
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMimic metalloenzymes with atomically dispersed Fe sites in covalent organic framework membranes for enhanced CO2 photoreduction†
Shuaiqi
Gao
a,
Xiao
Zhao
b,
Qian
Zhang
a,
Linlin
Guo
a,
Zhiyong
Li
 a,
Huiyong
Wang
*a,
Suojiang
Zhang
cd and
Jianji
Wang
*a
a,
Huiyong
Wang
*a,
Suojiang
Zhang
cd and
Jianji
Wang
*a
aKey Laboratory of Green Chemical Media and Reactions (Ministry of Education), Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China. E-mail: hywang@htu.edu.cn; jwang@htu.edu.cn
bScience and Technology on Aerospace Chemical Power Laboratory, Hubei Institute of Aerospace Chemotechnology, Xiangyang, 441003, P. R. China
cCollege of Chemistry and Molecular Sciences, Longzihu New Energy Laboratory, Henan University, Zhengzhou, Henan 450000, P. R. China
dBeijing Key Laboratory of Ionic Liquids Clean Process, State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 3rd December 2024
Abstract
The massive CO2 emissions from continuous increases in fossil fuel consumption have caused disastrous environmental and ecological crises. Covalent organic frameworks (COFs) hold the potential to convert CO2 and water into value-added chemicals and O2 to mitigate this crisis. However, their activity and selectivity are very low under conditions close to natural photosynthesis. In this work, inspired by the photosynthesis process in natural leaves, we successfully anchored atomically dispersed Fe sites into interlayers of the photoactive triazine-based COF (Fe–COF) membrane to serve as a mimic metalloenzyme for the first time. It is found that under gas–solid conditions and no addition of any photosensitizer and sacrificial reagent, the highly crystalline Fe–COF membrane shows a record high CO2 photoreduction performance with a CO production of 3972 μmol g−1 in a 4 h reaction, ∼100% selectivity of CO, and excellent cycling stability (at least 10 cycles). In such a remarkable photocatalytic CO2 conversion, the atomically dispersed Fe sites with high catalytic activity significantly reduce the formation energy barrier of key *CO2 and *COOH intermediates, the high-density triazine moieties supply more electrons to the iron catalytic center to promote CO2 reduction, and the homogeneous COF membrane greatly improves the electron/mass transport. Thus, this work opens a new window for the design of highly efficient photocatalysts and provides new insights into their structure–activity relationship in CO2 photocatalytic reduction.
Introduction
With the continuous increase in fossil fuel consumption, excessive CO2 emission has caused many environmental and ecological problems, such as global warming, disruption of weather patterns, and ocean acidification.1 Thus, for the sustainable development of modern civilization, carbon neutrality is becoming increasingly important around the world. Photocatalytic CO2 conversion to value-added chemicals is one of the technologies with the most potential for carbon neutrality. However, there are many problems to solve, including low yield, low selectivity, the use of extra photosensitizers and/or sacrificial agents, poor recycle and reuse performance, and difficult scaling up of the reaction systems. Recently, gas–solid reactions for photocatalytic CO2 reduction have been developed,2 where the photocatalyst particles are directly in contact with gaseous CO2 and H2O without using any photosensitizer, organic solvent, or sacrificial agent, which makes it easy to regenerate and reuse the systems for possible industrialization. Nevertheless, achieving a high yield and high selectivity of the target products is still a great challenge.Leaves of natural plants can directly convert CO2 and H2O into value-added chemicals by the gas–solid reaction with high efficiency and high selectivity, and they are also the largest factory for CO2 conversion and most important for the carbon cycle in nature.3 Basically, there are three main structural factors that determine such a highly efficient conversion in leaves: chloroplasts, metalloenzymes, and the electron transport chain and vein structure. Here, the chloroplast is responsible for light absorption to produce electrons and water photolysis to produce protons for CO2 reduction, and a metalloenzyme (such as ferredoxin) serves as the active center to catalyze the reactions,4 while the electron transport chain and vein structure provide highly rapid channels for electron and mass transport. These structural components are integrated into an organic unit to achieve highly efficient CO2 conversion synergistically.
Inspired by natural leaves, a photoactive center, mimic metalloenzyme, and electron/mass transport system have been combined in artificial photosynthesis for CO2 conversion.5–7 Among these factors, mimic metalloenzymes have attracted increasing attention in recent years.8–10 In this context, a mimic metalloenzyme is leveraged by the coordination interaction of active metal ions toward ligands (the primary coordination sphere) and the non-covalent interactions around the metal sites (the second coordination sphere), where the primary coordination sphere provides the catalytically active centers for CO2 reduction, while the second coordination sphere regulates the activity of the metal sites by weak interactions.11,12 Indeed, as an indispensable microenvironment, the second coordination sphere plays a vital role in mimicking metalloenzyme catalysis.13,14 However, in the previously reported design of mimic metalloenzymes, most of the investigations were focused on metal coordination, but not enough attention had been given to the second coordination sphere as well as the cooperation of a mimic metalloenzyme with a photoactive center and electron/mass transport system,15,16 which usually leads to low efficiency and selectivity.
Covalent organic frameworks (COFs) are a class of emerging porous materials with highly crystalline, regular porous structure and a π-conjugated system in the in-plane and stacking directions.17–19 This porous feature resembles veins and the electron transport chain in the natural leaves. Thus, COFs have been considered a potential platform to mimic natural metalloenzymes for CO2 conversion under gas–solid conditions in recent years. For example, for the first time, Lan et al.20 reported a kind of zincporphyrin-tetradentate tetrathiafulvalene COF (tetra-TTCOF-Zn), which combined a mimic metalloenzyme (zincporphyrin) and photosensitive center (porphyrin-tetrathiafulvalene) to perform CO2 conversion to CO, and the CO production was 12.33 μmol in a 60 h reaction. Recently, the same group21 developed a zincporphyrin-bidentate tetrathiofulvalenyl COF (bi-TTCOF-Zn) to enhance the photocatalytic activity, and bi-TTCOF-Zn exhibited a CO yield of 11.56 μmol g−1 h−1 and a selectivity of ∼100% due to the much stronger photoactivity of bidentate tetrathiafulvalene than that of tetradentate tetrathiafulvalene used in their previous work. Zhang and co-workers22 synthesized a new ionic liquid modified Zn-Salen-based triazine COF ([Emim][BF4]@Zn-S-COF) with Zn-Salen moieties as the mimic metalloenzyme and triazine moieties as the photosensitive center, and a much improved CO yield of 267.95 μmol g−1 h−1 was achieved. However, the yield of reduction products is too low to meet the needs of practical applications. After careful analysis of the above studies, we found that they generally suffer from the following problems: (i) CO2 catalytic activity of the Zn-based mimic metalloenzyme is low due to the low activity of Zn ions and the weak regulation of the second coordination sphere; (ii) the photoactivity of the photosensitive center is not strong enough except for triazine moieties; (iii) the COF catalysts used in such studies are all in powder form, which may lead to discontinuous electron/mass transfer and diffusion processes.23,24 Thus, to enhance the yield of products by COF-based catalysts, these bottlenecks must be addressed, at least in part, in the photocatalytic CO2 reduction reaction (CO2RR) to value-added chemicals and fuels.
In this work, for the first time, we designed and fabricated a highly efficient mimic metalloenzyme by anchoring atomically dispersed Fe sites into interlayers of the triazine-based COF membrane through N-coordination (Fig. 1). Here, the atomically dispersed Fe sites and imine moieties in the COF serve as the primary coordination sphere, and the benzene rings connected to the imines around Fe sites work as the second coordination sphere of the mimic metalloenzyme. Remarkably, the as-synthesized Fe–COF membrane achieved an impressive CO production of 3972 μmol g−1 in a 4 h reaction, approximately 100% selectivity, and high cycling stability (at least 10 cycles) toward CO2 photoreduction under gas–solid conditions without addition of any photosensitizer and sacrificial reagent, which outperforms all light-driven COF catalysts under gas–solid conditions reported so far. From X-ray absorption fine structure analysis and density functional theory (DFT) calculations, the superior photocatalytic performance is attributed to the synergy of the atomically dispersed Fe site and the triazine-based COF membrane host, where the atomically dispersed Fe sites decrease the reaction energy barriers for the formation of the key intermediates, and the high-density triazine moieties provide enough electrons to the Fe active center to promote CO2 reduction, while the unique physical form of homogeneous COF membranes behaves like a highway for charge/mass transport.
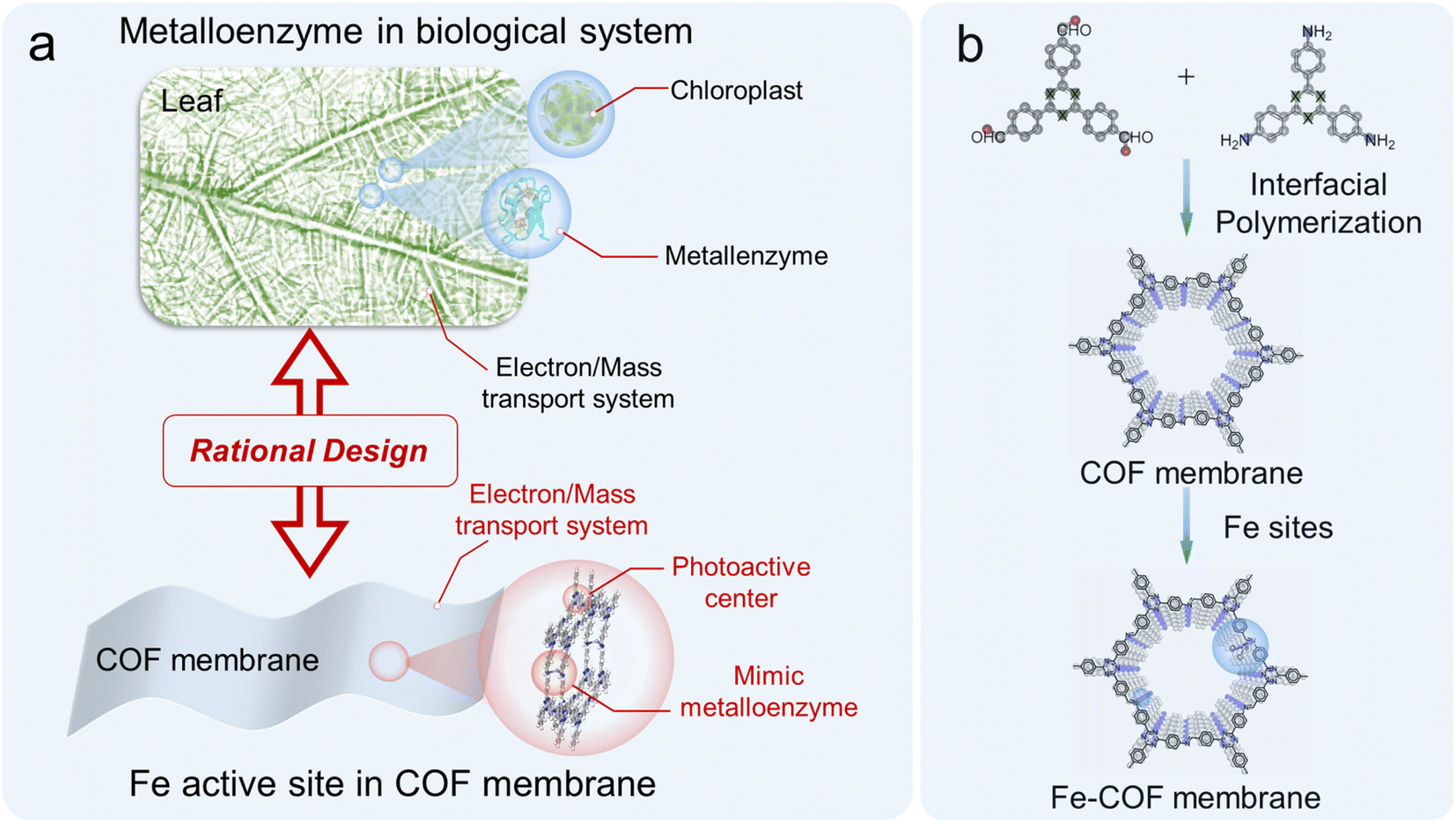 | ||
| Fig. 1 (a) Illustration of a metalloenzyme in a leaf and mimic metalloenzyme based on the Fe–COF membrane. (b) Schematic representation of the synthesis of Fe–COF membranes. | ||
Results and discussion
Structural characterization and analysis
As we recently reported,25 the ionic liquid–water interface is beneficial to prepare the highly crystalline COF membranes due to the dual-diffusion-controlled interfacial crystallization process. Thus, the freestanding triazine-based COF membrane was synthesized at the [C10Mim][Tf2N]–H2O interface by the condensation reaction of 2,4,6-tris(4-formylphenyl)-1,3,5-triazine (TTB) and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT), and no IL residual remains in the COF membrane after washing carefully. Then, different amounts of Fe(II) were anchored on the COF membrane to form Fe–COF membranes, and the synthetic details are shown in the ESI and Fig. S1.† The loading amount of atomically dispersed Fe atoms in these Fe–COF membranes was 0.8, 1.9, and 4.3 wt%, as determined by inductively coupled plasma mass spectrometry (ICP-MS). Unless otherwise specified, the Fe–COF membrane refers to the COF membrane with a Fe content of 1.9 wt%. Then, we applied Fourier transform infrared (FT-IR) spectroscopy, thermo-gravimetric analysis, X-ray diffraction (XRD) analysis, nitrogen adsorption–desorption isotherm, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) to understand the structures of these COF membranes. From the FT-IR spectrum, the observed characteristic peaks at 1400 and 1620 cm−1 are attributed to the stretching vibration of –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the pyridine ring and imine bond, respectively (Fig. S2†), which verify the successful synthesis of the targeted COF membrane.26 As shown in Fig. S3,† the thermogravimetric analysis reveals that both Fe–COF and pristine COF membranes present excellent thermal stability, which is also important for realistic catalytic applications.
C in the pyridine ring and imine bond, respectively (Fig. S2†), which verify the successful synthesis of the targeted COF membrane.26 As shown in Fig. S3,† the thermogravimetric analysis reveals that both Fe–COF and pristine COF membranes present excellent thermal stability, which is also important for realistic catalytic applications.
The XRD patterns of Fe–COF and pristine COF membranes (Fig. 2a and b) demonstrate that the strong diffraction peak at 3.97° is assigned to the (100) plane, and the other three peaks at 7.0, 8.1, and 10.5° are ascribed to the (110), (200), and (120) planes, respectively.27,28 The structural models for all these COF membranes were built and then optimized by using the experimental XRD data and the reflex module. The simulated AA eclipsed stacking models agree well with the experimental diffraction patterns. The nitrogen adsorption–desorption isotherm was evaluated to analyze the porosity of these COF membranes (Fig. 2c), and the BET surface areas of the pristine COF and Fe–COF membranes are measured to be 1012 and 651 m2 g−1, respectively. Moreover, the pore size of the COF membranes was calculated by the nonlocal density functional theory (NLDFT), and the results were found to be around 2.1 nm with pore volumes of 0.76 and 0.62 cm3 g−1 for pristine COF and Fe–COF membranes, respectively (Fig. S4†). The morphology details were investigated by SEM, where the surface roughness of pristine COF and Fe–COF membranes shows an unobvious difference (Fig. 2d and S5a†). The cross-sectional SEM images of each membrane exhibit a uniform thickness of 660 nm for the Fe–COF membrane and 650 nm for the pristine COF membrane (Fig. 2e and S5b†). Moreover, the high-resolution TEM images (Fig. 2f) and the two diffraction rings (Fig. 2g) also disclose the well-ordered crystallinity of the Fe–COF membrane.29 Specifically, the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images of the Fe–COF membrane show the ordered pores (Fig. 2h), and present bright dots (Fig. 2i), identifying atomically dispersed Fe atoms in the Fe–COF membrane. The above results confirm that homogeneous COF membranes with an ordered framework and atomically dispersed Fe atoms have been built, which may provide a suitable platform for the mimicking of metalloenzymes.
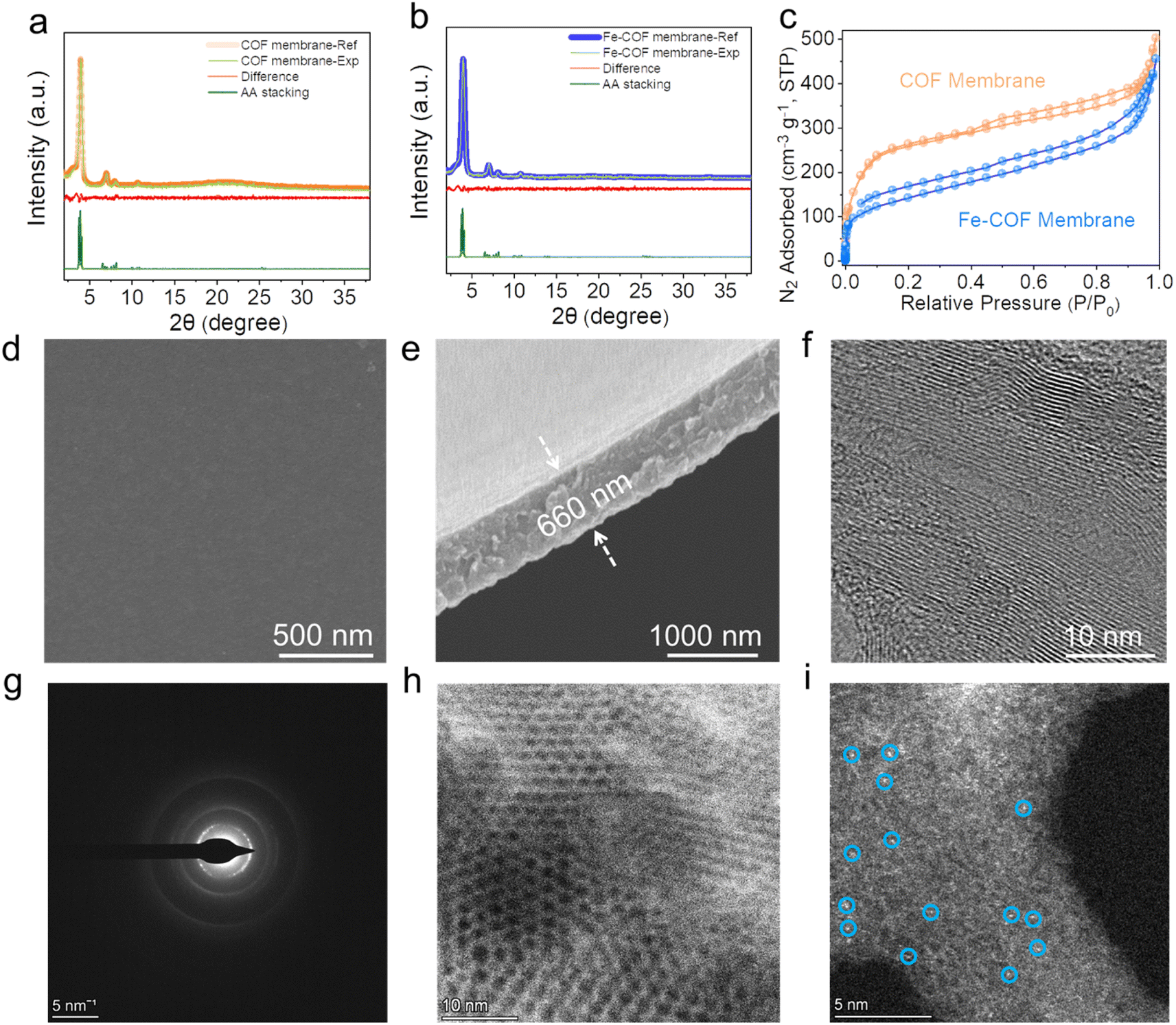 | ||
| Fig. 2 XRD patterns of the (a) Fe–COF membrane and (b) pristine COF membrane. (c) Nitrogen adsorption–desorption isotherm of different COF membranes. (d) Top-view SEM image and (e) cross-section SEM image of the Fe–COF membrane. (f) TEM image of the Fe–COF membrane. (g) Selected area electron diffraction (SAED) pattern of the Fe–COF membrane. (h and i) Aberration-corrected HAADF-STEM image of the Fe–COF membrane. | ||
The single atomic dispersion of the Fe site was investigated by energy-dispersive X-ray (EDX) mapping (Fig. 3a–c and S6†), which exhibited the uniform distribution of C, N, Cl, and Fe elements in the Fe–COF membrane. Then, the coordination environment of Fe atoms was investigated by X-ray absorption fine structure spectroscopy (XAFS). In the Fe K-edge X-ray absorption near-edge structure (XANES) spectrum (Fig. 3d), compared with Fe foil, FePc, FeO, and Fe2O3, the absorption edge position of the Fe–COF membrane is between that of FeO/FeAc and Fe2O3, but closer to that of Fe2O3, indicating that Fe species in the Fe–COF membrane mainly exist in trivalence state. In Fig. 3e, the Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) plots are exhibited, and the Fe–COF membrane shows a main peak at 1.62 Å, which belongs to the Fe–N bond.30–32 Moreover, there are no other peaks of the Fe–COF membrane in the range of 2.3–3.0 Å, such as those of Fe–O and Fe–Fe, suggesting that Fe atoms in the Fe–COF membrane are atomically dispersed.33 As shown in the wavelet transformation (WT) plot of the Fe–COF membrane (Fig. 3g–i), the WT maximum was detected at 5.7 Å−1, which is located between 5.2 Å−1 (characteristic peak of the Fe–N bond) and 6.3 Å−1 (characteristic peak of the Fe–Cl bond) and suggests that Fe–N and Fe–Cl bonds were present in the Fe–COF membrane. Quantitative EXAFS analysis (Fig. 3f and S7†) was conducted to obtain further structure information on the Fe atom in the Fe–COF membrane. The related fitting data are displayed in Table S1† and the fitting plots are consistent with the pristine data, thus demonstrating the reliability of fitting result of Fe–COF membrane.34 These results illustrate that Fe atoms are anchored into the COF membrane and surrounded by two N atoms and two Cl atoms with a Fe–N bond distance of 1.90 Å and a Fe–Cl bond distance of 2.16 Å, which builds a bridge to transfer electrons from the COF to Fe sites. Thus, the primary metal active sites have been successfully established.
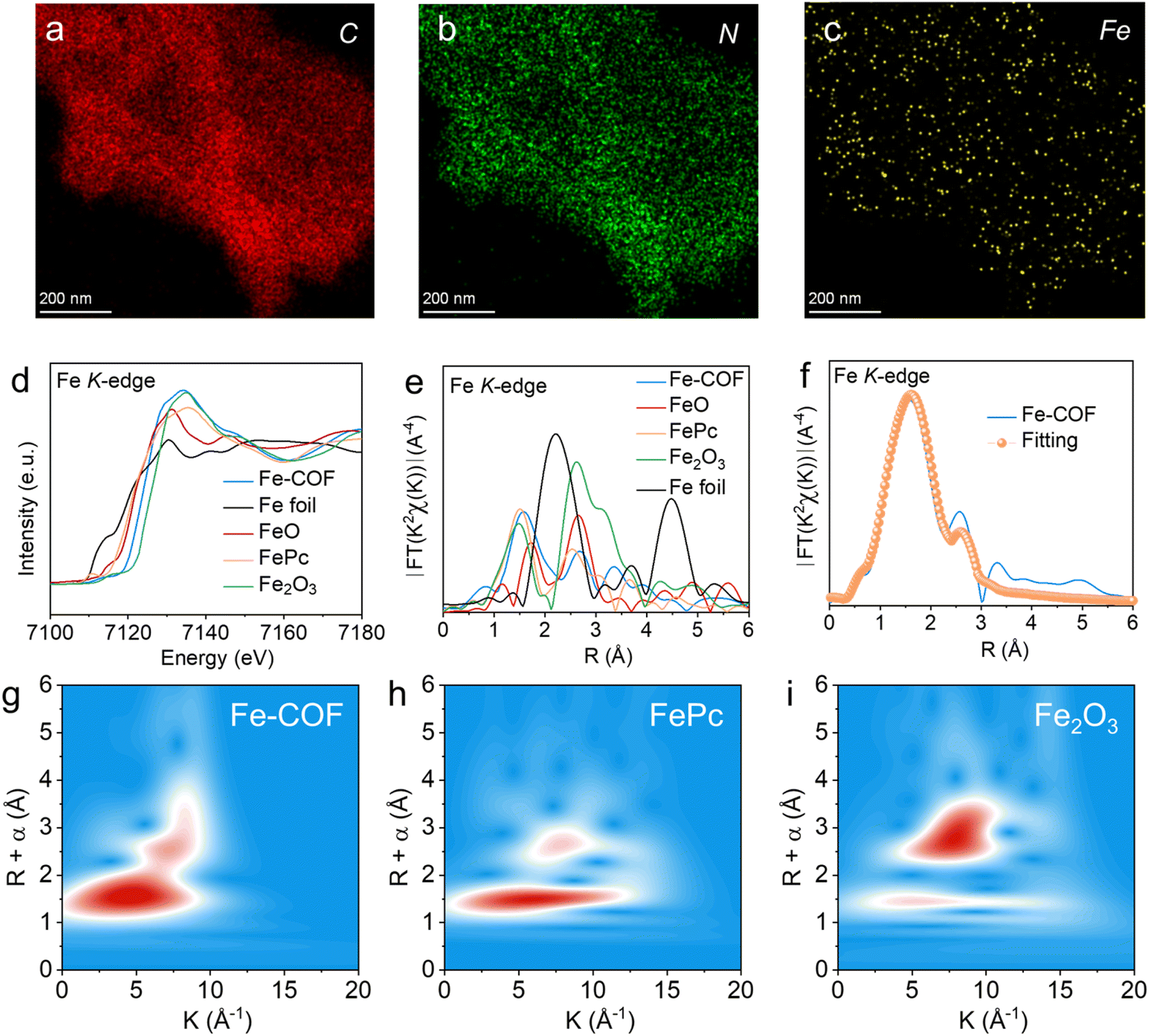 | ||
| Fig. 3 (a–c) Element mapping of the Fe–COF membrane. (d) Normalized XANES and (e) Fourier-transformed (FT) EXAFS spectra at the Fe K-edge. (f) The EXAFS fitting curves of the Fe–COF membrane in R space. The WT-EXAFS spectra of (g) the Fe–COF membrane, (h) FePc, and (i) Fe2O3. | ||
The coordination environment of Fe(II) in the Fe–COF membrane was also investigated by X-ray photoelectron spectroscopy, and this also exhibited the C, N, Cl, and Fe elements (Fig. S8a and b†). As shown in the N 1s XPS spectrum (Fig. S8c†), the N 1s signal of the imine-N in Fe–COF membrane (at 399.8 eV) is slightly shifted to higher binding energy compared to that in a pristine COF membrane, due to the coordination of N atoms to Fe(II).30,35 Additionally, in the high-resolution Fe 2p XPS spectrum (Fig. S8d†), the binding energy of Fe 2p3/2 and Fe 2p1/2 centered at 711.2 and 725.1 eV suggests the presence of Fe ions in the Fe–COF membrane rather than other Fe species such as metallic Fe.36 The coordinated environment of Fe atoms within the Fe–COF membrane was further investigated by DFT calculation (Fig. S9†), and the bond lengths of Fe–N and Fe–Cl were 1.91 Å and 2.19 Å, respectively, which means the Fe atom was coordinated by two N atoms in the imine moieties among the COF layers. Moreover, the bond length of Fe–N and Fe–Cl shown a good case with the above fitted EXAFS parameters (1.90 Å for Fe–N and 2.16 Å for Fe–Cl bonds, respectively). From the above results, we found that the Fe sites with interlayer coordination exhibited a significant difference with the reported mimic metalloenzyme based on the COF. Specifically, the metal sites in the reported COF mimic metalloenzymes are generally coordinated in layers,20–22 which results in the fact that each metal site can only accept electrons from at most two photoactive sites. However, in our case, the Fe sites with interlayer coordination may accept electrons from four photoactive sites. Thus, the Fe–COF membrane with a high density of triazine photocenters resulting from the specific Fe–N interlayer coordination is expected to boost CO2 reduction. In addition, the benzene rings connected to the imine moieties around Fe sites can be regarded as the second coordination sphere of the mimic metalloenzyme since it is an electron-donating moiety relative to imine, and may promote electron transfer to the Fe sites, and then enhance CO2 reduction.
Optical characters and photocatalytic performance
The influence of atomically dispersed Fe sites on the intrinsic light-harvesting capability and band structures of the membranes was studied by UV-vis diffuse reflectance spectroscopy (UV-vis DRS). As shown in Fig. 4a, the absorption edge of the Fe–COF membrane appears at ∼562 nm, which shows a red-shift compared with that of the pristine COF membrane (∼525 nm). This result shows that introducing Fe sites into the COF membrane could cause the vibronic broadening of the COF membrane and increased electron delocalization.26,37 The optical photograph exhibited the deeper color of the Fe–COF membrane compared with the pristine COF membrane (inset of Fig. 4a), which indicates that the Fe–COF membrane exhibits a stronger ability for the absorption of visible light. The band gap was determined using Tauc's relation,38,39 as shown in Fig. 4b, and the Fe–COF membrane possesses a smaller band gap (2.27 eV) than that of the pristine COF membrane (2.47 eV) due to the increased electron delocalization induced by the addition of single Fe(II). According to the Mott–Schottky curves (Fig. S10a and b†), the conduction band (CB) position was calculated for Fe–COF and the pristine COF membranes. Through the valence band (VB) XPS tests, the VB position of Fe–COF and the pristine COF membranes was also calculated (see Fig. S10c and d†). From the analysis of band gap structures (Fig. 4c), the CB position of the Fe–COF membrane is negative compared to that of the pristine COF membrane, suggesting stronger photocatalytic ability of the Fe–COF membrane toward CO2 reduction.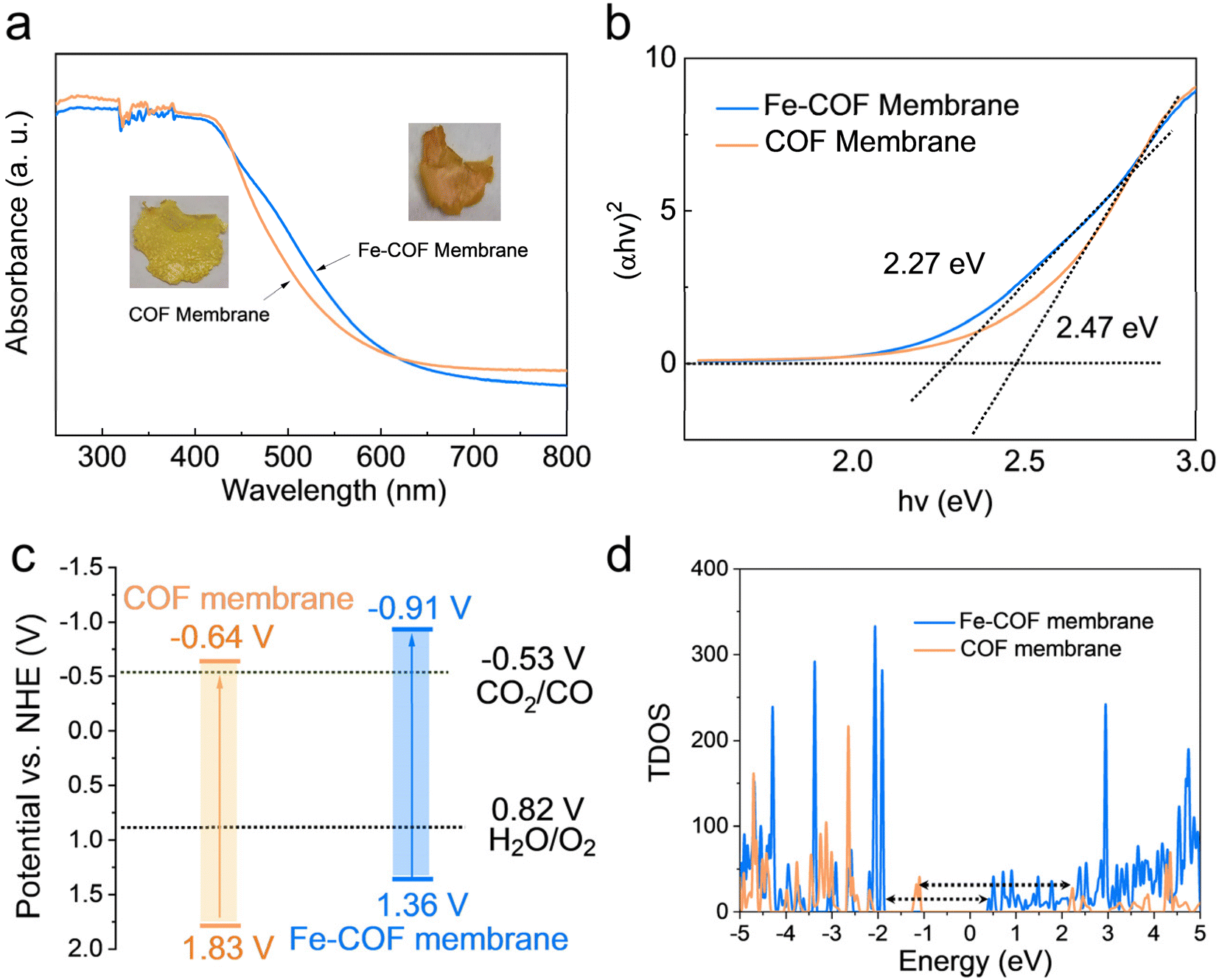 | ||
| Fig. 4 (a) UV-vis DRS spectra (inset: digital photographs) and (b) Tauc plots of Fe–COF and pristine COF membranes. (c) Energy band structure of Fe–COF and pristine COF membranes. (d) TDOS plots for Fe–COF and pristine COF membranes. | ||
Then, the electronic band structures of the Fe–COF and COF membranes were examined by DFT simulations, which also exhibited a narrower band gap for the Fe–COF membrane than that for the pristine COF membrane (Fig. S11†). As shown in Fig. 4d, the Fermi level (Ef) of the Fe–COF and pristine COF membranes was both in the region of density of states (DOS) = 0, predicting their semiconductor nature.30 Moreover, the total density of states (TDOS) at the valence band maximum and conduction band minimum was decreased by introducing atomically dispersed Fe atoms, leading to a narrower band gap than that of the pristine COF membrane, which caused easier photoinduced electron production and transfer.40 Moreover, the partial density of states (PDOS) of these COF membranes (Fig. S12a and b†) also showed a similar trend and indicated that the atomically dispersed Fe atom coordinated with N atoms in the COF membrane could enhance the electron transfer capability and the electron/hole separation performance.41 These results confirm that the introduction of atomically dispersed Fe sites into COF membranes is to the benefit of electron/hole separation and catalytic reactions.
Based on the above results, we performed CO2 photoreduction experiments at 25 °C with simulated solar irradiation under gas–solid conditions without the addition of any photosensitizer or sacrificial agent. First, a series of controlled experiments were conducted to determine the source of CO products. As shown in Fig. 5a, in the absence of CO2 or light illumination, the production of CO was not observed, verifying that the CO products were from the CO2 photoreduction. Subsequently, the photocatalytic performance of the Fe–COF membrane, Fe–COF powder, and pristine COF membrane was evaluated under the optimal conditions. It was shown that during the prolonged irradiation, the Fe–COF powder and the pristine COF membrane present a gradual increase in CO production although it is quite low. By contrast, the Fe–COF membrane shows an improved photocatalytic activity. After loading a tunable amount of the atomically dispersed Fe species (Fig. 5b), it is surprising to find that the Fe–COF membrane containing 1.9 wt% of Fe sites displays the highest CO production of 3968 μmol g−1 under 4 h light illumination, while the Fe–COF membranes containing 0.8 and 4.3 wt% of Fe exhibit a CO production of 1799 μmol g−1 and 2392 μmol g−1, respectively, showing that the loading content of Fe sites is not the more the better. These results suggest that an optimal loading amount of Fe species is also important for photocatalytic CO2 reduction, and a similar trend has been reported for other kinds of COFs in the literature.30 Clearly, under the optimal conditions, the CO yield of the Fe–COF membrane (1.9 wt% of Fe), 992 μmol g−1 h−1, is 3.2 times that of the pristine COF membrane and about 10.3 times that of the corresponding pristine COF powder (Fig. 5c), presenting a record high CO yield under gas–solid conditions among the COF catalysts reported previously (Table S2†). In addition, only CO and O2 were detected as the gaseous products by gas chromatography (Fig. S13†), and no liquid product was observed by 1H NMR spectroscopy (Fig. S14†). Meanwhile, the photocatalytic performance was still steady after 12 h, and the as-formed reduction product 13CO (m/z = 29), rather than CO (m/z = 28), originating from 13CO2 and the oxidation product 18O2 (m/z = 36) originating from H218O were detected by gas chromatography-mass spectrometry (Fig. S15†), which also verifies that the generated CO originates from CO2 rather than the decomposition of COF membranes.
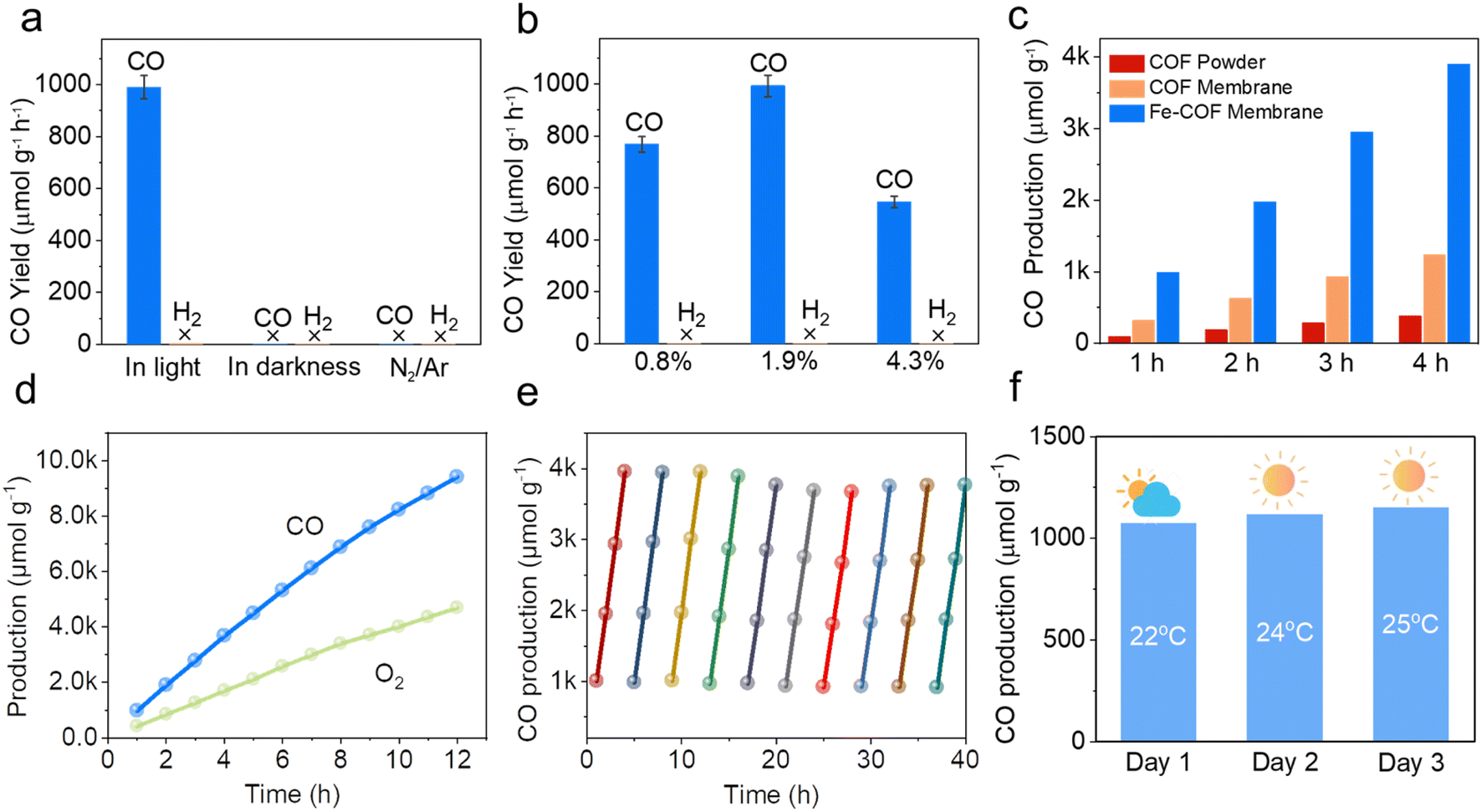 | ||
| Fig. 5 (a) Catalytic activity of the Fe–COF membrane under various reaction conditions. (b) Catalytic activity of different loadings of Fe atoms. (c) CO production in 4 hours catalyzed by the Fe–COF membrane, pristine COF membrane, and Fe–COF powder. (d) The CO production in 12 hours was catalyzed by the Fe–COF membrane. (e) Time courses of the photocatalytic activity and durability based on the Fe–COF membrane. Reaction conditions: catalyst, 1.00 mg; H2O, 3 mL; temperature, 25 °C; reaction time, 4 h for each cycle. (f) Practical performance tests based on the Fe–COF membrane under natural sunlight (4 h each day). | ||
The apparent quantum efficiency (AQE) of the Fe–COF membrane in photocatalytic CO2 reduction was measured at different wavelengths, and the results are shown in Table S3 and Fig. S16.† When the AQE values of the Fe–COF membrane were plotted against the absorption wavelength, the data points fell on its optical absorption spectrum curve.42 This result indicates that the Fe–COF membrane is responsible for the generation of excited electrons in the CO2RR. In addition, the long-term stability of photocatalysts was investigated due to their particular importance in practical applications (Fig. 5d). It was found that the CO yield of the Fe–COF membrane only slightly decreased to 937 μmol g−1 h−1 after 10 cyclic runs, demonstrating the excellent long-term cycling stability of the Fe–COF membrane (Fig. 5e). Furthermore, the unchanged PXRD patterns (Fig. S17†), FT-IR spectrum (Fig. S18†), and element mapping (Fig. S19†) of the Fe–COF membrane before and after recycling confirm its stable composition and crystal structure.
In terms of the practical application of the Fe–COF membrane-based mimic metalloenzyme, natural sunlight irradiation was also used for the photocatalytic CO2RR to CO. In detail, the Fe–COF membrane was mounted on a customized reaction tripod in a sealed glass reactor (250 mL) containing 3 mL of H2O at the bottom, which was then irradiated by direct sunlight under a CO2 atmosphere (1 atm) in the outdoor environment of the HNU (Henan Normal University) campus from 12:00 am to 4:00 pm on 28th April to 30th April in 2023 (the location is east longitude 113°54′46′′ and north longitude 35°19′46′′). As shown in Fig. 5f, the CO production is 1072 μmol g−1 in 4 hours of reaction on 30th April, and the resultant yield is as high as 287 μmol g−1 h−1 at an average temperature of 25 °C. At another time (say 28th April), the related CO yield is still above 263 μmol g−1 h−1 at a lower temperature of 22 °C. Without any help from the photosensitizer, and/or sacrificial agent, such a CO yield at much lower light intensity (less than 50 mW cm−2, Table S4†) is remarkable. This result indicates that with the advantages of the Fe–COF membrane as a mimic metalloenzyme, natural sunlight can also be efficiently utilized for photocatalytic CO2 reduction to CO.
A possible reaction mechanism
High accessibility of CO2 is important for high-performance catalysts for the CO2RR. As shown in Fig. S20a,† the Fe–COF membrane exhibited a high capacity for CO2 adsorption, especially at low pressure compared with the pristine COF membrane. This result reveals that the Fe–COF membrane had a stronger affinity towards CO2 molecules than the pristine COF membrane, which was confirmed by a temperature-programmed CO2 desorption experiment (Fig. S20b†). Moreover, after long-term use of the Fe–COF membrane, the change in CO2 adsorption capacity was very small (Fig. S21†), which also suggests the stable and reusable active sites in Fe–COF membranes. Then, in situ XPS spectroscopic measurement was performed to display the charge transfer process in the CO2RR, and the results are shown in Fig. 6a and b. It can be seen that the Fe 2p spectrum exhibits two binding energies of Fe 2p3/2 at 711.6 eV and Fe 2p1/2 at 725.3 eV. After simulated solar irradiation, they decreased to 711.3 and 724.9 eV, respectively. On the other hand, the binding energy of N 1s at 398.1 eV increased to 398.2 eV after simulated solar irradiation. We also determined the photocurrent density of the COF membranes with and without triazine moieties (Fig. S22†). The tiny photocurrent density of COF membranes without triazine moieties strongly suggests that triazine moieties are effective photoactive centers. Then, the electron density distribution of benzene rings in the pristine COF and Fe–COF membranes was calculated (Fig. S23†), and the results showed that the Bader charge of benzene rings in the pristine COF membrane and the Fe–COF membrane was −0.633e and −0.586e, respectively, which means that the partial electrons of benzene rings were transferred to Fe sites. These results clarify the electron donation properties of benzene rings in the COF membrane. Moreover, it is noted that the photoluminescence intensity (Fig. 6c), photoluminescence lifetime (Fig. 6d), photocurrent density (Fig. 6e) and charge transfer resistance (Fig. 6f) dramatically changed with addition of the atomically dispersed Fe sites in the COF membrane, where the higher transient photocurrent density reveals the better separation efficiency of photogenerated electrons and holes, the higher PL intensity implies the faster recombination rate of the electron–hole pairs, and the longer PL lifetime indicates the longer existing time of the photogenerated electrons. Due to the high density of photoactive sites resulting from the specific Fe–N interlayer coordination, the Fe–COF membrane presents the maximum photocurrent density, the lowest photoluminescence intensity, the longest photoluminescence lifetime, and the lowest charge transfer resistance. These results demonstrate that the rational design of the proportion between photoactive triazine moieties and the atomically dispersed Fe sites can strongly promote the separation of photogenerated electrons and holes.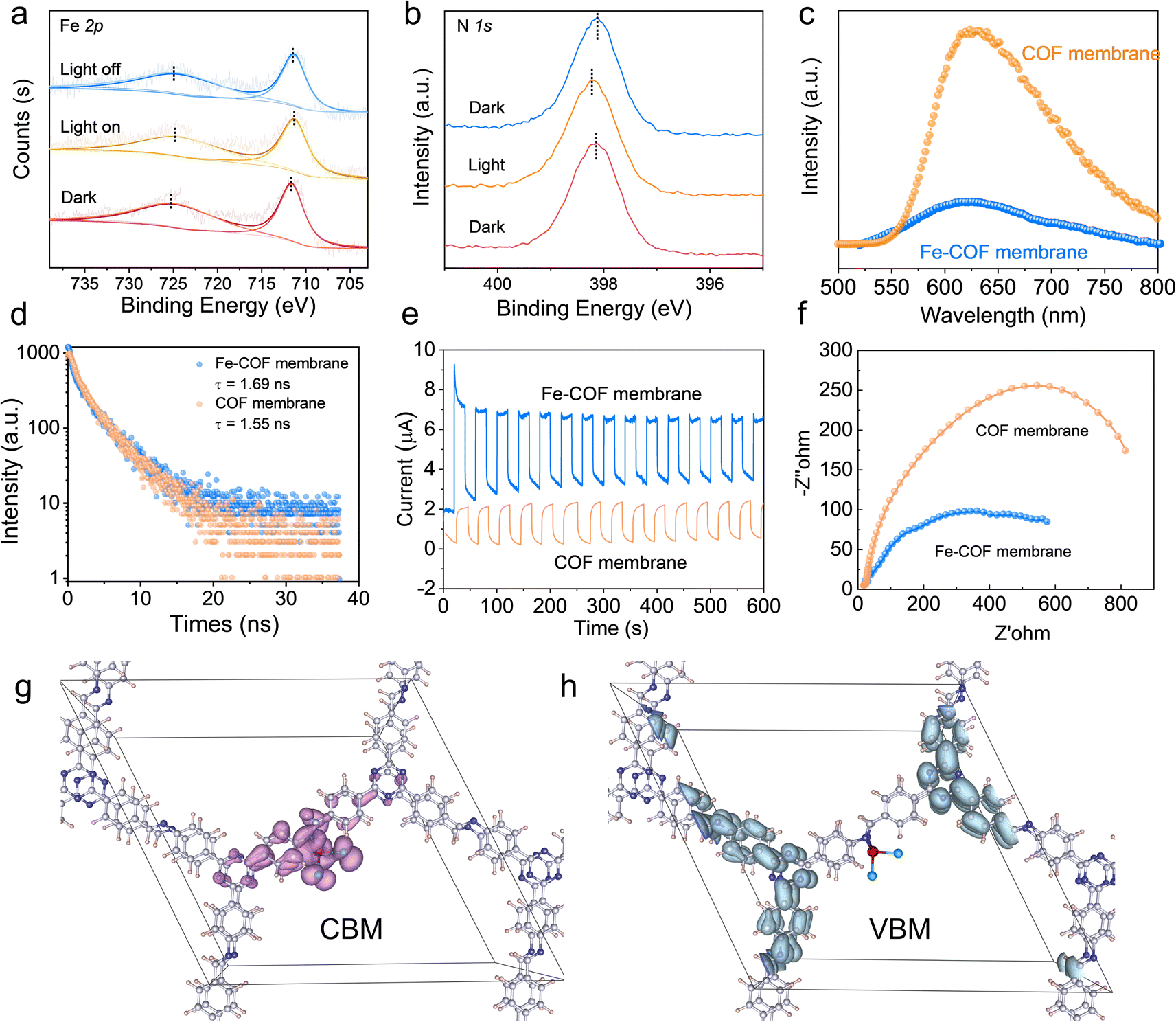 | ||
| Fig. 6 (a and b) Insitu high-resolution XPS spectra. (c) Photoluminescence intensity. (d) Photoluminescence lifetime. (e) Photocurrent density. (f) Charge transfer resistance of different COF membranes. (g) The CBM and (h) VBM of the Fe–COF membrane. | ||
Moreover, DFT calculations (Fig. 6g) show that the partial charge density of the conduction band minimum (CBM) for the Fe–COF membrane was delocalized across the imine moieties and atomically dispersed Fe sites, which means that the dispersed Fe sites are the electron acceptor. Nevertheless, the partial charge density of the valence band maximum (VBM), which serves as the electron donor, was mainly concentrated on triazine and the surrounding benzene moieties in the Fe–COF membrane (Fig. 6h). This result displays the highly efficient electron/hole separation in the Fe–COF membrane, which is superior to that in the pristine COF membrane (Fig. S24†). Combined with the photoelectric measurements and CBM–VBM results, it is also reasonable to deduce that the COF membrane with triazine moieties produced electrons after light irradiation, which were then transferred to dispersed Fe sites for CO2 photoreduction. That is to say, the COF membrane serves as a highway for efficient charge/mass transport.23 Moreover, the atomically dispersed Fe sites in the Fe–COF membrane shorten the electron transfer distance, accelerate the electron/hole separation, and promote electron accumulation for efficient CO2 reduction, thus endowing the mimic metalloenzyme with outstanding photocatalytic performance.
To uncover the catalytic CO2 photoreduction process of the Fe–COF membrane, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) studies were performed to disclose the intermediates involved in the photocatalytic CO2 reduction. As shown in Fig. 7a, the infrared peak intensity of the Fe–COF membrane shows remarkable changes with the extension of the irradiation time. The peak that appears at 1642 cm−1 is assigned to the characteristic *COOH intermediate, which could enhance the CO2 reduction of the Fe–COF membrane.39 Moreover, the gradually enhanced peak at 2146 cm−1 for the *CO intermediate also occurs in the photocatalytic process, which is another critical intermediate for the photocatalytic CO2RR to CO.26 The new peaks at 1450, 1278, and 1382 cm−1 are ascribed to *HCO3− (at 1450 and 1278 cm−1) and *CO32− (at 1382 cm−1), and these intermediates may reversibly transform into CO2 in the presence of gaseous H2O molecules.6
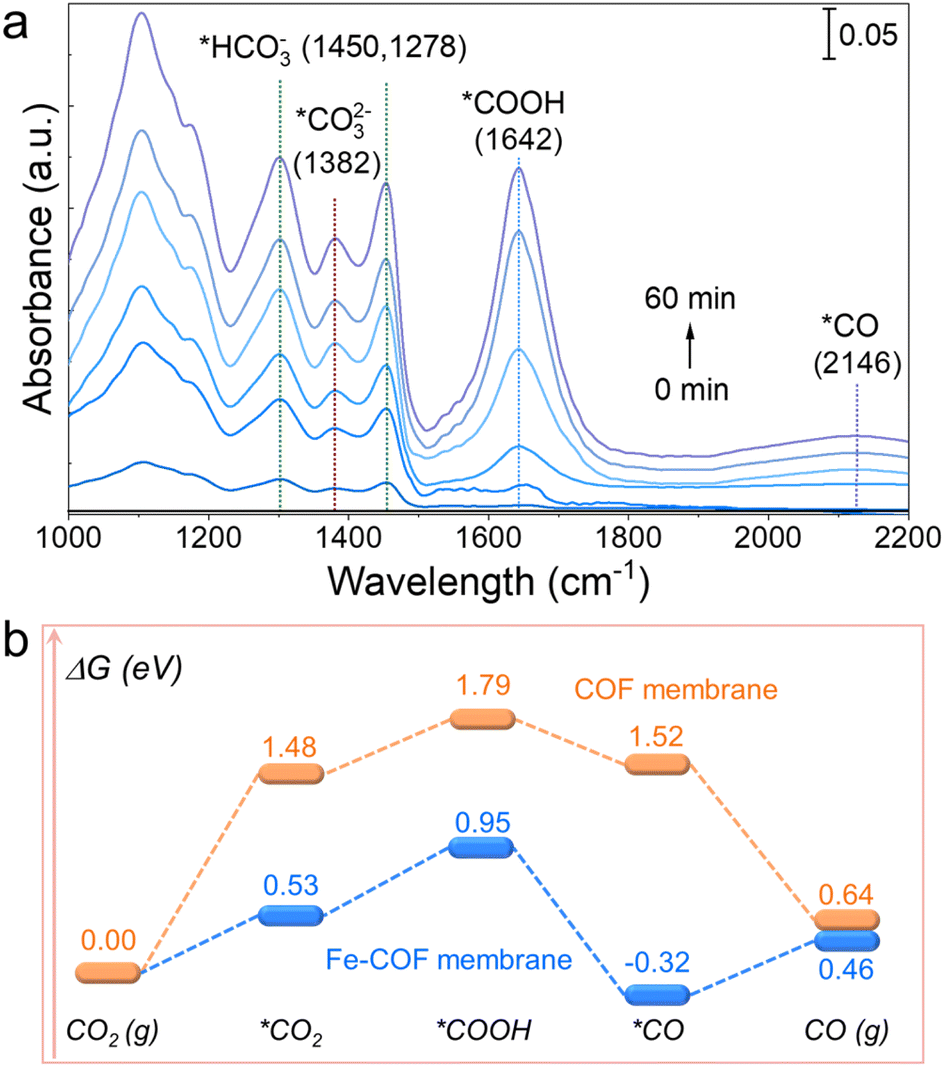 | ||
| Fig. 7 (a) In situ DRIFTS of the Fe–COF membrane during the CO2 photoreduction process. (b) Free energy profile of the CO2RR to CO catalyzed by the Fe–COF and the pristine COF membranes. | ||
Combined with DRIFTS analysis, we calculated the steps of CO2 photoreduction on the Fe–COF membrane. First, the membranes adsorb CO2 molecules to form *CO2, and then, the adsorbed *CO2 molecule interacts with the protons to gradually form the *COOH intermediates via an initial hydrogenation step, where the *COOH intermediates are further protonated and dehydrated to form the *CO product. The CO product as the last procedure depends on the desorption of adsorbed *CO from the catalyst surface. Furthermore, we calculated the change in Gibbs free energy (ΔG) for the steps of CO2 photoreduction. As shown in Fig. 7b and S25,† the energy barrier of CO2 adsorption in the Fe–COF membrane is much smaller (0.53 eV) than that of the pristine COF membrane (1.48 eV). Moreover, the formation energy barrier of the *COOH intermediate on the Fe–COF membrane is also much smaller than that of the pristine COF membrane (0.95 eV vs. 1.79 eV). The decreased formation energy barrier proves that the atomically dispersed Fe atoms promote the electron transfer from the photoactive center to the catalytically active center, which causes efficient conversion of the *COOH species on the electron-gathered Fe centers in the Fe–COF membrane, resulting in enhanced photocatalytic CO2 reduction. Moreover, the energy emission of the transition from *COOH to *CO intermediates on the Fe–COF membrane needs −0.32 eV, which is much lower than that on the pristine COF membrane (1.52 eV), indicating that the transition on the Fe–COF membrane is more thermodynamically profitable. Furthermore, the CO molecules are more conducive to the formation on the Fe–COF membrane than that on the pristine COF membrane due to the smaller energy barriers of CO (g) on the Fe–COF membrane.
According to the above analysis, we proposed a probable photocatalytic mechanism for the CO2RR over the Fe–COF membrane. First, the Fe–COF membrane effectively captures CO2 molecules due to the atomically dispersed Fe atoms and highly ordered COF membrane. Second, the photoactive triazine moieties in the Fe–COF membrane provide the enough electrons for atomically dispersed Fe centers through the unique physical form of the homogeneous COF membrane, thus achieving the highly efficient electron/hole separation of Fe–COF. Finally, the atomically dispersed Fe sites in the Fe–COF membrane significantly reduce the Gibbs free energy of *COOH and boost the photocatalytic CO2RR to CO.
Conclusions
Inspired by the natural photosynthesis in leaves, we successfully integrated the atomically dispersed Fe sites into interlayers of the pristine COF membrane through coordination with imine-N to serve as a mimic metalloenzyme. It was found that the mimic metalloenzyme based on the Fe–COF membrane exhibited excellent CO2 photoreduction performance under gas–solid conditions without the addition of any photosensitizer and sacrificial reagent. The CO production was 3972 μmol g−1 in a 4 h reaction, the selectivity was approximately 100%, and the cycling stability was more than 10 cycles, thus presenting a record high CO yield compared with the previously reported COF photocatalysts under gas–solid conditions. From the in/ex situ spectral studies and DFT calculations, it was revealed that the atomically dispersed Fe sites significantly reduce the energy barrier for the formation of key intermediates *CO2 and *COOH, the high-density triazine moieties around Fe sites supply enough electrons to the Fe sites, and the unique physical form of the homogeneous COF membrane provides efficient mass transfer and electron/proton transport, which work synergistically for the observed remarkable photocatalytic CO2 conversion to CO on the Fe–COF membrane. We believe that this proof-of-concept for the mimic metalloenzyme based on the Fe–COF membrane may motivate future research in this field and offer a useful guideline for the design of highly efficient catalysts in the photocatalytic CO2RR, which may help to promote sustainability of our environment and ecology.Data availability
The data that support the findings of this study are available in the paper and ESI.†Author contributions
W. J. J., W. H. Y., and S. Q. G. conceived the project and supervised the research. G. S. Q., W. J. J., and W. H. Y. led the writing of the paper. G. S. Q. and Z. Q. performed the experiments and analyzed the data. L. L. G. and Z. Y. L. completed some synthesis experiments. Z. X. provided the DFT calculation support. All authors analyzed the data and contributed to discussions.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 22233006, 22273018, and 22408089), the Natural Science Foundation of Henan province (No. 242300421206), and the 111 project (No. D17007).References
- B. Obama, Science, 2017, 355, 126–129 CrossRef CAS
.
- J.-X. Cui, L.-J. Wang, L. Feng, B. Meng, Z.-Y. Zhou, Z.-M. Su, K. Wang and S. Liu, J. Mater. Chem. A, 2021, 9, 24895–24902 RSC
- P. Gao, L. Zhong, B. Han, M. He and Y. Sun, Angew. Chem., Int. Ed., 2022, 61, e202210095 CrossRef CAS
- T. Watanabe, O. Pfeil-Gardiner, J. Kahnt, J. Koch, S. Shima and B. J. Murphy, Science, 2021, 373, 1151–1156 CrossRef CAS PubMed
- Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long, W. Zhang, Y. Yang, Y. Zhang, Y. Yao, H. Chi, J. Wang, Q. Shen, Y. Xiong, Z. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS
- D. Liu, H. Ma, C. Zhu, F. Qiu, W. Yu, L.-L. Ma, X.-W. Wei, Y.-F. Han and G. Yuan, J. Am. Chem. Soc., 2024, 146, 2275–2285 CrossRef CAS
- W. Wang, W. Zhang, C. Deng, H. Sheng and J. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202317969 CrossRef CAS
- S. Liang, M.-H. Li, M. Qi, L. Wang and Y.-W. Yang, Chem. Mater., 2023, 35, 8353–8370 CrossRef CAS
- P. Makam, S. S. R. K. C. Yamijala, V. S. Bhadram, L. J. W. Shimon, B. M. Wong and E. Gazit, Nat. Commun., 2022, 13, 1505 CrossRef CAS PubMed
- Z. Wang, P. Yeary, X. Feng and W. Lin, J. Am. Chem. Soc., 2023, 145, 8647–8655 CrossRef CAS
- N. F. Suremann, B. D. McCarthy, W. Gschwind, A. Kumar, B. A. Johnson, L. Hammarström and S. Ott, Chem. Rev., 2023, 123, 6545–6611 CrossRef CAS
- X. Yuan, X. Wu, J. Xiong, B. Yan, R. Gao, S. Liu, M. Zong, J. Ge and W. Lou, Nat. Commun., 2023, 14, 5974 CrossRef CAS PubMed
- S. Amanullah, P. Gotico, M. Sircoglou, W. Leibl, M. J. Llansola-Portoles, T. Tibiletti, A. Quaranta, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2024, 63, e202314439 CrossRef CAS
- G. Lan, Y. Fan, W. Shi, E. You, S. S. Veroneau and W. Lin, Nat. Catal., 2022, 5, 1006–1018 CrossRef CAS
- W. Xu, Y. Wu, W. Gu, D. Du, Y. Lin and C. Zhu, Chem. Soc. Rev., 2024, 53, 137–162 RSC
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC
- H. L. Nguyen and A. Alzamly, ACS Catal., 2021, 11, 9809–9824 CrossRef CAS
- Q. Yang, M. L. Luo, K. W. Liu, H. M. Cao and H. J. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS
- Q. Li, J.-N. Chang, Z. Wang, M. Lu, C. Guo, M. Zhang, T.-Y. Yu, Y. Chen, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2023, 145, 23167–23175 CrossRef CAS
- Y. Yang, H.-Y. Zhang, Y. Wang, L.-H. Shao, L. Fang, H. Dong, M. Lu, L.-Z. Dong, Y.-Q. Lan and F.-M. Zhang, Adv. Mater., 2023, 35, 2304170 CrossRef CAS
- S. Gao, Q. Zhang, X. Su, X. Wu, X.-G. Zhang, Y. Guo, Z. Li, J. Wei, H. Wang, S. Zhang and J. Wang, J. Am. Chem. Soc., 2023, 145, 9520–9529 CrossRef CAS PubMed
- Y. Yang, C. Schäfer and K. Börjesson, Chem, 2022, 8, 2217–2227 CAS
- S. Gao, Z. Li, Y. Yang, Z. Wang, Y. Wang, S. Luo, K. Yao, J. Qiu, H. Wang, L. Cao, Z. Lai and J. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 36507–36516 CrossRef CAS PubMed
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed
- F. Haase, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Mater. Chem. Front., 2017, 1, 1354–1361 RSC
- C. Dai, T. He, L. Zhong, X. Liu, W. Zhen, C. Xue, S. Li, D. Jiang and B. Liu, Adv. Mater. Interfaces, 2021, 8, 2002191 CrossRef CAS
- H. Zuo, B. Lyu, J. Yao, W. Long, Y. Shi, X. Li, H. Hu, A. Thomas, J. Yuan, B. Hou, W. Zhang and Y. Liao, Adv. Mater., 2024, 36, 2305755 CrossRef CAS
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed
- L. Su, P. Wang, X. Ma, J. Wang and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 21261–21266 CrossRef CAS PubMed
- C.-C. Hou, L. Zou, L. Sun, K. Zhang, Z. Liu, Y. Li, C. Li, R. Zou, J. Yu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 7384–7389 CrossRef CAS
- Y. Zang, D.-Q. Lu, K. Wang, B. Li, P. Peng, Y.-Q. Lan and S.-Q. Zang, Nat. Commun., 2023, 14, 1792 CrossRef CAS PubMed
- M. Zhou, Z. Wang, A. Mei, Z. Yang, W. Chen, S. Ou, S. Wang, K. Chen, P. Reiss, K. Qi, J. Ma and Y. Liu, Nat. Commun., 2023, 14, 2473 CrossRef CAS PubMed
- K. Sun, Y. Huang, Q. Wang, W. Zhao, X. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2024, 146(5), 3241–3249 CrossRef CAS
- Y. Zheng, X. Shen, M. Lin, M. Zhu, B. Yang, J. Yan, Z. Zhuang and Y. Yu, Small, 2024, 20, 2306836 CrossRef CAS
- W. Zhou, X. Wang, W. Zhao, N. Lu, D. Cong, Z. Li, P. Han, G. Ren, L. Sun, C. Liu and W.-Q. Deng, Nat. Commun., 2023, 14, 6971 CrossRef CAS
- L. Wang, L. Wang, Y. Xu, G. Sun, W. Nie, L. Liu, D. Kong, Y. Pan, Y. Zhang, H. Wang, Y. Huang, Z. Liu, H. Ren, T. Wei, Y. Himeda and Z. Fan, Adv. Mater., 2024, 36, 2309376 CrossRef CAS PubMed
- L. Zou, Z.-A. Chen, D.-H. Si, S.-L. Yang, W.-Q. Gao, K. Wang, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2023, 62, e202309820 CrossRef CAS PubMed
- B. B. Rath, S. Krause and B. V. Lotsch, Adv. Funct. Mater., 2024, 2309060 CrossRef CAS
- G. Fu, D. Yang, S. Xu, S. Li, Y. Zhao, H. Yang, D. Wu, P. S. Petkov, Z.-A. Lan, X. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 1318–1325 CrossRef CAS
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549–554 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05999a |
| This journal is © The Royal Society of Chemistry 2025 |