
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metathesis chemistry of inorganic cumulenes driven by B–O bond formation†
Jianqin
Tang
a,
Chenyang
Hu
ab,
Agamemnon E.
Crumpton
a,
Liam P.
Griffin
a,
Jose M.
Goicoechea
 *b and
Simon
Aldridge
*a
*b and
Simon
Aldridge
*a
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: simon.aldridge@chem.ox.ac.uk
bDepartment of Chemistry, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405, USA. E-mail: jgoicoec@iu.edu
First published on 18th December 2024
Abstract
The reaction chemistry of an unprecedented ‘inorganic cumulene’ – featuring a five-atom BNBNB chain – towards C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (and related) multiple bonds is disclosed. In marked contrast to related all-carbon systems, the intrinsic polarity of the BNBNB chain (featuring electron-rich nitrogen and electron-deficient boron centres) enables metathesis chemistry with electrophilic heteroallenes such as CO2 and with organic carbonyl compounds. Transfer of the borylimide unit to [CO], [CS], [PP{(NDippCH2)2}] and [C(H)Ph] moieties generates (boryl)NCX systems (X = O, S, PP{(NDippCH2)2}, C(H)Ph), driven thermodynamically by B–O bond formation. Pairwise exchange of O and {(HCDippN)2}BN fragments occurs via consecutive [2+2] cyclo-addition/cyclo-reversion steps. An isolable complex of stoichiometry K[(boryl)NB(O)OC(H)Ph], formed via [2+2] cycloaddition to [(boryl)NBO]− can be shown to be an intermediate in the formation of (boryl)NC(H)Ph, and provides corroborating evidence for a DFT-calculated mechanism proceeding via a ‘bora-Wittig’ mechanism.
O (and related) multiple bonds is disclosed. In marked contrast to related all-carbon systems, the intrinsic polarity of the BNBNB chain (featuring electron-rich nitrogen and electron-deficient boron centres) enables metathesis chemistry with electrophilic heteroallenes such as CO2 and with organic carbonyl compounds. Transfer of the borylimide unit to [CO], [CS], [PP{(NDippCH2)2}] and [C(H)Ph] moieties generates (boryl)NCX systems (X = O, S, PP{(NDippCH2)2}, C(H)Ph), driven thermodynamically by B–O bond formation. Pairwise exchange of O and {(HCDippN)2}BN fragments occurs via consecutive [2+2] cyclo-addition/cyclo-reversion steps. An isolable complex of stoichiometry K[(boryl)NB(O)OC(H)Ph], formed via [2+2] cycloaddition to [(boryl)NBO]− can be shown to be an intermediate in the formation of (boryl)NC(H)Ph, and provides corroborating evidence for a DFT-calculated mechanism proceeding via a ‘bora-Wittig’ mechanism.
Introduction
Metathesis constitutes a powerful and widely used methodology in synthetic chemistry, with applications spanning organometallics, pharmaceuticals, fine chemicals and materials science.1 While metathesis reactions utilising CC bonds predominate, the possibility of exploiting CO bonds in a similar fashion offers a strategy, for example, for the transformation of CO2 into useful chemical products.2 The use of oxophilic early transition metal complexes to accomplish CO to CNR transformations has been extensively reported,3,4 and similar chemistry using main group metal/metalloid derivatives has begun to be explored, driven, for example, by the formation of strong Al–O or Si–O bonds.5,6 Given the similar thermodynamic driver associated with B–O bond formation (559 kJ mol−1, cf. 595 kJ mol−1 for Al–O),7 we wondered whether boron-based non-metal systems could be employed to transform CO into CN bonds.8,9
We have recently developed high-yielding synthetic routes to ‘inorganic cumulenes’ (Scheme 1).10,11 These five-atom BNBNB chains analogous to [4]cumulenes can possess either linear or bent structures (i.e.1/2 or 3) depending on the nature of the groups attached to the boron termini. Chemical modification of diamido-terminated 1/2 to generate a linear system best described by a BNBNB resonance structure involves assimilation of the strong Lewis acid B(C6F5)3 to enhance the π-acidity of the end groups and conjugate the N-lone pairs. In addition, preliminary studies of the reactivity of K[(HCDippN)2BNBNB(NDippCH)2] (1) verify experimentally the intrinsic polarity within the BNBNB chain. The reaction of 1 with methyl iodide, for example, proceeds via nucleophilic attack by nitrogen to generate an iminoborane (4), featuring a formal BN triple bond.
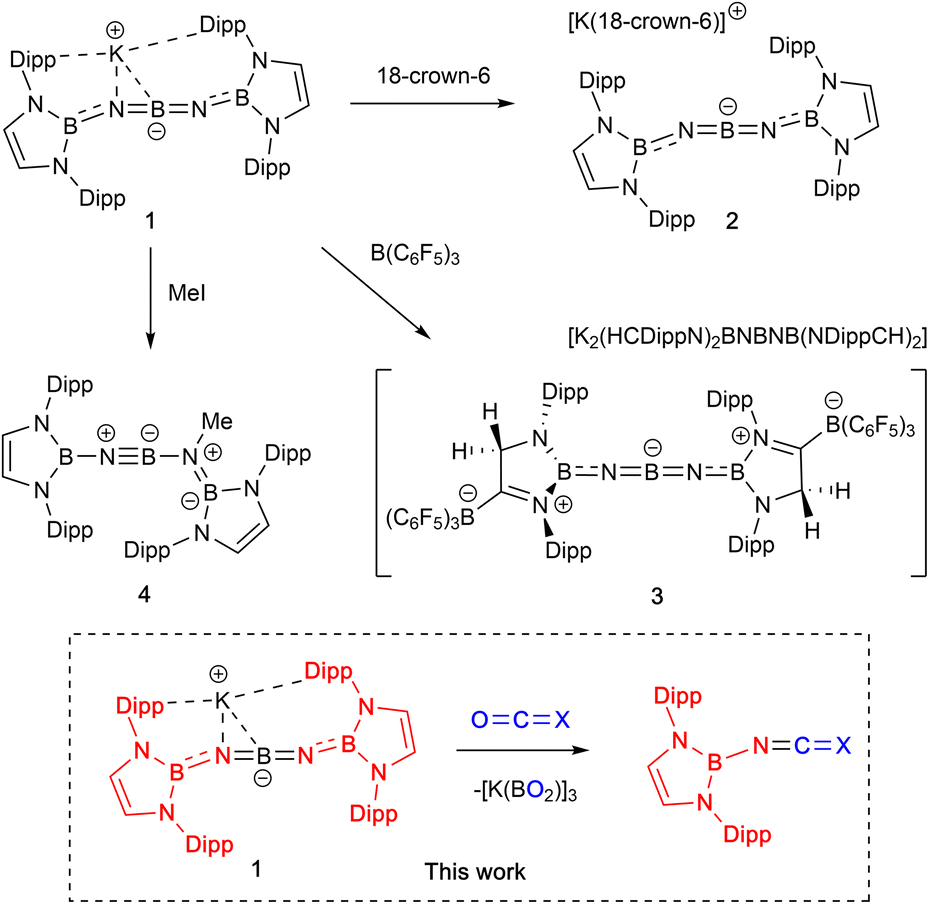 | ||
| Scheme 1 Preliminary studies of ‘inorganic cumulene’ 1.10 | ||
The central NBN core of 1 can also be viewed as being a diimidoboryl anion isoelectronic with CO2 itself, which prompted us to probe whether it might be capable of metathesis chemistry with O-containing heteroallenes, leading to exchange of borylimide/O atom fragments, driven thermodynamically by the formation of BO bond(s). We perceived that the previously demonstrated N-centred nucleophilicity of 1 might enable facile reactivity with substrates (such as CO2 itself) which react primarily as electrophiles.10
Results and discussion
The reactions of 1 with CO2, CS2 or the phosphaphosphinidene carbonyl {(H2CDippN)2}PPCO proceed via O/N(boryl) meta-thesis, leading to the formation of the iminoboryl compounds (boryl)NCE (E = O (5), S (6) and PP{(NDippCH2)2} (7); Scheme 2). 5 is a known compound, having recently been reported by Yuvaraj and Jones to be formed by N(boryl) transfer to CO2 from a boryl-substituted silaimine at 80 °C (in ca. 40% yield).5c In the chemistry reported here, 5 is formed in excellent yield (ca. 90%, based on the availability of two N(boryl) units per molecule of 1), while the heavier CE bonds in 6 (E = S) and 7 (E = P) are constructed in more modest yields (ca. 50% in each case). Both 6 and 7 are novel compounds, and each has been characterized by standard spectroscopic/analytical techniques and by X-ray crystallography (Fig. 2). The 11B NMR resonances determined for 6 and 7 (δB = 16.1 and 16.0 ppm) are similar to that measured for 5 (δB = 17.9 ppm), while the 31P signals measured for 7 (δP = −183.3 ppm, P![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) C; 165.6 ppm, PC; 1JP,P = 298.0 Hz) are shifted from those reported for the precursor {(H2CDippN)2}PPCO (δP = −245.6, 167.9 ppm; 1JP,P = 252.1 Hz)12 to an extent that is markedly greater for the exocyclic phosphorus atom. Structurally, 5–7 are also similar, (Fig. 1) featuring comparable NC distances (1.170(2), 1.179(7) and 1.184(3) Å), and a B–N–C angle which is marginally wider in the case of 7 (162.8(2)° vs. 159.4(1) and 156.8(4)° for 5 and 6, respectively).5c
C; 165.6 ppm, PC; 1JP,P = 298.0 Hz) are shifted from those reported for the precursor {(H2CDippN)2}PPCO (δP = −245.6, 167.9 ppm; 1JP,P = 252.1 Hz)12 to an extent that is markedly greater for the exocyclic phosphorus atom. Structurally, 5–7 are also similar, (Fig. 1) featuring comparable NC distances (1.170(2), 1.179(7) and 1.184(3) Å), and a B–N–C angle which is marginally wider in the case of 7 (162.8(2)° vs. 159.4(1) and 156.8(4)° for 5 and 6, respectively).5c
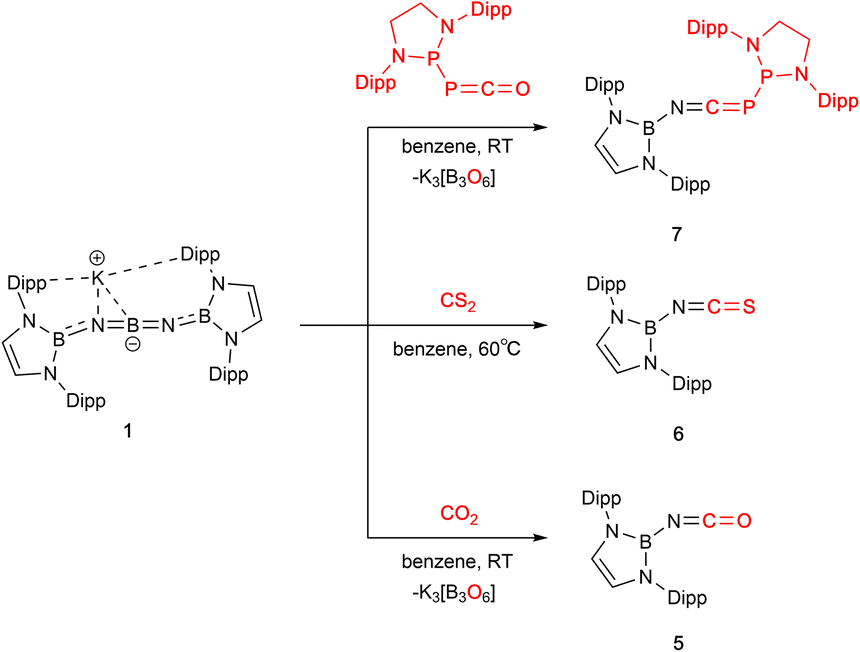 | ||
| Scheme 2 Metathesis reactions of compound 1 with CO2, CS2 and {(H2CDippN)2}PPCO. | ||
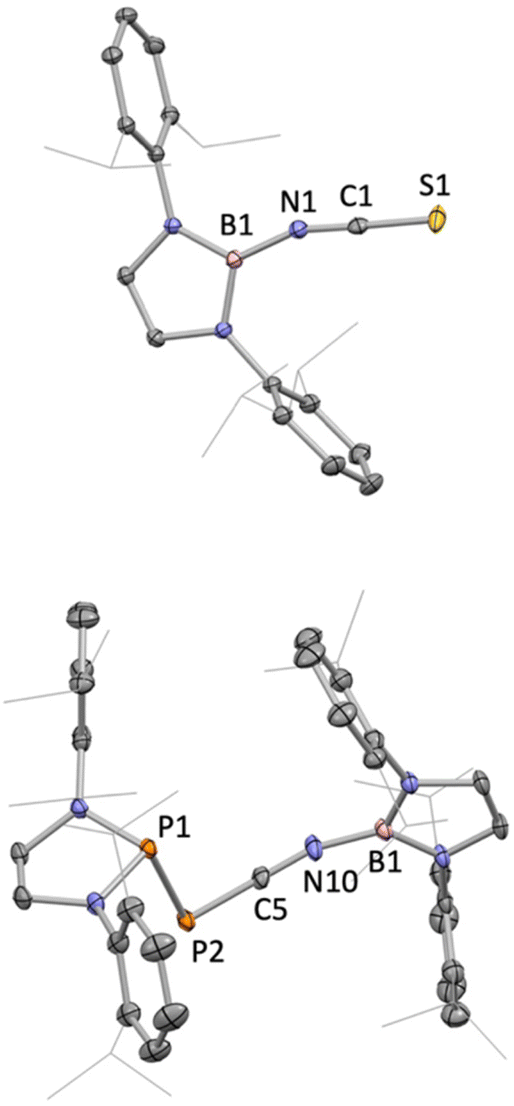 | ||
| Fig. 1 Molecular structures of 6 (upper) and one of the two independent molecules in the asymmetric unit of 7 (lower) in the solid state as determined by X-ray crystallography. Second disorder component and all hydrogen atoms omitted, and iPr groups shown in wireframe format for clarity; thermal ellipsoids shown at the 35% probability level. Key bond lengths (Å) and angles (°): (for 6) B1–N1 1.447(1), N1–C1 1.179(7), C1–S1 1.584(7), B1–N1–C1 156.8(4), N1–C1–S1 175.3(6); (for 7) B1–N10 1.430(3), N10–C5 1.184(3), C5–P2 1.668(2), P2–P1 2.299(1), B1–N10–C5 162.8(2), N10–C5–P2 178.7(2), C5–P2–P1 88.7(1). | ||
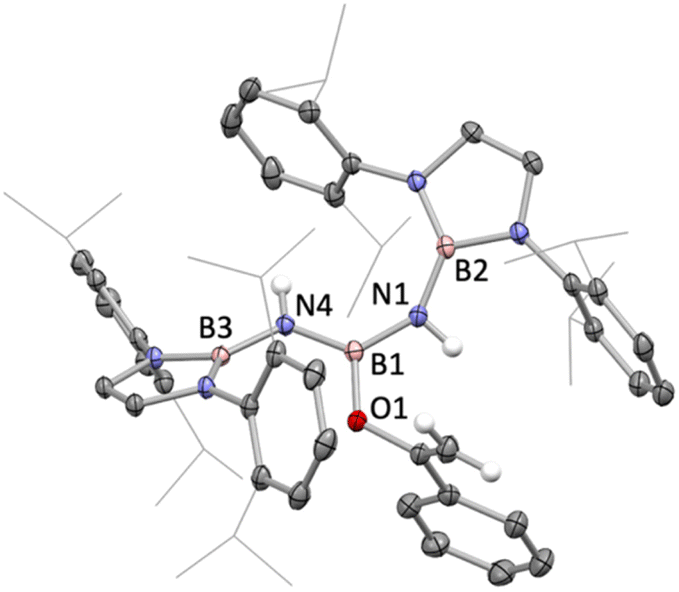 | ||
| Fig. 2 Molecular structure of 8 in the solid state as determined by X-ray crystallography. Most hydrogen atoms omitted and iPr groups shown in wireframe format for clarity; thermal ellipsoids shown at the 35% probability level. Key bond lengths (Å): B2–N1 1.438(2), N1–B1 1.426(2), B1–N4 1.420(1), N4–B3 1.451(1), B1–O1 1.398(1). | ||
The identity of the co-product derived from the central boron atom is difficult to establish unequivocally, although the formation of a white precipitate suggests that (in the cases of 5 and 7 at least) it is possibly the well-known cyclic metaborate K3[B3O6] (i.e. the trimeric aggregate of K[BO2]), consistent with its insolubility in organic solvents.13
With a view to exploring further both the scope and mechanism of this metathesis chemistry, we investigated the reactivity of 1 with organic carbonyl compounds. Ketones featuring enolizable hydrogen atoms, however, react with 1 in a different manner, reflecting the basic nature of the N centres, leading to α-deprotonation and assimilation of the corresponding enolate fragment at the central boron atom. Thus, the reaction of 1 with (excess) acetophenone leads to protonation of both nitrogen atoms, and the formation of a B-bound OC(Ph)CH2 moiety (Scheme 3 and Fig. 2).
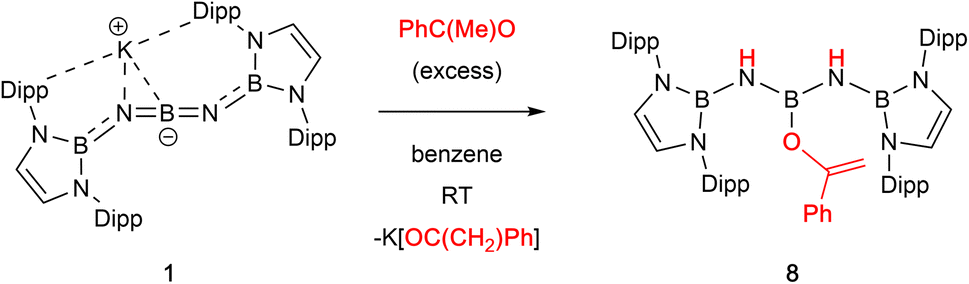 | ||
| Scheme 3 Reaction of 1 with acetophenone to generate boron enolate 8. | ||
Non-enolizable carbonyls such as benzaldehyde, PhCHO, on the other hand, react via CO bond cleavage in a manner similar to CO2 and {(H2CDippN)2}PPCO, and (in addition) allow further mechanistic details to be uncovered. Addition of excess benzaldehyde to a solution of 1 in benzene, leads to the clean formation (over a period of ca. 12 h at room temperature) of the borylimine, trans-(boryl)NC(H)Ph (10), which has been characterized structurally by X-ray crystallography (Fig. 3), and by standard spectroscopic/analytical methods. If the reaction is monitored in situ by 1H NMR spectroscopy in benzene-d6 solution, an intermediate species can be identified after 30 min (Fig. S10†), which is characterised by Dipp methine resonances at 3.61/3.89 ppm (each 2H), a boryl backbone signal (also 2H) at 6.05 ppm and a singlet (1H) at 5.28 ppm (cf. δH = 8.65 ppm for the imine proton of the final product 10). If volatiles are removed in vacuo after 1 h, and the reaction mixture recrystallized from pentane, a mixture of crystals is obtained, which includes not only 10, but also an intermediate K[(boryl)NB(O)OC(H)Ph], 9, which precedes the formation of the final imine product via elimination of the K[BO2] fragment (Scheme 4 and Fig. 3).
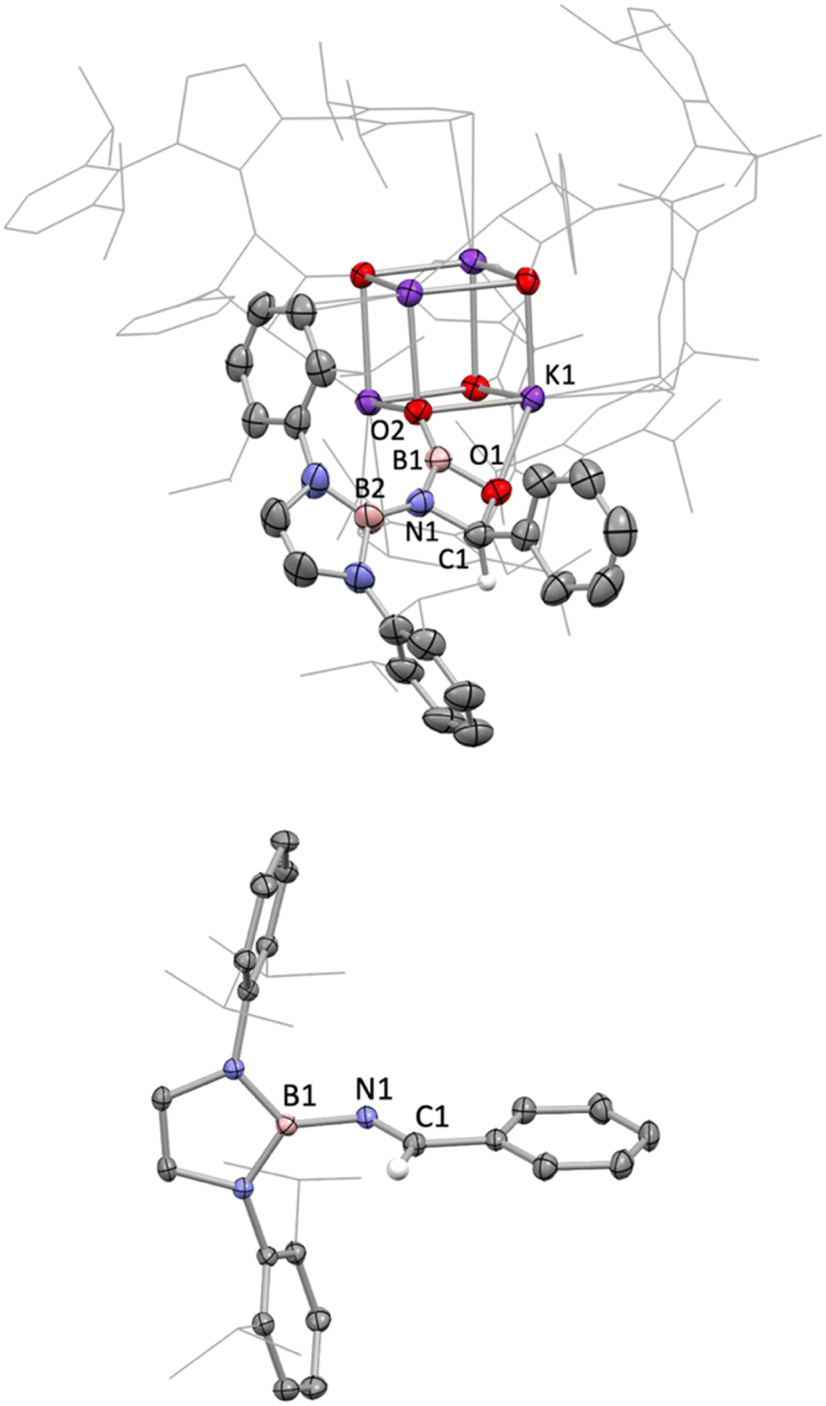 | ||
| Fig. 3 Molecular structures of compounds 9 (upper) and 10 (lower) in the solid state as determined by X-ray crystallography. Thermal ellipsoids are drawn at 50% probability level. Most H atoms are omitted for clarity. Key bond lengths (Å) and angles (°): (for 6) B2–N1 1.423(5), N1–C1 1.470(5), C1–O1 1.441(4), O1–B1 1.453(4), B1–N1 1.478(5), B1–O2 1.283(4); (for 7) B1–N1 1.448(3), N1–C1 1.254(3), B1–N1–C1 126.1(2), N1–C1–C2 123.5(2). | ||
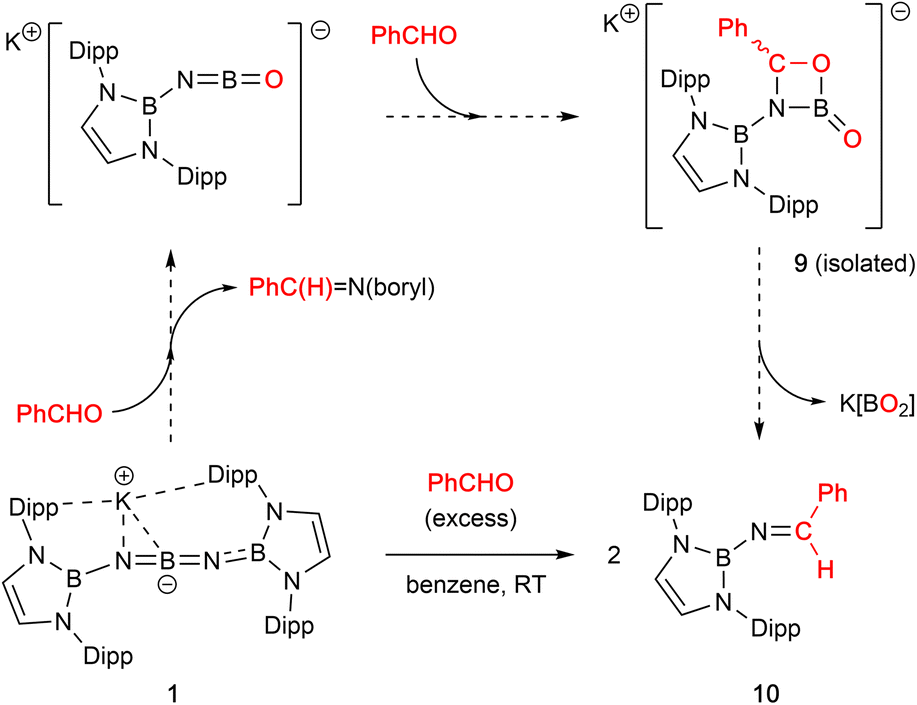 | ||
| Scheme 4 Cycloaddition reactivity of 1 with benzaldehyde, PhCHO, leading to the formation of borylimine 10. | ||
Analysis of crystalline 9 by X-ray diffraction reveals that the solid material contains a tetrameric potassium salt (based on a cubic K4O4 core), and an organic fragment featuring a four-membered CBNO ring. [2+2] cyclo-reversion via CO and BN bond cleavage then offers a route for the onward conversion of this species to borylimine 10.
Structurally, the organic component of 9 can be viewed as being derived from cycloaddition of the CO double bond of benzaldehyde across the NB double bond of an intermediate species of the type [(boryl)NBO]−. Geometrically, the bond length associated with the B–O linkage within the four-membered ring (1.453(4) Å) is consistent with a single bond; the associated O atom participates in a weak contact with a single K+ counterion, d(K⋯O) = 2.710(3) Å. By contrast, the exocyclic B–O bond is much shorter (1.283(4) Å), consistent with appreciable π-bonding character,14 and the associated O atom bridges three K+ cations (d(K⋯O) = 2.596(2), 2.604(2), 2.778(2) Å).
These structural and reactivity data suggest that 1 acts as a transfer agent for the [(boryl)N] group via a Wittig-type reaction involving [2+2] cyclo-addition/cyclo-reversion steps. To further probe the mechanism of the reaction with benzaldehyde, we examined the potential energy surface by density functional theory calculations (M06-2X/Def2-SVP//Def2-TZVP). The overall process involves two [2+2] cyclo-addition/reversion steps, each involving one of the BN bonds associated with the central boron atom of 1 (Fig. 4). The overall process is exergonic (by −20.5 kcal mol−1), and successive steps occur via transition states which are ca. 25–30 kcal mol−1 above the preceding intermediate. The first step in the reaction pathway is thought to be rate-determining, and involves [2+2] cyclo-addition of the CO bond of PhCHO across one BN unit of the anionic component of 1 to give IN1, which features an COBN four-membered ring bearing a pendant iminoboryl function. This step resembles the [2+2] cyclo-addition reported by Jones in the room temperature reaction of CO2 with the SiN bond of a silaimine.5c In the case of 1 it is only slightly exergonic (−0.7 kcal mol−1) and occurs via a barrier of +29.7 kcal mol−1.
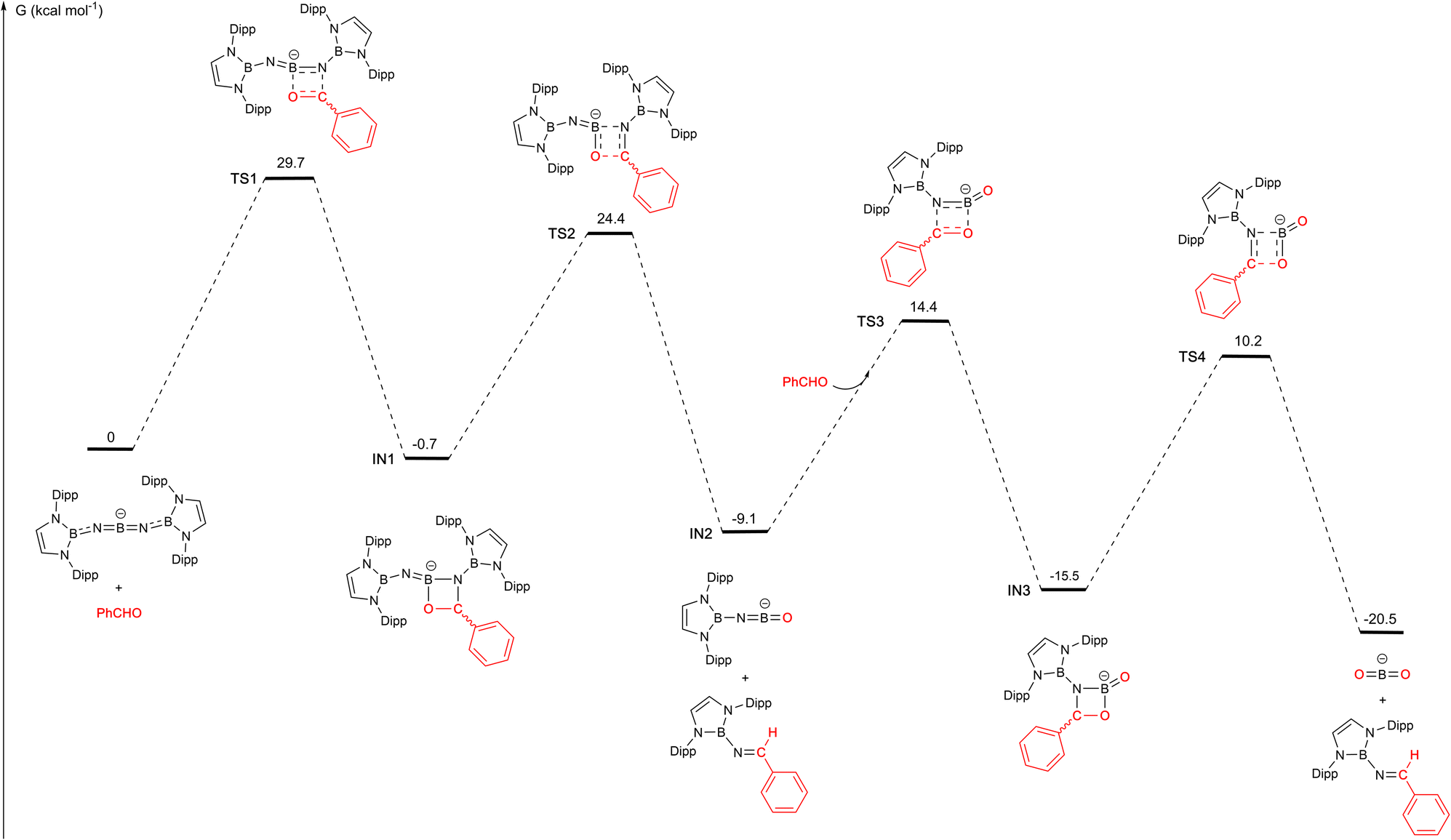 | ||
| Fig. 4 DFT calculated mechanism for the reaction of PhCHO with the anionic component of 1 (calculated at the M06-2X/Def2-SVP//Def2-TZVP level of theory with solvation modelled with smd, benzene; K+ counterions omitted for simplicity).15 | ||
Subsequent cyclo-reversion to release a first equivalent of trans-(boryl)NC(H)Ph, and generate IN2, [(boryl)NBO]−, involves traversing a slightly lower activation barrier of 25.1 kcal mol−1, and provides an extra 8.4 kcal mol−1 of thermodynamic stabilization. The uptake of a second equivalent of PhCHO occurs via a similar transition state to first generate a third intermediate IN3, akin to the structurally characterized system 9. [2+2] cyclo-reversion then yields the observed second equivalent of trans-(boryl)NC(H)Ph. The last two steps incur activation barriers of 23.5 and 25.7 kcal mol−1, respectively, and lead to a process which (overall) is exergonic to the tune of −20.5 kcal mol−1.15 We postulate, given the relatively low transition state barriers calculated beyond IN1, that the crystallization of compound 9 from solution relates to the crystallinity/insolubility of the tetrameric potassium salt, rather than to the intrinsic reaction barrier associated with its onward conversion to the final product (which is relatively low).
Finally, we set out to probe experimentally whether the differing geometric and electronic structures of bent BNBNB anion 1 and its linear counterpart 3 might lead to differences in reactivity towards electrophilic substrates. Surprisingly, however, the reaction of 3 with MeI can be shown to generate the same iminoborane product (HCDippN)2BNBN(Me)B(NDippCH)2 (4) as is formed in the reactions of 1/2 under comparable conditions (Scheme 1). The accompanying release of B(C6F5)3 can be confirmed by in situ11B and 19F NMR measurements (Fig. S13 and S14†). In similar fashion, the uptake of CO2 by 3 also yields boryl-isocyanate 5 in essentially quantitative conversion (Fig. S15†). These observations imply that the assimilation of the borane by the diazaborolyl heterocycle of 1/2 is chemically reversible – at least under these reactivity conditions.
Experimental
Included here are synthetic and characterising data for compounds 6, 7, 8 and 10. Data for all compounds, representative 1H NMR spectra, and details of crystallographic and computational studies are included in the ESI.†(HCDippN)2BNCS, 6. To a solution of 1 (100 mg 0.117 mmol) in benzene (10 mL) was added a drop of CS2 (ca. 0.02 mL, excess). The solution was heated at 60 °C overnight with a colour change from orange to red. Volatiles were removed in vacuo and the residue extracted with pentane (10 mL). The resulting solution was concentrated to ca. 2 mL and colourless crystals of 6 were obtained upon standing at room temperature which were suitable for X-ray crystallography. These were isolated by filtration and washed with cold pentane (2 × 1 mL). Yield: 51.3 mg (52.0%). 1H NMR (500 MHz, C6D6, 298 K): δH 1.17 (d, 3JH,H = 6.8 Hz, 12H, CH3 of Dipp), 1.29 (d, 3JH,H = 6.8 Hz, 12H, CH3 of Dipp), 3.12 (sept, 3JH,H = 6.8 Hz, 4H,C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)2 of Dipp), 5.91 (s, 2H, CH of boryl), 7.13 (m, 4H, m-ArH of Dipp), 7.20 (m, 2H, p-ArH of Dipp). 11B{1H} NMR (160 MHz, C6D6, 298 K): δB 16.1 (boryl). 13C{1H} NMR (126 MHz, C6D6, 298 K): δC 24.0, 24.5 (CH3 of Dipp), 28.9 (
(CH3)2 of Dipp), 5.91 (s, 2H, CH of boryl), 7.13 (m, 4H, m-ArH of Dipp), 7.20 (m, 2H, p-ArH of Dipp). 11B{1H} NMR (160 MHz, C6D6, 298 K): δB 16.1 (boryl). 13C{1H} NMR (126 MHz, C6D6, 298 K): δC 24.0, 24.5 (CH3 of Dipp), 28.9 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H(CH3)2 of Dipp), 118.6 (CH of boryl), 123.9 (m-Ar of Dipp), 128.6 (p-Ar of Dipp), 136.4 (CN of Dipp), 146.3 (o-Ar of Dipp), not observed (NCS). Elemental microanalysis: calc. for C27H36BN3S (%): C 72.80, H 8.15, N 9.43; meas. C 72.76, H 8.26, N 9.17.
H(CH3)2 of Dipp), 118.6 (CH of boryl), 123.9 (m-Ar of Dipp), 128.6 (p-Ar of Dipp), 136.4 (CN of Dipp), 146.3 (o-Ar of Dipp), not observed (NCS). Elemental microanalysis: calc. for C27H36BN3S (%): C 72.80, H 8.15, N 9.43; meas. C 72.76, H 8.26, N 9.17.
(HCDippN)2BNCPP(NDippCH2)2, 7. To a mixture of 1 (80 mg, 0.094 mmol) and (CH2DippN)2PPCO (69.3 mg, 0.148 mmol) in a 25 mL Schlenk flask was added 5 mL benzene. The resulting solution was stirred for 12 h with a colour change to dark green. Volatiles were removed in vacuo and the residue extracted with pentane (3 mL). The resulting solution was concentrated to ca. 1 mL and colourless crystals of 7 were obtained upon standing at room temperature which were suitable for X-ray crystallography. These were isolated by filtration and washed with cold pentane (2 × 1 mL). Yield: 88.6 mg (55.2%). 1H NMR (500 MHz, C6D6, 298 K): δH 1.05 (d, 3JH,H = 7.0 Hz, 12H, CH3 of (boryl)Dipp), 1.13 (d, 3JH,H = 7.0 Hz, 12H, CH3 of (boryl)Dipp), 1.19, 1.25, (d, 3JH,H = 6.6 Hz, 12H, CH3 of (phosphanyl)Dipp),1.31 (dd, 3JH,H = 6.6 Hz, 12H, CH3 of (phosphanyl)Dipp), 3.03(sept, 3JH,H = 7.0 Hz, 4H,C(CH3)2 of (boryl)Dipp), 3.11, 3.58 (m, 4H, CH2 of phosphanyl), 3.48, 3.76 (m, 4H,C(CH3)2 of (phosphanyl)Dipp), 5.86 (s, 2H, CH of boryl), 7.04 (m, 4H, m-ArH of (boryl)Dipp), 7.10 (m, 6H, ArH of (phosphanyl)Dipp), 7.18 (m, 2H, p-ArH of (boryl)Dipp). 11B{1H} NMR (160 MHz, C6D6, 298 K): δB 16.0 (boryl). 31P NMR (162 MHz, C6D6, 298 K): δP −183.3 (d, PC, 1JP,P = 298.0 Hz),165.6 (d, PC, 1JP,P = 298.0 Hz). 13C{1H} NMR (126 MHz, C6D6, 298 K): δC 23.9, 24.4 (CH3 of Dipp), 24.7, 25.2, 25.9 (d, CH3 of (phosphanyl)Dipp), 28.8 (H(CH3)2 of (boryl)Dipp), 28.9, 29.5 (d, H(CH3)2 of (phosphanyl)Dipp), 54.9 (d, CH2 of phosphanyl), 118.5 (CH of boryl), 123.6 (m-Ar of (boryl)Dipp), 123.9 (m-Ar of (phosphanyl)Dipp), 124.9 (p-Ar of (phosphanyl)Dipp), 127.5 (o-Ar of (boryl)Dipp), 136.8 (CN of (boryl)Dipp), 137.8 (d, CN of (phosphanyl)Dipp), 146.2 (o-Ar of (boryl)Dipp), 148.7, 150.4 (o-Ar of (phosphanyl)Dipp), not observed (NCP).
[{(HCDippN)2BNH}2B]OC(CH2)Ph, 8. 1 (15 mg 0.018 mmol) was dissolved in benzene (4 mL) and a drop of acetophenone (0.01 mL, excess) added. The reaction mixture was stirred for 1 h with an accompanying colour change to light yellow. Volatiles were removed in vacuo and the residue extracted with pentane (3 mL). The resulting solution was concentrated to ca. 0.5 mL and yellow crystals of 8 were obtained upon standing at room temperature which were suitable for X-ray crystallography. These were isolated by filtration and washed with cold pentane (2 × 1 mL). Yield: 7.8 mg (47.0%). 1H NMR (500 MHz, C6D6, 298 K): δH 1.16 (d, 3JH,H = 6.6 Hz, 24H, CH3 of Dipp), 1.18 (d, 3JH,H = 6.6 Hz, 24H, CH3 of Dipp), 2.56 (br, 2H, NH), 3.12, 4.43 (s, each 1H, CCH2), 3.28 (sept, 3JH,H = 6.8 Hz, 8H,C(CH3)2 of Dipp), 5.97 (s, 2H, CH of boryl), 7.07 (m, 17H, ArH of Dipp). 11B{1H} NMR (160 MHz, C6D6, 298 K): δB 22.4 (boryl). 13C{1H} NMR (126 MHz, C6D6, 298 K): δC 23.8, 25.5 (CH3 of Dipp), 28.8 (H(CH3)2 of Dipp), 97.7 (CH2), 118.6 (CH of boryl), 124.2 (m-Ar of Dipp), 125.3, 128.8, 136.7 (Ph), 127.4 (p-Ar of Dipp), 139.6 (CN of Dipp), 146.3 (o-Ar of Dipp), 154.4 (CH2). Elemental microanalysis: calc. for C60H81B3N6O (%): C 77.09 H 8.73 N 8.99; meas. C 76.89 H 8.51 N 9.12.
(HCDippN)2BN[κ2-(C,B)-C(H)PhBO2] 9 and (HCDippN)2BNC(H)Ph, 10. 1 (100 mg 0.117 mmol) was dissolved in benzene (10 mL) and a drop of benzaldehyde (0.02 mL, excess) added. The resulting solution was stirred for 30 min, with an accompanying colour change to yellow. Volatiles were removed in vacuo, and the residue extracted with pentane (5 mL). The resulting solution was concentrated to ca. 0.5 mL and a mixture of colourless crystals of 9 (unstable in solvent and briefly visible in NMR) and 10 were obtained which were suitable for X-ray crystallography.
(HCDippN)2BNCHPh, 10. 1 (100 mg 0.117 mmol) was dissolved in benzene (10 mL) and a drop of benzaldehyde (0.02 mL, excess) added. The resulting solution was stirred for 12 h with a colour change to light yellow. Volatiles were removed in vacuo and the residue extracted with pentane (5 mL). The resulting solution was concentrated to ca. 2 mL and colourless crystals of 10 were obtained which were suitable for X-ray crystallography. These were isolated by filtration and washed with cold pentane (2 × 1 mL). Yield: 16.6 mg (14.4%). 1H NMR (500 MHz, C6D6, 298 K): δH 1.27 (d, 3JH,H = 6.9 Hz, 12H, CH3 of Dipp), 1.29 (d, 3JH,H = 6.9 Hz, 12H, CH3 of Dipp), 3.43 (sept, 3JH,H = 6.9 Hz, 4H,C(CH3)2 of Dipp), 6.19 (s, 2H, CH of boryl), 6.82 (m, m-ArH of Ph), 6.89 (m, p-ArH of Ph), 7.17 (m, m-ArH of Dipp), 7.22 (m, p-ArH of Dipp), 7.46 (m, o-ArH of Ph), 8.65 (s, PhC(H)N). 11B{1H} NMR (160 MHz, C6D6, 298 K): δB 24.6 (boryl). 13C{1H} NMR (126 MHz, C6D6, 298 K): δC 24.2, 24.4 (CH3 of Dipp), 28.8 (H(CH3)2 of Dipp), 119.0 (CH of boryl), 123.7 (m-Ar of Dipp), 128.6 (p-Ar of Dipp),128.6 (o-Ar of Ph), 128.7 (m-Ar of Ph), 131.2 (p-Ar of Ph), 138.8 (CN of Dipp), 146.6 (o-Ar of Dipp), 167.8 (CN), overlap by solvent peak (Ph()–CN).
Conclusions
In conclusion, we report on the reaction chemistry of an unprecedented ‘inorganic cumulene’ – featuring a five-atom BNBNB chain – in particular with respect to species containing CO (and related) multiple bonds. The intrinsic polarity of the BNBNB chain (featuring electronic-rich nitrogen centres) is demonstrated by metathesis chemistry with electrophilic heteroallenes such as CO2, and with organic carbonyl compounds. Metathesis leads to {(HCDippN)2}BN transfer chemistry via consecutive [2+2] cyclo-addition/cyclo-reversion steps. Transfer of the imide unit to [CO], [CS], [PP{(NDippCH2)2}] and [C(H)Ph] moieties generates the (boryl)NCX systems (X = O, S, PP{(NDippCH2)2}, C(H)Ph), via processes driven thermodynamically by B–O bond formation. An isolable complex of stoichiometry K[(boryl)NB(O)OC(H)Ph] (9), formed via [2+2] cycloaddition of PhCHO to [(boryl)NBO]− can be shown to be an intermediate in the formation of (boryl)NC(H)Ph, and provides corroborating evidence for a DFT-calculated mechanism proceeding via consecutive bora-Wittig steps.
Data availability
The data on which this study is based are included in the ESI.†Author contributions
JT and CH carried out the syntheses and measured the characterizing data. AEC and LPG measured the single crystal X-ray diffraction data and solved the structures. AEC and JT carried out the quantum chemical calculations. SA and JT wrote the draft manuscript. SA and JMG managed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC Centre for Doctoral Training in Inorganic Chemistry for Future Manufacturing (OxICFM, EP/S023828/1; studentships to LPG and AEC), the Alexander von Humboldt Stiftung (postdoctoral fellowship, MD) and Indiana University (CH and JMG) for financial support.Notes and references
- See for example, (a) Handbook of Metathesis, Volume 1: Catalyst Development and Mechanism, ed. R. H. Grubbs and A. G. Wenzel, Wiley-VCH, Weinheim, 2nd edn, 2015 Search PubMed; (b) Handbook of Metathesis, Volume 2: Applications In Organic Synthesis, ed. R. H. Grubbs and D. J. O'Leary, Wiley-VCH, Weinheim, 2nd edn, 2015 Search PubMed; (c) Handbook of Metathesis, Volume 3: Polymer Synthesis, ed. R. H. Grubbs and E. Khosravi, Wiley-VCH, Weinheim, 2nd edn, 2015 Search PubMed.
- See for example, M. Aresta, A. Dibenedetto and E. Quaranta, Reaction Mechanisms in Carbon Dioxide Conversion, Springer, Berlin, 2016 Search PubMed.
- For transition metal oxide species formed from a d-block imide and CO2, see for example: (a) A. E. Guiducci, A. R. Cowley, M. E. G. Skinner and P. Mountford, J. Chem. Soc., Dalton Trans., 2001, 1392–1394 RSC; (b) S. R. Dubberley, A. Friedrich, D. A. Willman, P. Mountford and U. Radius, Chem.–Eur. J., 2003, 9, 3634–3654 CrossRef CAS; (c) C. L. Boyd, T. Toupance, B. R. Tyrrell, B. D. Ward, C. R. Wilson, A. R. Cowley and P. Mountford, Organometallics, 2005, 24, 309–330 CrossRef CAS; (d) C. L. Boyd, E. Clot, A. E. Guiducci and P. Mountford, Organometallics, 2005, 24, 2347–2367 CrossRef CAS; (e) S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed; (f) A. E. Guiducci, C. L. Boyd, E. Clot and P. Mountford, Dalton Trans., 2009, 5960–5979 RSC; (g) P. J. Tiong, A. Nova, L. R. Groom, A. D. Schwarz, J. D. Selby, A. D. Schofield, E. Clot and P. Mountford, Organometallics, 2011, 30, 1182–1201 CrossRef CAS; (h) A.-C. Schmidt, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2014, 53, 13142–13153 CrossRef CAS PubMed; (i) A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed; (j) A. J. Keane, W. S. Farrell, B. L. Yonke, P. Y. Zavalij and L. R. Sita, Angew. Chem. Int. Ed., 2015, 54, 10220–10224 CrossRef CAS PubMed . See also (k) ; (k) P.-J. Tiong, A. D. Schofield, J. D. Selby, A. Nova, E. Clot and P. Mountford, Chem. Commun., 2010, 46, 85–87 RSC; (l) J. Chu, E. Lu, Z. Liu, Y. Chen, X. Leng and H. Song, Angew. Chem., Int. Ed., 2011, 50, 7677–7680 CrossRef CAS; (m) P. J. Tiong, L. R. Groom, E. Clot and P. Mountford, Chem.–Eur. J., 2013, 19, 4198–4216 CrossRef CAS.
- For isolable transition metal carbamate complexes formed from an imide complex and CO2, see for example: (a) D. S. Glueck, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 2719–2721 CrossRef CAS; (b) D. S. Glueck, J. Wu, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1991, 113, 2041–2054 CrossRef CAS; (c) A. J. Blake, J. M. McInnes, P. Mountford, G. I. Nikonov, D. Swallow and D. J. Watkin, J. Chem. Soc., Dalton Trans., 1999, 379–392 RSC; (d) B. D. Ward, E. Clot, S. R. Dubberley, L. H. Gade and P. Mountford, Chem. Commun., 2002, 2618–2619 RSC; (e) Z. J. Tonzetich, R. R. Schrock and P. Müller, Organometallics, 2006, 25, 4301–4306 CrossRef CAS; (f) D. J. Mindiola, R. Waterman, V. M. Iluc, T. R. Cundari and G. L. Hillhouse, Inorg. Chem., 2014, 53, 13227–13238 CrossRef CAS PubMed; (g) M. Kinauer, M. Diefenbach, H. Bamberger, S. Demeshko, E. J. Reijerse, C. Volkmann, C. Würtele, J. van Slageren, B. de Bruin, M. C. Holthausen and S. Schneider, Chem. Sci., 2018, 9, 4325–4332 RSC.
- (a) F. M. Mück, J. A. Baus, A. Ulmer, C. Burschka and R. Tacke, Eur. J. Inorg. Chem., 2016, 1660–1670 CrossRef; (b) T. J. Hadlington, T. Szilvási and M. Driess, Chem. Commun., 2018, 54, 9352–9355 RSC; (c) K. Yuvaraj and C. Jones, Dalton Trans., 2019, 48, 11961–11965 RSC.
- (a) M. D. Anker, R. J. Schwamm and M. P. Coles, Chem. Commun., 2020, 56, 2288–2291 RSC; (b) A. Heilmann, P. Vasko, J. Hicks, J. M. Goicoechea and S. Aldridge, Chem.–Eur. J., 2023, 29, e202300018 CrossRef CAS.
- A. J. Downs and H.-J. Himmel, in The Chemistry of the Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, Wiley, Chichester, 2011, pp. 1–74 Search PubMed.
- For irreversible CO2 [2 + 2] cycloaddition at BN multiple bonds, without release of a CN-bonded product see, for example:
(a) L. Xie, J. Zhang, H. Hu and C. Cui, Organometallics, 2013, 32, 6875–6878 CrossRef CAS;
(b) F. Dahcheh, D. W. Stephan and G. Bertrand, Chem.–Eur. J., 2015, 21, 199–204 CrossRef CAS PubMed;
(c) M. Nitz, B. Borthakur, R. D. Dewhurst, A. Dießenberger, T. Dellermann, M. Schäfer, I. Krummenacher, A. K. Phukan and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef PubMed . See also:;
(d) G. A. Pierce, S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2007, 46, 2043–2046 CrossRef CAS;
(e) G. A. Pierce, N. D. Coombs, D. J. Willock, J. K. Day, A. Stasch and S. Aldridge, Dalton Trans., 2007, 4405–4412 RSC.
- For metathesis involving B–O bond formation from a MB unit see, for example:
(a) D. L. Kays (née Coombs), J. K. Day, L.-l. Ooi and S. Aldridge, Angew. Chem., Int. Ed., 2005, 44, 7457–7460 CrossRef;
(b) D. L. Kays (née Coombs), A. Rossin, J. K. Day, L.-l. Ooi and S. Aldridge, Dalton Trans., 2006, 399–410 RSC;
(c) H. Braunschweig, M. Burzler, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2007, 46, 8071–8073 CrossRef CAS;
(d) G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, A. L. Thompson, E. D. Jemmis, S. De and S. Aldridge, Organometallics, 2009, 28, 2947–2960 CrossRef CAS;
(e) S. De, G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, E. D. Jemmis and S. Aldridge, Organometallics, 2009, 28, 2961–2975 CrossRef CAS;
(f) J. Bauer, H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler and Q. Ye, J. Am. Chem. Soc., 2013, 135, 8726–8734 CrossRef CAS PubMed.
- J. Tang, C. Hu, A. E. Crumpton, D. Sarkar, L. P. Griffin, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2024, 146, 30778–30783 CrossRef CAS PubMed.
- For a recent review of cumulene chemistry see, for example: D. Wendinger and R. R. Tykwinski, Acc. Chem. Res., 2017, 50, 1468–1479 Search PubMed.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- W. H. Zachariasen, J. Chem. Phys., 1937, 5, 919–922 CrossRef CAS.
- Y. K. Loh, K. Porteous, M. A. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 Search PubMed.
- In the absence of K+ counterions (for computational simplicity) the trimerization of [BO2]− to [B3O6]3− makes little difference to the overall calculated free energy change shown in Fig. 4. This process is calculated to be close to thermo-neutral (ΔG = +0.5 kcal mol−1).
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and characterising data for all compounds, representative 1H NMR spectra, and details of crystallographic and computational studies are included in the supporting information. CCDC 2377740–2377745. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07487d |
| This journal is © The Royal Society of Chemistry 2025 |