
Bulk passivation and suppressing non-radiative recombination loss in a 3D all-inorganic CsPbIBr2 perovskite solar cell via a 2D layered perovskite framework†
Tapas
Das
 a,
Faisal
Farooq
b,
Parul
Garg
b,
Sakal
Singla
b,
Asim
Guchhait
*a and
Ashok
Bera
*b
a,
Faisal
Farooq
b,
Parul
Garg
b,
Sakal
Singla
b,
Asim
Guchhait
*a and
Ashok
Bera
*b
aDepartment of Physics, Prabhat Kumar College, Karkuli, Contai, West Bengal 721404, India. E-mail: guchhait.asim@gmail.com
bDepartment of Physics, Indian Institute of Technology Jammu, J & K 181221, India. E-mail: ashok.bera@iitjammu.ac.in
First published on 19th November 2024
Abstract
Improving perovskite film quality for reducing non-radiative recombination centers is one of the key aspects of designing efficient and stable perovskite solar cells (PSCs). In this work, we fabricated a high-performing and ambient stable CsPbIBr2-based PSC by incorporating a 2D perovskite framework within a 3D perovskite structure. An optimum amount of 2D doping can anchor the grain boundaries to improve the crystallinity and grain sizes and ultimately suppress non-radiative recombination centers within the perovskite. The solution-processed perovskite film with the structural formula ((PEA)2PbI4)X(CsPbIBr2)1−X for X = 0.02 exhibited an improved average grain size of 853.38 ± 0.18 nm in comparison to 350.43 ± 0.09 nm of pristine CsPbIBr2 thin films. The bulk passivation within the perovskite was supported by the X-ray diffraction, steady-state, and time-resolved photoluminescence results. We fabricated a PSC with the device structure FTO/c-TiO2/m-TiO2/(PEA)2PbI4)X(CsPbIBr2)1−X/Spiro-OMeTAD/Ag, and achieved a power conversion efficiency (PCE) of 10.13% under ambient conditions with X = 0.02 and only 8.08% PCE for the pristine 3D perovskite (X = 0) device. The devices with 2D incorporation showed excellent ambient stability without any encapsulation and retained 80% of their initial PCE (T80) after 500 hours of ambient storage, whereas the device with pure 3D perovskite retained only 20% of its initial PCE after 400 hours of ambient storage. Simulation results, in combination with the experimental data, show that a reduced density of recombination centers resulted in much improved device performance.
1 Introduction
Organic–inorganic hybrid perovskites, a class of crystalline compounds, are recognized as emerging materials in the thin film solar cell technology primarily because of their fascinating optoelectronic characteristics and their low-temperature, cost-effective solution processability.1–3 Although hybrid PSCs have achieved a remarkable record-breaking power conversion efficiency (PCE) of 26.7%,4 the Shockley–Queisser (S–Q) theory's radiative limit5 with an excellent long-term ambient device stability is still a challenge to the researchers. The use of multijunction two-terminal (monolithic) or four-terminal tandem solar cells is a promising way to reduce transmission and thermalization loss and reach performance beyond the S–Q theoretical limit. For the fabrication of perovskite tandem cells, a wide-bandgap (Eg) perovskite, typically with Eg ≥ 1.6 eV, is required for either perovskite/perovskite or perovskite/c-silicon tandem devices.6–12 The mixed-halide Cs-based all-inorganic perovskites stand out to achieve this aim since they can be tuned to have Eg between 1.7 and 2.3 eV by adjusting the I/Br ratio.13–15 Among these aforementioned perovskite families of CsPbIxBr3−x (where x = 1, 2, 3), researchers are trying to optimize CsPbIBr2 due to its high Eg of 2.05 eV, and better ambient phase stability.16 Several strategies have been adopted to improve the performance and the stability of the CsPbIBr2-based PSCs,17–20 but the synthesis of high-quality CsPbIBr2 thin film remains extremely difficult due to surface noticeable pinholes and large grain boundaries. More specifically, the grain boundaries may develop shallow defect states close to the band edge that trap the available carriers leading to non-radiative recombination in CsPbIBr2 layers. Ideally, every photogenerated carrier inside a solar cell should leave the cell to reach the S–Q limit. However, all varieties of PSCs do experience non-radiative recombination losses due to Auger recombination, electron-phonon coupling, defect-assisted recombination, and band-tail recombination.21 Furthermore, perovskite absorbers often degrade more quickly in the presence of a significant density of non-radiative recombination centers22,23 impeding the development of these materials for real-world applications. Reduction of all forms of non-radiative recombination losses to further push device performances and simultaneously improve long-term device photovoltaic (PV) stability is crucial. Defect state passivation is one of the key strategies to suppress the non-radiative recombination loss within the absorber layer.Recent studies have focused much emphasis on layered Ruddlesden–Popper (RP)-phase 2D perovskites because of less non-radiative recombination and their higher stability compared to 3D equivalents.24–26 The basic structural formula for a layered 2D RP phase perovskite is (RNH3)2An−1BnX3n+1, where n is the dimensional thickness of the inorganic layers and RNH3 is a big alkyl ammonium cation (organic spacer).27,28 2D perovskites containing RNH3 molecules are hydrophobic, which helps to increase moisture and air stability. However, compared to 3D-based PSCs, the solar cell performance of 2D-based PSCs is worse. This is expected since big cations obstruct out-of-plane charge-carrier movement, and organic spacers are insulators.29 Forming a layered 2D RP phase perovskite within 3D perovskites presents an effective way to reduce the non-radiative recombination losses within the perovskite, which may yield better performance. Generally, two types of passivation strategies have been reported: interfacial passivation and bulk passivation. Interfacial passivation on 3D CsPbIBr2 using a 2D RP phase layered perovskite is quite challenging because Cs+ has a strong interaction with Pb and halide ions, making it more difficult for the big ammonium cations.30–33 Conversely, bulk passivation within 3D CsPbIBr2via 2D RP phase perovskite doping requires no surface alteration caused by the organic electrically insulating group, which will cause the effective carrier transfer to decline, whereas the 2D perovskites can passivate the bulk and interface defects of 3D perovskites.34
Herein, our focus was to fabricate a uniform and pinhole-free all-inorganic CsPbIBr2 wide-Eg perovskite thin film via 2D (PEA)2PbI4 RP phase layered perovskite doping to reduce non-radiative recombination with better ambient phase stability. This 2D perovskite has better mechanical durability and ambient stability due to the longer aromatic carbon chain, making it more preferable compared to the other 2D perovskites like (BA)2PbI4, (C6H13NH3)2PbI4, and (C6H5C2H4NH3)2PbI4. Additionally, this (PEA)2PbI4 2D layered perovskite has a matching refractive index (real part of refractive index 2.100) and comparable average static dielectric constant (2.82) with the host CsPbIBr2 perovskite for better light management.35–37 There is also a preferable type II band alignment within the 3D:2D bulk heterojunction as the highest occupied molecular orbital (HOMO) energy level of CsPbIBr2 is −6.11 eV and the lowest unoccupied molecular orbital (LUMO) energy level is −4.08 eV and those for the 2D (PEA)2PbI4 one are −4.73 eV and −2.388 eV.38–40 This type II band offset charge would help in better charge separation. Further, we utilized our optimized perovskite thin film in an all-inorganic PSC with a device architecture of FTO/c-TiO2/m-TiO2/CsPbIBr2 and ((PEA)2PbI4)X(CsPbIBr2)1−X/Spiro-OMeTAD/Ag. At an optimum 2% doping concentration (x = 0.02), the average grain size increased from 350 nm to 854 nm; the photocarrier lifetime increased from 14 ns to 29 ns, resulting in an improved current density–voltage (J–V) performance of the device with a PCE enhancement from 8.08% to 10.13% with a reduced hysteresis index. The estimated Shockley–Read–Hall (S–R–H) recombination rate reduced by 3.8 × 1017 cm−3 s−1. We also found better ambient (relative humidity (RH) ≈ 60%, room temperature (RT) ≈ 24 °C) device stability with T80 > 500 hours and 100 hours of operational stability at 85 °C with 80% of their initial device performances, confirming an improved device performance and stability.
2 Experimental section
2.1 Materials
All the chemicals used in this report are commercially available unless specified otherwise. N,N-dimethylformamide (DMF, Sigma Aldrich, 99.9%), dimethyl sulfoxide (DMSO, Sigma Aldrich, 99.9%), chlorobenzene (CB, Sigma Aldrich, 99.99%), lead bromide (PbBr2, Sigma Aldrich, 99%), cesium iodide (CsI, Sigma Aldrich, 99.9%), spiro-OMeTAD (Sigma Aldrich, 99.99%), 4-tert-butylpyridine (tBP), bis(trifluoromethane) sulfonimide lithium salt (Li-TFSI, Sigma Aldrich, 99.99%), lead iodide (PbI2, TCI, 99.99%), phenethylammonium iodide (PEAI, Sigma Aldrich, 99.9%), titanium isopropoxide (Sigma Aldrich, 99.9%), hydrochloric acid (HCl, Sigma Aldrich, 99.999%), ethanol (Sigma Aldrich, 99.999%), [6,6]-phenylC61-butyric acid methyl ester (PCBM, TCI, 99.5%), fluorine doped tin oxide coated glass substrates (FTO, Sigma Aldrich, thickness 2.2 mm, sheet resistance 7 Ω sq−1), and titania paste (Sigma Aldrich, 99.99%).2.2 Device fabrication
The patterned FTO-coated glass substrates were cleaned ultrasonically through a sequential step using Hellmanex solution, DI water, acetone, and IPA for 15 minutes, carefully followed by oxygen plasma cleaning. A compact-titanium dioxide (c-TiO2) electron transport layer (ETL) was spin-coated on the precleaned FTO substrates at 3000 rpm for 30 s with 1500 acceleration per sec followed by annealing at 450 °C for 45 minutes. After that, mesoporous-TiO2 (m-TiO2) was spin-coated on the ETL at 5000 rpm for 40 s with 2500 acceleration per sec and annealed at 500 °C for 1 hour in a muffle furnace. Further, the absorber layer ((PEA)2PbI4)X(CsPbIBr2)1−X was spin-casted on the ETL at 1000 rpm for 10 s followed by 3000 rpm for 30 s with 200 μl of CB antisolvent treatment and annealed at 180 °C for 20 minutes directly in a controlled inert atmosphere, maintaining the humidity <0.1 ppm and O2 < 10 ppm. Then, the hole transport layer (HTL) spiro-OMeTAD solution was spin-coated on the pre-deposited perovskite layer at 4000 rpm for 40 s and stored in a dry and dark place overnight. We spin-coated n-type PCBM (20 mg ml−1 in CB) at 1500 rpm for 20 s for electron-only device fabrication. Finally, a 100 nm Ag electrode was thermally evaporated at 0.1 Å s−1 for the first 30 nm and at 0.5 Å s−1 for the remaining portion using a metal mask having an active area of 0.12 cm2.2.3 ETL solution preparation
The c-TiO2 was prepared using an acidic solution. 369 μl of titanium(IV) isopropoxide was diluted with 2.53 ml of ethanol and 170 μl of hydrochloric acid under vigorous stirring followed by filtration through a 0.45 μm polytetrafluoroethylene filter. The m-TiO2 solution was prepared by diluting titania paste in ethanol at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 W/V ratio, ultrasonicating for 30 minutes and stirring vigorously for 1 hour.
:5 W/V ratio, ultrasonicating for 30 minutes and stirring vigorously for 1 hour.
2.4 ((PEA)2PbI4)X(CsPbIBr2)1−X perovskite absorber solution preparation
First, we prepared a stock solution of (PEA)2PbI4 by mixing PEAI and PbI2 in a 2:1 molar ratio in 1.5 ml DMF followed by stirring for 2 hours at room temperature. Then, the CsPbIBr2 perovskite solution was prepared by mixing CsI, and PbBr2 in 1 ml DMSO by maintaining a 1:1 molar ratio. Then we mixed these two perovskite solutions under vigorous stirring stoichiometrically to maintain the 2D ratios of X = 0, 0.01, 0.02, 0.04, 0.08, 0.1, and 1.
2.5 Characterization
The X-ray diffraction (XRD) measurements were carried out at RT using a Bruker D8 Advance system with Cu Kα radiation. The morphological features of perovskite film were studied by field emission scanning electron microscopy (FESEM) (JEOL JSM 7900F) and atomic force microscopy (AFM) (Cypher S AFM – Asylum Research). The ultraviolet-visible (UV-vis) absorption spectrum was measured by using a UV-vis spectrophotometer (Cary 5000). X-ray photoelectron spectroscopy (XPS; Nexsa ThermoFisher) was carried out to estimate the chemical composition of the perovskite material. The J–V measurements were performed using a Keithley 2450 under AM 1.5 G illumination (100 mW cm−2) from a solar simulator (ABET 110005). The intensity of light was calibrated using a standard reference silicon cell. The external quantum efficiency (EQE) was determined in AC mode (ABET instrument). The photoluminescence (PL) and time-resolved PL spectra were measured using a customized confocal microscope (Witec Alpha 300). The electrochemical impedance spectroscopy (EIS) measurements were performed using a CHl 1150C, CH instrument with a 10 mW cm−2 intense 445 nm laser beam. The numerical simulations were carried out using solar cell capacitance simulator one dimension (SACPS 1D) software version 3.3.09.3 Results and discussion
At first, the quality of the deposited perovskite thin films was examined by analyzing the XRD pattern in the ambient environment, and the XRD patterns of the perovskite thin films are shown in Fig. 1a. The highly intense (100), (110), (200) planes corresponding to 2θ values of 15.40°, 21.59°, and 30.60° confirm the cubic α phase with space group Pm-3m of the 3D CsPbIBr2 perovskite phase formation (X = 0) matched with the ICDS number 97852, as reported previously.41 The (002), (004), (006), (008), (0010), and (0012) planes corresponding to 2θ values of 5.56°, 11.0°, 16.41°, 21.9°, 27.39°, 33.05° confirmed the formation of the 2D layered RP phase (PEA)2PbI4 perovskite.42 The peak intensity of the highly intense (200) crystal plane in the 3D perovskite thin film was enhanced for X = 0.02 and subsequently decreased with increasing 2D perovskite doping ratios. The position of the (200) plane shifted slightly to a higher angle as the X value increased from 0 to 0.04, showing an increase in lattice strain due to the incorporation of larger cations from the 2D perovskite. The peak intensities for the (200), (110), and (100) planes corresponding to 3D perovskite reduced significantly for X = 0.08, and an extra peak appeared at 11.54° (marked as # in Fig. 1a for X = 0.08), which might be due to the CsPb2I4Br platelet-like 2D structure, as reported previously.43,44 Furthermore, the reappearance of a strong PbI2 phase for X = 0.08 at 2θ = 12.97° (marked as * in Fig. 1a) supports the instability of the 3D perovskite phase with a high amount of 2D perovskite doping.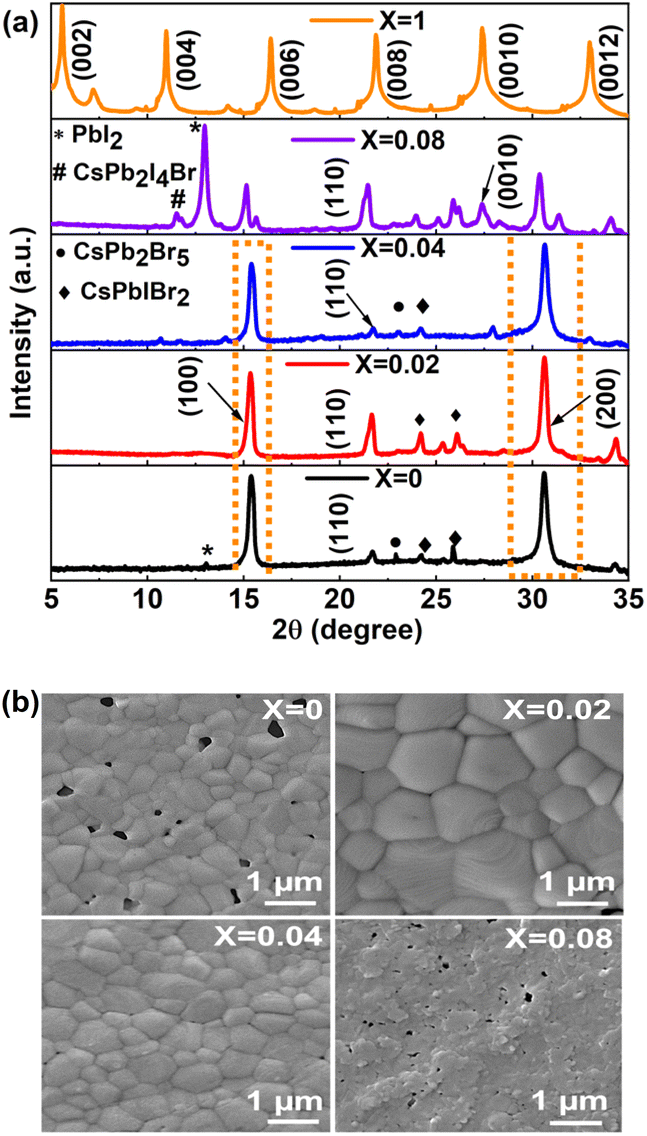 | ||
| Fig. 1 (a) XRD patterns of the synthesized ((PEA)2PbI4)X(CsPbIBr2)1−X perovskite thin films, #labelled peak corresponds to the CsPb2I4Br 2D phase and (b) top view FESEM morphological analysis of the fabricated ((PEA)2PbI4)X(CsPbIBr2)1−X perovskite thin films. | ||
We then studied the crystallite size (D) of the perovskite films using the Debye–Scherrer formula;45
 | (1) |
In addition to the good crystallinity of the perovskite materials, good morphology of the perovskite thin film is desirable for device application. We analyzed the morphology of the fabricated ((PEA)2PbI4)X(CsPbIBr2)1−X perovskite thin films using FESEM, as shown in Fig. 1b. We studied the statistical count of 40 grains from the FESEM top view images using ImageJ software of the fabricated perovskite thin films, as shown in Table S2 and Fig. S1.† The average grain size of the CsPbIBr2 (X = 0) 3D perovskite thin film was 350.43 ± 0.09 nm with a maximum grain size of 709 nm, while for X = 0.02, the average grain size was improved significantly to 853.38 ± 0.18 nm with a maximum grain size of 1.367 μm. Furthermore, with an increase in the X value up to 0.04, there are fewer large grains, and suddenly, the grains disappear with X = 0.08. This might be due to the increasing amount of long-chain PEA+ present in the 2D layered perovskite, which will not facilitate an increase in the grain size. The pin holes, which appeared to a large extent in the thin film with X = 0, were reduced significantly for the film with X = 0.02, which is helpful for improving the device performance. Similar improvement in the surface morphology was observed in AFM images in Fig. S2a and b,† in which the root mean square roughness of the CsPbIBr2 film decreases from 28.40 nm to 14.33 nm after (PEA)2PbI4 treatment. To confirm the incorporation of the 2D perovskite within the 3D CsPbIBr2 one, we performed the energy dispersive X-ray (EDX) analyses for the 2D doped (X = 0.02) perovskite thin film only, as shown in Fig. S3,† and the atomic percentages of the constituent elements are shown in Table S3.† The presence of 2.40% of N elements confirmed the incorporation of 2D perovskite within 3D CsPbIBr2 perovskite.
The XPS spectra of the fabricated perovskite films with and without 2D perovskite doping in bulk after applying an Ar+ ion beam etching for 120 s in Fig. 2a show the presence of Cs, Pb, Br, and I atoms. The high-resolution XPS spectra of the constituent elements of the perovskite are shown in Fig. 2b–f and S4.† The spin–orbit coupling splits the peaks associated with the d and f orbitals (Cs 3d, I 3d, Br 3d, and Pb 4f) into two components of d5/2 and d3/2 peaks for the d orbitals, and f7/2 and f5/2 peaks for the f orbitals. In the high-resolution Cs spectra, the peak positions of Cs 3d peaks (3d5/2 at 724.1 eV and 3d3/2 at 738 eV), Pb 4f peaks (4f7/2 at 138.6 eV and 4f5/2 at 143.5 eV), I 3d peaks (3d5/2 at 619.3 eV and 3d3/2 at 630.7 eV) and Br 3d peaks (3d5/2 at 68.7 eV and 3d3/2 at 69.7 eV) confirmed their corresponding oxidation states in CsPbIBr2 perovskite. The presence of the N 1s peak at 402 eV in the X = 0.02 perovskite thin film, as shown in Fig. 2f, supports the presence of PEA+ since there were no other organic cations and confirms the incorporation of the 2D framework within the bulk 3D perovskite. The reduced FWHM of the elements after doping, as given in Table S4,† shows an enhanced stable chemical environment within the perovskite structure. The improved peak intensity with 2D incorporation might result from the better morphology and pinhole-free film.
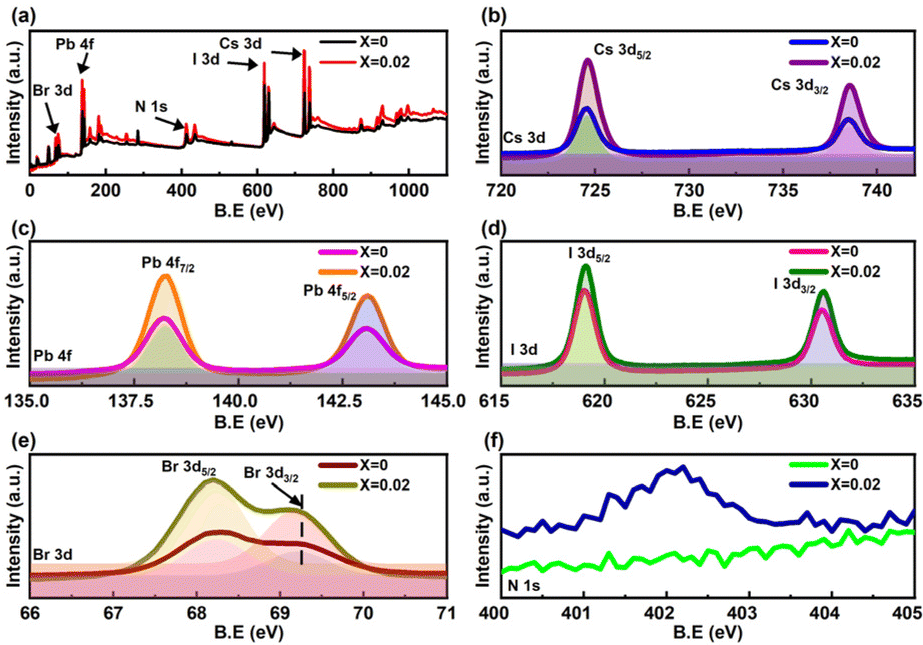 | ||
| Fig. 2 (a) XPS survey-spectra of the fabricated perovskite thin films with and without 2D perovskite doping. High-resolution XPS spectra corresponding to the analyses of (b) Cs 3d peak, (c) Pb 4f peak, (d) I 3d peak, (e) Br 3d peak, and (f) N 1s peak. | ||
The absorption spectra of the 3D all-inorganic mixed halide perovskite (AIMHP) CsPbIBr2 thin films doped with varying amounts of 2D layered perovskite material (PEA)2PbI4 with the structural formula ((PEA)2PbI4)X(CsPbIBr2)1−X were studied at RT in the wavelength range of 400–800 nm under ambient environmental conditions (RH ≈ 60%), as shown in Fig. 3a. The absorbance edge at ≈ 610 nm (Fig. 3a) corresponding to X = 0, confirms the 3D CsPbIBr2 perovskite phase formation, as reported previously. After increasing the 2D perovskite doping ratio (X values) from 0 to 0.02, there is no significant change in absorbance edge but a slight enhancement in the light energy absorbance in the lower wavelength region (400–573 nm). This lower wavelength light energy absorption is amplified with a blueshift in the absorbance edge when the X values are further increased to 0.1. The enhancement in light absorption with a blue shift confirms the lower dimension (2D) of the perovskite materials, which have more probability of electron confinement with a layered structure. We calculated the optical Eg of all fabricated thin films as shown in Fig. 3b, using the Tauc plot (αhν)2vs. hν, where α is the absorption coefficient and ν is the frequency of absorbed light.48 The calculated optical Eg of 2.063 eV from the Tauc plot (shown in Fig. 3b) confirms the 3D CsPbIBr2 perovskite phase (X = 0) as reported previously.46,47 These values increased from 2.063 eV to 2.066, 2.106, and 2.152 eV when the X values varied from 0 to 0.04, 0.08, and 0.1. The Eg was altered due to an increase in the valence band maximum alongside the conduction band minimum.49 The pure 2D perovskite exhibited an optical band energy of 2.342 eV, as depicted in Fig. 3b.
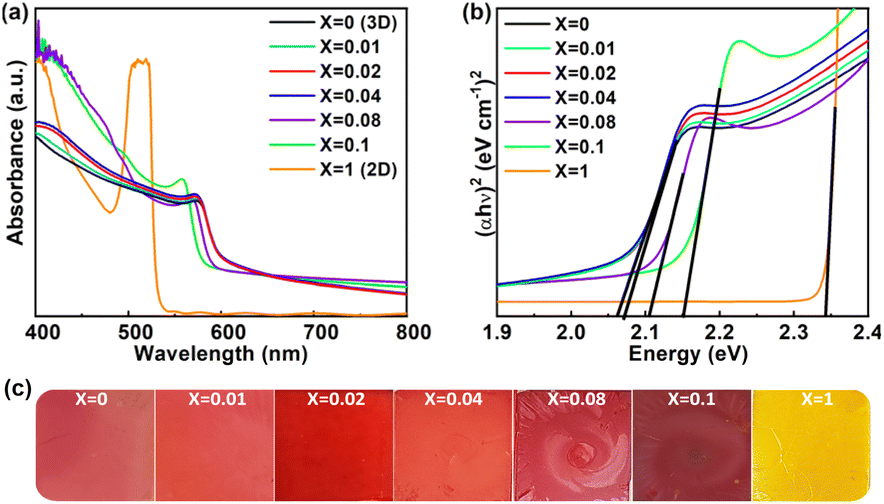 | ||
| Fig. 3 (a) UV-vis absorbance spectra of the perovskite thin films, (b) Tauc plot for optical Eg calculation, and (c) digital photographic images of the ((PEA)2PbI4)X(CsPbIBr2)1−X thin films deposited on the bare glass substrate. | ||
As we observed from FESEM, the incorporation of the 2D structure (with X = 0.02) improves the grain sizes significantly and likely leads to a bulk passivation of the trap states generated at the grain boundaries. This reduction of trap states could be supported by Urbach energy analysis, as shown in Fig. S5.† This analysis gives statistics of energetic disorder in these fabricated ((PEA)2PbI4)X(CsPbIBr2)1−X perovskite thin films and helps to understand the changes in absorbance spectra with varying the 2D content. The Urbach energy was determined using the formula:
 | (2) |
The passivation of the trap states within a light-absorbing material was further studied via PL measurements. Fig. 4a and b show the steady state PL (SSPL) and time-resolved PL (TRPL) of the fabricated pure 3D CsPbIBr2 film and the film doped with 2D (PEA)2PbI4 perovskite (X = 0.02) respectively excited using a 532 nm laser beam. The ≈581 nm emission peak with an enhanced intensity of the doped (X = 0.02) perovskite thin film compared to the pristine film confirmed the passivation in the 3D perovskite, as reported previously.30,57 The enhanced intensity defines the retardation in non-radiative recombination due to the reduced localized states just below the conduction band minima (CBM) of the perovskite layers. This suppressed non-radiative recombination in the X = 0.02 was again proved through the TRPL, as shown in Fig. 4b. The fitted graph revealed that the average photocarrier lifetime was enhanced from 14.05 ns to 29.62 ns in the 2D perovskite doped (X = 0.02) one.
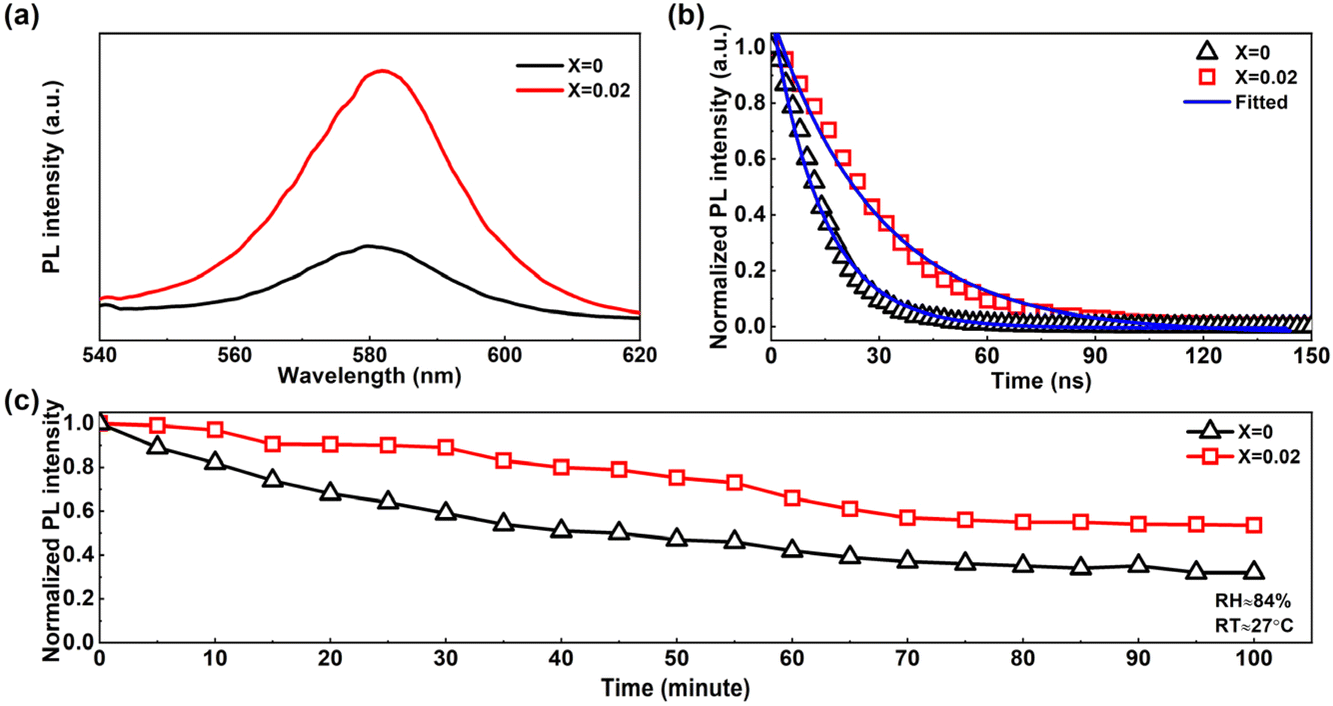 | ||
| Fig. 4 (a) The steady-state PL of the perovskite thin films, (b) time-resolved PL of the perovskite thin films, (c) PL degradation test of the fabricated perovskite thin films in a humid environment (RT ≈ 27 °C, RH ≈ 84%). | ||
We exposed the fabricated perovskite thin films (X = 0, 0.02) for 100 minutes in a highly humid environment with RH ≈ 84%, and RT ≈ 27 °C, and measured the SSPL emission of these thin films under 532 nm excitation. We found a decay in PL intensity over time for each kind of perovskite film. The normalized PL intensity of the films is shown in Fig. 4c, and the SSPL spectra are shown in Fig. S6.† The PL degradation rate was quite fast in the pure CsPbIBr2 perovskite thin film, compared to the doped one. This fast degradation supports poor ambient stability of the pure CsPbIBr2 perovskite, whereas 2D doping improves the ambient stability significantly. From the SSPL study of these continuous ambient exposed perovskite films, it was quite clear that the trap states generated with time in the bulk perovskite were much lower in concentration for CsPbIBr2 perovskite with 2D doping.
Motivated by the improved light energy absorption capability, better crystallinity, and improved average grain size distribution with fewer pinholes, we fabricated all-inorganic PSCs using FTO/c-TiO2/m-TiO2/((PEA)2PbI4)X(CsPbIBr2)1−X/Spiro-OMeTAD/Ag in the n-i-p architecture, where the X values are 0, 0.01, 0.02, 0.04, and 1. A schematic of the proposed device architecture is shown in Fig. 5a. The performance of the device mostly depends on the interfacial band bending, so we evaluated an estimated band bending at the interface using SCAPS 1D simulation, as shown in Fig. 5b. From this simulation study, we found that the conduction band offset (CBO)58 at the ETL/perovskite interface is 0.205 eV and −0.282 eV for X = 0 and X = 0.02 devices, respectively. The spike-like band59 at the ETL/perovskite interface for the X = 0 device, as shown in the inset of Fig. 5b, may restrict the electron flow. With 2D doping (X = 0.02) within the absorber, the electron charge transfer from the absorber to the interface layers was preferable for the perovskite absorber with the optimum amount of 2D doping (X = 0.02) within it due to the improvement in the cliff-like band.60 This optimum amount of 2D incorporation helps in shifting the band positions of the perovskite and makes a perfect band alignment. The quality of the actual device was further examined using a cross-sectional FESEM image, as shown in Fig. 5c, in which the deposited layers are easily visible with the absorber layer thickness of the order of 600 nm.
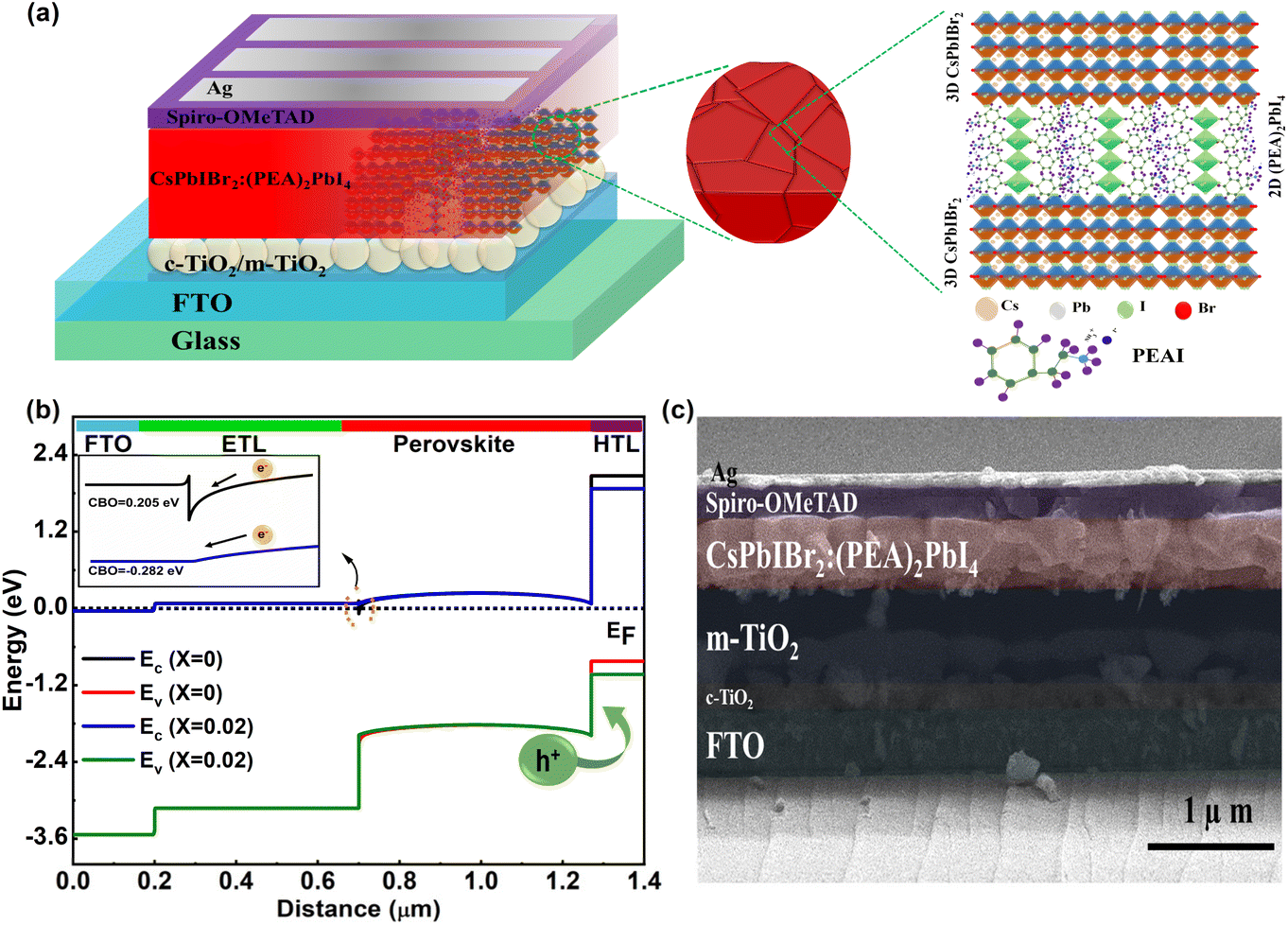 | ||
| Fig. 5 (a) Schematic of the fabricated PSC device architecture and the 3D perovskite bulk passivation mechanism through 2D perovskite doping, (b) energy band-diagram of the PSC using SCAPS 1D numerical simulation and, (c) cross-sectional FESEM image of the champion (X = 0.02) device. | ||
We analyzed the PV performance of the fabricated PSC under an AM 1.5 G, 100 mW cm−2 intense xenon light source under ambient environmental conditions at an average humidity of 60% using a dual voltage sweep in the forward and reverse scanning directions. The light-illuminated J–V plots of the champion devices with X = 0, 0.02 are shown in Fig. 6a, and those for the other devices are shown in Fig. S7.† The optical images of the fabricated devices are shown in the insets of Fig. 6a. From the J–V characteristics under illumination, we found a huge improvement in device performance with X = 0.02 compared to X = 0, as shown in Fig. 6a. The champion device with X = 0.02 achieved a short circuit current density (JSC) of 14.36 mA cm−2, a VOC of 1.12 V, a fill factor (FF) of 63%, and a PCE of 10.13%, while the device with X = 0 showed a JSC of 13.12 mA cm−2, a VOC of 1.10 V, a FF of 56% and a PCE of 8.08% in the reverse scanning direction. The PV performance details of all devices are given in Table 1. The device's PV performance and the J–V curve nature improved significantly with the 2D RP phase layered perovskite doping with a concentration of X = 0.02 but retarded slightly at a doping concentration of 0.04 and further reduced drastically on increasing 2D doping. Moreover, PV parameters of the PSC devices show variations during the dual voltage sweep, and the PV parameters are quite enhanced in the reverse scanning direction compared to the forward scanning direction due to the ion-migration61 effect in the all-inorganic perovskite. We analyzed this ion-migration effect through the hysteresis index (HI)62 defined as,
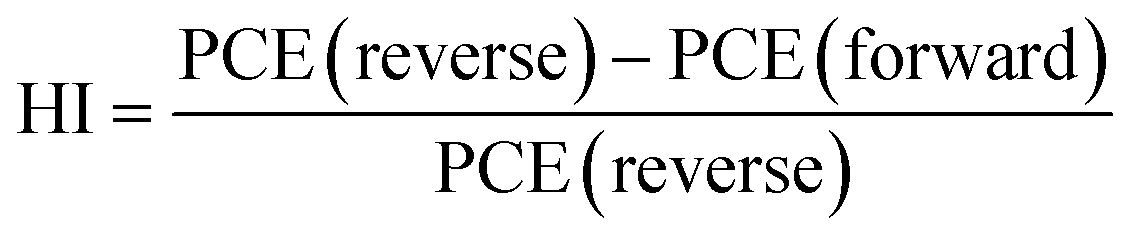 | (3) |
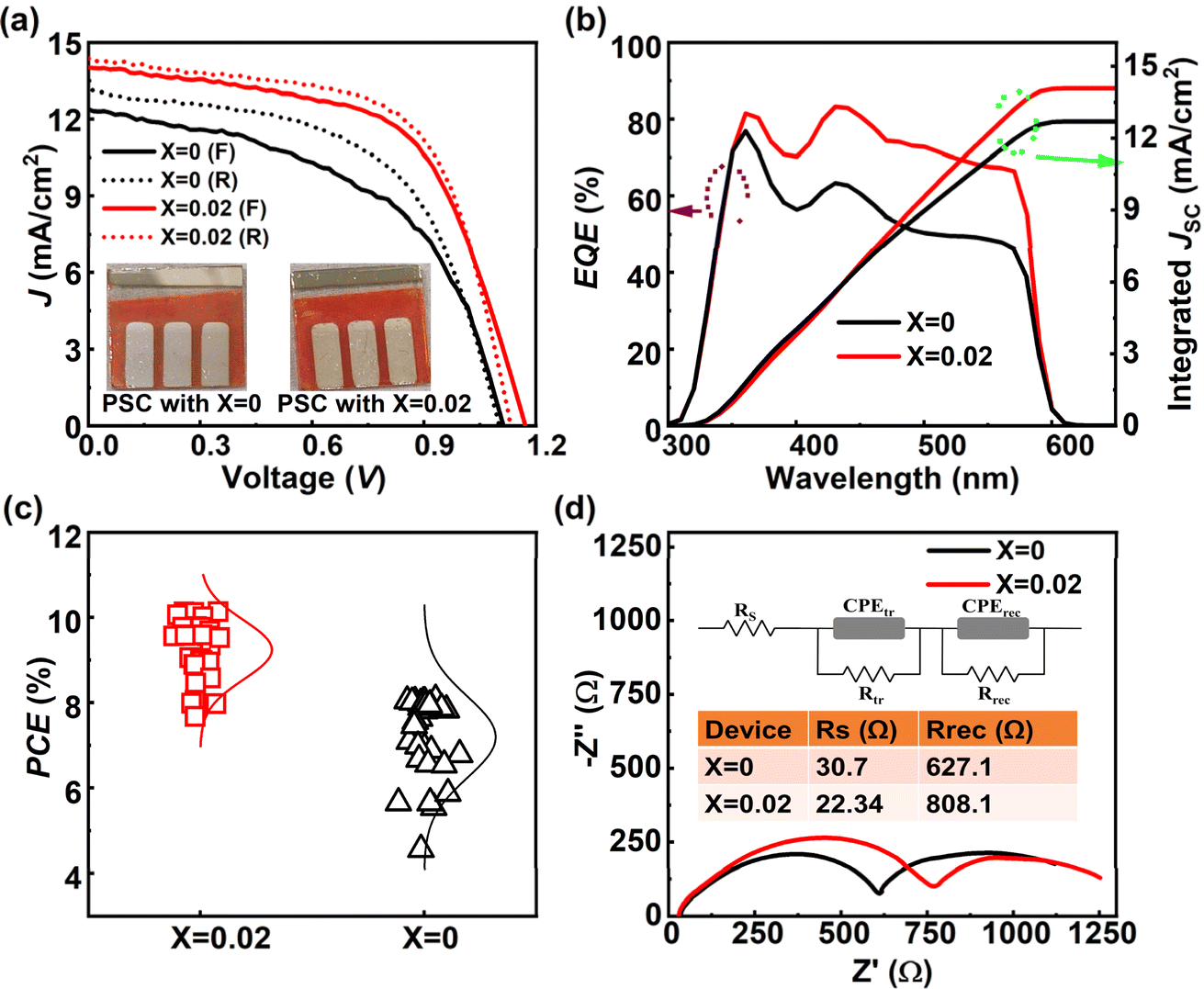 | ||
| Fig. 6 Light-illuminated J–V characteristics of the fabricated pristine and champion device. The real-time pictures of the fabricated champion devices are shown in the inset of the figure, (b) EQE vs. wavelength and integrated JSC characterization, (c) the device repeatability test of the fabricated devices concerning the PCE, and (d) device EIS study under a single wavelength irradiation. | ||
| The device with the scan direction | J SC (mA cm−2) | V OC (V) | FF (%) | PCE (%) |
|---|---|---|---|---|
| W/o-(F) | 12.30 | 1.11 | 0.52 | 7.09 |
| W/o-(R) | 13.12 | 1.10 | 0.56 | 8.08 |
| W(X = 0.01)-(F) | 13.35 | 1.13 | 0.62 | 9.35 |
| W(X = 0.01)-(R) | 14.06 | 1.14 | 0.63 | 10.09 |
| W(X = 0.02)-(F) | 14.01 | 1.16 | 0.59 | 9.58 |
| W(X = 0.02)-(R) | 14.36 | 1.12 | 0.63 | 10.13 |
| W(X = 0.04)-(F) | 13.12 | 1.10 | 0.55 | 7.93 |
| W(X = 0.04)-(R) | 13.72 | 1.10 | 0.59 | 8.90 |
| W(X = 1)-(F) | 4.12 | 0.79 | 0.41 | 1.33 |
| W(X = 1)-(R) | 3.74 | 0.84 | 0.57 | 1.97 |
and found that the HI improved to a value of 5.42% in the champion device (with X = 0.02) compared to the pristine device (X = 0) of 12.25%, as shown in Table S6.† This improvement is quite impressive compared to the recently reported articles related to the CsPbIBr2 3D perovskite.63,64
Furthermore, to compare the photon-to-electron conversion efficiency, we studied the pristine and champion device's (X = 0, 0.02) EQE, and the EQE vs. wavelength plot is shown in Fig. 6b. The overall EQE spectrum was improved with X = 0.02, showing fewer recombination losses; however no significant change in absorption edge was observed in the EQE spectrum. The associated JSC EQE in Fig. 6b shows very little JSC mismatch (ΔJSC)15,25 between the JSC JV, and JSC EQE, (and Table S7†). Furthermore, we checked the device repeatability test by fabricating two batches of devices (X = 0, 0.02), where each batch has 10 devices and 30 pixels. The device repeatability plot concerning PCE is shown in Fig. 6c, and the JSC, FF, and VOC are shown in Fig. S8 and S9.† We have calculated the statistical mean and maximum values with a standard deviation of the PV parameters from these sets of devices, as shown in Table S8.† The best-performing devices corresponding to X = 0.02 yielded the average value of the device parameters, a JSC of 13.53 ± 0.70 mA cm−2, a VOC of 1.12 ± 0.01 V, a FF of 0.60 ± 0.01%, and a PCE of 9.23 ± 0.69%, while control devices exhibited the average value of the parameters, a JSC of 12.40 ± 1.08 mA cm−2, a VOC of 1.04 ± 0.04 V, a FF of 0.55 ± 0.01%, and a PCE of 7.19 ± 0.95%. We also analysed the EIS measurements, shown in Fig. 6d, of the PSC devices (X = 0, 0.02) to study the charge transfer more comprehensively using a simplified equivalent circuit model (shown in the inset of Fig. 6d) by utilizing 10 mW cm−2 intense 445 nm laser beam irradiation. As shown in Fig. 6d, the first semicircle represents the high-frequency region corresponding to the charge transport resistance (Rct), while the second incomplete semicircle in the low-frequency region is mainly due to the charge recombination resistance (Rrec). The fitted values of the equivalent circuit parameters are shown in Table S9.† The device with the 2D perovskite doped CsPbIBr2 film (X = 0.02) had an Rct value of 924.4 Ω, whereas the device with only 3D perovskite (x = 0) had an Rct of 607 Ω. The high value of Rct in the case of 2D perovskite-doped devices is due to the lower conductivity of the 2D perovskite material itself. The values of Rrec of the devices for X = 0.0 and X = 0.02 are 627.1 Ω and 808.1 Ω, respectively. The high value of Rrec for the device with 2D perovskite doping resulted from the defect-free physical contact between the HTL and ETL, and the high-quality, fewer pinholes and uniform coverage of the CsPbIBr2 perovskite film, which led to suppression of carrier recombination.
We further studied the trap state defect density (ηtrap) using the space charge limited current (SCLC) method by fabricating electron-only devices using the FTO/ETL/CsPbIBr2 or ((PEA)2PbI4)0.02(CsPbIBr2)0.98/PCBM/Ag structure. In this method, we have analyzed the dark J–V plots, as shown in Fig. 7a and b. Log–log plots of dark J–V curves can be categorized into three regions: (1) the linear-ohmic region at low bias, (2) the SCLC region at moderate bias, and (3) the trap-filling limit (TFL) region. We have calculated the ηtrap from the trap-filled limit voltage (VTFL) using the following equation:
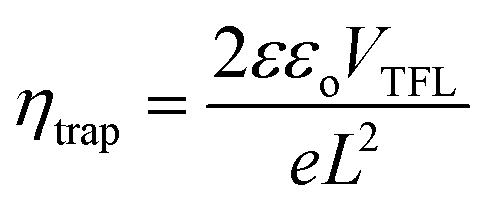 | (4) |
 | ||
| Fig. 7 Space charge-limited current (SCLC) versus voltage characteristic curve of the electron only (a) pristine and (b) best champion device, (c) numerical simulation of SRH recombination rate, and (d) Urbach analyses from the EQE spectra. | ||
We then determined the diode ideality factor (η) from the dark J–V curves to identify the dominant recombination type in both devices and calculated η from the semi-log dark J–V curve (shown in Fig. S10†) using the formula:
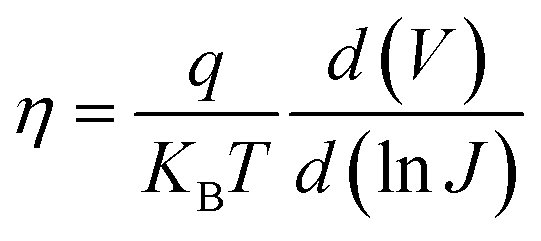 | (5) |
The values of η for the pristine and 2D perovskite doped champion PSCs were found to be 1.85 and 1.65, respectively, indicating a single non-radiative recombination process at the perovskite/transport layer (TL) interface. The lower η value for the 2D perovskite-doped PSC compared to that for the pure 3D perovskite-based PSC suggests a reduction in Shockley-Read-Hall (SRH) recombination at the perovskite/TL interface. Further, we confirmed the SRH recombination suppression after 2D bulk passivation in 3D AIMHP by numerically simulating the PSC device SRH recombination rate analyses through SCAPS 1D, as shown in Fig. 7c. The different parameters used in this numerical simulation are listed in Table S10,† and the absorption coefficient and defect trap densities were added from our experimental results. From Fig. 7c, a decrease in the SRH recombination rate in the case of 2D passivated perovskite compared to pure 3D perovskite from 2.40 × 1018 to 2.02 × 1018 cm−3 s−1 recommends a significantly reduced non-radiative recombination density of 3.8 × 1017 cm−3 s−1. To be more precise, we analyzed the localized defect states that accumulated at the near CBM of the perovskite from the EQE spectra through Urbach tail analyses, as given in Fig. 7d. We have taken the slope by fitting the graph just below the Eg value, which was calculated by plotting the derivative of EQE spectra w.r.t the incident photon energy (shown in Fig. S11†). Additionally, the higher derivative [d(EQE)/d(hν)] for the absorber doped with 2D perovskite compared to that for the pure 3D one showing a sharper transition indicates better band-edge absorption of photons, likely due to reduced defect density. For this Urbach model, a plot of ln(−ln(1 − (EQE))) versus the band gap energy was used, shown in Fig. 7d, to estimate the Urbach tail energy (EU EQE), with a lower value indicating fewer defects. We found the value of EU EQE for the 2D perovskite-doped PSC device to be approximately 15.72 meV and for the pure 3D perovskite-based PSC device to be approximately 21.27 meV, implying a significant reduction in defects within the perovskite absorber doped with 2D (PEA)2PbI4 perovskite.
The fabricated PSCs' stability assessment is a crucial issue to consider. We have evaluated the device stability using the ISOS-D-1 protocol33,65 in an ambient environment with an RH of approximately 65% and RT of around 26 °C by storing the pristine and doped devices in a dark and dry environment for approximately 576 hours (24 days) without any encapsulation. The device's ambient stability test concerning the PCE in Fig. 8 shows better ambient stability of the devices with 2D perovskite compared to that of the pure 3D CsPbIBr2-based PSC. The 2D passivated PSC retained 80% of its initial PCE (T80) after 500 hours of ambient storage, whereas the pure 3D CsPbIBr2-based PSC (pristine) degraded very fast, and retained approximately 20% of its initial PCE after 400 hours of ambient storage. We have also examined the operational stability of the devices by maintaining them at 85 °C under ambient conditions for over 100 hours. As shown in Fig. 8b, the doped devices maintained their PCE near 80%, but the PCE rapidly decreased for the pristine device, clearly showing that the ambient stability is improved by 2D perovskite doping.
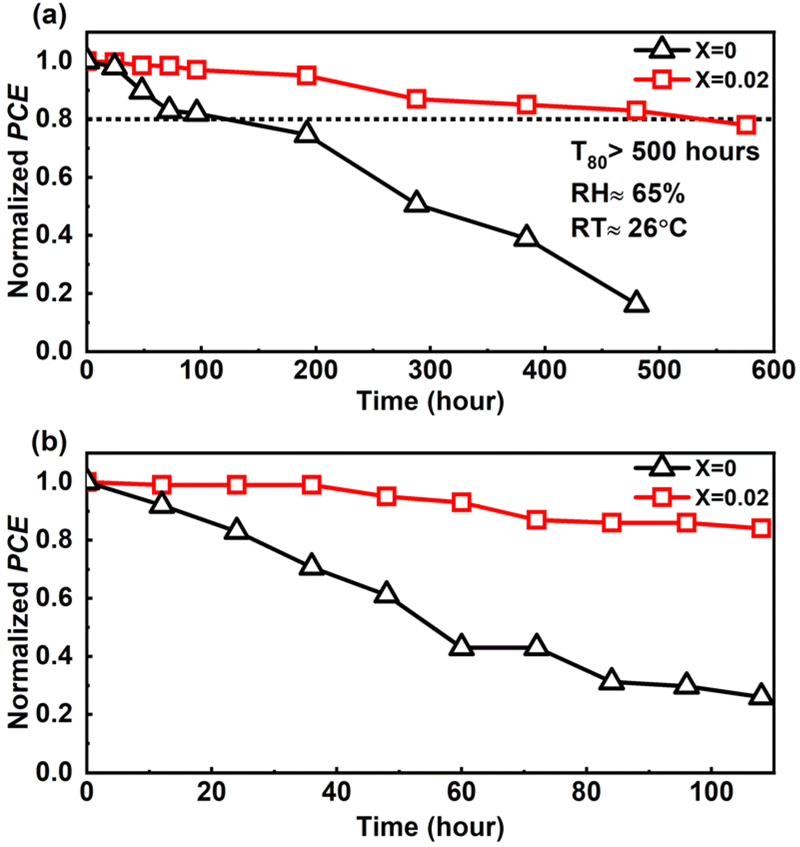 | ||
| Fig. 8 (a) Ambient stability test of the fabricated champion devices in terms of normalized PCE, and (b) operational ambient stability of the PSC devices below 85 °C corresponding to the normalized PCE. | ||
It is to be noted that the faster degradation of the pristine device compared to the doped one is consistent with the perovskite films’ stability characterized by the XRD measurements of the fabricated perovskite thin films with (X = 0.02) and without the 2D perovskite (X = 0) additive at different time intervals, as shown in Fig. S12a.† The intensity ratio between the diffraction peaks of the perovskite phase for the (100) and (200) planes decreased over time from day 1 to day 7 for the samples without the 2D perovskite additive. However, for the 2D perovskite passivated films, there was only a slight change in the intensity ratio between these diffraction peaks on the 7th day. Additionally, we captured digital photographs of the deposited perovskite thin films with and without 2D perovskite passivation (Fig. S12b and c†), which confirmed our findings. The 3D CsPbIBr2 perovskite film with doped 2D (PEA)2PbI4 perovskite retained its reddish colour even on the 7th day, whereas the film without 2D passivation started degrading. These observations clearly show that 2D (PEA)2PbI4-passivated 3D CsPbIBr2 perovskite thin films have better crystallinity, making them more stable in an ambient environment.
4 Conclusions
In this work, we successfully fabricated stable and efficient all-inorganic PSCs via 2D perovskite doping within 3D CsPbIBr2 perovskite. 2D incorporation significantly improved the phase stability and grain sizes of (PEA)2PbI4)X(CsPbIBr2)1−X, with the best crystallite size of 53.99 nm achieved with X = 0.02. The average grain size of the 3D (PEA)2PbI4)X(CsPbIBr2)1−X thin film was 350.43 ± 0.09 nm with a maximum grain size of 709 nm for X = 0, which increased to an average grain size of 853.38 ± 0.18 nm and a maximum grain size of 1.367 μm at X = 0.02. This increase in grain size reduces the defect-related charge trap density, which is responsible for non-radiative recombination leading to an improved photocarrier lifetime from 14.05 to 29.62 ns in the 2D passivated perovskite layers with a suppression in the SRH recombination rate corresponding to non-radiative recombination loss from 2.40 × 1018 to 2.02 × 1018 cm−3 s−1 which was confirmed through SSPL and SCAPS 1D numerical device simulations. This significant suppression in non-radiative recombination centres improved the band offset through the band energy alignment of the fabricated all-inorganic CsPbIBr2 PSCs, and we achieved a 25.37% enhancement in device PCE from 8.08% to 10.13%. With this excellent device performance, the 2D doped all-inorganic CsPbIBr2 PSCs showed an improved long-term ambient (RH ≈ 60 °C, RT ≈ 26 °C) device stability up to 500 hours and with 100 hours of operational stability at 85 °C (retained ≈ 84% of initial performance) without any encapsulation.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Tapas Das: conceptualization (supporting); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); writing – original draft (lead). Faisal Farooq: data curation (supporting); formal analysis (supporting); investigation (supporting); methodology (supporting); writing – original draft (supporting). Parul Garg: conceptualization (supporting); data curation (supporting); formal analysis (supporting); investigation (supporting); methodology (supporting); writing – original draft (supporting). Sakal Singla: data curation (supporting); formal analysis (supporting); investigation (supporting); methodology (supporting); writing – original draft (supporting). Asim Guchhait: conceptualization (lead); funding acquisition (lead); project administration (lead); resources (lead); supervision (lead); validation (lead); visualization (lead); writing – review & editing (lead). Ashok Bera: conceptualization (lead); funding acquisition (lead); project administration (lead); resources (lead); supervision (lead); validation (lead); visualization (lead); writing – review & editing (lead).Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
T.D. acknowledges the SVMCM scholarship (Grant No. WBP221666168821 of 2022) funded by the Government of West Bengal for research support. AG sincerely acknowledges the DST-FIST program, Govt. of India (SR/FST/College-2017/53 (C)), SERB, Govt. of India for the Start-up Research grant (SRG/2019/000318) and UGC-DAE CSR (CRS/2021-22/04/603) for supporting this work.References
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS.
- T. Das, R. Nag, N. K. Rana, M. Nayak, R. Paramanik, A. Bera, S. K. Saha and A. Guchhait, Energy Fuels, 2023, 37, 10642–10651 CrossRef CAS.
- R. He, S. Ren, C. Chen, Z. Yi, Y. Luo, H. Lai, W. Wang, G. Zeng, X. Hao, Y. Wang, J. Zhang, C. Wang, L. Wu, F. Fu and D. Zhao, Energy Environ. Sci., 2021, 14, 5723–5759 RSC.
- S. Yang, Y. Liu, X. Lian and J. Chu, Joule, 2022, 6, 2248–2250 CrossRef.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- M. Vasilopoulou, A. Fakharuddin, A. G. Coutsolelos, P. Falaras, P. Argitis, A. R. B. M. Yusoff and M. K. Nazeeruddin, Chem. Soc. Rev., 2020, 49, 4496–4526 RSC.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Science, 2022, 375, 434–437 CrossRef CAS PubMed.
- M. Kim, J. Jeong, H. Lu, T. K. Lee, F. T. Eickemeyer, Y. Liu, I. W. Choi, S. J. Choi, Y. Jo, H. B. Kim, S. I. Mo, Y. K. Kim, H. Lee, N. G. An, S. Cho, W. R. Tress, S. M. Zakeeruddin, A. Hagfeldt, J. Y. Kim, M. Grätzel and D. S. Kim, Science, 2022, 375, 302–306 CrossRef CAS.
- M. Jost, L. Kegelmann, L. Korte and S. Albrecht, Adv. Energy Mater., 2020, 10, 1904102 CrossRef CAS.
- J. Xu, C. C. Boyd, Z. J. Yu, A. F. Palmstrom, D. J. Witter, B. W. Larson, R. M. France, J. Werner, S. P. Harvey, E. J. Wolf, W. Weigand, S. Manzoor, M. F. A. M. Van Hest, J. J. Berry, J. M. Luther, Z. C. Holman and M. D. McGehee, Science, 2020, 367, 1097–1104 CrossRef CAS PubMed.
- M. I. Hossain, A. Mohammad, W. Qarony, S. Ilhom, D. R. Shukla, D. Knipp, N. Biyikli and Y. H. Tsang, RSC Adv., 2020, 10, 14856–14866 RSC.
- T. Das, N. kumar Rana, P. Garg, A. Bera and A. Guchhait, J. Phys. Chem. C, 2023, 127, 21504–21513 CrossRef CAS.
- F. He, W. Xu, M. Zhang, X. Zhang, B. Ding, G. Wei and F. Kang, RSC Adv., 2019, 9, 30534–30540 RSC.
- Z. Zhang, F. He, W. Zhu, D. Chen, W. Chai, D. Chen, H. Xi, J. Zhang, C. Zhang and Y. Hao, Sustainable Energy Fuels, 2020, 4, 4506–4515 RSC.
- J. Tong, Q. Jiang, F. Zhang, S. B. Kang, D. H. Kim and K. Zhu, ACS Energy Lett., 2021, 6, 232–248 CrossRef CAS PubMed.
- G. Wang, J. Bi, M. Lei, J. Chang and F. Meng, Energy Fuels, 2022, 36, 7755–7762 CrossRef CAS.
- Z. Zhu, J. Feng, X. Han, C. Bao, T. Yu, Z. Li and Z. Zou, Energy Fuels, 2022, 36, 15145–15153 CrossRef CAS.
- Q. Wang, Y. Xu, L. Zhang, A. Yang, T. Bai, F. Liu, M. Lyu and J. Zhu, ACS Appl. Energy Mater., 2022, 5, 3110–3118 CrossRef CAS.
- L. Gong, J. S. Zhang, C. Wang, H. Fu, Y. F. Lu, J. L. Chen, J. C. Fan and Z. S. Chao, ACS Appl. Energy Mater., 2021, 4, 4686–4694 CrossRef CAS.
- J. Chen and N. G. Park, Causes and Solutions of Recombination in Perovskite Solar Cells, Adv. Mater., 2019, 31, 1–56 Search PubMed.
- E. Aydin, M. De Bastiani and S. De Wolf, Defect and Contact Passivation for Perovskite Solar Cells, Adv. Mater., 2019, 31, 1900428 CrossRef.
- Y. Liu, S. Akin, L. Pan, R. Uchida, N. Arora, J. V. Milić, A. Hinderhofer, F. Schreiber, A. R. Uhl, S. M. Zakeeruddin, A. Hagfeldt, M. Ibrahim Dar and M. Grätzel, Sci. Adv., 2019, 5, 1–8 Search PubMed.
- X. Zhao, T. Liu and Y. L. Loo, Adv. Mater., 2022, 34, 1–19 Search PubMed.
- G. Wu, R. Liang, Z. Zhang, M. Ge, G. Xing and G. Sun, Small, 2021, 17, 2103514 CrossRef CAS.
- Y. Zheng, T. Niu, X. Ran, J. Qiu, B. Li, Y. Xia, Y. Chen and W. Huang, J. Mater. Chem. A, 2019, 7, 13860–13872 RSC.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- Y. Li, M. Dailey, P. J. Lohr and A. D. P. Printz, J. Mater. Chem. A, 2021, 9, 16281–16338 RSC.
- N. K. Tailor, Yukta, R. Ranjan, S. Ranjan, T. Sharma, A. Singh, A. Garg, K. S. Nalwa, R. K. Gupta and S. Satapathi, J. Mater. Chem. A, 2021, 9, 21551–21575 RSC.
- T. Liu, J. Zhang, M. Qin, X. Wu, F. Li, X. Lu, Z. Zhu and A. K. Y. Jen, Adv. Funct. Mater., 2021, 31, 1–9 Search PubMed.
- S. Tan, B. Yu, Y. Cui, F. Meng, C. Huang, Y. Li, Z. Chen, H. Wu, J. Shi, Y. Luo, D. Li and Q. Meng, Angew. Chem., Int. Ed., 2022, 61, e202201300 CrossRef CAS.
- Y. Wang, T. Zhang, M. Kan, Y. Li, T. Wang and Y. Zhao, Joule, 2018, 2, 2065–2075 CrossRef CAS.
- X. Liu, X. Wang, T. Zhang, Y. Miao, Z. Qin, Y. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 12351–12355 CrossRef CAS PubMed.
- A. Krishna, S. Gottis, M. K. Nazeeruddin and F. Sauvage, Adv. Funct. Mater., 2019, 29, 1–20 CrossRef.
- S. B. Saadatmand, S. Shokouhi, V. Ahmadi and S. M. Hamidi, ACS Omega, 2024, 9, 24925–24932 CrossRef CAS PubMed.
- M. I. Hossain, M. Shahiduzzaman, J. H. Rafij, A. Tamang, M. Akhtaruzzaman, A. Hamad, J. Uddin, N. Amin, J. M. Nunzi and T. Taima, Mater. Horiz., 2024, 11, 4329–4337 RSC.
- D. Li, D. Li, A. Yang, H. Zhang, X. Lai and C. Liang, ACS Omega, 2021, 6, 20877–20886 CrossRef CAS.
- H. Zhai, F. Liao, Z. Song, B. Ou, D. Li, D. Xie, H. Sun, L. Xu, C. Cui and Y. Zhao, ACS Appl. Energy Mater., 2021, 4, 13482–13491 CrossRef CAS.
- J. Kistner-Morris, A. Shi, E. Liu, T. Arp, F. Farahmand, T. Taniguchi, K. Watanable, V. Aji, C. H. Lui and N. Gabor, Nat. Commun., 2024, 15, 4075 CrossRef CAS.
- J. Lu, S. C. Chen and Q. Zheng, ACS Appl. Energy Mater., 2018, 1, 5872–5878 CrossRef CAS.
- J. Lin, M. Lai, L. Dou, C. S. Kley, H. Chen, F. Peng, J. Sun, D. Lu, S. A. Hawks, C. Xie, F. Cui, A. P. Alivisatos, D. T. Limmer and P. Yang, Nat. Mater., 2018, 17, 261–267 CrossRef CAS PubMed.
- J. Wang, J. Zhu, Y. Jiang, M. Li, K. Yu and G. P. Wang, Nanophotonics, 2021, 10, 4009–4017 CrossRef CAS.
- K. H. Wang, L. Wu, L. Li, H. B. Yao, H. S. Qian and S. H. Yu, Angew. Chem., Int. Ed., 2016, 55, 8328–8332 CrossRef CAS.
- C. Wang, Y. Wang, X. Su, V. G. Hadjiev, S. Dai, Z. Qin, H. A. Calderon Benavides, Y. Ni, Q. Li, J. Jian, M. K. Alam, H. Wang, F. C. Robles Hernandez, Y. Yao, S. Chen, Q. Yu, G. Feng, Z. Wang and J. Bao, Adv. Mater., 2019, 31, 1–10 Search PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- W. Li, M. U. Rothmann, A. Liu, Z. Wang, Y. Zhang, A. R. Pascoe, J. Lu, L. Jiang, Y. Chen, F. Huang, Y. Peng, Q. Bao, J. Etheridge, U. Bach and Y. B. Cheng, Adv. Energy Mater., 2017, 7, 1–8 Search PubMed.
- C. Liu, W. Li, J. Chen, J. Fan, Y. Mai and R. E. I. Schropp, Nano Energy, 2017, 41, 75–83 CrossRef CAS.
- Ł. Haryński, A. Olejnik, K. Grochowska and K. Siuzdak, Opt. Mater., 2022, 127, 112205 CrossRef.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, 2D J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS.
- F. Urbach, Phys. Rev., 1953, 92, 1324 CrossRef CAS.
- A. Sadhanala, F. Deschler, T. H. Thomas, E. Dutton, K. C. Goedel, F. C. Hanusch, M. L. Lai, U. Steiner, T. Bein, P. Docampo, D. Cahen and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1–5 CrossRef.
- J. Jean, T. S. Mahony, D. Bozyigit, M. Sponseller, J. Holovský, M. G. Bawendi and V. Bulović, ACS Energy Lett., 2017, 2, 2616–2624 CrossRef CAS.
- C. Zhang, S. Mahadevan, J. Yuan, J. K. W. Ho, Y. Gao, W. Liu, H. Zhong, H. Yan, Y. Zou, S. W. Tsang and S. K. So, ACS Energy Lett., 2022, 7, 1971–1979 CrossRef CAS.
- M. Ledinsky, A. Vlk, T. Schonfeldova, J. Holovsky, E. Aydin, H. X. Dang, Z. Hajkova, L. Landova, J. Valenta, A. Fejfar and S. de Wolf, J. Phys. Chem. C, 2020, 124, 27333–27339 CrossRef CAS.
- E. Ugur, M. Ledinský, T. G. Allen, J. Holovský, A. Vlk and S. De Wolf, Life on the Urbach Edge, J. Phys. Chem. Lett., 2022, 13, 7702–7711 CrossRef CAS PubMed.
- Y. Liu, J. P. Banon, K. Frohna, Y. H. Chiang, G. Tumen-Ulzii, S. D. Stranks, M. Filoche and R. H. Friend, ACS Energy Lett., 2023, 8, 250–258 CrossRef CAS PubMed.
- J. Lu, S. C. Chen and Q. Zheng, ACS Appl. Energy Mater., 2018, 1, 5872–5878 CrossRef CAS.
- D. N. Q. Agha and Q. T. Algwari, Results Opt., 2022, 9, 100291 CrossRef.
- A. Tara, V. Bharti, S. Sharma and R. Gupta, Mater. Today Commun., 2022, 33, 104546 CrossRef CAS.
- N. Singh, A. Agarwal and M. Agarwal, Opt. Mater., 2022, 125, 112112 CrossRef CAS.
- Y. Zhao, W. Zhou, Z. Han, D. Yu and Q. Zhao, Phys. Chem. Chem. Phys., 2021, 23, 94–106 RSC.
- S. N. Habisreutinger, N. K. Noel and H. J. Snaith, ACS Energy Lett., 2018, 3, 2472–2476 CrossRef CAS.
- J. He, Q. Wang, Y. Xu, X. Guo, L. Zhou, J. Su, Z. Lin, J. Zhang, Y. Hao and J. Chang, Small, 2023, 19, 1–11 Search PubMed.
- H. Wang, S. Cao, B. Yang, H. Li, M. Wang, X. Hu, K. Sun and Z. Zang, Sol. RRL, 2020, 4, 1–8 Search PubMed.
- M. O. Reese, S. A. Gevorgyan, M. Jørgensen, E. Bundgaard, S. R. Kurtz, D. S. Ginley, D. C. Olson, M. T. Lloyd, P. Morvillo, E. A. Katz, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. Zhang, M. Andersson, K. Tvingstedt, M. Lira-Cantu, D. Laird, C. McGuiness, S. Gowrisanker, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. Delongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J. B. Bonekamp, A. J. J. M. Van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2011, 95, 1253–1267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01437e |
| This journal is © The Royal Society of Chemistry 2025 |