
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic polymerization of vinyl ethers using trifluoromethyl sulfonate/solvent/ligand to access well-controlled poly(vinyl ether)s†
Liangyu
Chen
,
Zhihao
Wang
,
En
Fang
,
Zhiqiang
Fan
 and
Shaofei
Song
*
and
Shaofei
Song
*
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: songsf@zju.edu.cn
First published on 9th December 2024
Abstract
Cationic polymerization of vinyl ethers to access poly(vinyl ether) polymeric materials has been challenging due to stringent polymerization conditions and inevitable chain transfer. Herein we introduce a protocol using trifluoromethyl sulfonates to catalyze the polymerization of a series of vinyl ethers. These trifluoromethyl sulfonates are fully commercially available and can be stored under ambient conditions. Solvents and ligands have profound influences on the polymerization process, and poly(vinyl ether)s with different molecular weights, molecular weight distributions, and tacticities were obtained. A few combinations of trifluoromethyl sulfonate/solvents/O^O type ligands were explored. They showed high activities and afforded poly(vinyl ether)s with well-controlled tacticity, of which the isotacticity can be up to 81% m. Poly(vinyl ether)s with high tacticities exhibit crystallization behaviors with melting points. We also probed the cationic reversible addition–fragmentation chain transfer (RAFT) polymerization of ethyl vinyl ether employing a RAFT chain transfer agent. Low molecular weight distributions (Đs) around 1.1 can be realized. Since trifluoromethyl sulfonates can be fed at a remarkably low catalyst loading and other chemicals are cheap and easily available, the poly(vinyl ether) polymeric materials are promisingly prepared on a large scale.
1. Introduction
Cationic polymerization using vinyl ether monomers to access poly(vinyl ether)s has exhibited many attributes.1–6 For example, it can well control the molecular weights similar to radical polymerization or anionic polymerization of vinyl monomers. The shortcomings, however, come from the poorly controlled chain propagation process due to the high reactivity of the active cationic species, and chain transfer to monomers, solvents or impurities easily occurs, which would lower the molecular weights and complicate chain structures.1 For the earlier strategies to suppress undesirable chain transfer during the propagation, people conducted cationic polymerization at quite low temperatures, which can stabilize the active cationic species. Living cationic polymerization of vinyl monomers was discovered for vinyl ether and isobutene in the 1980s.7,8 Since then, scientists developed approaches to prepare polymers with higher molecular weights (Scheme 1A).9–44 To provide more alternative protocols to acquire poly(vinyl ether)s with narrow molecular weight distributions, Kamigaito and the group recently developed the cationic reversible addition–fragmentation chain transfer (RAFT) polymerization strategy.18 Based on the high reactivity between carbocationic species and sulfur atoms, they employed a series of thiocarbonylthio compounds to conduct the conventional cationic polymerizations of iso-butyl vinyl ether (iBVE) in the presence of a small amount of a strong Lewis acid. They revealed that cationic polymerization proceeded through reversible chain transfer of the propagating carbocationic species to the dormant thioester bond. Then living polymerization to afford polymers and block copolymers with well-controlled molecular weights and low dispersity was realized. Fors and coworkers reported another metal-free catalyst, pentacarbomethoxycyclopentadiene (PCCP) (Scheme 1B).24 This electron-deficient cyclopentadiene has a quite low pKa and is widely used in small molecules transformations. Fors and coworkers employed it as single-component initiating organic acid and a series of vinyl ethers were polymerized in a controlled manner under ambient conditions. The tight ion complex formed between the PCCP anion and the oxocarbenium ion chain end was the critical factor preventing the chain-transfer reaction and allowing for living polymerization. Consequently, poly(vinyl ether)s with narrow distributions and molecular weights which were well consistent with theoretical values were made. Moreover, this catalyst promisingly enabled scale-up preparation of poly(vinyl ether) materials.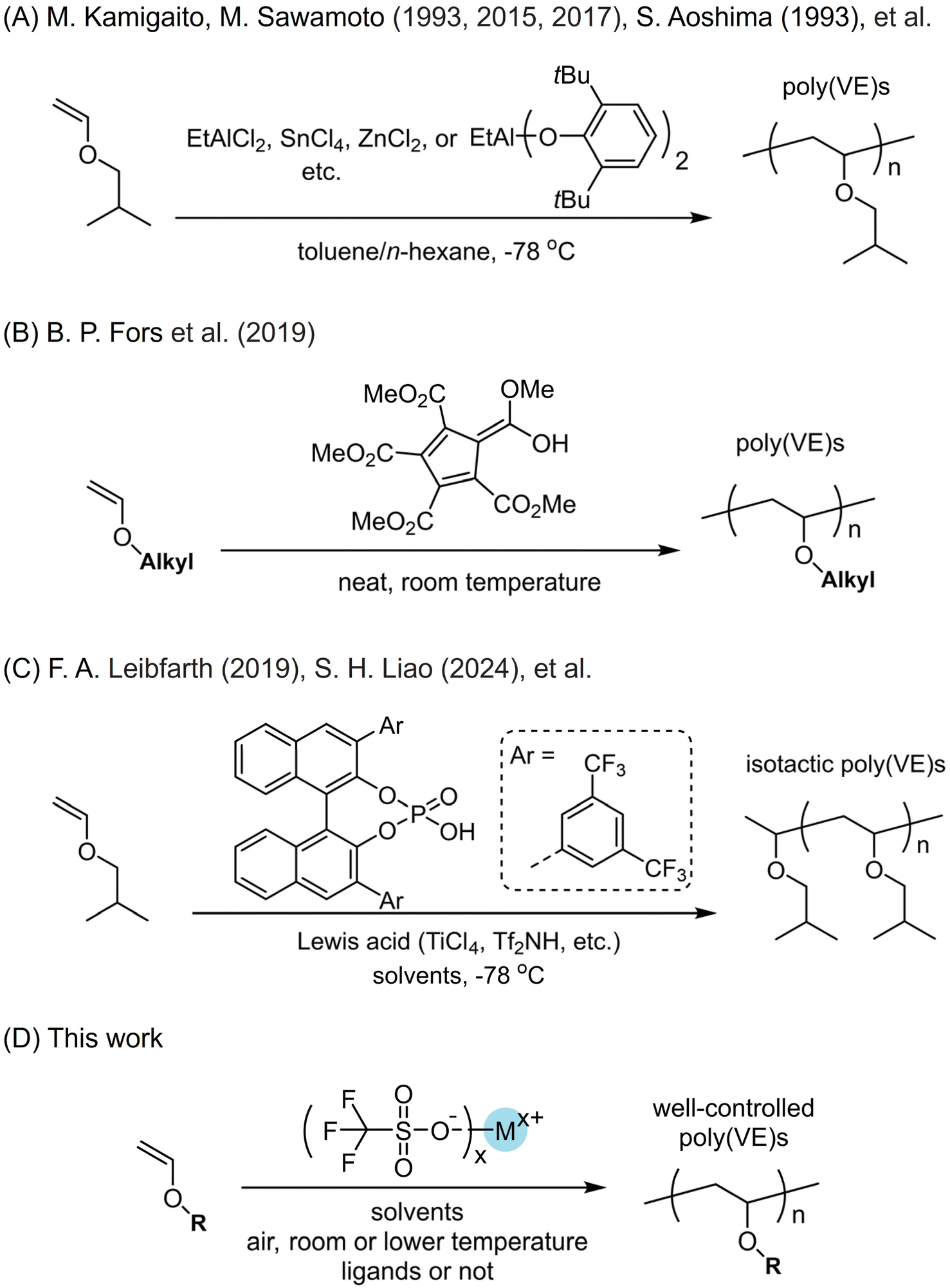 | ||
| Scheme 1 Approaches to synthesize poly(vinyl ether)s. | ||
For vinyl monomers and their polymers, tacticity has a substantial influence on their physical properties.45 For instance, isotactic polypropylene (iPP) is a widely used plastic with production at a scale of >50 million metric tons annually. However, the isomers syndiotactic PPs and atactic PPs show few applications.46,47 For vinyl ether polymers, the similar issue appears. Since poly(vinyl ether)s are promisingly employed as engineering materials due to their readily available feedstocks, studies on their stereoselective cationic polymerization become increasingly necessary. Recently, accompanied by considerable efforts having been made to circumvent the undesirable chain transfer, for some elaborately designed catalysts, poly(vinyl ether)s with high content of isotacticity were obtained. They showed similar catalysis performance to transition metal complexes catalyzing coordination polymerization of olefins.48 Leibfarth and coworkers recently developed an approach to conduct the catalyst-controlled stereoselective cationic polymerization of vinyl ethers (Scheme 1C).25 Lewis acids such as TiCl4 as well as their complexes with tetrahydrofuran were employed. They proposed the chiral counterion strategy where they chose 1,1′-bi-2-naphthol (BINOL)-based phosphoric acids and leveraged the modularity of them, which can effectively tune the reactivity and chain-end interactions of chiral counterions. At −78 °C and in hexane/toluene mixed solvents, a few poly(vinyl ether)s, e.g. poly(iso-butyl vinyl ether), with number-average molecular weights from 25 to 106 kg mol−1 and up to 93% degrees of isotacticity were made. The stereoenriched poly(iso-butyl vinyl ether) showed a melting transition around 138 °C and comparable thermomechanical strength to the commercial low-density polyethylene. There are also other studies to employ chiral catalysts to perform stereocontrolled cationic polymerization of vinyl ether monomers and to afford poly(vinyl ether)s with high stereoselectivity. After conducting intensive studies on stereoselective cationic polymerization,29,33,39 Liao and the group discovered an asymmetric ion-pairing photoredox catalyst based on photoredox cations and the chiral anion N,N′-bis(triflyl)phosphoramidimidates.42 Pyrylium and acridinium tetrafluoroborates were employed to access photoredox cations. N,N′-Bis(triflyl)phosphoramidimidates helped afford isotactic poly(vinyl ether)s with isotacticity up to 91% m. This strategy can be utilized for photocontrolled stereoselective cationic RAFT polymerization of a family of vinyl ethers such as iso-butyl vinyl ether, n-propyl vinyl ether, n-butyl vinyl ether, benzyl vinyl ether and so forth.
In principle, vinyl ether monomers are derived from an inexpensive and underused feedstock, and poly(vinyl ether)s feature a polar ether functionality in each repeat unit, which are rarely commercially used when compared to polyolefins and polyesters; therefore, extensive studies and efforts to develop simple preparation protocols are requested. Generally, when a polymeric material is subject to industrial production, besides the readily available feedstocks and the catalysts, the polymerization process should be easily conducted. The ideal protocol is to conduct the cationic polymerization under ambient conditions without an inert atmosphere, too low temperatures, or strict purification of reagents including monomers, solvents and other agents. To stabilize the active cationic species and thus to suppress the chain transfer, low temperatures such as <−60 °C are generally needed, but if RAFT agents were added to purposely make use of the chain transfer, the reaction temperatures can be a little higher. If the catalysts can be commercially acquired, it will greatly lower the production cost of poly(vinyl ether) materials. When some catalysts have to be elaborately synthesized, however, it is still practical if they are fed at a remarkably low catalyst loading.
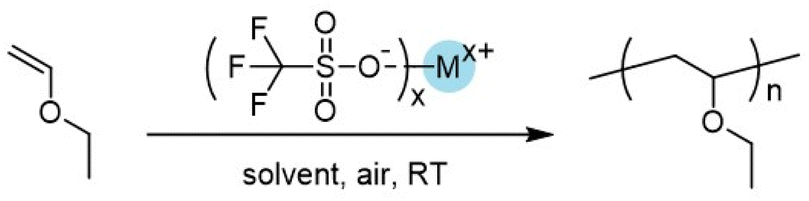
We herein probed the trifluoromethyl sulfonate-catalyzed cationic polymerization of a series of vinyl ethers under moderately mild conditions (Scheme 1D). Even though there had been a few studies on cationic polymerization induced by trifluoromethyl sulfonates (triflates),49–53 due to their explosive polymerization behaviors and uncontrolled chain transfer in bulk,49 systematic studies especially explorations on how to obtain well-controlled polymerization, to the best of our knowledge, were not reported. We commenced our studies by conducting cationic polymerization at room temperature. Ethyl vinyl ether was chosen due to its much easier availability than other vinyl ethers. Trifluoromethyl sulfonates with up to 17 kinds of different ions and 15 kinds of solvents were employed. Most of the trifluoromethyl sulfonates can afford polymer products. To suppress the chain transfer, we also lowered the polymerization temperature e.g. −78 °C. Poly(vinyl ether)s with molecular weights which are close to theoretical values were obtained. N^N, N^O, and O^O type ligands were employed. It was found that N^N and N^O type ligands cannot afford polymers under predetermined conditions. O^O type ligands facilitated the formation of homogeneous reactions, affording polymers with relatively narrow molecular weight distributions. For 1,1′-bi-2-naphthol (BINOL)-derived ligands, improvement of isotacticity of poly(ethyl vinyl ether) at −60 °C was realized. The resulting polymers with high isotacticity 81% m exhibited semicrystallization behavior with melting points. We also probed the preparation of poly(n-octadecyl vinyl ether) (P18VE) on an 80 g scale simply employing trifluoromethyl sulfonate and commonly used solvents. Due to its side chain crystallization, poly(n-octadecyl vinyl ether) can be used as plastic with ether units as functional components along the backbone.
2. Results and discussion
2.1 Probing the polymerization conditions
All chemicals employed in this work are commercially available without further synthesis, modification or purification. The synthesis route is presented in Scheme 1D. All trifluoromethyl sulfonates (MOTfs) were used as purchased. To intuitively learn the performance in catalysis, we herein showed the structures and the difference between the ionic diameters in these chemicals in Scheme 2.54 It can be seen that the Al3+ exhibits the smallest diameter while K+, Ag+, Ba2+ and NH4+ show much larger diameters than other ions. The pyridinium cation is an arylene-based ion; thus it is of little significance to compare its diameter with that of others. We chose monomers based on their commercial availabilities. The solvents used in this work are presented in the scheme including commonly used reagents such as toluene, hexane, halogenated alkanes and aromatic hydrocarbons, ethers, dimethyl sulfoxide (DMSO), acetonitrile (AN), etc.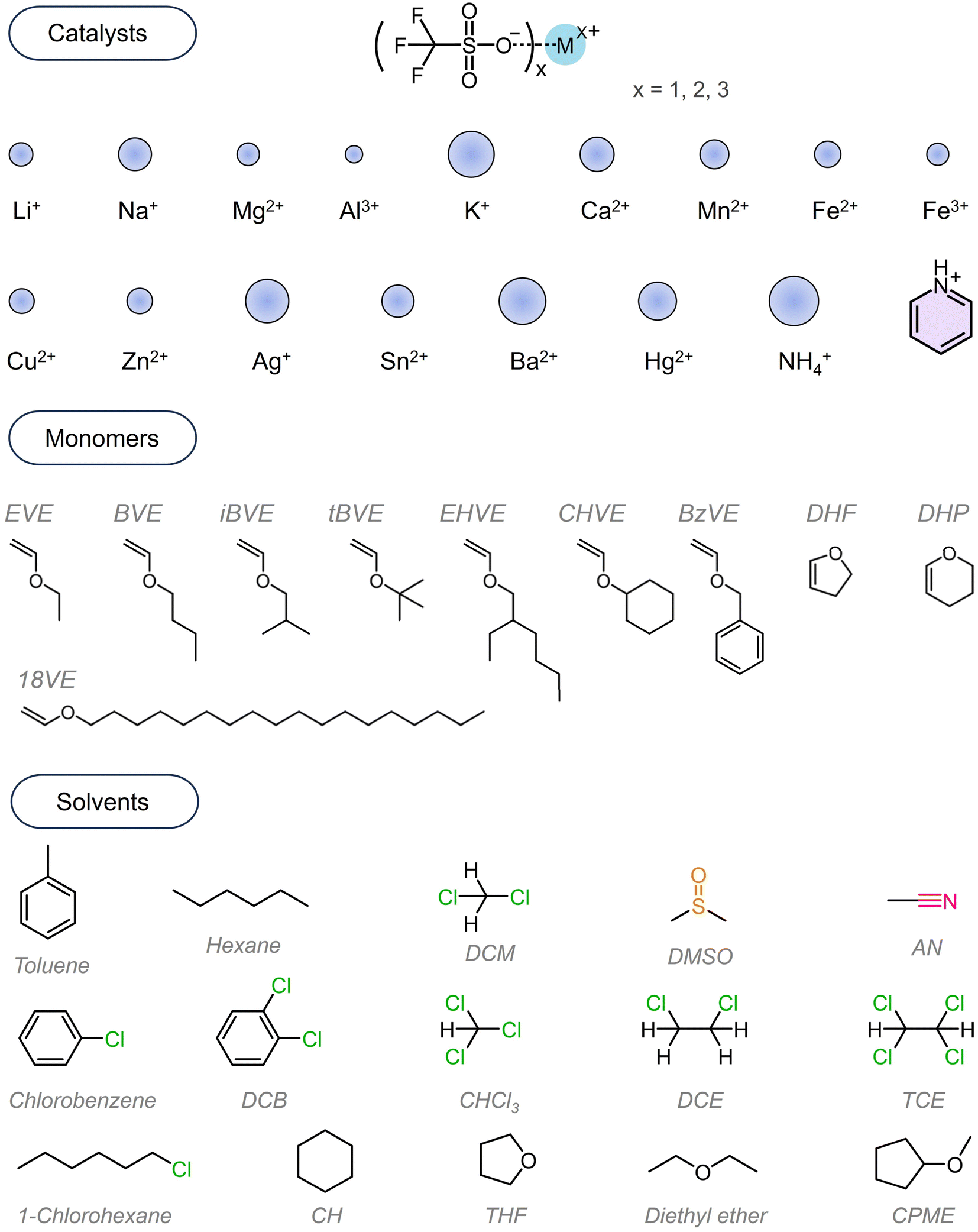 | ||
| Scheme 2 Structures of the catalysts, monomers and solvents employed in this work. | ||
We added the trifluoromethyl sulfonates e.g. 5 μmol into the solvent e.g. 10 mL and dissolved them by stirring at room temperatures for a while and then removed the insoluble components by filtration. After recalculation, the filtrate afforded the catalyst concentration ranging from 0.47 mM to 0.5 mM. The filtrate was taken into flasks according to predetermined volume and molar ratio followed by dropwise addition of the monomers (molar ratio of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to catalyst). Initially, the reaction temperature was set at room temperature (RT) by placing the reaction flasks into a water bath. Ethyl vinyl ether (EVE) was employed as the monomer, and different trifluoromethyl sulfonates and solvents were chosen and collocated. The polymerization was proceeded for 6 h to enable monomers to react as fully as possible. Diethylamine or methanol was employed to quench the reaction. Subsequently, the mixture was added dropwise into methanol to precipitate the polymer products. The product was collected and characterized by NMR and GPC. It should be noted that iso-butyl vinyl ether (iBVE) has been extensively studied, and a few literature studies revealed that it is more difficult to afford well-controlled PEVE including its molecular weights and molecular weight distributions and the stereoselectivity of EVE during polymerization,27,55 so we herein concentrate our attention first on cationic polymerization of EVE. Then the optimal polymerization conditions were extended to other monomers.
:1 to catalyst). Initially, the reaction temperature was set at room temperature (RT) by placing the reaction flasks into a water bath. Ethyl vinyl ether (EVE) was employed as the monomer, and different trifluoromethyl sulfonates and solvents were chosen and collocated. The polymerization was proceeded for 6 h to enable monomers to react as fully as possible. Diethylamine or methanol was employed to quench the reaction. Subsequently, the mixture was added dropwise into methanol to precipitate the polymer products. The product was collected and characterized by NMR and GPC. It should be noted that iso-butyl vinyl ether (iBVE) has been extensively studied, and a few literature studies revealed that it is more difficult to afford well-controlled PEVE including its molecular weights and molecular weight distributions and the stereoselectivity of EVE during polymerization,27,55 so we herein concentrate our attention first on cationic polymerization of EVE. Then the optimal polymerization conditions were extended to other monomers.
The polymerization results were collected as shown in Tables 1 and S1–S16.† We also showed the GPC curves illustrating the monomodal features with medium polydispersity indices e.g. 2.0 (ESI Fig. S1–S14†). Not all trifluoromethyl sulfonates/solvents/EVE can afford polymer products under the conditions mentioned above. For the solvent DMSO used (Scheme 2), we found that all trifluoromethyl sulfonates showed no catalytic activities. The problems should not be attributed to trifluoromethyl sulfonates since we also did the experiment employing them to catalyze the bulk polymerization of EVE (Table S1†) and the polymers with broad molecular weights and low isotacticities (∼50% m) were obtained. To probe the reason, we removed the solvents and unreacted monomers after the polymerization. And then we determined the catalyst residue and found that they were the trifluoromethyl sulfonates themselves. We utilized it to catalyze the bulk polymerization of EVE and also obtained the polymers. Therefore, it can be concluded that DMSO, a strong Lewis base, presumably turned them into a dormant state. It was found that LiOTf, NaOTf, KOTf, Mg(OTf)2, Ca(OTf)2, and Ba(OTf)2 cannot initiate the polymerization of EVE in the monomer's bulk state and in solvents except in 1,1,2,2-tetrachloroethane (TCE) (Table S11†). However, if they were exposed to aqueous vapor for a while, then EVE was added, the solutions became dark and almost explosive polymerization occurred. A few brown viscous polymer products were observed after precipitating into methanol. We attributed the occurring reaction to the newly generated trifluoromethanesulfonic acid (HOTf) from the partial hydrolysis of MOTfs in an aqueous vapor atmosphere. More experiments concerning this need to be conducted to present accurate conclusions in a separate study.
|
|
||||||
|---|---|---|---|---|---|---|
| Ion (Mx+) | Solvent | Conversionb (%) | M theon (kDa) | M GPCn (kDa) | Đ | m%e |
| a Polymerization conditions: trifluoromethyl sulfonate, 5 μmol; solvent, 10 mL; monomer, 2.5 mmol; 6 h, 23 °C (RT). b Determined via1H NMR spectroscopy in CDCl3. c Theoretical molecular weights calculated as [monomer/trifluoromethyl sulfonate feed ratio] × MW of monomer × conversion. d Determined via GPC (1 mL min−1 in tetrahydrofuran), 3‰ concentration, using polystyrene calibration. e Determined via13C NMR. | ||||||
| Fe2+ | Toluene | 99 | 35.7 | 3.6 | 2.10 | 56 |
| Zn2+ | Toluene | 99 | 35.7 | 6.3 | 1.99 | 59 |
| Fe2+ | Hexane | 99 | 35.7 | 6.4 | 1.75 | 61 |
| Al3+ | DCM | 99 | 35.7 | 3.6 | 1.97 | 59 |
| Ag+ | AN | 99 | 35.7 | 1.7 | 1.42 | 59 |
| Fe2+ | Chlorobenzene | 99 | 35.7 | 7.4 | 1.96 | 62 |
| Ag+ | DCB | 99 | 35.7 | 9.9 | 1.87 | 60 |
| Cu2+ | CHCl3 | 99 | 35.7 | 3.7 | 2.10 | 54 |
| Py+ | DCE | 78 | 28.1 | 1.9 | 1.38 | 62 |
| NH4+ | TCE | 73 | 26.3 | 11.6 | 1.75 | 55 |
| Hg2+ | CH | 99 | 35.7 | 1.7 | 1.90 | 56 |
| Py+ | THF | 87 | 31.4 | 1.8 | 1.54 | 59 |
| Zn2+ | Diethyl ether | 99 | 35.7 | 4.5 | 1.91 | 62 |
| Py+ | CPME | 92 | 33.2 | 4.1 | 1.57 | 60 |
Fortunately, a few trifluoromethyl sulfonates showed good catalytic activities and afforded high monomer conversions. Viscous colorless polymers with low molecular weights i.e. several thousands of Dalton and polydispersity indices (Đs) ranging from 1.38 to ∼2.20 were obtained. We presented the representative polymerization results in Table 1. Given the initial molar ratio between monomers and catalysts (500:1) and the theoretical molecular weights, it is believed that serious chain transfer occurred. These low-molecular-weight PEVEs may find their applications in lubricants and adhesives due to the high monomer conversions and good fluidity.
Based on these results, in the following studies, we paid our attention to how to suppress chain transfer and aimed to control the polymerization process to obtain lowly dispersed polymers. The first strategy was to lower the polymerization temperature. We selected a few trifluoromethyl sulfonates and solvents that had exhibited good catalysis performance i.e. (CF3SO3)3Al, (CF3SO3)2Fe, CF3SO3Ag, and CF3SO3Py. Toluene, hexane, DCM, and diethyl ether were employed as solvents. The solvents were used without further purification. At low temperature i.e. −20 °C and −60 °C, the trifluoromethyl sulfonates became decreasingly soluble even with 5 μmol in 10 mL solvent. To resolve the problem, a few molecules containing O^O, N^O, and N^N type groups were utilized to improve their solubility as shown in Scheme 3.
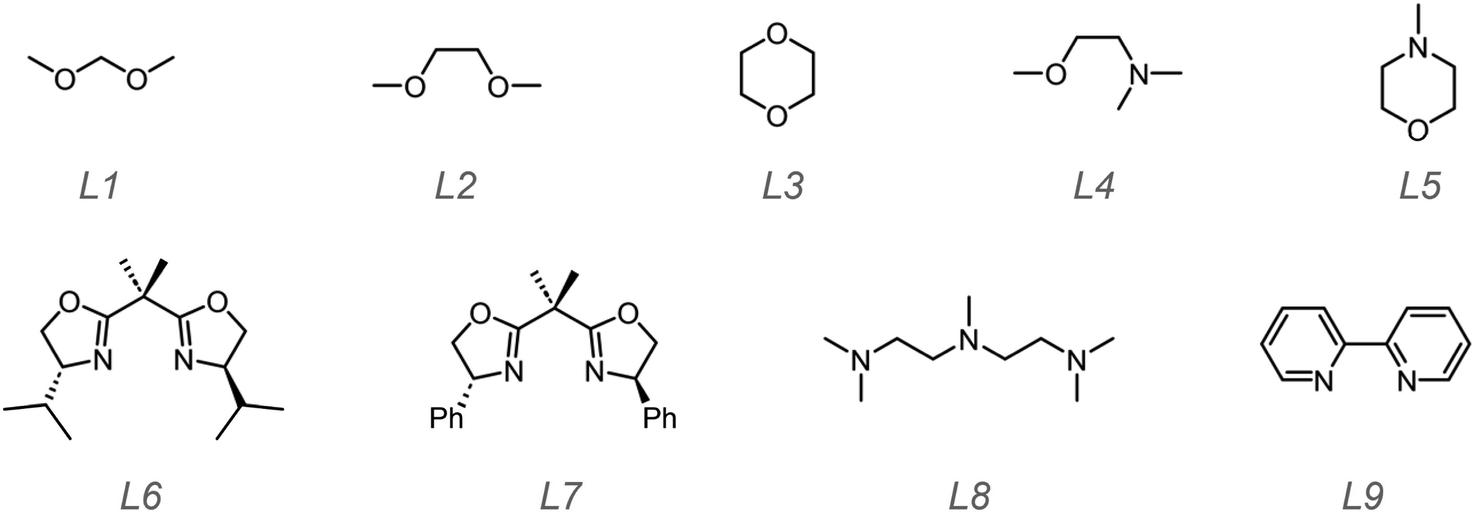 | ||
| Scheme 3 Structures of the ligands employed in this work. All of them are commercially available. | ||
Generally, homogeneous catalyst systems can provide a stable, controlled and predictive catalysis environment. Conversely, heterogeneous catalysts such as MgCl2-supported Ziegler–Natta catalysts may afford quite polydisperse products.56 It is believed that the molecules in Scheme 3 can be viewed as ligands to coordinate the ions in trifluoromethyl sulfonates, thus improving their solubility in poor solvents e.g. hexane. Trifluoromethyl sulfonates (5 μmol) and ligands (>5 μmol) were weighed and dissolved into the solvent (25 mL) by stirring at room temperatures for a while. Then the mixture was placed into a low temperature bath (−78 °C). After it was cooled to the preset temperature, monomer EVE was added dropwise via a syringe. During the polymerization, an aliquot was acquired for monomer conversion and molecular weight information every 0.5 h by 1H NMR and GPC. After 6 h, the polymerizations were quenched and precipitated to obtain polymers. We collected the results in Tables 2 and S17–S19.†
|
|
||||||
|---|---|---|---|---|---|---|
| Ion (Mx+) | Ligand | Conversionb (%) | M theon (kDa) | M GPCn (kDa) | Đ | m%e |
| a Polymerization conditions: trifluoromethyl sulfonate, 5 μmol; ligand, >5 μmol; toluene, 25 mL; monomer, 2.5 mmol; 8 h, −78 °C. b Determined via1H NMR spectroscopy in CDCl3. c Theoretical molecular weights calculated as [monomer/trifluoromethyl sulfonate feed ratio] × MW of monomer × conversion. d Determined via GPC (1 mL min−1 in tetrahydrofuran), 3‰ concentration, using polystyrene calibration. e Determined via13C NMR. | ||||||
| Al3+ | L1 | 99 | 35.7 | 33.1 | 1.17 | 55 |
| Al3+ | L2 | 99 | 35.7 | 30.5 | 1.15 | 57 |
| Al3+ | L3 | 99 | 35.7 | 31.4 | 1.12 | 57 |
| Fe2+ | L1 | 99 | 35.7 | 35.6 | 1.18 | 62 |
| Fe2+ | L2 | 99 | 35.7 | 39.5 | 1.15 | 62 |
| Fe2+ | L3 | 99 | 35.7 | 32.0 | 1.23 | 64 |
| Ag+ | L1 | 99 | 35.7 | 40.2 | 1.13 | 57 |
| Ag+ | L2 | 99 | 35.7 | 33.5 | 1.19 | 62 |
| Ag+ | L3 | 99 | 35.7 | 31.8 | 1.17 | 60 |
| Py+ | L1 | 77 | 27.8 | 34.6 | 1.10 | 62 |
| Py+ | L2 | 80 | 28.8 | 35.3 | 1.14 | 59 |
| Py+ | L3 | 83 | 29.9 | 36.4 | 1.09 | 57 |
For O^O type ligands L1–L3 (Scheme 3), polymer products were obtained. Their molecular weights showed 31 kDa to 40 kDa which were very close to the theoretical value (35.7 kDa) with narrow molecular weight distributions ranging from 1.09 to 1.19. We employed 13C NMR to measure the isotacticities and found no remarkable improvement compared to that of polymers obtained at RT. For N^O type ligands L4 and L5 and N^N type ligands L6–L9, we did not obtain polymer products. The mixtures remained clear after quenching. We then conducted this polymerization at RT; however, still no polymer products were prepared. This is presumably due to that N^N type ligands inhibited the catalytic activities of (CF3SO3)3Al, (CF3SO3)2Fe, CF3SO3Ag, and CF3SO3Py, given that polymers can be made without these ligands (Table 1). We had attempted to prepare single crystals using the ligands and MOTfs to learn the possible structures of the complexes formed, but it failed probably due to the stringent growth requirements. We will continue to probe the optimized conditions for formation of single crystals. Since tetrahydrofuran can be ring-opened using TfOH and triflate esters to afford linear polyethers based on the cationic polymerization mechanism,57 we employed NMR to trace whether ring-opening polymerization of the ligand L3 occurred or not. However, no signals in 1H NMR were detected for the polymer i.e. linear polyether. To reconfirm that trifluoromethyl sulfonates cannot induce polymerization of these cyclic ethers under the conditions in this work, we also conducted the bulk polymerization of L3 using all trifluoromethyl sulfonates in Scheme 2. Like before, no polymer products were obtained under the conditions in this work. Therefore, these cyclic ethers e.g.L3 did not act as polymerizable monomers; instead, they only assisted in the solubility of MOTfs.
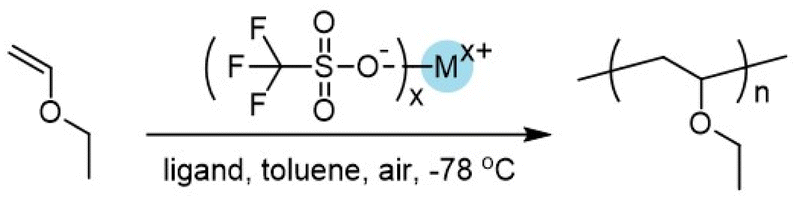
In Fig. 1, we showed kinetic studies on the polymerization. The monomer conversion exhibited nearly linear dependance on the polymerization time (Fig. 1A). With the increase of monomer conversion, molecular weights of the PEVEs increased linearly and agreed well with the theoretical values (Fig. 1B); meanwhile, the molecular weight distributions remained around 1.10–1.19, suggesting the characteristics of a living polymerization process. The GPC traces also revealed that molecular weights increased with polymerization time and always showed monomodal features (Fig. 1C). Representative 1H and 13C NMR spectra are presented in Fig. 1D. It was found that there were no significant changes in isotacticity with or without the ligands as shown in Scheme 3, indicating that these ligands displayed no influences on the propagating fashion.
 | ||
| Fig. 1 (A) Monomer conversion-time for the polymerization of EVE catalyzed by (CF3SO3)2Fe in toluene at −78 °C. (B) Increase in molecular weight with monomer conversion. The line refers to the theoretical molecular weight. (C) GPC traces of PEVE at different monomer conversions as shown in (B). (D) 1H and 13C NMR spectra of PEVE obtained. CDCl3 as deuterated solvent (25 °C). | ||
2.2 Chiral ligands assist to afford well-controlled poly(vinyl ether)s
Polymers with high isotacticities have more applications; for instance, isotactic PP is a commercially and widely used material in plastic, packaging, structural materials and engineering fields while syndiotactic PP and atactic PP show limited applications. In this section, we focused our attention on improvement of isotacticities. In the work reported before, it has been revealed that transition metal catalysts can catalyze the stereoselective polymerization of olefins.58,59 Organic ligands play a vital role in regular chain propagation. Therefore, organic ligands are worth employing to assist stereoselective polymerization of vinyl ethers.60 In the last section, we showed that L1–L3 can promote solubility of trifluoromethyl sulfonates but showed no effects on the chain propagating fashion. This encouraged us to probe other organic molecules structurally based on them. It is reported that 1,1′-bi-2-naphthol (BINOL)-derived Lewis acids can initiate the stereoselective polymerization of vinyl ethers and produce PVEs with high isotacticities.25,29,30,33 Inspired by these studies, we herein employed BINOL-derived molecules with ether substituents as ligands to probe whether they can assist to afford stereocontrolled PVEs. The structures of BINOL O^O type ligands are shown in Scheme 4. | ||
| Scheme 4 Structures of the 1,1′-bi-2-naphthol (BINOL)-derived O^O type ligands employed in this work. All of them are commercially available. | ||
Similar procedures were followed. (CF3SO3)2Fe (5 μmol) and ligands L10–L15 (>5 μmol) were weighed and dissolved into toluene (25 mL) by stirring at RT for 20 minutes. The mixture was placed into the low temperature bath (−60 °C), and then the monomer EVE (2.5 mmol) was added dropwise via a syringe pump. After 8 h, the polymerization was quenched and the polymer product was collected by purification through a short silica plug, precipitating into methanol and drying to constant weight. Polymer's structure and molecular weight information determined by NMR and GPC were collected and are shown in Table 3, Fig. 2 and S15.†
| Ligand | Conversionb (%) | M theon (kDa) | M GPCn (kDa) | Đ | m%e |
|---|---|---|---|---|---|
| a Polymerization conditions: (CF3SO3)2Fe, 5 μmol; ligand, >5 μmol; toluene, 25 mL; EVE 2.5 mmol; 8 h, −60 °C. b Determined via1H NMR spectroscopy in CDCl3. c Theoretical molecular weights calculated as [monomer/trifluoromethyl sulfonate feed ratio] × MW of monomer × conversion. d Determined via GPC (1 mL min−1 in tetrahydrofuran), 3‰ concentration, using polystyrene calibration. e Determined via13C NMR. | |||||
| L10 | 99 | 35.7 | 32.3 | 1.16 | 73 |
| L11 | 99 | 35.7 | 35.8 | 1.14 | 75 |
| L12 | 99 | 35.7 | 37.6 | 1.13 | 79 |
| L13 | 99 | 35.7 | 34.4 | 1.12 | 81 |
| L14 | 99 | 35.7 | 31.9 | 1.14 | 75 |
| L15 | 99 | 35.7 | 33.3 | 1.19 | 69 |
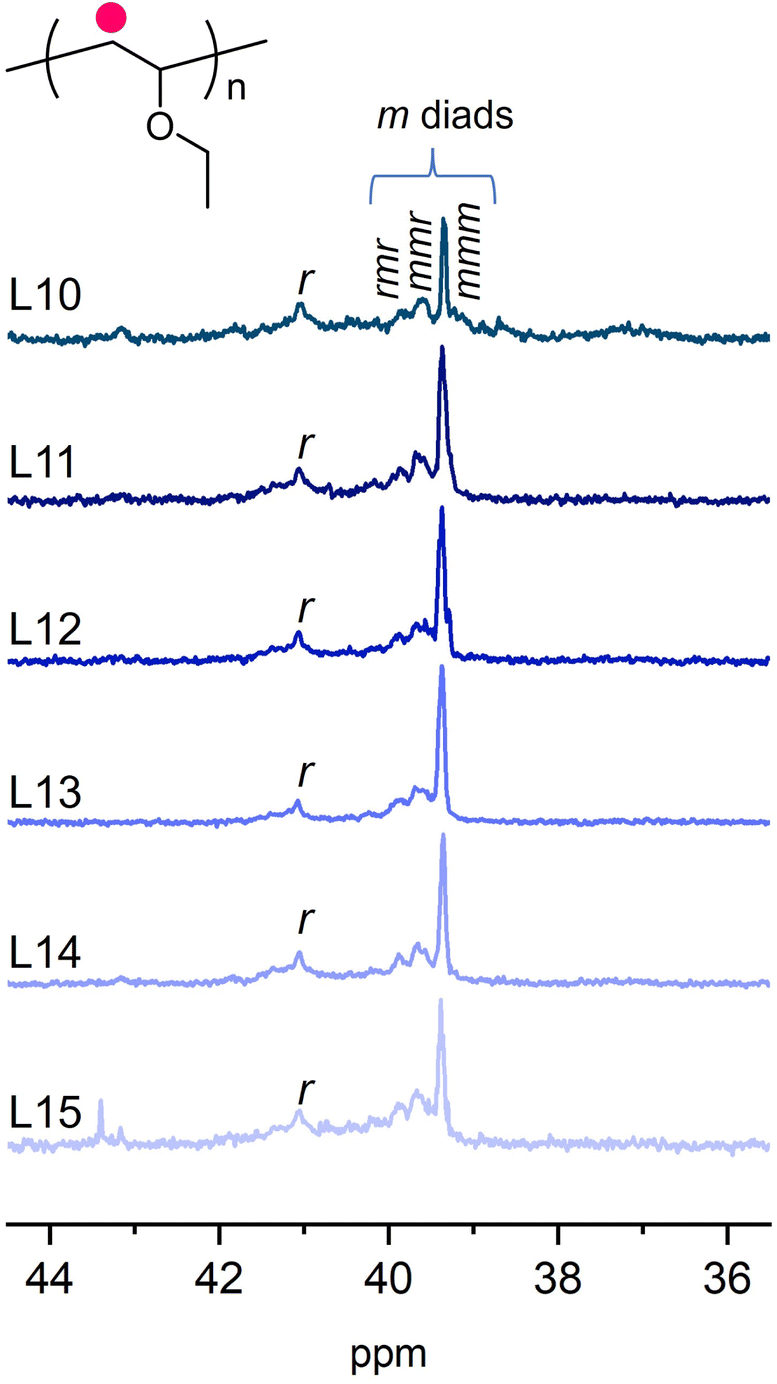 | ||
| Fig. 2 13C NMR spectra around 35–45 ppm of PEVEs obtained using (CF3SO3)2Fe/ligands (L10–L15) in toluene at −60 °C. CDCl3 as deuterated solvent (25 °C). | ||
The molecular weights were close to the theoretical values (36 kDa), and molecular weight distributions remained similarly low compared to those in Tables 1 and 2. Determined by 13C NMR, the tacticities of these polymers were specified. By comparing the m% values in Tables 2 and 3, it was found that isotacticities increased from 62% up to 81%. This significant improvement in tacticity transformed the polymer from a viscous state into a solid substance. We employed DSC to measure their thermal properties and presented the results in Fig. S16.† Poly(ethyl vinyl ether) with 62% m exhibited a glass transition at ∼40 °C; however, PEVE with 81% m by (CF3SO3)2Fe/L13 showed several melting temperatures around 51 °C and 28 °C, respectively. These results indicated that these BINOL ligands exhibited significant influence on the chain propagating fashion, thus emerging as a useful scaffold to develop stereoselective cationic polymerizations. As shown in Fig. 2, we found that all BINOL ligands can increase the isotacticities to a certain degree. For instance, L10 with methoxy and L15 with ester units in 2,2′-positions promoted a few improvements to 73% m and 69% m, respectively. L11, L12 and L14 with different groups in 2,2′-positions and 3,3′-positions exhibited similar promotions of tacticity up to 75% m and 79% m. We had tried to learn the structure of the possible complex according to crystallography but it was difficult to get the crystals. Further efforts will be made for that. In addition, due to the limited numbers of these commercially available BINOL molecules, more of them would be needed to conclude on the specific influences of different functional groups in 2,2′-positions and 3,3′-positions including steric and electronic effects on the isotacticities. Nevertheless, the present work provides more evidence and experimental demonstrations for further development of coordination ligands used for cationic polymerization and motivates the discovery of synthetic methods to access stereoregular derivatives of trifluoromethyl sulfonates, thus creating an opportunity for effectively preparing isotactic poly(vinyl ether) materials.
2.3 Block copolymers by the cationic copolymerization
To suppress the chain transfer reaction and to obtain lowly dispersed polymer products, trifluoromethyl sulfonate-catalyzed cationic polymerization is generally needed to be conducted at low temperature i.e. −78 °C. It sometimes required cryogenic equipment. Accordingly, we herein turned our attention to cationic RAFT polymerization under relatively mild conditions. (CF3SO3)2Fe and the RAFT chain transfer agent, i.e. methyl piperidine-1-carbodithioate, were utilized to conduct cationic RAFT polymerization and then to prepare block copolymers. (CF3SO3)2Fe (5 μmol) was weighed and dissolved into toluene (10 mL) by stirring at RT for 5 minutes. Then the mixture was placed into the low temperature bath (−48 °C). After 20 minutes methyl piperidine-1-carbodithioate (10 μmol) in toluene (5 mL) was injected and the monomer EVE (2.5 mmol) in toluene (10 mL) was added dropwise via a syringe pump. For block copolymerization, EVE and 18VE were dissolved in toluene/DCM (7/3 v/v) mixed solvent and added in sequence. The addition of DCM was to inhibit precipitation of 18VE due to its strong crystallization at low temperature. The polymerization was quenched with methanol and then polymer products were further purified and dried. 18VE as the second monomer following EVE was added to synthesize block copolymers. We characterized the polymers by NMR and GPC and presented the results in Table 4, Fig. 3A and B.|
|
|||||
|---|---|---|---|---|---|
| Monomer | Conversionb (%) | M theon (kDa) | M GPCn (kDa) | Đ | m%e |
| a Polymerization conditions: (CF3SO3)2Fe, 5 μmol; RAFT agent, 10 μmol; toluene, 25 mL, for 18VE, 30 v/v% DCM is needed to keep it soluble at low temperature; monomer(s), 2.5 mmol for EVE and 1.25 mmol for 18VE; 6 h + 6 h, −40 °C. b Determined via1H NMR spectroscopy in CDCl3. c Theoretical molecular weights calculated as [monomer/trifluoromethyl sulfonate feed ratio] × MW of monomer × conversion. d Determined via GPC (1 mL min−1 in tetrahydrofuran), 3‰ concentration, using polystyrene calibration. e Determined via13C NMR. | |||||
| EVE | 99 | 17.8 | 17.3 | 1.13 | 55 |
| EVE + 18VE | 93 | 52.3 | 41.6 | 1.18 | 59 |
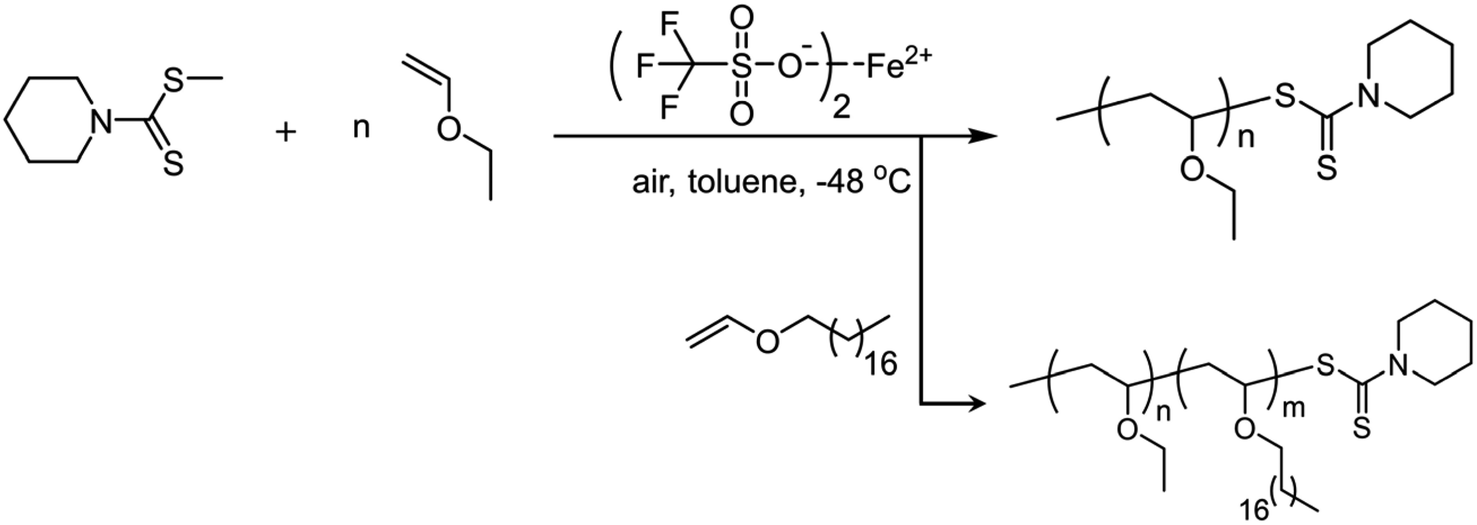
 | ||
| Fig. 3 (A) GPC traces of the block copolymer PEVE-b-P18VE (RI signal) and PEVE (RI (blue) and UV (purple) signals). (B) 1H NMR spectrum of PEVE-b-P18VE obtained using cationic RAFT polymerization. CDCl3 as deuterated solvent (25 °C). (C) TEM image of assemblies formed by crystallization-driven self-assembly (CDSA) of PEVE-b-P18VE in hexane (0.1 mg mL−1). | ||
The monomer conversions were still high and the molecular weights were very approximate to the theoretical values (17.8 kDa). For radical RAFT polymerization, polymers with narrow molecular weight distributions are generally prepared. As for cationic RAFT polymerization of vinyl ethers, it also afforded lowly dispersed polymers (Đ = 1.13 and 1.18). The UV-detected GPC curve in Fig. 3A confirmed that PEVE contained the RAFT agent terminals. After addition of the second monomer 18VE, the molecular weight increased a lot (Fig. 3A). The RAFT agent cannot assist in improvement of isotacticity since both the PEVE homopolymer and PEVE-b-P18VE exhibit low isotacticities (55% m and 59% m) (Table 4 and Fig. S17†). According to similar cationic RAFT polymerization studies,18 no driving forces creating a chain-end stereochemical environment during the chain propagation should be the critical reason to afford lowly isotactic polymers. For the block copolymer, the RAFT agent terminal can be detected by 1H NMR as shown in Fig. 3B. The methine adjacent to O and S atoms at the chain terminal was found at 6.1–6.2 ppm whereas methylene adjacent to the N atom of the RAFT agent was at ∼4.0 ppm. The other peaks assigned to terminal groups were overlapped by that of monomer units. DOSY spectra were acquired and reconfirmed both units i.e. EVE and 18VE that resided in the block copolymer backbone by comparing the DOSY spectrum of PEVE-b-P18VE and that of the PEVE and P18VE mixture (Fig. S18†).
The block copolymer has an amorphous block and a crystalline block; therefore, we thought it would be suitable for crystallization-driven self-assembly (CDSA). We previously performed CDSA using semicrystalline diblock copolymers and prepared many nanomaterials.61–64 Crystallization provides a strong force to induce formation of regular structures. Following the corresponding procedures, we then performed the self-assembly in hexane. 0.5 mg PEVE-b-P18VE was placed into 5 mL hexane and the mixture was sealed and heated to 70 °C for 30 minutes. Subsequently, it was cooled to RT and aged for 24 hours. TEM was employed to observe the structures formed after being stained using ruthenium tetroxide. The image is presented in Fig. 3C and S19.† The nanoscale assemblies formed, suggesting that the block copolymers via cationic RAFT polymerization can find applications in nanomaterials and drive innovations in method development using poly(vinyl ether)s. Further studies focusing on preparation of regular and well-controlled nanostructures will be made.
2.4 Other poly(vinyl ether)s from more vinyl ether monomers
Since vinyl ethers can only be effectively initiated and polymerized via the cationic mechanism, cationic polymerization promisingly becomes a prominent complement to other polymerization approaches and could contribute more polymeric materials that cannot be prepared by other means. Based on the studies made above, we then turned to cationic polymerization of other vinyl ether monomers using trifluoromethyl sulfonate i.e. (CF3SO3)2Fe. Ligand L13 was chosen to improve the isotacticity and toluene was employed to ensure that the ligand and polymer products can dissolve well. (CF3SO3)2Fe and L13 were added into toluene and dissolved to afford a clear mixture by stirring for 10 min, and the solution was cooled to −60 °C. Then the monomer was added dropwise. Similar quenching and purifying steps to prepare PEVE were followed. NMR, GPC and DSC were used to characterize the polymers and the data are collected in Table 5, Fig. 4 and S20–S38.†| Monomer | Conversionb (%) | M theon (kDa) | M GPCn (kDa) | Đ | m%e |
|---|---|---|---|---|---|
|
a Polymerization conditions: trifluoromethyl sulfonate, 5 μmol; ligand, >5 μmol; toluene, 25 mL; monomer, 2.5 mmol; 8 h, −60 °C; for 18VE, 30 v/v% DCM is needed to keep it soluble at low temperature, and 80 g scale polymerization (catalyst/ligand/monomer 1:1:500) was made.
b Determined via1H NMR spectroscopy in CDCl3.
c Theoretical molecular weights calculated as [monomer/trifluoromethyl sulfonate feed ratio] × MW of monomer × conversion.
d Determined via GPC (1 mL min−1 in tetrahydrofuran), 3‰ concentration, using polystyrene calibration.
e Determined via13C NMR.
f Not applicable.
|
|||||
| BVE | 99 | 49.6 | 45.2 | 1.17 | 72 |
| iBVE | 99 | 49.6 | 47.3 | 1.21 | 79 |
| tBVE | 99 | 49.6 | 42.8 | 1.16 | 77 |
| EHVE | 99 | 77.4 | 63.1 | 1.13 | 75 |
| CHVE | 99 | 62.5 | 58.3 | 1.10 | 78 |
| BzVE | 99 | 66.4 | 56.5 | 1.15 | 75 |
| DHF | 99 | 34.7 | 33.4 | 1.29 | —f |
| DHP | 13 | 5.4 | 7.8 | 1.33 | — |
| 18VE | 90 | 133.4 | 112.3 | 1.14 | 75 |
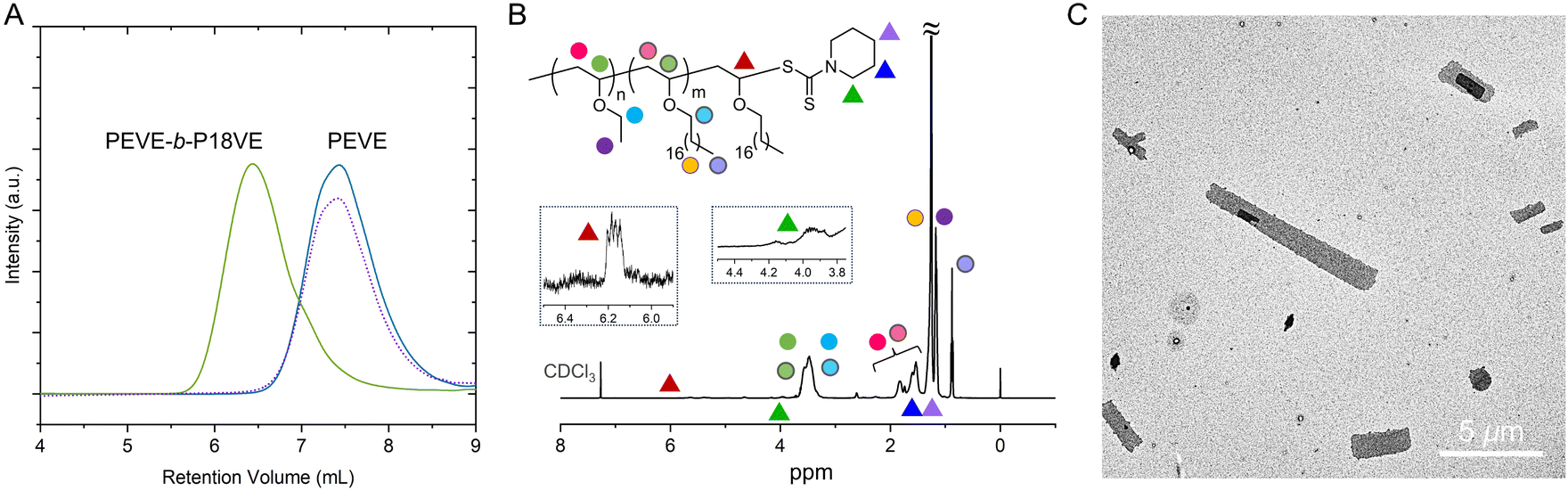
 | ||
| Fig. 4 (A) and (B) DSC results of poly(vinyl ether) polymers obtained. PEVE with 81% m was tested. Data are rescaled and shifted vertically for clarity. (C) Stress vs. strain curves for P18VE with 75% m and LDPE. (D) XRD pattern of the P18VE film. | ||
Nine poly(vinyl ether) polymers were successfully prepared, suggesting that trifluoromethyl sulfonates in this work can be applicable to the family of vinyl ethers. The monomer conversions show nearly complete stoichiometry except for the monomer DHP and 18VE. DHP is a stable six-membered ring and displays low activity in ring-opening polymerization. 18VE is semicrystalline as well as poorly soluble at low temperatures, which implies that it is difficult to keep the active chain ends throughout the whole polymerization process. However, for most monomers employed, they can form polymers with high molecular weights, ensuring the realization of properties especially mechanical performance. The molecular weights were acquired from GPC, which may account for that they show a little lower than theoretical values. The molecular weight distributions (Đs) also were maintained at narrow ranges, suggesting that the chain transfer was significantly suppressed. We employed 13C NMR to calculate the isotacticities of these PVEs, which displayed up to 79% m. Then we measured their thermal properties using DSC. All the samples obtained were processed through a heating–cooling–heating procedure to eliminate thermal history. The DSC nonisothermal crystallization curves at a cooling rate of 10 °C min−1 and the subsequent melting curves are collected. We showed the second heating run i.e. melting curves in Fig. 4A and B. It showed that polymer PEVE (81% m), PiBVE, PtBVE and P18VE exhibited melting points at 51 °C (28 °C), 83 °C, −2 °C, and 49 °C respectively. Other PVEs are amorphous with glass transition temperatures at −55 °C for PBVE, −30 °C for PEHVE, 54 °C for PCHVE, 59 °C for PBzVE, 120 °C for PDHF, and 103 °C for PDHP. For appearance of multiple melting peaks for PEVE and hysteresis of the melting endotherm for PiBVE, we attributed that to the uneven distributions of mmm diads along the backbone. The r diads would restrict regular chain packing and afford various lamellae with different thicknesses, leading to multiple or broad melting behaviors.
Semicrystalline poly(vinyl ether) polymers may find their applications in fields of packaging materials, self-assembly, additive manufacturing and engineering materials. We chose P18VE to learn its tensile mechanical properties and crystallization behavior, since the polymerization of 18VE was conducted on an 80 g scale which thus allowed its macroscopic mechanical performance to be learnt. The specimens were made by subjecting materials to hot compression using a plate vulcanizing machine and stainless-steel die molds to prepare dog bone samples. The compression was made at 150 °C under 10 MPa pressure for 15 min. Then we cooled the specimens to 23 °C. Uniaxial tensile elongation testing was carried out at 20 °C and with a tensile rate of 50 mm min−1. As shown in Fig. 4C, we found that the tensile behaviors of P18VE were similar to those of low-density polyethylene (LDPE), with yield points at approximately 7 MPa and a strain at break of 740%, suggesting a polar thermal plastic material. The wide angle X-ray diffraction (WAXD) pattern of P18VE is shown in Fig. 4D. It exhibited a single diffraction peak at 2θ 21.68°, corresponding to the orthorhombic crystal structure. The polyethylene oligomer-like side chains of P18VE let it display similar diffraction behavior to linear polyethylene.65 The difference was that the crystal growth along the (200) direction in P18VE was severely suppressed, which should be attributed to obstacles from the main chains on side molecular chain's packing.
Last but not least, we would like to present the possible polymerization mechanism using trifluoromethyl sulfonate/solvent/ligand, especially to depict the role ligands may play. As discussed before, solvent exhibited no significant influences on the molecular weights and isotacticities at the same polymerization temperatures (Tables S1–S16†) except for some strong Lewis bases such as DMSO. This should be attributed to that solvents cannot effectively prevent chain transfer. When the trifluoromethyl sulfonates were well soluble in the solution, temperatures dominated the stabilities of active cationic species. At lower temperature i.e. −60 °C, cationic species can remain in a controlled state and induce polymerization in a determinable fashion. Unlike catalysts containing chiral counterions providing stereoselective polymerizations,25,29,30,33 trifluoromethyl sulfonates themselves cannot afford well-controlled isotactic poly(vinyl ether)s, since no driving forces create a chain-end stereochemical environment during cationic polymerization. Drawing inspiration from the concept of chiral counterion catalysis and coordination polymerization using transition metals and organic ligands to prepare isotactic polyolefins, we herein chose a series of BINOL-derived molecules as ligands to assist formation of a chain-end stereochemical environment and then afford poly(vinyl ether)s with increased isotacticities. For now, even though we have no more details on the precise structures including single crystals formed by the catalysts and ligands, the sure thing is that BINOL ligands showed unignorable effects on the propagating process. We summarize the mechanism in Scheme 5 and present the mechanistic hypothesis to reveal the polymerization process. At the beginning, trifluoromethyl sulfonate induced monomers to form active cationic species, where the process may be assisted by the HOTf from hydrolysis of trifluoromethyl sulfonate. HOTf reacted with a vinyl ether forming an ionization complex promoted by the same trifluoromethyl sulfonate after hydrolysis. Then in the chain-end stereochemical environment provided by coordination between BINOL ligands and trifluoromethyl sulfonate, other monomers inserted and promoted the chain growth and finally formed high-molecular-weight well-controlled polymers. Of note, given the commercially available chemicals including solvents in this work, the trace of moisture inherent in these reagents would possibly facilitate the hydrolysis of trifluoromethyl sulfonates into HOTf. However, it also should not be excluded that the trifluoromethyl sulfonate can induce the direct initiation process.49 Due to the potentially complex process, a more systematic study will be needed.
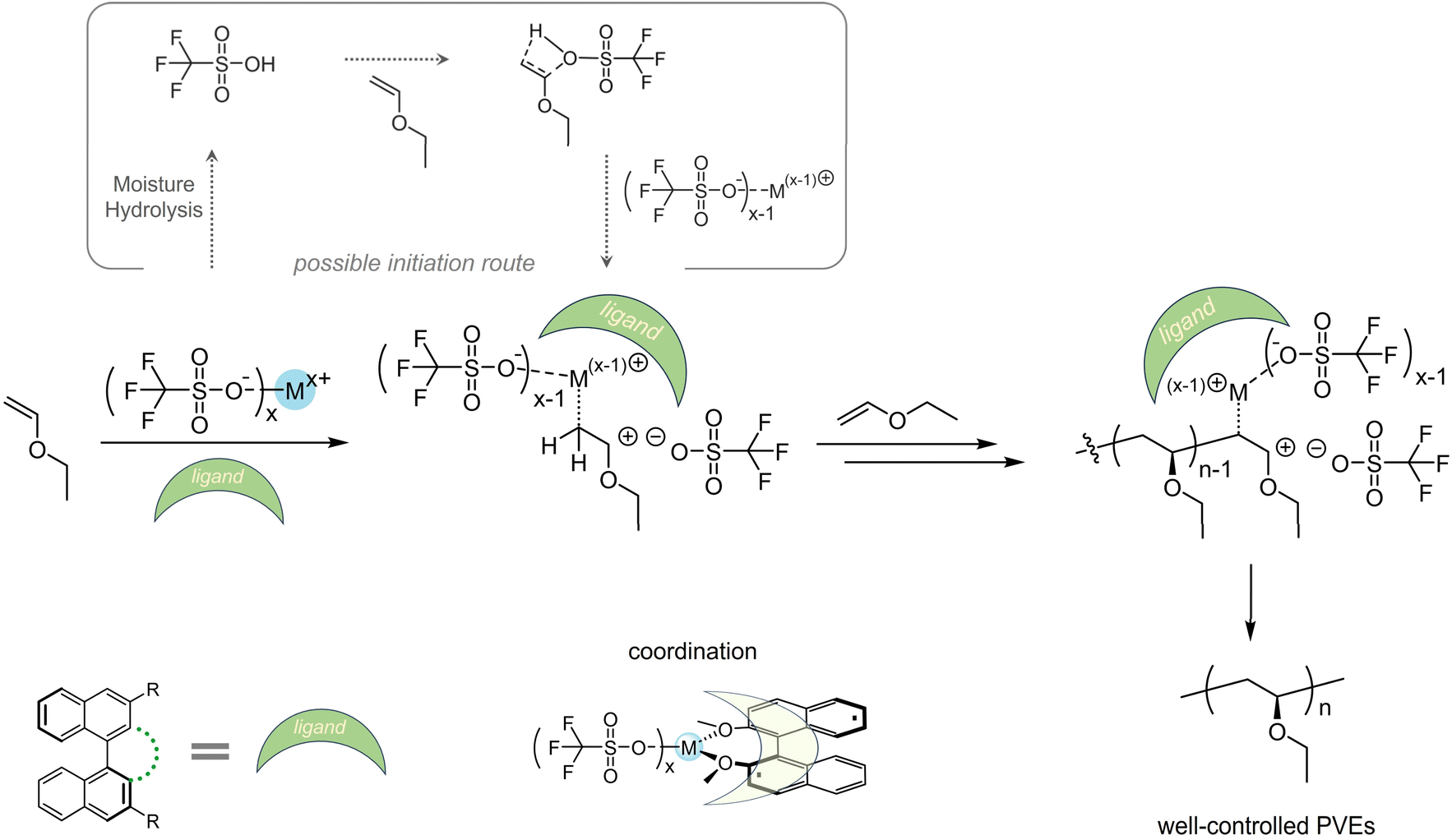 | ||
| Scheme 5 Proposed mechanism of cationic polymerization of EVE initiated by trifluoromethyl sulfonates/BINOL ligands. L10 was employed as an instance for coordination. | ||
3. Summary and conclusions
The cationic polymerization of vinyl ethers by employing trifluoromethyl sulfonates, which are commercially available, air-stable and applicable in many solvents, was successfully conducted. At room temperature, low-molecular-weight polydisperse poly(vinyl ether) (PVE) polymers were obtained due to chain transfer led by active cationic species. By lowering the polymerization temperatures and employing O^O type ether ligands, lowly dispersed polymers were prepared. With BINOL ligands, well-controlled PVEs with high isotacticities up to 81% m were acquired. Highly isotactic PEVEs would exhibit semicrystallization behaviors with melting behaviors. Then this work turned to subjecting these trifluoromethyl sulfonates to cationic RAFT polymerization, which can effectively access lowly dispersed PVEs and block copolymers. The resulting crystalline block copolymer found its application in crystallization-driven self-assembly (CDSA) to afford nanomaterials. After probing the optimal combinations of trifluoromethyl sulfonate/solvents/ligands and polymerization conditions, it was found that more vinyl ethers can be catalyzed to form more PVEs. Preparation of some crystalline PVEs was realized. We made poly(n-octadecyl vinyl ether) (P18VE) on an 80 g scale and found its similar mechanical performance to LDPE, the commonly used plastic packaging material.In conclusion, trifluoromethyl sulfonates, with the assistance of solvents and ligands, can be widely used to prepare PVE materials from inexpensive and underused feedstocks. Highly isotactic semicrystalline vinyl ether homo- and copolymers are synthesized and used for their applications. For PVEs, mechanical properties can be improved when both molecular weights and stereoselectivities are considered to be increased through well controlling chain transfer and stereoselective chain structures during polymerization of vinyl ethers. There are other valuable studies on the details of possible complexes formed by trifluoromethyl sulfonate/BINOL ligands and we are extending our scope to afford poly(vinyl ether)s under much milder conditions.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. C. and S. S. conceived the project and designed the experiments. L. C. carried out the experiments of synthesis and other characterization studies. Z. W. and E. F. conducted some characterization studies (DSC and TEM). S. S. and L. C. analyzed the data and co-wrote the manuscript with input from Z. F.; S. S. supervised the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 52473011) and International Scientific Research Cooperation Seed Fund of International Campus (Zhejiang University) is gratefully acknowledged.References
- S. Aoshima and S. Kanaoka, A Renaissance in Living Cationic Polymerization, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- Y. Chen, L. Zhang, Y. Jin, X. Lin and M. Chen, Recent Advances in Living Cationic Polymerization with Emerging Initiation/Controlling Systems, Macromol. Rapid Commun., 2021, 42, 2100148 CrossRef CAS.
- C. C. Sorensen, C. T. Kozuszek, M. A. Borden and F. A. Leibfarth, Asymmetric Ion-Pairing in Stereoselective Vinyl Polymerization, ACS Catal., 2023, 13, 3272–3284 CrossRef CAS.
- L. Wu, B. Rondon, S. Dym, W. Wang, K. Chen and J. Niu, Regulating Cationic Polymerization: From Structural Control to Life Cycle Management, Prog. Polym. Sci., 2023, 145, 101736 CrossRef CAS.
- X. Feng, R. Liu, L. Liu, Y. Jin, Q. Shi, P. Yan and Y. Wu, Recent Advances on Visible Light Induced Cationic Polymerization, J. Polym. Sci., 2023, 61, 2411–2425 CrossRef CAS.
- S. Singha, S. Pan, S. S. Tallury, G. Nguyen, R. Tripathy and P. De, Recent Developments on Cationic Polymerization of Vinyl Ethers, ACS Polym. Au, 2024, 4, 189–207 CrossRef CAS.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Living Polymerization of Isobutyl Vinyl Ether with Hydrogen Iodide/Iodine Initiating System, Macromolecules, 1984, 17, 265–268 CrossRef CAS.
- R. Faust and J. P. Kennedy, Living Carbocationic Polymerization, Polym. Bull., 1986, 15, 317–323 CrossRef CAS.
- M. Kamigaito, Y. Maeda, M. Sawamoto and T. Higashimura, Living Cationic Polymerization of Isobutyl Vinyl Ether by Hydrogen Chloride/Lewis Acid Initiating Systems in the Presence of Salts: In-Situ Direct NMR Analysis of the Growing Species, Macromolecules, 1993, 26, 1643–1649 CrossRef CAS.
- S. Aoshima, Y. Ito and E. Kobayashi, Stereoregularity of Poly(Vinyl Ether)s with a Narrow Molecular Weight Distribution Obtained by the Living Cationic Polymerization, Polym. J., 1993, 25, 1161–1168 CrossRef CAS.
- M. Kamigaito, M. Sawamoto and T. Higashimura, Alkoxy-Substituted Titanium(IV) Chlorides as Lewis Acid Activators for Living Cationic Polymerization of Isobutyl Vinyl Ether. Control of Lewis Acidity in the Design of Initiating Systems, Macromolecules, 1995, 28, 5671–5675 CrossRef CAS.
- T. Kawaguchi, F. Sanda and T. Masuda, Polymerization of Vinyl Ethers with Transition-Metal Catalysts: An Examination of the Stereoregularity of the Formed Polymers, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3938–3943 CrossRef CAS.
- L. Sipos, P. De and R. Faust, Effect of Temperature, Solvent Polarity, and Nature of Lewis Acid on the Rate Constants in the Carbocationic Polymerization of Isobutylene, Macromolecules, 2003, 36, 8282–8290 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Major Progress in Catalysts for Living Cationic Polymerization of Isobutyl Vinyl Ether: Effectiveness of a Variety of Conventional Metal Halides, Macromolecules, 2009, 42, 3965–3972 CrossRef CAS.
- P. Sudhakar and K. Vijayakrishna, Polymerization of Vinyl Ethers Using Titanium Catalysts Containing Tridentate Triamine Ligand of the Type N[CH2CH(Ph)(Ts)N]22-, Polymer, 2009, 50, 783–788 CrossRef CAS.
- P. Sudhakar and K. Vijayakrishna, Highly Stereoselective Living Polymerization of Vinyl Ethers at Ambient Temperature Mediated by Chiral Titanium Complexes, ChemCatChem, 2010, 2, 649–652 CrossRef CAS.
- H. Kalita, S. Selvakumar, A. Jayasooriyamu, S. Fernando, S. Samanta, J. Bahr, S. Alam, M. Sibi, J. Vold, C. Ulven and B. J. Chisholm, Bio-Based Poly(Vinyl Ether)s and Their Application as Alkyd-Type Surface Coatings, Green Chem., 2014, 16, 1974–1986 RSC.
- M. Uchiyama, K. Satoh and M. Kamigaito, Cationic RAFT Polymerization Using ppm Concentrations of Organic Acid, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS PubMed.
- V. Kottisch, Q. Michaudel and B. P. Fors, Cationic Polymerization of Vinyl Ethers Controlled by Visible Light, J. Am. Chem. Soc., 2016, 138, 15535–15538 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, A Phosphonium Intermediate for Cationic RAFT Polymerization, Polym. Chem., 2016, 7, 1387–1396 RSC.
- Q. Michaudel, V. Kottisch and B. P. Fors, Cationic Polymerization: From Photoinitiation to Photocontrol, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- M. Uchiyama, K. Satoh, T. G. McKenzie, Q. Fu, G. G. Qiao and M. Kamigaito, Diverse Approaches to Star Polymers via Cationic and Radical RAFT Cross-Linking Reactions Using Mechanistic Transformation, Polym. Chem., 2017, 8, 5972–5981 RSC.
- K. Takagi, K. Yamauchi and H. Murakata, Halogen-Bonding-Mediated and Controlled Cationic Polymerization of Isobutyl Vinyl Ether: Expanding the Catalytic Scope of 2-Iodoimidazolium Salts, Chem.–Eur. J., 2017, 23, 9495–9500 CrossRef CAS.
- V. Kottisch, J. O'Leary, Q. Michaudel, E. E. Stache, T. H. Lambert and B. P. Fors, Controlled Cationic Polymerization: Single-Component Initiation under Ambient Conditions, J. Am. Chem. Soc., 2019, 141, 10605–10609 CrossRef CAS.
- A. J. Teator and F. A. Leibfarth, Catalyst-Controlled Stereoselective Cationic Polymerization of Vinyl Ethers, Science, 2019, 363, 1439–1443 CrossRef CAS PubMed.
- A. J. Teator, T. P. Varner, P. E. Jacky, K. A. Sheyko and F. A. Leibfarth, Polar Thermoplastics with Tunable Physical Properties Enabled by the Stereoselective Copolymerization of Vinyl Ethers, ACS Macro Lett., 2019, 8, 1559–1563 CrossRef CAS PubMed.
- J. Li, M. Zhang, X. Pan, Z. Zhang, S. Perrier, J. Zhu and X. Zhu, Visible Light Induced Controlled Cationic Polymerization by in situ Generated Catalyst From Manganese Carbonyl, Chem. Commun., 2019, 55, 7045–7048 RSC.
- T. P. Varner, A. J. Teator, Y. Reddi, P. E. Jacky, C. J. Cramer and F. A. Leibfarth, Mechanistic Insight into the Stereoselective Cationic Polymerization of Vinyl Ethers, J. Am. Chem. Soc., 2020, 142, 17175–17186 CrossRef CAS.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, Metal-free Cationic Polymerization of Vinyl Ethers with Strict Temporal Control by Employing an Organophoto catalyst, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS.
- P. C. Knutson, A. J. Teator, T. P. Varner, C. T. Kozuszek, P. E. Jacky and F. A. Leibfarth, Bronsted Acid Catalyzed Stereoselective Polymerization of Vinyl Ethers, J. Am. Chem. Soc., 2021, 143, 16388–16393 CrossRef CAS PubMed.
- V. Kottisch, J. Jermaks, J.-Y. Mak, R. A. Woltornist, T. H. Lambert and B. P. Fors, Hydrogen Bond Donor Catalyzed Cationic Polymerization of Vinyl Ethers, Angew. Chem., Int. Ed., 2021, 60, 4535–4539 CrossRef CAS PubMed.
- M. Chen, J. J. Li, K. Q. Ma, G. Q. Jin, X. Q. Pan, Z. B. Zhang and J. Zhu, Controlling Polymer Molecular Weight Distribution through a Latent Mediator Strategy with Temporal Programming, Angew. Chem., Int. Ed., 2021, 60, 19705–19709 CrossRef CAS.
- X. Zhang, Z. Yang, Y. Jiang and S. Liao, Organocatalytic, Stereoselective, Cationic Reversible Addition–Fragmentation Chain-Transfer Polymerization of Vinyl Ethers, J. Am. Chem. Soc., 2022, 144, 679–684 CrossRef CAS PubMed.
- S. W. Spring, J. H. Hsu, R. J. Sifri, S.-M. Yang, C. S. Cerione, T. H. Lambert, C. J. Ellison and B. P. Fors, Poly(2,3-Dihydrofuran): A Strong, Biorenewable, and Degradable Thermoplastic Synthesized via Room Temperature Cationic Polymerization, J. Am. Chem. Soc., 2022, 144, 15727–15734 CrossRef CAS PubMed.
- S. L. Shankel, T. H. Lambert and B. P. Fors, Moisture Tolerant Cationic RAFT Polymerization of Vinyl Ethers, Polym. Chem., 2022, 13, 5974–5979 RSC.
- M. Uchiyama, Y. Murakami, K. Satoh and M. Kamigaito, Synthesis and Degradation of Vinyl Polymers with Evenly Distributed Thioacetal Bonds in Main Chains: Cationic DT Copolymerization of Vinyl Ethers and Cyclic Thioacetals, Angew. Chem., Int. Ed., 2023, 62, e202215021 CrossRef CAS.
- Y. P. Xu, L. Wang, C. S. Chen, P. Huang, H. J. Dai, W. F. Jiang and Y. F. Zhou, Living Cationic Polymerization of ε-Caprolactone Catalyzed by a Metal-free Lewis Acid of Trityl Tetrafluoroborate, Macromolecules, 2023, 56, 501–509 CrossRef CAS.
- Y. Mishima, A. Kanazawa and S. Aoshima, Photoinduced Superacceleration of Metal-Free Living Cationic Polymerization Using Diaryliodonium Salts as Organic Lewis Acid Catalysts, Macromolecules, 2023, 56, 6941–6950 CrossRef CAS.
- J. Chen, J. Lin, Z. Yang, Y. Wu and S. Liao, The Development of Highly Efficient Monophosphonium Photocatalysts for the Visible Light-Regulated Metal-Free Cationic Polymerization of Vinyl Ethers, Polym. Chem., 2023, 14, 486–491 RSC.
- B. M. Hosford, W. Ramos and J. R. Lamb, Combining Photocontrolled-Cationic and Anionic-Group-Transfer Polymerizations using a Universal Mediator: Enabling Access to Two- and Three-Mechanism Block Copolymers, Chem. Sci., 2024, 15, 13523–13530 RSC.
- Z. Wei, W. He, Z. Liu, Y. Lin, M. Wang, L. Li, C. Wu, S. Yang, G. Liu and R. Yang, Orthogonal Radical and Cationic Single-Unit Monomer Insertions for Engineering Polymer Architectures, Angew. Chem., Int. Ed., 2024, 63, e202402265 CrossRef CAS PubMed.
- Z. Yang, Y. Liao, Z. Zhang, J. Chen, X. Zhang and S. Liao, Asymmetric Ion-Pairing Photoredox Catalysis for Stereoselective Cationic Polymerization under Light Control, J. Am. Chem. Soc., 2024, 146, 6449–6455 CrossRef CAS.
- Y. Eguchi, A. Kanazawa and S. Aoshima, Metal-Free, Photoinitiated Cationic Terpolymerization of Vinyl Ethers, Oxiranes, and Ketones: Simultaneous Control of Monomer Sequence and Molecular Weight by the Formation of Long-Lived Propagating Species, Macromolecules, 2024, 57, 3346–3357 CrossRef CAS.
- Y. Zhou, Y. Sun, Y. Zhang and M. Hong, One-Step Synthesis of a Degradable Thermoplastic Elastomer with Gradient Sequence via Chiral Brønsted Acid-Mediated Cationic Copolymerization of Vinyl Ether Monomer Mixtures, Macromolecules, 2024, 57, 7227–7236 CrossRef CAS.
- K. Satoh and M. Kamigaito, Stereospecific Living Radical Polymerization: Dual Control of Chain Length and Tacticity for Precision Polymer Synthesis, Chem. Rev., 2009, 109, 5120–5156 Search PubMed.
- C. De Rosa and F. Auriemma, Structure and Physical Properties of Syndiotactic Polypropylene: A Highly Crystalline Thermoplastic Elastomer, Prog. Polym. Sci., 2006, 31, 145–237 CrossRef CAS.
- V. Busico, Giulio Natta and the Development of Stereoselective Propene Polymerization, Adv. Polym. Sci., 2013, 257, 37–58 CrossRef CAS.
- G. W. Coates, Precise Control of Polyolefin Stereochemistry Using Single-Site Metal Catalysts, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed.
- J. Collomb, A. Gandini and H. Cheradame, Cationic Polymerisation Induced by Metal Salts, 2 A Preliminary Communication on Homogeneous Systems, Makromol. Chem., Rapid Commun., 1980, 1, 489–491 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, Controlled cationic polymerization of p-methoxystyrene in aqueous media with Yb(OTf)3, Macromolecules, 1999, 32, 3827–3832 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, Lanthanide triflates-mediated emulsion cationic polymerization of p-alkoxystyrenes in aqueous media, Macromolecules, 2000, 33, 4660–4666 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, Metal triflates and tetrafluoroborates as water-tolerant lewis acids for cationic polymerization in aqueous media, Macromolecules, 2000, 33, 5836–5840 CrossRef CAS.
- K. Satoh, M. Kamigaito and M. Sawamoto, Sulfonic acids as water-soluble initiators for cationic polymerization in aqueous media with Yb(OTf)3, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2728–2733 CrossRef CAS.
- M. Rahm, R. Hoffmann and N. W. Ashcroft, Atomic and Ionic Radii of Elements 1–96, Chem. Eur. J., 2016, 22, 14625–14632 CrossRef CAS.
- M. Ouchi, M. Kamigaito and M. Sawamoto, Stereoregulation in Cationic Polymerization by Designed Lewis Acids. II. Effects of Alkyl Vinyl Ether Structure, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1060–1066 CrossRef CAS.
- S. F. Song, X. Y. Liu, H. Zhang, Z. S. Fu, J. T. Xu and Z. Q. Fan, Trans-1,4-Stereospecific Copolymerization of Ethylene and Isoprene Catalyzed by MgCl2-Supported Ziegler–Natta Catalyst, J. Polym. Sci., Part A:Polym. Chem., 2018, 56, 2715–2722 CrossRef CAS.
- C. Wu, Y. Liu, C. Xiao, C. Hu, L. Chen, X. Pang and X. Chen, A New Radical Initiation for the Ring-Opening Polymerization of Tetrahydrofuran with a Radical Substitution Mechanism, Polymer, 2024, 300, 126984 CrossRef CAS.
- C. Wu, B. Liu, F. Lin, M. Wang and D. M. Cui, Cis-1,4-Selective Copolymerization of Ethylene and Butadiene: A Compromise between Two Mechanisms, Angew. Chem., Int. Ed., 2017, 56, 6975–6979 CrossRef CAS.
- X. Y. Wang, Y. S. Gao and Y. Tang, Sustainable Developments in Polyolefin Chemistry: Progress, Challenges, and Outlook, Prog. Polym. Sci., 2023, 143, 101713 CrossRef CAS.
- E. Y. X. Chen, Coordination Polymerization of Polar Vinyl Monomers by Single-Site Metal Catalysts, Chem. Rev., 2009, 109, 5157–5214 CrossRef CAS PubMed.
- S. F. Song, X. Liu, E. Nikbin, J. Y. Howe, Q. Yu, I. Manners and M. A. Winnik, Uniform 1D Micelles and Patchy & Block Comicelles via Scalable, One-step Crystallization-Driven Block Copolymer Self-assembly, J. Am. Chem. Soc., 2021, 143, 6266–6280 CrossRef CAS PubMed.
- S. F. Song, H. Zhou, I. Manners and M. A. Winnik, Block Copolymer Self-assembly: Polydispersed Corona-Forming Blocks Leading to Uniform Morphologies, Chem, 2021, 7, 2800–2821 CAS.
- S. F. Song, J. J. Jiang, E. Nikbin, J. Y. Howe, I. Manners and M. A. Winnik, The Role of Cooling Rate in Crystallization-driven Block Copolymer Self-assembly, Chem. Sci., 2022, 13, 396–409 RSC.
- S. F. Song, Q. Yu, H. Zhou, G. Hicks, H. Zhu, C. K. Rastoogi, I. Manners and M. A. Winnik, Solvent Effects Leading to a Variety of Different 2D Structures in the Self-Assembly of a Crystalline-Coil Block Copolymer with an Amphiphilic Corona-Forming Block, Chem. Sci., 2020, 11, 4631–4643 RSC.
- B. Inci, I. Lieberwirth, W. Steffen, M. Mezger, R. Graf, K. Landfester and K. B. Wagener, Decreasing the Alkyl Branch Frequency in Precision Polyethylene: Effect of Alkyl Branch Size on Nanoscale Morphology, Macromolecules, 2012, 45, 3367–3376 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06181k |
| This journal is © The Royal Society of Chemistry 2025 |