
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient construction of high-quality sulfonated porous aromatic frameworks by optimizing the swelling state of porous structures†
Lulu
Yang‡
a,
Zhen
Zhan‡
b,
Lin
Zhao
a,
Chengxin
Zhang
ac,
Shaolei
Wang
*a,
Wei
Hu
*a and
Guangshan
Zhu
 *a
*a
aKey Laboratory of Polyoxometalate and Reticular Material Chemistry of Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, China. E-mail: wangsl030@nenu.edu.cn; huw848@nenu.edu.cn; zhugs100@nenu.edu.cn
bDepartment of Applied Physics, The Hong Kong Polytechnic University, Kowloon, 999077, Hong Kong SAR, China
cSchool of Materials Science and Engineering, Shenyang Ligong University, Shenyang, 110159, China
First published on 27th November 2024
Abstract
Conventional post-modification methods usually face the fundamental challenge of balancing the high content of functional groups and large surface area for porous organic polymers (POPs). The reason, presumably, stems from ineffective and insufficient swelling of the porous structure of POP materials, which is detrimental to mass transfer and modification of functional groups, especially with large-sized ones. It is important to note that significant differences exist in the porous structures of POP materials in a solvent-free state after thermal activation and solvent swelling state. Herein, we propose that the improvement of the swelling state of the porous structure of POP materials is more conducive to obtaining high-quality sulfonated POP materials, and employ a one-pot modification strategy for preparing sulfonated porous aromatic frameworks (PAFs) to prove the proposal. These results show that the specific surface area of the resulting polymer is 580 m2 g−1 with a sulfur content of up to 13.2 wt%, which is superior to most sulfonated porous materials and the control sample. More importantly, we have also shown that the same conclusion is reached by performing similar treatments on hyper-crosslinked polymers (HCPs) and conjugated microporous polymers (CMPs), proving that our hypothesis is effective and feasible when compared to the conventional post-sulfonation method. The excellent hydrophilicity, rich content of sulfonic acid groups, high specific surface area and hierarchical pore structure make the resulting polymer an ideal adsorbent for micro-pollutants in water. The maximum adsorption capacities for Rhodamine B (RhB), Methylene Blue (MB), Tetracycline (TC) and Ciprofloxacin (CIP) are 1075 mg g−1, 1020 mg g−1, 826 mg g−1 and 1134 mg g−1, respectively. This study not only demonstrates the preparation of efficient sulfonated porous adsorbents for the efficient removal of cationic dyes and antibiotics but also illustrates an effective method for constructing high-quality functional POP materials by optimizing the swelling state of the porous structure.
Introduction
Porous organic polymers (POPs) are highly attractive materials due to their high specific surface area, designable structure, diverse synthesis strategies and stable physical and chemical properties.1 These features make POP materials particularly beneficial for the introduction of functional groups, the exposure of more catalytic sites and the mass transfer of guest molecules. Consequently, these properties of POP materials contribute to their broad application prospects in the fields of separation and adsorption, catalysis, sensing, energy storage, energy conversion, etc.2,3 Among the functional POP materials, the modification with highly polar and large-sized sulfonic acid groups (ca. 0.45 nm) drastically alters surface properties, such as hydrophilicity and hydrophobicity, while also affecting the electronic and porous distributions within the frameworks of sulfonated POP materials.4,5 To date, sulfonated POP materials have been widely used in various fields including pollutant adsorption,6 heavy metal ion capture,7 heterogeneous catalysis,8 proton conduction,9 photocatalysis,10 battery materials,11 water vapor and ammonia adsorption,12,13 as well as microfiltration and nanofiltration membranes.14,15Under ideal conditions, a high content of sulfonic acid groups combined with a huge surface area enhances the performance of sulfonated POP materials. However, the sulfonated POP materials often display a specific pattern where they tend to exhibit either a high specific surface area with a low content of sulfonic acid groups or a low specific surface area with a high content of sulfonic acid groups. This phenomenon is reminiscent of the trade-off law of separation membranes.16–18 For example, Senker et al. reported the synthesis and post-synthetic sulfonation of a porous aromatic framework with varying degrees of sulfonation, resulting in a significant decrease in specific surface area from 1692 to 124 m2 g−1 with an increasing degree of sulfonation.19 Moreover, sulfonated POP materials obtained from in situ construction also present challenges. For instance, a sulfonated anionic covalent organic framework (TpPa-SO3H) with a surface area of 56 m2 g−1 is successfully synthesized via a Schiff-base condensation reaction comprising 1,3,5-triformylphloroglucinol (Tp) and 2,5-diaminobenzenesulfonic acid (Pa-SO3H).14 This highlights the inherent challenge in balancing porous structural properties with the incorporation of sulfonic acid groups to further improve the overall performance of sulfonated POP materials.
Unlike other functional groups, the bulky and polar nature of sulfonic acid groups can only be grafted onto the pore surface, inevitably leading to pore narrowing, clogging, and adversely affecting the mass transfer and the modification of sulfonation reagents for sulfonated POP materials.14,20,21 This limitation explains why sulfonated POP materials obtained from in situ construction or post-sulfonation show a trade-off phenomenon. Therefore, the pore structure becomes a key issue in achieving high-quality sulfonated POP materials, necessitating that POP materials as precursors should possess larger pore structures. Different from covalent organic frameworks, the formation of porous structures is not only affected by the choice and configuration of monomers, but also more influenced by the ineffective accumulation of polymer networks after solvent removal for amorphous POP materials.3,22,23 What's worse, porous structures after solvent removal often cannot be effectively and thoroughly swelled during post-modification progress for amorphous POP materials as precursors, which is usually overlooked in most post-modification methods. This detail is detrimental to the mass transfer and modification of reagents, especially those with larger functional groups, thus affecting the synthesis of high-quality functional POP materials. Notably, significant differences in the pore structure, specific surface area, and pore volume have been observed between the solvent-free state after thermal activation and the solvent-swelling state of porous structures, confirming that supercritical CO2 processing can be employed for the activation of POP materials.24,25
To address this issue, we propose that the improvement of the swelling state of the porous structure of POP materials is more conducive to obtaining high-quality sulfonated POP materials, and employ a one-pot strategy for preparing sulfonated porous aromatic frameworks (PAFs) to validate this proposal. The advantage of the one-pot strategy is that the porous structure remains in a fully swelling state before sulfonation. The results indicate that the specific surface area of the resulting polymer reaches 580 m2 g−1 with a sulfur content of up to 13.2 wt%, suggesting that each phenyl ring in the framework contains 0.89 molar multiples of sulfonic acid groups. The control sample prepared via the conventional post-sulfonation strategy exhibited a surface area of 342 m2 g−1 and a sulfur content of 10.1 wt%. More importantly, similar treatments were applied to hyper-crosslinked polymers (HCPs) and conjugated microporous polymers (CMPs), which confirmed the effectiveness and feasibility of the proposed method compared to the conventional post-sulfonation method. Moreover, the removal rates for Rhodamine B (RhB), Methylene blue (MB), Tetracycline (TC), and Ciprofloxacin (CIP) exceeded 94% within 5 s, achieving k2 values of 26.80, 25.39, 9.18, and 22.59 g mg−1 min−1 at 25 ppm, respectively. The maximum adsorption capacities for RhB, MB, TC and CIP are 1075 mg g−1, 1020 mg g−1, 826 mg g−1 and 1134 mg g−1, respectively, outperforming the performance of most reported adsorbents. This work not only prepares low-cost and efficient sulfonated porous adsorbents for the removal of cationic dyes and antibiotics, but also offers a simple and effective solution to the limitations of conventional post-modification methods. In particular, it enables the construction of high-quality functional POP materials through optimization of the swelling state of the porous structure.
Experimental section
Synthesis of PAF-215
Firstly, the dissolution system consisting of 0.1 g anhydrous FeCl3 and 10 mL 1,2-dichloroethane (DCE) was stirred vigorously for 10 min and allowed to stand for 2 h, and then 2 mL of the supernatant was collected and mixed with 6 mL DCE to obtain FeCl3 diluents. Secondly, 0.5 mL divinylbenzene (DVB), 6 mL DCE, and 0.5 mL FeCl3 diluents were sequentially added into a 25 mL round-bottom flask and stirred for 12 h, and then an additional 6 mL DCE and 0.3 g FeCl3 were added into the reaction system and continuously stirred for another 12 h to obtain a porous precursor. Subsequently, 6 mL ClSO3H was added into the reaction system in an ice water bath, followed by stirring at room temperature for 1 day to complete sulfonation. Finally, the post-sulfonation reaction was quenched with ice water, and the resulting polymer was washed with distilled water until neutral. The resulting polymer was further extracted with methanol using Soxhlet extraction for 2 days and dried under vacuum at 60 °C, yielding the final product, named PAF-215.Synthesis of the porous precursor and the control sample
Firstly, 0.5 mL DVB, 6 mL DCE, and 0.5 mL FeCl3 diluents were sequentially added into a 25 mL round-bottom flask and stirred for 12 h. An additional 6 mL DCE and 0.3 g FeCl3 were introduced into the reaction system and continuously stirred for another 12 h. The resulting precipitate was washed three times with methanol, followed by 24 h of Soxhlet extraction with methanol. The resulting product was then dried under vacuum at 60 °C for 24 h to yield a porous precursor. Subsequently, 0.2 g porous precursor and 12 mL DCE were added to a 25 mL round-bottom flask to thoroughly swell the polymer framework for 2 days. 6 mL ClSO3H was added to the system in an ice water bath, followed by stirring at room temperature for 1 day to ensure complete sulfonation. Finally, the post-sulfonation reaction was quenched in ice water, and the control sample was washed with distilled water until neutral. The control sample was subsequently extracted with methanol using a Soxhlet extractor for 2 days and dried under vacuum at 60 °C.Results and discussion
To demonstrate the feasibility of this hypothesis, the resulting polymer was synthesized via a one-pot method that involved three reactive processes of pre-crosslinking, deep crosslinking, and subsequent sulfonation. DVB was employed as a self-crosslinking monomer, while anhydrous FeCl3 and ClSO3H served as the polymerization catalyst and sulfonating reagent, respectively (Fig. 1a). Moreover, the control sample was prepared through the conventional post-sulfonation method, where the porous structure of POP materials typically underwent two processes of thermal activation under vacuum and re-swelling. The porous precursor of the resulting polymer and control sample is identical, but the porous precursor exhibits significant volumetric changes before and after thermal activation due to the collapse and accumulation of the porous structure after solvent removal (Fig. 2a). This phenomenon highlights the substantial differences in the pore structure, specific surface area, and pore volume between solvent-free and solvent-swelling states of the porous structure.24,25 Meanwhile, the re-swelling of the porous structure after thermal activation is often incomplete and inadequate during the post-modification process. These details would reduce the accessibility of reagents to the interior of pores, resulting in less efficient modification and notable differences in the overall performance of the resulting polymer and control sample.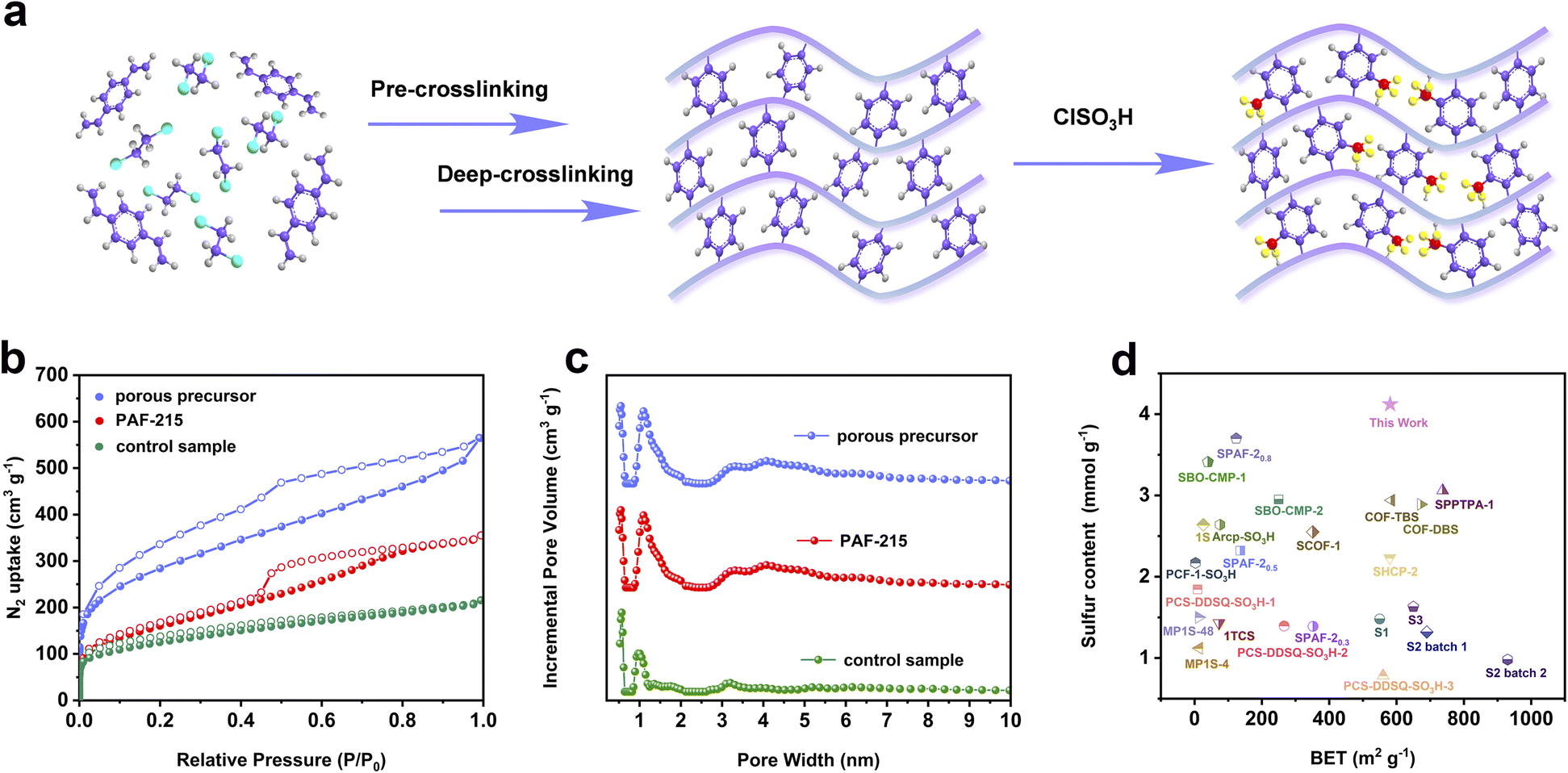 | ||
| Fig. 1 (a) Synthetic procedure of the porous aromatic framework with a high sulfonation degree by a one-pot method (grey, purple, red, and yellow represent hydrogen, carbon, sulfur and oxygen elements, respectively). (b) N2 adsorption and desorption isotherms of samples at 77.3 K. (c) Pore size distributions calculated by using density functional theory (DFT) methods (slit pore models and differential pore volumes). (d) Comparison of sulfur content with BET surface area for the resulting polymer with other sulfonated POP materials.7,10,16–19,26–32 | ||
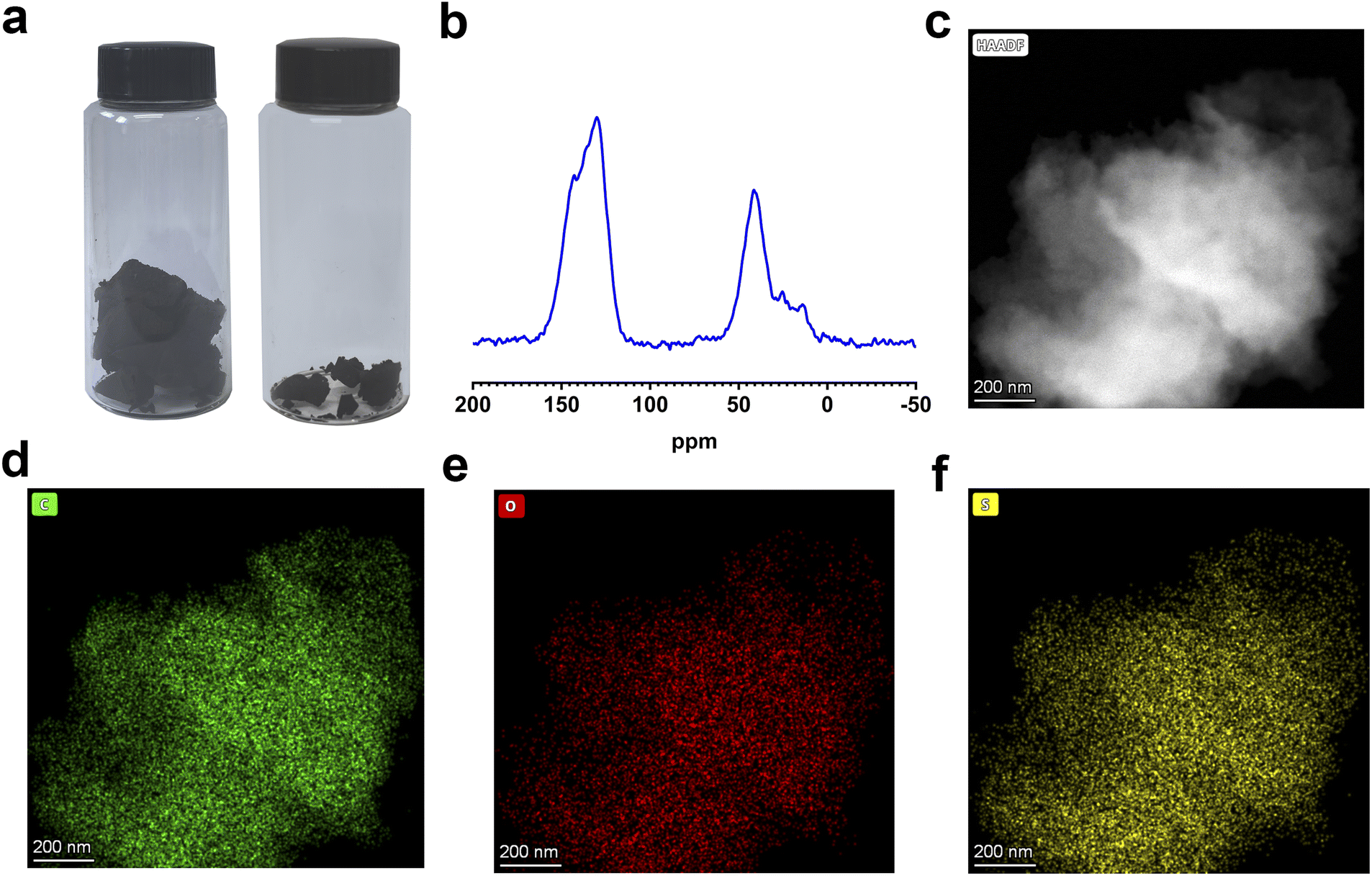 | ||
| Fig. 2 (a) Optical photos of volume change for porous precursor before (right) and after (left) thermal treatment. (b) Solid-state 13C CP-MAS NMR spectrum of the resulting polymer. (c–f) High-angle annular dark field (HAADF) mapping images of the resulting polymer. | ||
The chemical structure of the resulting polymer was characterized by Fourier transform infrared (FT-IR) spectroscopy, solid-state 13C CP/MAS nuclear magnetic resonance (NMR) spectroscopy and X-ray photoelectron spectroscopy (XPS). The peaks observed near 1620 cm−1 and 1452 cm−1 indicate the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration peaks of aromatic carbon,33 while the broad peaks at 2800–3000 cm−1 correspond to saturated C–H vibration peaks of alkyl groups. The appearance of these peaks implies the successful linkage of vinyl groups of monomers, forming polymer networks (Fig. S7†). Meanwhile, two new peaks near 1168 cm−1 and 1035 cm−1 are ascribed to the OSO asymmetric and symmetric stretching vibrations respectively,34 while other peaks at around 897 cm−1 and 615 cm−1 are attributed to S–OH bonds and C–S bonds, respectively (Fig. S7†).35 As shown in Fig. 2b, the resonance peaks near 136 ppm and 149 ppm are ascribed to aromatic carbon substituted by alkyl groups and aromatic carbon substituted by sulfonic acid groups, respectively. The resonance peak around 130 ppm is linked to unsubstituted aromatic carbon. The carbon signal of ethylene linkers appears near 41 ppm stemming from the self-crosslinking of vinyl groups.36 In addition, the C 1s, O 1s, S 2s and S 2p peaks appear near 282.4 eV, 531.7 eV, 231.4 eV and 168.2 eV, respectively (Fig. S8†). The high-resolution XPS spectrum reveals two peaks at 168.1 eV and 169.3 eV, corresponding to the OSO bonds of S 2p1/2 and S 2p3/2, respectively (Fig. S9†).35 These findings from FT-IR, XPS, and solid-state 13C NMR analysis sufficiently confirm the effectiveness of the one-pot strategy for constructing polymer networks from DVB and introducing sulfonic acid groups into the porous framework.18
C vibration peaks of aromatic carbon,33 while the broad peaks at 2800–3000 cm−1 correspond to saturated C–H vibration peaks of alkyl groups. The appearance of these peaks implies the successful linkage of vinyl groups of monomers, forming polymer networks (Fig. S7†). Meanwhile, two new peaks near 1168 cm−1 and 1035 cm−1 are ascribed to the OSO asymmetric and symmetric stretching vibrations respectively,34 while other peaks at around 897 cm−1 and 615 cm−1 are attributed to S–OH bonds and C–S bonds, respectively (Fig. S7†).35 As shown in Fig. 2b, the resonance peaks near 136 ppm and 149 ppm are ascribed to aromatic carbon substituted by alkyl groups and aromatic carbon substituted by sulfonic acid groups, respectively. The resonance peak around 130 ppm is linked to unsubstituted aromatic carbon. The carbon signal of ethylene linkers appears near 41 ppm stemming from the self-crosslinking of vinyl groups.36 In addition, the C 1s, O 1s, S 2s and S 2p peaks appear near 282.4 eV, 531.7 eV, 231.4 eV and 168.2 eV, respectively (Fig. S8†). The high-resolution XPS spectrum reveals two peaks at 168.1 eV and 169.3 eV, corresponding to the OSO bonds of S 2p1/2 and S 2p3/2, respectively (Fig. S9†).35 These findings from FT-IR, XPS, and solid-state 13C NMR analysis sufficiently confirm the effectiveness of the one-pot strategy for constructing polymer networks from DVB and introducing sulfonic acid groups into the porous framework.18
The morphology of the resulting polymer was investigated using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images confirm that the resulting polymer has an irregular and amorphous morphology with a rough surface which has been confirmed by TEM images (Fig. S10†). Besides, TEM images show that the resulting polymer possesses an amorphous structure with disordered pores. Elemental distribution mapping demonstrates that three constituent elements are uniformly distributed throughout the structure (Fig. 2c–f and S11†). The hydrophilicity of the resulting polymer was assessed through water vapor adsorption and water contact angle measurements. Notably, the resulting polymer shows strong hydrophilic properties, and water droplets can completely infiltrate and cause significant volume expansion within 0.56 s (Fig. S12†). Moreover, the water vapor adsorption capacity reaches 190 cm3 g−1 at low pressure (P/P0 = 0.3), with a maximum value of 864 cm3 g−1 at P/P0 = 1 (Fig. 3a). These results indicate that the introduction of sulfonic acid group endows the excellent wettability and hygroscopicity to the resulting polymer, making it suitable for use as an atmospheric water harvester in arid areas. Additionally, the resulting polymer shows excellent thermostability in air and nitrogen atmosphere (Fig. S13 and S14†), and the rapid decrease of the residual quantity of the resulting polymer near 400 °C could be attributed to the higher content of sulfonic acid groups compared to the control sample and porous precursor.
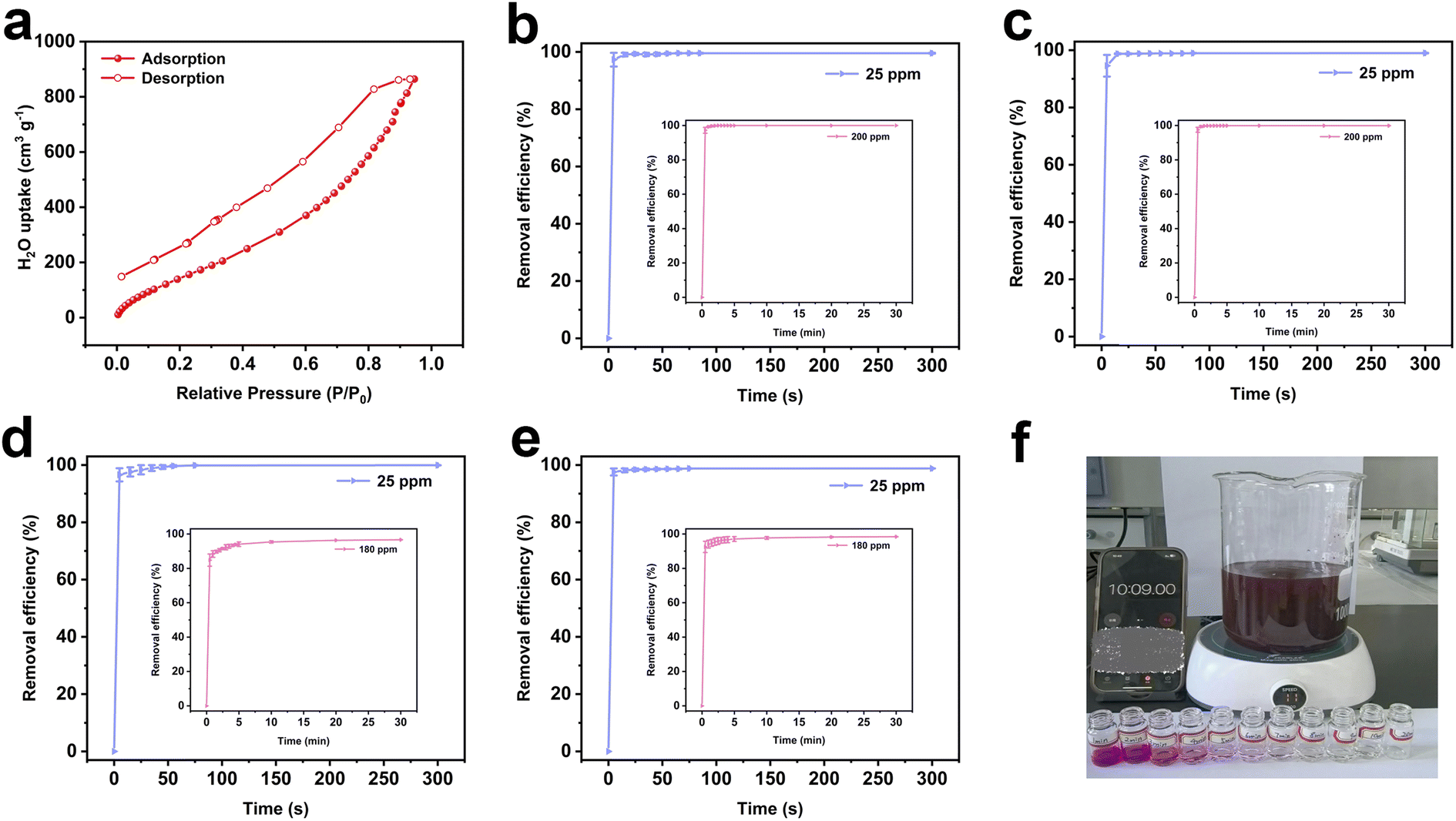 | ||
| Fig. 3 (a) Water vapor adsorption and desorption isotherms of the resulting polymer at 298 K. The time-dependent adsorption of MB (b), RhB (c), TC (d) and CIP (e) at different concentrations by the resulting polymer. (f) Optical photos of RhB removal efficiencies at different times by the resulting polymer (180 ppm). | ||
The permanent porosity of the samples was evaluated using nitrogen adsorption–desorption isotherms at 77.3 K. As shown in Fig. 1b, the rapid increase of nitrogen adsorption capacity in the low-pressure region indicates that the resulting polymer has abundant micropores and a high specific surface area based on the International Union of Pure and Applied Chemistry (IUPAC) classification. Meanwhile, the appearance of hysteresis loops and the increase of nitrogen adsorption capacity in the high-pressure region suggest that the resulting polymer possesses certain mesopores and macropores. The pore size distribution further confirms that the resulting polymer exhibits a hierarchical pore structure (Fig. 1c). The specific surface area of the resulting polymer is found to be 580 m2 g−1, with a predominant pore distribution at 1.19 nm. In contrast, the nitrogen adsorption–desorption isotherms of the control sample show a low nitrogen adsorption capacity in the low-pressure region, implying a lower surface area and micropore volume (Fig. 1b). The specific surface area and pore volume of the control sample are 342 m2 g−1 and 0.29 cm3 g−1, respectively. Thus, the introduction of sulfonic acid groups via the one-pot method does not cause significant pore blockage and an intense decrease of specific surface area compared to the control sample obtained from the conventional post-sulfonation method (Table 1).
| Samples | S BET (m2 g−1) | Pore volumeb (cm3 g−1) | Sulfur content (wt%) |
|---|---|---|---|
| a Surface area calculated from nitrogen adsorption isotherms at 77.3 K using the BET equation. b Pore volume calculated from nitrogen isotherms at 77.3 K and P/P0 = 0.995. c SKAP-1 obtained by the one-pot method. d SKAP-1 obtained by the conventional post-sulfonation method. e SCMP-1 obtained by an improved post-sulfonation strategy. f SCMP-1 obtained by the conventional post-sulfonation method. | |||
| Porous precursor | 1017 | 0.79 | 0 |
| Control sample | 342 | 0.29 | 10.1 |
| PAF-215 | 580 | 0.51 | 13.2 |
| KAP-1 | 1211 | 1.53 | 0 |
| SKAP-1c | 739 | 0.92 | 8.5 |
| SKAP-1d | 234 | 0.29 | 7.4 |
| CMP-1 | 1077 | 0.59 | 0 |
| SCMP-1e | 164 | 0.23 | 9.3 |
| SCMP-1f | 53 | 0.06 | 7.5 |
Subsequently, the sulfur content of the samples was examined using energy-dispersive X-ray spectroscopy (EDS). The analysis reveals that the sulfur content of the resulting polymer reaches 13.2 wt%, suggesting that each phenyl ring contains 0.89 molar multiples of sulfonic acid groups in the frameworks (Table 1). Conversely, the sulfur content of the control sample is noted to be only 10.1 wt%. The porosity and sulfur content of the resulting polymer are superior to those of most sulfonated POP materials obtained through the conventional post-sulfonation strategy such as SCOF-1,17 PCS-DDSQ-SO3H-2,18 and PCF-1-SO3H-1 (ref. 28) (Fig. 1d). The aforementioned study demonstrates that the proposed post-sulfonation strategy offers significant advantages over the conventional sulfonation approach. This is attributed to the different treatment strategies of two post-sulfonation methods applied to porous precursors before sulfonation. Following the conventional post-sulfonation method, the porous structure of precursors usually undergoes two treatment processes of thermal activation and re-swelling, and the re-swelling of the porous structure is often incomplete and inadequate before sulfonation. In contrast, the porous structure of precursors always remains in the solvent swelling state in the proposed post-sulfonation strategy. This is beneficial for the effective diffusion and modification of sulfonation reagents throughout the porous structure without causing pore blockage, resulting in a complete and thorough sulfonation reaction. Therefore, the resulting polymer achieves a much higher degree of sulfonation and an increased specific surface area by holding the swelling state of the porous structure.
More importantly, HCPs and CMPs, two typical classes of amorphous POP materials, were further used to validate the universality of our hypothesis. The BET surface area, pore volume and sulfur content of sulfonated KAP-1 obtained by the one-pot method are 739 m2 g−1, 0.92 cm3 g−1 and 8.5 wt%, respectively. However, the BET surface area, pore volume and sulfur content of sulfonated KAP-1 obtained by the conventional post-sulfonation method are 234 m2 g−1, 0.29 cm3 g−1 and 7.4 wt%, respectively (Fig. S15† and Table 1). Moreover, similar trends are observed for sulfonated CMP materials. Compared to CMP-1, the BET surface area and pore volume of two sulfonated CMP-1 samples decrease to varying extents, of which sulfonated CMP-1 prepared by the conventional post-sulfonation method exhibits more pronounced changes (Fig. S16† and Table 1). These results not only serve as additional evidence to confirm that the porous structure of precursors with sufficient swelling is more beneficial for post-sulfonation, but also, more importantly, highlight the excellent universality of our hypothesis for amorphous POP materials including HCPs, PAFs, CMPs etc. Drawing from previous studies, the present findings identify an effective method to prepare high-quality sulfonated POP materials by optimizing the swelling state of the porous structure, which is anticipated to be equally effective for other types of post-modification strategies for amorphous POP materials.
The as-prepared polymer exhibits excellent hydrophilicity, rich sulfonic acid groups, high specific surface area and a hierarchical pore structure, making it an ideal adsorbent for micro-pollutants found in natural water. The adsorption performance for micro-pollutants was assessed using two dyes (MB and RhB) and two antibiotics (CIP and TC) as model pollutants, long-term accumulation of which would pose severe risks to the environment and human health. The changes of UV-visible spectra at different time intervals are illustrated in Fig. S17.† Inspired by these significant advantages, the resulting polymer shows rapid adsorption of micro-pollutant models at extremely low concentrations (Fig. 3b–e and Table S1†). The removal efficiencies for MB and RhB reach 97.31% and 94.57% within 5 s, and ultimately stabilize at 99.18% and 98.85% within 45 s, respectively. The removal efficiencies for TC and CIP achieve 96.57% and 97.58% within 5 s, rapidly reaching adsorption equilibrium within 75 s. Subsequently, the adsorption performance of the resulting polymer was further investigated at high concentrations of two cationic dyes (200 ppm) and two antibiotics (180 ppm). The resulting polymer could effectively and swiftly remove MB, RhB, TC, and CIP from high-concentration solutions, achieving adsorption equilibrium within 10 min (Fig. 3b–f, Video S1 and S2†).
The fitting results derived from the adsorption kinetics curves demonstrate that the pseudo-second-order kinetic model provides a more precise description of the adsorption processes, as evidenced by the linearity and R2 values. This suggests that chemisorption plays a major role in micro-pollutant adsorption processes (Fig. S19–22, Tables S2 and S3†).37,38 Notably, the pseudo-second-order rate constants for RhB, MB, TC, and CIP are 26.80 g min−1, 25.39 g min−1, 9.18 g min−1 and 22.59 g min−1, respectively, which are much higher than those of most porous adsorbents, suggesting that the resulting polymer exhibits a faster adsorption rate (Table S4†). The adsorption process and the maximum adsorption capacity (qm) of the resulting polymer were estimated through adsorption isotherm experiments. The R2 value of the Langmuir isotherm model is higher than that of the Freundlich isotherm model (Fig. 4a–d and S23†), signifying that the adsorption process aligns more closely with the Langmuir model, characterized by monolayer and uniform adsorption behavior.39 Based on the Langmuir adsorption equation, the theoretical maximum adsorption capacities for RhB, MB, TC, and CIP are calculated to be 1075 mg g−1, 1020 mg g−1, 826 mg g−1, and 1134 mg g−1, respectively (Table S5†). It bears mentioning that these capacities surpass those of most porous micro-pollutant adsorbents such as TP-POPs-SO3H,34 CaPy,40 COF-Z2,41 and Tb@TpPa-SO3H42 (Fig. 4e and f).
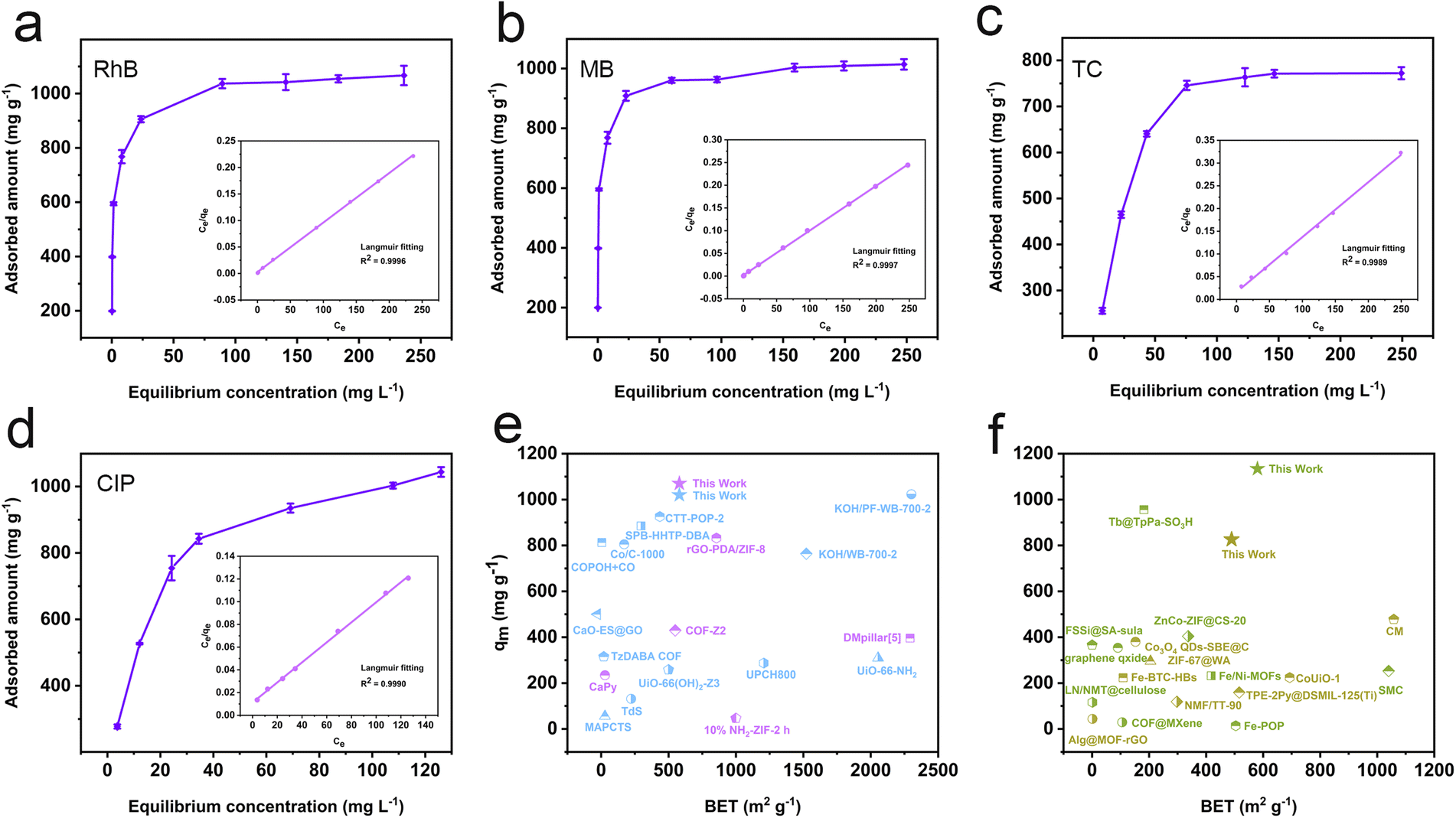 | ||
| Fig. 4 Relationship between equilibrium adsorption capacities and equilibrium concentrations and Langmuir isotherm model fitting curves of RhB (a), MB (b), TC (c) and CIP (d); (e and f) comparison of maximum adsorption capacities of RhB (purple), MB (blue), TC (yellow) and CIP (green) with BET surface area for the resulting polymer with other POP materials based on Tables S6–S9.† | ||
Given the crucial role of environmental factors, it is essential to consider the effects of pH and temperature on adsorption performance. The adsorption capacities for RhB, MB, TC, and CIP show minimal variation across a broad pH range (pH = 2–7), with optimal adsorption performance observed at a pH of 5 (Fig. S24†). The rationale lies in the fact that most sulfonic acid groups exist in their deprotonated form (–SO3−), which facilitates electrostatic interactions between negatively charged binding sites and ionized micro-pollutants.43,44 The adsorption capacities for RhB, MB, and TC increase with increasing temperature, whereas the CIP shows the opposite trend. This difference may stem from the different adsorption heats between the adsorbent and micro-pollutants during the process (Fig. S25†). Meanwhile, the resulting polymer shows high cycle stability and maintains superior adsorption performance for micro-pollutant models even after five cycles (>98%) (Fig. S26†). Moreover, no significant changes of chemical or morphological structure are found after repeated use for the resulting polymer (Fig. S27 and S28†). Collectively, these findings prove that the resulting polymer not only achieves an extremely high removal rate of micro-pollutants, but also possesses high adsorption capacity and strong interaction with these micro-pollutants.
Conclusion
To sum up, the similar results of the sulfonated PAFs, HCPs, and CMPs obtained by the two post-sulfonation methods validate our hypothesis that the optimization of the swelling state of the porous structure is more effective and feasible than the conventional post-sulfonation method. The porous structure of POP materials remains sufficiently in the swelling state, facilitating the diffusion and modification of sulfonation reagents to all parts of the porous structure without causing pore blockage. This enhances the completeness of the sulfonation reaction, making it easier to prepare high-quality sulfonated POP materials. Additionally, the resulting polymer shows excellent hydrophilicity, high content of sulfonic acid groups, large specific surface area and hierarchical pore structure, which provide abundant adsorption sites and promote efficient mass transfer, thereby resulting in excellent adsorption performance. Notably, the removal rates for RhB, MB, TC, and CIP exceed 94% within 5 s, with k2 values of 26.80 g min−1, 25.39 g min−1, 9.18 g min−1, and 22.59 g min−1, respectively. The maximum adsorption capacities for RhB, MB, TC, and CIP are noted to be 1075 mg g−1, 1020 mg g−1, 826 mg g−1, and 1134 mg g−1, respectively, outperforming those of most reported porous adsorbents. Hence, this study introduces a straightforward and viable approach to prepare sulfonated POP materials with a high degree of sulfonation and large surface area. It also identifies a universal, effective and feasible method for constructing high-quality functional POP materials by optimizing the swelling state of porous structures.Data availability
The data supporting this article have been uploaded as part of the ESI,† and are also available from the corresponding authors upon reasonable request.Author contributions
Lulu Yang: writing – original draft and conceptualization. Zhen Zhan and Lin Zhao: methodology, formal analysis and data curation. Chengxin Zhang and Wei Hu: validation, investigation, writing – review & editing. Shaolei Wang and Guangshan Zhu: resources, writing – review &editing, project administration, funding acquisition, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (No. 52003041 and 22475034) and the Fundamental Research Funds for the Central Universities (2412022QD010).Notes and references
- Q. Hao, Y. Tao, X. Ding, Y. Yang, J. Feng, R. L. Wang, X. M. Chen, G. L. Chen, X. Li, H. OuYang, X. Hu, J. Tian, B. H. Han, G. Zhu, W. Wang, F. Zhang, B. Tan, Z. T. Li, D. Wang and L. J. Wan, Porous organic polymers: a progress report in China, Sci. China:Chem., 2023, 66, 620–682 CAS.
- S. E. Neumann, J. Kwon, C. Gropp, L. Ma, R. Giovine, T. Q. Ma, N. Hanikel, K. Y. Wang, T. F. Y. Chen, S. Jagani, R. O. Ritchie, T. Xu and O. M. Yaghi, The propensity for covalent organic frameworks to template polymer entanglement, Science, 2024, 383, 1337–1343 CrossRef CAS.
- J. S. M. Lee and A. Cooper, Advances in Conjugated Microporous Polymers, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS.
- M. Furtmair, J. Timm and R. Marschall, Sulfonation of porous materials and their proton conductivity, Microporous Mesoporous Mater., 2021, 312, 21 CrossRef.
- S. S. Liu, Q. Q. Liu, S. Z. Huang, C. Zhang, X. Y. Dong and S. Q. Zang, Sulfonic and phosphonic porous solids as proton conductors, Coord. Chem. Rev., 2022, 451, 32 CrossRef.
- W. Zhao, Y. Z. Jiao, R. R. Gao, L. P. Wu, S. Y. Cheng, Q. Zhuang, A. M. Xie and W. Dong, Sulfonate-grafted conjugated microporous polymers for fast removal of cationic dyes from water, Chem. Eng. J., 2020, 391, 8 Search PubMed.
- A. M. James, S. Harding, T. Robshaw, N. Bramall, M. D. Ogden and R. Dawson, Selective Environmental Remediation of Strontium and Cesium Using Sulfonated Hyper-Cross-Linked Polymers (SHCPs), ACS Appl. Mater. Interfaces, 2019, 11, 22464–22473 CrossRef CAS PubMed.
- Y. X. Liao, L. C. Guo, L. L. Gong, Q. Y. Zhang, D. Zhao, Y. Z. Jia, R. Hua and F. Luo, Regulating Benzene Ring Number as Connector in Covalent Organic Framework for Boosting Photosynthesis of H2O2 from Seawater, Nano Lett., 2024, 24, 3819–3825 CrossRef CAS.
- O. Hubert, N. Todorovic, L. M. R. González, E. Costagliola, A. Blocher, A. Mautner, R. T. Woodward and A. Bismarck, Sulfonated hypercrosslinked polymer enhanced structural composite supercapacitors, Compos. Sci. Technol., 2023, 242, 9 CrossRef.
- C. F. Zhang, Q. Zhuang, H. Wang, X. T. Ying, R. Y. Ji, D. H. Sheng, W. Dong and A. M. Xie, Constructing an acidic microenvironment by sulfonated polymers for photocatalytic reduction of hexavalent chromium under neutral conditions, J. Colloid Interface Sci., 2023, 630, 235–248 CrossRef CAS.
- L. Cao, I. C. Chen, X. W. Liu, Z. Li, Z. Y. Zhou and Z. P. Lai, An Ionic Diode Covalent Organic Framework Membrane for Efficient Osmotic Energy Conversion, ACS Nano, 2022, 16, 18910–18920 CrossRef PubMed.
- P. Schweng, F. Mayer, D. Galehdari, K. Weiland and R. T. Woodward, A Robust and Low-Cost Sulfonated Hypercrosslinked Polymer for Atmospheric Water Harvesting, Small, 2023, 19, 8 CrossRef.
- X. Kan, Z. Q. Liu, F. J. Liu, F. Y. Li, W. Chen, X. F. Yi, A. M. Zheng, L. L. Jiang and F. S. Xiao, Sulfonated and ordered mesoporous polymers for reversible adsorption of ammonia: Elucidation of sequential pore-space diffusion, Chem. Eng. J., 2023, 451, 9 CrossRef.
- X. Liu, J. R. Wang, Y. X. Shang, C. T. Yavuz and N. M. Khashab, Ionic Covalent Organic Framework-Based Membranes for Selective and Highly Permeable Molecular Sieving, J. Am. Chem. Soc., 2024, 146, 2313–2318 CrossRef CAS PubMed.
- C. Y. Fan, L. Cao, C. Yang, Q. X. Xiao, X. D. You, X. Y. Wang, Y. Kong, H. Wu, Y. W. Liu and Z. Y. Jiang, Charged nanochannels endow COF membrane with weakly concentration-dependent methanol permeability, J. Membr. Sci., 2022, 645, 10 CrossRef.
- M. Du, A. M. Agrawal, S. Chakraborty, S. J. Garibay, R. Limvorapitux, B. Choi, S. T. Madrahimov and S. T. Nguyen, Matching the Activity of Homogeneous Sulfonic Acids: The Fructose-to-HMF Conversion Catalyzed by Hierarchically Porous Sulfonic-Acid-Functionalized Porous Organic Polymer (POPS) Catalysts, ACS Sustainable Chem. Eng., 2019, 7, 8126–8135 CrossRef CAS.
- L. P. Zhai, Y. Z. Yao, B. W. Ma, M. M. Hasan, Y. X. Han, L. W. Mi, Y. K. Nagao and Z. P. Li, Accumulation of Sulfonic Acid Groups Anchored in Covalent Organic Frameworks as an Intrinsic Proton-Conducting Electrolyte, Macromol. Rapid Commun., 2022, 43, 7 CrossRef.
- X. H. Zhao, Q. Z. Wang, R. Kunthom and H. Z. Liu, Sulfonic Acid-Grafted Hybrid Porous Polymer Based on Double-Decker Silsesquioxane as Highly Efficient Acidic Heterogeneous Catalysts for the Alcoholysis of Styrene Oxide, ACS Appl. Mater. Interfaces, 2023, 15, 6657–6665 CrossRef CAS.
- S. F. Winterstein, A. F. Privalov, C. Greve, R. Siegel, B. Pötzschner, M. Bettermann, L. Adolph, J. Timm, R. Marschall, E. A. Rössler, E. M. Herzig, M. Vogel and J. Senker, Ultrafast Proton Conduction in an Aqueous Electrolyte Confined in Adamantane-like Micropores of a Sulfonated, Aromatic Framework, J. Am. Chem. Soc., 2023, 145, 27563–27575 CrossRef CAS.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjeet, Mechanochemical Synthesis of Chemically Stable Isoreticular Covalent Organic Frameworks, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS.
- W. W. Zou, G. X. Jiang, W. F. Zhang, L. H. Zhang, Z. M. Cui, H. Y. Song, Z. X. Liang and L. Du, Hierarchically Macro-Microporous Covalent Organic Frameworks for Efficient Proton Conduction, Adv. Funct. Mater., 2023, 33, 8 Search PubMed.
- L. X. Tan and B. Tan, Hypercrosslinked porous polymer materials: design, synthesis, and applications, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- N. B. McKeown, The synthesis of polymers of intrinsic microporosity (PIMs), Sci. China:Chem., 2017, 60, 1023–1032 CrossRef CAS.
- R. K. Totten, Y. S. Kim, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, Enhanced Catalytic Activity through the Tuning of Micropore Environment and Supercritical CO2 Processing: Al (Porphyrin)-Based Porous Organic Polymers for the Degradation of a Nerve Agent Simulant, J. Am. Chem. Soc., 2013, 135, 11720–11723 CrossRef CAS.
- R. K. Totten, L. L. Olenick, Y. S. Kim, S. Chakraborty, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, A dual approach to tuning the porosity of porous organic polymers: controlling the porogen size and supercritical CO2 processing, Chem. Sci., 2014, 5, 782–787 RSC.
- D. W. Kang, K. S. Lim, K. J. Lee, J. H. Lee, W. R. Lee, J. H. Song, K. H. Yeom, J. Y. Kim and C. S. Hong, Cost-Effective, High-Performance Porous-Organic-Polymer Conductors Functionalized with Sulfonic Acid Groups by Direct Postsynthetic Substitution, Angew. Chem., Int. Ed., 2016, 55, 16123–16126 CrossRef CAS.
- Y. Zang, Y. Y. Yu, Y. L. Chen, M. Y. Fan, J. J. Wang, J. Liu, L. Xu, H. G. Jia and S. B. Dong, Synthesis of conjugated microporous polymers rich in sulfonic acid groups for the highly efficient adsorption of Cs, Chem. Eng. J., 2024, 484, 13 CrossRef.
- P. Samanta, A. V. Desai, B. Anothumakkool, M. M. Shirolkar, A. Karmakar, S. Kurungot and S. K. Ghosh, Enhanced proton conduction by post-synthetic covalent modification in a porous covalent framework, J. Mater. Chem. A, 2017, 5, 13659–13664 RSC.
- S. Mondal, J. Mondal and A. Bhaumik, Sulfonated Porous Polymeric Nanofibers as an Efficient Solid Acid Catalyst for the Production of 5-Hydroxymethylfurfural from Biomass, ChemCatChem, 2015, 7, 3570–3578 CrossRef CAS.
- D. W. Kang, M. Kang, M. Moon, H. Kim, S. Eom, J. H. Choe, W. R. Lee and C. S. Hong, PDMS-coated hypercrosslinked porous organic polymers modified Vis double postsynthetic acidifications for ammonia capture, Chem. Sci., 2018, 9, 6871–6877 RSC.
- Y. Xu, Z. W. Yu, Q. Y. Zhang and F. Luo, Sulfonic-Pendent Vinylene-Linked Covalent Organic Frameworks Enabling Benchmark Potential in Advanced Energy, Adv. Sci., 2023, 10, 10 Search PubMed.
- S. J. Yang, X. S. Ding and B. H. Han, Conjugated Microporous Polymers with Dense Sulfonic Acid Groups as Efficient Proton Conductors, Langmuir, 2018, 34, 7640–7646 CrossRef CAS.
- X. Q. He, Y. Y. Cui and C. X. Yang, Thiol-Yne Click Postsynthesis of a Sulfonate Group-Enriched Magnetic Microporous Organic Network for Efficient Extraction of Benzimidazole Fungicides, ACS Appl. Mater. Interfaces, 2021, 13, 39905–39914 CrossRef CAS.
- Y. C. Zhu, R. Ding, C. Shenghuang, X. Y. Qu, Y. Z. Yang and X. M. Zhang, Three-dimensional sulfonic-functionalized porphyrin-based porous organic polymer for high-performance methylene blue and ciprofloxacin capture, Sep. Purif. Technol., 2024, 333, 12 Search PubMed.
- Y. Z. Zhang, Y. Zhang, X. B. Luo, Q. Gao, Y. X. Liu, X. Wang, S. Y. Zhou, D. F. Wang, P. Y. Gu and Z. Y. Li, Superfast removal of dyes and herbicides with triphenylamine-based porous organic polymers by one-step sulfonation and carboxylation, Sep. Purif. Technol., 2023, 327, 11 Search PubMed.
- S. L. Wang, C. X. Zhang, Q. S. Liu and B. E. Tan, Unprecedented Processable Hypercrosslinked Polymers with Controlled Knitting, Macromol. Rapid Commun., 2022, 43, 6 CrossRef.
- Y. Xiao, J. Azaiez and J. M. Hill, Erroneous Application of Pseudo-Second-Order Adsorption Kinetics Model: Ignored Assumptions and Spurious Correlations, Ind. Eng. Chem. Res., 2018, 57, 2705–2709 CrossRef CAS.
- J. M. Liu, H. H. Cui, J. H. Li and M. C. Chen, A research on the cadmium ions adsorption of Sulfhydryl- and sulfo-functionalized UIO-66 with silica layer from water, J. Environ. Chem. Eng., 2021, 9, 11 Search PubMed.
- N. Ayawei, A. N. Ebelegi and D. Wankasi, Modelling and Interpretation of Adsorption Isotherms, J. Chem., 2017, 2017, 11 Search PubMed.
- L. C. Tian, S. Y. Zhou, J. J. Zhao, Q. F. Xu, N. J. Li, D. Y. Chen, H. Li, J. H. He and J. M. Lu, Sulfonate-modified calixarene-based porous organic polymers for electrostatic enhancement and efficient rapid removal of cationic dyes in water, J. Hazard. Mater., 2023, 441, 13 Search PubMed.
- J. Zhao, X. Y. Shen, Y. F. Liu and R. Y. Zou, (3,3)-Connected Triazine-Based Covalent Organic Frameworks for Efficient CO2 Separation over N2 and Dye Adsorption, Langmuir, 2023, 39, 16367–16373 CrossRef CAS.
- W. Jiang, W. R. Cui, R. P. Liang and J. D. Qiu, Difunctional covalent organic framework hybrid material for synergistic adsorption and selective removal of fluoroquinolone antibiotics, J. Hazard. Mater., 2021, 413, 9 Search PubMed.
- A. Y. Jia, F. Y. Zhang, Y. S. Zhao, Z. Liu, L. Xin, M. Hong and Y. X. Li, Facile fabrication of shapeable three-component hydrogen-bonded covalent-organic aerogel as permeable reactive barrier material for efficient in situ fluoroquinolone contaminated groundwater remediation, Sep. Purif. Technol., 2024, 334, 12 CrossRef.
- C. A. Igwegbe, S. N. Oba, C. O. Aniagor, A. G. Adeniyi and J. O. Ighalo, Adsorption of ciprofloxacin from water: A comprehensive review, J. Ind. Eng. Chem., 2021, 93, 57–77 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05329j |
| ‡ Lulu Yang and Zhen Zhan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |