
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcentration-dependent kinetic study of graphene oxide (GO) reduction using biophenol and electrochemical analysis†
Saad
Zafar‡
,
Sanjana
Krishna Mani‡
,
Monisha
Monisha
 and
Bimlesh
Lochab
*
and
Bimlesh
Lochab
*
Materials Chemistry Laboratory, Department of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence, Gautam Buddha Nagar, Uttar Pradesh 201314, India. E-mail: bimlesh.lochab@snu.edu.in
First published on 18th November 2024
Abstract
Graphene, a two-dimensional material, has garnered significant interest among researchers globally due to its exceptional characteristics, including a substantial surface area, remarkable chemical stability, elevated electron mobility, and electrical conductivity. The present study explored the synthesis of reduced graphene oxide (rGO), a derivative of graphene materials, by the utilization of gallic acid as a green reducing agent. The successful reduction of graphene oxide (GO) was assessed by X-ray diffraction, UV-vis spectroscopy, Raman, Transmission Electron Microscopy (TEM), and X-ray photoelectron spectroscopy. Cyclic voltammetry (CV), galvanostatic charge/discharge (GCD), and electrochemical impedance spectroscopy (EIS) investigations were employed to analyze the electrochemical and capacitive performance of reduced graphene oxide (rGO). The specific capacitance of rGO was determined to be 301.7 F g−1 at a current density of 1 A g−1. The electrode exhibits an energy density of 121.1 W h kg−1 at a power density of 853.2 W kg−1, and has an exceptional cycle stability of 91% after undergoing 2000 cycles. This green reduction technique is environmentally friendly and shows promising reduction of graphene oxide (GO) into reduced graphene oxide (rGO). Additionally, the prepared rGO exhibited improved electrochemical and capacitive properties showcasing its potential use in supercapacitor applications.
Sustainability spotlightThe present work highlights the optimal use of renewable biophenol as a safer alternative to toxic and hazardous hydrazine for the sustainable and industrially viable production of reduced graphene oxide (rGO) from graphite. The quality of rGO produced depends on the amount of gallic acid used. Even a small weight percentage of this lignin-sourced bioreductant is efficient in producing highly reduced rGO, providing excellent electrochemical properties. The sustainable synthesis of nanomaterials and their application in meeting the growing demand for clean energy solutions is the way forward to supplement the sustainable growth of this industry. |
1 Introduction
Sustainable synthesis of nanomaterials and their application in meeting the growing demand for clean energy solutions have garnered significant attention in recent years.1,2 In particular, to address the increasing energy demand, extensive efforts have been devoted to developing energy storage devices.3–5 Rapid advancement of electric vehicles and portable electronic devices has further driven the exploration of abundant, low-cost, and safer chemistries for producing more sustainable raw materials in providing coveted energy resources. Electrochemical capacitors, also known as supercapacitors or ultracapacitors, have received significant attention over a decade due to their exceptional energy and power density, cost-effectiveness, and long-lasting durability.6 Supercapacitors are frequently combined with batteries and fuel cells to provide high power and energy harvesting capabilities. Supercapacitors are primarily categorized as electrical double layered capacitors (EDLCs) and pseudocapacitors (redox) based on their energy storage mechanism.7 Nevertheless, the practical uses of supercapacitors can be advanced and realized when their energy storage capacity approaches that of batteries.7–9 Thus, approaches to enhance the specific capacitance and energy density while maintaining the life cycle are gaining impetus.In this regard, one of the widely used nanomaterials is graphene, a single layer of carbon atoms arranged in a honeycomb lattice structure, which is sourced from graphite. It is considered as an excellent electrode material for supercapacitors due to its excellent electrical conductivity (106 S m−1), appreciable mechanical properties, high theoretical surface area (2600 m2 g−1), and electric double-layer capacitor (EDLC) nature.10–12 Extensive use of this nanomaterial in a wide variety of applications demands cost-effective technologies for easy large-scale production of graphene and this has become an area of great interest. There are many methods for the synthesis of graphene such as mechanical exfoliation, chemical vapor deposition, ball milling, laser-induced graphene and solution processing techniques.13–16 Chemical synthesis is a simple and cost-effective method and this route has vastly attracted a vast arena of applications, including supercapacitors, compared to other procedures.17 Chemical reduction of oxidized graphite, graphene oxide (GO),18 using reducing agents such as hydrazine hydrate,19 hydroiodic acid,20,21 agricultural byproducts,22 and sodium borohydride23 to form reduced graphene oxide (rGO) is well reported. However, these reactions cannot be scaled up safely due to their hazardous and explosive properties. Alternatively, organic reagents such as hydroquinone,24 pyrogallol,25 and sodium hydroxide26 were also explored. However, these reagents remained in suspension and showed inferior reduction ability than the former reducing agents. Further, rising environmental concerns have attracted the use of greener reducing agents such as L-ascorbic acid,25 gallic acid (GA),27–29 sugars (glucose and fructose),30 tannic acid,31 and plant extracts.32–34 GA is a naturally sourced antioxidant and showed promising reducing ability. To date, the GA method achieved a maximal reduction of GO (GO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GA, 1:10 w/w) to form rGO and that too with remarkable dispersibility.27 However, this excessive use of GA poses a significant purification challenge of rGO as residual GA bound strongly to the graphene framework via π–π stacking interactions leading to the formation of the amorphous nature of rGO. As a result, the conductivity of rGO decreases and hampers energy storage applications.30 Additionally, it is worth gaining some insights into understanding the reduction mechanism of GO with GA, leading to the exfoliation of the stacked graphene layers due to the introduction of functional groups. rGO synthesized under high pH conditions from GO showed a better restoration of the sp2 graphitic lattice, and a pH-dependent effect on the properties of rGO is observed.35,36
:GA, 1:10 w/w) to form rGO and that too with remarkable dispersibility.27 However, this excessive use of GA poses a significant purification challenge of rGO as residual GA bound strongly to the graphene framework via π–π stacking interactions leading to the formation of the amorphous nature of rGO. As a result, the conductivity of rGO decreases and hampers energy storage applications.30 Additionally, it is worth gaining some insights into understanding the reduction mechanism of GO with GA, leading to the exfoliation of the stacked graphene layers due to the introduction of functional groups. rGO synthesized under high pH conditions from GO showed a better restoration of the sp2 graphitic lattice, and a pH-dependent effect on the properties of rGO is observed.35,36
In this study, we aim to develop and optimize the concentration of gallic acid (GA), a biophenol, as a greener reductant for the synthesis of reduced graphene oxide (rGO). We investigate the GA-mediated reduction of graphene oxide (GO) using various weight ratios of GO to GA to determine the extent of GO reduction. The highly reduced graphene oxide (rGO) sample obtained was thoroughly characterized and assessed for its electrochemical performance. This work provides a strategy for the efficient, scalable, and sustainable chemical synthesis of rGO, contributing to the development of sustainably sourced 2D graphene materials and their applications in materials science.
2 Experimental details
2.1 Materials
Graphite powder (Alfa Aesar), ammonium hydroxide (NH4OH, 50%, Alfa Aesar), gallic acid (Sigma-Aldrich), sodium nitrate (NaNO3, Fisher scientific), sulfuric acid (H2SO4, Fisher scientific), potassium permanganate (KMnO4, Fisher scientific), ethanol (Emsure), hydrogen peroxide (H2O2, 30% w/v, Finar), and methanol (CH3OH, Finar). All the reagents were used as received.2.2 Synthesis of graphene oxide
In a typical synthesis, graphite powder (3.0 g, 1 eq.) was added in concentrated H2SO4 (69.0 mL) along with NaNO3 (1.5 g, 0.5 eq.), and the mixture was cooled to 0 °C using an ice bath. To the above stirred reaction mixture, KMnO4 (9.0 g, 3 eq.) was added in small amounts maintaining temperature below 20 °C. The color of the suspension turned from black to dark green. Thereafter, the ice bath was removed, and the reaction mixture was stirred at room temperature for 30 min. To this, water (∼120 mL) was added slowly (careful: highly exothermic reaction) and the color changed to brown. The reaction temperature was maintained (∼98 °C) and stirred for 15 min. Finally, the reaction mixture was allowed to cool down to room temperature, and additional water (∼270 mL) and H2O2 (3.0 mL) were added. The reaction mixture was allowed to stand undisturbed overnight to allow gravity settling of GO. The supernatant was decanted, and the brown residue was taken in Falcon tubes and centrifuged at 9000 rpm, 5 °C for 20 min. The residue was collected and repeatedly washed with distilled water until the pH of the supernatant becomes neutral. The final suspension was dried using a rotary evaporator (with water bath temperature at 60 °C) in a vacuum. The solid obtained was further dried in a vacuum oven at 60 °C overnight to obtain GO as brown colored flakes (1.74 g, 58%).2.3 Synthesis of reduced graphene oxide (rGO)
The method was followed from previously reported literature with some modification.27 GO (100.0 mg) was dispersed in distilled water (40.0 mL) using a probe sonicator for 5 min. To this dispersion, liquor ammonia (100 μL) was added to adjust the pH at 12 to promote the colloidal stability of the graphene oxide sheets through electrostatic repulsion. Varied amounts of GA (50/75/100/200/300 mg), with GO:GA ratios of 1:0.5, 1:0.75, 1:1, 1:2, 1:3 w/w, were added to the suspension followed by stirring under reflux conditions under a nitrogen atmosphere. Aliquots were taken from the reaction mixture at regular intervals till 10 h and the progress of the reaction was monitored using the UV-vis absorption spectrum. The mixture was allowed to cool down to room temperature and rGO was filtered using vacuum filtration. Samples are abbreviated as rGOx, where x is the amount of GA used. The resultant samples are abbreviated as rGO50, rGO75, rGO100, rGO200, and rGO300, respectively.
2.4 pH-study protocol
The pH of the GA and aqueous NH4OH mixture containing varied amounts of GA was recorded. Typically, 10 μL of 50% NH4OH (fresh sample) was added to 10 mL of H2O. The pH of this mixture was recorded as shown in Fig. S1.† To this solution, 5 mg of GA was added to the mixture (to obtain rGO50). UV-vis spectra and the pH of the mixture were recorded. Similarly, the readings were taken after sequential addition of GA as 2.5 mg, 2.5 mg, 10 mg, and 10 mg, to mimic the reaction suspensions as rGO75, rGO100, rGO200, and rGO300, respectively. Similarly, the pH of the bare GO dispersion (control sample) without adding distilled water was monitored.2.5 Characterization
FTIR spectra in the range of 4000–400 cm−1 were recorded on a Nicolet iS5 spectrometer equipped with an attenuated total reflectance diamond accessory (iD5-ATR, Thermoscientific, USA). Absorbance measurements were carried out on a Thermo Scientific Evolution 201 UV-visible spectrophotometer (Thermoscientific, USA). A small amount of sample (200 μL) was collected at regular intervals, purified by centrifugation, and repeatedly washed with deionized water to remove residual gallic acid. Thermogravimetric analysis (TGA) was performed using a Mettler Toledo instrument under a N2 atmosphere (40 mL min−1) at a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD, Billerica, MA, USA) patterns were recorded on a Bruker D8-Discover using Cu-Kα radiation (λ = 0.154 nm) in the scattering range (2θ) of 7–50° at room temperature. Raman spectroscopy (Kotoku, TYO, Japan) measurements were carried out with an STR500 Airix microscope using a 532 nm laser at a power of 3 mW. Transmission electron microscopy (TEM, Akishima, TYO, Japan) images were recorded using an HRTEM, JEOL-2100F instrument at an accelerating voltage of 200 kV. Samples for TEM analysis were prepared by dispersing 1 mg of the material in 5 mL of ethanol solution followed by ultrasonication for 5 min and dropwise deposited onto a copper grid. X-ray photoelectron spectroscopy (XPS) measurements were conducted on an XPS Omicron Multiprobe surface analyzer equipped with a monochromatized Al-Kα (1486.7 eV) radiation source. The obtained spectra were charge corrected at a binding energy of the carbon 1s peak at 284.6 eV and plotted using the CasaXPS software.2.6 Electrochemical measurements
All the electrochemical analyses of the electrode materials were performed on a CHI 700E electrochemical workstation through cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) measurements. A three-electrode system was used to investigate the electrochemical properties of individual electrodes, which were used as working electrodes, with a high purity graphite rod as the counter electrode and Ag/AgCl as a reference electrode. The three-electrode testing was performed using 0.5 M H2SO4 solution. The working electrode was prepared by mixing rGO100 (80 wt%), acetylene black (SuperP, 10 wt%), and polyvinylidene fluoride (PVDF, 10 wt%) in N-methy-2-pyrrolidone solvent. The slurry was then cast on carbon cloth and dried at 70 °C for 12 h under vacuum. The specific capacitance (C, F g−1) of the electrode was calculated from GCD (Csp) curves according to eqn (1):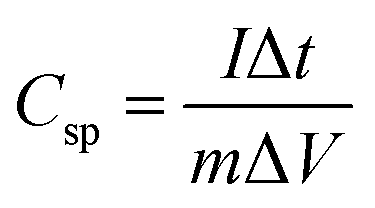 | (1) |
3 Results and discussion
A facile method for the synthesis of reduced graphene oxide (rGO) using gallic acid is discussed here. The reaction involves a reduction of graphene oxide (GO) which can be easily followed with color change. The color of the reaction mixture gradually progressed from brown (GO suspension) to black in rGO indicating restoration of the π-conjugation due to the reduction of oxo-functionalities in GO.27 The UV-vis absorption spectrum of GO showed a maximum around 230 nm and a shoulder at 310 nm corresponding to the π to π* and n to π* transitions respectively.37 Upon reduction, the shoulder band vanished, and an intense red-shifted absorption is observed. Since the extent of reduction of oxo-functionalities is closely correlated with the absorption peak position, the progress of the reaction could be easily monitored by UV-vis absorption spectroscopy to determine the effect of GA on the reduction reaction of GO. The GA absorption maximum appeared at 263 nm (Fig. S2†), which is far beyond the rGO absorption peak. To rule out any discrepancy in data analysis due to residual GA, reaction aliquots were washed extensively to remove any unconsumed GA. A concentration dependent reduction of GO by GA at different GO:GA ratios is followed by recording time-dependent UV-vis. spectra. From Fig. 1a–e, it was found that in all weight ratios of GO:GA, the characteristic GO absorption peak at 228 nm corresponding to the n to π* transition red shifted with time, suggesting that the reduction is affected by the variation in the concentration of GA. Notably, λmax altered relatively fast up to a certain time, and thereafter insignificantly varied despite keeping the reaction for a longer interval (>10 h), confirming successful completion of reduction. It is evident from the UV-vis plots, even though all absorption peak patterns are similar, however rGO100 showed the highest λmax value compared to the other ratios. This indicates that the rGO100, with a GO:GA of 1:1, w/w, facilitated reduction efficiently. Fig. 1f shows variation in λmaxvs. time at different feed-in ratios of GA. It can be observed that the plateau is reached relatively fast, whereas for other samples, the curve changes only slowly before reaching the plateau. The ratios 1:0.5 to 1:1 showed a faster increase in λmax while ratios >1:1 i.e., 1:2 and 1:3 showed a slower initial increase, indicating faster reaction in the former ratios. Interestingly, 1:1 showed a maximum red shift in λmax by 50 nm within 2 h. Unexpectedly, a high GA amount (rGO300) neither leading to a significantly reduced rGO sample (as indicated by low λmax) nor facilitating the reduction of GO in a time-efficiently manner (an additional 3 h is required to achieve similar λmax to other ratios).
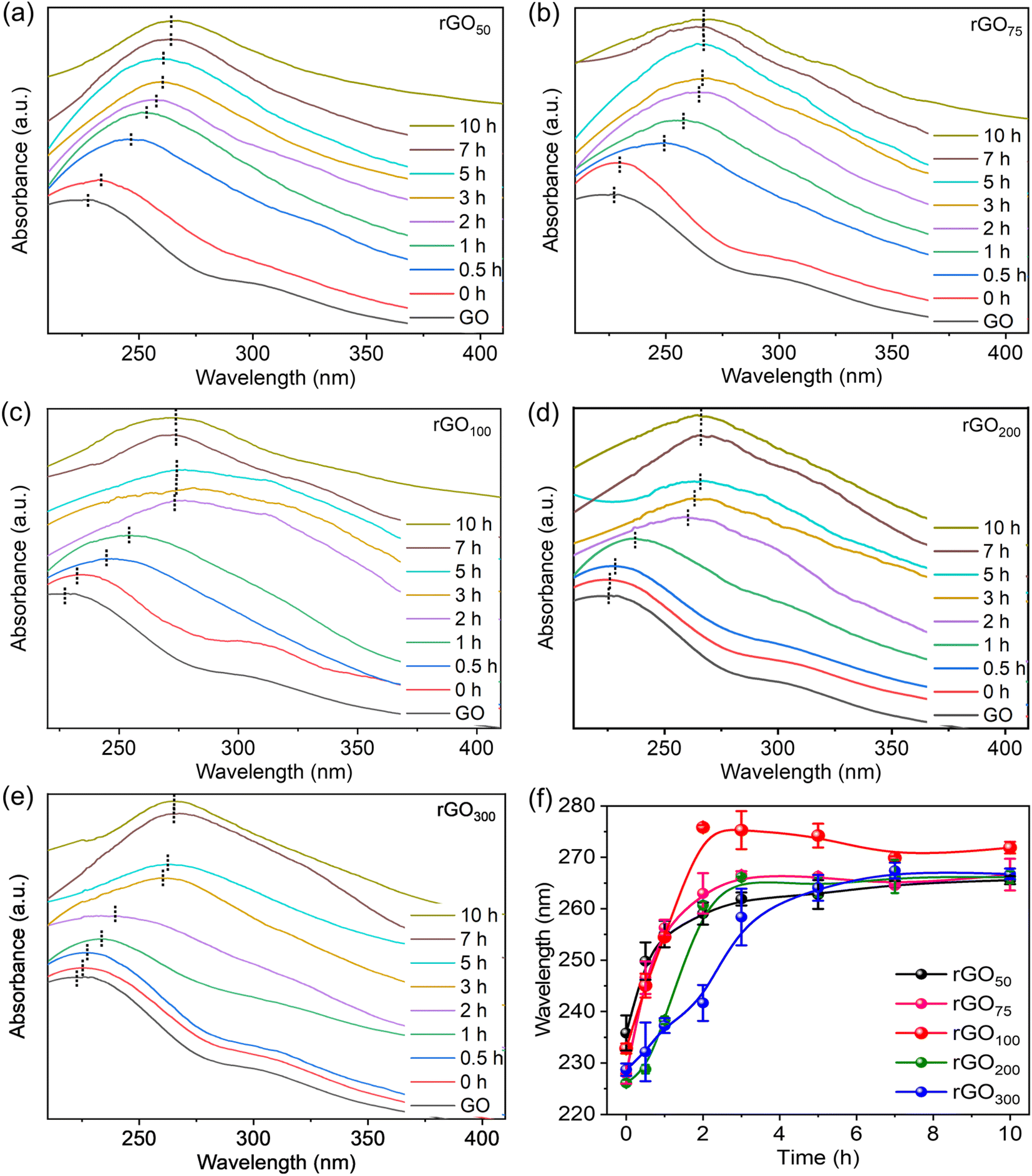 | ||
| Fig. 1 Progress of the reduction reaction is monitored by UV-vis spectra as a function of reaction time for (a) rGO50, (b) rGO75, (c) rGO100, (d) rGO200, and (e) rGO300, and (f) variation of absorption wavelength maximum with time. | ||
A concentration dependent variation in λmax achieved at different GO:GA contents is presented in Fig. 2a. A bell-shaped curve with the maximum observed for the 1:1 ratio indicates maximal reduction in the case of rGO100. Fig. 2b represents the initial change in slope values i.e. change in λmax in the first 1 h (data are summarized as Table S1†) at rGO75.
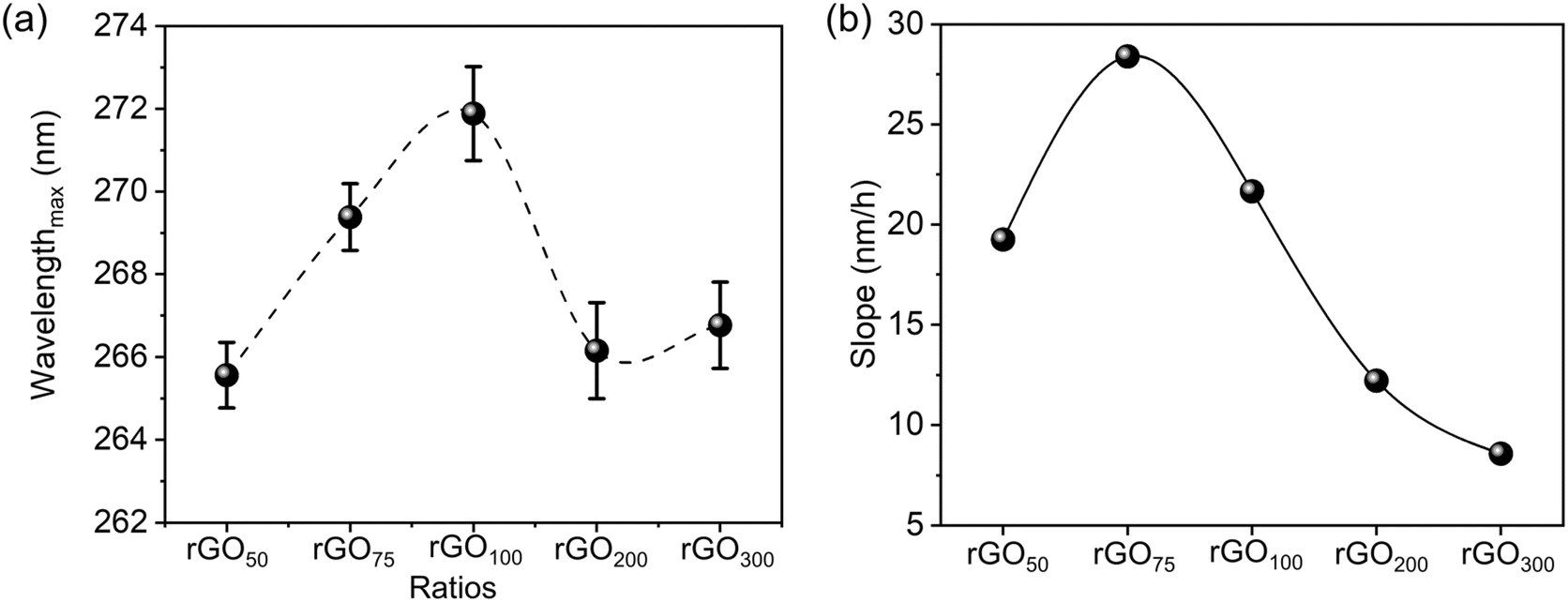 | ||
| Fig. 2 Variation of the GA ratio in GO: (a) plot of λmax (π to π* transition) of synthesized rGOs, and (b) kinetics of the reduction reaction (change in wavelength within the initial 1 h of the reaction). | ||
Bi-phenols, such as gallic acid (GA) and epigallocatechin gallate (EGCG), are used as reducing agents for converting GO to rGO.38–40 The reduction mechanism involves the reaction of epoxy groups on GO with the phenolic-OH groups in GA forming a six-membered transition state, which subsequently converted into a galloyl-derived ortho-quinone with the release of water as a byproduct, to form a double bond. This chemical process restores the sp2 carbon network and promotes the re-establishment of extended π-conjugation via reduction of epoxide functionalities at the basal plane, which is essential for the improved electronic properties of rGO (Fig. 3).
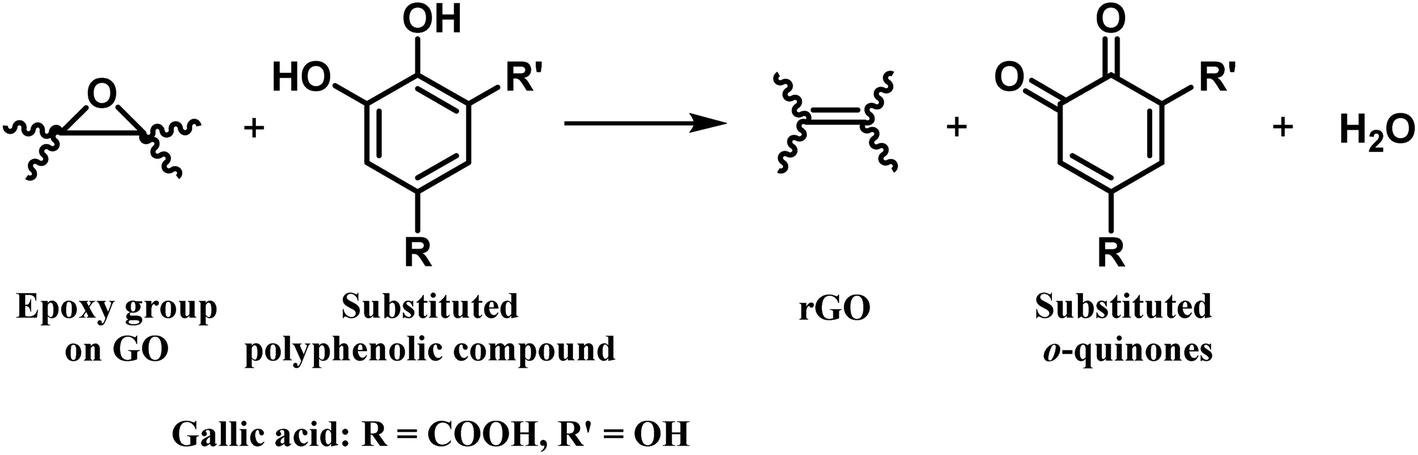 | ||
| Fig. 3 Probable reaction for the formation of rGO using polyphenols as reducing agents. | ||
We hypothesize that the reduction of graphene oxide (GO) to reduced graphene oxide (rGO) using gallic acid (GA) occurs through a specific mechanism influenced by the GA concentration. An increase in GA concentration (from GO ratios of 1:0.5 to 1:1) correlates with a greater extent of GO reduction. However, at ratios exceeding 1:1 (e.g., 1:2 and 1:3), the reduction extent diminishes. This behavior suggests that at high GA concentrations, GA may preferentially adsorb onto the graphene oxide/rGO surfaces via π–π stacking and polar interactions, which inhibits its role in reduction reactions. The high surface area of the 2D graphene sheets enhances this adsorption, limiting the availability of GA for further reduction. Furthermore, the physical binding of GA can obstruct adjacent reactive sites, sterically hindering interactions with other GA molecules and impeding effective reduction kinetics. In contrast, at lower GA concentrations, π–π stacking interactions are less pronounced, allowing for more efficient interaction with GO and resulting in faster reduction rates. Therefore, using GA beyond a certain concentration is undesirable when a high purity of rGO is desired.
The interlayer spacing is the most significant parameter to access the structural information of graphene. XRD is a technique which deals with the layer-to-layer space and crystalline structure of graphene. In this study, we studied the variation in the interlayer d-spacing of GO before and after reduction. In comparison to GO, rGO is related to the removal of oxo-functionalities including basal plane epoxy functionalities. The powder XRD spectrum of graphite, Fig. 4a, revealed an intense and sharp peak at 2θ ∼26.7° corresponding to a d-spacing (calculated using Bragg's equation) of 0.34 nm. After graphite oxidation to GO, the disappearance of a sharp and strong graphitic peak and appearance of a new broad peak at 2θ ∼11.2° are observed. This also confirmed that the GO sample is free from the graphite impurity. Further, this increase in the d-spacing to 0.8 nm infers successful oxidation of graphite and formation of oxo-functionalities at the basal plane enabling separation of layers. Post-reduction reaction of GO using GA, this XRD peak of GO vanished, and a broad prominent diffraction peak at 2θ ∼26° is observed, confirming successful restoration of the graphitic network as in rGO.
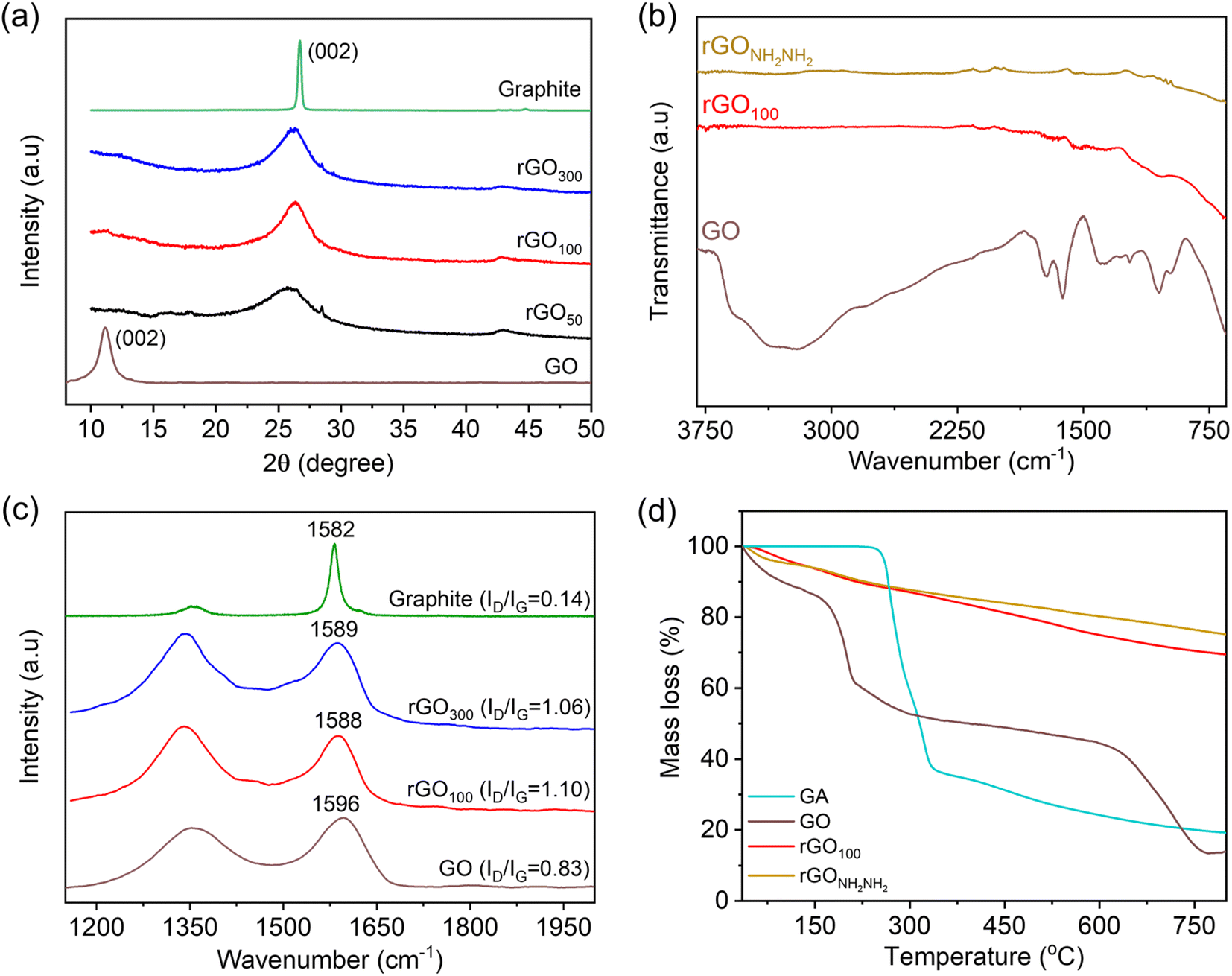 | ||
| Fig. 4 (a) XRD spectra of GO, graphite, rGO300, rGO100 and rGO50, (b) FT-IR spectra of GO, rGO100 and rGONH2NH2, (c) Raman spectra of graphite, synthesized GO, rGO100 and rGO300, and (d) thermogravimetric curves of GA, GO, rGO100 and rGONH2NH2. | ||
ATR-FTIR spectroscopy was employed to understand the nature of oxo-functionalities in GO and the role of GA in reducing these functional groups, Fig. 4b. Various IR absorption bands are due to the O–H stretching (3400 cm−1), carbonyl and carboxylic acid groups (∼1723 cm−1), aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and O–H bending (1617 cm−1), phenolic groups (1260 cm−1), and C–O–C epoxy (1224 cm−1 and 1056 cm−1) groups.41,42 The transmission intensity of these bands significantly reduced after reduction with GA. Interestingly, IR bands related to GA (Fig. S3†) are absent in rGO100, inferring the sample is free from the GA impurity. In contrast, reported FTIR spectra of rGO produced employing a higher amount of gallic acid clearly showed its remnants.27 NH2NH2 has been reported previously to be an efficient reducing agent for GO reduction, and the rGO obtained is free from the residual reducing agent on the graphene surface.19 For the comparison purpose, rGO was obtained through sustainable (GA as the reducing agent) and non-sustainable (hydrazine as the reducing agent) routes, and we synthesized rGO using hydrazine hydrate, abbreviated as rGONH2NH2. The IR spectrum of rGO100 became featureless due to the formation of a highly conjugated π-framework, which is similar to that of rGONH2NH2, indicating GA has a similar reducing potential to hydrazine.
C bonds and O–H bending (1617 cm−1), phenolic groups (1260 cm−1), and C–O–C epoxy (1224 cm−1 and 1056 cm−1) groups.41,42 The transmission intensity of these bands significantly reduced after reduction with GA. Interestingly, IR bands related to GA (Fig. S3†) are absent in rGO100, inferring the sample is free from the GA impurity. In contrast, reported FTIR spectra of rGO produced employing a higher amount of gallic acid clearly showed its remnants.27 NH2NH2 has been reported previously to be an efficient reducing agent for GO reduction, and the rGO obtained is free from the residual reducing agent on the graphene surface.19 For the comparison purpose, rGO was obtained through sustainable (GA as the reducing agent) and non-sustainable (hydrazine as the reducing agent) routes, and we synthesized rGO using hydrazine hydrate, abbreviated as rGONH2NH2. The IR spectrum of rGO100 became featureless due to the formation of a highly conjugated π-framework, which is similar to that of rGONH2NH2, indicating GA has a similar reducing potential to hydrazine.
Raman spectra of the samples were recorded to determine the difference in the relative intensities of G (arising from the first order scattering of E2g phonons or sp2 domains) and D (arising from a breathing mode of A1g symmetry) bands.30 Hence, comparison of as prepared-rGO using gallic acid will provide an understanding of the quality of graphene obtained. Graphite showed the G band at 1582 cm−1 and a weak D band at 1355 cm−1, corresponding to the strong in-plane vibration of the graphite lattice and very few defects, respectively in Fig. 4c.43 Upon chemical oxidation, the peak position of G (1596 cm−1) and D (1345 cm−1) bands is altered due to the introduction of oxo-functionalities in the conjugated framework and formation of isolated double bonds that resonate at higher frequencies. The D band also became prominent due to the introduction of these defects in the graphitic plane.30 After the reduction of GO with GA, the G and D bands shifted to lower wavenumbers confirming the effectiveness of GA as a reducing agent.44 Further, both rGO100 and rGO300 showed similar resonance frequencies. Additional information on the reduction potential of GA was obtained from the intensity ratio D/G (denoted as ID/IG).19 The ID/IG value calculated for GO, rGO100 and rGO300 was found to be 0.83, 1.10 and 1.06, respectively.45 This suggests reestablishment of a conjugated structure along with development of more crystalline domains, which are successfully achieved in rGO100 as a result of significant lowering in defects, even at a relatively low amount of GA.46
The thermal stability of rGO50, rGO100, rGO300 and rGONH2NH2 from GO was performed by TGA. From the TG curves Fig. 4d, GO showed mass loss around 100 °C attributed to the loss of adsorbed water molecules. Both the rGOs showed a similar mass loss pattern, a negligible weight loss at ∼100 °C followed by a slow, steady mass loss from 200–800 °C, attributed to the loss of non-reduced oxygen functional groups.47 Also, the residual masses obtained for both rGOs at 800 °C were close (∼80%), which is expectedly very high compared to GO (13%). There is no mass loss associated with GA as can be seen in the TG curve of rGO100, signifying no bound GA on rGO. Similar thermal stability of both rGOs suggested that GA is equally effective as hydrazine along with additional attractive features is safe to use, especially during the scale up reaction.
To investigate the morphology and crystallinity of the rGO100, TEM (Transmission Electron Microscopy) and SAED (Selected Area Electron Diffraction) recorded. Both GO and rGO sheets showed corrugations, wrinkles, and folds similar to the earlier work.30,43 The SAED patterns of the GO are diffuse while rGO shows hexagonal symmetry with well-defined diffraction spots (inset Fig. 5b and d), illustrating the aromatic configuration was restored after reduction.
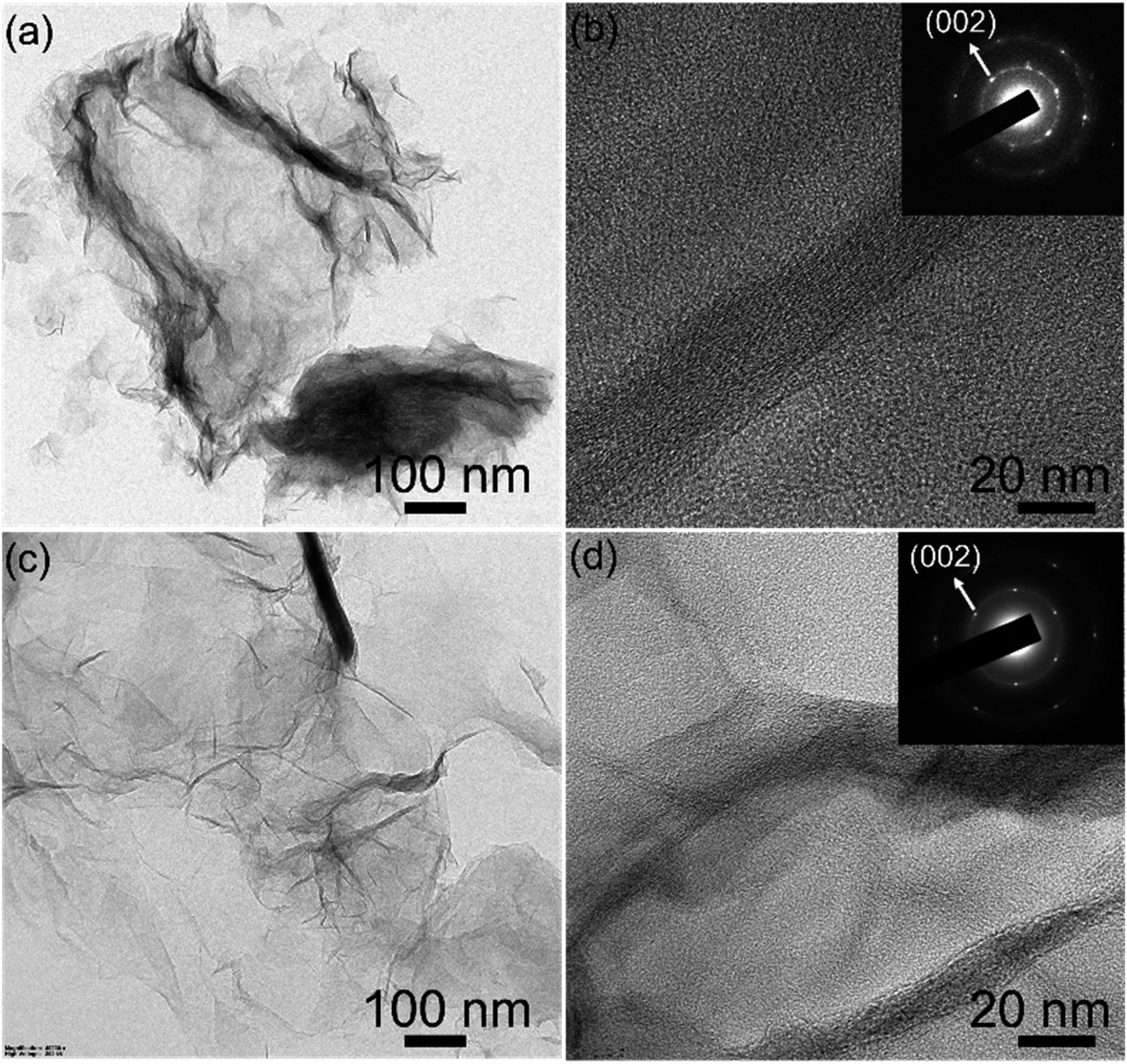 | ||
| Fig. 5 TEM and SAED patterns (inset) of (a and b) GO and (c and d) rGO100 respectively. | ||
From X-ray photoelectron spectroscopy (XPS), the relative amounts of carbon, oxygen and other functional groups present in the GO and rGOx were determined. From wide survey spectra, Fig. S4a,† all the prepared materials were found to be rich in C and O and no other atomic impurities were observed. The relative intensity of C/O provided an insight into the extent of reduction in rGO as shown in Fig. S4b.† GO showed the lowest C/O ratio compared to the other prepared rGO samples. Furthermore, the C/O ratio of rGO100 is the highest compared to the other rGO>100, indicating the formation of a maximally reduced graphene structure in the former. From the deconvoluted spectra, the characteristic peaks obtained can be used to elucidate the type of functional groups present.46,48 GO showed four peaks centered at 284.5, 285.7, and 287.4, 291.5 eV (Fig. 6a), corresponding to CC/C–C/C–H in aromatic rings, C–O/C–OH (epoxy and hydroxyl), >CO and COOH groups, and a satellite peak, respectively.25,49 After reduction, the intensities of carbon–oxygen bonds decreased substantially indicating the effectiveness of GA as a bioreductant in reducing oxo-functionalities in GO (Fig. 6b–d).
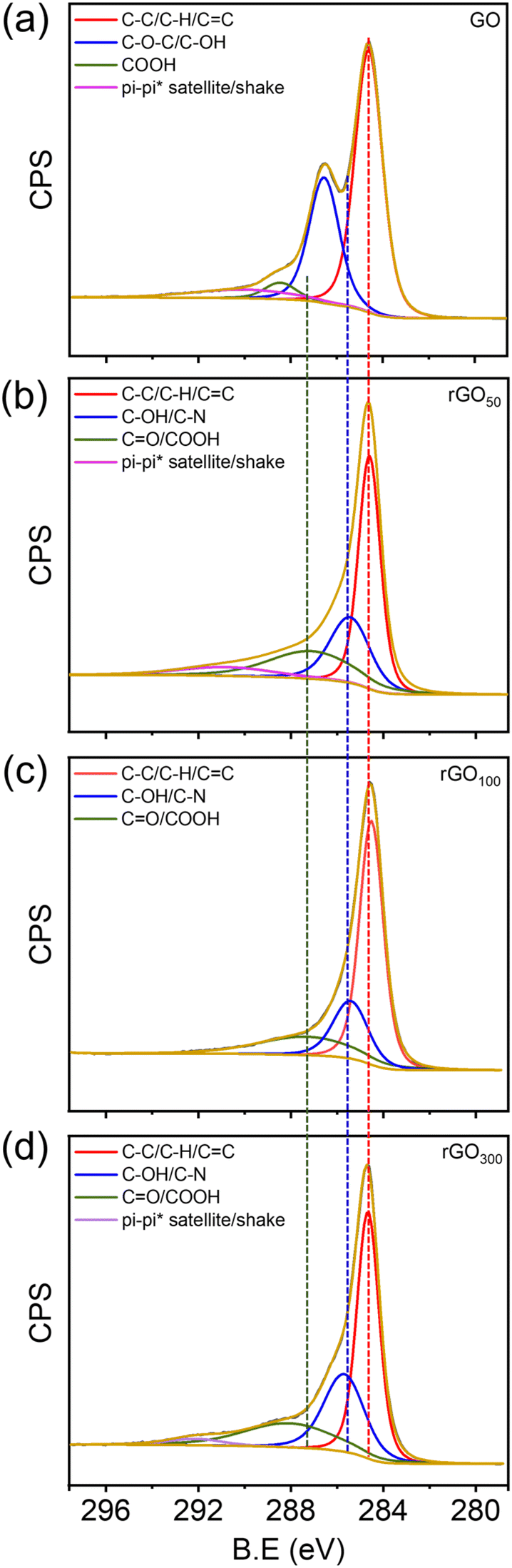 | ||
| Fig. 6 Deconvoluted XPS spectra of the carbon 1s peak of GO (a) and rGO (b–d) samples. | ||
Electrochemical properties of GO and rGO100 were assessed using cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS). The CV curve was obtained within a potential window of −0.5–1.0 V for GO and −0.6–1.1 V for rGO100 at scan rates of 20, 50, 100, 200, 400, 600, and 800 mV s−1. The quasi-rectangular shape of the CV curve resembles the EDLC (Electric Double Layer Capacitor) characteristic commonly found in carbon materials.50 Typically, the capacitance of a supercapacitor is determined by comparing the area under the CV curve at a given potential range.
As depicted in Fig. 7a, rGO100 exhibited the largest area under the CV curve, indicating a higher capacitance compared to GO. GO CV curves displayed a minimal current response across various scan rates (Fig. 7b). Conversely, the area under the CV curve for rGO100 (Fig. 7d) increased, indicating a higher current response in a 0.5 M H2SO4 electrolyte at different scan rates. Fig. 7c and e illustrate the GCD curves of GO and rGO100 electrodes at different current densities. The GCD analyses were conducted at varying current densities of 1, 2, 3, 5, and 10 A g−1. Both GO and rGO100 displayed nearly triangular-shaped GCD curves. The electrode specific capacitance can be determined using the GCD data. The specific capacitance values for GO and rGO100 at various current densities, calculated from the galvanostatic discharge information, are tabulated in Table 1. As the applied current density increased, the specific capacitance decreased. This decrease is attributed to the limitations in the kinetic diffusion process at higher current densities and the decreased number of active sites available for electrochemical reactions.51,52 Additionally, the average crystal size significantly impacts the electrochemical performance. A decrease in crystal size can increase the transport of oxygen vacancies. Since oxygen vacancies act as charge carriers and possess high mobility, which can result in higher specific capacitance.53,54 The plot depicting specific capacitance against current density is displayed in Fig. S5a.† At a current density of 1 A g−1, the calculated specific capacitances for GO and rGO100 were 26.6 and 301.7 F g−1, respectively. A considerable enhancement in the specific capacitance of rGO100 is attributed to the swift electron transport during the charge–discharge process, associated with the higher conductivity of rGO100 compared with GO.
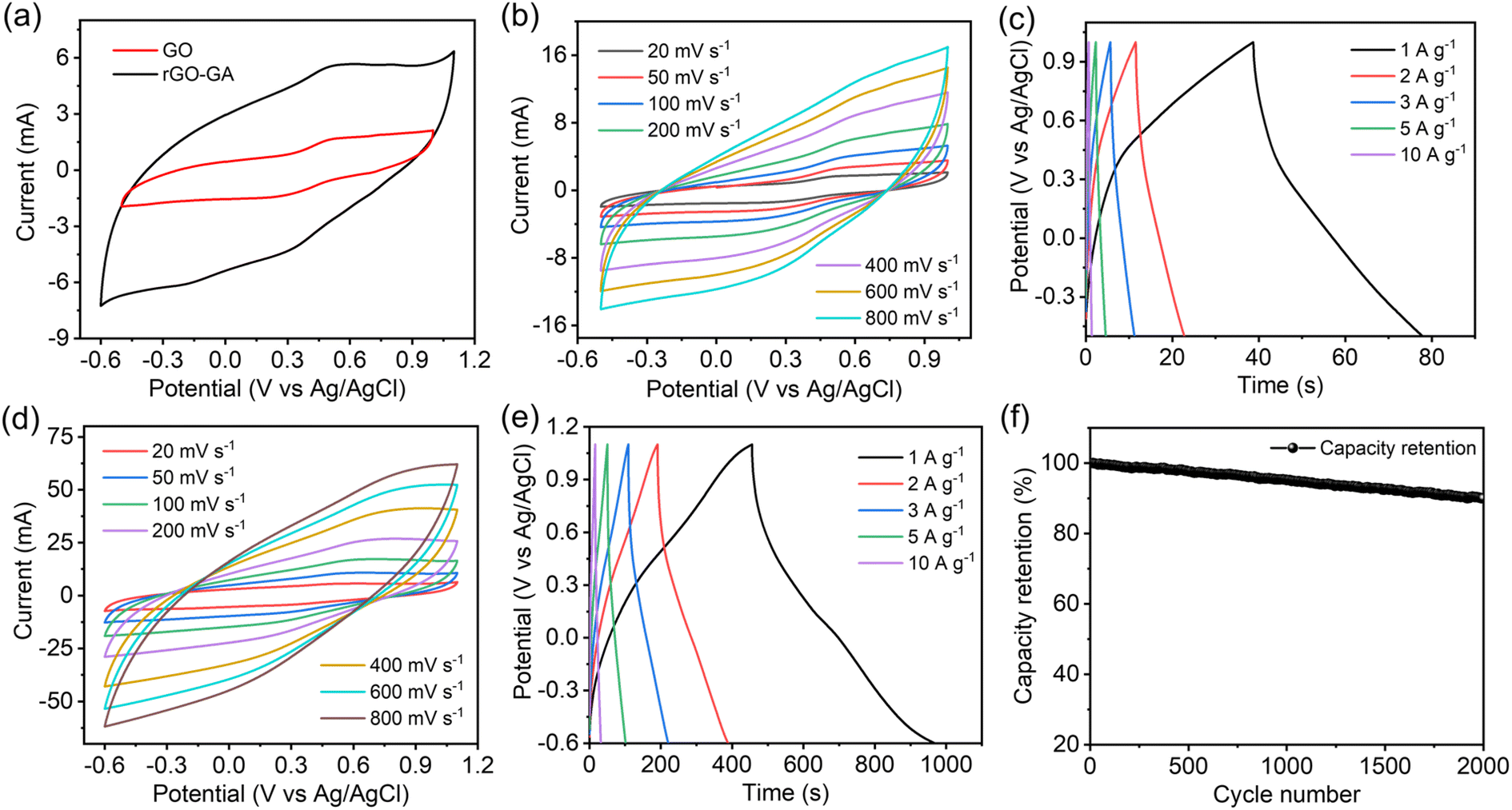 | ||
| Fig. 7 (a) CV curves of GO and rGO100 at 20 mV s−1, (b) CV curves of GO at different scan rates, (c) GCD curves of GO, (d) CV curves of rGO100 at different scan rates, (e) GCD curve of rGO100, and (f) capacity retention of rGO100. | ||
| Sample | Current density (A g−1) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 5 | 10 | |
| a *S.D. = standard deviation in parentheses. | |||||
| GO (F g−1) | 26.6 (1.2) | 16.2 (0.6) | 12.6 (0.4) | 8.0 (0.4) | 6.3 (0.4) |
| rGO100 (F g−1) | 301.7 (3.6) | 233.5 (2.1) | 197.7 (2.2) | 156.4 (2.5) | 93.7 (1.7) |
A comparison of the specific capacitance values obtained in this study with earlier investigations is presented in Table S2.† In comparison to these prior studies, the specific capacitance of rGO obtained in this work demonstrates a far superior value. The use of an acidic electrolyte such as H2SO4 has notably enhanced the capacitive behavior of rGO due to the small size of H+ ions, facilitating their rapid mobility to reach the active sites within the electrode materials.
The energy density (E) and power density (P) stand as critical parameters when assessing the performance of energy storage devices, both of which are anticipated to demonstrate high values. These metrics can be computed using the subsequent eqn (2) and (3):
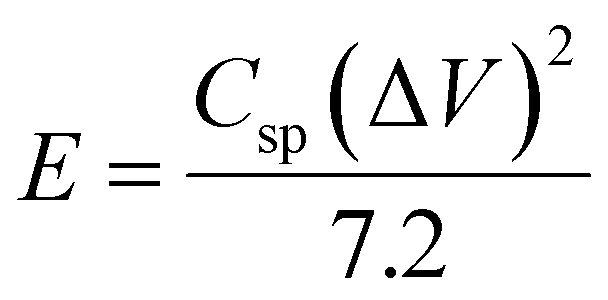 | (2) |
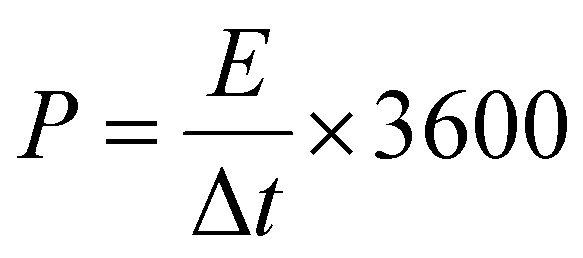 | (3) |
4 Conclusions
Graphene oxide was reduced effectively using GA as a reducing agent with ten times less gallic acid than the literature reported value. A GO:GA ratio of 1:1 (rGO100) facilitated higher reduction of oxo-functionalities and helped in faster and efficient purification than when performed at a higher amount of GA. The lower ratios of GO:GA (≤1:1) resulted in a faster reaction rate than that obtained with higher ratios (>1:1). The properties of rGO obtained using a greener phenolic reductant are similar to those observed using hydrazine. rGO obtained showed a capacitive value of 301 F g−1 and a power density of 853.2 W kg−1. Our present work illustrates a safe reduction and an easier purification process to enable efficient use of a bioreductant and with a potential to apply for a scale up process for graphene synthesis.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors are thankful to the financial and infrastructural support from Shiv Nadar Foundation. SZ and MM are supported by fellowships and SKM work was funded through the Opportunities for Undergraduate Research (OUR) scheme from Shiv Nadar Institution of Eminence. BL acknowledges the Indian Council of Medical Research (ICMR) grant (35/10/2022-NANO/BMS).References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS.
- S. Zafar, A. Thomas, S. N. Mahapatra, N. Karmodak, H. S. Arora and B. Lochab, J. Mater. Chem. A, 2023, 11, 21263–21271 RSC.
- S. Zafar, M. Sharma, K. Shai Mp, N. Karmodak, S. K. Singh and B. Lochab, J. Mater. Chem. A, 2024 10.1039/D4TA01656D.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 RSC.
- F. Ahmad, M. Zahid, H. Jamil, M. A. Khan, S. Atiq, M. Bibi, K. Shahbaz, M. Adnan, M. Danish, F. Rasheed, H. Tahseen, M. J. Shabbir, M. Bilal and A. Samreen, J. Energy Storage, 2023, 72, 108731 CrossRef.
- T. Chen and L. Dai, J. Mater. Chem. A, 2014, 2, 10756–10775 RSC.
- N. Xiao, H. Tan, J. Zhu, L. Tan, X. Rui, X. Dong and Q. Yan, ACS Appl. Mater. Interfaces, 2013, 5, 9656–9662 CrossRef CAS.
- C. Wang, H. Chen, W. Dong, J. Ge, W. Lu, X. Wu, L. Guo and L. Chen, Chem. Commun., 2014, 50, 1202–1204 RSC.
- V. B. Mbayachi, E. Ndayiragije, T. Sammani, S. Taj, E. R. Mbuta and A. U. khan, Results Chem., 2021, 3, 100163 CrossRef CAS.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- V. Agarwal and P. B. Zetterlund, Chem. Eng. J., 2021, 405, 127018 CrossRef CAS.
- P. Zaccagnini and A. Lamberti, Appl. Phys. Lett., 2022, 120, 100501 CrossRef CAS.
- A. Ghosh, S. Kaur, G. Verma, C. Dolle, R. Azmi, S. Heissler, Y. M. Eggeler, K. Mondal, D. Mager, A. Gupta, J. G. Korvink, D.-Y. Wang, A. Sharma and M. Islam, ACS Appl. Mater. Interfaces, 2024, 16, 40313–40325 CrossRef CAS.
- V. S. Abhilash and P. Meshram, Carbon Lett., 2022, 32, 653–669 CrossRef.
- P. R. Wallace, Phys. Rev., 1947, 71, 622 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H.-M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CrossRef CAS.
- A. E. D. Mahmoud, Mater. Chem. Phys., 2020, 253, 123336 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim and J. Y. Choi, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS.
- M. J. Fernández-Merino, L. Guardia, J. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Funct. Mater., 2008, 20, 4490–4493 CAS.
- J. Li, G. Xiao, C. Chen, R. Li and D. Yan, J. Mater. Chem. A, 2013, 1, 1481–1487 RSC.
- Z. Gan, T. Wang, C. Zhai, J. Ma, R. Cao and Y. Wang, ACS Appl. Electron. Mater., 2023, 5, 1148–1155 CrossRef CAS.
- M. Moreno, A. Sánchez Arribas, S. Royano, Y. Izquierdo and M. Chicharro, FlatChem, 2024, 44, 100605 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS.
- Y. Wang, Z. Shi and J. Yin, ACS Appl. Mater. Interfaces, 2011, 3, 1127–1133 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Carbon, 2012, 50, 914–921 CrossRef CAS.
- Q. Yu, J. Jiang, L. Jiang, Q. Yang and N. Yan, Nano Res., 2021, 14, 3724–3743 CrossRef CAS.
- G. B. Besinella, J. E. Padilha, F. B. Scheufele, L. J. Gasparrini, C. E. Borba and H. J. Alves, Adv. Mater., 2021, 2, 403–412 RSC.
- D. E. Glass and G. K. Surya Prakash, Electroanalysis, 2018, 30, 1938–1945 CrossRef CAS.
- C.-J. Shih, S. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Langmuir, 2012, 28, 235–241 CrossRef CAS PubMed.
- B. Badhani, N. Sharma and R. Kakkar, RSC Adv., 2015, 5, 27540–27557 RSC.
- X. Wang, S. Huang, L. Zhu, X. Tian, S. Li and H. Tang, Carbon, 2014, 69, 101–112 CrossRef CAS.
- S. X. Xu C, A. Ji, L. Shi, C. Zhou and Y. Cui, PLoS One, 2015, 19, 0144842 Search PubMed.
- M. F. Abdullah, R. Zakaria and S. H. S. Zein, RSC Adv., 2014, 4, 34510–34518 RSC.
- A. Mathkar, D. Tozier, P. Cox, P. Ong, C. Galande, K. Balakrishnan, A. Leela Mohana Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS.
- H. Guo, X. Wang, Q. Qian, F. Wang and X. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS.
- M. Aunkor, I. Mahbubul, R. Saidur and H. Metselaar, RSC Adv., 2016, 6, 27807–27828 RSC.
- A. J. Glover, M. Cai, K. R. Overdeep, D. E. Kranbuehl and H. C. Schniepp, Macromolecules, 2011, 44, 9821–9829 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- O. C. Compton, B. Jain, D. A. Dikin, A. Abouimrane, K. Amine and S. T. Nguyen, ACS Nano, 2011, 5, 4380–4391 CrossRef CAS PubMed.
- A. E. D. Mahmoud, A. Stolle and M. Stelter, ACS Sustainable Chem. Eng., 2018, 6, 6358–6369 CrossRef.
- S. Zafar, S. K. Singh and B. Lochab, Mater. Adv., 2023, 4, 2425–2436 RSC.
- R. Hadi, K. Rahimpour, E. Payami and R. Teimuri-Mofrad, Appl. Organomet. Chem., 2020, 34, e5946 CrossRef CAS.
- X. W. Wang, Q. Q. Zhu, X. E. Wang, H. C. Zhang, J. J. Zhang and L. F. Wang, J. Alloys Compd., 2016, 675, 195–200 CrossRef CAS.
- K. S. Lau, R. T. Ginting, S. T. Tan, S. X. Chin, S. Zakaria and C. H. Chia, J. Mater. Sci.: Mater. Electron., 2019, 30, 19182–19188 CrossRef CAS.
- T. N. Vinuth Raj, P. A. Hoskeri, H. B. Muralidhara, C. R. Manjunatha, K. Yogesh Kumar and M. S. Raghu, J. Electroanal. Chem., 2020, 858, 113830 CrossRef CAS.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef CAS PubMed.
- M. Handayani, Y. Mulyaningsih, M. Aulia Anggoro, A. Abbas, I. Setiawan, F. Triawan, N. Darsono, Y. Nugraha Thaha, I. Kartika, G. Ketut Sunnardianto, I. Anshori and G. Lisak, Mater. Lett., 2022, 314, 131846 CrossRef CAS.
- R. I. J. Jaidev, A. K. Mishra and S. Ramaprabhu, J. Mater. Chem. A, 2011, 21, 17601–17605 RSC.
- W. Raza, F. Ali, N. Raza, Y. Luo, K.-H. Kim, J. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Nano Energy, 2018, 52, 441–473 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: pH variation of the GO and NH4OH solution with the sequential addition of gallic acid to obtain rGO50 to rGO300; UV-vis and plot FT-IR spectrum of gallic acid; variation of the slope in plot within the initial first hour of the reduction reaction; XPS wide survey spectra of GO and rGOs, and their respective carbon to oxygen ratio; comparative specific capacitance values reported for rGO obtained using different reducing agents; plot depicting specific capacitance against current density; Ragone plot; EIS spectra of GO and rGO100. See DOI: https://doi.org/10.1039/d4su00430b |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |