
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic regulation of single-atomic Ti sites on metal hydroxide for boosting photocatalytic CO2 reduction†
Ning-Yu
Huang‡
a,
Bai
Li‡
c,
Duojie
Wu
d,
Di
Chen
 a,
Yu-Tao
Zheng
a,
Bing
Shao
a,
Wenjuan
Wang
a,
Meng
Gu
d,
Lei
Li
*c and
Qiang
Xu
*ab
a,
Yu-Tao
Zheng
a,
Bing
Shao
a,
Wenjuan
Wang
a,
Meng
Gu
d,
Lei
Li
*c and
Qiang
Xu
*ab
aShenzhen Key Laboratory of Micro/Nano-Porous Functional Materials (SKLPM), SUSTech-Kyoto University Advanced Energy Materials Joint Innovation Laboratory (SKAEM-JIL), Department of Chemistry, Department of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: xuq@sustech.edu.cn
bInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: xu.qiang@icems.kyoto-u.ac.jp
cShenzhen Key Laboratory of Micro/Nano-Porous Functional Materials (SKLPM), Department of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: lil33@sustech.edu.cn
dDepartment of Materials Science and Engineering, Guangdong Provincial Key Laboratory of Energy Materials for Electric Power, Southern University of Science and Technology, Shenzhen 518055, China
First published on 5th December 2024
Abstract
Photocatalytic CO2 reduction is considered a sustainable method to address energy and environmental issues by converting CO2 into fuels and chemicals, yet the performance is still unsatisfactory. Single atom catalysts hold promising potential in photocatalysis, but the selection of metal species is still limited, especially in early transition metals. Herein, inspired by the structure of anatase TiO2, single Ti sites were successfully incorporated into a metal hydroxide support for the first time via cationic defects, significantly enhancing the photocatalytic performance by more than 30 times (from 0.26 to 8.09 mmol g−1 h−1). Based on the theoretical calculation and in situ characterization, the enhancement of photocatalytic performance can be attributed to the regulation of the electronic structure by the introduction of atomically dispersed Ti sites, leading to stronger binding with intermediates and enhanced charge transfer.
Introduction
The overuse of non-renewable fossil fuels has caused excessive atmospheric CO2 emission, which has become one of the urgent topics globally.1–6 Photocatalytic CO2 reduction, utilizing sustainable solar energy to convert CO2 into fuels and chemicals (e.g., carbon monoxide, formic acid, and methane), is considered a promising method to address the energy crisis and environmental issues.7–16 Because of the inertness of CO2 molecules and insufficient utilization of active sites in photocatalysts, the current performances for photocatalytic CO2 reduction are far from satisfactory towards practical applications.17–20 Therefore, developing novel catalysts with efficient active sites is of great significance but challenging as well. Single-atom catalysts (SACs), uniformly dispersing metal atoms on appropriate substrates, have shown high catalytic efficiency and product selectivity towards CO2 reduction because of their open active sites and unique electronic properties.21–29 Among reported literature, late transition metals (e.g., Cu, Co, Pt, Ir, and Pd) have been well-studied as active metal sites for various catalytic applications.30–33 However, very few early transition metals (groups 3–7) have been considered for the construction of SACs, which normally possess variable oxidation states, strong metal-carrier interaction and electron transfer efficiency.34 Developing SACs with early transition metal species can open up new opportunities for the exploration of efficient photocatalysts in CO2 reduction.Because of its abundance, non-toxicity and unique redox ability, titanium (Ti) has been widely applied as a component of many classical photocatalysts (e.g., TiO2, SrTiO3, etc.) for decades.35–37 Among them, anatase and rutile, as two different phases of TiO2, have different coordination configurations and electronic structures, leading to distinct catalytic properties. Compared to rutile TiO2, the more distorted TiO6 octahedron and different structural rearrangements in anatase TiO2 can offer a larger band gap and stronger redox driving force, as well as higher surface area, more active sites (such as oxygen vacancies) and more efficient charge separation, thus exhibiting better photocatalytic activity.38 Therefore, transplanting the isolated TiO6 octahedron of anatase TiO2 in suitable supports to construct SACs can trigger the modification of electronic structures and enhance the performance in various solar driven applications.39 For example, Shi and coworkers anchored Ti single atoms on reduced graphene oxide for tuning the electronic properties and minimizing the charge resistance, resulting in high-performance perovskite solar cells and encouraging further exploration of Ti-based SACs.40
Although Ti-based semiconductors have been widely studied in the field of photocatalysis, there is only one example of Ti SACs for photocatalytic CO2 reduction, in which the single Ti site is coordinated by N atoms of graphitic carbon nitride (g-C3N4).41 Herein, taking advantage of unique electronic properties, we incorporate single-atomic Ti sites into defective nickel hydroxide substrates to afford an earth-abundant SAC, significantly promoting the performance of photocatalytic CO2 reduction to CO (Fig. 1a). Nickel hydroxide (Ni(OH)2), as a typical transition metal hydroxide, exhibits a two-dimensional structure and the distorted NiO6 octahedron coordination mode similar to anatase TiO2, making it suitable for the construction of Ti SACs.42–44 By creating cationic defects, Ti atoms tend to coordinate with dangling O atoms to fill the vacancies and the single metal sites can be atomically dispersed into Ni(OH)2 with a high loading amount, which is a feasible method for preparing SACs.
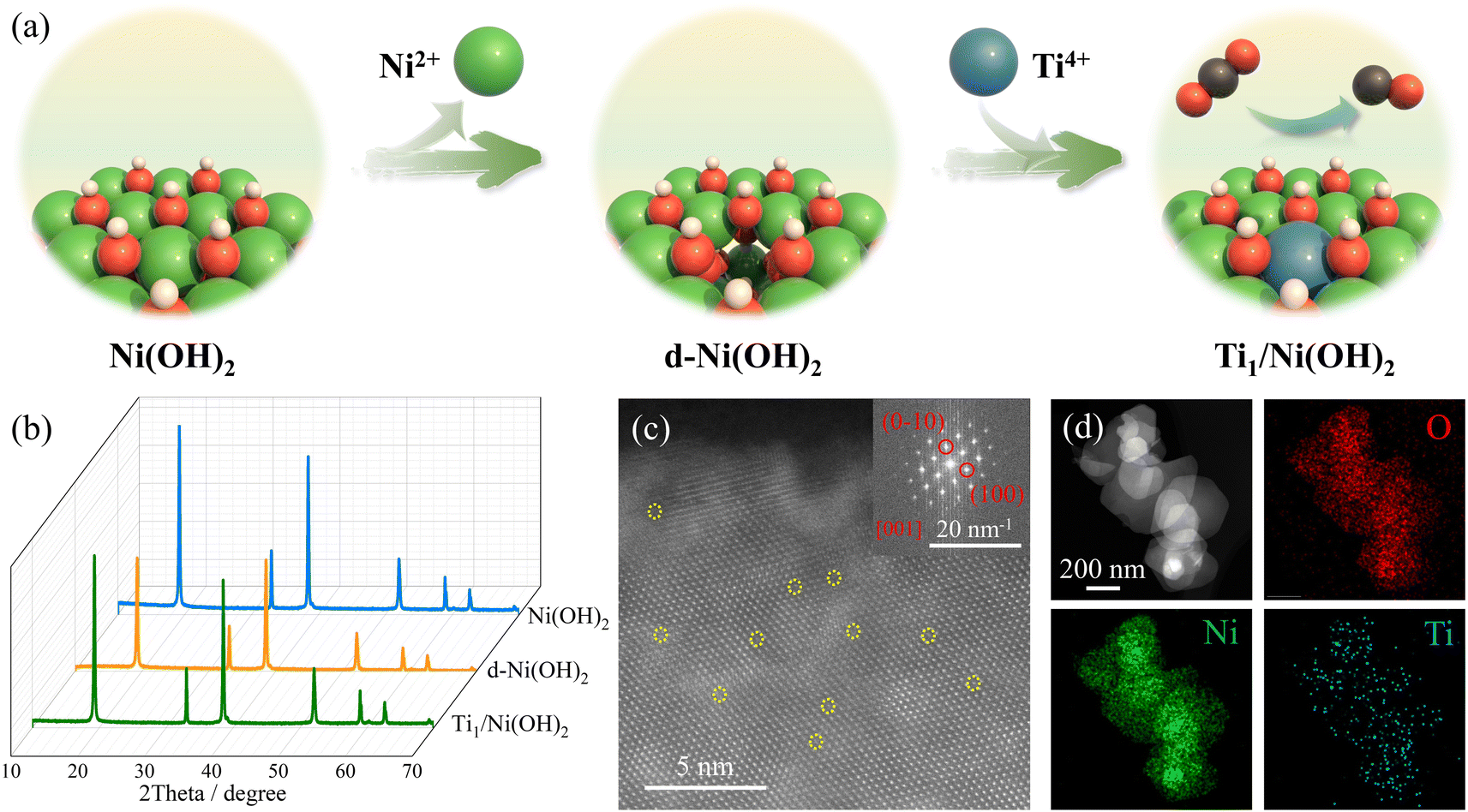 | ||
| Fig. 1 (a) Schematic illustration of the incorporation of single Ti sites on Ni(OH)2. (b) PXRD patterns of Ni(OH)2, d-Ni(OH)2 and Ti1/Ni(OH)2. (c) Atomic resolution HAADF-STEM image of Ti1/Ni(OH)2. The dark spots representing Ti atomic sites were highlighted. The inset shows an FFT pattern corresponding to Ni(OH)2. (d) HAADF-STEM image of Ti1/Ni(OH)2 and the corresponding elemental mapping images of O, Ni and Ti. | ||
Results and discussion
According to the previous literature, the metal ions of metal hydroxides can be dissolved out by coordinating with appropriate ligands or solvents, inducing cationic vacancies in the pristine materials.45 Therefore, treated in a mixed solution of N,N-dimethylformamide (DMF)/ethanol/H2O, defective Ni(OH)2 (d-Ni(OH)2) was synthesized from Ni(OH)2 nanosheets via a simple solvothermal method, showing lower peak intensity of PXRD patterns compared to pristine Ni(OH)2 without additional compounds (Fig. 1b and S1†). As shown in the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. S2 and S3†), d-Ni(OH)2 exhibited sheet-like morphology like Ni(OH)2 with cavities of 1–2 nm, indicating the creation of defects after solvothermal treatment. Moreover, the dissolved Ni2+ ions in mother liquor were determined by inductively coupled plasma mass spectrometry (ICP-MS, Table S1†), in which ca. 0.18% of Ni ions in pristine Ni(OH)2 was dissolved. Based on the X-ray photoelectron spectroscopy (XPS), the peaks in the Ni 2p spectrum (Fig. S4†) of d-Ni(OH)2 shifted to higher binding energy compared to pristine Ni(OH)2, consistent with the result from Ni K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. S5a†), which can be assigned to the increased valence state of Ni caused by the existence of cationic defects. More importantly, the Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra showed longer Ni–Ni bond length for d-Ni(OH)2 compared to pristine Ni(OH)2 (Fig. S5b†), which was attributed to the local extension of the neighboring Ni sites due to the existence of Ni2+-based cationic defects. Furthermore, zeta potential measurements revealed that compared to positively charged Ni(OH)2, d-Ni(OH)2 got negatively charged after solvothermal treatment (Fig. S6†), further suggesting the successful construction of defective Ni(OH)2 as a substrate for anchoring Ti sites.With the help of the strong interaction between Ti ions and dangling oxo species, the Ti1/Ni(OH)2 SACs were prepared via a simple impregnation strategy by stirring in the solution of organic Ti salt. The PXRD pattern of Ti1/Ni(OH)2 showed enhanced intensity compared to d-Ni(OH)2 without additional peaks of TiO2 or other Ti-related materials (Fig. 1b). Raman spectra indicated the successful anchoring of Ti sites, showing obvious peaks at 556 and 664 cm−1 in Ti1/Ni(OH)2 corresponding to the Ti–O bonds (Fig. S7†). The loading content of Ti in Ti1/Ni(OH)2 was determined to be ca. 2.06 wt% by using ICP-MS (Table S1†), which is relatively high among the reported SACs. After introducing higher-valent Ti ions, the Ni 2p XPS spectrum of Ti1/Ni(OH)2 shifted to higher binding energy compared to that of d-Ni(OH)2 (Fig. S4†), which can be attributed to the effect of electron delocalization, leading to a higher valence state and binding energy for Ni ions. The O 1s XPS spectra showed an observable shift toward higher binding energy for d-Ni(OH)2 (531.1 eV) and even further for Ti1/Ni(OH)2 (531.5 eV), which can be ascribed to a synergy effect of oxygen vacancies and Ti sites. Moreover, according to the electron paramagnetic resonance (EPR) spectra (Fig. S8†), although a small amount of oxygen vacancies appeared (peak at g = 2.00) during the construction of cationic defects in d-Ni(OH)2, the oxygen vacancies were mostly filled in Ti1/Ni(OH)2 accompanied by the introduction of Ti species (peak at g = 1.98).46,47 As shown in the SEM images (Fig. S2†), the sheet-like morphology of Ni(OH)2 remained unchanged after modification and no other Ti particles were formed, further suggesting the atomic dispersion of Ti species. The uniform distribution of Ti atoms was confirmed by the energy-dispersive X-ray spectroscopy (EDS) analysis and elemental mapping (Fig. 1d and S9†), showing a Ti loading amount of 1.66 wt%, which is consistent with the ICP-MS result. Because of the lower atomic number of Ti, the darker spots in the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) image (yellow circles in Fig. 1c) could be attributed to the Ti single atoms. More importantly, the brightness intensity profiles along different lines all showed single spots with lower intensity corresponding to Ti atomic sites (Fig. S10†), further confirming the presence of Ti single atoms in Ni(OH)2. To better illustrate the single-atomic nature, other group 4 metals (Zr and Hf) were also employed to construct SACs on Ni(OH)2, namely Zr1/Ni(OH)2 and Hf1/Ni(OH)2. The uniform dispersion of metal sites was confirmed by PXRD patterns and EDX mapping (Fig. S11–S13†). As shown in the HAADF-STEM images (Fig. S12 and S13†), observable bright spots corresponding to the single-atomic sites in Zr1/Ni(OH)2 and Hf1/Ni(OH)2 further demonstrated the general synthesis of early transition metal SACs.
To further investigate the local coordination environment of the Ti atom, XPS and X-ray absorption spectroscopy (XAS) were conducted. According to the Ti 2p XPS spectra of Ti1/Ni(OH)2 (Fig. 2a), the characteristic peaks at a binding energy of 458.5 and 464.4 eV were assigned to Ti 2p3/2 and Ti 2p1/2, respectively, indicating an approximate +4 valence state of Ti ions in Ti1/Ni(OH)2, which is further determined to be +3.5 by the linear fitting results based on the corresponding XANES spectra (Fig. 2b and S14†). More importantly, Ti1/Ni(OH)2 displayed distinct peaks in the pre-edge region of the Ti XANES spectrum compared to anatase TiO2 (the inset of Fig. 2b), indicating the absence of anatase TiO2 in this material. Furthermore, the FT-EXAFS spectra of Ti1/Ni(OH)2 exhibited the main peak located at ca. 1.56 Å (Fig. 2c), which can be attributed to the Ti–O scattering path similar to that of TiO2. In the meantime, the signal of Ti–Ti at ca. 2.52 Å was significantly lower compared to that of both TiO2 and Ti foil, further demonstrating the high dispersion of Ti sites. Based on the EXAFS fitting analysis for Ti1/Ni(OH)2 (Fig. 2d and Table S2†), the coordination number of Ti atoms in Ti1/Ni(OH)2 was determined to be 5.7, suggesting that the Ti atom is octahedrally coordinated in place of the Ni atom. Compared to those of Ti foil and TiO2, the wavelet transformation (WT) plot of Ti1/Ni(OH)2 showed the WT maximum at 1.3 Å−1, ascribed to Ti–O bonding, while no intensity maximum was observed for Ti–Ti (Fig. 2e–g). Overall, combined with XAS, HAADF-STEM and a series of characterization methods, it was demonstrated that the monodisperse Ti sites were successfully anchored on the defective Ni(OH)2 substrate created by the solvothermal method.
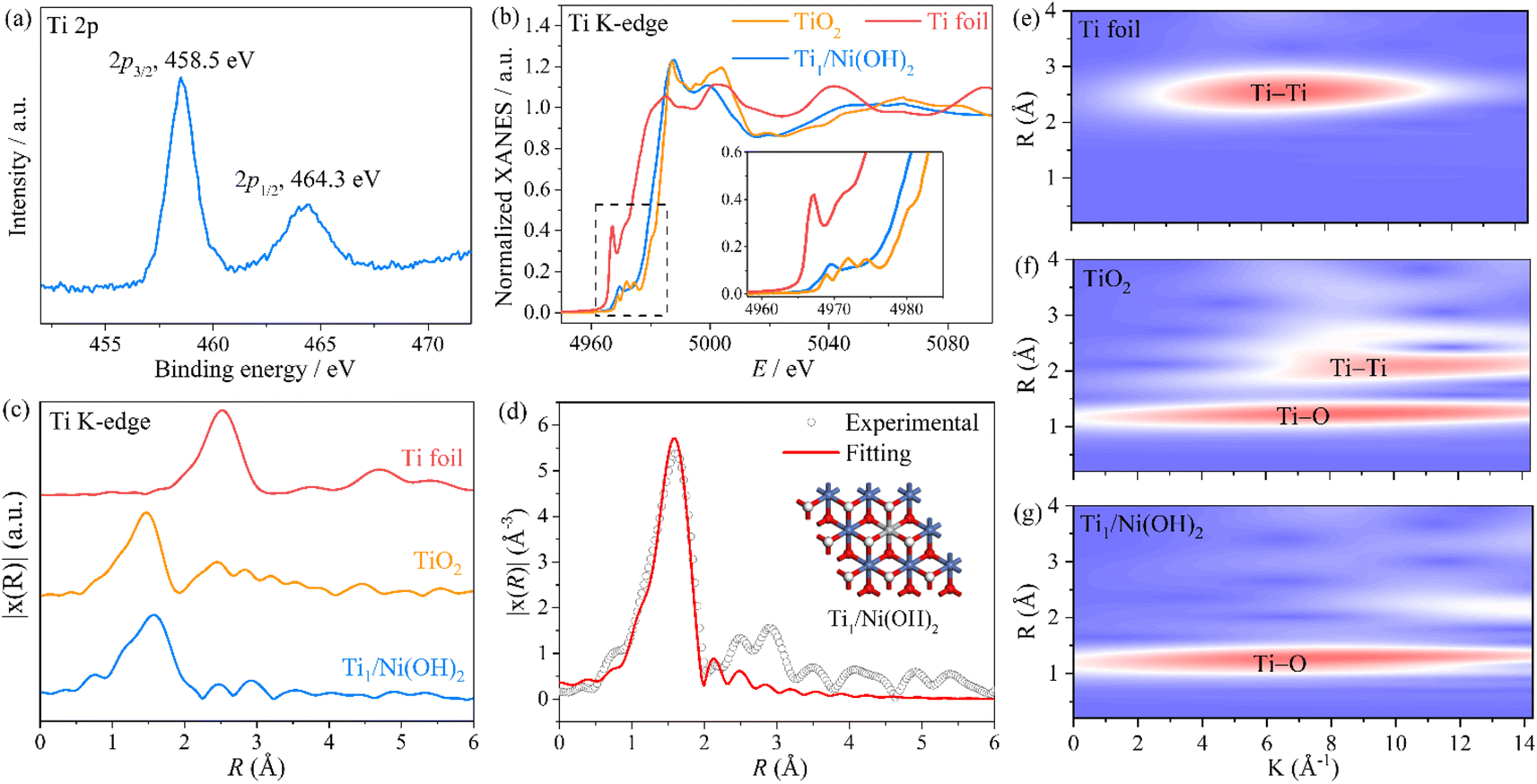 | ||
| Fig. 2 (a) Ti 2p XPS spectrum of Ti1/Ni(OH)2. (b) Ti K-edge XANES spectra of Ti1/Ni(OH)2, TiO2 and Ti foil. The inset highlights the near-edge absorption energy. (c) Ti K-edge EXAFS spectra of Ti1/Ni(OH)2, TiO2 and Ti foil and (d) the corresponding EXAFS fitting curve at R-space and the structural model (inset) of Ti1/Ni(OH)2. (e–g) WT-EXAFS contour plots of Ti foil, TiO2 and Ti1/Ni(OH)2, respectively. | ||
To evaluate the critical role of single Ti sites, photocatalytic CO2 reduction experiments were conducted under LED irradiation (λ > 410 nm), using 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as a sacrificial agent and [Ru(phen)3]Cl2 (phen = 1,10-phenanthroline) as a photosensitizer. The gaseous and liquid products evolved in the reaction system were detected by gas chromatography (GC) and 1H nuclear magnetic resonance (1H NMR), respectively. After light irradiation, CO and H2 were detected as the main products and no other liquid-phase product was detected in the catalytic system (Fig. S15 and S16†). Prior to the detailed evaluation of photocatalytic performance, the best performance of Ti1/Ni(OH)2 was optimized by varying its synthetic conditions, including the amount of Ti precursor, synthetic time and temperature of d-Ni(OH)2 (Fig. S17†). During 4 h of light irradiation, the amount of CO and H2 increased linearly with time by using Ti1/Ni(OH)2 as a catalyst (Fig. 3a), showing a CO production rate of 8.09 mmol g−1 h−1 with excellent CO selectivity as high as 96.5%, which is comparable to the reported benchmark performance (Table S3†). In comparison, the CO production rates of Ni(OH)2 and d-Ni(OH)2 were 0.80 and 0.26 mmol g−1 h−1, respectively (Fig. 3b). The photocatalytic activity of Ti1/Ni(OH)2 was more than 30 times higher than that of pristine Ni(OH)2, strongly suggesting that the introduction of single Ti sites played a significant role in promoting the photocatalytic performance. Moreover, Ti1/Ni(OH)2 greatly outperformed anatase TiO2 or the physical mixture of Ni(OH)2 and TiO2 (Fig. S18†), further demonstrating the importance of the synergistic effect in this Ti-metal hydroxide system.
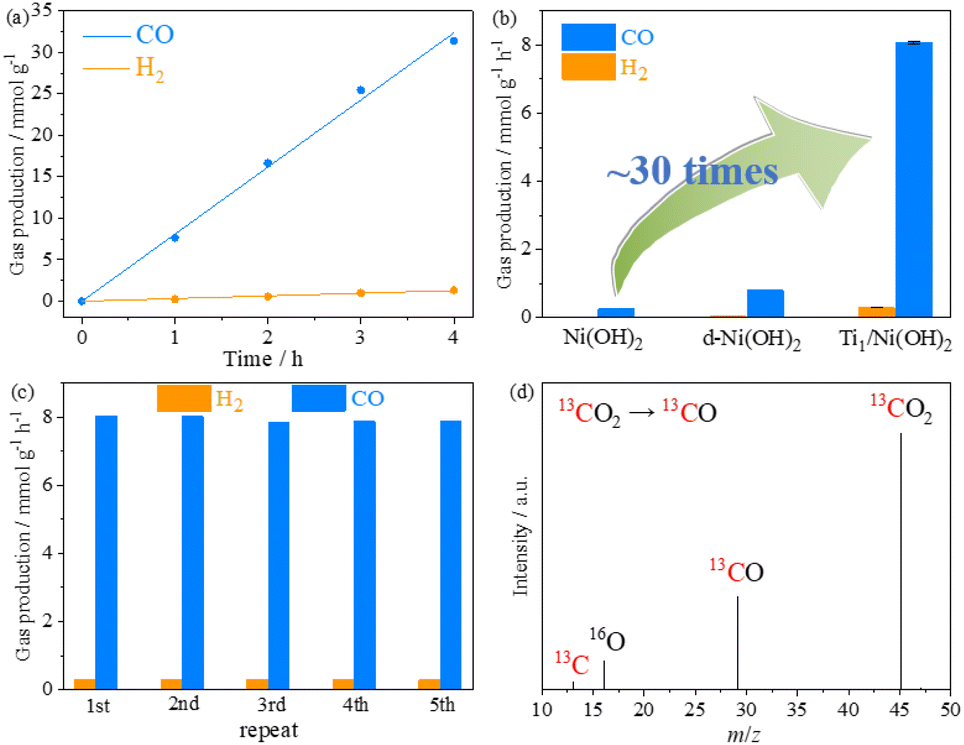 | ||
| Fig. 3 (a) CO and H2 production of photocatalytic CO2 reduction using the catalyst Ti1/Ni(OH)2. (b) Comparison of the gas production for Ni(OH)2, d-Ni(OH)2 and Ti1/Ni(OH)2. (c) Recycle experiments of Ti1/Ni(OH)2. (d) Mass spectra of the gaseous products of photocatalytic CO2 reduction by using 13CO2 as the gas source. | ||
During the cyclic experiments, Ti1/Ni(OH)2 can maintain 98% of the initial performance after 5 consecutive runs (Fig. 3c), indicating its excellent photocatalytic stability, which was also confirmed by the PXRD patterns, SEM images and ICP-MS measurements of the sample after photocatalysis (Fig. S19 and Table S1†). According to the control experiments, only a negligible product can be observed in the absence of the catalyst, the photosensitizer, the sacrificial agent, CO2, H2O and light, indicating the necessity of these conditions for the photocatalytic process (Fig. S20†). To further confirm the carbon source, isotope labeling experiments were conducted by using 13CO2 as the feedstock gas. As shown in gas chromatography-mass spectrometry (GC-MS, Fig. 3d), the signal of 13CO (m/z = 29) was clearly observed, demonstrating that CO was generated from the CO2 feedstock instead of the photosensitizer, the sacrificial agent or the catalyst itself.
According to the UV-vis spectra (Fig. S21†), after anchoring single Ti sites, Ti1/Ni(OH)2 exhibited enhanced light absorption in the region of 300–400 nm compared to d-Ni(OH)2 and Ni(OH)2. To better reveal the charge transfer behavior, photoluminescence (PL) spectra and electrochemical impedance spectroscopy (EIS) spectra were measured for Ti1/Ni(OH)2 and its control samples. Under the excitation at 465 nm, a characteristic emission peak of the excited Ru(phen)3Cl2 can be observed at ca. 570 nm (Fig. S22†), which can be effectively quenched by adding either Ni(OH)2, d-Ni(OH)2 or Ti1/Ni(OH)2. In particular, the PL intensity with Ti1/Ni(OH)2 is significantly lower than those with Ni(OH)2 and d-Ni(OH)2, indicating the most efficient charge transfer process for Ti1/Ni(OH)2. Furthermore, according to the EIS spectra, Ti1/Ni(OH)2 exhibited the smallest semicircle in the Nyquist plots (Fig. S23†), suggesting the lowest charge transfer resistance and more efficient separation of photogenerated charge carriers, which can well elucidate the best photocatalytic performance for Ti1/Ni(OH)2.
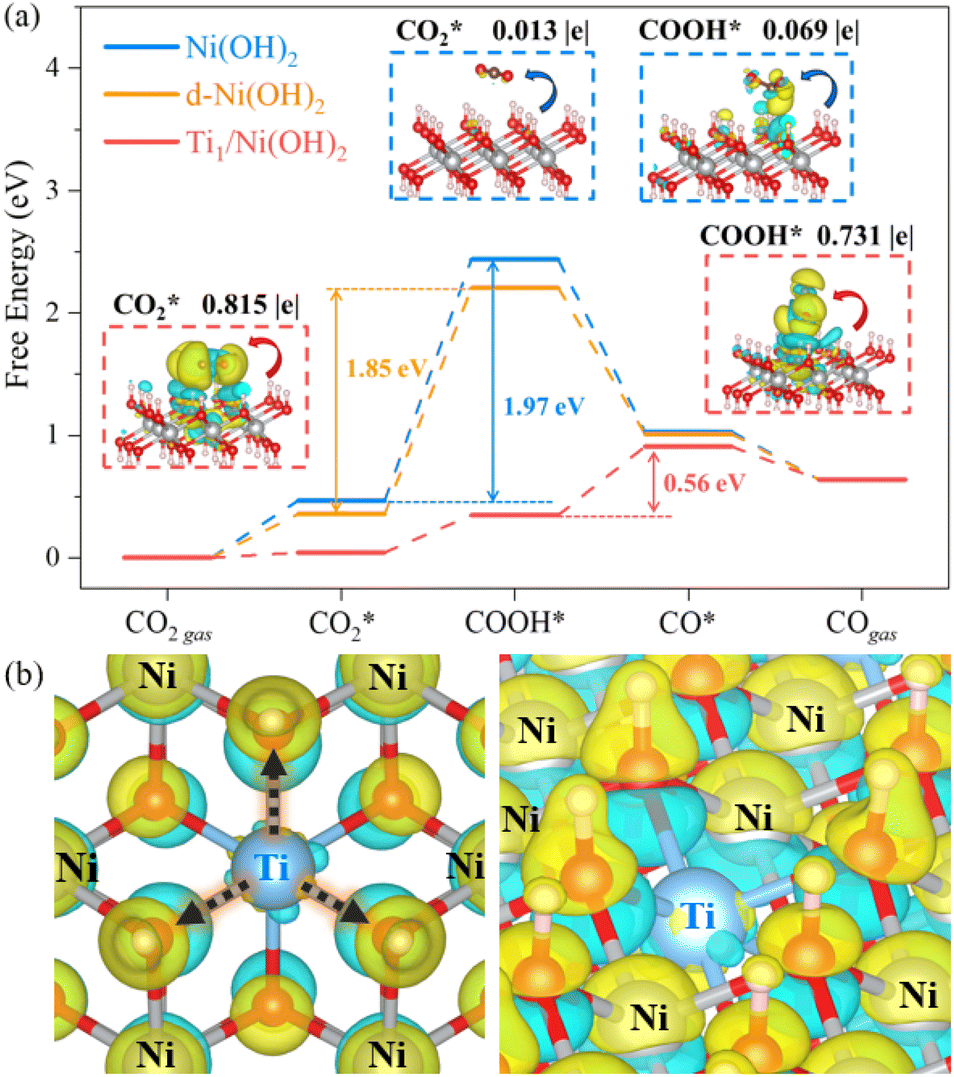
To gain insight into the enhanced performance of the Ti single atom in the photocatalytic CO2 reduction, the density functional theory (DFT) method was employed to calculate the free-energy profiles for CO2 reduction on pristine Ni(OH)2, d-Ni(OH)2 and Ti1/Ni(OH)2 (Fig. S24†). For both Ni(OH)2 and d-Ni(OH)2, the rate determining steps (RDSs) were the protonation of the CO2 molecule  , with free energy changes (DG) of 1.97 eV and 1.85 eV, respectively (Fig. 4a). Anchoring a single Ti site into Ni(OH)2 significantly enhances the adsorption of key intermediates
, with free energy changes (DG) of 1.97 eV and 1.85 eV, respectively (Fig. 4a). Anchoring a single Ti site into Ni(OH)2 significantly enhances the adsorption of key intermediates 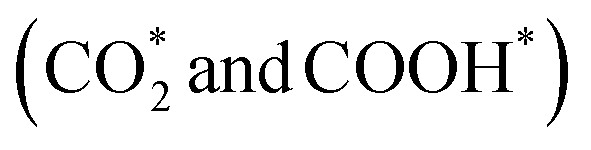 , as indicated by the shorter vertical distance upon adsorption (Fig. S25†). In particular, the strong interaction between CO2 and Ti1/Ni(OH)2 leads to a bent adsorption configuration of CO2 (Fig. S25†), activating C–O bonds and significantly lowering the free energy of corresponding reaction intermediates. Therefore, the RDS for Ti1/Ni(OH)2 was altered to the dehydration of COOH* (COOH* + H+ + e− → CO* + H2O), of which the ΔG remarkably decreased to 0.56 eV, resulting in increased CO production in photocatalytic CO2 reduction. To probe the key intermediates and verify the reaction mechanism, in situ Fourier transform infrared spectroscopy (FTIR) measurements were conducted using Ti1/Ni(OH)2 as the photocatalyst (Fig. S26†). Under light irradiation, the peaks at 1542 and 1647 cm−1 corresponding to the key intermediate COOH* increased with the accumulation of CO32− at 1515 cm−1,48,49 while no observable signals can be detected in the dark, indicating the process of photocatalytic CO2 reduction. More critically, the characteristic peak of CO* at 2073 cm−1 can be clearly identified,50 demonstrating the altered RDS and the efficient formation of CO.
, as indicated by the shorter vertical distance upon adsorption (Fig. S25†). In particular, the strong interaction between CO2 and Ti1/Ni(OH)2 leads to a bent adsorption configuration of CO2 (Fig. S25†), activating C–O bonds and significantly lowering the free energy of corresponding reaction intermediates. Therefore, the RDS for Ti1/Ni(OH)2 was altered to the dehydration of COOH* (COOH* + H+ + e− → CO* + H2O), of which the ΔG remarkably decreased to 0.56 eV, resulting in increased CO production in photocatalytic CO2 reduction. To probe the key intermediates and verify the reaction mechanism, in situ Fourier transform infrared spectroscopy (FTIR) measurements were conducted using Ti1/Ni(OH)2 as the photocatalyst (Fig. S26†). Under light irradiation, the peaks at 1542 and 1647 cm−1 corresponding to the key intermediate COOH* increased with the accumulation of CO32− at 1515 cm−1,48,49 while no observable signals can be detected in the dark, indicating the process of photocatalytic CO2 reduction. More critically, the characteristic peak of CO* at 2073 cm−1 can be clearly identified,50 demonstrating the altered RDS and the efficient formation of CO.
 | ||
| Fig. 4 (a) Energy profiles of CO2-to-CO conversion on Ni(OH)2, d-Ni(OH)2 and Ti1/Ni(OH)2. Insets show the electron density difference for *CO2 and *COOH adsorption on Ni(OH)2 (blue frame) and Ti1/Ni(OH)2 (red frame). Yellow and cyan represent electron accumulation and loss, respectively. (b) Top view and side view for the electron density difference of Ti1/Ni(OH)2. | ||
Furthermore, we confirmed that the intensified interaction between Ti1/Ni(OH)2 and intermediates results from the substantial charge transfers from the catalyst to the adsorbate (0.815|e| for 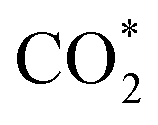 and 0.731|e| for COOH*), which were significantly larger than those observed in Ni(OH)2 (0.013|e| for
and 0.731|e| for COOH*), which were significantly larger than those observed in Ni(OH)2 (0.013|e| for 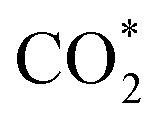 and 0.069|e| for COOH*, insets of Fig. 4a). According to the electron density difference, the introduction of single Ti sites led to electron donation to the adjacent –OH groups (Fig. 4b and S27†), thus increasing their electron density and resulting in amplified charge transfer from the –OH group to the adsorbates. Overall, the introduction of single Ti sites in Ti1/Ni(OH)2 effectively regulates the electronic structure and enhances intermediate adsorption on catalysts, thereby decreasing ΔG of the RDS and eventually leading to boosted performance in photocatalytic CO2 reduction.
and 0.069|e| for COOH*, insets of Fig. 4a). According to the electron density difference, the introduction of single Ti sites led to electron donation to the adjacent –OH groups (Fig. 4b and S27†), thus increasing their electron density and resulting in amplified charge transfer from the –OH group to the adsorbates. Overall, the introduction of single Ti sites in Ti1/Ni(OH)2 effectively regulates the electronic structure and enhances intermediate adsorption on catalysts, thereby decreasing ΔG of the RDS and eventually leading to boosted performance in photocatalytic CO2 reduction.
Conclusions
In conclusion, through defect engineering, a Ti SAC was successfully constructed on a metal hydroxide support for the first time. By transplanting the TiO6 octahedron unit from anatase TiO2, the obtained Ti1/Ni(OH)2 exhibited significantly enhanced photocatalytic CO2 reduction activity by more than 30 times compared to pristine Ni(OH)2. According to the mechanistic study, the promoted photocatalytic performance can be attributed to the enhanced affinity to key intermediates and modified electronic structures brought by isolated Ti sites. This work has broadened the scope of single-atom catalysts in early transition metals and provided a useful strategy in photocatalyst design and modification with enhanced performance for CO2 reduction.Data availability
The data that support the findings of this study are included in the paper and the ESI.†Author contributions
Q. X. designed the research. N.-Y. H. performed syntheses and measurements. N.-Y. H., D. C. and Y.-T. Z. performed basic characterization. B. S. performed XAS analysis. W. W. performed EPR measurements. B. L. and L. L. conducted DFT calculations. D. W. and M. G. performed HAADF-STEM measurement. N.-Y. H., B. L., L. L. and Q. X. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Project (2023YFA1506601), Guangdong Grants (2021ZT09C064), Shenzhen Key Laboratory of Micro/Nano-Porous Functional Materials (SKLPM) (ZDSYS21210709112802010) and Training Program of the Major Research Plan of the National Natural Science Foundation of China (92270103). The authors acknowledge the assistance of the Pico Center at the Southern University of Science and Technology Core Research Facilities. The theoretical calculations were supported by Center for Computational Science and Engineering at Southern University of Science and Technology. The in situ FTIR measurements were supported by Prof. Pei-Qin Liao and Hui-Ying Chen at Sun Yat-sen University.Notes and references
- D. I. Armstrong McKay, A. Staal, J. F. Abrams, R. Winkelmann, B. Sakschewski, S. Loriani, I. Fetzer, S. E. Cornell, J. Rockström and T. M. Lenton, Science, 2022, 377, eabn7950 CrossRef PubMed.
- L. Al-Ghussain, Environ. Prog. Sustain., 2019, 38, 13–21 CrossRef CAS.
- D.-D. Zhou, X.-W. Zhang, Z.-W. Mo, Y.-Z. Xu, X.-Y. Tian, Y. Li, X.-M. Chen and J.-P. Zhang, EnergyChem, 2019, 1, 100016 CrossRef.
- C. Zhou, J. Chen, J. Zhao, Y. Meng, Z. Li, X. Meng and J. Wang, Renewables, 2024, 2, 89–110 CrossRef.
- N.-Y. Huang, Z.-Y. Chen, F.-L. Hu, C.-Y. Shang, W. Wang, J.-R. Huang, C. Zhou, L. Li and Q. Xu, Nano Res., 2023, 16, 7756–7760 CrossRef CAS.
- N.-Y. Huang, Y.-T. Zheng, D. Chen, Z.-Y. Chen, C.-Z. Huang and Q. Xu, Chem. Soc. Rev., 2023, 52, 7949–8004 RSC.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS PubMed.
- Y. Wang, E. Chen and J. Tang, ACS Catal., 2022, 12, 7300–7316 CrossRef CAS PubMed.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- J. Di, G. Hao, G. Liu, J. Zhou, W. Jiang and Z. Liu, Coord. Chem. Rev., 2023, 482, 215057 CrossRef CAS.
- Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS.
- D. Feng, X. Li, Y. Liu, X. Chen and S. Li, Renewables, 2023, 1, 485–513 CrossRef.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74 CrossRef CAS PubMed.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W.-L. Man, S.-M. Yiu, K.-C. Lau, T.-C. Lau and M. Robert, Nat. Catal., 2019, 2, 801–808 CrossRef CAS.
- N.-Y. Huang, B. Li, D. Wu, Z.-Y. Chen, B. Shao, D. Chen, Y.-T. Zheng, W. Wang, C. Yang, M. Gu, L. Li and Q. Xu, Angew. Chem., Int. Ed., 2024, 63, e202319177 CrossRef CAS PubMed.
- S. Dai, T. Kajiwara, M. Ikeda, I. Romero-Muñiz, G. Patriarche, A. E. Platero-Prats, A. Vimont, M. Daturi, A. Tissot, Q. Xu and C. Serre, Angew. Chem., Int. Ed., 2022, 61, e202211848 CrossRef CAS PubMed.
- P. M. Stanley, V. Ramm, R. A. Fischer and J. Warnan, Nat. Syn., 2024, 3, 307–318 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- Y. Zhang, H. Liu, F. Gao, X. Tan, Y. Cai, B. Hu, Q. Huang, M. Fang and X. Wang, EnergyChem, 2022, 4, 100078 CrossRef CAS.
- N.-Y. Huang, H. He, S. Liu, H.-L. Zhu, Y.-J. Li, J. Xu, J.-R. Huang, X. Wang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 17424–17430 CrossRef CAS.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS.
- P. Liu, Z. Huang, X. Gao, X. Hong, J. Zhu, G. Wang, Y. Wu, J. Zeng and X. Zheng, Adv. Mater., 2022, 34, 2200057 CrossRef CAS.
- J.-R. Huang, X.-F. Qiu, Z.-H. Zhao, H.-L. Zhu, Y.-C. Liu, W. Shi, P.-Q. Liao and X.-M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202210985 CrossRef CAS PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS.
- H. Shi, Y. Liang, J. Hou, H. Wang, Z. Jia, J. Wu, F. Song, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2024, e202404884 CAS.
- H. Ou, G. Li, W. Ren, B. Pan, G. Luo, Z. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 22075–22082 CrossRef CAS PubMed.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS.
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- Z. Wang, H. Niu, T. Wu, S. Ding, B. Yu Xia and Y. Su, Renewables, 2024, 2, 213–221 CrossRef.
- X. Jia, Q. Meng, R. Zheng, X. Liu, Y. Zhao, C. Liu, M. Xiao and W. Xing, Renewables, 2023, 1, 694–719 CrossRef.
- L.-J. Yuan, X.-L. Sui, H. Pan and Z.-B. Wang, Renewables, 2023, 1, 514–540 CrossRef.
- S. Kaushik, D. Wu, Z. Zhang, X. Xiao, C. Zhen, W. Wang, N.-Y. Huang, M. Gu and Q. Xu, Adv. Mater., 2024, 36, 2401163 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- S. Kreft, D. Wei, H. Junge and M. Beller, EnergyChem, 2020, 2, 100044 CrossRef.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- Y. Wang, H. Liu, Q. Shi, Z. Miao, H. Duan, Y. Wang, H. Rong and J. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202404911 CrossRef CAS.
- C. Zhang, S. Liang, W. Liu, F. T. Eickemeyer, X. Cai, K. Zhou, J. Bian, H. Zhu, C. Zhu, N. Wang, Z. Wang, J. Zhang, Y. Wang, J. Hu, H. Ma, C. Xin, S. M. Zakeeruddin, M. Grätzel and Y. Shi, Nat. Energy, 2021, 6, 1154–1163 CrossRef CAS.
- S. Tang, X. Yin, G. Wang, X. Lu and T. Lu, Nano Res., 2019, 12, 457–462 CrossRef CAS.
- A. Pei, G. Li, L. Zhu, Z. Huang, J. Ye, Y.-C. Chang, S. M. Osman, C.-W. Pao, Q. Gao, B. H. Chen and R. Luque, Adv. Funct. Mater., 2022, 32, 2208587 CrossRef CAS.
- X. Chen, J. Wan, J. Wang, Q. Zhang, L. Gu, L. Zheng, N. Wang and R. Yu, Adv. Mater., 2021, 33, 2104764 CrossRef CAS.
- J. Zhang, X. Wu, W.-C. Cheong, W. Chen, R. Lin, J. Li, L. Zheng, W. Yan, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 1002 CrossRef.
- Y.-j. Wu, J. Yang, T.-x. Tu, W.-q. Li, P.-f. Zhang, Y. Zhou, J.-f. Li, J.-t. Li and S.-G. Sun, Angew. Chem., Int. Ed., 2021, 60, 26829–26836 CrossRef CAS PubMed.
- Z. Chen, L. Liang, H. Yuan, H. Liu, P. Wu, M. Fu, J. Wu, P. Chen, Y. Qiu, D. Ye and L. Chen, Appl. Catal., B, 2021, 298, 120507 CrossRef CAS.
- M. D'Arienzo, M. V. Dozzi, M. Redaelli, B. D. Credico, F. Morazzoni, R. Scotti and S. Polizzi, J. Phys. Chem. C, 2015, 119, 12385–12393 CrossRef.
- Y.-N. Gong, J.-H. Mei, W.-J. Shi, J.-W. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2024, 63, e202318735 CrossRef CAS PubMed.
- X. Zu, Y. Zhao, X. Li, R. Chen, W. Shao, L. Li, P. Qiao, W. Yan, Y. Pan, Q. Xu, J. Zhu, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202215247 CrossRef CAS PubMed.
- H. Huang, J. Zhao, B. Weng, F. Lai, M. Zhang, J. Hofkens, M. B. J. Roeffaers, J. A. Steele and J. Long, Angew. Chem., Int. Ed., 2022, 61, e202204563 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and characterization data. See DOI: https://doi.org/10.1039/d4sc07257j |
| ‡ N.-Y. Huang and B. Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |