
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCollective total synthesis of chartreusin derivatives and bioactivity investigations†
Hong-Zhou
Yi‡
ab,
Shu-Min
Liang‡
a,
Jing-Jing
Li
*c,
Hui
Liu
a,
Jin-Xi
Liao
*a,
De-Yong
Liu
a,
Qing-Ju
Zhang
a,
Ming-Zhong
Cai
 *a and
Jian-Song
Sun
*ab
*a and
Jian-Song
Sun
*ab
aNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, 99 Ziyang Avenue, Nanchang 330022, China. E-mail: sunjiansong@jiangnan.edu; mzcai@jxnu.edu.cn; jinxiliao@jxnu.edu.cn
bSchool of Life Science and Health Engineering, Jiangnan University, 1800 Lihu Avenue, Wuxi 214122, China
cAffiliated Hospital of Shandong Secondary Medicinal University, 4948 Shengli East Street, Weifang 261042, China. E-mail: li_jingjing@sdsmu.edu.cn
First published on 2nd December 2024
Abstract
Capitalizing on Hauser annulation and Yu glycosylation, the chemical synthesis of chartreusin-type aromatic polycyclic polyketide glycosides has been investigated, culminating in the successful establishment of chemical approaches toward chartreusin derivatives with intricate chemical structures but promising bioactivities. Based on the chemical synthesis strategy, the first and collective chemical syntheses of chartreusin, D329C, and elsamicins A and B have been accomplished. The chemical strategy was featured by two complementary routes to secure chartarin 10-O-monosaccharide glycosides, the key intermediates in chartreusin derivative synthesis, as well as the highly stereoselective construction of the difficult glycosidic linkages. Through the synthetic investigations, viable donors and acceptors of 3-C-methyl-branched sugars were determined for the first time. Moreover, facilitated by the established chemical synthetic strategy, the cytotoxic activities of chartreusin derivatives against human cancer cell lines were assessed and profound antineoplastic effects for chartreusin and elsamicins A and B were recorded. Based on RNA-seq analysis, the underlying working mechanisms against ES-2 cells were investigated, and the appended sugar chain-determined function mechanisms were disclosed.
Introduction
Aromatic polycyclic polyketide (APP) glycosides constitute a large family of natural products.1 Represented by doxorubicin2 and landomycins,3 APP glycosides have attracted considerable attention both from pharmacologists and from chemists due to their impressive biological activities and intricate chemical scaffolds. Led by chartreusin (Fig. 1), isolated from soil-dwelling bacterium Streptomyces chartreusis,4 a small sub-family of APP glycosides is formed (5 naturally occurring members have been isolated and characterized).5 Featured by the unusual benzonaphthopyranone aglycone with a bislactone scaffold (chartarin), the chartreusin-type APP glycosides are distinguished from other congeners. More noticeable is that chartreusin-type APP glycosides exhibit promising antitumoral activities: chartreusin remarkably inhibits the proliferation of various tumor cell lines including murine ascetic P388, L1210 leukemia, and B16 melanoma cells,6 while elsamicin A (elsamitrucin), produced by actinomycete,7 and IST-622, the prodrug of chartreusin, have entered phase II clinical trials for tumor treatment.8,9 Preliminary function mechanism investigations reveal that chartreusin-type APP glycosides exert their antineoplastic activities by DNA intercalation, which can cause radical-mediated single strand breaks of DNA, as well as topoisomerase II inhibition.10 An alternative function mechanism related to ROS generation was also disclosed recently.11 In addition, the antibiotic activities of chartreusin derivatives are also conspicuous as they can be used as lead compounds for the discovery of novel antibiotics against pathogenic microorganisms especially antibiotic-resistant bacteria.12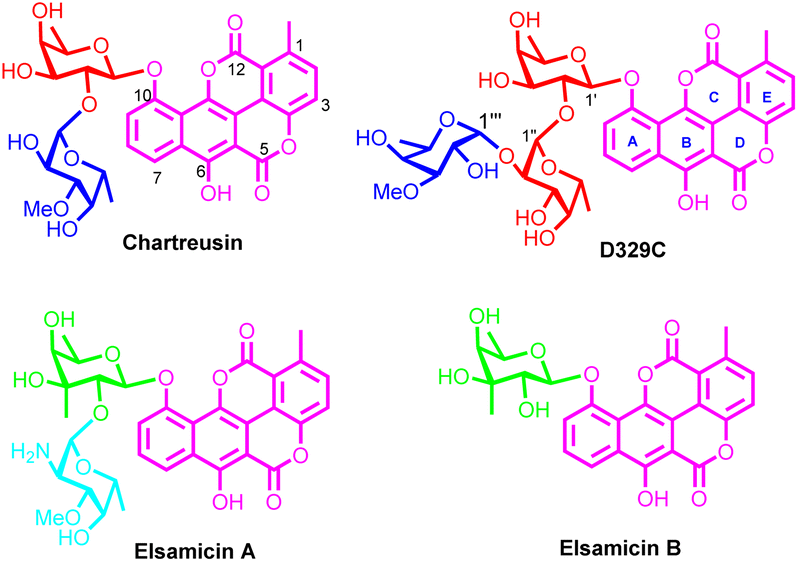 | ||
| Fig. 1 The chemical structures of chartreusin-type APP glycosides. | ||
The ideal lead compound properties have dramatically boosted the endeavors in pursuing robust strategies for easy and highly efficient access to chartreusin derivatives. Under such circumstances, biosynthesis13 and chemobiosynthesis14,15 approaches have been devised, resulting in the access of a series of analogues with modified chartarin aglycones and facilitating the preliminary structure–activity-relationship (SAR) studies. However, given the limited availability of bio-manufacturers coupled with the scalability problem, the established strategies cannot fulfil the need of acquiring diverse and ample amounts of chartreusin derivatives for chartreusin-based pharmaceutical development. Thus, alternative highly efficient approaches, chemical synthesis methods for instance, have to be built up.
Theoretically, chemical synthesis methods could tackle the shortcomings of the existing synthetic strategies. Nevertheless, the unique and intricate chemical structures make chartreusin and their analogues challenging target molecules; therefore none of them has succumbed to chemical total synthesis so far. While methods for the construction of chartarin aglycone architecture either by Hauser–Kraus annulation16,17 or by thermally induced fragmentation of cyclopropanated indanone18 already exist, the preparation of chartarin derivatives suitable for C10-OH glycosylation has not yet been reported because overcoming the unfavorable factors of chartarin as a glycosylation acceptor, such as extremely low solubility in generally applied glycosylation solvents owing to the planar pentacyclic benzonaphthopyranone skeleton,19 the electron-deficient core induced by the bislactone functionality,20 and C10-OH-involved intramolecular H-bond formation,21 is by no means a trivial problem. Meanwhile, the phenolic glycosidic linkage appending the sugar residues to the chartarin core is envisioned to be intractable since even simple phenols are inferior acceptors for glycosylation reactions,22 let alone the complex chartarin with various deleterious properties as mentioned above. Additionally, the difficulty associated with C10 phenolic glycosidic linkage formation can be further exacerbated by the appended rare sugars, especially 3-C-methyl-branched sugars embedded in elsamicins A and B, whose analogues have been proven invalid glycosyl donors in the synthesis of repeating subunits of lipopolysaccharide.23 Moreover, all intra-sugar-chain glycosidic linkages in chartreusin derivatives are 1,2-cis glycosidic linkages, which are notorious for unsatisfactory stereo-control and tedious purification processes.24 Finally, the diversity of the rare monosaccharide components of the sugar chain, including D-fucose, 3-O-methyl-D-fucose (digitalose), 2-deoxy-2-amino-D-digitalose, and 3-C-methyl-branched D-fucose, inevitably entails tedious routes for building block preparation, adding additional complexity to the chemical synthesis of chartreusin derivatives. It should be pointed out that the synthesis of 3-C-methyl-branched D-fucosyl donors has been accomplished by Magauer and co-workers.25
In line with our continuous interest in the chemical synthesis of bioactive glycoconjugates,26,27 we decided to launch a program towards chartreusin-type APP glycosides aiming to establish robust and efficient chemical synthesis approaches. The endeavor culminated in the successful establishment of the first chemical synthesis strategy to secure chartreusin derivatives, whereby the collective and highly efficient syntheses of chartreusin, D329C, and elsamicins A and B were achieved. The chemical synthesis protocols are featured on one hand by two disparate routes to secure the key chartarin 10-O-monosaccharide glycoside intermediate to exquisitely balance the flexibility and convergence of the synthetic protocols and on the other hand by good to excellent stereocontrol in challenging glycosidic linkage construction either by the Pico-involved intermolecular H-bond mediated intramolecular aglycone delivery (HAD) strategy and favorable steric effect or by Lev-mediated remote participation effect. To further corroborate the antineoplastic effects of chartreusin derivative, the cytotoxicity of the obtained compounds against a series of human cancer cell lines was also assessed and the working mechanisms against the ES-2 cell line were systematically investigated by RNA-seq analysis.
Results and discussion
Chemical synthesis investigations
Being aware of the origins of challenges in the chemical synthesis of chartreusin-type APP glycosides, we realized that the successful development of a chemical synthesis strategy would highly rely on efficient synthesis of chartarin 10-O-monosaccharide glycoside. As such, retrosynthetically, chartreusin (1), D329C (2), and elsamicins A (3) and B (4) could be traced back to monosaccharide glycoside A (Fig. 2), which in turn could be assembled by two different routes. Route a, involving Hauser–Kraus annulation between glycosylated phthalide B and coumarin C, was characterized by arranging the construction of the challenging phenolic glycosidic linkage prior to the fabrication of the complete chartarin aglycone core, so as to bypass the direct construction of the thorny chartarin 10-O-glycosidic linkage and improve the flexibility of the synthetic approach as well. Meanwhile, encouraged by the significant advances in carbohydrate chemistry,28 direct glycosylation of chartarin acceptor E with monosaccharide Yu donor D would also be explored in order to enhance the convergence of the synthetic method (route b). The choice of the Hauser–Kraus reaction for aglycone skeleton formation and the Yu donor29 for glycosidic linkage construction is due to the mild reaction conditions and fully testified synthetic potential.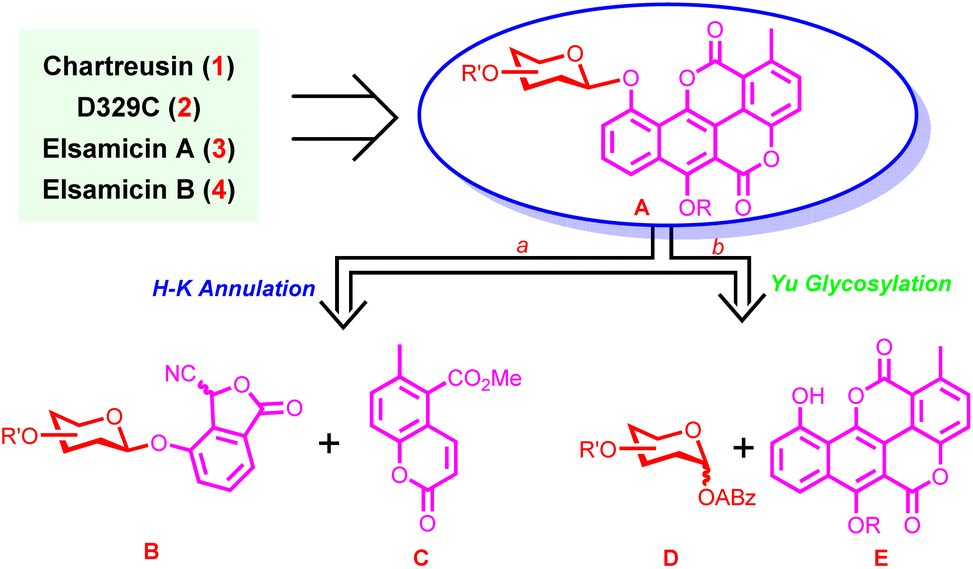 | ||
| Fig. 2 Retrosynthetic analysis. | ||
Route a with chartarin 10-O-glycosidic linkage construction prior to the fabrication of the complete aglycone skeleton was exploited first.30 The endeavor commenced with the synthesis of glycosylated phthalide intermediates (Scheme 1A). Condensation between peracetylated β-galactoside 5 with 2-methyl-3-methoxycarbonyl phenol 6 under the effect of BF3·Et2O delivered phenol glycoside 7 (95%), which was then converted to 8via a sequence of deacetylation, acetonide formation, open-chain ketal cleavage, and Appel reaction (89%, 4 steps). Under radical conditions, iodide 8 proceeded deiodination smoothly to generate phenyl D-fucoside 9 (93%). Selective bromination of the methyl group attached to the phenyl ring was subsequently carried out under UV irradiation. Although mild conditions were adopted (0 °C), the isopropylidenyl protecting group in 9 was partially cleaved. Thus, re-isopropylidenation was needed to afford bromide 10 (82%, 2 steps). Afterwards, intermediate 10 was subjected successively to modified Kornblum oxidation,31 Et3N-catalyzed O-trimethylsilyl cyanohydrin formation,32 and acid-mediated lactonization,33 to furnish fucosylated phthalide 11 (75%, 2 steps) as an inseparable mixture of diastereomers.
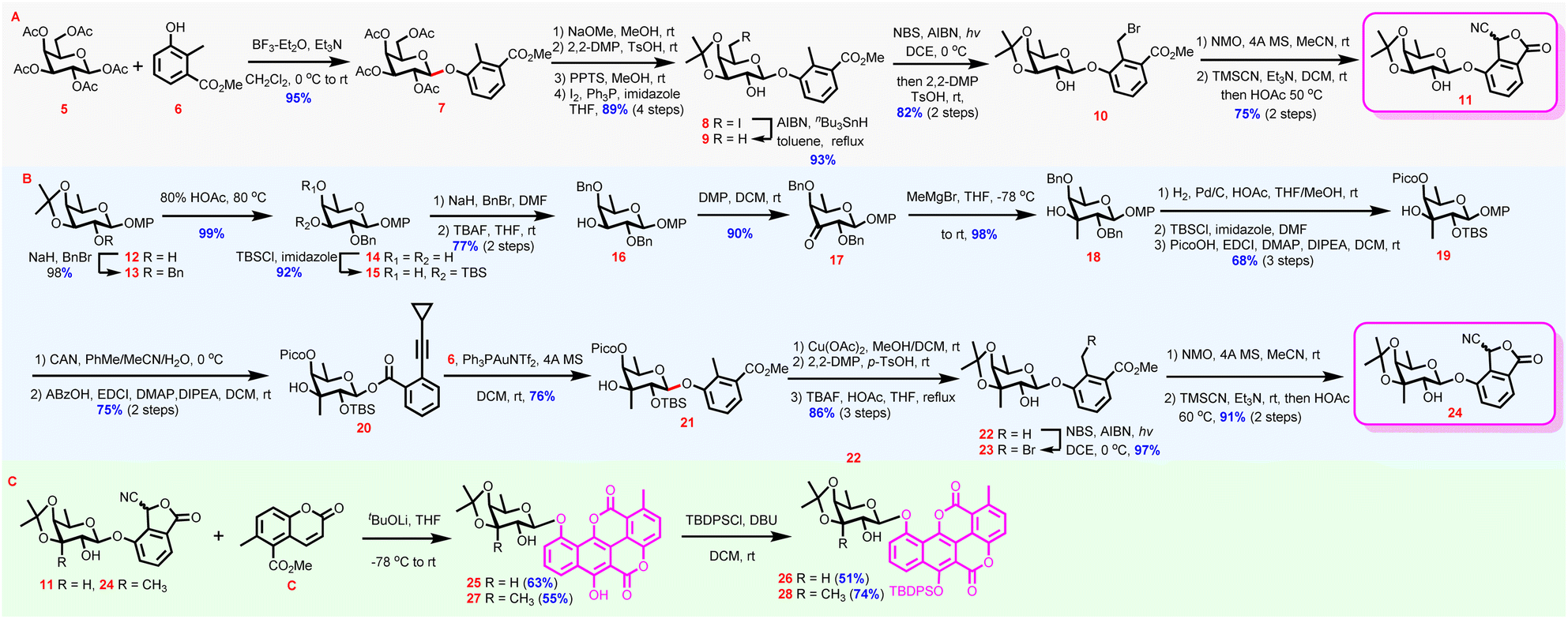 | ||
| Scheme 1 Synthesis of chartarin 10-O-monosaccharide glycosides 26 and 28via route a. | ||
Synthesis of phthalide with 3-C-methyl-branched fucosyl residue started with fucosyl derivative 12 (Scheme 1B), which was benzylated to provide 13 (98%). Acid-mediated de-isopropylidenation converted 13 to diol 14 (99%), whose equatorial C3-OH was then selectively blocked as a silyl ether to afford 15 (92%). After the remaining axial C4-OH was protected with a benzyl group, the temporary C3-OTBS was removed, delivering the alcohol intermediate 16 (77%, 2 steps) primed for 3-C-methyl branch installation. Oxidation of 16 was followed by methyl Grignard reagent addition to the resulting ketone group to provide tertiary alcohol 18via17 (88%, 2 steps). The addition of methyl Grignard reagent to the C3-ketone of 17 took place axially disposing the nascent tertiary OH in the equatorial position exclusively, which was assigned by the NOE experiment.34 Hydrogenolysis of 18 to cleave the two benzyl groups coupled with regioselective C2-OH silylation and C4-OH picoloylation furnished 19 (68%, 3 steps). The installation of TBS and Pico groups at C2 and C4 OHs, respectively, was aimed at steering the ensuing glycosylation to proceed β-stereoselectively both by steric hindrance (from the TBS group) and by the HAD strategy (from the Pico group).35 Oxidative cleavage of the anomeric p-methoxyphenyl group yielded the hemiacetal intermediate, which was further subjected to o-cyclopropylethynylbenzoylation under dehydrative conditions to generate Yu donor 20 β-stereoselectively (75%, 2 steps). In sharp contrast to the results from the Kim group,23 and, pleasantly, the coupling between 6 and the elaborately designed Yu donor 20 proceeded smoothly under the effect of Au(I) complex providing 21 stereoselectively and efficiently (76%, for the anomeric proton: 5.00 ppm, d, J = 8.0 Hz).36 It should be pointed out that although the neighboring-group participation effect has been widely accepted as a reliable means for stereoselective 1,2-trans glycosidic linkage construction, the robust effect proved invalid in 3-C-methyl fucosyl donors, as the o-cyclopropylethynylbenzoate or trifluoroacitimidate37 donors protected in 2-O-acyl-3,4-O-isopropylidenyl pattern afforded α glycosylation products predominantly, if not exclusively, although the α-glycosides suffered from severe 1,3-syn-diaxial repulsion. Moreover, the 1,2-trans-steering power of the C2-O-positioned 2,2-dimethyl-2-(o-nitrophenyl)acetyl (DMNPA) group that proved to exhibit reinforced anchimeric effect by a dual participation mechanism38 was demonstrated, even weaker than that of the TBS group relying solely on the bulkiness effect (Table S1 in the ESI†).34 The crowded α-space imposed by the axial C3-branched methyl group, which seriously destabilized the 1,2-dioxonium intermediate required for 2-O-acyl participation,39 might be invoked to account for the high difficulty in 1,2-trans-glycosylation of C3-methyl-branched fucosyl donors, which was further aggravated by the axially oriented C4-OH. After completing the formation of the challenging glycosidic linkage, now the stage was set for the construction of the cyanophthalide moiety. Before this, protecting group adjustments were conducted via a sequence of Pico group removal, actonization, and desilylation to give 22 (86%, 3 steps). Eventually, following a similar sequence to that used for the synthesis of 11 from 9, 22 was converted to cyanophthalide 24via intermediate 23 uneventfully (88%, 3 steps).
Coumarin C was efficiently obtained on a gram-scale by slightly modifying Mal's procedure.16,34 With phthalides 11 and 24 as well as coumarin C in hand, the pivotal Hauser–Kraus annulation was examined (Scheme 1C). Despite the high application frequency of the Hauser–Kraus reaction in total synthesis, annulations entailing complex glycosylated partners have only been reported once.40 After extensive trials (Table S2 in the ESI†), it was revealed that the annulation reaction was highly steric hindrance sensitive, and MOM or TBS protected 11 only gave low yields of the desired annulation products. With 11 as a substrate the optimal result was obtained when tBuOLi (1.1 eq) was selected as the base (25, 63%).34 A large excess of tBuOLi or prolonged reaction time was detrimental to the desired annulation. LiHMDS could also promote the annulation reaction but gave much inferior yield in comparison to tBuOLi, presumably because the free OHs in 11 and 24 may cause transesterification side reactions under strong basic conditions. Under the optimized conditions, the more challenging annulation between 24 and C proceeded smoothly, presenting 27 in a respectable 55% yield. Selective protection of the C6-OHs of 11 and 24 with the TBDPS group afforded 26 and 28, thus achieving the successful synthesis of chartarin 10-O-monosaccharide glycosides, the key intermediate for the chemical synthesis of chartreusin derivatives (51% and 74% yield, respectively).
Encouraged by the successful synthesis of 26 and 28via route a, synthesis of the key chartarin 10-O-monosaccharide glycoside intermediates via route b featuring direct glycosylation of the stubborn chartarin-derivative acceptors was attempted subsequently. Preparation of chartarin derivatives with ameliorated acceptor properties was explored first (Scheme 2). With 29 as the starting material, silylation of C6-OH was conducted under the effect of DBU/TBDPSCl to yield 30 (91%). The choice of TBDPS as the protecting group for C6-OH was based on the assumption that its electron-donating capability could ameliorate the electron-deficient nature of the dilactonized benzonaphthopyranone core of chartarin meanwhile, the considerable bulkiness of TBDPS was expected to distort the planarity of the aglycone and accordingly enhance the solubility in glycosylation media. After C10-OMOM cleavage,41 chartarin acceptor 31 was secured (95%, Scheme 2A). Indeed, 31 possesses reasonable solubility in dichloromethane, the preferred solvent for glycosylation reactions. With appropriate acceptor being ready, a series of Yu donors in different protecting patterns were designed, synthesized, and examined considering that chartarin-type acceptors are extremely resistant to stereoselective and efficient glycosylation. Eventually, it turned out that fucosyl o-cyclopropylethynylbenzoate 34 and 3-C-methyl-branched fucosyl o-cyclopropylethynylbenzoate 40 were viable donors for acceptor 31 (Scheme 2B). Synthesis of donor 34 commenced with regioselective benzylation of 14via the in situ generated stannylene acetal to give 32 (94%),42 whose remaining free OH was then blocked with a picoloyl group to afford 33 (97%). Then swap of the anomeric MP with an o-cyclopropylethynylbenzoyl group was achieved by oxidative MP removal and dehydrative esterification with ABzOH, delivering β-34 exclusively (60%, 2 steps, Scheme 2B). With 35 as the starting material, successive benzylation and acid-mediated deisopropylidenation delivered diol 37via36 (62%, 2 steps). The diol 37 was then subjected to Bu2SnO-mediated regioselective benzylation of the tertiary C3-OH to afford 38 (78%) at elevated temperature, which was then equipped with a picoloyl group under dehydrative conditions to give 39 (93%). Following the sequence of anomeric hydroxyl group release under oxidative conditions and dehydrative esterification, 39 was converted to donor 40 β-stereoselectively (51%, 2 steps). In combination with β-20, the β-stereoselective formation of 34 and 40 from fucosyl lactols bearing the C4-OH appended Pico group is noteworthy, since stereoselective derivatization of glycosyl lactols, especially esterification43 and glycosylation44 to furnish glycosyl esters and trehalose derivatives, is extremely difficult owing to the easy epimerization. For fucosyl lactols with 4-OPico groups, the exclusive β-stereoselectivity in esterification to form Yu donors presumably could be attributed to the intramolecular H-bond between anomeric OH and the C4-OH appended Pico group which forces the anomeric OH to orientate cis to the C4-OH. As Pico groups were installed in donors 34 and 40, the ensuing glycosylations were expected to proceed smoothly and stereoselectively by virtue of the HAD strategy. Indeed, under the catalysis of Ph3PAuOTf,45 the couplings of 31 with 34 and 40 proceeded smoothly, furnishing 41 and 42 in yields of 52% and 66%, respectively, with moderate to good β-stereoselectivity (41: α/β = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8 42: α/β = 1:6.7, Scheme 2C). The β-stereoselectivity was determined from 1H NMR spectra (anomeric protons: 5.46 ppm, d, J = 7.6 Hz for 41β 5.53 ppm, d, J = 7.8 Hz for 42β). Surprisingly, although 34 and 40 could couple with 31, donors with varied protection patterns including 20 failed to afford the desired glycosylation product, further verifying the inertness of chartarin derivatives as glycosylation acceptors (Table S3 in the ESI†).34 Also, the glycosylation results clearly imply that C4-OPico and C3-OBn are crucial for successful glycosylation of 31 and the less favorable steric hindrance from C2-OBn should be responsible for the compromised stereoselectivity of the glycosylation.
:2.8 42: α/β = 1:6.7, Scheme 2C). The β-stereoselectivity was determined from 1H NMR spectra (anomeric protons: 5.46 ppm, d, J = 7.6 Hz for 41β 5.53 ppm, d, J = 7.8 Hz for 42β). Surprisingly, although 34 and 40 could couple with 31, donors with varied protection patterns including 20 failed to afford the desired glycosylation product, further verifying the inertness of chartarin derivatives as glycosylation acceptors (Table S3 in the ESI†).34 Also, the glycosylation results clearly imply that C4-OPico and C3-OBn are crucial for successful glycosylation of 31 and the less favorable steric hindrance from C2-OBn should be responsible for the compromised stereoselectivity of the glycosylation.
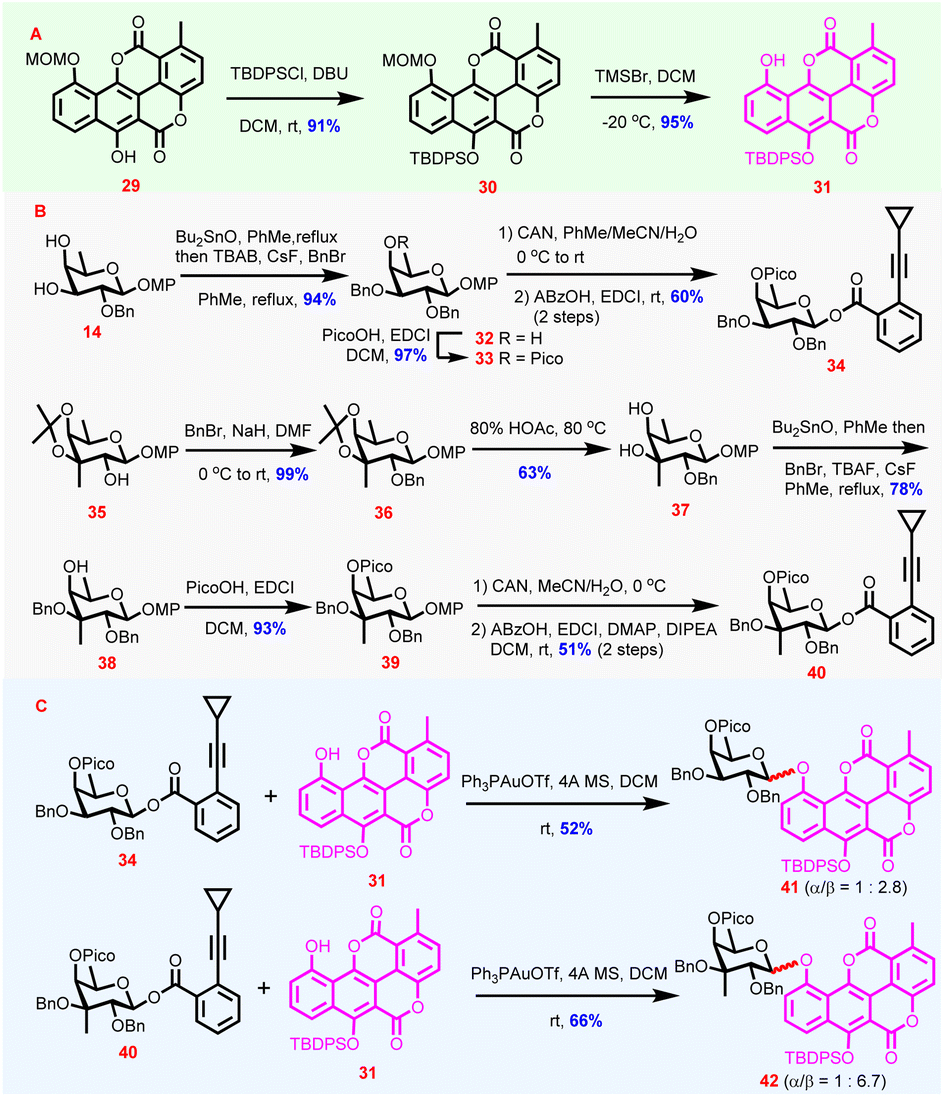 | ||
| Scheme 2 Synthesis of chartarin 10-O-monosaccharide glycosides 41 and 42via route b. | ||
Hitherto, the syntheses of chartarin 10-O-monosaccharide glycosides, the key intermediates for the chemical synthesis of chartreusin derivatives, have been accomplished via two different routes. In the first route (route a), the subunit of chartarin aglycone was used as the glycosylation acceptor lowering the difficulty in forging the pivotal and challenging glycosidic linkage between chartarin aglycone and C10-OH-appended sugar residue and guaranteeing the flexibility of the synthetic protocol. In the second route (route b), the chartarin derivative was used directly as the acceptor, ensuring the convergence and conciseness of the synthetic approach.
The successful access to 26, 28, 41, 42 laid a firm foundation for achieving the first total synthesis of chartreusin derivatives. Thus, syntheses of chartreusin 1, D329C 2, and elsamicins A 3 and B 4 were subsequently investigated. The sugar chains of chartreusin derivatives are distinguished by 1,2-connected 1,2-cis intra-sugar-chain glycosidic linkages. Thus, glycosyl donors have to be well designed and meanwhile a linear tactic will be adopted during the elongation of the sugar chain to ensure the desired 1,2-cis-stereoselectivity.
As the levulinoyl group has been demonstrated to be powerful in stereocontrol of glycosylation reactions via the remote participation effect,46D-fucosyl donors carrying the C4-O-levulinoyl group were designed to fulfil 1,2-cis-glycosidic linkage construction (Scheme 3). Synthesis of the digitalosyl donor commenced with 14, which was subjected successively to stannylene acetal-involved regioselective methylation and levulinoylation to provide 44 through intermediate 43 (82%, 2 steps). Compound 44 was then transformed to donor 45a through oxidative cleavage of the anomeric MP and the following dehydrative esterification of the resulting hemiacetal (63%, 2 steps, Scheme 3A). Similarly, after levulinoylation and anomeric substituent manipulations, 14 was converted to donor 45bvia46 (66%, 3 steps, Scheme 3B). Synthesis of 2-deoxy-2-azido-digitalosyl donor 45c from 12 required azido group installation first, which was achieved by C2-OH inversion, activation by triflate formation, and substitution with TMSN3 (Scheme 3C). The C2-OH inversion was accomplished by oxidation under Ley conditions (47, 82%)47 and reduction to provide 48 (88%). After being triflated, the C2-OTf in 48 was subsequently substituted with TMSN3 to finish azido group installation (49, 90%, 2 steps). Intermediate 49 then underwent deacetonation (50), regioselective methylation (51), and levulinoylation to provide 52 (97%, 3 steps). Likewise, the desired donor 45c was obtained smoothly from 52 as the sole β-isomer following the oxidative MP cleavage and dehydrative acylation sequence (67%, 2 steps).
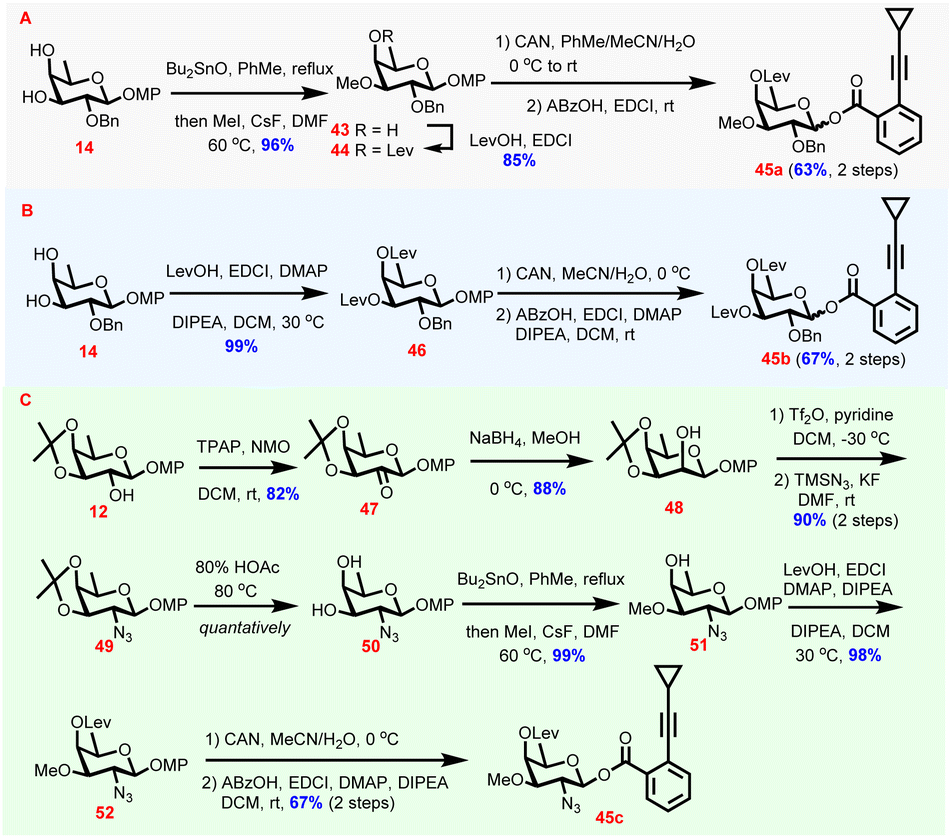 | ||
| Scheme 3 Preparation of monosaccharide donors 45a–c required for the synthesis of naturally occurring chartreusin derivatives. | ||
With 45a–c as well as chartarin 10-O-monosaccharide glycosides in hand, now the stage was set for the completion of the synthesis of the four target molecules (Scheme 4). Expectedly, with 45a as the donor and 26 as the acceptor, the condensation proceeded uneventfully under the catalysis of Ph3PAuOTf to afford disaccharide glycoside 53 (88%) α-stereoselectively. The configuration of the newly forged glycosidic linkage was assigned according to the corresponding anomeric proton (6.15 ppm, d, J = 2.8 Hz). Desilylation and cleavage of the levulinoyl group yielded intermediate 54 (88%, 2 steps), which was further exposed to debenzylation/deactonation conditions to provide chartreusin 1 (76%, 2 steps). In fact, chartarin monosaccharide glycoside 25 could also serve as a viable acceptor to couple with 45b, affording 55α (89%) exclusively, as determined by the anomeric proton of the newly incorporated sugar residue (6.14 ppm, d, J = 4.0 Hz). After blocking the phenolic C6-OH with a Lev group (56, 75%), the benzyl group in 56 was cleaved to deliver disaccharide acceptor 57 (82%), primed for further sugar chain elongation. Again, glycosylation of 57 with 45a proceeded α-stereoselectively to furnish the fully protected D329C 58 (92%), as verified by H′′′-1 which resides at 5.14 ppm (d, J = 3.6 Hz). Global deprotection of 58 was accomplished by debenzylation, deisopropylidenation, and delevulinoylation to provide D329C 2 (88%, 3 steps), the most complex member of chartreusin-type APP glycosides known thus far.48
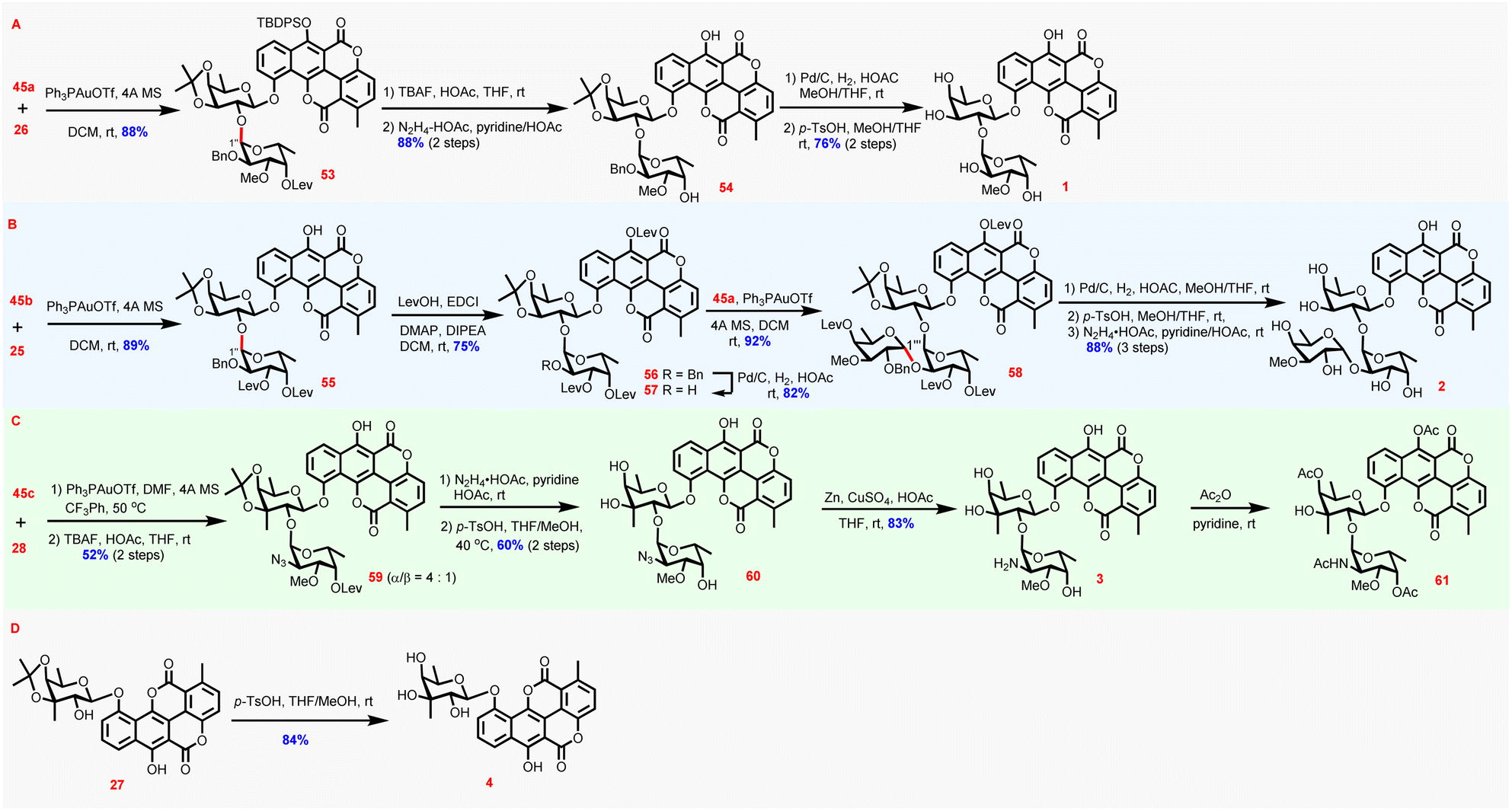 | ||
| Scheme 4 Completion of the synthesis of the target molecules 1–4. | ||
Synthesis of elsamicin A entailed glycosylation of 2-OH of 3-C-methyl-branched fucoside, which might be problematic because of the severe steric hindrance. Indeed, the glycosylation of 28 with 45c suffered from compromised stereoselectivity and efficiency even under the optimized conditions [DMF as additive49 in CF3Ph at 50 °C],34 and partial desilylation was also observed (Table S4 in the ESI†). Thus, the glycosylation step and the following desilylation step were conducted continuously without isolation of the glycosylation product intermediate to afford 59 as an easily separable mixture of α/β isomers favoring the desired α isomer (α/β = 4:1) in 52% yield. 59α was then subjected to delevulinoylation and deisopropylidenation to provide 60 (60%) ready for azido reduction. Surprisingly, the reduction of the azido group proved to be much more difficult than what was anticipated, and conventional reduction conditions including Staudinger reduction, Pd/C-mediated hydrogenation, Raney Ni-promoted reduction, and propanedithiol reduction could not afford the desired amine product 3 (Table S5 in the ESI†).34 Eventually, it was found that the combination of Zn/CuSO4/HOAc could efficiently effect the reduction, thus affording elsamicin A 3 successfully (83%). For thorough chemical structure determination by comparison with reported data, 3 was acetylated to produce 61 (85%).50 Finally, synthesis of elsamicin B 4 was accomplished by deisopropylidenation of 27 (84%). The NMR spectra of all synthesized compounds 1–4 were in good accordance with those reported in the literature (for elsamicin 3 comparison of the data of 61 with those in the literature), confirming the correctness of the chemical structures of 1–4 obtained by the novel chemical synthesis strategy.
With commercially available 5 as the starting material, the syntheses of chartreusin 1 and D329C 2 have been accomplished in 9% and 14% overall yields through 15- and 17-step longest linear sequences (LLSs). Similarly, starting with easily available 12, elsamicins A 3 and B 4 were secured in 2% and 8% overall yields through 27- and 21-step LLSs, respectively. The successful synthesis of 1–4 ends the era without chemical synthesis routes to chartareusin derivatives.
Bioactivity investigations
With 1–4 in hand, the cytotoxic activities against a series of human cancer cell lines including HCT116, BxPC3, T47D, and ES-2 were assessed (Table 1). Both elsamicins A (3) and B (4) showed profound cytotoxic effects against all four human tumor cell lines with the corresponding IC50 values being less than 31.0 μM. While the prohibitory effects of chartreusin (1) against HCT116, BxPC3, and ES-2 were also significant (IC50 below 13 μM), the corresponding effect against T47D was less effective. Even with the most complex chemical structure, D329C (2) displayed negligible cytotoxic effects against all tested cell lines.| Tumor cellsb | IC50 [μM] | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| a The CCK-8 method was used to determine the IC50 values. b HCT116 = human colon cancer cell line, BxPC3 = human pancreatic cancer cell line, T47D = human bread cancer cell line, ES-2 = human ovarian cancer cell line. c No cytotoxic activity was detected. (All the cell lines were purchased from American Type Culture Collection (ATCC) and tested negative for mycoplasma contamination). | ||||
| HCT116 | 12.93 | —c | 6.99 | 6.55 |
| BxPC3 | 3.35 | 751.32 | 4.04 | 4.28 |
| T47D | 911.20 | —c | 4.72 | 15.05 |
| ES-2 | 5.70 | —c | 1.00 | 30.99 |
The clinical application of the existing chartreusin derivatives has been hampered by unfavorable pharmacokinetics due to the rapid biliary excretion and slow gastrointestinal absorption.51 However, various available drug administration manners for clinical treatment of human ovarian cancers, such as intraperitoneal infusion, may largely solve the problems encountered for the target molecules administrated by i.v. infusion, potentially opening an alternative way for clinical use of the obtained chartreusin derivatives. Thus the impressive prohibitory effects of 1, 3 and 4 against ES-2 deserve further investigations to decipher the underlying functional mechanism. As such, RNA sequencing analysis of chartreusine derivative-treated ES-2 cells was performed to globally reveal the transcriptional profile alternations, based on which the underlying functional mechanisms were deduced (Fig. 3). The heatmap showed that chartreusin derivatives 1, 3 and 4 altered the globally transcriptional profiles of ES-2 dramatically, with differentially expressed genes (DEGs) being 380 for 1 and 1186 and 1867 for 3 and 4, respectively (Fig. 3A).34 Encouraged by these results, the most significant pathway enrichments by the Kyto Encyclopedia of Genes and Genomes (KEGG) analysis were conducted to shed light on the underlying functional mechanisms (Fig. 3B–D). For chartreusin 1, the downregulated oxidative phosphorylation (OXPHOS) pathway was enriched, which is indispensable for the survival of certain types of cancer cells.52 Although differing only by the ending sugar residue, the cytotoxic effects of elsamicin A were attributed primarily to motor proteins, growth hormone synthesis/secretion/action, and homologous recombination pathways, while the Hippo signaling pathway was enriched for elsamicin B to exert inhibitory effects against ES-2 cells. The lowered levels of motor proteins and growth hormones would inevitably result in the decelerated tumor cell proliferation,53 whereas the decreased homologous recombination would lead to DNA lesions, including double strand breaks (DSBs) and interstrand cross-links (ICLs),54 all of which aligns well with the obtained cytotoxic results. Moreover, the Hippo signaling pathway has been proven to contribute greatly to cancer growth and metastasis.55 Interestingly, the data clearly showed that although sharing the common aglycone, the underlying enriched pathways for compounds 1, 3, and 4 varied dramatically, demonstrating the crucial roles played by the appended sugar chains. Furthermore, the sugar-chain dependent working mechanisms of chartreusin derivatives afford the promise of developing chartreusin-type antineoplastic agents which could overcome the potentially developed drug resistance mutually.
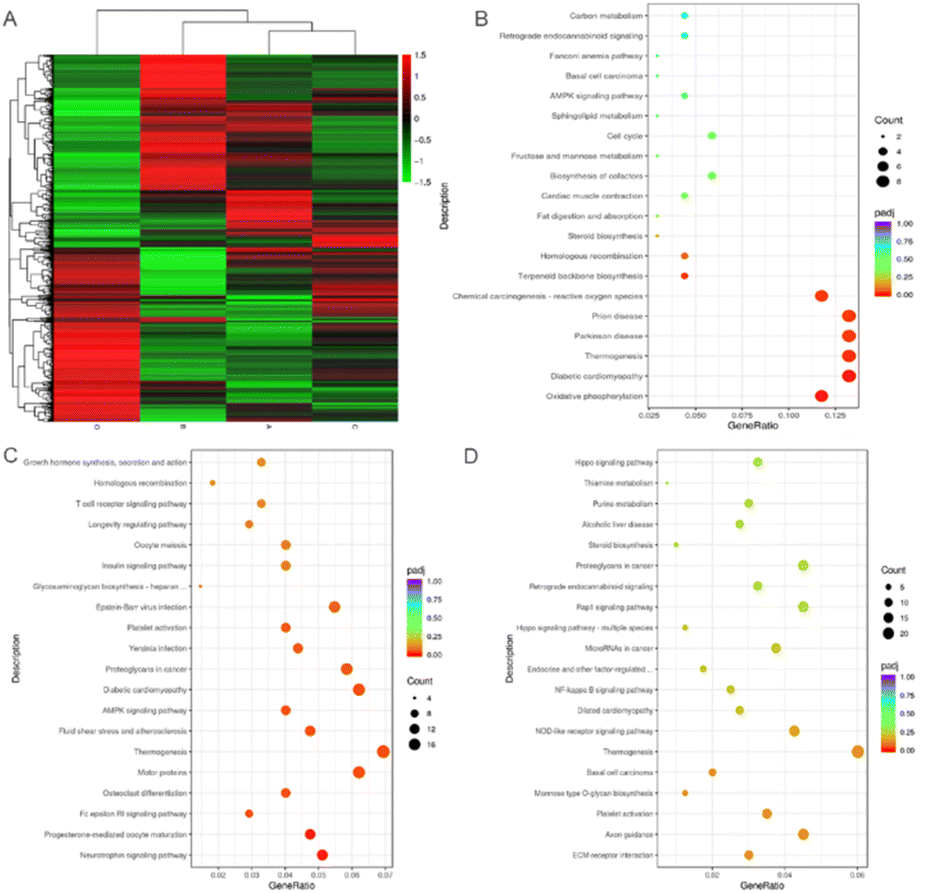 | ||
| Fig. 3 RNA-sequencing analysis. (A) Heat map of differentially expressed genes: red, upregulated; green, downregulated. (B) KEGG enrichment analysis of downregulated genes after 1 (5.70 μM) treatment (C) KEGG enrichment analysis of downregulated genes after 3 (1.00 μM) treatment. (D) KEGG enrichment analysis of downregulated genes after 4 (30.99 μM) treatment. | ||
Conclusions
In summary, an efficient strategy for the chemical preparation of chartreusin-type APP glycosides has been established, whereby the first and collective chemical syntheses of chartreusin, D329C, and elsamicins A and B have been successfully achieved. To juggle the flexibility and convergence of the chemical synthetic strategy, two complementary routes to chartreusin 10-O-monosaccharide glycosides, the key intermediate in the chemical synthesis of chartreusin derivatives, have been disclosed, differing in the time when the pivotal and challenging glycosidic linkage between the sugar chain and chartarin 10-OH was forged. In the first route (route a) the key glycosidic linkage was constructed before the chartarin skeleton was fabricated, thus bypassing the direct forging of the notorious chartarin 10-O-glycosidic linkages and improving the flexibility of the whole synthetic approach. In the second route (route b), the construction of the key glycosidic linkage was postponed after the chartarin core was built, thus enhancing the convergence and conciseness of the overall synthetic approach. Hauser annulation was applied to forge the chartarin core owing to the mildness and high efficiency while the Yu glycosylation protocol was adopted to forge all glycosidic linkages, including the pivotal and challenging glycosidic linkage between chartarin and the sugar chain and intra-sugar-chain glycosidic linkages. The Pico-involved HAD strategy and favorable steric hindrance from the 2-OH protecting groups of donors were used to steer 1,2-trans-glycosylation whereas the stereoselective construction of 1,2-cis-glycosidic linkages was realized by the efficacious remote participation effect of the levulinonyl group. Through the synthetic investigations, viable donors and acceptors derived from 3-C-methyl branched sugars were determined for the first time, which may find broad applications in the synthesis of challenging but bioactive glycosides containing 3-C-methyl-branched monosaccharide moieties. Moreover, fueled by the efficiency of the chemical synthesis strategy, the cytotoxic effects of the obtained chartreusin derivatives against human cancer cell lines HCT116, BxPC3, T47D, and ES-2 were assessed, and excellent bioactivities especially for elsamicins A and B were recorded. Based on RNA-seq analysis, the function mechanisms of chartreusin as well as elsamicins A and B against ES-2 cells were investigated, and the appended sugar chain-determined working mechanisms were disclosed.The establishment of the chemical synthetic strategy will dramatically ease the availability of chartreusin-type APP glycosides, thus accordingly accelerating the identification of chartreusin derivatives with dramatically improved drugability. The synthesis of chartreusin analogues to form a compound library and bioactivity studies are underway in our lab and the corresponding results will be reported in the near future.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Design of the research and experiment: J.-S. S.; synthetic work: H.-Z. Y. and S.-M. L.; bioactivity investigation: J.-J. L.; NMR investigation: D.-Y. L.; supervision: J.-S. S., H. L., J.-X. L., Q.-J. Z., M.-Z. C.; writing – original draft: J.-S. S., J.-X. L., and H.-Z. Y.; writing – review & editing: J.-S. S., J.-X. L., and H.-Z. Y.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22177042, 82104289, and 22377038). The generous support from the Beijing Municipal Commission of Science and Technology (L212015) was also highly appreciated.Notes and references
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 RSC.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS.
- R. T. Crow, B. Rosenbaum, R. Smith III, Y. Guo, K. S. Ramos and G. A. Sulikowski, Bioorg. Med. Chem. Lett., 1999, 9, 1663–1666 CrossRef CAS.
- B. E. Leach, K. M. Calhoun, L. E. Johnson, C. M. Teeters and W. G. Jackson, J. Am. Chem. Soc., 1953, 75, 4011–4012 CrossRef CAS.
- C. D. Donner, Nat. Prod. Rep., 2015, 32, 578–604 RSC.
- J. P. McGovren, G. L. Neil, S. L. Crampton, M. I. Robinson and J. D. Douros, Cancer Res., 1977, 37, 1666–1672 CAS.
- M. Konishi, K. Sugawara, F. Kofu, Y. Nishiyama, K. Tomita, T. Miyaki and H. Kawaguchi, J. Antibiot., 1986, 39, 784–791 CrossRef CAS.
- G. Asai, N. Yamamoto, M. Toi, E. Shin, K. Nishiyama, T. Sekine, Y. Nomura, S. Takashima, M. Kimura and T. Tominaga, Cancer Chemother. Pharmacol., 2002, 49, 468–472 CrossRef CAS.
- J. Verweij, J. Wanders, A. L. Nielsen, N. Pavlidis, F. Calabresi, W. ten Bokkel Huinink, U. Bruntsch, M. Piccart, H. Franklin and S. B. Kaye, Ann. Oncol., 1994, 5, 375–376 CrossRef CAS PubMed.
- F. Barcelo and J. Portugal, FEBS Lett., 1991, 292, 223–228 CrossRef.
- A. Domenech-Carbo, G. Cebrian-Torrejon, N. Montoya, N. Ueberschaar, M. T. Scotti and C. Hertweck, RSC Adv., 2017, 7, 45200–45210 RSC.
- H. Uchida, Y. Nakakita, N. Enoki, N. Abe, T. Nakamura and M. Munekata, J. Antibiot., 1994, 47, 648–654 CrossRef CAS.
- H. Uchida, Y. Nakakita, N. Enoki, N. Abe, T. Nakamura and M. Munekata, J. Antibiot., 1994, 47, 655–667 CrossRef CAS PubMed.
- N. Ueberschaar, H. M. Dahse, T. Bretschneider and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 6185–6189 CrossRef CAS.
- N. Ueberschaar, Z. Xu, K. Scherlach, M. Metsa-Ketela, T. Bretschneider, H.-M. Dahse, H. Gorls and C. Hertweck, J. Am. Chem. Soc., 2013, 135, 17408–17416 CrossRef CAS.
- D. Mal, A. Patra and H. Roy, Tetrahedron Lett., 2004, 45, 7895–7898 CrossRef CAS.
- X. Huang, T. Zhu, Z. Huang, Y. Zhang, Z. Jin, G. Zanoni and Y. R. Chi, Org. Lett., 2017, 19, 6188–6191 CrossRef CAS.
- T. A. Unzner, A. S. Grossmann and T. Magauer, Angew. Chem., Int. Ed., 2016, 55, 9763–9767 CrossRef CAS.
- N. Ueberschaar, F. Meyer, H.-M. Dahse and C. Hertweck, Chem. Commun., 2016, 52, 4894–4897 RSC.
- R. Kobayashi, K. Hanaya, M. Shoji, S. Ohba and T. Sugai, Biosci., Biotechnol., Biochem., 2013, 77, 810–813 CrossRef CAS.
- J. Yu, J. Sun, Y. Niu, R. Li, J. Liao, F. Zhang and B. Yu, Chem. Sci., 2013, 4, 3899–3905 RSC.
- M. Jacobsson, J. Malmberg and U. Ellervik, Carbohydr. Res., 2006, 341, 1266–1281 CrossRef CAS PubMed.
- D. B. Fulse, H. B. Jeon and K. S. Kim, J. Org. Chem., 2007, 72, 9963–9972 CrossRef CAS.
- S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC.
- L. Roder, S. T. Venegas, K. Wurst and T. Magauer, Tetrahedron Lett., 2024, 140, 155041 CrossRef.
- T. Liu, J. Liao, Y. Hu, Y. Tu and J. Sun, J. Org. Chem., 2017, 82, 4170–4178 CrossRef CAS PubMed.
- Z. Qiao, H. Liu, J. Sui, J. Liao, Y. Tu, R. R. Schmidt and J. Sun, J. Org. Chem., 2018, 83, 11480–11492 CrossRef CAS PubMed.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed.
- B. Yu, J. Sun and X. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed.
- W. P. Griffith, M. Jolliffe, S. V. Ley, K. F. Springhorn and P. D. Tiffin, Synth. Commun., 1992, 22, 1967–1971 CrossRef CAS.
- A. Baeza, C. Najera, M. de G. Retamosa and J. M. Sansano, Synthesis, 2005, 2787–2797 CAS.
- K. C. Nicolaou, D. Das, Y. Lu, S. Rout, E. N. Pitsinos, J. Lyssikatos, A. Schammel, J. Sandoval, M. Hammond, M. Aujay and J. Gavrilyuk, J. Am. Chem. Soc., 2020, 142, 2549–2561 CrossRef CAS PubMed.
- See (ESI).†.
- J. P. Yasomanee and A. V. Demchenko, J. Am. Chem. Soc., 2012, 134, 20097–20102 CrossRef CAS.
- 3-C-Branched donors have been used in C-glycosides synthesis but only in low to moderate yields, see: F. Wu, J. Zhang, F. Song, S. Wang, H. Guo, Q. Wei, H. Dai, X. Chen, X. Xiao, X. Liu, L. Zhang, J.-Q. Yu and X. Lei, ACS Cent. Sci., 2020, 6, 928–938 CrossRef CAS.
- B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC.
- H. Liu, T. Hansen, S. Zhou, G. Wen, X. Liu, Q. Zhang, J. D. C. Codee, R. R. Schmidt and J. Sun, Org. Lett., 2019, 21, 8713–8717 CrossRef CAS.
- S. Son, Y. Byun and N. Sakairi, Org. Lett., 2019, 21, 9368–9371 CrossRef CAS PubMed.
- S. Charkraborty and D. Mal, J. Org. Chem., 2018, 83, 1328–1339 CrossRef PubMed.
- S. Hanessian, D. Delore and Y. Dufresne, Tetrahedron Lett., 1984, 25, 2515–2518 CrossRef CAS.
- S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS.
- Z. Liu, D. Liu, D. Zhu and B. Yu, Org. Lett., 2023, 25, 5372–5377 CrossRef CAS PubMed.
- K. C. Nicolaou, F. L. van Delft, S. R. Conley, H. J. Mitchell, Z. Jin and R. M. Rodriguez, J. Am. Chem. Soc., 1997, 119, 9057–9058 CrossRef CAS.
- H. Liu, J. Liao, Y. Hu, Y. Tu and J. Sun, Org. Lett., 2016, 18, 1294–1297 CrossRef CAS PubMed.
- Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Angew. Chem., Int. Ed., 2021, 60, 12597–12606 CrossRef CAS PubMed.
- S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639–666 CrossRef.
- H. Uchida, Y. Nakakita, N. Enoki, N. Abe, T. Nakamura and M. Munekata, J. Antibiot., 1993, 46, 1611–1615 CrossRef CAS.
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS.
- K. Sugawara, M. Tsunakawa, M. Konishi, H. Kawaguchi, B. Krishnan, C. H. He and J. Clardy, J. Org. Chem., 1987, 52, 996–1001 CrossRef CAS.
- J. Rohr and C. Hertweck, in Natural products structural diversity-I secondary metabolites: organization and biosynthesis, Comprehensive natural products II: Chemistry and Biology, ed. Liu H.-W. and Monder L., Elsevier Ltd, 2010, pp. 228–293 Search PubMed.
- Y. Xu, D. Xue, A. Bankhead III and N. Neamati, J. Med. Chem., 2020, 63, 14276–14307 CrossRef CAS PubMed.
- K. G. Reddie, D. R. Roberts and T. M. Dore, J. Med. Chem., 2006, 49, 4857–4860 CrossRef CAS PubMed.
- F. Huang, O. M. Mazina, I. J. Zentner, S. Cocklin and A. V. Mazin, J. Med. Chem., 2012, 55, 3011–3020 CrossRef CAS.
- K. Wang, F. Ma, S. Arai, Y. Wang, A. Varkaris, L. Poluben, O. Voznesensky, F. Xie, X. Zhang, X. Yuan and S. P. Balk, Cancer Res., 2023, 83, 1016–1030 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05629a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |