
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Absolute standard hydrogen electrode potential and redox potentials of atoms and molecules: machine learning aided first principles calculations
Ryosuke
Jinnouchi
*a,
Ferenc
Karsai
b and
Georg
Kresse
bc
aToyota Central R&D Labs., Inc., Yokomichi 41-1, Nagakute, Aichi, Japan. E-mail: jryosuke@mosk.tytlabs.co.jp
bVASP Software GmbH, Berggasse 21, A-1090 Vienna, Austria
cUniversity of Vienna, Faculty of Physics, Kolingasse 14-16, A-1090 Vienna, Austria
First published on 21st May 2025
Abstract
Correction for ‘Absolute standard hydrogen electrode potential and redox potentials of atoms and molecules: machine learning aided first principles calculations’ by Ryosuke Jinnouchi et al., Chem. Sci., 2025, 16, 2335–2343, https://doi.org/10.1039/D4SC03378G.
The authors regret that in the original manuscript, a systematic error was present in the calculation of vibrational quantum corrections for the solvated proton.
Specifically, in the classical harmonic oscillator model used to evaluate the nuclear quantum contribution to the free energy via eqn (S24), base-10 logarithms were mistakenly used instead of natural logarithms. The corrected version of eqn (S24) is shown below.
Ac,vib = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(hνi/kBT) (S24) ln(hνi/kBT) (S24) |
This error resulted in an overestimation of the free energy of the solvated proton by approximately 0.1 eV, which in turn caused an upward shift in both the real potential and the absolute standard hydrogen electrode potential (ASHEP), as presented in Table 1.
Although this correction also leads to minor changes in the plot of the redox potential for the 2H+/H2 couple and the RMSE bar in Fig. 4, the visual differences are subtle and not easily discernible. The main conclusion of the study remains unchanged.
The corrected values for the real potential, ASHEP, and the vibrational quantum corrections are provided in the revised versions of Tables 1 and S5 shown below.
 (eV), ASHEP (V) and relevant free energies (eV) calculated by five exchange–correlation functionals (RPBE+D3, PBE0, PBE0+D3, HSE06 and B3LYP) compared with the experimental values recommended by the International Union of Pure and Applied Chemistry (IUPAC).1 ΔatE and ΔatG0 represent the atomization energy and dissociation free energy of the H2 molecule, respectively. ΔionG0 is the ionization potential of an H atom in vacuum. MLFF denotes the machine-learned force field trained on the RPBE+D3 data. The specified modelling error bars correspond to 2σ, estimated by block averaging analysis.2 The corrected values are highlighted in bold for clarity
(eV), ASHEP (V) and relevant free energies (eV) calculated by five exchange–correlation functionals (RPBE+D3, PBE0, PBE0+D3, HSE06 and B3LYP) compared with the experimental values recommended by the International Union of Pure and Applied Chemistry (IUPAC).1 ΔatE and ΔatG0 represent the atomization energy and dissociation free energy of the H2 molecule, respectively. ΔionG0 is the ionization potential of an H atom in vacuum. MLFF denotes the machine-learned force field trained on the RPBE+D3 data. The specified modelling error bars correspond to 2σ, estimated by block averaging analysis.2 The corrected values are highlighted in bold for clarity
| ΔatE | ΔatG0 | ΔionG0 |
|
ASHEP | |
|---|---|---|---|---|---|
| MLFF | 4.58 | 4.04 | 13.75 | −11.09 ± 0.05 | −4.68 ± 0.05 |
| RPBE+D3 | 4.58 | 4.04 | 13.75 | −11.12 ± 0.06 | −4.65 ± 0.05 |
| PBE0 | 4.53 | 3.99 | 13.64 | −11.15 ± 0.09 | −4.48 ± 0.09 |
| PBE0+D3 | 4.53 | 3.99 | 13.64 | −11.21 ± 0.09 | −4.42 ± 0.09 |
| HSE06 | 4.53 | 3.99 | 13.63 | −11.15 ± 0.09 | −4.47 ± 0.09 |
| B3LYP | 4.78 | 4.25 | 13.67 | −11.02 ± 0.08 | −4.77 ± 0.09 |
| Exp. | 4.73 | 4.21 | 13.62 | −11.28 ± 0.02 | −4.44 ± 0.02 |

Table S5 Nuclear quantum effects on the free energies of H2O and H3O+ isolated in vacuum estimated as the difference between the quantum oscillator model and the harmonic oscillator model. The estimation using the experimental vibrational frequencies of solvated proton is also listed. Units of the free energy and vibrational frequencies are eV and cm−1, respectively. The corrected values are highlighted in bold for clarity.
| Species | Property | RPBE+D3 | PBE0 | PBE0+D3 | Exp. |
|---|---|---|---|---|---|
| H2O | ν i | 3831 | 4020 | 4020 | |
| 3702 | 3886 | 3885 | |||
| 1592 | 1611 | 1611 | |||
| A q,vib | 0.566 | 0.590 | 0.590 | ||
| A c,vib | 0.201 | 0.204 | 0.204 | ||
| A q−c | 0.365 | 0.386 | 0.386 | ||
| ZPE | 0.566 | 0.590 | 0.590 | ||
| H3O+ | ν i | 3599 | 3761 | 3761 | 3020 |
| 3598 | 3760 | 3760 | |||
| 3482 | 3650 | 3649 | |||
| 1647 | 1669 | 1669 | 1760 | ||
| 1638 | 1652 | 1651 | 1250 | ||
| 802 | 688 | 688 | |||
| A q,vib | 0.916 | 0.941 | 0.941 | 0.374 | |
| A c,vib | 0.360 | 0.361 | 0.360 | 0.170 | |
| A q−c | 0.556 | 0.580 | 0.580 | 0.204 | |
| ZPE | 0.916 | 0.942 | 0.942 | 0.374 | |
Correction to 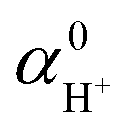 |
0.190 | 0.194 | 0.194 | 0.204 | |
| ZPE[H3O+]–ZPE[H2O] | 0.350 | 0.351 | 0.351 | 0.374 |
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
Notes and references
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, 1987 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |