
Impact of rheology on formation of oil-in-liquid metal emulsions†
Shreyas
Kanetkar
a,
Sai P.
Peri
 a,
Husain
Mithaiwala
a,
Febby
Krisnadi
b,
Michael D.
Dickey
b,
Matthew D.
Green
a,
Robert Y.
Wang
*a and
Konrad
Rykaczewski
*a
a,
Husain
Mithaiwala
a,
Febby
Krisnadi
b,
Michael D.
Dickey
b,
Matthew D.
Green
a,
Robert Y.
Wang
*a and
Konrad
Rykaczewski
*a
aSchool for Engineering of Matter, Transport and Energy, Arizona State University, Tempe, AZ 85287, USA. E-mail: rywang@asu.edu; konradr@asu.edu
bDepartment of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, North Carolina 27695, USA
First published on 29th November 2024
Abstract
To quantify how the viscosities of silicone oil (SO) and liquid metal (LM) relate to emulsion-formation (LM-in-SO versus SO-in-LM), a process was developed to produce LM pastes with adjustable viscosity and minimal oxide and bubbles. Increased LM viscosity allows greater silicone oil intake and/or intake of higher-viscosity silicone oils.
Gallium-based liquid metals (LM), known for a variety of established and emerging applications,1–5 typically break up into micro-droplets when mixed with other liquids. For example, mixing LM with silicone oils (SO) leads to the formation of LM-in-SO emulsions.6–11 A notable exception to this behavior has been reported over the last few years, in that LM made with pure Ga, GaIn, or GaInSn can incorporate up to 40% by volume (vol%) of SO with viscosity ranging from 0.01–1 Pa s (10–1000 cSt), forming SO-in-LM emulsions.6,12,13 Phase inversion occurring during addition of large volume fractions of pure GaInSn into uncured polydimethylsiloxane (PDMS) leading to PDMS-in-LM emulsion formation has also been reported.14 Our previous work demonstrated that the formation of SO-in-LM emulsions requires the presence of oxygen, which enables the growth of 1–3 nm, chemically asymmetric,15 gallium oxide around the oil micro-droplets, indicating that the emulsions form by internalizing new SO capsules rather than replacing air in existing foam pockets.12,13 The oxide shells also prevent direct SO pocket to pocket contact and coalescence, and thereby resist spontaneous phase separation and create SO-in-LM emulsions that remain highly stable over time.12 Even with the maximum volume fraction of the oil, the SO-in-LM emulsions exhibit a high thermal conductivity (∼10 W m−1 K−1).6,13 In addition, at this composition, the presence of a 0.1 to 1 μm thin exterior oil film12 on the SO-in-LM emulsions prevents gallium-induced degradation of contacting metals.6,13 These two characteristics make SO-in-LM emulsions uniquely suited for next-generation thermal interface materials. However, the complex nature of LM foams has posed challenges in investigating the impact of viscosities on the mixing outcome of two liquids (i.e., formation of LM-in-SO versus SO-in-LM emulsions) and the maximum achievable SO volume fraction, which are factors known to be significant in emulsification and phase inversion of other liquid–liquid systems.16–18
The fabrication processes used in creating LM foams result in a complex multiphase composition, structure, and rheology, making it difficult to quantify the impact of the continuous metallic phase viscosity on the outcome of mixing the two liquids (SO with various viscosities are readily available). LM foams are produced by stirring the liquid, either alone or with solid particles, in air.19–21 Other foaming processes exist,22–26 but result in even more complex compositions and have not been employed in SO-in-LM emulsion studies. Stirring of the pure liquid metal in air internalizes oxide flakes formed at the air–liquid interface. This increases viscosity and is correlated with the onset of air bubble capture.19 The addition of solid particles can alter foaming, with an increase in particle content and a decrease in their size, enhancing air bubble capture.21 In both foaming processes, oxide formation around the air bubbles is required, which also leads to the internalization of numerous crumpled oxide flakes that may contain nanoscale air bubbles.19,21 Although the density of the oxide flakes is nearly identical to that of pure LM, we observed that the entrapment of air bubbles of various sizes results in buoyancy-induced segregation of the LM foams, creating a buoyant, air-rich phase at the top and a denser LM-rich phase at the bottom.19,21 If the sample is not foamed completely, viscosity measurements reflect an effective value for the two layers, which after the initial drop associated with bubble internalization increases as the stirring process continues.19 However, our measurements of the isolated top layer show that its viscosity remains nearly constant at about 1000 to 2000 Pa s (at 1 s−1 shear rate, see ESI†), regardless of the stirring duration. Therefore, foams produced with different stirring times are not suitable for studying the impact of LM viscosity on emulsion type. While the viscosity of the LM can also be adjusted by adding particles,27,28 we have demonstrated that incorporating most particles into LM requires oxide formation,29 which leads to air bubble entrapment (and, therefore, possible layer segregation).21,30 Accordingly, an alternative method for increasing LM viscosity without air entrapment is needed.
Here, we introduce a simple method to create homogenous Ag-in-Ga pastes that are nearly air bubble- and oxide-free and use them to study the impact of the viscosities of the two liquids on the outcome of their mixing (i.e., the formation of LM-in-SO vs. SO-in-LM emulsions) and maximum SO volume fraction achievable prior to the continuous LM phase inversion. We selected to add Ag (silver powder, APS 4–7, 99.9%, Thermo Scientific Chemicals, see electron micrographs in the ESI†) into Ga (Rotometals) because the two materials alloy rapidly into Ag–Ga nanoneedles.10,11,31–33 The schematic in Fig. 1a shows that to minimize LM oxide formation and air entrapment, we mixed the two materials in a nitrogen environment. The electron micrographs in Fig. 1b show that the Ag-in-Ga mixture is mostly air bubble-free (some air is inherently present in Ga samples due to the handling history of the Ga) and has a homogenous distribution of Ag–Ga nanoneedles throughout the volume of the mixture (i.e., the particles do not settle). Dependent on the viscosity of the silicone oil and particle volume content, mixing of 10 to 40 vol% of SO (Sigma-Aldrich 0.01, 0.1, 0.5, and 1 Pa s (equivalent to 10, 100, 500, 1000 cSt)) into the Ag-in-Ga results in either inversion of the continuous phase and the breakup of the LM mixture into LM-in-SO emulsion (see Fig. 1c and d) or internalization of the SO into LM and formation of SO-in-LM emulsion (see Fig. 1e–h).
 | ||
| Fig. 1 (a) Fabrication schematic and (b) cross-sectional electron micrographs of the Ag-in-Ga mixture highlighting uniform distribution of the Ag–Ga nanoneedles, including in the top and bottom regions; (c) fabrication schematic of and (d) image of phase inversion when the Ag-in-LM breaks up into droplets when mixed with SO, forming LM-in-SO emulsions; (e)–(h): (e) fabrication schematic, (f) images of early stage mixing process (0–1.5 min) as well as final emulsion after 15 min of mixing, (g) overview and (h) close-up cross-sectional electron micrographs of the SO-in-LM emulsions showing that the microscopic pockets of the SO with irregular and near-spherical shapes are distributed across the entire emulsion volume. | ||
The cross-sectional environmental scanning electron microscopy (see the ESI† for imaging details) of the SO-in-LM emulsions demonstrates that microscopic pockets of the silicone oil are distributed across the entire emulsion volume. The SO features have irregular or spherical shapes with dominant dimensions ranging from around 20 to 300 μm (see Fig. 1g and h). The SO pockets with distorted shape are a result of the SO and Ag-in-Ga mixing dynamics and the formation of the oxide shell. In particular, as highlighted in Fig. 1e and f, slugs of SO form during the mixing process in between temporarily separated parts of the metallic phase. The metallic phase keeps on separating and combining for the first few minutes of the mixing process. This process absorbs slugs of SO, whose shape is preserved by formation of the oxide shell. The SO pockets are then further broken up and distorted by continued mixing, resulting in many highly irregularly shaped and sized SO features along with classical spherical SO droplets (see Fig. 1h). A new oxide shell can grow on these further distorted features, preserving their shape.12 Besides allowing for presence of non-spherical shapes, the oxide shell also promotes emulsion stability by preventing direct liquid contact and coalescence of the droplets. Therefore, the SO and Ag-in-Ga liquid–liquid system allows exploration of the emulsion formation dependance on liquid rheology. To proceed, we first measure how the viscosity of Ag-in-Ga changes with particle content.
We fabricated Ag-in-Ga mixtures with 0.25 vol%, 0.5 vol%, 0.75 vol%, 1 vol%, and 1.5 vol% of Ag by mixing the particles with melted Ga for 3 min, using a mortar and pestle in a glove bag continuously purged with house nitrogen. The native oxide was scraped off the molten Ga using a cotton swab before adding the Ag particles. While the Ag and Ga alloying process is very rapid, we kept the melted samples in nitrogen for 2 h to ensure complete conversion of Ag to Ag–Ga alloy. Subsequently, we measured viscosities while keeping the samples at 45 °C to ensure that Ga remains in liquid form (see ESI† for details). Fig. 2a shows that the viscosity of the Ag-in-Ga mixtures decreases with increasing shear rate (see all three repetitions per composition in ESI†), but increases with the volume fraction of Ag particles. To quantify the latter, we selected to compare the viscosity values at a shear rate of 1 s−1, which is approximately comparable to the manual stirring rate during mixing with the SO. Fig. 2b shows that the mean viscosity of pure Ga is 98 Pa s, and that it increases with silver content to average values of 209–7200 Pa s for Ag of 0.25–1.5 vol%, respectively. We note that in contrast to the Ag-in-Ga mixtures, the viscosities of the SO did not change in the measured shear rate range and were close to the nominal values (see measurements in the ESI†). Next, we explore how the viscosities and volume fractions of the two liquids relate to the outcome of their mixing (LM-in-SO versus SO-in-LM emulsions).
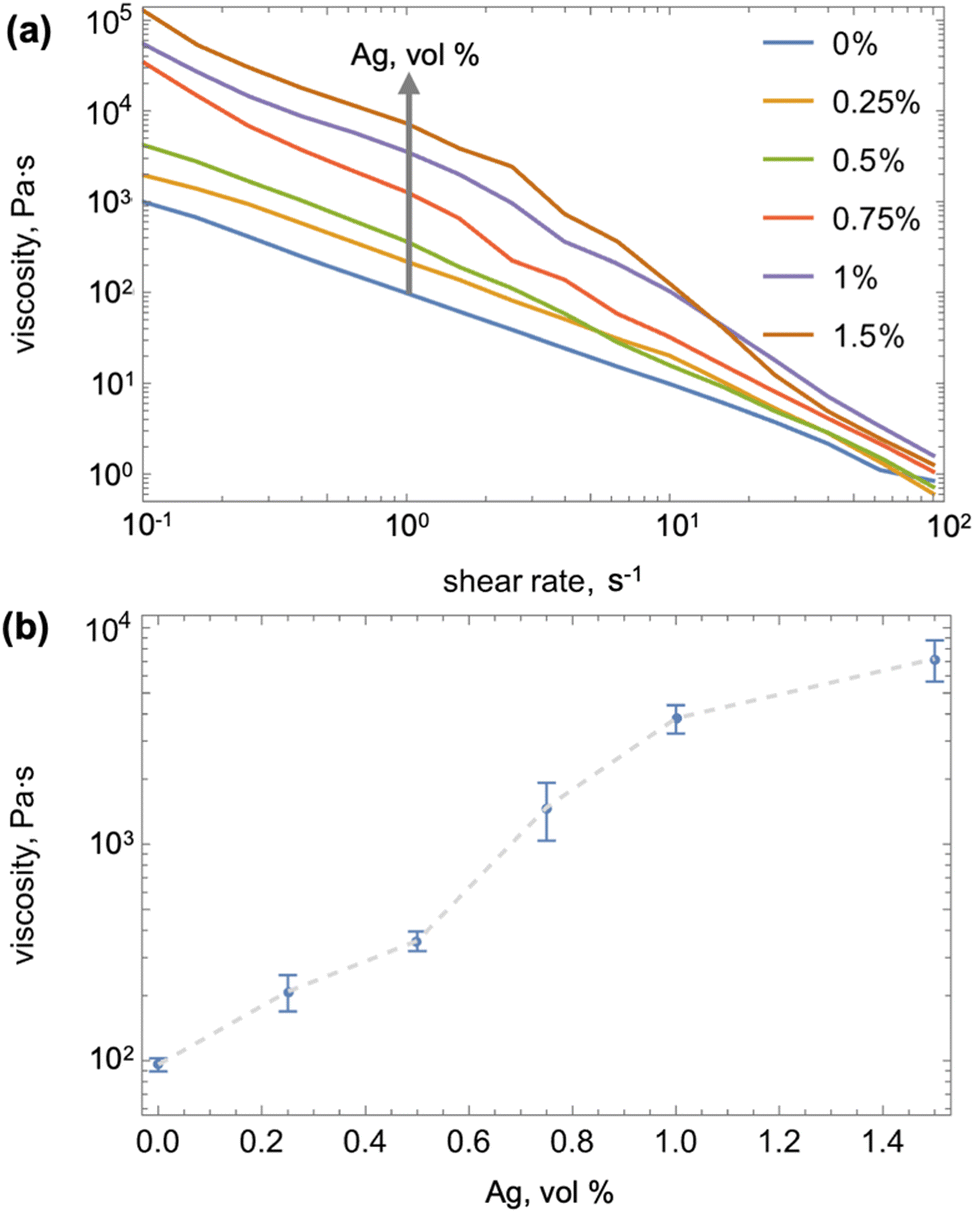 | ||
| Fig. 2 (a) Viscosity of Ag-in-Ga measured for varying volume percentages of the Ag particles and varied shear rates (example curves are shown, all three repetitions per composition are shown in the ESI†) and (b) the viscosity of Ag-in-Ga mixture at 1 s−1 (indicated with gray arrow in (a)) as a function of volume percentage of Ag particles (error bars correspond to one standard deviation). | ||
We visually quantified the outcome of mixing (i.e., SO-in-LM emulsion or phase inversion into LM-in-SO) SO with four viscosities (0.01, 0.1, 0.5, and 1 Pa s) into Ag-in-Ga with the six viscosities (98 ± 7, 209 ± 40, 358 ± 37, 1480 ± 442, 3820 ± 570, and 7200 ± 1555 Pa s (± one standard deviation)), resulting in 24 viscosity combinations. The formation of LM-in-SO emulsion versus SO-in-LM emulsion can be readily visually determined. An LM-in-SO emulsion will rapidly switch from a single reflective LM entity in SO into discontinuous LM droplets within SO, where the LM droplet size depends on mixing time and oil viscosity (Fig. 1d).6,12,13 Conversely, a SO-in-LM emulsion will visually retain its structure as a single entity at the end of the mixing period (throughout the mixing period it might break into 2–3 components that recombine while absorbing slugs of SO, see Fig. 1f). If we observe that the majority of the input SO is no longer visible within the mixing container (see image of the final emulsion after 15 min of mixing in Fig. 1f), our prior work has demonstrated that the formation of SO-in-LM emulsion is highly likely.6,12,13 We ensure that the silicone oil is not concentrated within large (>0.5 mm) voids within the LM by freezing, cross-sectioning, and inspecting the samples under optical microscope.
For each combination of the SO and Ag-in-LM viscosities, we conducted mixing experiments starting with 10 vol% of SO and proceeded in steps of 10 vol% until inversion of the phases was observed (Fig. 3a). Each experiment was repeated three times, and if some discrepancy was observed (e.g., one inversion at 20 vol% and two at 10 vol%), we conservatively report the lower inversion threshold. As commonly observed,6–11 mixing in just 10 vol% of any of the SO into pure gallium resulted in phase inversion. When the viscosity of the continuous phase increased from about 98 Pa s to 209 Pa s, we were only able to form SO-in-LM emulsion with 10 vol% of the lowest viscosity SO (0.01 Pa s). For this SO, the maximum volume fraction allowable in the LM prior to phase inversion increased gradually to 20 vol%, 30 vol%, and 40 vol% with increasing Ag-in-Ga viscosity to about 1480, 3820, and 7200 Pa s, respectively (see Fig. 3a). We observed the same trend of SO capacity increasing with the viscosity of continuous phase (Ag-in-Ga) for the higher viscosity oils, but with shifted thresholds. In particular, to intake 10 vol% of the SO with 0.1 Pa s viscosity, the Ag-in-Ga had to have at least 1480 Pa s viscosity.
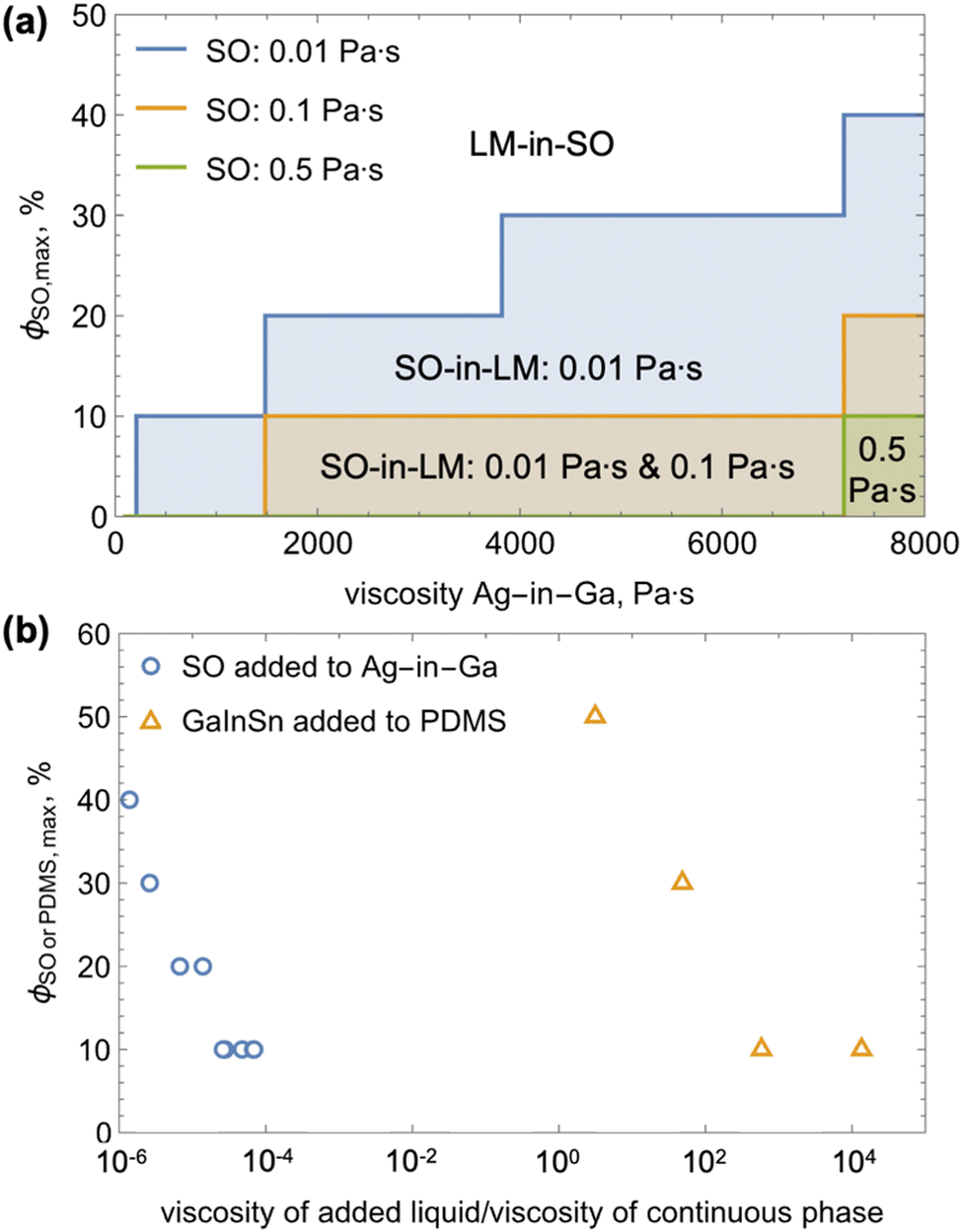 | ||
Fig. 3 (a) Plot of the maximum volume percentage of silicone oil (SO), ϕSO,max, of varied viscosities (see legend) that can be added to form SO-in-LM emulsion to Ag-in-Ga of varied viscosity. The colored regions indicate SO-in-LM emulsion, whereas the white region indicates LM-in-SO phase inversion; (b) the results from (a) along with those of Koh et al.14 who added GaInSn to uncured PDMS in terms of the maximum volume percentage of silicone oil (SO) or PDMS, ϕSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) orPDMS,max, versus the viscosity ratio of the added liquid to continuous phase. orPDMS,max, versus the viscosity ratio of the added liquid to continuous phase. | ||
The threshold continuous phase viscosity for the onset of SO intake increased about proportionately with the SO viscosity. The threshold Ag-in-Ga viscosity increased from about 209 to 1480 Pa s with a SO viscosity increase from 0.01 to 0.1 Pa s. Similarly, a Ag-in-Ga viscosity increase from about 1480 to 7205 Pa s was required to enable intake of SO with viscosity increasing from 0.1 to 0.5 Pa s. As in the case of the lowest viscosity oil, we observed that the maximum capacity for the 0.1 Pa s SO increased with viscosity of the continuous phase (to 20 vol% at Ag-in-Ga viscosity of 7205 Pa s). Mixing of the most viscous SO (1 Pa s) with any of the Ag-in-Ga compositions resulted in continuous phase inversion. We expect that making the continuous phase more viscous would allow for SO-in-LM emulsion formation even with the most viscous oil, however we were not able to demonstrate this due to practical reasons (adding more than 1.5 vol% Ag into Ga produced very hard composites that could not be reliably mixed with SO). In all, our experiments demonstrate a general trend of an increase in continuous phase viscosity enabling the intake of more viscous SO and increasing the LM capacity for the oil (i.e., the maximum SO volume fraction before inversion).
When plotted in terms of the relative viscosity of the SO to the Ag-in-Ga, our results for all observed SO-in-LM emulsions collapse on a single trend shown in Fig. 3b. Furthermore, when plotted in general terms of variation of the silicone oil or PDMS maximum volume fraction versus the ratio of the added liquid to continuous liquid's viscosity (Ag-in-Ga in our case and PDMS in prior literature), the data on PDMS-in-GaInSn from prior literature14 display the same, albeit substantially shifted, trend. The trend in both cases implies that the threshold silicone volume fraction for phase inversion in silicone and LM systems increases with the viscosity of the continuous phase, which agrees with the rudimentary scaling from Stoke's sedimentation law.16 We note that in contrast to the PDMS-in-LM data,14 our results do not agree with the theoretical formula proposed by Steinmann et al.17,18 However, the departure of our data from one of the models of phase inversion is not surprising as Perazzo et al.16 pointed out in a recent review that despite over a century of work, satisfactory models for the mechanisms governing phase inversion in two liquid systems are still not available.
As in the case of other liquid–liquid systems where surfactants play a major role in emulsification, the presence of the gallium oxide on the SO droplets might also alter the dynamics of SO-in-LM emulsion formation and explain the impact of the continuous phase viscosity. In particular, since the oxide growth progresses in a non-uniform (not layer-by-layer) but rather fractal like fashion (i.e., oxide is not a complete film and is weaker during growth),34 higher viscosity LM phase might promote emulsion formation by allowing for growth of a more complete oxide shell during mixing. In a more viscous surrounding, SO pockets are slower and therefore take longer to come in contact with other fragments of the oil or bulk of the liquid. This additional time might enable growth of a complete and stronger oxide shell around droplets that will provide an effective barrier to SO coalescence with other pockets and the bulk of the liquid. The presence of the Ag–Ga particles in the continuous liquid phase could also potentially also cause departure from pure liquid–liquid system behavior. However, in contrast to particles accumulating at bubble-LM interfaces during foaming,21 we did not observe any significant accumulation on silicone oil–LM interfaces of either spherical silica microparticles in our prior work13 or the Ag–Ga nanoneedles in current system. Thus, the primary role of the Ag–Ga nanoneedles in the emulsion formation process relates to increasing the LM phase viscosity.
In summary, to study how rheology impacts the outcome of mixing SO and LM, we introduced a new method for creating homogenous Ag-in-Ga pastes that are nearly free of air bubbles and excess oxide flakes. The viscosity of these pastes can be increased about 75 times by increasing the volume fraction of added Ag, which rapidly alloys with Ga into Ag–Ga nanoneedles. By mixing six compositions of the Ag-in-Ga mixture with silicone oils with four viscosities, we quantified how the rheology of both the liquids influences the outcome of their mixing (i.e., formation of LM-in-SO versus SO-in-LM emulsions). In particular, we identified the maximum volume fraction of the SO before the continuous phase inversion occurred. Our results show that increasing the viscosity of the Ag-in-Ga mixture increases the capacity to intake more silicone oil and/or silicone oil of higher viscosity. A comparison with prior data from the literature on PDMS-in-LM emulsions with our results indicates a general trend: higher continuous phase viscosity allows for greater silicone oil (or uncured PDMS) fraction and shifts phase inversion thresholds. This observation provides insights into increasing the range of possible compositions of SO-in-LM emulsions that can be applied to create a new generation of highly thermally conductive and corrosion inhibiting thermal interface materials.
Author contributions
Conceptualization: KR, RYW, MDD; data curation: SK, SPP, HM, KR; formal analysis: KR; funding acquisition: KR and MDD; investigation, SK, SPP, HM, KR; methodology: SK, SPP, HM, FK, MDD, RYW, KR; project administration and supervision: KR and RYW; visualization: SK and KR; writing – original draft SK, KR, RYW; writing – review and editing: all authors.Data availability
All data are contained within the manuscript or ESI.†Conflicts of interest
MDG is co-founder of NuAria, LLC and owns 50% equity interest in the company. The work of this manuscript is not directly related to the activities of the company. Other authors have no conflicts to declare.Acknowledgements
We acknowledge the use of facilities within the Eyring Materials Center at Arizona State University supported in part by NNCI-ECCS-2025490. This research was funded by National Science Foundation Division of Civil, Mechanical and Manufacturing Innovation grant 2032415. M. D. G. and H. M. gratefully acknowledge support by the NSF CBET award# 1836719 and the Global Center for Water Treatment which is part of the Arizona Water Innovation Initiative funded from the State of Arizona.References
- T. Daeneke, K. Khoshmanesh, N. Mahmood, I. A. de Castro, D. Esrafilzadeh, S. J. Barrow, M. D. Dickey and K. Kalantar-Zadeh, Chem. Soc. Rev., 2018, 47, 4073–4111 RSC.
- S. Chen, H.-Z. Wang, R.-Q. Zhao, W. Rao and J. Liu, Matter, 2020, 2, 1446–1480 CrossRef.
- M. H. Malakooti, M. R. Bockstaller, K. Matyjaszewski and C. Majidi, Nanoscale Adv., 2020, 2, 2668–2677 RSC.
- S. Chen, Z. Deng and J. Liu, Nanotechnology, 2020, 32, 092001 CrossRef PubMed.
- S. Handschuh-Wang, F. J. Stadler and X. Zhou, J. Phys. Chem. C, 2021, 125, 20113–20142 CrossRef CAS.
- N. U. H. Shah, W. Kong, N. Casey, S. Kanetkar, R. Y.-S. Wang and K. Rykaczewski, Soft Matter, 2021, 17, 8269–8275 RSC.
- R. Tutika, S. Kmiec, A. B. M. T. Haque, S. W. Martin and M. D. Bartlett, ACS Appl. Mater. Interfaces, 2019, 11, 17873–17883 CrossRef CAS PubMed.
- M. D. Bartlett, N. Kazem, M. J. Powell-Palm, X. Huang, W. Sun, J. A. Malen and C. Majidi, Proc. Natl. Acad. Sci., 2017, 114, 2143–2148 CrossRef CAS PubMed.
- N. Kazem, T. Hellebrekers and C. Majidi, Adv. Mater., 2017, 29, 1605985 CrossRef PubMed.
- A. Uppal, W. Kong, A. Rana, R. Y. Wang and K. Rykaczewski, ACS Appl. Mater. Interfaces, 2021, 13, 43348–43355 CrossRef CAS PubMed.
- A. Uppal, W. Kong, A. Rana, J. S. Lee, M. D. Green, K. Rykaczewski and R. Y. Wang, Adv. Mater. Interfaces, 2023, 10, 2201875 CrossRef CAS.
- N. U. H. Shah, S. Kanetkar, A. Uppal, M. D. Dickey, R. Y. Wang and K. Rykaczewski, Langmuir, 2022, 38, 13279–13287 CrossRef CAS PubMed.
- S. Kanetkar, N. U. H. Shah, F. Krisnadi, A. Uppal, R. M. Gandhi, M. D. Dickey, R. Y. Wang and K. Rykaczewski, J. Phys.: Condens. Matter, 2024, 36, 425104 CrossRef CAS PubMed.
- A. Koh, J. Sietins, G. Slipher and R. Mrozek, J. Mater. Res., 2018, 33, 2443–2453 CrossRef CAS.
- W. Jung, M. H. Vong, K. Kwon, J. U. Kim, S. J. Kwon, T. Kim and M. D. Dickey, Adv. Mater., 2024, 2406783 CrossRef CAS PubMed.
- A. Perazzo, V. Preziosi and S. Guido, Adv. Colloid Interface Sci., 2015, 222, 581–599 CrossRef CAS PubMed.
- S. Steinmann, W. Gronski and C. Friedrich, Rheol. Acta, 2002, 41, 77–86 CrossRef CAS.
- S. Steinmann, W. Gronski and C. Friedrich, Polymer, 2002, 43, 4467–4477 CrossRef CAS.
- W. Kong, N. U. H. Shah, T. V. Neumann, M. H. Vong, P. Kotagama, M. D. Dickey, R. Y. Wang and K. Rykaczewski, Soft Matter, 2020, 16, 5801–5805 RSC.
- X. Wang, L. Fan, J. Zhang, X. Sun, H. Chang, B. Yuan, R. Guo, M. Duan and J. Liu, Adv. Funct. Mater., 2019, 1907063 CrossRef CAS.
- S. Kanetkar, N. U. H. Shah, R. M. Gandhi, A. Uppal, M. D. Dickey, R. Y. Wang and K. Rykaczewski, ACS Appl. Eng. Mater., 2023, 1, 1504–1513 CrossRef CAS.
- H. Wang, B. Yuan, S. Liang, R. Guo, W. Rao, X. Wang, H. Chang, Y. Ding, J. Liu and L. Wang, Mater. Horiz., 2018, 5, 222–229 RSC.
- Y. Xu, Z. Lin, K. Rajavel, T. Zhao, P. Zhu, Y. Hu, R. Sun and C.-P. Wong, Nano-Micro Lett., 2022, 14, 1–15 CrossRef PubMed.
- J. Gao, J. Ye, S. Chen, J. Gong, Q. Wang and J. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 17093–17103 CrossRef CAS PubMed.
- F. Krisnadi, S. Kim, S. Im, D. Chacko, M. H. Vong, K. Rykaczewski, S. Park and M. D. Dickey, Adv. Mater., 2024, 2308862 CrossRef CAS PubMed.
- M. Zhang, L. Liu, C. Zhang, H. Chang, P. Zhang and W. Rao, Adv. Mater. Interfaces, 2021, 2100432 CrossRef CAS.
- U. Daalkhaijav, O. D. Yirmibesoglu, S. Walker and Y. Mengüç, Adv. Mater. Technol., 2018, 1700351, 1–9 Search PubMed.
- F. Carle, K. Bai, J. Casara, K. Vanderlick and E. Brown, Phys. Rev. Fluids, 2017, 2, 13301 CrossRef.
- W. Kong, Z. Wang, M. Wang, K. C. Manning, A. Uppal, M. D. Green, R. Y. Wang and K. Rykaczewski, Adv. Mater., 2019, 31, 1904309 CrossRef CAS PubMed.
- H. Chang, R. Guo, Z. Sun, H. Wang, Y. Hou, Q. Wang, W. Rao and J. Liu, Adv. Mater. Interfaces, 2018, 5, 1800571 CrossRef.
- M. M. Yazdanpanah, S. A. Harfenist, A. Safir and R. W. Cohn, J. Appl. Phys., 2005, 98, 073510 CrossRef.
- M. Tavakoli, M. H. Malakooti, H. Paisana, Y. Ohm, D. Green Marques, P. Alhais Lopes, A. P. Piedade, A. T. De Almeida and C. Majidi, Adv. Mater., 2018, 30, 1801852 CrossRef PubMed.
- J. Crawford, M. A. Sayeed and A. P. O’Mullane, Colloids Surf., A, 2021, 623, 126750 CrossRef CAS.
- J. M. Chabala, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 11346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01361a |
| This journal is © The Royal Society of Chemistry 2025 |