Ag-12BT, and (c) Ag-12BT-OMe. ΔAg represents the difference in metal participation before and after excitation. The centroid distance between holes and electrons of (d) Ag-12BT-CN, (e) Ag-12BT, and (f) Ag-12BT-OMe. The distributions and centroids of holes and electrons are colored blue and red, respectively. | ||
The key intermediate (MAC)AgCl was prepared according to the procedures reported in the literature.38l The amide ligands of 12BT-CN and 12BT-OMe were synthesized via a two-step route including Pd-catalyzed C–N coupling and Pd-catalyzed intramolecular cyclization reactions (Scheme S1†). The target Ag(I) complexes were obtained by a nucleophilic reaction between the key intermediate of (MAC)AgCl and the respective amide ligands. 1H/13C nuclear magnetic resonance and 1H–1H COSY spectroscopy, high-resolution mass spectroscopy and elemental analysis were conducted to characterize chemical structures of the Ag(I) complexes. Furthermore, the single crystal of Ag-12BT-OMe was obtained by layering of CH2Cl2 solution with n-hexane. As depicted in Fig. S7 and Tables S2, S3,† Ag-12BT-OMe exhibited similar C–Ag and Ag–N bond lengths of 2.103(5) and 2.082(4) Å, respectively, together with a small ligand–ligand dihedral angle of ∼14.3° and a nearly linear ∠C–Ag–N angle of 171.85(17)°.42 These structure features are basically consistent with the theoretical results. Furthermore, Ag-12BT-OMe delivered the Ag⋯S distance of 3.44 Å, smaller than the sum of van der Waals radii of the Ag and S atoms (3.52 Å).43 This pinpoints the existence of secondary metal–ligand interactions between the Ag atom and amide ligand in Ag-12BT-OMe. Atom in molecular (AIM) and natural bonding orbital (NBO) calculations further verified that the nature of such secondary metal–ligand interactions is attractive noncovalent interactions between the n orbital of the sulfur atom and the s orbital of the silver nucleus (Fig. S8†).44 Such interactions can stabilize the flattened conformations of these complexes to some extent and thus can be favorable to maintain high oscillator strength and suppress excited state reorganization.41
All the complexes underwent amide-ligand attributed irreversible oxidation processes (Fig. S9†). According to the half-wave potentials (calibrated versus ferrocenium/ferrocene), the HOMO levels of these complexes gradually increased in the order of −5.64 eV (Ag-12BT-CN) < −5.44 eV (Ag-12BT) < −5.27 eV (Ag-12BT-OMe), in accordance with the simulated results. Combined with the optical bandgaps, the LUMO levels of these complexes were estimated to be from −2.84 to −2.86 eV.
As pictured in Fig. 3a, despite the similar ligand-contributed absorption below 400 nm, the LLCT absorption peaks of these complexes were gradually red-shifted, following the trend of 405 nm (Ag-12BT-CN) < 424 nm (Ag-12BT) < 430 nm (Ag-12BT-OMe). Moreover, their LLCT absorption bands had enhanced intensity and broadened width with the gradually increasing π-donating strength of the amide ligands (Fig. 3b). Accordingly, the experimental S1–S0 oscillator strength of these complexes was determined to be 0.022 for Ag-12BT-CN, 0.037 for Ag-12BT, and 0.049 for Ag-12BT-OMe. Similar to the sequence of the LLCT absorption bands, these complexes displayed tunable emission profiles spanning from 527 to 608 nm in solution and from 521 to 571 nm in thin films [30 wt% in mCP (N,N-dicarbazolyl-3,5-benzene)] (Table 1, Fig. 3a and c). To gain insight into the photophysical behaviors of these Ag(I) complexes, transient photoluminescence (PL) curves were obtained. Inspiringly, Ag-12BT and Ag-12BT-OMe delivered shorter emission lifetimes of 70 and 11 ns, respectively, compared with that of Ag-12BT-CN (210 ns). Under aerated conditions, the lifetimes of these complexes were significantly shortened, implying the participation of triplet excitons (Fig. S10†). Benefitting from the extremely short emission lifetime, Ag-12BT-OMe has a large kr constant of 9.1 × 106 s−1, among the best values for Ag(I) complexes.16–25,35,38f,l In the rigid mCP host matrix, the emission lifetimes of Ag-12BT and Ag-12BT-OMe were still as short as 330 and 144 ns, respectively, yet Ag-12BT-CN delivered a longer lifetime of 3.9 μs (Fig. 3e and Table 1). Moreover, Ag-12BT-CN and Ag-12BT delivered significantly higher ΦPL of 60% and 72% compared with that of Ag-12BT-OMe (ΦPL = 23%). This result matches well with the limited excited state reorganization of Ag-12BT-CN and Ag-12BT with respect to Ag-12BT-OMe in the above theoretical study, indicating that the ΦPL of these Ag(I) complexes are mainly influenced by nonradiative processes rather than their radiative probabilities (related to oscillator strength). In this context, Ag-12BT demonstrated the highest kr of 2.2 × 106 s−1 in the film state. Temperature-dependent lifetimes of these complexes were conducted to determine their emission origins. As illustrated in Fig. S11 and S12,† when cooled down to 77 K, all the complexes delivered nearly one order of magnitude longer average decay lifetimes than those at 300 K (Table S4†), establishing the TADF nature of these Ag(I) complexes. Moreover, the local triplet excited states (3LE) of the amide ligands are energetically higher than the CT-featured S1 states of the corresponding complexes, preventing the direct phosphorescence from the amide ligands (Fig. S2†). Subsequently, the ΔEST of these complexes was determined to be 96/80 meV for Ag-12BT-CN, 24/33 meV for Ag-12BT, and 22/28 meV for Ag-12BT-OMe, according to the Boltzmann- and Arrhenius-type fittings (Fig. 3f and S12†). In this regard, the smallest ΔEST is one of the main reasons for the shortest TADF lifetime of Ag-12BT-OMe, which originated from the longer-range LLCT (Fig. 2). To reveal the influence of the central Ag ions on the spin-flip processes between S1 and T1 states, the SOC constants between S1 and T1 were evaluated for these complexes. As listed in Table S1,† the SOC constants evolved in the order of 0.06 cm−1 (Ag-12BT-CN) < 1.53 cm−1 (Ag-12BT) < 1.60 cm−1 (Ag-12BT-OMe), basically opposite to the trend of TADF lifetimes. We also noted that this series of Ag(I) emitters exhibited smaller ΦPL and shorter emission lifetimes with respect to their Cu(I) counterpart Cu-12BT in our previous work.41 This phenomenon roots from the larger atomic radii of silver, which lead to longer metal–ligand bonds. On one hand, the longer C–Ag and Ag–N bonds enable more separated HOMO–LUMO distributions and smaller ΔEST values, thus rendering shorter emission lifetimes. On the other hand, the longer coordinate bonds reduce the steric hindrance between the carbene and amide ligands, allowing higher ligand–ligand rotation/twist freedom. This could result in higher nonradiative rates and lower ΦPL. Considering that the singlet radiative rate (kr,s) is another critical rate-determining step for the TADF process, we estimated kr,s based on a two-level Boltzmann dynamic model. As shown in Fig. S12,† Ag-12BT and Ag-12BT-OMe exhibited larger kr,s of 2.2 × 107 s−1 and 6.9 × 107 s−1 than Ag-12BT-CN (kr,s = 7.3 × 106 s−1) in the film state, which originated from their larger S1–S0 oscillator strength according to the Einstein relations of radiative lifetime (Fig. 3b).45 In this sense, the enhanced π-donating strength of the amide ligands not only reduces the ΔEST for these Ag(I) emitters but also accelerates the S1–S0 radiative rate via enhancing the oscillator strength.
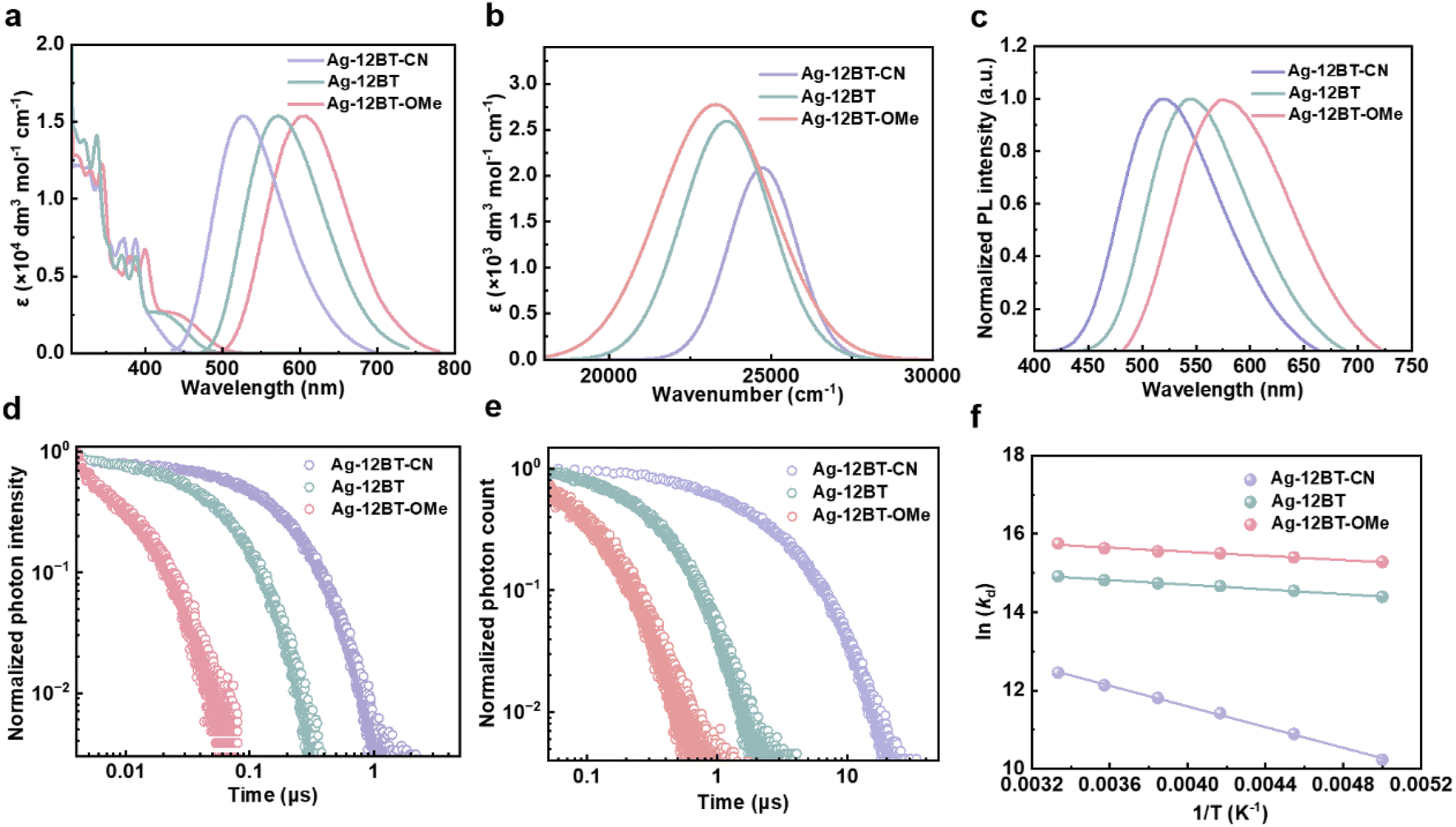 | ||
| Fig. 3 (a) UV-vis absorption and normalized fluorescence spectra of Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe in toluene solutions (10−4 M, 300 K). (b) LLCT band of Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe in toluene solutions. The spectra were extracted by the Gaussian-type peak analysis. (c) Normalized fluorescence spectra of Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe in mCP-doped films with a 30 wt% doping concentration, following excitation at 350 nm. Transient PL decay curves of Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe (d) in toluene solutions (10−4 M, 300 K) and (e) mCP-doped films with a 30 wt% doping concentration, following excitation at 375 nm. (f) Arrhenius-type fit of the temperature-dependent lifetime data (symbols) to eqn S2† (line) for the doped films of Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe. | ||
| Complex | λ PL [nm] | Φ PL [%] | τ d [μs] | k r [106 s−1] | k nr [106 s−1] | ΔESTa/b [meV] |
|---|---|---|---|---|---|---|
| a Fitted from the Boltzmann-type dynamic model. b Fitted from the Arrhenius-type dynamic model. | ||||||
| In toluene (10 −4 M) | ||||||
| Ag-12BT-CN | 527 | 25 | 0.21 | 1.2 | 3.6 | — |
| Ag-12BT | 571 | 32 | 0.070 | 4.6 | 9.8 | — |
| Ag-12BT-OMe | 608 | 10 | 0.011 | 9.1 | 91 | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| 30 wt% mCP-doped film | ||||||
| Ag-12BT-CN | 521 | 60 | 3.9 | 0.2 | 0.1 | 96/80 |
| Ag-12BT | 545 | 72 | 0.33 | 2.2 | 0.9 | 24/33 |
| Ag-12BT-OMe | 571 | 23 | 0.14 | 1.6 | 6.7 | 22/28 |
To further picture the exciton dynamics of these complexes, fs- and ns-scale transient absorption (TA) spectroscopy studies were conducted in chlorobenzene solution (2 mg mL−1). In the first 2 ps, the positive signals of photoinduced absorption (PIA) for all the complexes were significantly increased, which can be assigned to the formation of charge transfer S1 excited states. After that, the S1 conformation reorganization occurred with time constants of 1.2–6.6 ps. For Ag-12BT-CN, in the time range of 5 ps–3 ns, the amplitude of the PIA signal at 500–600 nm gradually decreased, accompanied by its increase at 600–625 nm, with a time constant of 333 ps (Fig. 4a and S13a†), which could be attributed to the ISC process. This value is consistent with the previously reported ISC rates (tens to hundreds of ps) for CMA complexes in the literature.38g–l,39,40 Comparatively, the ISC process of Ag-12BT occurred within 1 ns with a faster time constant of 186 ps (Fig. 4b and S13b†). For Ag-12BT-OMe, the PIA signals at 500–650 nm and 700–800 nm increased and dropped, respectively, within 30 ps, while all the signals dropped significantly after 30 ps. This may be caused by the PIA signal overlaps of singlet and triplet excited states. The ISC time constant of Ag-12BT-OMe was roughly determined as 147 ps according to the dynamic curves (Fig. S13c†). In the ns-scale time range, the amplitude of the PIA signals in all detecting wavelengths decreased significantly for all these complexes, indicating the decay of T1 excitons (including reverse ISC and nonradiative processes) (Fig. 4d–f and S14†). Similar to the sequence of ISC rates, the lifetimes of T1 excitons followed the trend of 200 ns (Ag-12BT-CN) > 38.9 ns (Ag-12BT) > 7.5 ns (Ag-12BT-OMe), well consistent with the emission lifetimes of these complexes recorded in transient PL in solutions (Fig. 3d and Table 1). Combined with the above PL studies, Fig. 4g–i depicts the simplified excited-state dynamics of these complexes. Basically, all the Ag(I) complexes experienced the fast ISC process with rate constants of 3.0–6.8 × 109 s−1, followed by a fast reverse ISC (RISC) process and further singlet radiation. In particular, Ag-12BT and Ag-12BT-OMe underwent the ultrafast RISC process with the rate constants of ca. 108 s−1, among one of the highest values for TADF emitters.
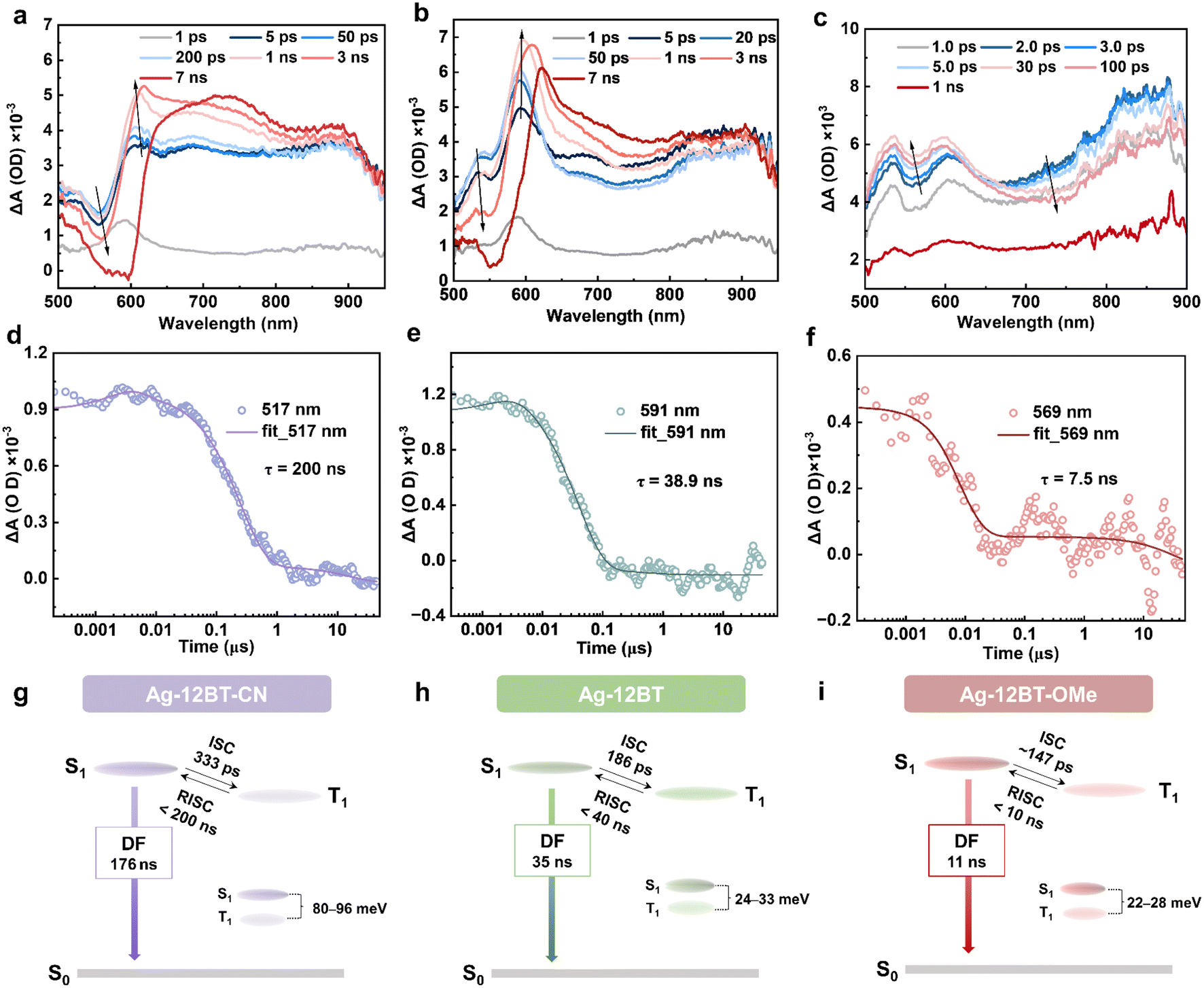 | ||
| Fig. 4 Fs-TA spectra of (a) Ag-12BT-CN, (b) Ag-12BT, and (c) Ag-12BT-OMe in chlorobenzene solution (2 mg mL−1), following excitation at 400 nm. The ns-transient kinetics of (d) Ag-12BT-CN at 819 nm, (e) Ag-12BT at 591 nm, and (f) Ag-12BT-OMe at 569 nm, following excitation at 400 nm. Schematic diagram of exciton dynamics of (g) Ag-12BT-CN, (h) Ag-12BT, and (i) Ag-12BT-OMe based on TA and transient PL experiments. | ||
In view of the outstanding optical properties with high ΦPL and short TADF lifetimes, solution-processed OLEDs were fabricated to explore the electroluminescence (EL) performance of these Ag(I) complexes. Before the device characterization, we used atomic force microscopy to study the film-forming ability of the Ag(I) emitters. The spin-coated doped films of the Ag(I) emitters afforded smooth surfaces with root-mean-square roughness values of ∼0.3 nm (Fig. S15†), which are feasible for the fabrication of solution-processed OLEDs. The device structure consisted of indium tin oxide (ITO)/poly-(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) (35 nm)/emissive layer (40 nm)/dibenzo[b,d]furan-4,6-diylbis(diphenylphosphine oxide) (PPF) (10 nm)/1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) (50 nm)/8-hydroxyquinolinolato-lithium (Liq) (1 nm)/Al (100 nm) (Fig. 5a and b). The Ag(I)-CMA complexes were doped into the typical host mCP with an optimal 30 wt% concentration. As shown in Fig. 5c and Table 2, the devices based on Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe displayed green to yellow EL emissions peaking at 512, 528, and 558 nm, along with the corresponding CIE coordinates of (0.26, 0.45), (0.33, 0.53), and (0.41, 0.53), respectively. As illustrated in Fig. 5d, all the devices had low turn-on voltages of 3.2 V, implying the balanced charge carrier injection and transport in the devices. Due to the higher ΦPL of Ag-12BT-CN and Ag-12BT, the resulting devices delivered higher EQEs of 12.8% and 16.2% at 10–15 cd m−2, respectively, compared with 4.4% EQE of the Ag-12BT-OMe-based device (Table 2). To the best of our knowledge, the EQE of 16.2% represents the state-of-the-art EL efficiency for Ag(I) emitters (excluding Ag clusters containing Au and/or Pt atoms) (Table S5†).35,36,46 Moreover, the Ag-12BT-based device afforded an alleviated efficiency roll-off value of 17.2% together with the high EQE of 13.4% at the practical luminance of 1000 cd m−2. This roots from the short emission lifetimes and ultrafast spin-flip processes of the emitter, benefiting the triplet harvesting meanwhile preventing the triplet-involved annihilation at high current densities. Interestingly, the champion emitter Ag-12BT supported its devices exhibiting remarkably high EQEs of 12.2–16.2% with the doping concentrations in the range of 20–50 wt% (Fig. S16 and Table S6†), highlighting the suppressed concentration-caused emission quenching of Ag-12BT.
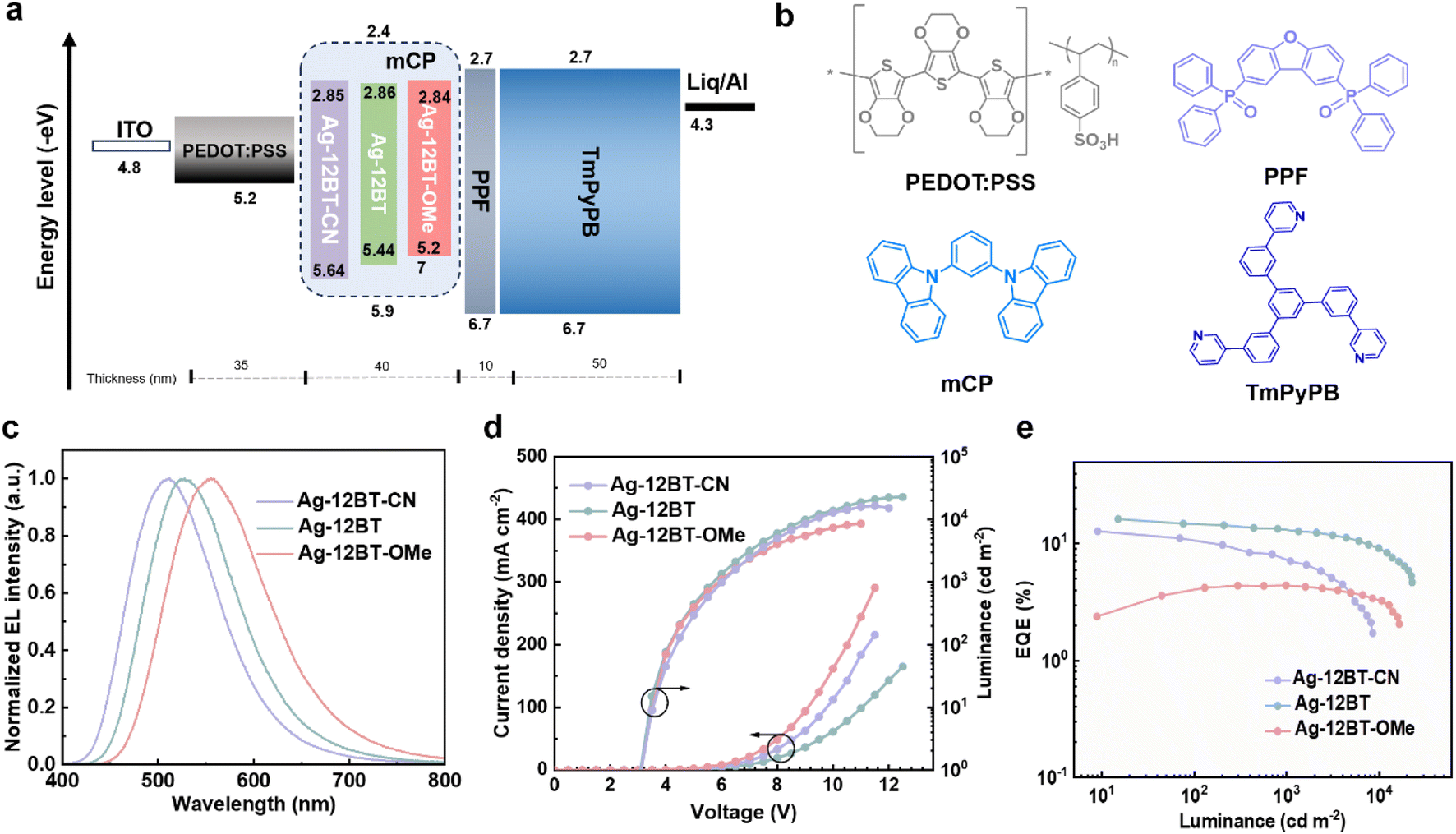 | ||
| Fig. 5 (a) Device structure of the optimal devices based on Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe. (b) Molecular structures of the functional materials used in the devices. (c) Normalized EL spectra and (d) current density–voltage–luminance curves of the devices based on Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe. (e) External quantum efficiency as a function of luminance for the devices based on Ag-12BT-CN, Ag-12BT, and Ag-12BT-OMe. | ||
| Emitter | V on [V] | CEb [cd A−1] | PEc [lm W−1] | EQEd [%] | ELpeak [nm] | CIE (x, y) |
|---|---|---|---|---|---|---|
| a The turn-on voltage recorded at a luminance of 1 cd m−2. b Current efficiency. c Power efficiency. d External quantum efficiency at the maximum, 100 and 1000 cd m−2. | ||||||
| Ag-12BT-CN | 3.2 | 35.0, 21.6, 19.0 | 31.4, 14.2, 10.0 | 12.8, 10.6, 7.1 | 512 | 0.26, 0.45 |
| Ag-12BT | 3.2 | 48.7, 44.3, 39.9 | 43.7, 34.8, 22.8 | 16.2, 14.8, 13.4 | 528 | 0.33, 0.53 |
| Ag-12BT-OMe | 3.2 | 12.8, 12.3, 12.7 | 6.5, 4.8, 3.3 | 4.4, 4.1, 4.3 | 558 | 0.41, 0.53 |
Conclusions
In summary, we have developed a series of two-coordinate Ag(I) complexes bearing the carbene-metal-amide (CMA) motif. By tuning the π-donating strength of the amide ligands, these complexes delivered tunable emission colors with a large radiative rate constant of ∼107 s−1 as well as distinct thermally activated delayed fluorescence (TADF) properties with short emission lifetimes of down to 11 ns in solution. The transient absorption spectroscopies disclosed that Ag-12BT and Ag-12BT-OMe in solution experience ultrafast spin-flip dynamics with large forward and reverse intersystem crossing (ISC) rate constants of up to ∼109 s−1 and ∼108 s−1, respectively. Inspiringly, Ag-12BT exhibits the best emission performance in the thin film state, having the high ΦPL of 72%, the short lifetime of 330 ns, and the fast radiative rate of 2.2 × 106 s−1. The resultant solution-processed OLEDs based on these complexes achieved record external quantum efficiencies of up to 16.2%, representing the state-of-the-art efficiency performance for the OLEDs based on Ag(I) complexes. This work not only unlocks the large potential of Ag(I)-CMA complexes in developing high-efficiency OLEDs but also opens a new avenue for the design of short-lived TADF materials.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
A. Y. and N. L. contributed equally to this work. S. G. conceived and designed the project. A. Y. synthesized the complexes and measured photophysical properties. N. L. and C. Y. fabricated and characterized the devices. X. C. and J. X. performed transient absorption spectroscopy. A. Y. conducted the quantum chemical calculations. S. G. supervised this research. A. Y. and S. G. co-wrote the manuscript. All the authors discussed the results and contributed to the manuscript.Conflicts of interest
S. G. and A. Y. are inventors on a patent application related to this work (CN patent application no. 202310604157.4) filed by Wuhan University. The authors declare no other competing interests.Acknowledgements
S. G. gratefully acknowledges financial support from the National Natural Science Foundation of China (52022071). The numerical calculations in this paper were done on the supercomputing system in the Supercomputing Center of Wuhan University. The authors thank Prof. Cheng Zhong (Wuhan University) for the helpful suggestion on theoretical calculations. The authors thank the Core Facility of Wuhan University for atomic force microscopy measurements.References
- (a) C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Brase, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS; (b) H. Madec, F. Figueiredo, K. Cariou, S. Roland, M. Sollogoub and G. Gasser, Chem. Sci., 2023, 14, 409–442 RSC.
- (a) Y. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2018, 375, 514–557 CrossRef CAS; (b) R.-Q. Xia, Z.-N. Liu, Y.-Y. Tang, X. Luo, R.-J. Wei, T. Wu, G.-H. Ning and D. Li, Chem. Sci., 2024, 15, 14513–14520 RSC.
- X. Li, Y. Xie and Z. Li, Chem.–Asian J., 2021, 16, 2817–2829 CrossRef CAS PubMed.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- A. Ramírez-Solís, V. Vallet, C. Teichteil, T. Leininger and J. P. Daudey, J. Chem. Phys., 2001, 115, 3201–3207 CrossRef.
- (a) R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed; (b) K. A. Spence, J. V. Chari, M. Di Niro, R. B. Susick, N. Ukwitegetse, P. I. Djurovich, M. E. Thompson and N. K. Garg, Chem. Sci., 2022, 13, 5884–5892 RSC.
- J. R. Kirchhoff, R. E. Gamache Jr, M. W. Blaskie, A. A. Del Paggio, R. K. Lengel and D. R. McMillin, Inorg. Chem., 1983, 22, 2380–2384 CrossRef CAS.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS PubMed.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed.
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS PubMed.
- A. Schinabeck, J. Chen, L. Kang, T. Teng, H. H. H. Homeier, A. F. Suleymanova, M. Z. Shafikov, R. Yu, C.-Z. Lu and H. Yersin, Chem. Mater., 2019, 31, 4392–4404 CrossRef CAS.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- C. E. Housecroft and E. C. Constable, J. Mater. Chem. C, 2022, 10, 4456–4482 RSC.
- N. Aizawa, Y. Harabuchi, S. Maeda and Y.-J. Pu, Nat. Commun., 2020, 11, 3909 CrossRef CAS.
- K. Matsumoto, T. Shindo, N. Mukasa, T. Tsukuda and T. Tsubomura, Inorg. Chem., 2010, 49, 805–814 CrossRef CAS.
- M. Osawa, I. Kawata, R. Ishii, S. Igawa, M. Hashimoto and M. Hoshino, J. Mater. Chem. C, 2013, 1, 4375 RSC.
- J. Chen, T. Teng, L. Kang, X.-L. Chen, X.-Y. Wu, R. Yu and C.-Z. Lu, Inorg. Chem., 2016, 55, 9528–9536 CrossRef CAS PubMed.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Chem. Mater., 2017, 29, 1708–1715 CrossRef CAS.
- X.-M. Gan, R. Yu, X.-L. Chen, M. Yang, L. Lin, X.-Y. Wu and C.-Z. Lu, Dalton Trans., 2018, 47, 5956–5960 RSC.
- M. Z. Shafikov, A. F. Suleymanova, A. Schinabeck and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 702–709 CrossRef CAS.
- A. V. Artem'ev, M. Z. Shafikov, A. Schinabeck, O. V. Antonova, A. S. Berezin, I. Y. Bagryanskaya, P. E. Plusnin and H. Yersin, Inorg. Chem. Front., 2019, 6, 3168–3176 RSC.
- M. Klein, N. Rau, M. Wende, J. Sundermeyer, G. Chengz, C.-M. Che, A. Schinabeck and H. Yersin, Chem. Mater., 2020, 32, 10365–10382 CrossRef CAS.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508–3535 CrossRef CAS.
- M. Z. Shafikov, R. Czerwieniec and H. Yersin, Dalton Trans., 2019, 48, 2802–2806 RSC.
- C.-W. Hsu, C.-C. Lin, M.-W. Chung, Y. Chi, G.-H. Lee, P.-T. Chou, C.-H. Chang and P.-Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085–12099 CrossRef CAS.
- F. So, C. Adachi, H. Yersin, M. J. Leitl and R. Czerwieniec, in Organic Light Emitting Materials and Devices XVIII, 2014, vol. 9183, pp. 43–53 Search PubMed.
- M. Osawa, M. Hashimoto, I. Kawata and M. Hoshino, Dalton Trans., 2017, 46, 12446–12455 RSC.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Inorg. Chem., 2017, 56, 13274–13285 CrossRef CAS PubMed.
- J.-H. Jia, D. Liang, R. Yu, X.-L. Chen, L. Meng, J.-F. Chang, J.-Z. Liao, M. Yang, X.-N. Li and C.-Z. Lu, Chem. Mater., 2019, 32, 620–629 CrossRef.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- J. Jayabharathi, V. Thanikachalam and S. Thilagavathy, Coord. Chem. Rev., 2023, 483, 215100 CrossRef CAS.
- H.-T. Mao, G.-F. Li, G.-G. Shan, X.-L. Wang and Z.-M. Su, Coord. Chem. Rev., 2020, 413, 213283 CrossRef CAS.
- A. S. Romanov, S. T. E. Jones, L. Yang, P. J. Conaghan, D. Di, M. Linnolahti, D. Credgington and M. Bochmann, Adv. Opt. Mater., 2018, 6, 1801347 CrossRef.
- T. Teng, K. Li, G. Cheng, Y. Wang, J. Wang, J. Li, C. Zhou, H. Liu, T. Zou, J. Xiong, C. Wu, H.-X. Zhang, C.-M. Che and C. Yang, Inorg. Chem., 2020, 59, 12122–12131 CrossRef CAS PubMed.
- W. Brütting, J. Frischeisen, T. D. Schmidt, B. J. Scholz and C. Mayr, Phys. Status Solidi A, 2013, 210, 44–65 CrossRef.
- (a) D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS; (b) S. Shi, M. C. Jung, C. Coburn, A. Tadle, D. Sylvinson, M. R, P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS; (c) J. Li, L. Wang, Z. Zhao, X. Li, X. Yu, P. Huo, Q. Jin, Z. Liu, Z. Bian and C. Huang, Angew. Chem., Int. Ed., 2020, 59, 8210–8217 CrossRef CAS; (d) A. S. Romanov, S. T. E. Jones, Q. Gu, P. J. Conaghan, B. H. Drummond, J. Feng, F. Chotard, L. Buizza, M. Foley, M. Linnolahti, D. Credgington and M. Bochmann, Chem. Sci., 2020, 11, 435–446 RSC; (e) T.-Y. Li, J. Schaab, P. I. Djurovich and M. E. Thompson, J. Mater. Chem. C, 2022, 10, 4674 RSC; (f) C. N. Muniz, J. Schaab, A. Razgoniaev, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2022, 144, 17916–17928 CrossRef CAS; (g) M. Gernert, L. Balles-Wolf, F. Kerner, U. Müller, A. Schmiedel, M. Holzapfel, C. M. Marian, J. Pflaum, C. Lambert and A. Steffen, J. Am. Chem. Soc., 2020, 142, 8897–8909 CrossRef CAS; (h) R. Tang, S. Xu, T.-L. Lam, G. Cheng, L. Du, Q. Wan, J. Yang, F.-F. Hung, K.-H. Low, D. L. Phillips and C.-M. Che, Angew. Chem., Int. Ed., 2022, 61, e202203982 CrossRef CAS; (i) H.-J. Wang, Y. Liu, B. Yu, S.-Q. Song, Y.-X. Zheng, K. Liu, P. Chen, H. Wang, J. Jiang and T.-Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217195 CrossRef CAS; (j) Q. Gu, F. Chotard, J. Eng, A. P. M. Reponen, I. J. Vitorica-Yrezabal, A. W. Woodward, T. J. Penfold, D. Credgington, M. Bochmann and A. S. Romanov, Chem. Mater., 2022, 34, 7526–7542 CrossRef CAS PubMed; (k) A. Ying, Y. Ai, C. Yang and S. Gong, Angew. Chem., Int. Ed., 2022, 61, e202210490 CrossRef CAS PubMed; (l) R. Hamze, S. Shi, S. C. Kapper, D. S. Muthiah Ravinson, L. Estergreen, M. C. Jung, A. C. Tadle, R. Haiges, P. I. Djurovich, J. L. Peltier, R. Jazzar, G. Bertrand, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 8616–8626 CrossRef CAS PubMed; (m) A. Ying, L. Zhan, Y. Tan, X. Cao, C. Yang and S. Gong, Sci. China: Chem., 2023, 66, 2274–2282 CrossRef CAS; (n) J. Feng, A.-P. M. Reponen, A. S. Romanov, M. Linnolahti, M. Bochmann, N. C. Greenham, T. Penfold and D. Credgington, Adv. Funct. Mater., 2021, 31, 2005438 CrossRef CAS.
- (a) A. Ying and S. Gong, Chem.–Eur. J., 2023, 29, e2023018 Search PubMed; (b) T.-Y. Li, S.-J. Zheng, P. I. Djurovich and M. E. Thompson, Chem. Rev., 2024, 124, 4332–4392 CrossRef CAS; (c) Y. Tan, A. Ying, J. Xie, G. Xie and S. Gong, Chem. Sci., 2024, 15, 11382–11390 RSC.
- D. S. M. Ravinson and M. E. Thompson, Mater. Horiz., 2020, 7, 1210–1217 RSC.
- A. Ying, Y. Tan and S. Gong, Adv. Opt. Mater., 2024, 12, 2303333 CrossRef CAS.
- A. Ying, CCDC Experimental Crystal Structure Determination, 2024, p. 2305500, DOI:10.5517/ccdc.csd.cc2hd1zy.
- A. V. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS; (b) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS; (c) M. Liu, X. Han, H. Chen, Q. Peng and H. Huang, Nat. Commun., 2023, 14, 2500 CrossRef CAS.
- R. C. Hilborn, Am. J. Phys., 1982, 50, 982–986 CrossRef CAS.
- Z. Jiao, M. Yang, J.-Y. Wang, Y.-Z. Huang, P. Xie and Z.-N. Chen, J. Mater. Chem. C, 2021, 9, 5528–5534 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04607b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |