
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroformylation, hydroaminomethylation and related tandem reactions of bio-derived renewable olefins: a step closer to sustainability
Rupali S.
Prajapati
and
Bhalchandra M.
Bhanage
 *
*
Department of Chemistry, Institute of Chemical Technology, Matunga, Mumbai 400019, Maharashtra, India. E-mail: bm.bhanage@ictmumbai.edu.in
First published on 9th December 2024
Abstract
The rapidly growing population and increased energy consumption are leading to the depletion of non-renewable sources, thus posing a great threat of resource unavailability to future generations. This problem can be tackled using sustainable and renewable sources and by practicing the principles of green chemistry. Hydroformylation, which has applications in various industries, is a highly commercialised, transition metal-catalysed process that is used to produce tonnes of chemicals globally. In this process, the employment of bio-renewable starting materials is a great step toward sustainability. This review highlights the hydroformylation, hydroaminomethylation, and associated tandem reactions of natural olefins, such as terpenes, allyl/propenyl benzene derivatives, oleo-compounds, and steroids. This review intends to provide a clear picture of the research reported to date, encouraging further research and advancement of sustainable practices, environmental friendliness, and application of green chemistry principles in this field.
 Rupali S. Prajapati | Rupali S. Prajapati received her Bachelor's degree in Chemistry in 2016 from Jai Hind College, Mumbai. Subsequently, she received her Master's degree in Organic Chemistry in 2018 from the University of Mumbai. In the same year, she joined Wilson College, Mumbai as an Assistant Professor. In 2022, she joined the research group of Prof. Bhalchandra M. Bhanage in the Institute of Chemical Technology, Mumbai as a PhD research scholar through the UGC-NET Fellowship. Since then, she has been working on the transition metal-catalysed hydroformylation of olefins using homogeneous and heterogeneous systems. |
 Bhalchandra M. Bhanage | Prof. Bhalchandra M. Bhanage received his PhD degree in 1996 from the National Chemical Laboratory and Pune University. He is a Senior Professor at the Institute of Chemical Technology, Mumbai. He is working in the field of homogeneous catalysis, hydroformylation reactions, carbonylation reactions, C–H activation, CO2 fixation, asymmetric synthesis, enzyme catalysis and catalytic application of nanomaterials. |
Sustainability spotlightThe high consumption of non-renewable resources is leading to their depletion, thus demanding a shift toward renewable resources to satisfy the needs for energy, goods and chemicals globally. The production of tonnes of chemicals is necessitated to accommodate the requirements of the huge world population. Aldehydes and their derivatives find wide applications in flavours, fragrances, pharmaceuticals and other industries. They are produced through hydroformylation reaction, one of the most significant processes in industrial catalysis. Hence, the use of bio-derived renewable starting materials in this industrial process innovates a greener and sustainable approach. The sustainability spotlight of this review focuses on the United Nations Sustainable Development Goals (SDGs), specifically SDG 12 (responsible consumption and production) and SDG 9 (industry, innovation and infrastructure). |
1 Introduction
1.1 History and significance
The rapidly increasing population demands a variety of products, ranging from goods to satisfy basic needs to luxury items, making the high production of chemicals inevitable. At the same time, the exploitation and overuse of non-renewable sources have raised concerns regarding the depletion of these sources, necessitating the shift toward sustainable practices, such as the use of renewable biological feedstocks for accessing chemically significant goods. Bio-renewable starting materials serve as sustainable, convenient, inexpensive and naturally abundant supplies for synthesising valuable end-products through chemical manipulation.1 Biomass has been utilized in the production of value-added chemicals, such as aldehydes, alcohols and nitrogenous compounds, through innovative techniques, including one-pot processes and light-assisted technologies.1 The use of the renewable sources is in alignment with the United Nations Sustainable Development Goals, particularly SDG 12, which is “Responsible consumption and production”.One of the most widely utilised processes in industrial catalysis is hydroformylation, which is used globally to produce tonnes of chemicals each year.2 The use of bio-renewable olefins as the starting material in this process can act as a significant step in developing a sustainable society because the use of renewable starting materials reduces the dependence on non-renewable materials, hence preventing their depletion and ensuring the survival of future generations.
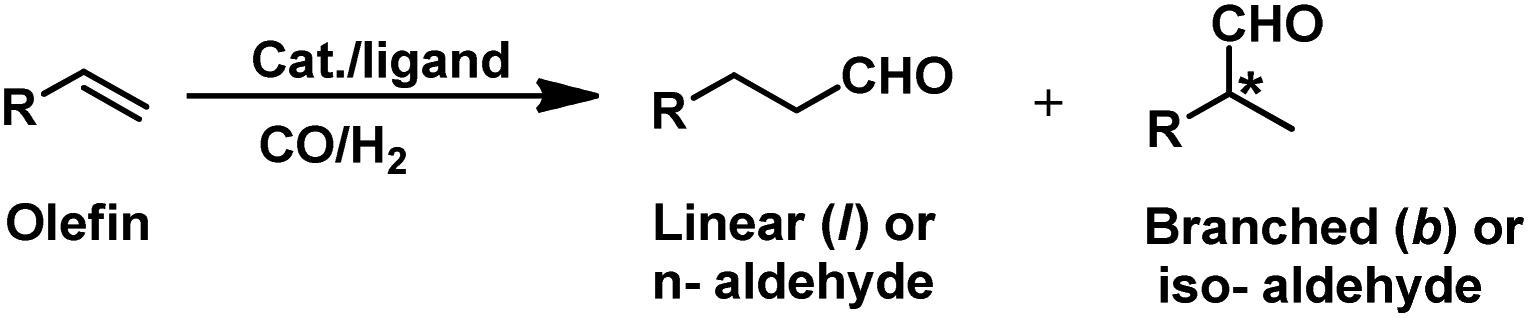
The hydroformylation reaction, also called as the oxo process, was a chance discovery by the German chemist Dr Otto Roelen in 1938 in an attempt to study the oxygenated products of the Fischer–Tropsch synthesis using a cobalt catalyst.3 This transition metal-catalysed procedure involves the addition of a mixture of carbon monoxide and hydrogen gas (generally referred to as synthesis gas or syngas) over the double bond of an olefin, resulting in the incorporation of a hydro and a formyl group (hence the name “hydroformylation”), giving the corresponding aldehyde with an additional carbon (Scheme 1).4,5 Usually, rhodium and cobalt-based metal precursors together with different ligands (preferably phosphorus derived) are employed for this process, which under suitable reaction conditions give a mixture of linear and branched aldehydes. Oxo synthesis occupies an important position in industrial organic synthesis given that the products obtained are further utilised in industries based on flavours, fragrances, cosmetics, soaps, detergents, pharmaceuticals and fine chemicals to produce high value end products.2
 | ||
| Scheme 1 General hydroformylation reaction. | ||
Hydroformylation is a pure addition reaction, and thus it works in alignment with the principles of green chemistry of atom economy and waste prevention.6 Furthermore, this process becomes more energy efficient with the use of catalysts, which allows it to proceed under mild conditions.7 The emergence of next-generation rhodium-based catalysts overpowered the first-generation cobalt-catalysed hydroformylation due to its higher selectivity and activity.8,9 This reaction is regioselective, and usually the linear aldehyde is the desired product, which acts as an intermediate for the production of many other commercially valuable chemicals.10 However, the branch-selective hydroformylation produces a chiral center next to the carbonyl group, bringing into picture the concept of stereoselectivity (Scheme 1).11 The branch-selective aldehydes can be further converted to their chiral derivatives.
Naturally occurring olefins have been proven to be a sustainable source for the synthesis of these chemicals through hydroformylation, employing the principles of green chemistry of renewable feedstock utilisation, smart catalysis, designing safer chemicals, waste prevention and atom economy.12 The design of efficient one-pot processes enjoys the advantages of less time, labour and resources given that it eliminates the requirement of work-up, product separation and purification.13 One-pot processes employing hydroformylation together with other steps such as hydroformylation–acetalisation,14–16 hydroformylation–lactolisation/hemiacetalisation,17–19 hydroformylation–cyclisation,20,21 hydroformylation–reduction22 and hydroformylation–reductive amination23–29 can yield versatile end products in a green manner. In the past, several accounts summarising the hydroformylation process have been published.30–32 However, only a few discussed the hydroformylation of sustainable renewable resources,28,33 and extensive research has been reported since then. Thus, a detailed up-to-date account of the work on the transition metal-catalysed hydroformylation of natural olefins and the conditions utilised for this process can be very beneficial for further research in this area for promoting the responsible consumption of resources and development of innovative sustainable practices.
1.2 Mechanism
Scheme 2 depicts the generally accepted mechanism for hydroformylation reactions involving unmodified cobalt and rhodium catalytic precursors.9,34,35 The first step constitutes the formation of active rhodium hydride species 2 from rhodium precursor 1. Subsequently, this species coordinates to the substrate alkene through rhodium to form π complex 3. Upon hydride insertion in 3, regio- and stereo-selectivity comes into the picture as linear 4 or branched 8 metal–alkyl intermediates are generated. As visible by their structures, stereoselectivity is possible only in the branched intermediates (in case of terminal olefins) due to the presence of a chiral centre. Species 4 and 8 form rhodium carbonyl intermediates 5 and 9, respectively, by the coordination of carbon monoxide through rhodium. Rhodium-acyl species 6 and 10 are made by the alkyl migration to CO in 5 and 9. The formation of linear 7 and branched 11 aldehydes together with the regeneration of active rhodium hydride species 2 takes place in the final step through the oxidative addition of H2, sequentially followed by reductive elimination. All the steps in this catalytic cycle are reversible (alkyl insertion in the Rh–H bond is irreversible at room temperature), except for the final step of aldehyde formation. In conclusion, this means that once the aldehydes are formed, they cannot be reverted to other species. However, the other species maintain sensitive equilibria with each other and can be converted back and forth.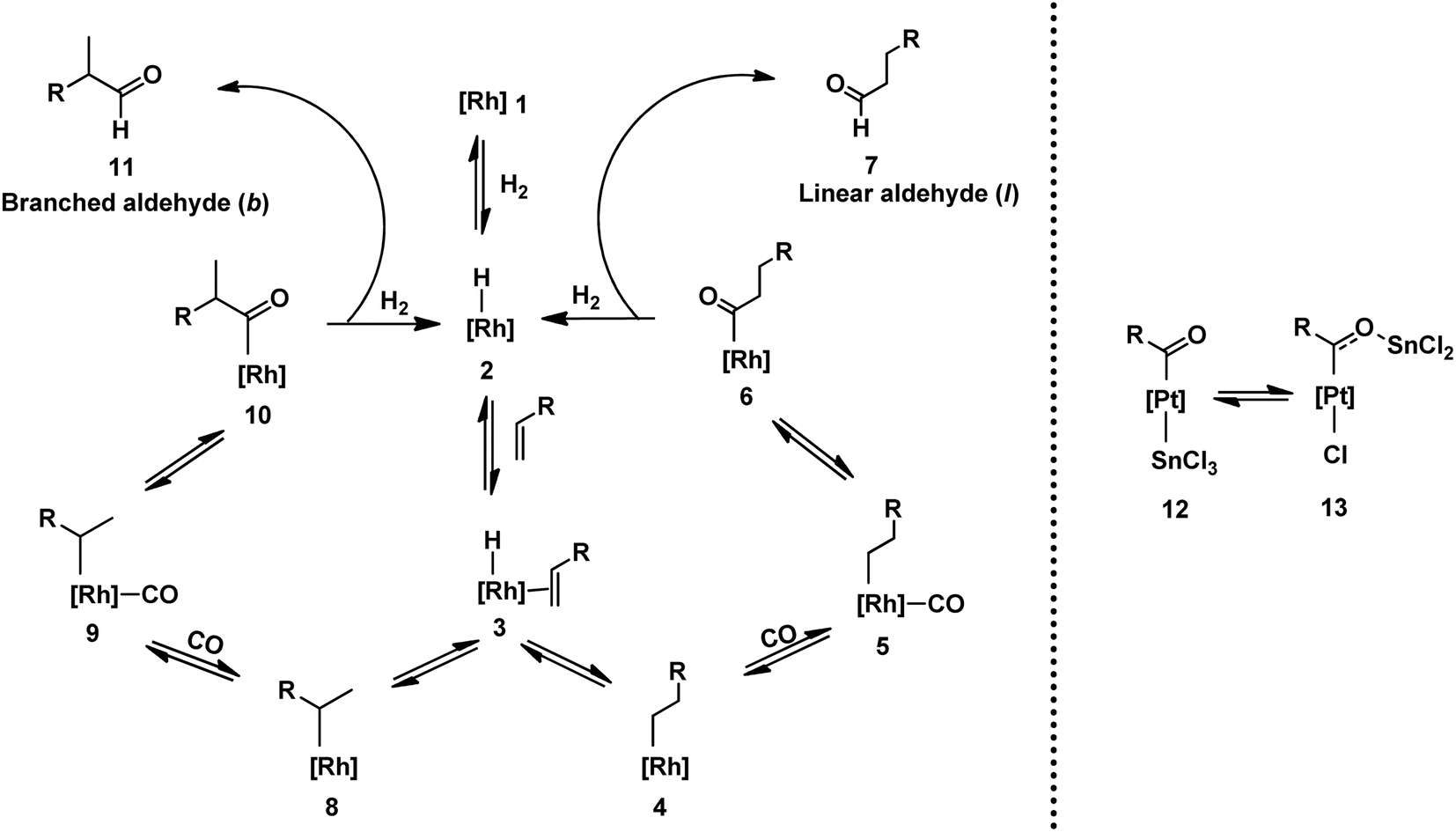 | ||
| Scheme 2 General mechanism for hydroformylation. | ||
The active catalytic species, selectivity, l/b ratio, rate and rate-determining step vary in the presence or absence of ligands, where in the latter case, the nature of the ligands and the reaction conditions such as pressure and temperature also have an effect.36
In platinum/tin-catalysed systems, SnCl3− may interact in various ways with platinum catalysts.31 Besides imparting its Lewis acid character, tin chloride may coordinate as a ligand or act as a counterion. In the coordination mode of tin chloride, after CO migration into the platinum–alkyl bond, two complexes, 12 and 13, are developed (Scheme 2).
1.3 Catalytic systems
To date, the most widely utilised transition metal complexes for hydroformylation reactions are those derived from rhodium metal.37 Second to rhodium, cobalt complexes are employed, which require harsh reaction conditions and provide poor chemo-, regio- and stereo-selectivity in the system.38 Alternatively, rhodium-based catalysts lead to much better selectivity, even in the absence of harsh conditions.9,39 However, there is a growing need to search for efficient non-rhodium catalysts due to the increasing cost of the precious rhodium metal. Thus, to address this problem, numerous investigations have been reported on the catalytic capabilities of alternative transition metal-based complexes including palladium,40 iridium,41 ruthenium,42 cobalt,35,43 platinum44 and iron.45 The results in this regard have been elaborately summarised in two reviews.31,46Homogeneous unmodified and modified rhodium-based complexes have been employed for hydroformylating the double bonds present in olefins. The introduction of different phosphorus47 and diphosphorus48 ligands allows the reaction parameters to be tuned, achieving high yield and selectivity. Tolman's cone angle concept49 is useful for monophosphine ligands, while for diphosphine ligands, the concept of bite angles is considered. Diphosphines adopting bite angles higher than 100° are found to favour linear aldehydes over branched aldehydes in rhodium-catalysed systems.50 The effect of the bite angle of diphosphines on the regio- and stereo-selectivity of the system has been overviewed in the literature.51 Moreover, phosphite ligands are introduced in enantioselective hydroformylation to increase the enantiomeric excess.52 Several P-stereogenic and backbone-chiral phosphorus ligands have been studied in asymmetric hydroformylations to increase the stereoselectivity and obtain a high enantiomeric excess.53 Previously, various review articles focused on asymmetric hydroformylation using chiral ligands.4,54–56 Nitrogen-based ligands have also been explored for manipulating the system toward high activity in hydroformylation and associated hydroformylation-derived one-pot reactions.57
Economically, the recovery and recyclability of the catalytic system have major significance considering that oxo synthesis is a highly industrially utilised process.58 Homogeneous catalysis suffers from the loss of catalyst and contamination of the product due to the inability to separate it from the catalyst. These problems resulted in the discovery and establishment of the aqueous biphasic Ruhrchemie/Rhône-Poulenc process in 1984.59 In this system, one phase is formed by the rhodium complex derived from the water-soluble ligand triphenyl phosphine-3,3′,3′′-trisulfonate (TPPTS) dissolved in water, whereas the other phase consists of substrates and products. However, higher alkenes usually show poor solubility in water, and hence mass transfer agents are used for this purpose. One of the options is to apply a co-solvent that will enhance the solubility of the substrate in the aqueous phase by increasing the lipophilicity of this phase, thus facilitating the substrate–catalyst interactions.60 Another measure is to introduce surfactants to assist in the attraction of the alkenes toward the catalyst. This happens because the surfactant forms an interface of micelles between the aqueous and organic phases, where these micelles possess a hydrophilic exterior and a hydrophobic core. The alkene gets solubilised in the core of the micelles, boosting the rate of the reaction.61 Cationic surfactants such as cetyltrimethylammonium bromide (CTAB) are considered the most suitable given that they are electrostatically attracted toward the anionic sulfonate group of the Rh/TPPTS system.62 Polyethylene glycol (PEG) is a beneficial additive due to its rate-enhancing property, together with the ability to prevent the catalyst from leaching into the organic phase.63 Several cyclodextrins such as α-CD, β-CD and γ-CD have been employed, among which β-CD is the most suitable.64,65 Furthermore, cyclodextrins can be chemically alternated to obtain randomly methylated α-CD (RAME-α-CD), randomly methylated β-CD (RAME-β-CD) and randomly methylated γ-CD (RAME-γ-CD), increasing their efficiency.65 The reason for the acceleration of the reaction with modified CDs is their accumulation in the biphasic interface, forming inclusion complexes with the substrate, and thus encouraging substrate–catalyst interactions.66 Activated carbon is an efficient mass-transfer agent, which also works by bringing the substrate in the vicinity of the catalyst at the interface.67 By using these agents, the catalytic system can be recycled and reused for multiple reaction cycles without remarkable loss in its activity. Different ligand systems involving amphiphilic ligands,68 polymer-supported ligands69 and supported aqueous phase catalysts (SAPC)70 have been studied in the past to improve the results of aqueous biphasic hydroformylation. Hydroformylation using different aqueous biphasic systems has been overviewed in the literature.71
Besides water/organic biphasic systems, ionic liquid/organic biphasic systems have also been investigated and ionic liquids have been found to increase the stability and productivity of the catalyst.72,73 In comparison to aqueous systems, non-aqueous biphasic systems consisting of ionic liquids can be applied to longer-chain olefins.72,74 Accounts on hydroformylation in ionic liquids with varying parameters are available.75 Methodologies have been developed to carry out hydroformylation in solventless systems, given that they will lead to a more environmentally friendly system based on the principles of green chemistry.76 The employment of nanoparticles in place of complexes of metals such as cobalt, rhodium and copper has also been reported.77 Heterogeneous rhodium-based systems such as immobilised catalysts on solid supports and polymer-supported catalysts have been studied and the reports have been summarised.78
Tandem hydroaminomethylation requires the same conditions as that of hydroformylation together with the addition of substrate amines to produce the corresponding product amines.79 Multiple reviews have discussed this process.80
The aforementioned catalytic systems are utilised in the hydroformylation and hydroaminomethylation of natural olefins and are discussed in the following sections.
This review is mainly focussed on the hydroformylation of sustainable, renewable olefins, which deals with UN's Sustainable Development Goals, including Responsible Consumption and Production (SDG 12) and Industry, Innovation and Infrastructure (SDG 9). The use of renewable starting materials such as terpenes including limonene and pinene, which are naturally available from plants and their components, is an approach to responsibly consume and produce chemicals through innovative strategies. This when extended to the industrial scale through the building of a sustainable infrastructure, will prevent the depletion of non-renewable resources. The effects of the metal precursor and ligand modifications on the regioselectivity of the aldehyde have also been discussed. A brief section is dedicated separately to the hydroaminomethylation of these olefins. The examples available in the literature for natural renewable olefins grouped as terpenes, allyl/propenyl benzene derivatives, oleo compounds and steroids are provided. The purpose of this review is to elaborate a roadmap of the research work performed to date in the field of hydroformylation and hydroaminomethylation of naturally occurring olefins to boost the research in this field, further leaning toward sustainability.
2 Hydroformylation of natural olefins
2.1 Terpenes
Terpenes are an important family of natural products.81 Generally, terpenic compounds are an important biomass-derived sustainable and renewable feedstock in the food, flavour, fragrance and cosmetic industries as well as in aromatherapy.82The terminal double bond of limonene preferably undergoes hydroformylation, while its endocyclic, internal double bond is usually resistant toward the addition of a carbonyl group due to steric and electronic factors.94 Also, phosphorus-based ligands and CO compete with limonene for the binding site on rhodium catalysts. Therefore, the rate of this reaction decreases with an increase in the concentration or basicity of the phosphine ligand and increase in the concentration of CO pressure at a constant H2 pressure. However, an increase in the H2 pressure by maintaining a constant CO pressure has no effect on the reaction rate. This implies that the hydroformylation of limonene follows zero-order kinetics in hydrogen and a negative order with respect to CO. The rate of hydroformylation of natural limonene is 35-times higher when the bulky phosphite ligand tris(o-tbutylphenyl)phosphite is used instead of triphenyl phosphine with Rh(COD)OAc as the metallic precursor.95
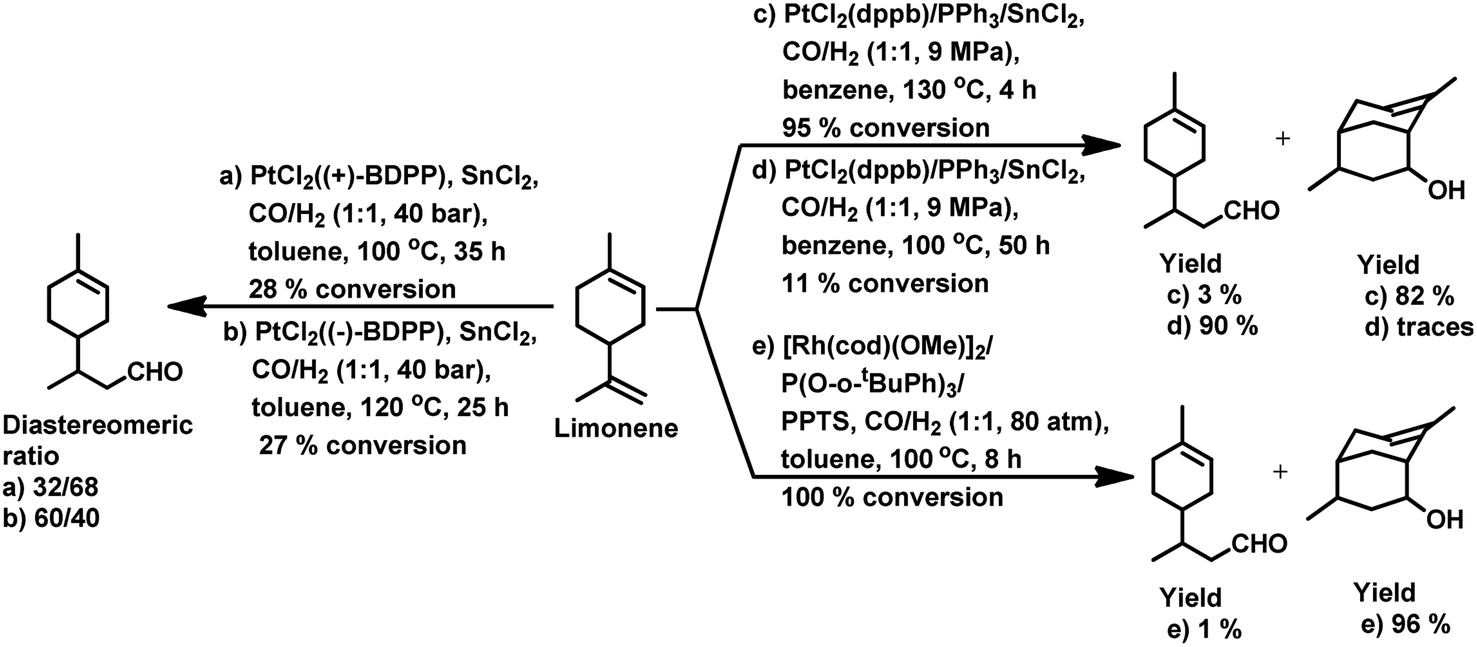
In the hydroformylation of (+)-R-limonene, a bimetallic catalyst system based on Pt/Sn was shown to yield the corresponding linear aldehyde exclusively with no traceable amounts of branched products although the conversion was low.84 The asymmetric carbon centre of the substrate influences the diastereoselectivity of the product when a specific enantiomer of a chiral phosphine ligand, such as (+)-BDPP and (−)-BDPP (BDPP = 2,4-bis(diphenylphosphino)pentane), is employed in PtCl2-bisphosphine-SnCl2 catalytic systems (Scheme 3a and b).
 | ||
| Scheme 3 Hydroformylation/cyclisation of limonene. | ||
When the PtCl2 (dppb = 1,4-bis(diphenylphosphino)butane)/PPh3/SnCl2 catalytic system was employed, limonene underwent a one-pot conversion to a bicyclic alcohol, 4,8-dimethyl-bicyclo[3.3.1]non-7-en-2-ol, involving hydroformylation as the first step, followed by the stereospecific intramolecular cyclisation of the hydroformylated product (Scheme 3c and d).85,86 In this case, the bicyclic alcohol was the major product at a temperature of 130 °C, while the hydroformylated product was favoured with a decrease in the temperature to 100 °C but suffered from low conversion. Moreover, a monometallic system lacking either Pt complex or SnCl2 resulted in no cyclisation under similar reaction conditions, showing that the bimetallic species is synergistically involved in the formation of the bicyclic alcohol. An excess of Sn, which acts as a Lewis acid, led to the undesirable isomerisation of the double bonds in limonene, leading to the formation of α-terpinolene and γ-terpinene in this bimetallic system. When the Rh/[PPh3 or P(O-o-tBuPh)3]/PPTS (pyridinium p-toluenesulphonate) system was subjected to hydroformylation conditions in the presence of [Rh(COD)(OMe)]2 as the catalyst precursor and toluene as the solvent, the bicyclic alcohol was formed quantitatively and stereoselectively in a much shorter time and at relatively lower catalyst concentrations (Scheme 3e).21 Replacing PPh3 with the P(O-o-tBuPh)3 ligand enhanced the efficiency and rate of both the hydroformylation and the cyclisation steps, yielding about 90% of the bicyclic alcohol in 8 h, whereas the former requires 24 h for the same result under similar reaction conditions. This is because the bulkier phosphite ligand disfavours the binding of a second phosphite ligand to the rhodium species, making room for limonene, and also it makes the dissociation of CO easier due to its weak σ-basicity and strong π-acidity. Both ligands give two diastereomers of 4,8-dimethyl-bicyclo[3.3.1]non-7-en-2-ol, one from each of the two diastereomers of the aldehyde intermediate. The use of the acid co-catalyst PPTS is crucial given that it accelerates the cyclisation step significantly and its absence leads to a low conversion of aldehyde to the cyclised product.
Employing a water/toluene biphasic system comprised of [Rh(COD)(μ-OAc)]2 and TPPTS as the ligand, limonene underwent hydroformylation even in the absence of a surfactant.96 However, the cationic surfactant cetyltrimethylammonium chloride (CTAC) acted as a promoter up to a CTAC/TPPTS ratio of 1/10; beyond which it started to act as an inhibitor. The inhibition effect was seen at increased concentrations given that the limonene molecules have to pass through the layer of accumulated surfactant at the interface region to access the catalyst site.
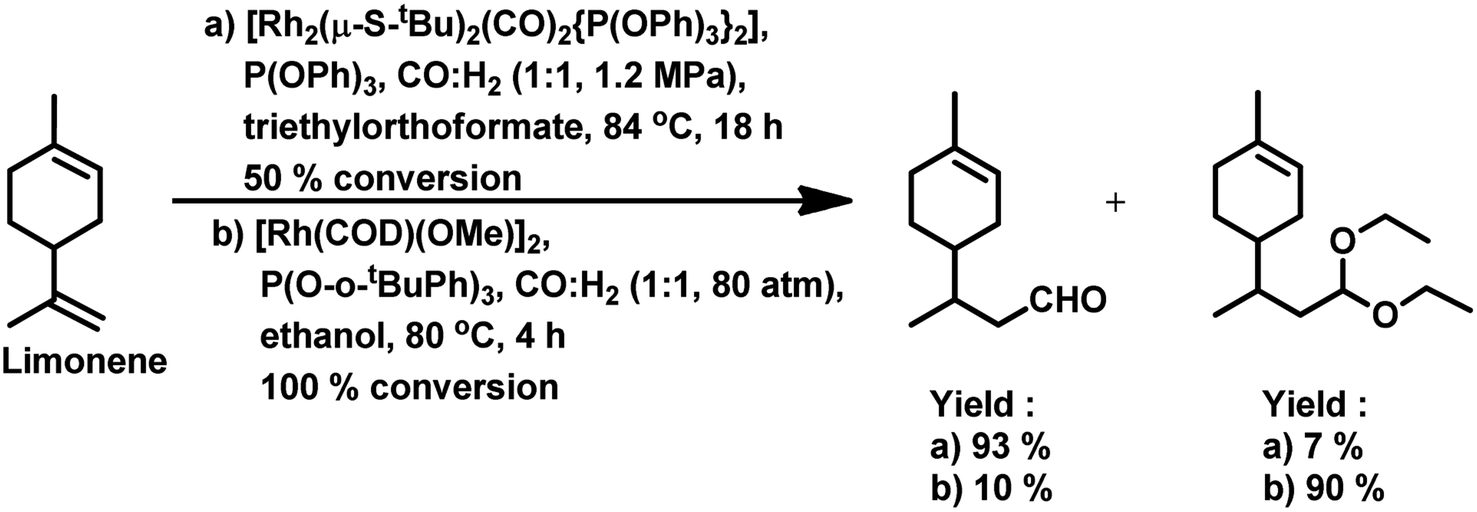
The hydroformylation of this substrate with the use of [{Rh(μ-StBu)(CO)P(OPh)3}2] in triethylorthoformate as the solvent yielded 93% of aldehyde together with 7% of the corresponding acetal, albeit the conversion was only 50% (Scheme 4a).15 The rate of both hydroformylation and acetalisation increased upon the replacement of PPh3 with P(O-o-tBuPh)3, where the latter gave up to 90% yield of acetal and a total conversion of 100% at 80 °C in an ethanolic one-pot acetalisation without any co-catalyst (Scheme 4b).16
 | ||
| Scheme 4 One-pot hydroformylation/acetalisation of limonene. | ||
The rhodium-catalysed hydroformylation of (R)-limonene using the bidentate phosphine ligands BDPP and BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) in γ-valerolactone as a benign and environmentally friendly solvent gave better chemo- and regio-selectivity but lower catalytic activities compared to the conventional and toxic solvent toluene.97
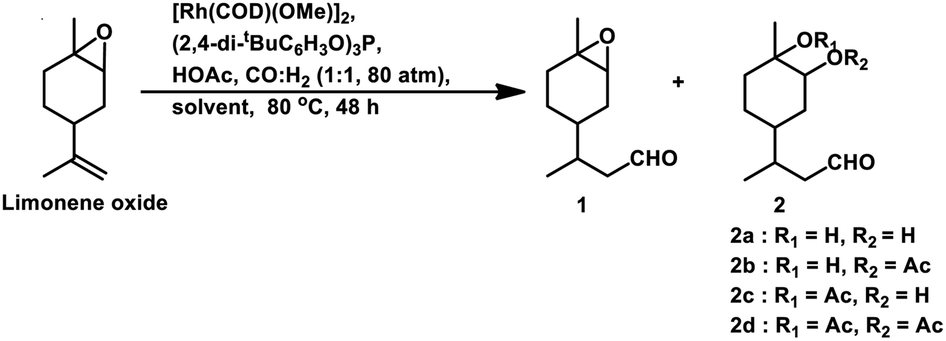
Limonene oxide, an oxygenated derivative of limonene, is a naturally occurring monoterpene. The one-pot hydroformylation and simultaneous epoxy ring cleavage of commercial (+)-limonene oxide (mixture of cis and trans) occurred using [Rh(COD)(OMe)]2 as the catalyst precursor together with PPh3 or (2,4-di-tBuC6H3O)3P as an auxiliary ligand in toluene, ethanol, anisole, dimethylcarbonate (DMC) or diethylcarbonate (DEC).98 The acetylation agent for the opening of the oxirane ring can be acetic anhydride or acetic acid, generating the corresponding acetoxy derivatives of the diols. Acetic acid is less efficient than acetic anhydride given that it leads to reduced aldehyde selectivity, undesirable isomerisation of limonene oxide and the inability to convert monoacetates to diacetates. Compared to PPh3, the use of (2,4-di-tBuC6H3O)3P not only results in better selectivity but also faster conversion to the aldehyde. The conversion rate and selectivity for aldehyde are comparable in DMC, approximately the same in DEC and higher in anisole compared to that in toluene (Table 1). The products obtained from the hydroformylation/O-acylation of this substrate has olfactory features and can be used in fragrance formulations.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Solvent | Conversion (%) | Yield (%) | |||||
| 1 | 2a | 2b | 2c | 2d | 2 (total) | ||
| Toluene | >99 | 4 | 2 | 44 | 34 | 0 | 80 |
| DMC | >99 | 13 | 9 | 30 | 20 | 2 | 61 |
| DEC | >99 | 9 | 4 | 27 | 24 | 1 | 56 |
| Anisole | >99 | 5 | 5 | 40 | 28 | 2 | 75 |
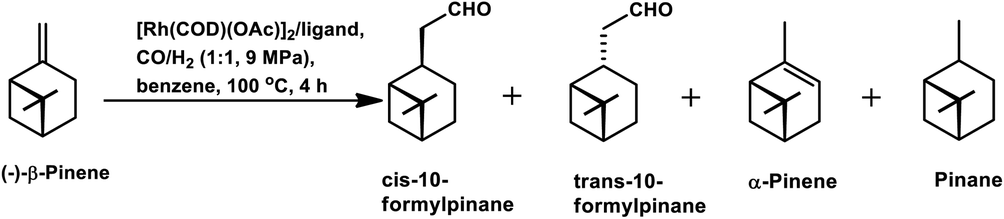
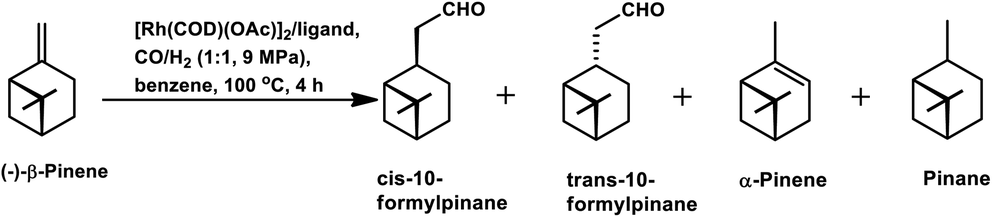
β-Pinene isomerises easily to α-pinene under unmodified cobalt and rhodium-catalysed oxo conditions. [Rh6(CO)16], [Co2(CO)8] and dinuclear [{Rh(μ-S-tBu)(CO)P(OPh)3}2] catalyzed the hydroformylation of β-pinene to yield cis-10-formylpinane, trans-3-formylpinane and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis-10- and trans-3-formylpinane as the major products, respectively.19,90,101 However, these catalysts showed low activities and high rates for the isomerisation of β- to α-pinene. On the contrary, very high catalytic activity was shown by both [RhCI(CO)(PPh3)2] and [HRh(CO)(PPh3)3] in the presence of free phosphines. An increase in the phosphine PPh3/rhodium ratio increased the stereoselectivity of [HRh(CO)(PPh3)3], yielding cis-10-formylpinane as the major product, whereas only a marginal increase in the stereoselectivity was obtained with [RhCI(CO)(PPh3)2]. The choice of tris(2-tbutylphenyl)phosphite as the ligand instead of triphenyl phosphine reduced the catalytic activity and gave trans-10-formylpinane as the major product, which was usually obtained in low quantities.100 The two faces of β-pinene only slightly differ from each other due to their similar steric hindrance, leading to low diastereoselectivity toward the hydroformylation of the exocyclic double bond. Substituted and unsubstituted metal carbonyl centres both attack this olefin from the least hindered site (anti to the isopropylidene bridge), while the addition of ancillary ligands leads to the preferential attack of this olefin from the most hindered site (syn to the isopropylidene bridge), yielding cis-10-formylpinane and trans-10-formylpinane as the major products, respectively.19,89
:1 mixture of cis-10- and trans-3-formylpinane as the major products, respectively.19,90,101 However, these catalysts showed low activities and high rates for the isomerisation of β- to α-pinene. On the contrary, very high catalytic activity was shown by both [RhCI(CO)(PPh3)2] and [HRh(CO)(PPh3)3] in the presence of free phosphines. An increase in the phosphine PPh3/rhodium ratio increased the stereoselectivity of [HRh(CO)(PPh3)3], yielding cis-10-formylpinane as the major product, whereas only a marginal increase in the stereoselectivity was obtained with [RhCI(CO)(PPh3)2]. The choice of tris(2-tbutylphenyl)phosphite as the ligand instead of triphenyl phosphine reduced the catalytic activity and gave trans-10-formylpinane as the major product, which was usually obtained in low quantities.100 The two faces of β-pinene only slightly differ from each other due to their similar steric hindrance, leading to low diastereoselectivity toward the hydroformylation of the exocyclic double bond. Substituted and unsubstituted metal carbonyl centres both attack this olefin from the least hindered site (anti to the isopropylidene bridge), while the addition of ancillary ligands leads to the preferential attack of this olefin from the most hindered site (syn to the isopropylidene bridge), yielding cis-10-formylpinane and trans-10-formylpinane as the major products, respectively.19,89
E. Gusevskaya et al. revealed that the basicity of the auxiliary ligands highly affects the activity, chemo- and regio-selectivity of rhodium-based catalytic systems for the hydroformylation of β-pinene, wherein more basic ligands show lower activity and higher chemo- and diastereo-selectivity toward the cis product.102 With an increase in temperature, the trans product becomes more and more favoured given that the intermediate involved in its formation is thermodynamically more stable, while lower temperatures favour the cis product given that it involves the formation of a kinetically stable intermediate with low activation energy. The introduction of phosphine, diphosphine and phosphite ligands increases the selectivity for the cis product (Tables 2 and 3). Besides, in the case of ligands with cone angles in the range of 128–165°, there is no direct relation between their bulkiness and the diastereoselectivity of the system.
|
|
||||||
|---|---|---|---|---|---|---|
| Ligand | Conversion (%) | Selectivity (%) | Product distribution (%) | |||
| Cis | Trans | α-Pinene | Pinane | |||
| None | 98 | 46 | 5 | 41 | 52 | 2 |
| PPh3 | 68 | 89 | 73 | 16 | 6 | 5 |
| dppe | 9 | 90 | 85 | 5 | 9 | 1 |
| dppb | 12 | 93 | 86 | 7 | 7 | — |
| NAPHOS | 10 | 91 | 81 | 10 | 9 | — |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Ligand | Cone angle (°) | Conversion (%) | Selectivity (%) | Product distribution (%) | |||
| Cis | Trans | α-Pinene | Pinane | ||||
| PPh3 | 145 | 68 | 87 | 69 | 18 | 10 | 3 |
| P(p-OCH3Ph)3 | 145 | 43 | 95 | 81 | 14 | 5 | — |
| P(CH2Ph)3 | 165 | 92 | 87 | 75 | 12 | 13 | — |
| P(n-Bu)3 | 132 | 61 | 96 | 86 | 10 | 4 | — |
| Pcy3 | 170 | 42 | 94 | 90 | 4 | 6 | — |
| P(C6F5)3 | 184 | 98 | 46 | 4 | 42 | 51 | 3 |
| P(o-CH3Ph)3 | 194 | 97 | 42 | 5 | 37 | 57 | 1 |
| P(O-o-tBuPh)3 | 175 | 99 | 62 | 26 | 32 | 38 | — |
| P(Oph)3 | 128 | 95 | 78 | 48 | 30 | 22 | — |
The application of density functional theory (DFT) on β-pinene revealed the diastereofacial selectivity for the trans isomer of 10-formylpinane based on the energetic, electronic and structural factors with the use of [HRh(CO)3] for unmodified rhodium catalytic systems in hydroformylation.103 β-Pinene coordinates through the less sterically hindered face, yielding the cis aldehyde, which is thermodynamically preferred by 3.5 kcal mol−1. However, the coordination through the more sterically hindered face, yielding the trans aldehyde, gives a lower activation energy (8.5 kcal mol−1) for the olefin insertion reaction.
The rate of hydroformylation and the trans product selectivity significantly increased upon the employment of bimetallic cobalt–rhodium catalytic systems such as preformed [Co2Rh2(CO)12] and stoichiometric mixtures of [Co2(CO)8] and [Rh4(CO)12] or [Rh6(CO)16] and chloro-rhodium systems such as [N(PPh3)2]Cl/[Rh4(CO)12] compared to simple homometallic catalytic systems (Table 4).104,105 However, the conversion is usually below 35% and the presence or absence of free phosphines in these bimetallic systems does not cause a significant variation in the results. Alternatively, the homometallic system [Rh4(CO)12] shows a switch from trans to cis selectivity and a drop in the isomerisation of β-pinene to α-pinene. The change from PPh3 (unidentate phosphine ligand) to PCy3 (unidentate phosphine ligand with a large cone angle) and dppe (1,2-bis(diphenylphosphino)ethane, a bidentate ligand) resulted in enhanced selectivity toward the cis isomer (Table 4).106
|
|
|||||
|---|---|---|---|---|---|
| Catalyst precursor | Temp. (°C) | Time (h) | Conversion (%) | Product distribution (%) | |
| Cis | Trans | ||||
| [Co2Rh2(CO)12] | 100 | 1 | 32 | 1.6 | 95 |
| [Rh4(CO)12] + [Co2(CO)8] | 100 | 1 | 31 | 1.6 | 92 |
| [Rh4(CO)12] + [Co2(CO)8] | 125 | 0.5 | 32 | 0.6 | 76 |
| [Rh4(CO)12]/2 Cl− | 125 | 8 | 30 | 7.4 | 64 |
| [Rh4(CO)12] + PPh3 | 125 | 5 | 96 | 94 | 4 |
| [Rh4(CO)12] + Pcy3 | 125 | 5 | 51 | 85 | 12 |
| [Rh4(CO)12] + dppe | 150 | 5 | 15 | 93 | 4 |
The stereo- and regio-selective hydroformylation of (−)-β-pinene occurred with the application of a Pt/Sn bimetallic system in the presence of phosphine ligands, yielding exclusively the linear aldehyde and up to 98% diastereoselectivity for the thermodynamically stable, trans product.85,86 The hydroformylation of (−)-β-pinene with PtCl2 (phosphine or diphosphine)/SnCl2/PPh3, where the phosphine is PPh3 and the diphosphines are dppe, dppp (1,3-bis(diphenylphosphino)propane), and dppb, resulted in the formation of a diastereomeric mixture of cis and trans aldehydes, corresponding alcohols due to the reduction of the aldehydes, hydrogenation to pinane, isomerisation of the double bond to α-pinene, skeletal isomerisation to α-terpinolene and γ-terpinene, skeletal isomerisation followed by the addition of chloride, giving bornyl and fenchyl chlorides. Among them, the best catalytic system was the one with dppb, wherein the selectivity was 85% for aldehydes and 5% for alcohol, although the conversion was only 31% (Table 5). However, an increase in the temperature to 130 °C increased the conversion to 70% and decreased the selectivity to a total of 84% for aldehydes and alcohol. The system with only PPh3 showed the highest activity for the formation of alcohols but the lowest activity for pinane, and also lower selectivity for the hydroformylated products compared to the system with dppb. Stannous chloride is a Lewis acid that forms a stable 1:1 complex with PtCl2. Therefore, when used in higher concentrations, that is, Sn/Pt > 1, it deactivates the catalyst, leading to the isomerisation of β-pinene.
|
|
||||||
|---|---|---|---|---|---|---|
| Ligand | SnCl2·2H2O (mmol) | Temp. (°C) | Conversion (%) | Selectivity (%) | Product distribution (%) | |
| Aldehydes (cis/trans) | Alcohol | |||||
| PPh3 | 0.05 | 100 | 25 | 81 | 51 (3/97) | 30 |
| dppe | 0.05 | 100 | 17 | 74 | 70 (3/97) | 4 |
| dppp | 0.05 | 100 | 45 | 81 | 75 (2/98) | 6 |
| dppb | 0.05 | 100 | 31 | 90 | 85 (3/97) | 5 |
| dppb | 0.05 | 130 | 70 | 84 | 81 (3/97) | 3 |
| dppb | 0.10 | 100 | 28 | 85 | 81 (2/98) | 4 |
| dppb | 0.25 | 100 | 25 | 39 | 33 (3/97) | 6 |
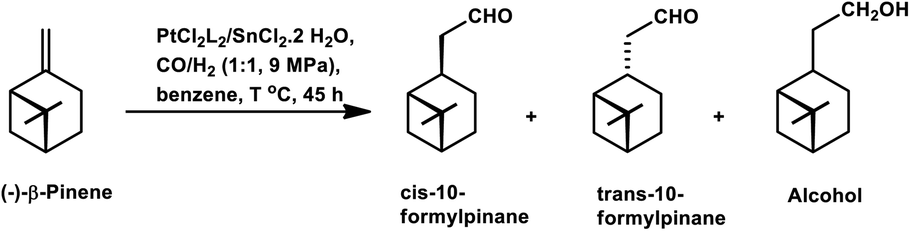
The chemo- and diastereo-selective acetalisation of β-pinene occurred upon the use of the dinuclear rhodium complex [Rh2(μ-2)2(CO)4][CF3SO3]2 in triethyl orthoformate with 80% conversion and 71% yield for acetals (Scheme 5a).15 α-Pinene and β-pinene underwent tandem hydroformylation–acetalisation with [Rh(COD)(Ome)]2 as the catalyst precursor and P(O-o-tBuPh)3 or PPh3 as an auxiliary ligand in the environmentally friendly solvent ethanol.107 This tandem reaction is more feasible in case of β-pinene with an exocyclic double bond than in the case of α-pinene, which has a sterically hindered endocyclic trisubstituted double bond. β-Pinene underwent this reaction even with PPh3, though its replacement with P(O-o-tBuPh)3 tripled the rate of the reaction. On the contrary, α-pinene reacted under mild conditions only in the presence of P(O-o-tBuPh)3 to give 93% conversion with a total of 82% acetals (Scheme 6). Under the optimised conditions, β-pinene showed 100% conversion with a total of 39% aldehydes and 49% main acetals (Scheme 5b).
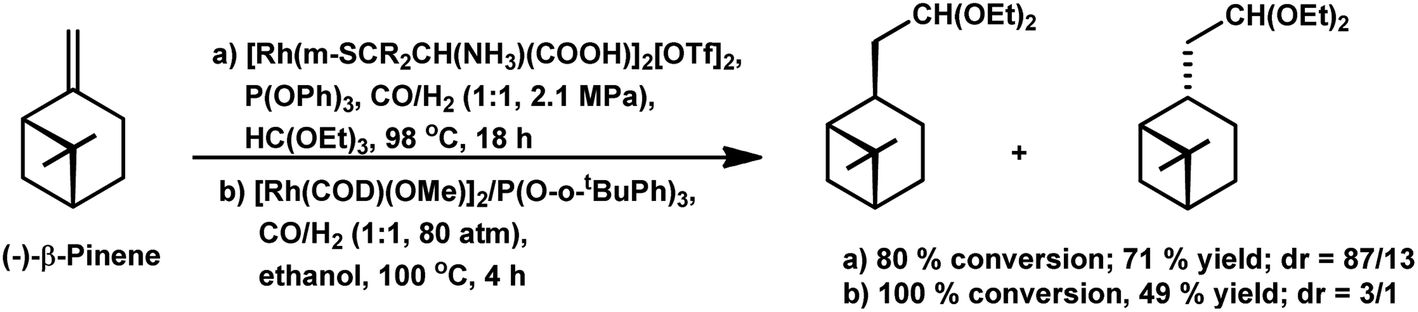 | ||
| Scheme 5 Tandem hydroformylation/acetalisation of (−)-β-pinene. | ||
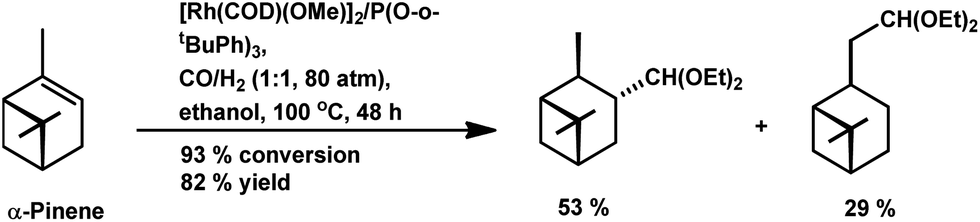 | ||
| Scheme 6 Tandem hydroformylation/acetalisation of α-pinene. | ||
β-Pinene also underwent a two-step domino hydroformylation/arylation process on reaction with arylboronic acids and rhodium-based catalyst to give an alcohol as the final product in 20% yield (Scheme 7).108
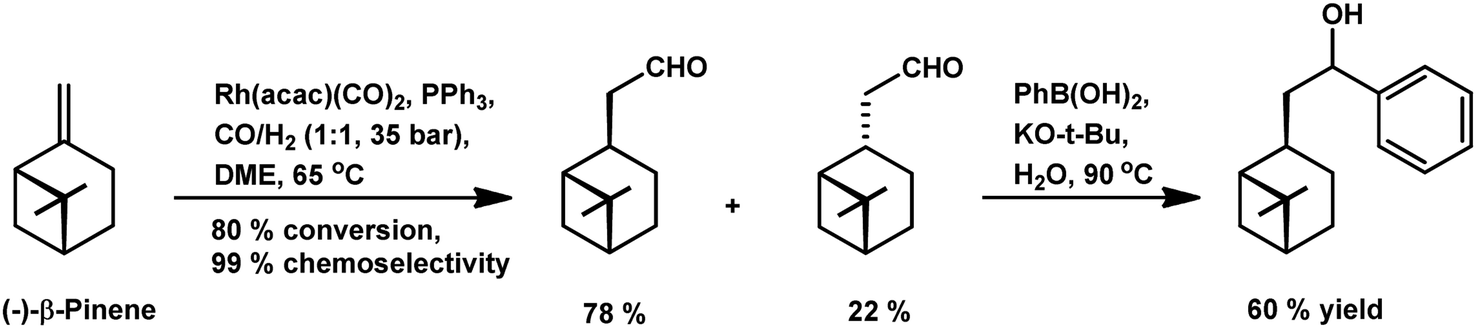 | ||
| Scheme 7 Tandem hydroformylation/arylation of β-pinene. | ||
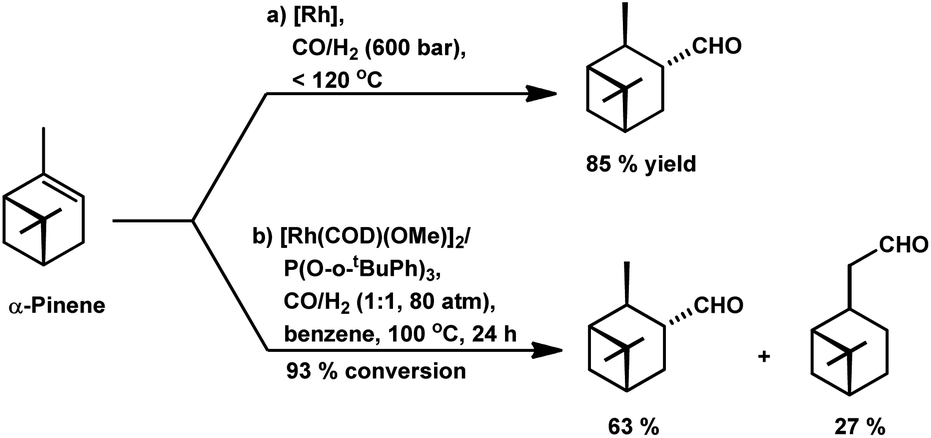
Way back in 1980, Siegel et al. found that α-pinene converted to (+)-3-pinanecarbaldehyde with up to 85% selectivity in a rhodium-catalysed system with 600 bar pressure of syngas and temperature of around 120 °C (Scheme 8a).109 α-Pinene incorporated the formyl group successfully with the employment of the bulky phosphite ligand, P(O-o-tBuPh)3 together with [Rh(COD)(Ome)]2 through 93% conversion and 63% of trans-3-formylpinane. The bulky phosphite ligand ensured easy CO dissociation and prevented inactive multi(ligand) rhodium species (Scheme 8b).110
 | ||
| Scheme 8 Hydroformylation of α-pinene. | ||
 | ||
| Scheme 9 Hydroformylation of camphene. | ||
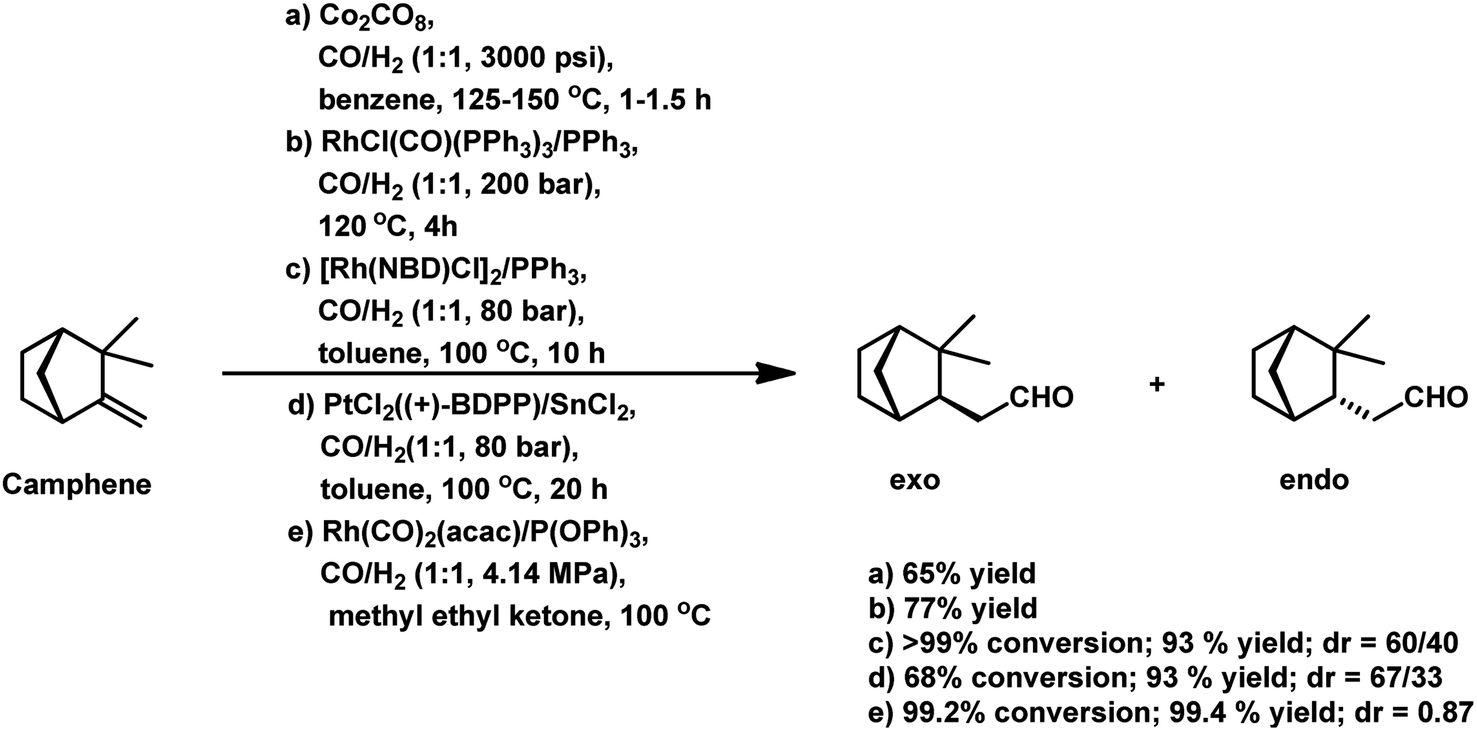
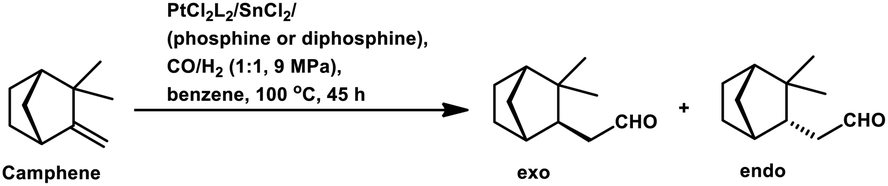
Employing PtCl2((+)-BDPP)/SnCl2, a conversion of 68% was obtained together with a yield of 93% for aldehydes.87 Alternatively, [Rh(NBD)CI]2/PPh3 appears to show nearly complete conversion with the same yield of aldehydes. However, despite its high activity, the latter resulted in a lower diastereoselectivity than the former (Scheme 9c and d). In addition, the PtCl2(PPh3 or diphosphine)/SnCl2/PPh3 system, where the diphosphines are dppe, dppp and dppb, led to the carbonylation of camphene with exclusive regioselectivity for the linear aldehyde, but poor yield.85 Moreover, the diastereoselectivity is also low; the cause of which is the similar steric hindrance on both faces of this substrate. The use of the PtCl2(PhCN)2/SnCl2/(chiral diphosphines) system increased the conversion and the diastereoselectivity for the exo aldehyde (Table 6).86,113 The diastereomeric excess of the exo product in these systems was up to 60% when the chiral diphosphines were (S,S)-DIOP (DIOP = (2,2-dimethyl-1,3-dioxolane-4,5-diyl)bis(methylene)bis(diphenylphosphine)), (R,R)-DIOP, ®-BINAP and (S)-BINAP.113 This fact simply means that the chiral, steric and electronic features of these ligands make it possible to distinguish between the two faces of camphene despite the small difference in their steric crowding.
|
|
||||
|---|---|---|---|---|
| Ligand (L) | Phosphine or diphosphine | Conversion (%) | Yield (%) | Ratio of diastereomers (exo/endo) |
| PPh3 | PPh3 | 40 | 89 | 52/42 |
| Dppe | PPh3 | 52 | 96 | 58/42 |
| dppp | PPh3 | 53 | 89 | 55/45 |
| Dppb | PPh3 | 49 | 92 | 57/43 |
| PhCN | — | 65 | 4 | — |
| Ph | ®(R)-BINAP | 92 | 84 | 80/20 |
| PhCN | (S)-BINAP | 88 | 80 | 74/26 |
| PhCN | (S,S)-DIOP | 100 | 33 | 70/30 |
| PhCN | (R,R)-DIOP | 100 | 34 | 62/38 |
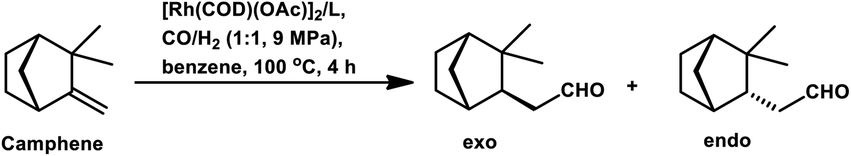
Differing from Pt/Sn systems, the modified homogeneous rhodium-catalysed systems show the dominance of the thermodynamically less stable endo aldehyde given that it is formed through a thermodynamically stable intermediate with an exo/endo ratio of approximately 1/1.5.102 The difference in the steric and electronic factors of the different phosphine and phosphite ligands does not have a significant impact on the diastereomeric excess of the endo aldehyde (Table 7).
|
|
||||
|---|---|---|---|---|
| Ligand | Cone angle (°) | Conversion (%) | Product distribution (%) | |
| Exo | Endo | |||
| None | — | 100 | 48 | 52 |
| PPh3 | 145 | 100 | 41 | 59 |
| P(p-OCH3Ph)3 | 145 | 44 | 41 | 59 |
| P(CH2Ph)3 | 160 | 81 | 39 | 61 |
| PCy3 | 170 | 16 | 40 | 60 |
| P(O-o-tBuPh)3 | 175 | 100 | 38 | 62 |
| P(C6F5)3 | 184 | 90 | 47 | 53 |
The surfactant CTAC acted as an inhibitor in the water/toluene biphasic system comprised of [Rh(COD)(μ-OAc)]2 and TPPTS even at concentrations as low as 2.5 × 10−3 M (Table 8).96 With an increase in the concentration of CTAC, the conversion decreased, while favouring the formation of more endo aldehyde. This inhibition can be attributed to the fact that camphene is a bulky substrate, which fails to assume the required rod-like or planar conformation, and thus its permeation through the surfactant array would result in an energetically unfavourable pathway.
|
|
|||
|---|---|---|---|
| [CTAC] (×10−3 M) | Conversion (%) | Product distribution (%) | |
| Exo | Endo | ||
| — | 71 | 43 | 57 |
| 2.5 | 65 | 36 | 64 |
| 12 | 50 | 35 | 65 |
| 25 | 39 | 37 | 63 |
| 35 | 35 | 33 | 67 |
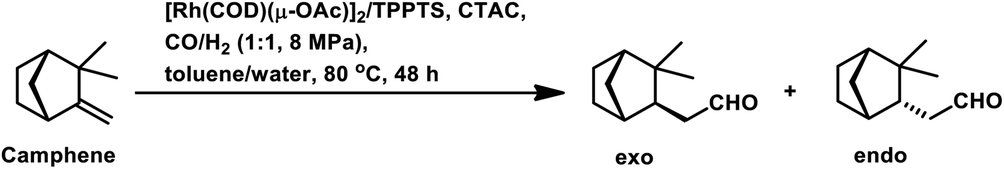
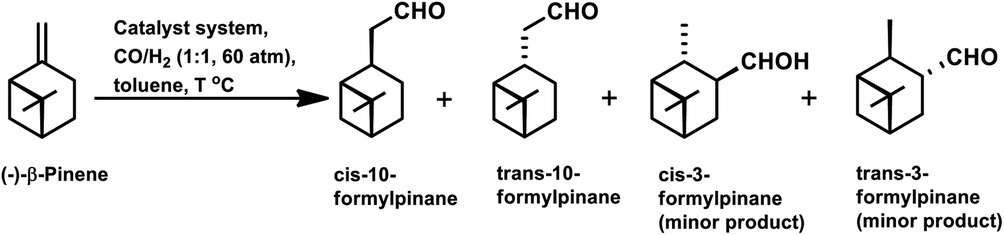
The hydroformylation of (−)-camphene occurred with 99.2% conversion and 99.4% selectivity for aldehydes with the Rh(CO)2(acac)/p(OPh)3 system in methyl ethyl ketone (Scheme 9e).114 In the temperature range of 90–110 °C, the rate is first order with respect to the substrate and the catalyst concentration, while complex orders and partial orders were found with respect to the pressure of CO and H2, respectively.115
The hydroformylation of the para-menthenic terpenes, α-terpinene, γ-terpinene and α-terpinolene (δ-terpinene) has seldom been proven to be an easy task due to the presence of sterically crowded double bonds. This reaction is less troublesome in the case of α-terpinene with conjugated double bonds than for γ-terpinene and α-terpinolene, which have non-conjugated double bonds. The endocyclic double bond of α-terpinolene reacted at 44 bar of syngas pressure with RhHCO(PPh3)3 as the catalyst precursor to give the corresponding aldehyde in a low yield of 25% (Scheme 10b).93 On replacing the catalyst with [Rh(2-ethylhexanoate)]/PPh3 and increasing the pressure to 260 bar, the aldehyde yield increased significantly in just 12 h (Scheme 10c).117 γ-Terpinene gave ca. 45% conversion and only 28% of aldehyde selectivity with Rh(CO)2(acac)/P(OPh)3.114
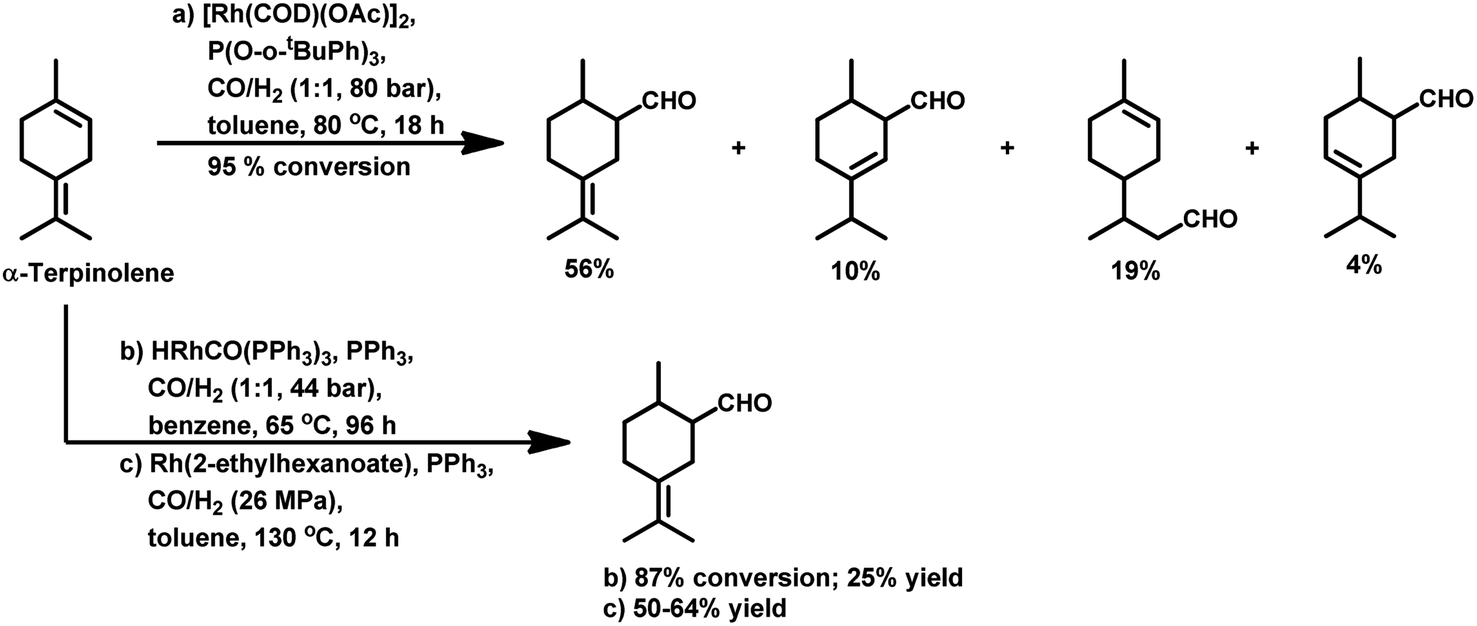 | ||
| Scheme 10 Hydroformylation of α-terpinolene. | ||
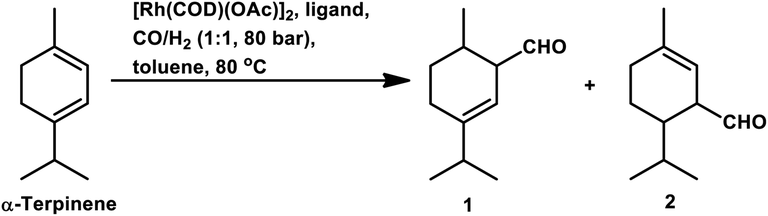
With the use of the [Rh(cOD)(OAc)]2/PPh3 catalytic system at 80 °C and 80 atm, 70% reaction conversion of α-terpinene occurred to yield 97% of two major aldehydes in 24 h when that P/Rh is equal to 20.94 α-Terpinolene is a cyclic conjugated diene and control of its hydroformylation is complicated given that it undergoes a slow, almost irreversible conversion of the active η1-allyl rhodium species to the highly stable, inactive η3-allyl rhodium complex. The η1-allyl rhodium complex yields either the aldehydes or the substrate hydrogenation product depending on the amount of P-donor auxiliary ligands (Scheme 11). Aldehyde formation is preferential at P/Rh ≥ 10, wherein, the CO insertion is faster than the reaction with H2. In contrast, at a low ligand concentration, the extent of hydrogenation of the substrate increases. Moreover, at low ligand concentrations, the reaction conversion is low but the selectivity for aldehydes is high, while it is the opposite at high ligand concentrations. Also, this substrate shows a product preference based on the cone angle of the ligand instead of its basicity; the proof being provided by the amount of substrate hydrogenation, which was 79% for PCy3, 23% for PBz3 and 7% for PPh3 (Table 9). Under the same reaction conditions, γ-terpinene and α-terpinolene only underwent 9% and 10% conversion, respectively, with a complex mixture of products from substrate isomerisation, substrate hydrogenation and hydroformylation.94
 | ||
| Scheme 11 Mechanism of the hydroformylation of α-terpinene. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Ligand | P/Rh | Time (h) | Conversion (%) | Selectivity (%) | ||
| 1 | 2 | Hydrogenated products | ||||
| PPh3 | 10 | 24 | 80 | 68 | 21 | 7 |
| PPh3 | 20 | 24 | 70 | 75 | 22 | 1 |
| PBz3 | 10 | 10 | 95 | 62 | 15 | 23 |
| PCy3 | 10 | 10 | 96 | 17 | 2 | 79 |
| P(O-o-tBuPh)3 | 20 | 7 | 96 | 70 | 17 | 10 |
Replacement of PPh3 with P(O-o-tBuPh)3 under the same conditions for the α-terpinene, γ-terpinene and α-terpinolene substrates gave nearly complete conversions with reduced substrate hydrogenation and 80–90% combined selectivity for aldehydes.118 α-Terpinene (Table 9) resulted in the formation of two main aldehydes due to the hydroformylation of its two conjugated endocyclic double bonds, while γ-terpinene (Scheme 12) and α-terpinolene (Scheme 10a) formed four aldehydes through the hydroformylation of their double bonds as well as their isomerisation. P(O-o-tBuPh)3 provided efficient results for all three substrates, given that it also prevented the formation of the η3-allyl rhodium species; thus, preventing the deactivation of the catalyst.
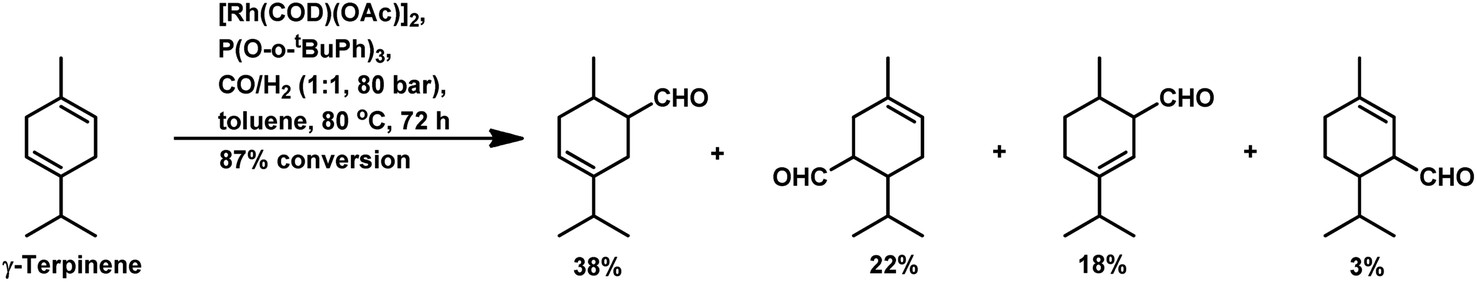 | ||
| Scheme 12 Hydroformylation of γ-terpinene. | ||
Under the same conditions in ethanolic solutions, the formation of diethyl acetals from the primarily formed aldehydes was observed in a one-pot process.16 As expected from previous reports, the reaction is much faster with P(O-o-tBuPh)3 than with PPh3 as the P-donor ligand. Furthermore, ethanol significantly accelerates both the hydroformylation and acetalisation of these three substrates compared to toluene. α-Terpinene (Scheme 13) showed a selectivity of 11% for aldehydes and 79% for acetals with nearly full conversion in ethanolic solutions. γ-Terpinene (Scheme 14) and α-terpinolene (Scheme 15) showed a selectivity of up to 80% and 70% for the associated acetals, respectively. α-Terpinol reacted with unmodified Rh(CO)2(acac) catalyst at 69 bars to yield a mixture of three hydroxy aldehydes (Scheme 16).119
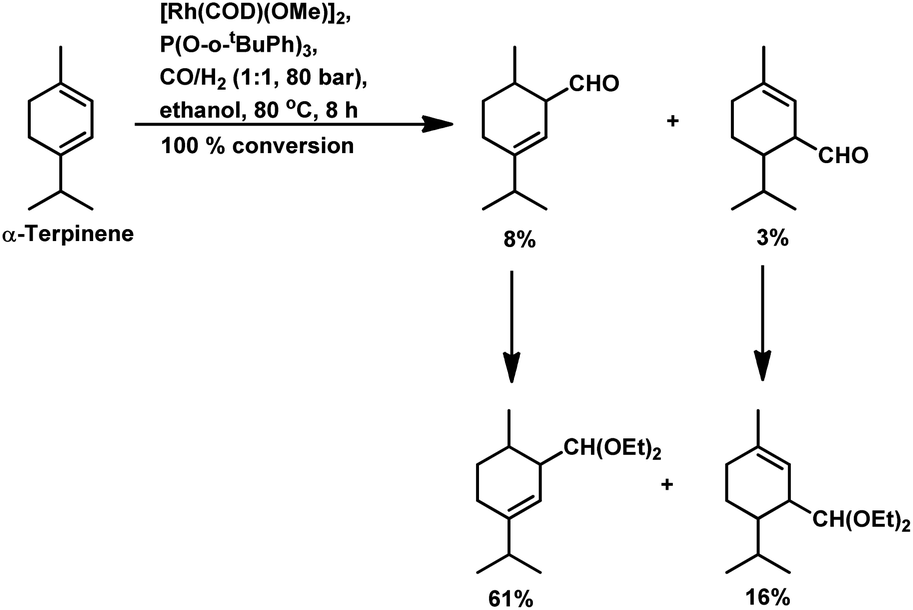 | ||
| Scheme 13 Hydroformylation/acetalisation of α-terpinene. | ||
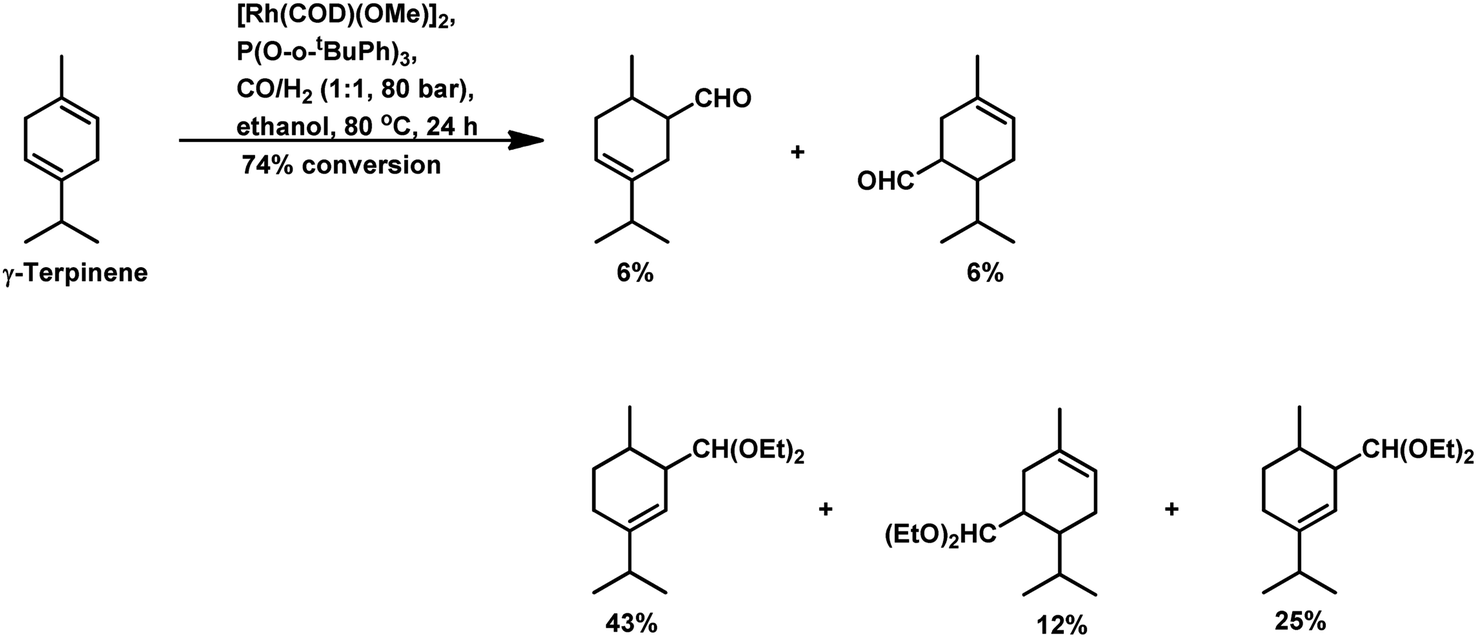 | ||
| Scheme 14 Hydroformylation/acetalisation of γ-terpinene. | ||
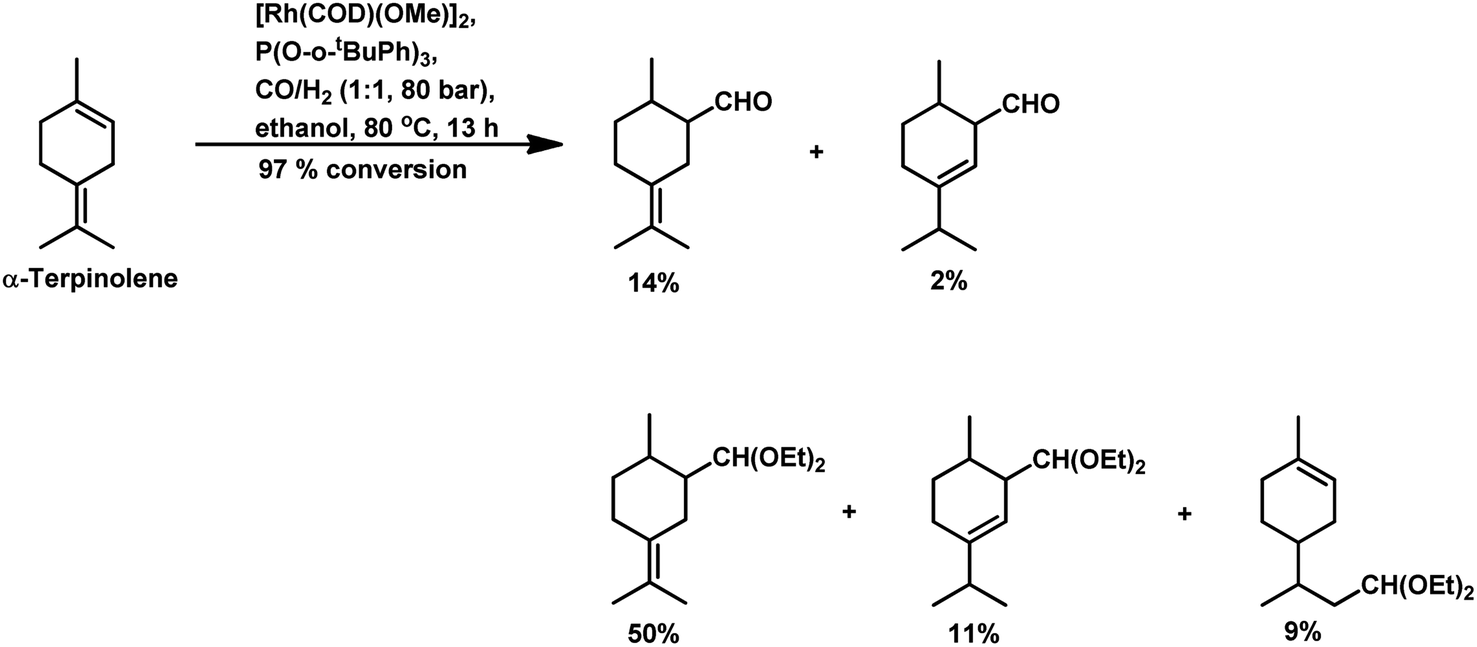 | ||
| Scheme 15 Hydroformylation/acetalisation of α-terpinolene. | ||
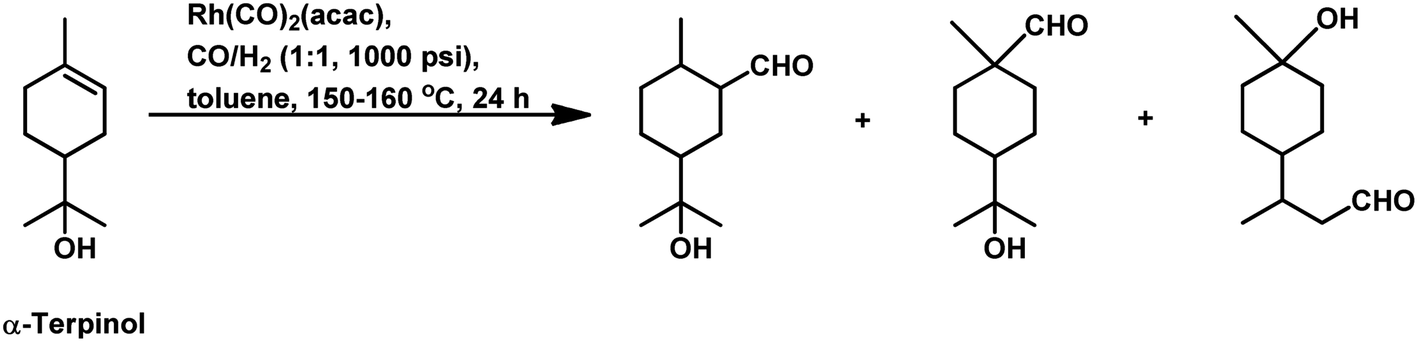 | ||
| Scheme 16 Hydroformylation of α-terpinol. | ||
Benaissa et al. performed the kinetic study of linalool hydroformylation using SAPC with [Rh2(μ-S-tBu)2(CO)2(TPPTS)2] supported on silica DS50 as the catalytic system in the temperature range of 333–363 K in toluene solutions.123 Under these conditions, the rate was observed to be first order with respect to the concentration of linalool and the catalyst and the syngas pressure together with 14.5 kcal mol−1 activation energy.
On employing [Rh(cOD)(OAc)]2 as a catalyst precursor with different phosphines, linalool underwent hydroformylation, followed by concomitant hemiacetal formation through the intramolecular cyclisation of the initially formed hydroxy-aldehyde.124 The unmodified system showed an extremely low conversion of 12%; however, PPh3 and diphosphines resulted in high conversion with good chemo- and stereo-selectivity for the cis and trans isomers of hemiacetal. The reason for this is that the substrate forms chelates due to the simultaneous interaction of the double bond and the hydroxy group with the rhodium species, while the presence of ligands leads to the breaking of these chelates. On increasing the P/Rh ratio and temperature, the cis isomer increases, while an increase in the hydrogen pressure or the total pressure favours the trans isomer. The diphosphine ligands with small bite angles, namely, dppe, dppp, and dppb, preferentially form the cis isomer, while PPh3 or the absence of ligands prefer the trans isomer (Table 10).
|
|
||||
|---|---|---|---|---|
| Ligand | Time (h) | Conversion (%) | Selectivity (%) | Cis/trans |
| None | 6 | 12 | 85 | 1/2.2 |
| PPh3 | 6 | 100 | 93 | 1/1.3 |
| dppe | 12 | 100 | 73 | 2.0/1 |
| dppp | 12 | 100 | 70 | 3.1/1 |
| dppb | 12 | 90 | 60 | 2.2/1 |
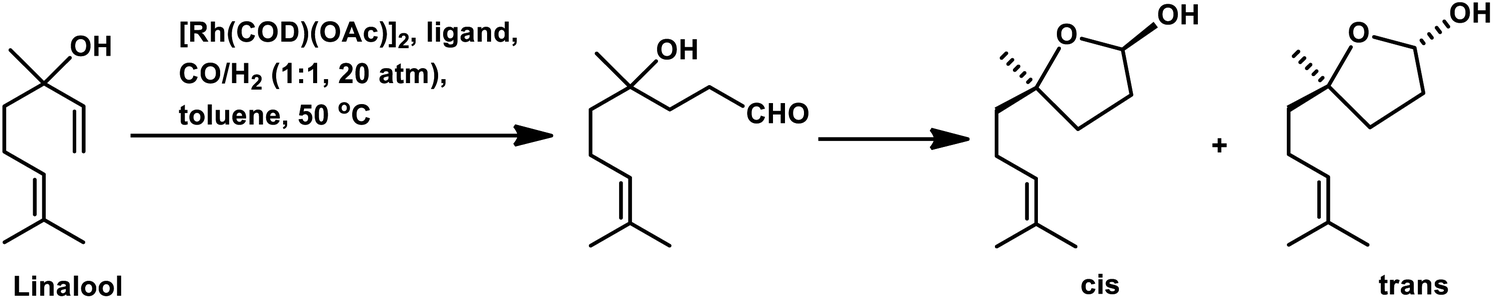
Also, when P(O-o-tBuPh)3 was used in place of PPh3 as the ligand with [Rh(COD)(OMe)]2 as the catalyst precursor, the rate increased by ca. 10 times in toluene and ca. 5 times in ethanol together with a switch in the diastereoselectivity, wherein the former favoured the cis isomer, while the latter favoured the trans isomer in toluene at 40 °C and 20 bar of pressure.125 Also, a shift from the conventional solvent toluene to the sustainable solvent ethanol accelerated the reaction by ca. 2 times under similar reaction conditions. With an increase in the phosphine/phosphite concentration, the reaction was accelerated up to the P/Rh ratio of 10 in toluene, above which the rate either remained constant or started declining due to the ligand-substrate competition for the active site of the metal. The exclusively obtained product in ethanolic solutions was a mixture of cyclic acetals instead of the hemiacetals obtained in the toluene solutions (Scheme 17). This acetal is supposed to be formed from etherification of the initially formed hemiacetal in ethanol without any additional acid as a co-catalyst. Similar results were found with the use of the Rh/TPPTS/CTAC aqueous biphasic catalytic system, wherein the hemiacetal was formed as the major product via the intramolecular cyclisation of the hydroxy aldehyde.126
 | ||
| Scheme 17 Tandem hydroformylation/acetalisation of linalool. | ||
The kinetics of the hydroformylation of myrcene contradicts with the trends observed in the case of simple olefins.94 With an increase in the concentration and basicity of the phosphorous ligand and the pressure of CO and hydrogen, the rate of hydroformylation increased, revealing that the most significant step in this mechanism is the formation of η1-complexes from the less reactive η3-allylrhodium species.
In 1991, Chalchat et al. reported the hydroformylation of myrcene using RhHCO(PPh3)3 as the catalyst and PPh3 as the ligand to obtain 3-ethyl-7-methyloct-6-enal as the major product from the reaction of the disubstituted double bond.93 It is rather surprising that myrcene preferentially undergoes hydroformylation at the disubstituted double bond instead of the otherwise expected monosubstituted double bond. In the [Rh(COD)OAc]2/PPh3 catalytic system, an increase in the phosphine ligand increased the reaction rate, making the reaction feasible with mild conditions of temperature and pressure. On increasing the P/Rh ration from 2 to 40, the reaction time decreased by four times, while the conversion was pushed toward completion.91
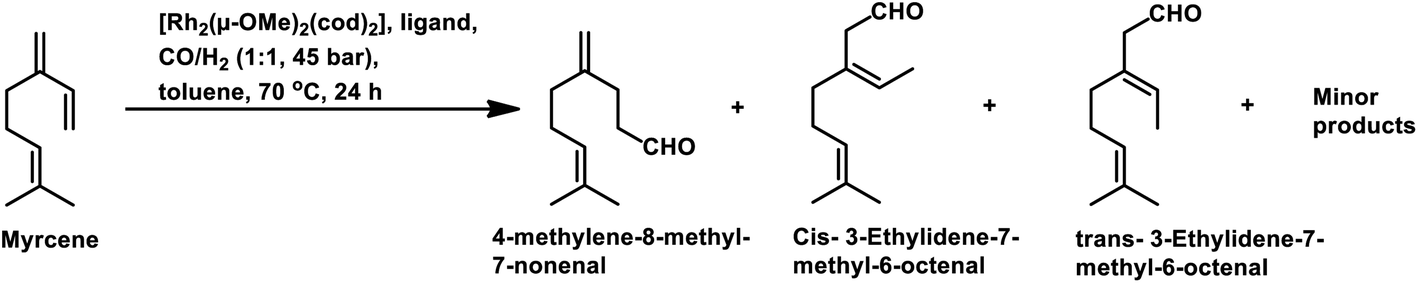
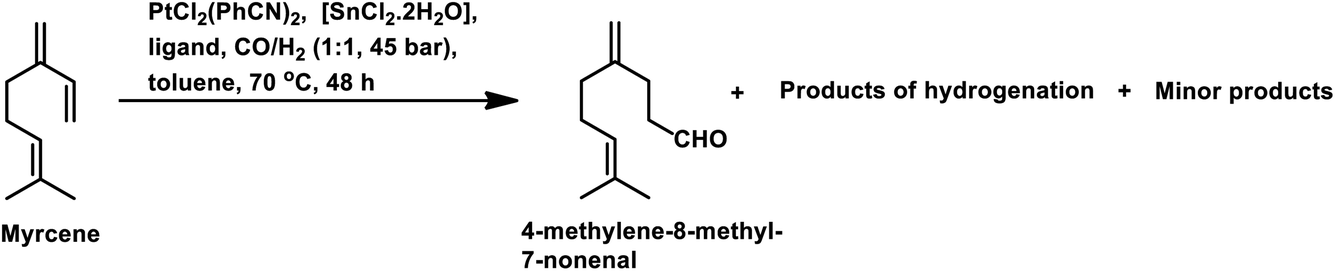
The Rh- and Pt/Sn-catalysed hydroformylation of this substrate using various phosphine and diphosphine ligands gives nine major products, usually mono- and di-aldehydes through the formation of n-alkyl and η3-allyl intermediates (Scheme 18).129 The Rh/Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) system yielded 4-methylene-8-methyl-7-nonenal as the major product but with BISBI (2,2′-bis(diphenylphosphino)methyl)-1,1′-biphenyl), which has a more flexible backbone, cis- and trans-3-ethylidene-7-methyl-6-octenal were formed as the major products. In the presence of diphosphine ligands such as dppp, dppe, dppb and monophosphine ligand, PPh3 majorly formed cis- and trans-3-ethylidene-7-methyl-6-octenal in a rhodium-catalysed system (Table 11). The Pt/Sn system also gave 4-methylene-8-methyl-7-nonenal as the major product with no trace of cis- or trans-3-ethylidene-7-methyl-6-octenal, irrespective of the type of phosphine ligand used. PPh3 gave a higher rate of reaction and less conversion to hydrogenated products than diphosphine ligands, dppe, dppp, dppb and NAPHOS (2,2-bis[(diphenylphosphino)methyl]-1,1-binaphthyl) (Table 12). A synergistic effect was observed between Pt and SnCl2 given that no transformation occurred for the platinum complexes in the absence of SnCl2.
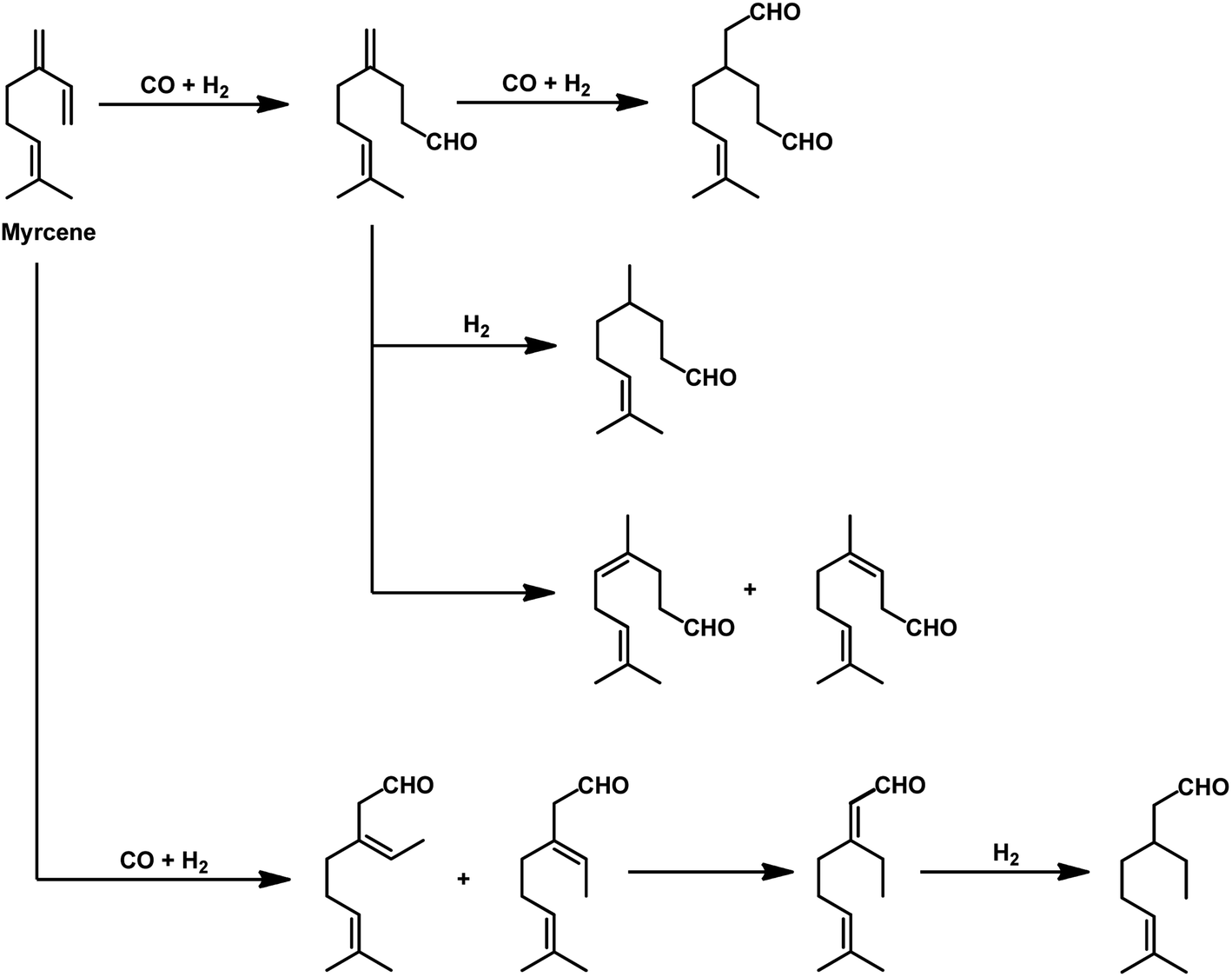 | ||
| Scheme 18 Products in the hydroformylation of myrcene. | ||
|
|
|||
|---|---|---|---|
| Ligand | Conversion (%) | 4-Methylene-8-methyl-7-nonenal | Cis- and trans-3-ethylidene-7-methyl-6-octenal |
| PPh3 | 99 | 6 | 73 |
| Dppe | 94 | 9 | 73 |
| Dppp | 43 | 12 | 78 |
| Dppb | 75 | 31 | 63 |
| BISBI | 99 | 17 | 77 |
| Xantphos | 64 | 62 | 28 |
|
|
|||
|---|---|---|---|
| Ligand | Conversion (%) | 4-Methylene-8-methyl-7-nonenal | Products of hydrogenation (%) |
| PPh3 (80 °C) | 55 | 74 | 7 |
| Dppe | 28 | 21 | 62 |
| Dppp | 30 | 32 | 47 |
| Dppb | 28 | 34 | 47 |
| NAPHOS (80 °C) | 15 | 54 | 16 |
The aqueous biphasic system using the surfactant CTAC provided 96% conversion and reduced yields of hydrogenated products compared to the aforementioned homogeneous systems in the hydroformylation of myrcene (Scheme 19).96 The reaction only worked in the presence of CTAC and the rate of conversion increased with an increase in CTAC concentration up to 2.5 × 10−3 M, and then decrease with a further increase in its concentration.
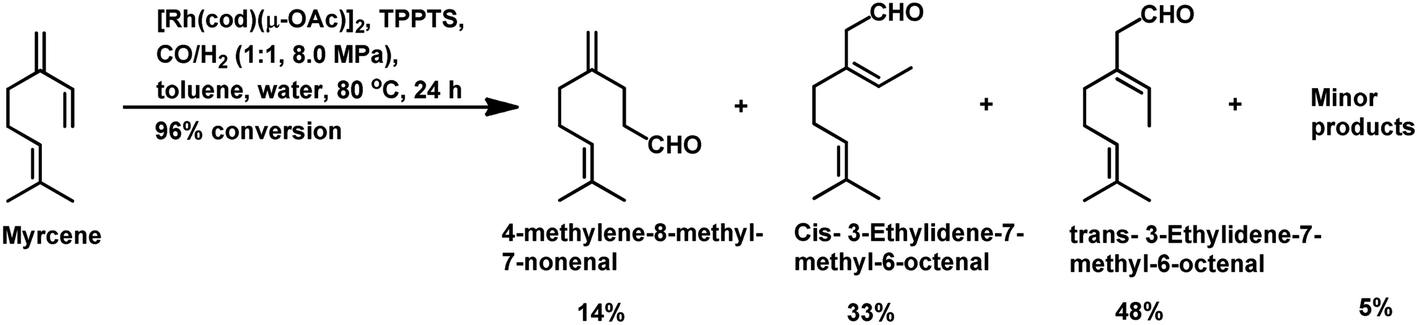 | ||
| Scheme 19 Hydroformylation of myrcene in aqueous biphasic system. | ||
Citronellene was shown to undergo rhodium-catalysed hydroformylation at the terminal double bond at a high syngas pressure of 600 bar to exclusively form the corresponding linear aldehyde (Scheme 20a).109 In 2000, Waldvogel et al. reported the regioselective hydroformylation of citronellene with 95% selectivity for the linear aldehyde by using the novel bidentate diphosphine ligand N,N-bis-(P-(phosphabicyclo[3.3.1]nonan)-methyl)aniline with Rh(COD)BF4 as the catalyst system in toluene at 100 bar pressure of syngas (Scheme 20b).130
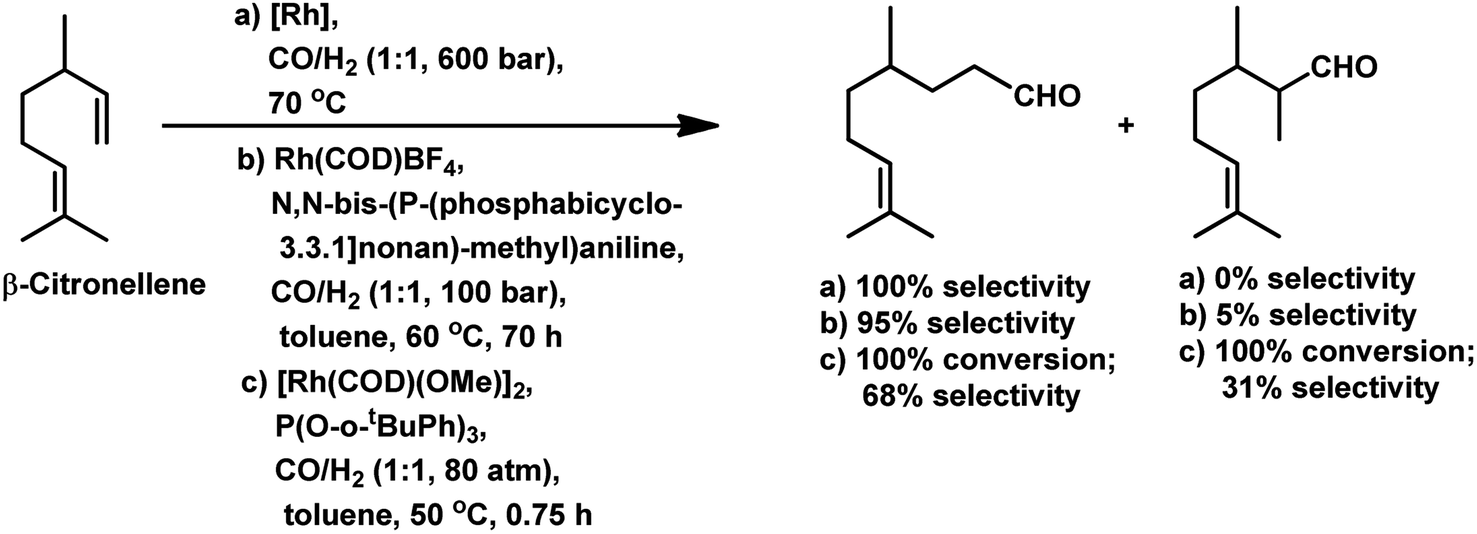 | ||
| Scheme 20 Hydroformylation of β-citronellene. | ||
The hydroformylation of β-citronellene resulted in the formation of a linear aldehyde and a quantitative mixture of two diastereomers of the branched aldehyde in toluene solutions. The unmodified catalyst system resulted in faster conversion but a lower selectivity.125 In contrast, PPh3 resulted in a lower conversion rate but higher selectivity (Scheme 20c). This problem was fixed with the employment of the bulky phosphite ligand P(O-o-tBuPh)3, which accelerated the reaction by 2.5 times compared to the unmodified system and more than 10 times compared to PPh3. The reason for this is the large cone angle of this phosphite ligand, which supported the formation of only monoligand rhodium species, and the special electronic factors, leading to the easy dissociation of CO. On replacement of toluene with ethanol, either exclusively aldehydes or aldehyde-acetal mixture was formed based on the reaction temperature. This reaction was twice as fast in ethanol than in toluene (Scheme 21). Also, it proceeded in an aqueous biphasic system with the use of a water-soluble ligand, TPPTS, and a phase transfer agent, CTAC, to give up to 87% conversion and 100% aldehyde selectivity with an n/iso ratio of 9.126
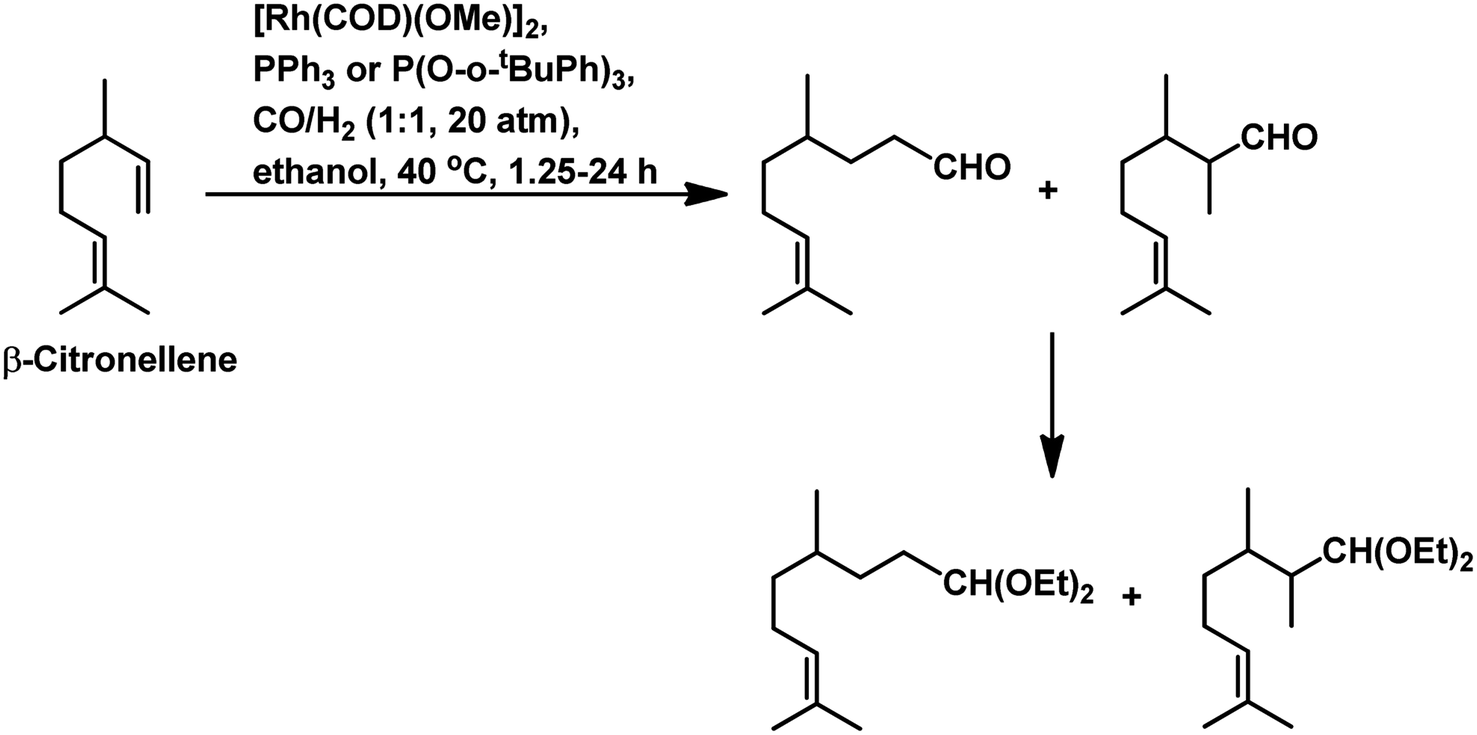 | ||
| Scheme 21 One-pot hydroformylation/acetalisation of β-citronellene. | ||
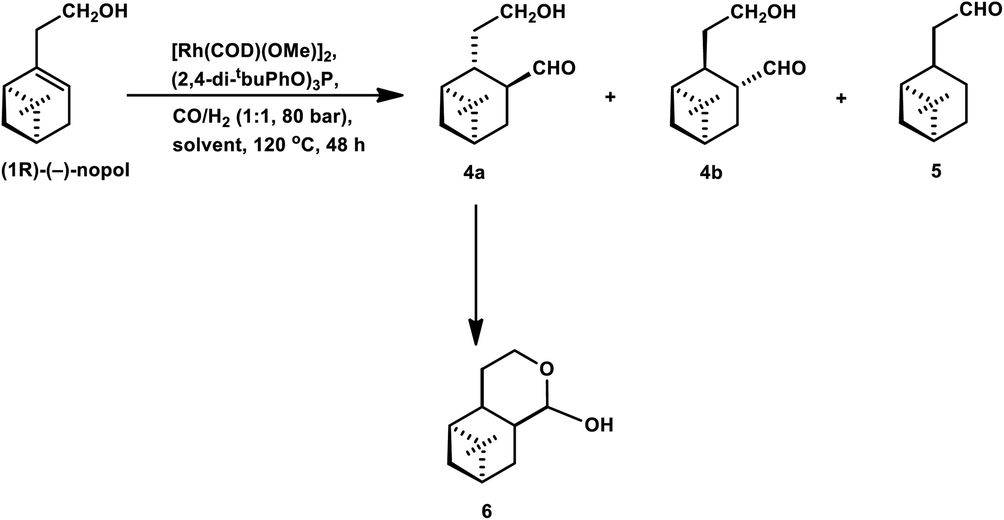
The hydroformylation of the hydroxyolefins (1R)-(−)-myrtenol and (1R)-(−)-nopol is quite challenging given that double bonds in both substrates are sterically hindered together with a close-lying hydroxy group, hindering their interaction with the rhodium catalyst system.132 Also, both substrates undergo isomerisation to form the corresponding saturated aldehydes on exposure to hydroformylation conditions.
(−)-Myrtenol catalysed by [Rh(μ-S-tBu)(CO){P(OPh)3}]2 yielded a hydroxy aldehyde, which underwent intramolecular hemiacetalisation to form a tetrahydrofuran derivative upon flash chromatography on silica gel (Scheme 22).18 In the hydroformylation of myrtenol, no conversion occurred with the unmodified rhodium catalyst or with PPh3 as the ligand. Nevertheless, the reaction proceeded with the use of the bulky phosphite ligand (2,4-di-tBuPhO)3P with a large cone angle given that it discouraged the formation of bis- or tris-phosphite rhodium species, providing an opportunity to the olefin for binding on the active site of the metal. Additionally, this phosphite ligand is weakly electron donating but strongly electron withdrawing, supporting the dissociation of CO from and association of the olefin to the catalyst. Myrtenol produced two diastereomers, resulting from the hydroformylation of the olefin with formyl group at the least sterically hindered carbon (as the major product), together with myrtanal (from isomerisation) and a small amount of cyclic acetal obtained from the acetalisation of the major product. This hydroformylation is also feasible in environmentally friendly solvents such as DEC, ethanol and MeTHF, with DEC proving to be the best alternative to toluene and providing a higher yield of the total hydroformylated products (Table 13).132
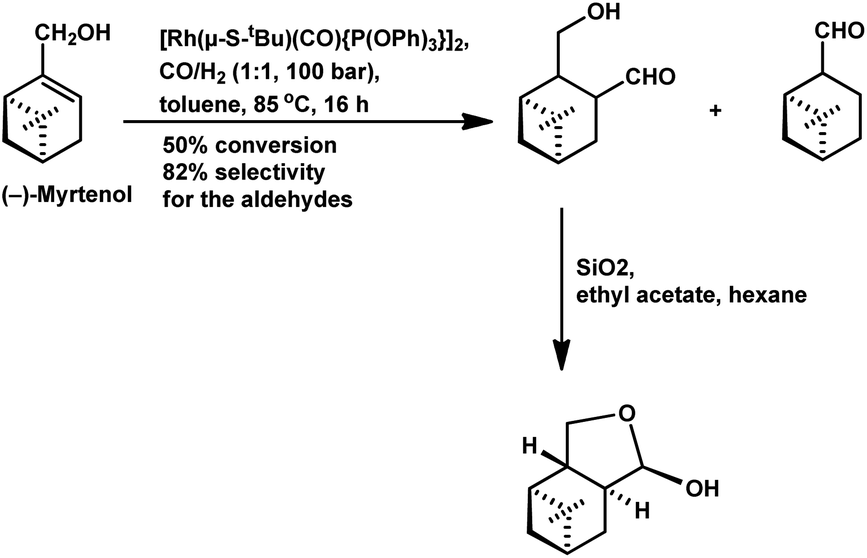 | ||
| Scheme 22 Hydroformylation of (−)-myrtenol. | ||
|
|
||||
|---|---|---|---|---|
| Solvent | Conversion (%) | Selectivity (%) | ||
| 1 (1a + 1b) | 2 | 3 | ||
| Toluene | 58 | 56 | 42 | 2 |
| DEC | 58 | 65 | 29 | 5 |
| Ethanol | 50 | 30 | 19 | 1 (+50% diethylacetals) |
| MeTHF | 63 | 31 | 69 | 0 |
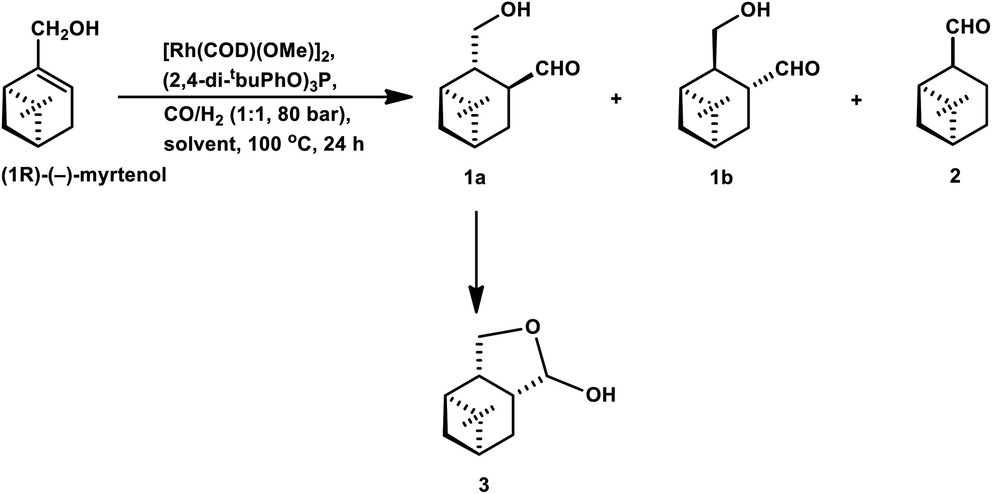
Similar results were obtained in the hydroformylation of nopol, albeit its reactivity is even lower than myrtenol.132 Also, nopol generated diastereomeric aldehyde (from hydroformylation) as the major product together with the saturated aldehyde (from isomerisation) and a small amount of cyclic acetal. DEC gave a higher rate of conversion than toluene (Table 14).
|
|
||||
|---|---|---|---|---|
| Solvent | Conversion (%) | Selectivity (%) | ||
| 1 (1a + 1b) | ||||
| Toluene | 46 | 70 | 22 | 8 |
| DEC | 57 | 73 | 16 | 10 |
| Ethanol | 47 | 22 | tr | 13 (+65% of diethylacetals) |
| MeTHF | 39 | 52 | 27 | 15 |
The concomitant hydroformylation/acetalisation of myrtenol and nopol occurred when Rh/(2,4-di-tBuPhO)3P/Ac2O was employed at a high syngas pressure of 800 bar, giving the corresponding acylated aldehydes (Scheme 23).133
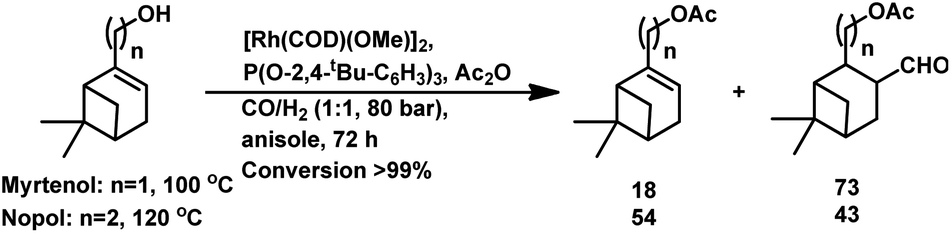 | ||
| Scheme 23 Hydroformylation/acetalisation of myrtenol and nopol. | ||
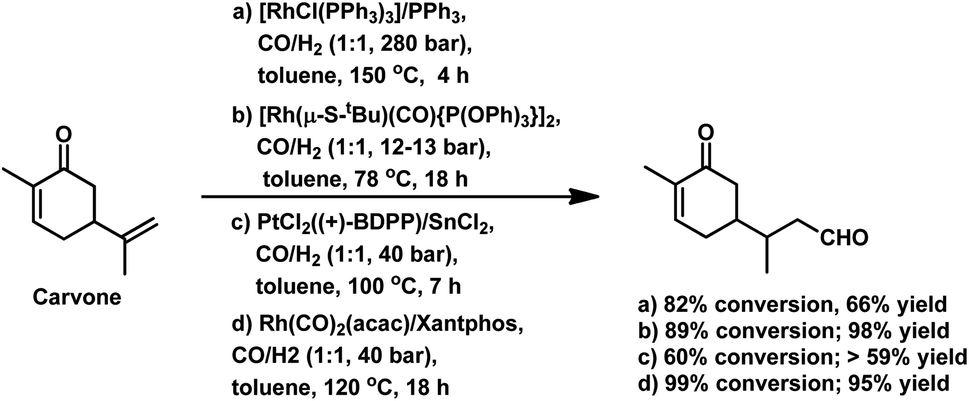
The monometallic rhodium-catalysed hydroformylation of carvone occurs under both harsh and mild conditions to preferentially produce the corresponding linear aldehyde with different conversions and yields (Scheme 24a and b), respectively.135,136 Dihydrocarvone is hydroformylated under similar harsh conditions (Scheme 25).135 With ®-carvone in a Rh/Xantphos catalytic system, the conversion reached as high as 99% and the yield reached 95% (Scheme 24d).137
 | ||
| Scheme 24 Hydroformylation of carvone. | ||
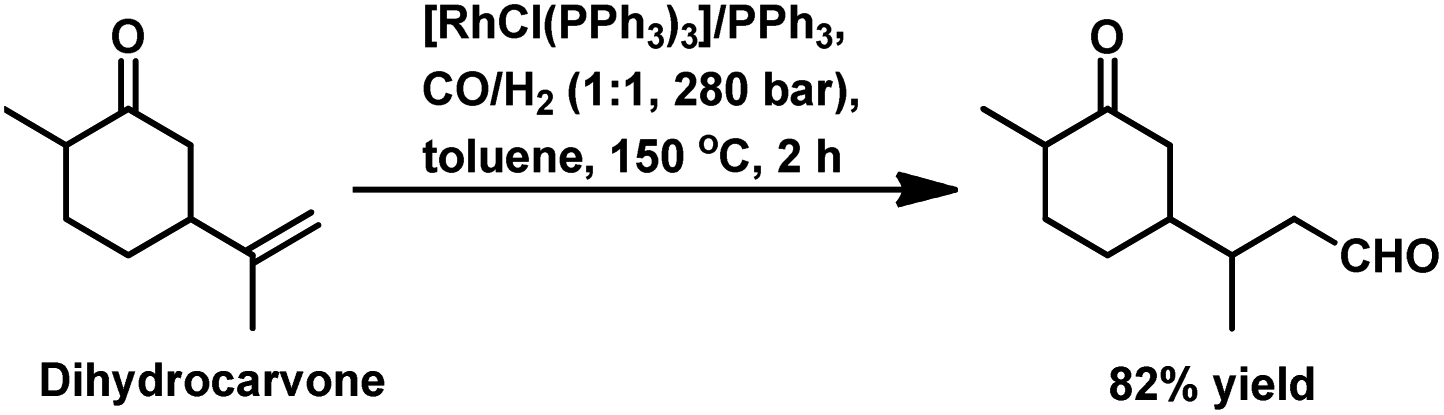 | ||
| Scheme 25 Hydroformylation of dihydrocarvone. | ||
The bimetallic Pt/Sn catalytic system with different biphosphines was found to be highly regioselective for the hydroformylation of (−)-R-carvone, exclusively yielding the corresponding linear aldehyde with no trace of the branched aldehyde and a conversion of up to 60% (Scheme 24c).84 The diastereoselectivity of the system depends on the isomer of chiral biphosphines used, in which case (+)-BDPP shows selectivity toward one regioisomer, while (−)-BDPP shows selectivity toward the other regioisomer of the product.
The synthesis of 1,5-dimethylhexahydro-1H-inden-4(2H)-one utilised in the formation of the sesquiterpene alcohol tamariscol was reported by Bhagade et al. to take place fr®(R)-carvone and dihydrocarvone through a sustainable domino multistep process.138 The first step in this process is the hydroformylation of the terminal double bond of the substrate, followed by the hydrogenation of the endocyclic double bond, which undergoes an intramolecular keto-aldol condensation catalysed by PPTS, forming an enone. Subsequently, the newly formed α,β-unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the enone gets hydrogenated to give the final product (Scheme 26). An undesired side-product in this process is the complete hydrogenation of the alkene double bonds in the substrate to give tetrahydrocarvone. The use of the acidic catalyst PPTS and dppp as the phosphine ancillary was found to be crucial for the activity of this system. Dihydrocarvone shows a slow conversion rate given that it retards the catalytic activity for hydroformylation due to its less planar carbon backbone, which generates steric hindrance.
C double bond in the enone gets hydrogenated to give the final product (Scheme 26). An undesired side-product in this process is the complete hydrogenation of the alkene double bonds in the substrate to give tetrahydrocarvone. The use of the acidic catalyst PPTS and dppp as the phosphine ancillary was found to be crucial for the activity of this system. Dihydrocarvone shows a slow conversion rate given that it retards the catalytic activity for hydroformylation due to its less planar carbon backbone, which generates steric hindrance.
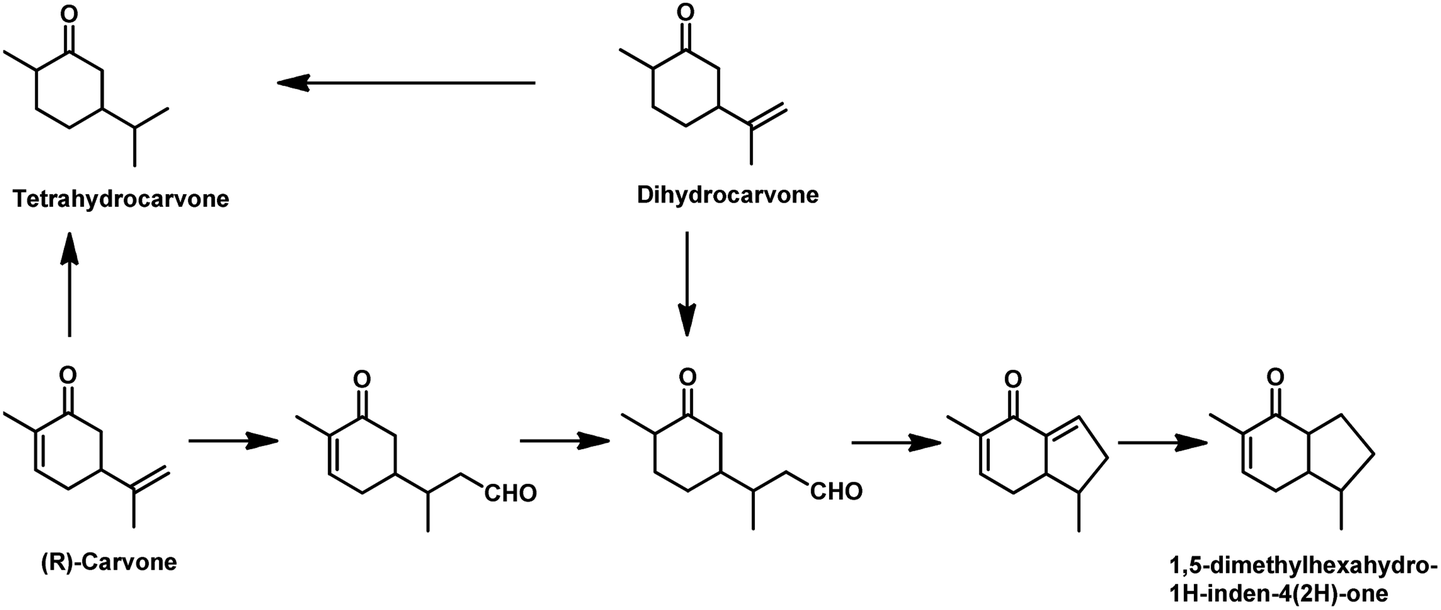 | ||
| Scheme 26 Synthesis of 1,5-dimethylhexahydro-1H-inden-4(2H)-one®om (R)-carvone. Reaction conditions: Rh(CO)2(acac), dppp, PPTS, CO/H2 (1:1, 650 psi), toluene, 120 °C, 22 h. | ||
There are only a few reports available on the hydroformylation of carenes given that they contain sterically encumbered endocyclic double bonds, making them reluctant toward this reaction. The initial studies carried out by Siegel et al. disclosed that (+)-3-carene undergoes hydroformylation at extreme syngas pressures to yield (+)-2-caranecarbaldehyde with 60–65% selectivity (Scheme 27e).109 Nevertheless, the hydroformylation of both 2-carene and 3-carene is found to be possible under mild conditions of pressure and temperature, with the former reacting faster than the latter.110 In unmodified systems, the reaction proceeds to give a mixture of aldehydes and alcohols with a low conversion of only 47%. The use of diphosphines as well as phosphines such as P(OPh)3 and PPh3 decreases the substrate isomerisation but simultaneously also retards the catalytic activity for the hydroformylation. Replacing these ligands with P(O-o-tBuPh)3 prevents isomerisation and favourably accelerates hydroformylation. The reason for this is its weak electron-donating and strong electron-withdrawing nature, facilitating the dissociation of CO, and thus making room for the binding of the alkene with the rhodium species. This ligand also works successfully at high P/Rh ratios of up to 20 without forming inactive bis and tris(ligand) rhodium species due to its large cone angle of 175°. This system yielded products with high regio- and chemo-selectivity and ca. 100% diastereoselectivity toward the trans isomers (Scheme 27c and d). On replacing ethanol with benzene under similar reaction conditions, a tandem hydroformylation/acetalisation reaction occurred, yielding a mixture of aldehydes and the corresponding diethyl acetals without the need for a co-catalyst (Scheme 27a and b).107
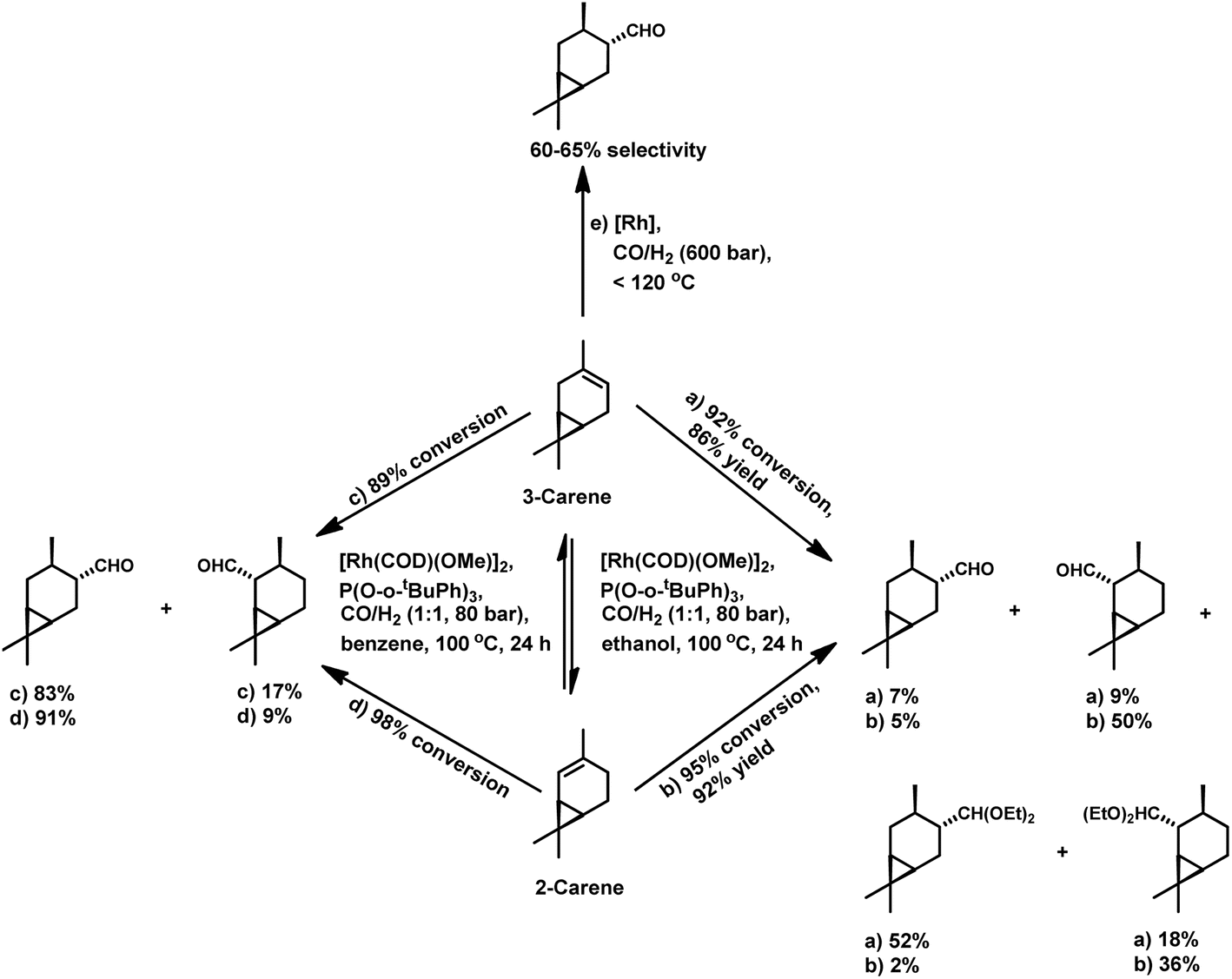 | ||
| Scheme 27 Hydroformylation/hemiacetalisation and hydroformylation/acetalisation of nerolidol. Reaction conditions: [Rh(COD)(OMe)]2/(PPh3 or P(O-o-tBuPh)3), CO/H2 (1:1, 20 atm), toluene or ethanol, 40–50 °C, 1–24 h. | ||
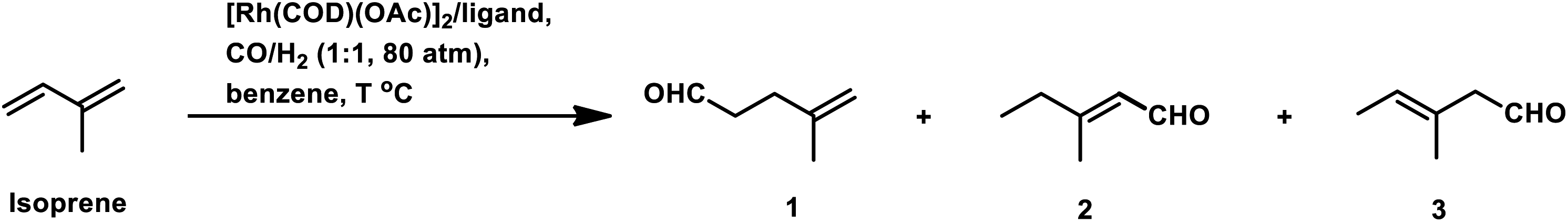
Isoprene was hydroformylated at high pressures of 3000–4000 psi with Co2(CO)8 catalyst to give a complicated mixture of products (Scheme 28a).143 This reaction was retarded by the formation of π-allyl complexes with cobalt species.144 The use of the rhodium-based chiral system HRhCO(PPh3)3/(−)-DIOP resulted in (S)-3-methylpentanal as the major product with 32% optical yield, together with other side-products (Scheme 28b).145 Cocondensated mesitylene-rhodium vapour/dppe as the catalytic system provided 70% conversion and 65% yield of the β,γ-unsaturated monoaldehydes (Scheme 28c).146 However, the appropriate tuning of the ratio of phosphine ligand to that of the rhodium catalyst precursor led to a high conversion percentage and rate, together with high selectivity for the aldehydes even under mild reaction conditions.91 Using the [Rh(COD)(OAc)]2/PPh3 catalytic system, the optimum P/Rh ratio at 80 °C was 40, resulting in full conversion and high chemoselectivity.
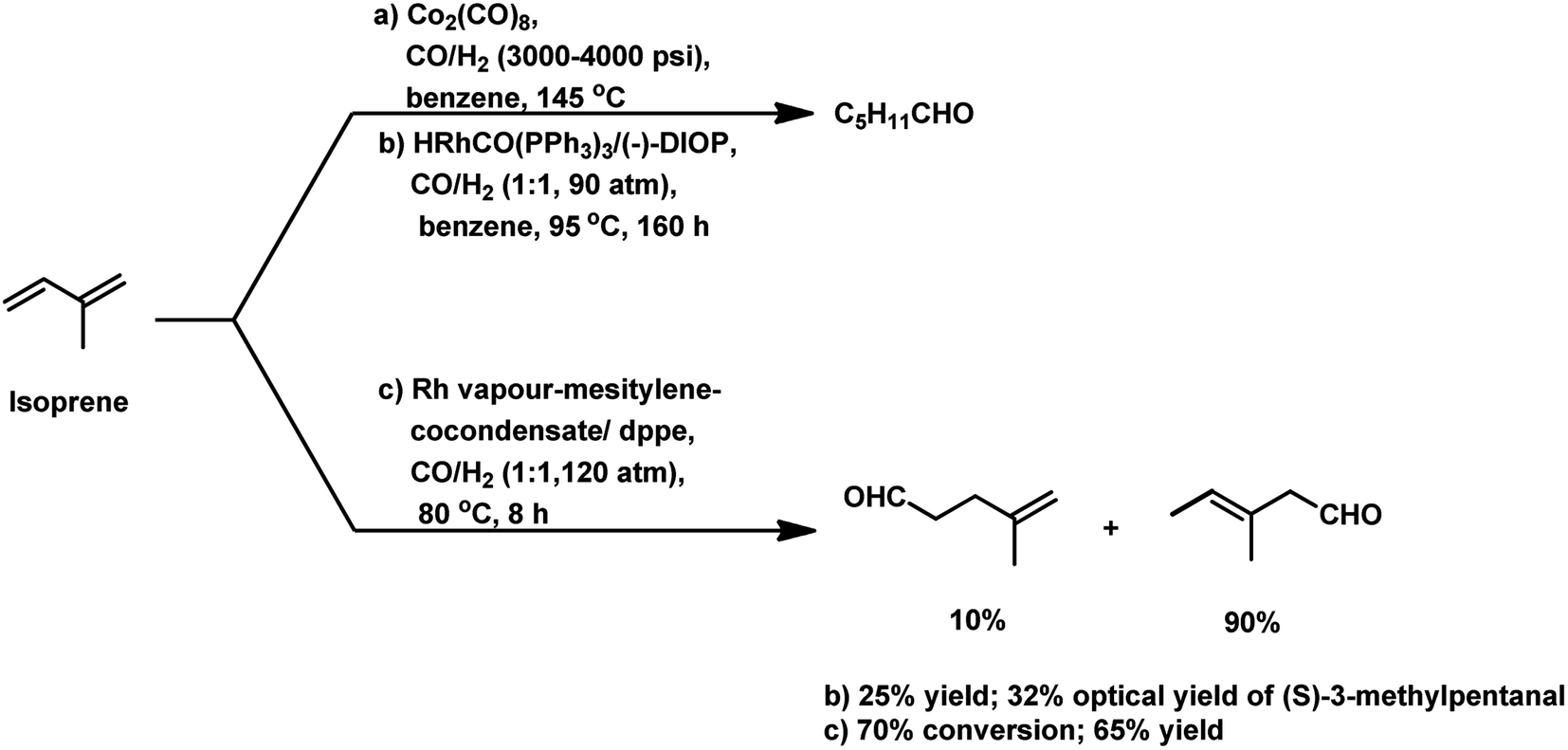 | ||
| Scheme 28 Hydroformylation of isoprene. | ||
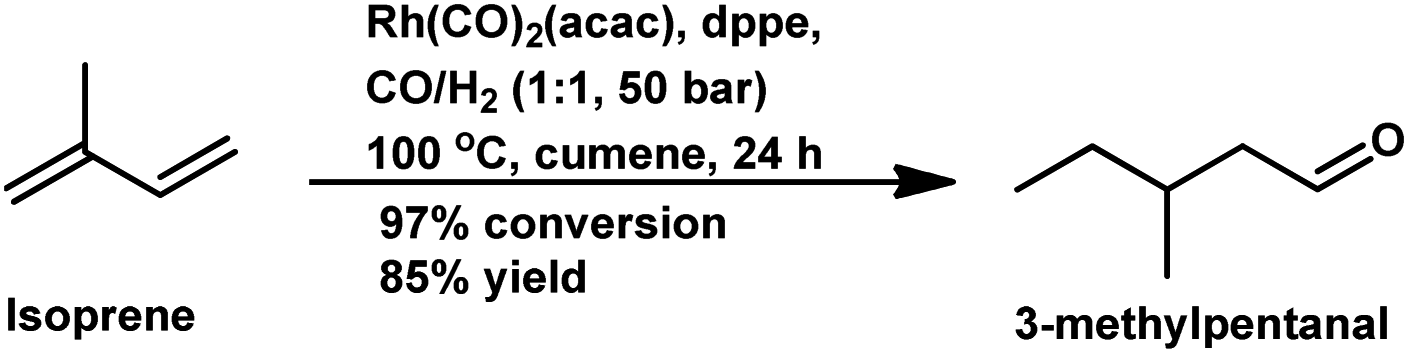
Besides the ligand concentration, the ligand basicity and syngas pressure also affect the hydroformylation of isoprene.147 By replacing PPh3 with diphosphine ligands such as dppe and dppb, the optimum P/Rh ratio was reduced to 10 although the selectivity was comparable (Table 15). Inconsistent with the usual trend, isoprene hydroformylation is first order with respect to both, H2 and CO. A hydroformylation/hydrogenation tandem reaction is observed with the Rh/dppe catalytic system to yield 3-methylpentanal in 85% yield with 50 bar pressure of syngas and cumene as the solvent at 100 °C (Scheme 29).148 Some stable (π-allyl)-closo-rhodacarboranes are extremely active in the hydroformylation of this substrate via CH3⋯Rh agnostic interactions to endowing the system with high conversion and selectivity.149
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Ligand | P/Rh | Temp. (°C) | Time (h) | Conversion (%) | Yield of aldehydes (%) | Product distribution (%) | ||
| 1 | 2 | 3 | ||||||
| PPh3 | 40 | 80 | 5 | 100 | 97 | 17 | 49 | 31 |
| PPh3 | 20 | 100 | 4 | 100 | 96 | 17 | 48 | 31 |
| dppe | 10 | 100 | 4 | 90 | 96 | 12 | 48 | 30 |
| dppb | 10 | 100 | 4 | 90 | 96 | 25 | 44 | 27 |
 | ||
| Scheme 29 Hydroformylation/hydrogenation of isoprene. | ||
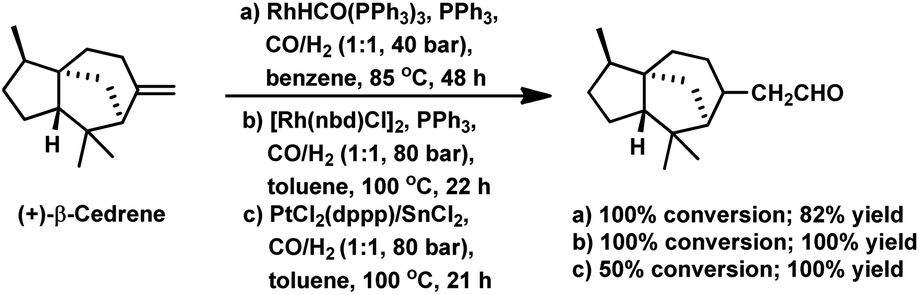
The hydroformylation of α- and β-cedrene was carried out by Chalchat et al. with RhHCO(PPh3)3 and it was found that the former was totally unreactive, while the latter gave full conversion with 82% yield (Scheme 30a).93 β-Cedrene gets hydroformylated with chemo- and regio-specificity, yielding exclusively the linear aldehyde upon the use of either, bimetallic Pt/Sn or rhodium catalytic systems (Scheme 30c).87 However, the Pt/Sn system showed a low conversion, while the Rh catalytic system showed full conversion.
 | ||
| Scheme 30 Hydroformylation of β-cedrene. | ||
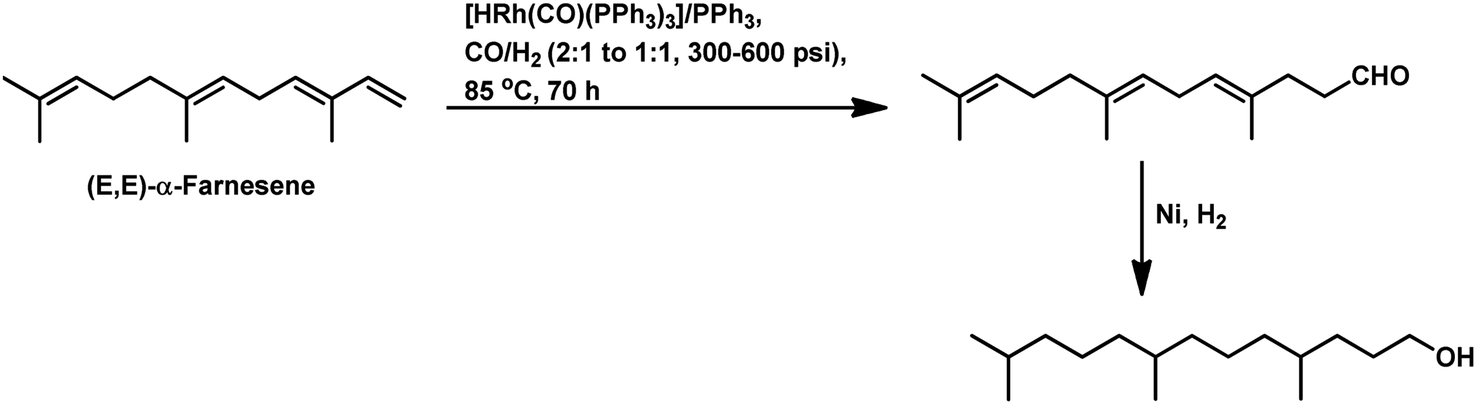 | ||
| Scheme 31 Hydroformylation of α-farnesene. | ||
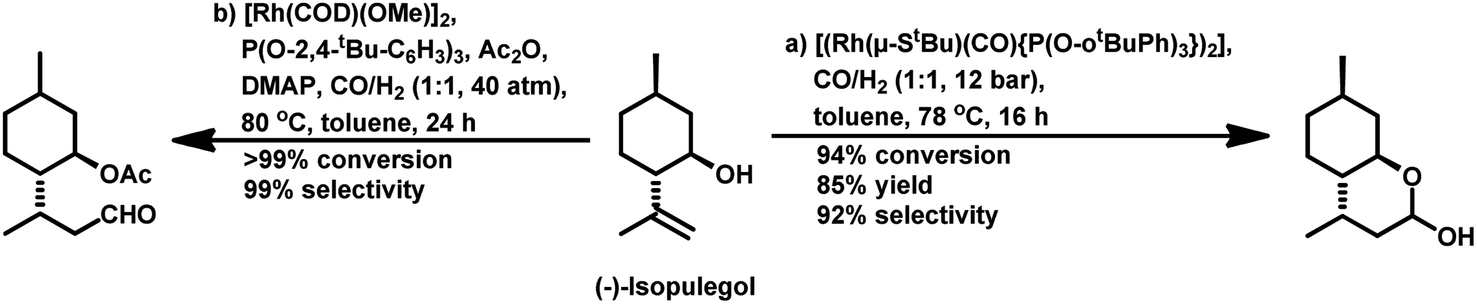 | ||
| Scheme 32 Hydroformylation/hemiacetalisation and hydroformylation/acetalisation of (−)-isopulegol. | ||
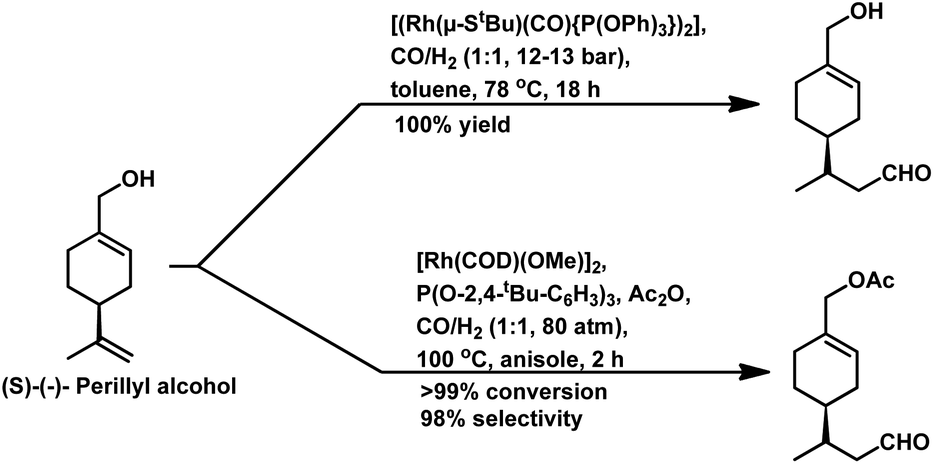 | ||
| Scheme 33 Hydroformylation of perillyl alcohol. | ||
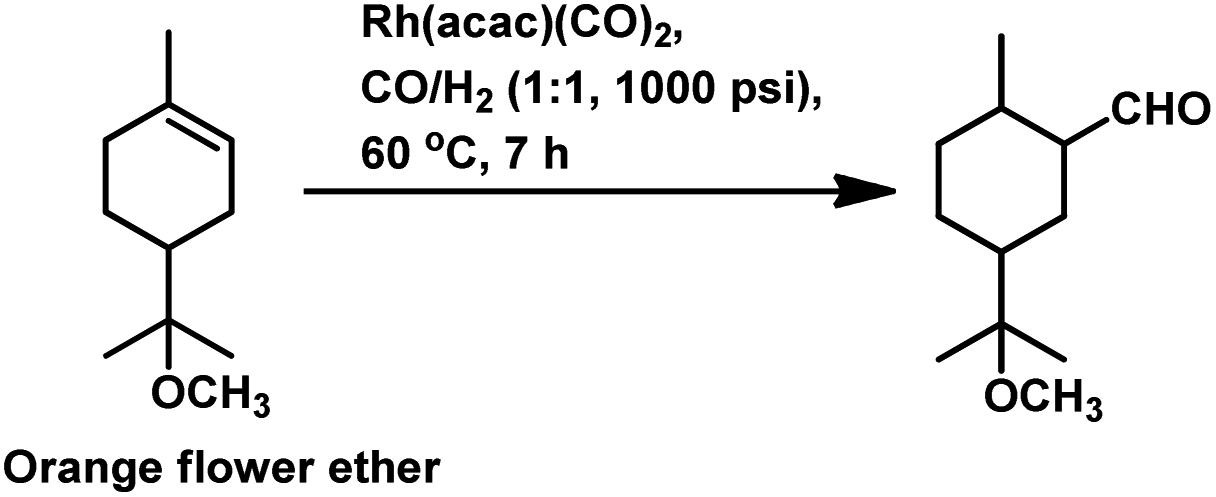 | ||
| Scheme 34 Hydroformylation of orange flower ether. | ||
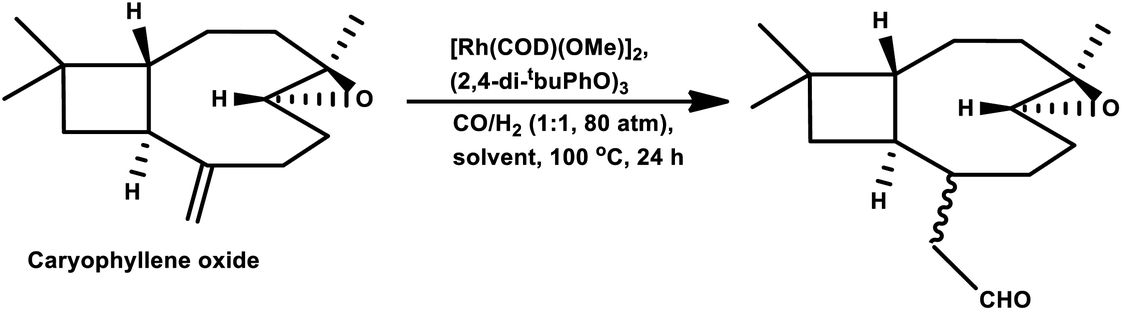
Under the harsh reaction conditions of 120 °C and 120 bar with a modified rhodium catalyst, a four aldehyde mixture was obtained in the hydroformylation of (−)-β-caryophyllene, only two of which proved to be identifiable by IR spectroscopy (Scheme 35).89 The main product was obtained from the exocyclic double bond reacting to yield the corresponding aldehyde.
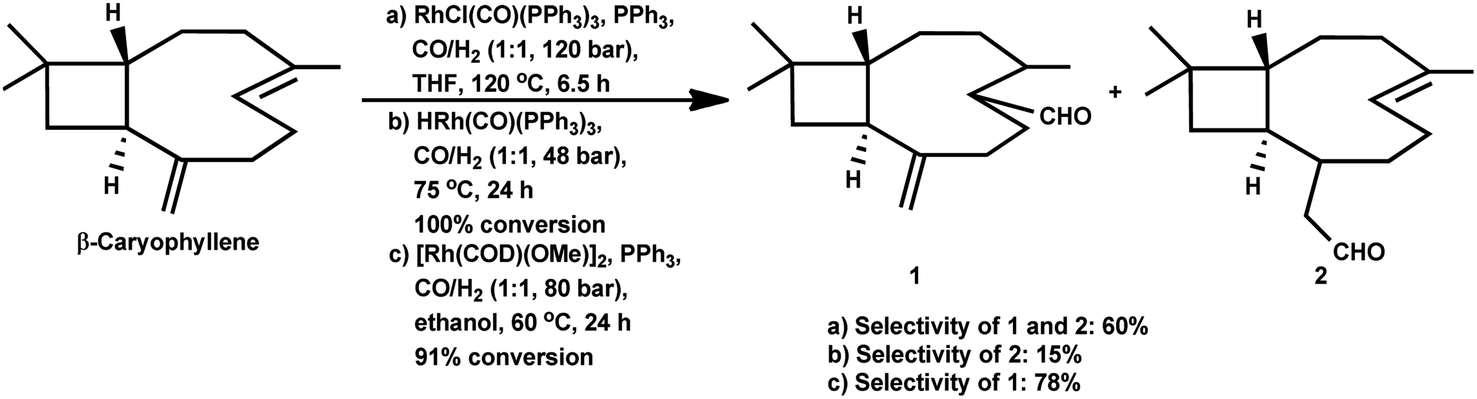 | ||
| Scheme 35 Hydroformylation of β-caryophyllene. | ||
Using the RhHCO(PPh3)3/PPh3 catalytic system and under mild reaction conditions, α-caryophyllene proved to be unreactive with no conversion in the temperature range of 70–100 °C.93 Alternatively, β-caryophyllene underwent full conversion under similar conditions, though a very low selectivity of ca. 15% was witnessed for the major product, which was derived from the exocyclic double bond (Scheme 35).
In 2016, Oliveira et al. reported the efficient rhodium-catalysed hydroformylation of β-caryophyllene using the cheaply available phosphine ligand PPh3 as the promoter and the bio-renewable solvent ethanol.159 No hydroformylation was observed in the non-promoted systems and PPh3 is required to break the chelates arising due to the interaction of both double bonds with the rhodium species. In contrast with the earlier reports on the hydroformylation of β-caryophyllene, this work revealed that the major product is formed through the rhodium species-endocyclic bond interaction and the rhodium species-exocyclic double bond interaction. Also, no traces of the monoaldehyde derived from the exocyclic double bond was found, although the formation of dialdehydes and acetals was noticed in small amounts. The optimum temperature of 60 °C was found to be useful, whereas higher temperatures or the use of bulky monophosphites such as (2-tBuPhO)3P and (2,4-di-tbuPhO)3P led to the undesired substrate decomposition with an increase in the isomerisation of the substrate and its consequential hydroformylation.
In 1991, Chalchat et al. showed that the hydroformylation of the oxygenated derivative of caryophyllene, caryophyllene oxide, gives a slower conversion but a better yield relative to β-caryophyllene in the RhHCO(PPh3)3/PPh3 system.93 Faria et al. found that the hydroformylation of caryophyllene oxide takes place even in non-promoted systems, whereas the use of the aforementioned bulky monophosphites accelerated the reaction.160 Another point to note is that no conversion occurred under the optimum conditions for the β-caryophyllene hydroformylation and product formation was observed only at 100 °C, giving rise to diethylacetal formation in ethanolic solutions. Hence, other green solvents, namely, DMC, DEC, p-cymene and MeTHF, have been explored with p-cymene being as efficient as toluene for this reaction (Table 16). The hydroformylation of β-caryophyllene was also studied in different green solvents, wherein, p-cymene gives comparable results with toluene. The selective double-hydroformylation of β-caryophyllene was carried out using a two-step process to obtain the dialdehyde (Scheme 36).
|
|
||
|---|---|---|
| Solvent | Conversion (%) | Selectivity (%) |
| Toluene | 98 | 92 |
| Ethanol | 100 | 28 |
| DMC | 99 | 91 |
| DEC | 100 | 95 |
| p-Cymene | 98 | 92 |
| MeTHF | 98 | 83 |
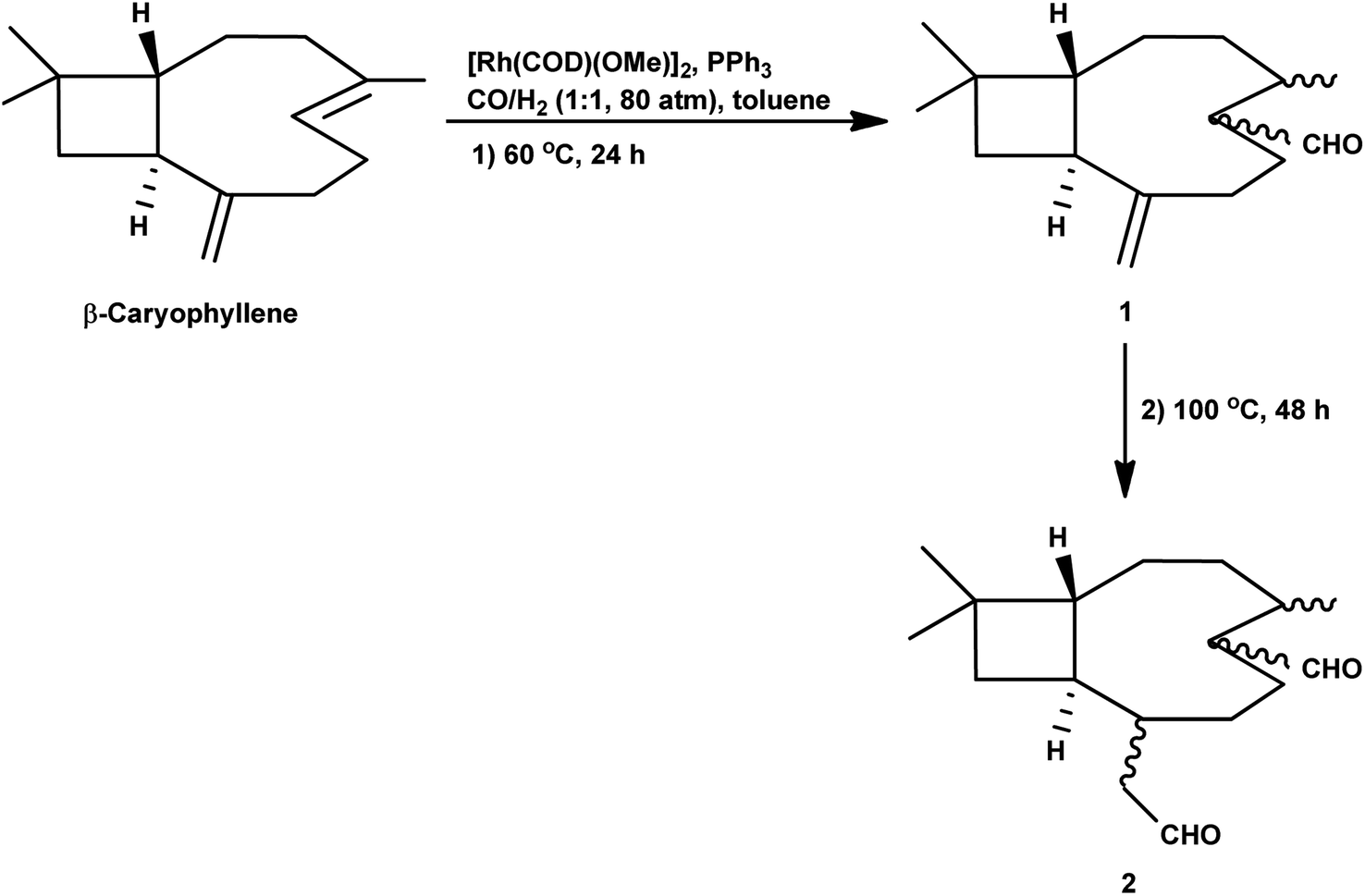 | ||
| Scheme 36 Double hydroformylation of β-caryophyllene. | ||
Nerolidol undergoes a one-pot hydroformylation/acetalisation with the use of the Rh/PPh3 or Rh/P(O-o-tBuPh)3 catalytic system in either toluene or ethanol solutions.163 The type of ligand and solvent has a remarkable effect on the rate and product of the reaction, respectively. The replacement of PPh3 with P(O-o-tBuPh)3 accelerated the reaction by ten-fold. Conversely, toluene gave cyclic hemiacetals, while ethanol gave cyclic acetals as the major product. In this case, the hemiacetals/acetals are produced by the intramolecular cyclisation of the initially formed hydroxy aldehyde. In toluene solutions, the reaction stops at the hemiacetal formation stage, while in ethanol the hemiacetals react further to form the acetals (Scheme 37). These systems resulted in full conversion with up to 98% of selectivity and a predominance to the cis product isomer. Similar results were obtained when an aqueous biphasic Rh/TPPTS/CTAC system was used, wherein TPPTS is a water soluble ligand, while CTAC acts as a phase transfer agent to contribute the cyclic hemiacetals.126
 | ||
| Scheme 37 Hydroformylation/hemiacetalisation and hydroformylation/acetalisation of nerolidol. | ||
2.2 Allyl/propenyl benzene derivatives
Allylbenzene derivatives are made from allyl and phenyl groups attached together, wherein other positions of the benzene ring may occupy different functional groups. A frequent by-reaction is the isomerisation of the allylic double bond to give the corresponding trans-propenyl benzene derivatives.164 Many of them are sustainable, renewable olefins, which are used as starting materials in the hydroformylation process as discussed below.The hydroformylation of eugenol and isoeugenol was carried out by H. Siegel et al. in 1980 under harsh conditions of pressure and varying temperatures, showing that the regioselectivity of the system is dependent on the reaction temperature (Table 17).109 The isomerisation of both, eugenol and isoeugenol was shown to increase as the temperature increased from 70 °C to 130 °C. Hence, the selectivity for the product with the formyl group at carbon-a and that at carbon-c continued increasing for eugenol and isoeugenol, respectively, as the temperature increased. The catalytic system comprised of [Rh(μ-S-tBu)2(CO)2L2] (L = phosphine/phosphite ligand) gives different conversion rates, yields and selectivity for the substrates eugenol and eugenol methyl ether depending on the nature of the ligands and solvents.167 PPh3, which is more basic than P(OMe)3 and P(OPh)3, gives a faster conversion but a lower selectivity for the linear aldehydes than these phosphite ligands. Also, non-coordinating polar solvents such as trichlorotrifluoroethane give higher rates, while the coordinating polar solvent DMF (dimethylformamide) slows down the reaction given that it coordinates with the metal centre, hence competing with the alkene.
|
|
||||||
|---|---|---|---|---|---|---|
| Temp. (°C) | Eugenol | Isoeugenol | ||||
| a (%) | b (%) | c (%) | a (%) | b (%) | c (%) | |
|
a Reaction conditions: [RhCI(1,5-cyclooactadiene)]2, CO/H2 (1:1, 600 bar).
|
||||||
| 70 | 0 | 48 | 52 | 95 | 5 | 0 |
| 80 | 3 | 45 | 52 | 90 | 10 | 0 |
| 100 | 11 | 37 | 52 | 50 | 45 | 5 |
| 130 | 41 | 24 | 35 | 40 | 50 | 10 |

It was later reported by A. C. da Silva et al. that eugenol undergoes hydroformylation under mild conditions with bis[(μ-acetate)(1,5-cyclooctadiene)rhodium(I)] as the catalyst precursor and the regioselectivity of the system is highly influenced by the auxiliary ligands.168 In the case of monophosphine ligands, the regioselectivity of the system only depends on the basicity and not on the steric factors of the ligand. Therefore, less basic ligands such as PPh3 and tribenzylphosphine (P(CH2Ph)3) with different cone angles favour the linear aldehyde with an n/iso ratio of 2.1, while more basic ligands such as tri(n-butyl)phosphine (P(n-Bu)3) and tricyclohexylphosphine (P(Cy)3) give a lower n/iso ratio of 1.1 and 1.2, respectively. A more basic phosphine ligand leads to greater back-donation of electron density from the metal centre to the carbonyl group, making the Rh–CO bond stronger and less susceptible to dissociation, and thus lowering the reaction rate. Alternatively, the regioselectivity of Rh-diphosphine systems show a strong correlation with the ligand bite angles. Diphosphine ligands such as dppe and dppp with a bite angle of around 90° give the branched aldehydes with the formyl group at the β-position in 60–70% selectivity. However, diphosphine ligands with a large bite angle of around 120°, such as NAPHOS and BISBI, preferably yield the linear aldehyde with 98% selectivity and n/iso ratio of 49.0 (Table 18). These diphosphine ligands with different bite angles differ in their mode of coordination to the metal. The ligands with small and large bite angles coordinate in an axial–equatorial and equatorial–equatorial fashion, respectively, in which the former has the hydride in the trans position to that of the phosphine ligand. Hence, in the case of diphosphines with large bite angles, the hydride is less basic, which disfavours its interaction with the terminal carbon, leading to the preferred formation of the linear aldehyde. The hydroformylation of isoeugenol is much slower than that of eugenol; the reason for this being the higher steric hindrance of the internal double bond than that of the terminal double bond, which makes the catalyst binding difficult. However, the hydroformylation of isoeugenol in the Rh–NAPHOS system favours the branched aldehyde with the formyl group in the sterically demanding α-position. This reaction also operates smoothly when diphenyl-based Buchwald phosphine ligands are used, granting 90% selectivity to the linear aldehyde.169
|
|
||||
|---|---|---|---|---|
| Ligand | Conversion (%) | Selectivity (%) | ||
| n | iso | n/iso | ||
| PPh3 | 100 | 68 | 31 | 2.1 |
| P(CH2Ph)3 | 100 | 68 | 32 | 2.1 |
| P(n-Bu)3 | 100 | 52 | 48 | 1.1 |
| P(Cy)3 | 100 | 55 | 45 | 1.2 |
| dppe | 55 | 38 | 62 | 0.6 |
| dppp | 74 | 31 | 69 | 0.5 |
| dppb | 99 | 66 | 34 | 1.9 |
| BISBI | 81 | 98 | 2 | 49.0 |
| NAPHOS | 90 | 98 | 2 | 49.0 |
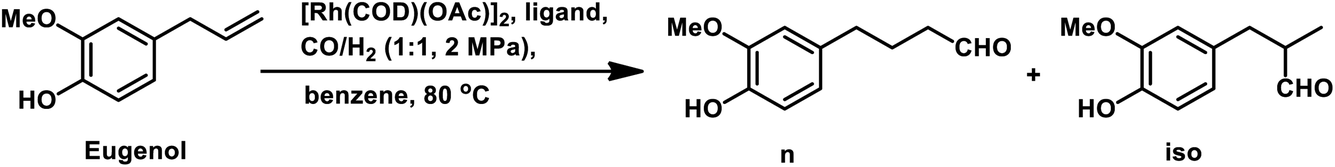
The synthesis of the 5,6-dihydronaphthalene derivative of eugenol occurred through a tandem hydroformylation/cyclisation process in the Rh(CO)2acac/ultranox626/H3PO4/CH2Cl2 catalytic system.170 Replacement of either ultranox626 with other ligands (Table 19) or H3PO4 with other acid additives (Table 20) highly influenced the selectivity and conversion by this system; however, the amount of acid used had little importance. The cyclised product is believed to be formed from the linear aldehyde through an intramolecular reaction in which a tetrahydronaphthalene alcohol is formed as an intermediate, which then undergoes dehydration to give the double bond in conjugation with the phenyl ring (Scheme 38). Similar results were obtained when eugenol methyl ether was used as the substrate in place of eugenol.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Ligand | Conversion (%) | D (%) | E (%) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
| A | B | C | |||||
|
a Reaction conditions: Rh(CO)2acac, ligand, H3PO4 (85%), CO/H2 (1:2, 300 psi), CH2Cl2, 110 °C, 2 h.
|
|||||||
| PPh3 | 98 | 21 | 55 | 22 | 4 | 96 | 0 |
| Ultranox626 | 98 | 4 | 65 | 29 | 4 | 86 | 10 |
| (PhO)3P | 97 | 16 | 52 | 29 | 5 | 95 | 0 |
| dppp | 3 | 1 | 0 | 2 | 0 | 100 | 0 |
| dppb | 9 | 1 | 3 | 5 | 0 | 45 | 55 |
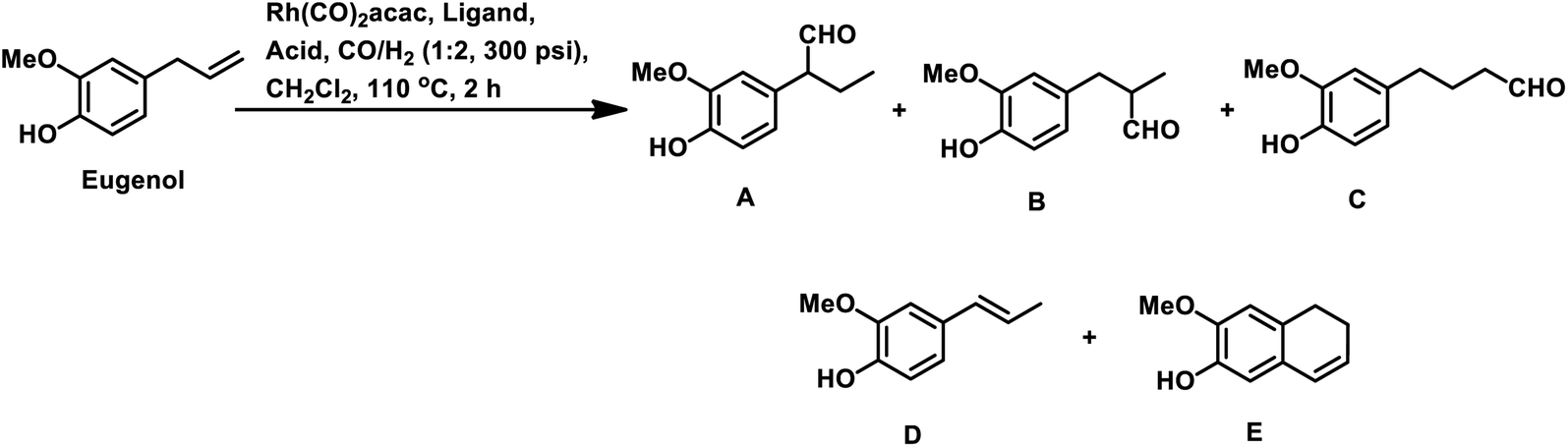
| Acid | Conversion (%) | D (%) | E (%) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
|---|---|---|---|---|---|---|---|
| A | B | C | |||||
|
a Reaction conditions: Rh(CO)2acac, ultranox626, CO/H2 (1:2, 300 psi), CH2Cl2, 110 °C, 2 h.
|
|||||||
| — | 99 | 3 | 0 | 96 | 0 | 19 | 81 |
| H3PO4 (85%, 0.07 mmol) | 99 | 6 | 59 | 34 | 3 | 84 | 13 |
| H3PO4 (85%, 0.14 mmol) | 98 | 4 | 65 | 29 | 4 | 86 | 10 |
| H3PO4 (85%, 0.28 mmol) | 99 | 6 | 65 | 28 | 4 | 92 | 4 |
| H3PO3 (42%, 0.14 mmol) | 91 | 11 | 54 | 26 | 0 | 100 | 0 |
| CH3COOH (99.8%, 0.14 mmol) | 97 | 10 | 0 | 87 | 1 | 14 | 85 |
| H2SO4 (98%, 0.14 mmol) | 80 | 28 | 11 | 41 | 0 | 89 | 11 |
| p-TsOH (0.14 mmol) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
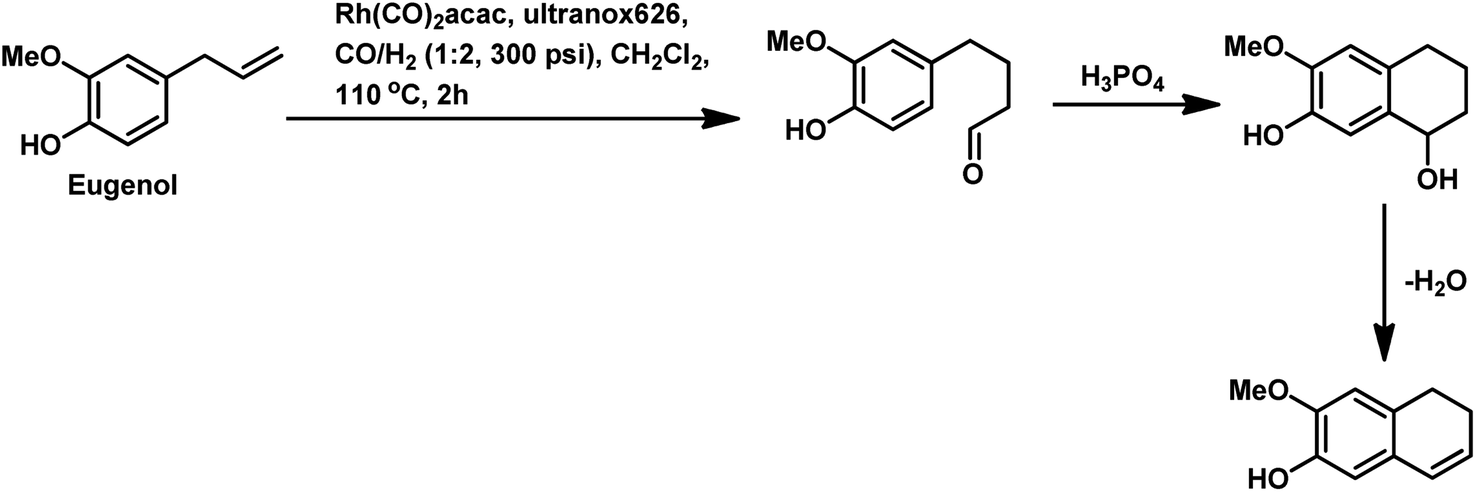 | ||
| Scheme 38 Synthesis of 5,6-dihydronaphthalene derivative of eugenol through hydroformylation and intramolecular cyclisation. | ||
The rhodium-catalysed hydroformylation of eugenol in an aqueous biphasic system occurs in the presence of water-soluble phosphines such as TPPTS, bisdiphenylphosphinoethanetetra-sulphonate (BDPPETS), bisdiphenylphosphinopropanetetrasulphonate (BDPPPTS) and diphosphane 2,2-bis(diphenylphosphinomethyl)-1,1′-biphenyl disulphonate (BISBIS), with or without the use of a surfactant.171 However, the introduction of the cationic surfactant CTAB accelerates the reaction given that it increases the interfacial area consisting of micelles and its positive charge attracts the anionic Rh complex to the biphasic interface through electrostatic interactions with the negatively charged sulfonate group of the TPPTS ligand, thus facilitating its reaction with the substrate. However, a very high concentration of surfactant decelerates the reaction due to the creation of microemulsions and because the counter ions (bromide ions) of CTAB compete with eugenol for the active site on the metal. The replacement of the water soluble sulfonated monophosphine TPPTS (l/b ≈ 2.5) with the water soluble sulfonated diphosphines BDPPETS and BDPPPTS possessing small bite angles preferentially yielded the branched aldehyde (l/b ≈ 0.6), while BISBIS having a large bite angle preferred the linear aldehyde (l/b ≈ 12) (Table 21). This catalytic system was recyclable for a minimum of five cycles without serious degradation in its activity or selectivity. Eugenol undergoes this reaction in rhodium- and ruthenium-catalysed aqueous biphasic systems even with CTAC as a phase transfer agent.172
|
|
|||||
|---|---|---|---|---|---|
| Ligand | Time (min) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
| A | B | C | |||
| BDPPETS | 105 | 99 | 0 | 58 | 42 |
| BDPPPTS | 150 | 94 | 3 | 60 | 37 |
| BISBIS | 30 | 92 | 1 | 7 | 92 |
| TPPTS | 20 | 95 | 3 | 27 | 70 |
In 2017, Bhanage and co-workers carried out this reaction in an Rh/TPPTS/CDs/H2O aqueous biphasic, recyclable system by screening various cyclodextrins as a mass transfer promoter. α-CD, β-CD, γ-CD, RAME-α-CD, RAME-β-CD and hydroxyl propyl β-cyclodextrin (hp-β-CD) were screened, and among them, RAME-β-CD was found to be the best phase transfer agent, resulting in 97% conversion and 87% selectivity toward the linear aldehyde under the optimised conditions (Table 22).173 This system works by eugenol binding to the hydrophobic inner region, while the catalytic system interacts with the hydrophilic outer portion of the RAME-β-CD. The catalytic system was reusable for four consecutive cycles, retaining its activity and providing unaltered conversion and selectivity.
|
|
|||||
|---|---|---|---|---|---|
| CDs | Conversion (%) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
| A | B | C | |||
| — | 68 | 60 | 14 | 34 | 52 |
| α-CD | 84 | 72 | 15 | 20 | 65 |
| β-CD | 92 | 72 | 06 | 16 | 78 |
| γ-CD | 79 | 64 | 14 | 16 | 70 |
| RAME-α-CD | 84 | 69 | 19 | 21 | 60 |
| RAME-β-CD | 97 | 89 | 03 | 10 | 87 |
| hp-β-CD | 86 | 85 | 12 | 20 | 68 |
Recently, our group introduced the novel aqueous biphasic catalytic system, Rh/PTABS (4-(1,3,5-triaza-7-phosphaadamantan-1-ium-1-yl)butane-1-sulfonate)/Xantphos/CTAB, through the highly chemo- and regio-selective hydroformylation of eugenol.174 Herein, PTABS is a water-soluble ligand, while Xantphos acts as a water-insoluble promoter ligand, which accelerates the reaction, and simultaneously also improves the regioselectivity of the system. In this case, the conversion was 100%, the chemoselectivity reached 93% and the l/b ratio was up to 24 (Scheme 39a). This system was found to be active for up to four runs.
 | ||
| Scheme 39 Hydroformylation and one-pot hydroformylation/O-acylation of eugenol. | ||
The non-aqueous biphasic hydroformylation of eugenol was achieved with the binuclear complex [Rh(COD)(μ-OMe)]2 as the catalyst precursor, TPPTS as the ligand, BMI-BF4 (1-butyl-3-methylimidazolium tetrafluoroborate) as an ionic liquid with hydrophilic characteristics and toluene as the second phase.175 This system gave 99% conversion with 94% selectivity for the corresponding aldehydes without any phase transfer agents such as surfactants. The addition of the surfactant CTAB to the system retarded the hydroformylation reaction given that it forms an interface and interacts with the ionic liquid, inhibiting its interaction with the catalyst system. This system was shown to provide good results for up to four cycles.
Recently, the homogeneous one-pot hydroformylation/O-acylation of eugenol and isoeugenol was reported to occur under hydroformylation conditions in the presence of acetic anhydride as an acylating agent.176 When [Rh(COD)(OMe]2 acts as the metal precursor and Xantphos is the ligand, eugenol successfully undergoes this reaction to regioselectively produce the linear aldehyde with an acetate group (from the acylation of the phenolic group) (Scheme 39b). In the presence of acetic anhydride, the hydroformylation step is slower than it is when only toluene is present given that the by-product of the acylation step is acetic acid, which promotes the isomerisation of eugenol to isoeugenol. This conversion is much slower for isoeugenol given that it has a double bond in a sterically compromising position. However, when P(O-2,4-tBu-C6H3)3 is the auxiliary ligand, the branched aldehyde at the α-position to the phenyl ring is formed along with the acyl group at its para-position (Scheme 40). Both eugenol and isoeugenol undergo this reaction in anisole as a sustainable solvent, in place of toluene, with comparable conversion and selectivity. A delicate balance between various factors exists in the tandem two-step hydroformylation/hydrogenation/O-acylation of eugenol in the formation of the corresponding diacetate esters.177 The first step is hydroformylation/hydrogenation, which gives the corresponding alcohol, while the subsequent addition of acetic anhydride in the second step leads to the O-acylation of the newly formed hydroxy group as well as the already existing phenolic group (Scheme 41).
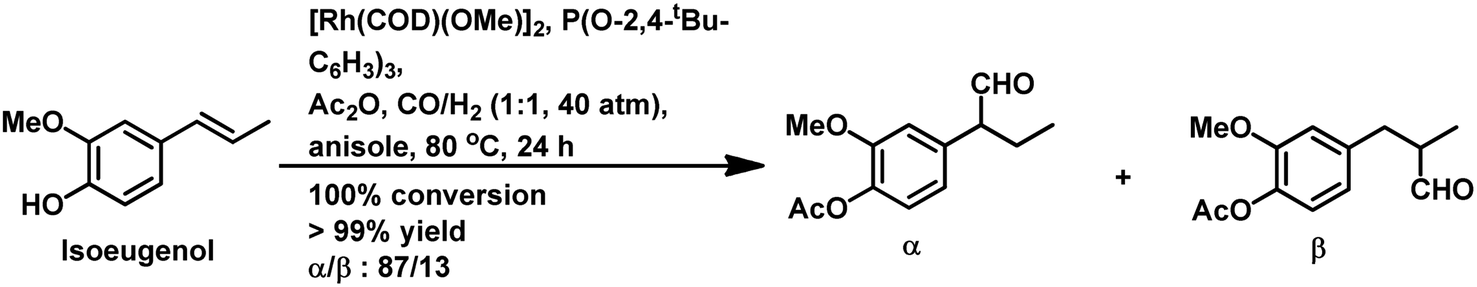 | ||
| Scheme 40 One-pot hydroformylation/O-acylation of isoeugenol. | ||
 | ||
| Scheme 41 One-pot hydroformylation/hydrogenation/O-acylation. | ||
Recently, it has been shown that the sequential hydroformylation/acetalisation of eugenol to the corresponding acetals can be realised with the use of multi-stage flow reactors, enabling the recovery and recycling of the solid catalysts for several uses, and thus reducing the use of solvents and increasing the energy efficiency of the process.178 Eugenol and methyl eugenol undergo a regioselective transfer hydroformylation upon the use of [Rh(COD)Cl]2/rac-BINAP/Nixantphos (4,6-bis(diphenylphosphino)-10H-phenoxazine)/formaldehyde system without the requirement of CO/H2, resulting in an aldehyde yield of 81% and 86%, respectively.179
Estragole undergoes hydroformylation with [Rh(μ-S-tBu)2(CO)2L2] (L = phosphine/phosphite ligand) as the catalytic system, wherein the conversion rate and the regioselectivity of the system shows a strong dependence on the type of ligands and solvents.167 Non-coordinating polar solvents such as DMF are preferred as coordinating polar solvents, which decelerate the reaction by coordinating unfavourably with the metal catalyst, hence blocking the substrate–catalyst binding. The regioselectivity for the linear aldehyde is favoured with phosphite ligands, which are less basic than phosphine ligands. Consequently, the yield of the linear aldehyde is 77% with P(OPh)3 and 68% with PPh3. The rhodium-diphosphite system efficiently performed the hydroformylation of trans-anethole and estragole with high regioselectivity in the former and low regioselectivity in the latter.181 When Buchwald phosphine ligands were used for this purpose, a selectivity of 75% for the linear aldehyde and 65% for the branched aldehyde were obtained in case of estragole and anethole, respectively.169
The hydroformylation of estragole and anethole takes place with [Rh(nbd)Cl]2/PPh3 (or DIOP) and PtCl2(bdpp)/SnCl2 catalysts. In the Pt/Sn system, α-, β- and γ-formyl derivatives were formed in case of both substrates, indicating that isomerisation of the double bond occurs in each of them (Tables 23 and 24).182 However, using a rhodium catalyst, no γ-formyl derivative was obtained for anethole, while all the three derivatives were obtained for estragole. This means that the internal double bond of anethole does not isomerise to the terminal position (forming estragole), but the terminal double bond of estragole isomerises to the internal position (forming anethole) to some extent in rhodium catalytic systems. The major product for anethole hydroformylation is the α-formyl derivative, while that for estragole is the γ-formyl derivative (β-formyl derivative with Pt/Sn). The conversions and chemoselectivity for both substrates are higher in the rhodium catalytic system than the bimetallic Pt/Sn system. Another catalytic system for the carbonylation of estragole is Rh(CO)2(acac)/1,3-bis((diphenylphosphino)oxy)-1,3-diphenylpropane, which gives 99% conversion and 100% yield (Scheme 42a).183
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst precursor | Time (h) | Conversion (%) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
| α | β | γ | ||||
| PtCl2 + SnCl2 | 27 | 8.5 | 82 | 28.5 | 43 | 28.5 |
| [Rh(nbd)Cl]2 + PPh3 | 7 | 97 | 89 | 24 | 35 | 41 |
| [Rh(nbd)Cl]2 + DIOP | 7 | 99.5 | 94 | 24 | 34 | 42 |
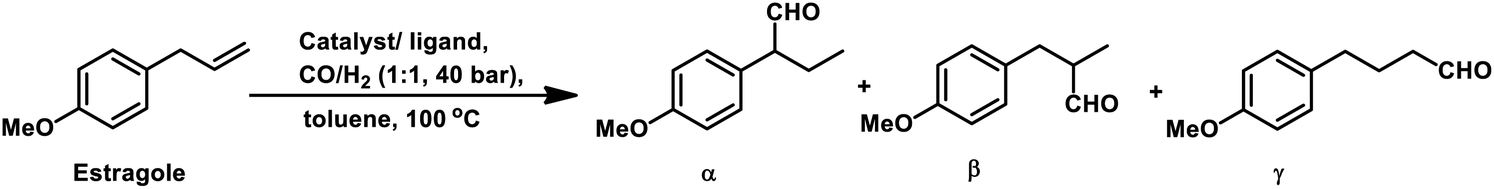
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst precursor | Time (h) | Conversion (%) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
| α | β | γ | ||||
| PtCl2 + SnCl2 | 21 | 44.5 | 94 | 52.5 | 33.5 | 14 |
| [Rh(nbd)Cl]2 + PPh3 | 7 | 84 | 99 | 85 | 15 | 0 |
| [Rh(nbd)Cl]2 + DIOP | 14 | 51 | 100 | 86 | 14 | 0 |
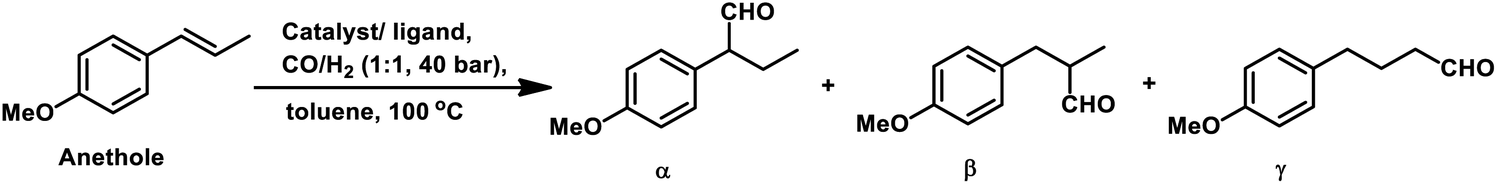
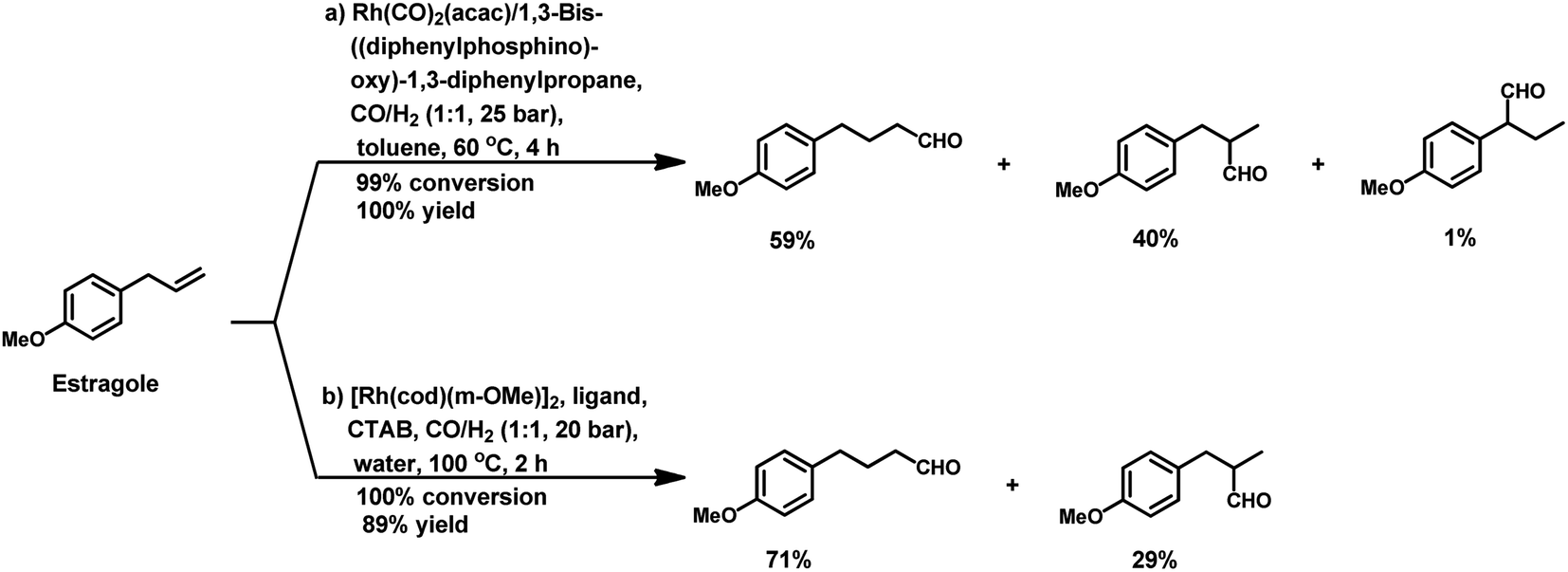 | ||
| Scheme 42 Hydroformylation of estragole. | ||
Almeida et al. disclosed a cascade hydroformylation/arylation reaction for anethole using arylboronic acids and a single catalyst to give alcohols as the final product.108 Anethole upon hydroformylation gives two branched aldehydes, which in the second step undergo arylation with phenylboronic acid, forming the corresponding alcohols with pharmacological importance (Scheme 43).
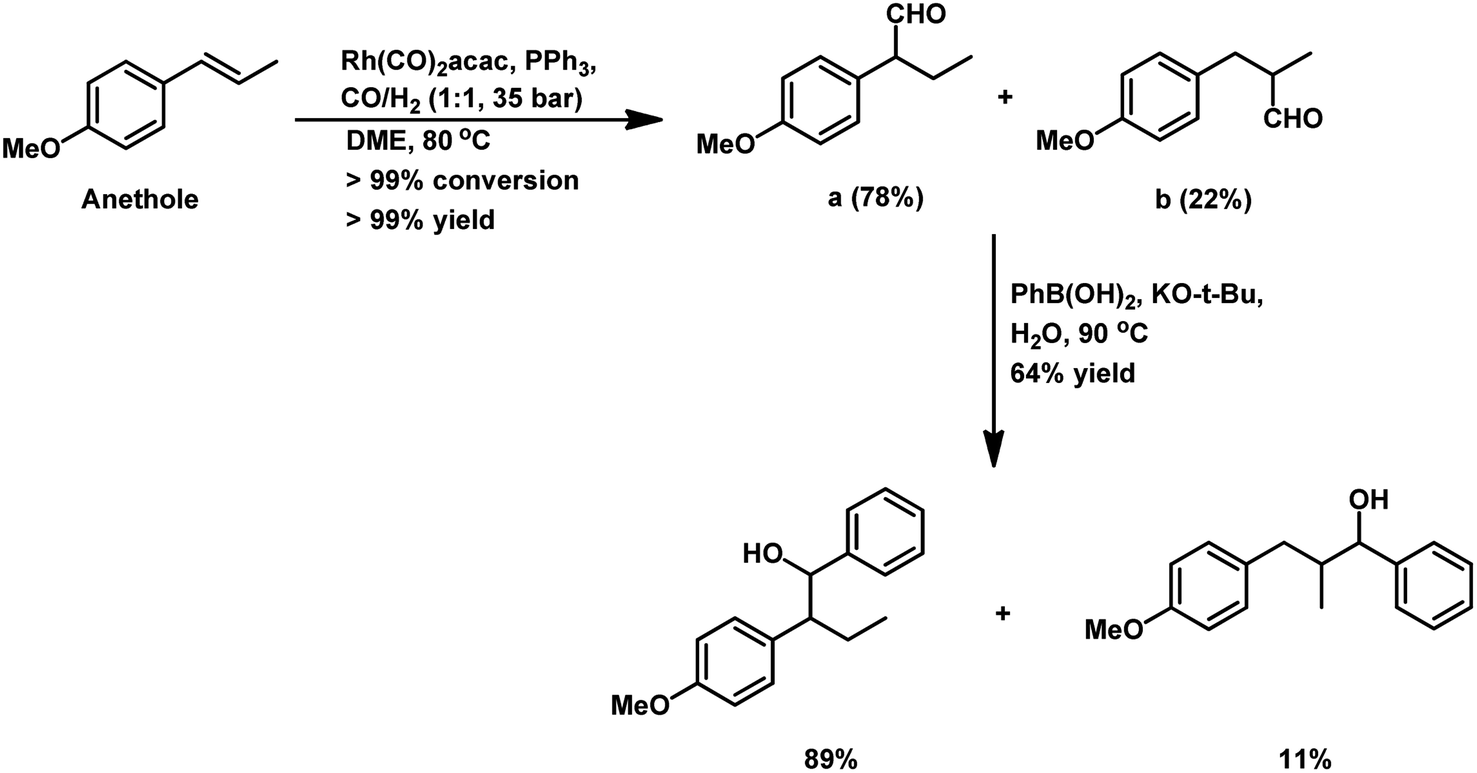 | ||
| Scheme 43 Tandem hydroformylation/arylation of anethole. | ||
Estragole was also hydroformylated in a rhodium-based aqueous biphasic system with TPPTS as the water-soluble phosphine and CTAB as the mass transfer agent to give full conversion with 89% yield of aldehydes (Scheme 42b).171 This reaction with eugenol is faster than with estragole given that the former has an additional hydroxy group, which hydrogen bonds with water, making it more soluble in aqueous medium. Estragole as well as anethole undergo this reaction in rhodium- and ruthenium-based water soluble catalysts even with CTAC as a mass transfer agent.172
The aqueous biphasic hydroformylation of estragole and anethole was also achieved using the Rh/TPPTS/RAME-β-CD/H2O recyclable system.173 Estragole gave a higher conversion and yield of the aldehydes under milder conditions than anethole (Table 25) given that the former has a terminal double bond, which is more susceptible to hydroformylation than the internal double bond in the latter. Branched aldehyde is the major product in case of anethole, while estragole forms the linear aldehyde preferentially. Also, both substrates yield three aldehydes, which means that isomerisation of the double bond occurs in both of them.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Substrate | Temp. (°C) | Pressure (bar) | Time (h) | Conversion (%) | Yield of aldehydes (%) | Selectivity of aldehydes (%) | ||
| α | β | γ | ||||||
| Estragole | 80 | 25 | 6 | 98 | 89 | 13 | 12 | 75 |
| Anethole | 90 | 40 | 10 | 94 | 84 | 30 | 55 | 15 |
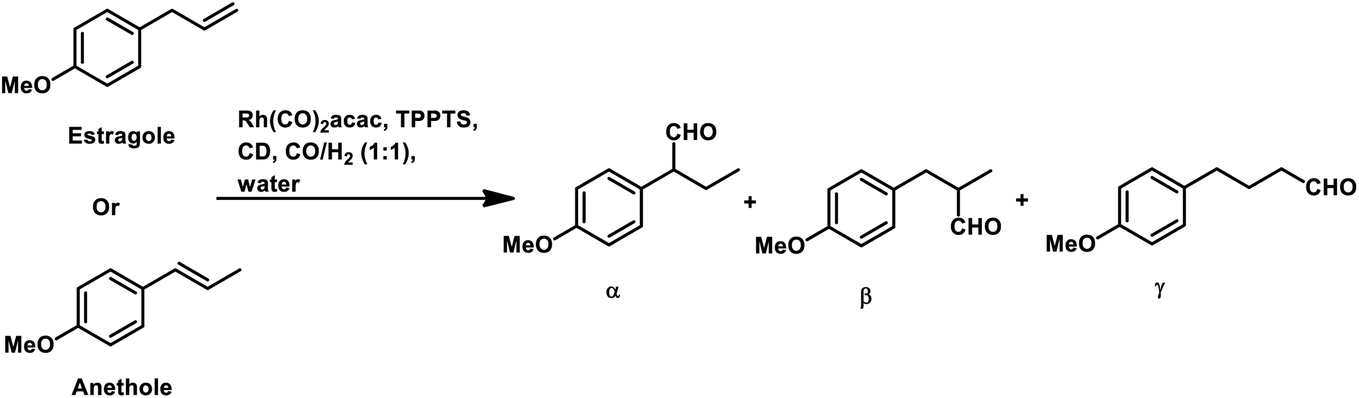
The binuclear complex [Rh(COD)(μ-OMe)]2 acts as the catalyst precursor in the non-aqueous biphasic hydroformylation of estragole using the TPPTS ligand that is soluble in the ionic liquid, BMI-BF4, which has hydrophilic nature.175 A high conversion of 99% was obtained together with 95% of aldehyde selectivity (l/b = 2.0). The regioselective transfer hydroformylation of estragole occurred with the incorporation of formaldehyde as a syngas surrogate in the [Rh(COD)Cl]2/rac-BINAP/Nixantphos system, providing 89% of aldehyde with an n/iso ratio of 9.179
The catalytic system [Rh(μ-S-tBu)2(CO)2L2] (L = PPh3/P(OPh)3/P(OMe)3) can be used to hydroformylate safrole to majorly obtain the linear aldehyde in up to 88% in DMF.167 The rhodium-catalysed hydroformylation of safrole gives different conversions and regioselectivities when the diphosphines dppp and NAPHOS are used.168 NAPHOS, which has a wide bite angle, gives linear aldehyde as the major product. However, replacement of NAPHOS with the small bite angle ligand dppp switches the regioselectivity in favour of the branched aldehyde (Table 26).
|
|
|||
|---|---|---|---|
| Ligand | Conversion (%) | Yield of aldehydes (%) | |
| Branched | Linear | ||
| dppp | 62 | 66 | 33 |
| NAPHOS | 100 | 5 | 93 |
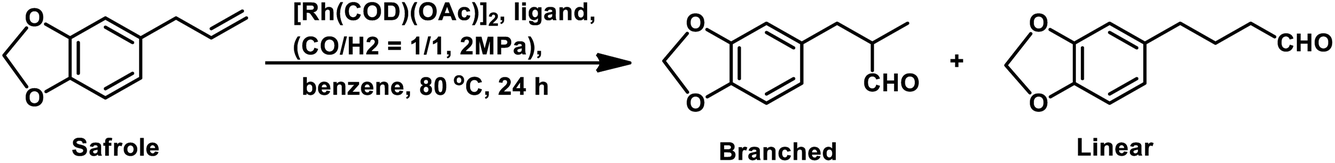
The employment of the Rh(CO)2acac/ultranox626/H3PO4/CH2Cl2 catalytic system with safrole gives rise to a tandem hydroformylation/cyclisation process to yield the 5,6-dihydronaphthalene derivative of safrole with 99% conversion and 62% selectivity together with 31% aldehydes (Scheme 44).170 This reaction proceeded through a cascade mechanism, wherein the linear aldehyde forms a tetrahydronaphthalene alcohol derivative as an intermediate, which gives the cyclised product upon the elimination of a water molecule (similar to eugenol, Scheme 38).
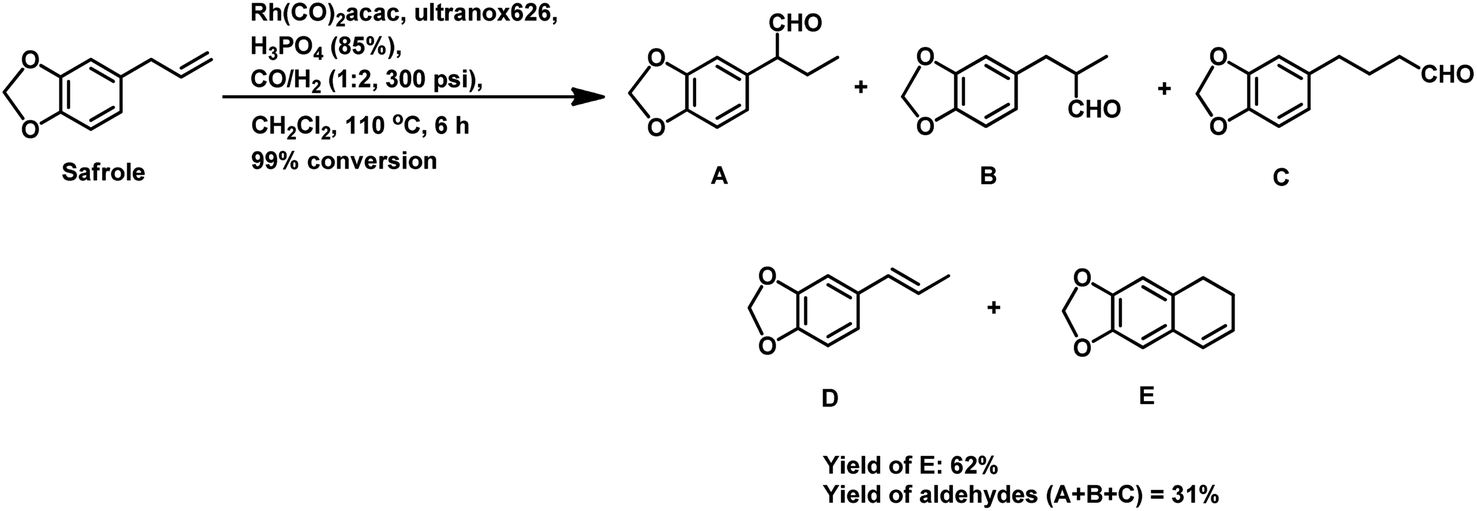 | ||
| Scheme 44 One-pot hydroformylation/cyclisation of safrole. | ||
The efficient rhodium-catalysed hydroformylation of safrole took place in an aqueous biphasic medium using [Rh(COD)(μ-OMe)]2 as the metal precursor, TPPTS as the water-soluble phosphine ligand and CTAB as the mass transfer promoter to give full conversion and 85% yield of aldehydes.171 This conversion is also regioselective with respect to the linear aldehyde (Scheme 45, entry 1). However, it is ca. 10 times slower for safrole compared to eugenol given that the latter possesses a hydroxy group, which makes it more soluble in the aqueous medium, while the former lacks it. This is possible even by making use of CTAC as a phase transfer agent using water-soluble catalysts.172
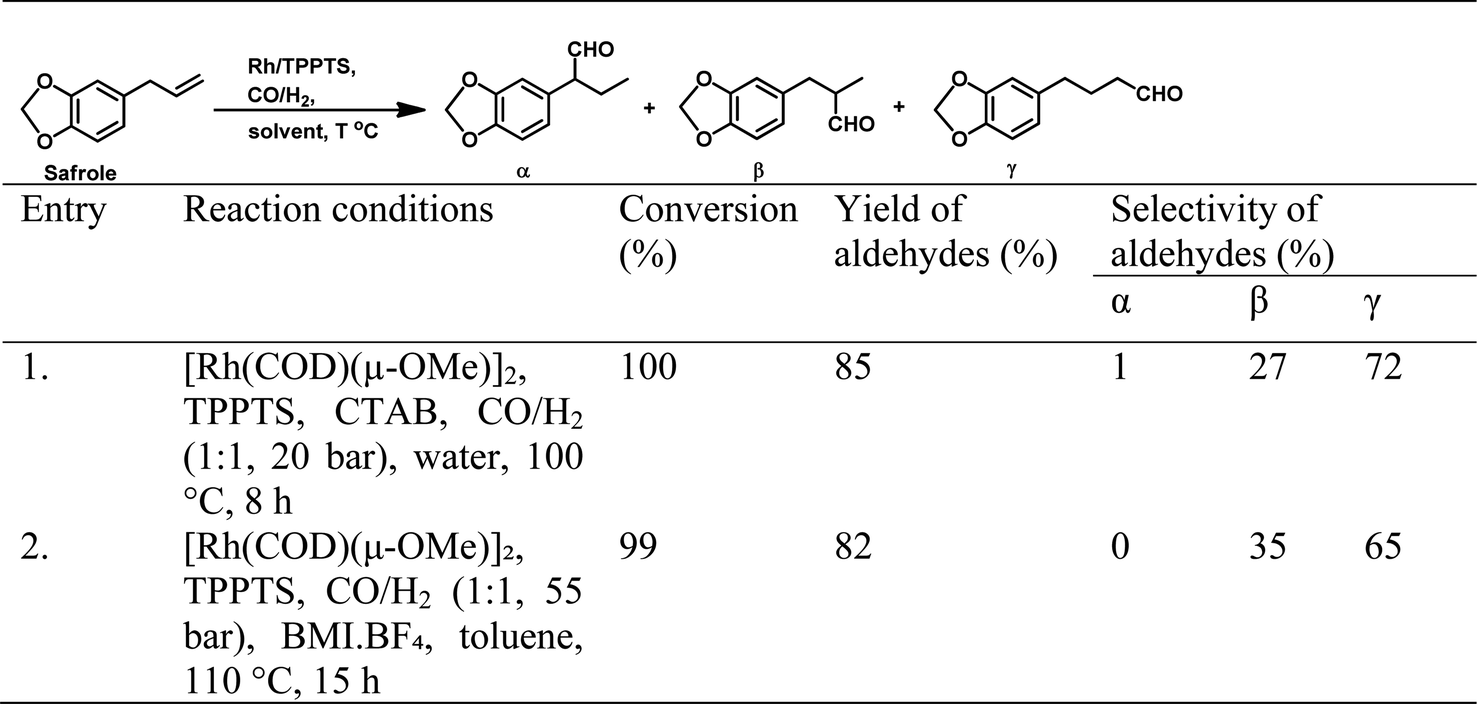 | ||
| Scheme 45 Hydroformylation of safrole in aqueous and non-aqueous biphasic systems. | ||
Baricelli et al. performed the systematic rhodium-catalysed hydroformylation of safrole in a BMI-BF4/toluene non-aqueous biphasic system without the use of phase transfer promoters, resulting in 99% conversion and 82% aldehyde selectivity (l/b = 1.9) (Scheme 45, entry 2).175 Applying formaldehyde as a replacement for syngas in the catalytic system, [Rh(COD)Cl]2/rac-BINAP/Nixantphos led to 85% selectivity toward the aldehyde and an n/iso ratio of 11.5.179
2.3 Hydroformylation of natural oleo compounds
Oils and fats derived from vegetable and animal sources possess great industrial applications. Three units of fatty acids upon condensation with a glycerol molecule gives the corresponding triesters called triglycerides, which form the class of oils and fats. The hydroformylation of renewables such as naturally derived unsaturated oleo compounds provides aldehydes that have commercial significance. The products of the hydroformylation of oils and fats find major application in the production of polyurethanes, which are utilised in the manufacture of viscoelastic foams and coatings.185The first report on the hydroformylation of these compounds dates back to 1948 in the patent from DU PONT.186 The early studies on the hydroformylation of fatty acid esters such as methyl oleate were done using cobalt-based catalytic systems (Scheme 46).187 The hydroformylation of vegetable oils and unsaturated fatty esters using Co2(CO)8 gives different major products under different conditions of temperature.188 In the temperature range of 100–110 °C, fatty aldehydes are formed, while an increase in temperature to 175–190 °C yields fatty alcohols as the main product. A mixture of several isomers of formyl and hydroxy products are obtained given that this catalytic system brings about the isomerisation of the double bond in the substrate and reduction of the aldehydes into the corresponding alcohols, thus affecting the chemoselectivity of the system. The same result was witnessed with a tributylphosphine/cobalt carbonyl complex, wherein the hydroxy esters predominated. On the contrary, a system comprised of rhodium-supported on C, CaCO3 or Al2O3 was found to be effective by Frenkel to carry out the hydroformylation of methyl elaidate and oleic acids, esters and triglycerides (Scheme 47) to formyl products with no or trace amounts of side products.189 In the presence of triphenylphosphine and high pressures of 1000–2000 psi, it forms a rhodium carbonyl triphenylphosphine complex as the active catalyst, which is inactive toward common side reactions such as reduction and isomerisation of the double bonds in the substrate and the reduction of aldehydes to alcohols. The hydroformylation of methyl oleate with this rhodium-based system yields majorly the 9- and 10-formylstearate even at the high temperature of 180 °C as opposed to that of the cobalt-carbonyl system, wherein many branched products form due to the isomerisation of the unsaturated bonds and conversion of formyl to hydroxy products at high temperatures. The same reaction with an activated rhodium catalyst-triphenyl phosphite system and methyl oleate as the substrate is feasible at pressures as low as 200 psi.190 The process of hydroformylation of unsaturated fats and esters yield hydroxymethyl stearates, which are used to synthesise acyl esters. These acyl esters find applications as plasticisers for polyvinyl chloride.191 On application of rhodium-triphenylphosphine and palladium chloride-triphenylphosphine catalytic systems in polyunsaturated fatty acids, functionality can be introduced at each unsaturated site to produce polyformyl- and polycaboxy-stearates.192 The products obtained from the hydroformylation of unsaturated fatty compounds can be further subjected to other reactions such as oxidation, reduction, acetylation, reductive amination, aminolysis and Tollens condensation to obtain products with diverse applications.193 Koen et al. showed that a bulky tris(2-tert-butyl-4-methylphenyl)phosphite-modified rhodium catalyst is much more active than the typical triphenylphosphine-modified rhodium catalyst in the oxo process of unsaturated fatty acid esters.194
 | ||
| Scheme 46 Hydroformylation of methyl oleate. | ||
 | ||
| Scheme 47 Hydroformylation of triglyceride. | ||
Kandanarachchi and co-workers studied the hydroformylation of various vegetable compounds and model compounds with Rh(CO)2(acac) as the catalyst precursor and PPh3 or P(OPh)3 as the catalyst modifier, which revealed that this phosphite ligand shows poor reactivity compared to the phosphine ligand.195 This is in contrast to the general trend, in which case bulky phosphite ligands show higher reactivity than phosphine ligands. Another important aspect is that the π-allyl intermediate is formed with rhodium when methyl esters of fatty acids are used as polyolefins. Nevertheless, the reactivity of triglycerides is not influenced by the type of olefin. The kinetics and mechanism involved in the hydroformylation of soybean oil also suggest the difference in the reactivity of these two ligands, wherein the activation energies are (61.1 ± 0.8 kJ mol−1) and (77.4 ± 5.0 kJ mol−1) in the presence of PPh3 and P(OPh)3, respectively.196 Soybean oil and methyl oleate have been found to undergo hydroformylation in the presence of the [RhH(CO)(PPh3)3] metal precursor with or without the use of a ligand, resulting in full conversion with 80–91% selectivity for aldehydes.197 In the case of soybean oil, trans isomers and conjugated dienes are obtained as side products. When [RhH(CO)(PPh3)3] is replaced by [RhCl3·3H2O] as the metal precursor with soybean oil as the substrate, conjugated dienes are formed as the only product. Moreover, the introduction of triphenylphosphine in this system leads to the formation of an inactive metal complex, [Rh(Cl)(CO)(PPh3)2].198 The hydroformylation of fatty acid esters such as oleic acid ester and linoleic acid ester takes place at 115 °C with 20 bar of syngas pressure. With both substrates, saturated fatty compounds are formed in high quantities given that hydrogenation is favoured, while hydroformylation is difficult as the ester group disfavours functionalisation in its locality. The maximum yield of linear aldehyde is as low as 26% and 34% for oleic acid ester and linoleic acid ester, respectively.
The reactivation and recycling of the supported rhodium-on-alumina catalyst is possible for up to 9 cycles without remarkable loss in its activity in the hydroformylation of methyl oleate to methyl formylstearate.199 This can significantly reduce the cost of production on a plant scale. Fell et al. showed that short chain fatty acids such as methyl 10-undecenoate get hydroformylated in aqueous medium using the Rh/TPPTS system without the need for a surfactant (Scheme 48).200 Contrary to this, long chain fatty acids and their derivatives such as linolenic fatty ester suffer from phase transfer limitations due to their hydrophobic nature, which necessitates the addition of a surfactant to carry out an efficient hydroformylation process. The hydroformylation of linseed oil also occurs efficiently in a micellar two phase system. Several sulfonated trialkyl phosphines and sulfonated dialkyl aralkyl phosphines were developed by Tulchinsky, Abatjoglou, Hefner and Peterson to facilitate the hydroformylation of long chain olefinic substrates in a biphasic system.201 The use of these aqueous phase soluble phosphines ensures the recovery and recyclability of the costly metal catalysts. The hydroformylation of linolenic methyl esters occurred using rhodium sulfate as the metal precursor and triphenylphosphinemonosulfonate (TPPMS) as the ligand in methanol at 120 °C and 200 bar.202 The recyclability of this system was demonstrated by Fell. The utilisation of heterogenised cobalt carbonyl and rhodium carbonyl catalysts in the hydroformylation of fatty acids also forms a recyclable system.203 Davis and Hanson developed a new category of heterogeneous catalysts, which was demonstrated by the hydroformylation of oleyl alcohol through supported aqueous phase catalysis.204
 | ||
| Scheme 48 Aqueous biphasic hydroformylation of methyl 10-undecenoate. | ||
Hydroformylation of methyl soyate gives a mixture of hyperbranched polyols with the number of hydroxy groups ranging from zero to three, with two being the most common.205 Hydroformylation of unsaturated fatty esters in aqueous phase takes place with the use of activated carbon, namely, Nuchar® WV-B as the mass transfer promoter.206 It is more efficient than co-solvents and cyclodextrins as a mass transfer promoter and gives up to 90% yield of aldehydes when applied to methyl oleate. The use of 1-butanol as a co-solvent in the aqueous biphasic rhodium-catalysed hydroformylation of methyl 10-undecenoate was proven to be beneficial in rendering a highly efficient system.207 The turnover frequency with the iridium-biphephos system was approximately five-fold lower than that of the rhodium-biphephos system in the hydroformylation of 10-undecenitrile.208 Nevertheless, the former proved to be more efficient in the controlled hydroformylation-isomerisation of terminal fatty alkenes. On the contrary, the ruthenium-biphephos system was 1–2 times less active and comparatively less capable of isomerizing internal double bonds to terminal double bonds than the rhodium-biphephos system.209 Methyl oleate undergoes linear selective isomerisation/hydroformylation in a tandem catalytic system comprised of a palladium-based catalyst for double bond isomerisation and a rhodium-based catalyst for hydroformylation to give aldehyde yields of up to 74% and n/iso ratio of 91/9.210 Hydroformylation of canola fatty acid methyl esters takes place with doped ruthenium supported on hyper crosslinked polystyrene to give industrially important aldehydes.211 Recently, methyl oleate and methyl 10-undecenoate were hydroformylated regioselectively using a continuous jet-loop reactor, wherein yields above 80% were obtained upon more than 55 h of operation.212
2.4 Hydroformylation of steroids
Steroids, which play diverse roles in biological functions, are made up of four fused rings structurally.213 Many steroids occur naturally and have the potential to act as sustainable sources of starting materials in the hydroformylation process.The initial reports on the hydroformylation of steroids, published in 1959, dealt with the reaction of a double bond on the six membered ring of the substrate nucleus.214 3β,20β-diacetoxy-pregn-5-ene and 3β-acetoxy-pregn-5-ene-20-one, when subjected to this process in the presence of Co2(CO)8 and under harsh conditions of pressure and temperature, resulted in the corresponding 6α-hydroxymethylpregnanes. The vinylation of androsta-16-ene-17-yl iodide led to the formation of a steroid, which under hydroformylation conditions yielded the corresponding formyl derivatives (Scheme 49).215 Unsaturated steroids can be hydroformylated by making use of [Rh(diene)Cl]2 as a catalyst precursor and tertiary phosphines as ligands.216 The syntheses of (20R)-3β, 20β-dihydroxy-16α-formyl-pregn-5-ene and various other formyl-androstane derivatives showed that only the double bond in the five-membered D ring of the substrate undergoes the hydroformylation reaction, while that in the six-membered B ring remains intact even under high temperatures and pressures (Scheme 50).217 When amines are introduced in this system, hydroaminomethylation takes place to give the corresponding aminomethyl steroids (Scheme 51). The hydroformylation of vinyl derivative of estrone takes place equally well in both rhodium- and platinum-catalysed systems with various phosphines (Scheme 52).218 (R)-PROPHOS (1,2-bis(diphenylphosphino)propane) acts as a good regioselective ligand and branched aldehydes are formed regioselectively in both the systems. Upon switching from the rhodium to the platinum system, the regioselectivity of the system decreases.
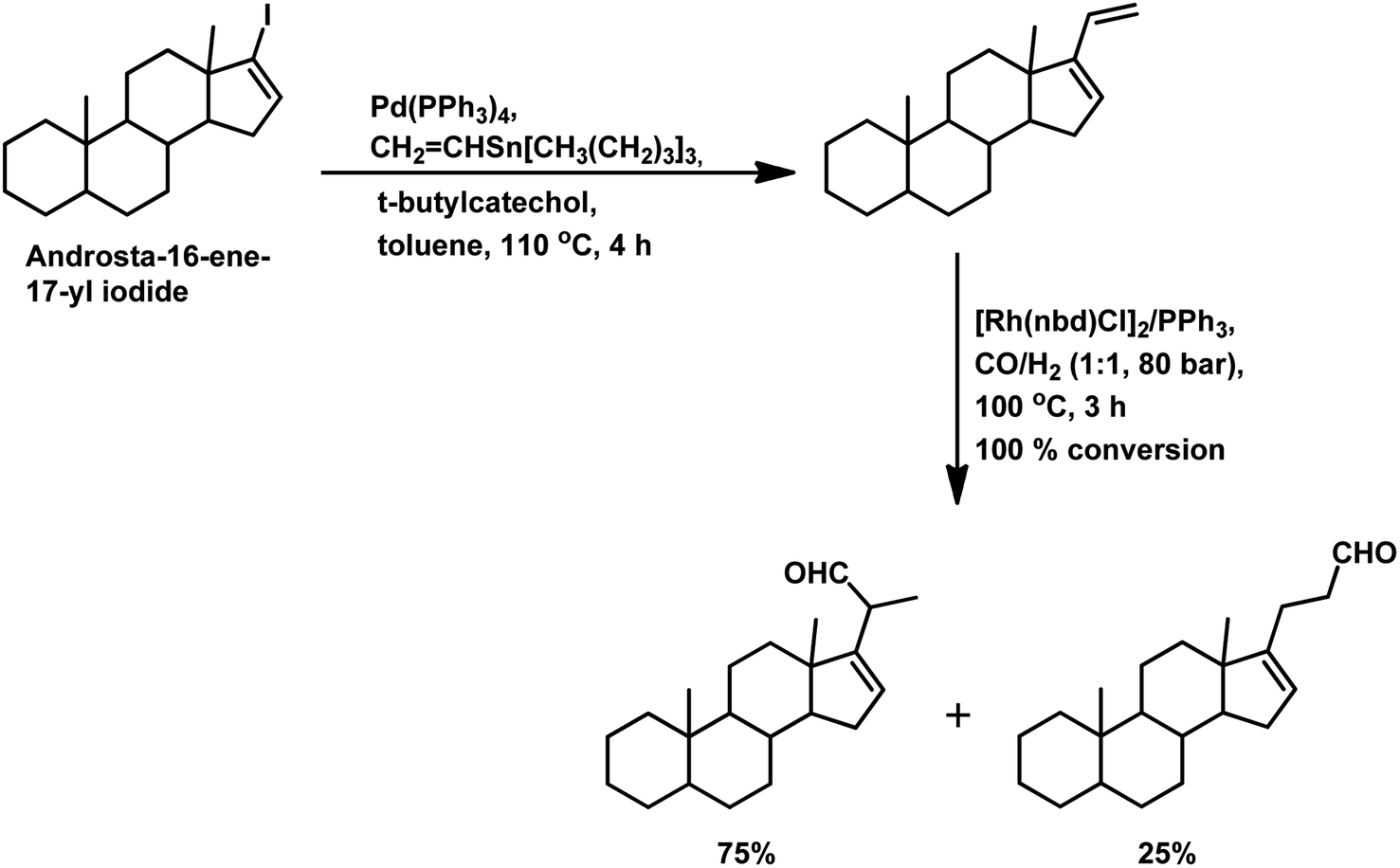 | ||
| Scheme 49 Two-step vinylation/hydroformylation of androsta-16-ene-17-yl iodide. | ||
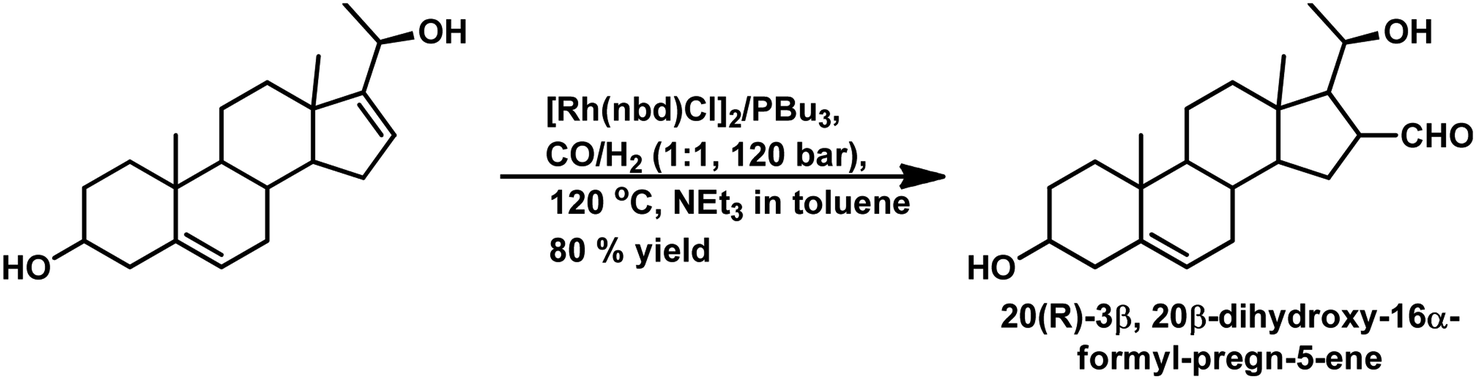 | ||
| Scheme 50 Synthesis of (20R)-3β, 20β-dihydroxy-16α-formyl-pregn-5-ene. | ||
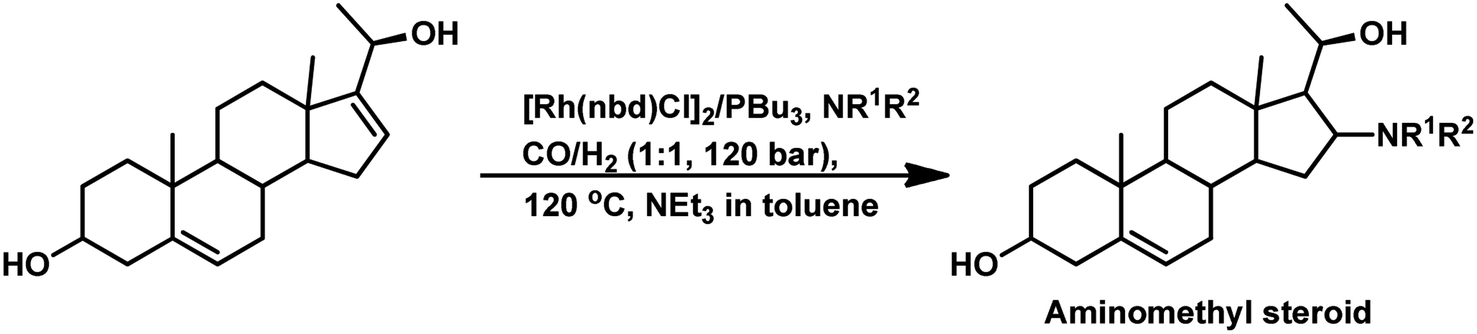 | ||
| Scheme 51 Synthesis of aminomethyl steroids through hydroaminomethylation. | ||
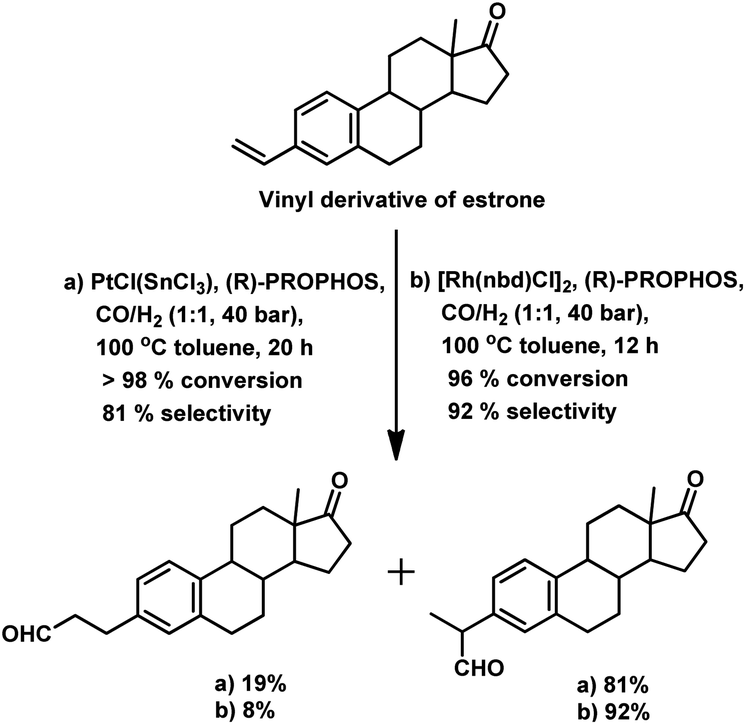 | ||
| Scheme 52 Hydroformylation of vinyl derivative of estrone. | ||
The hydroformylation of Δ4-steroids, namely, 17β-acetoxyandrost-4-ene and 3β,17β-diacetoxyandrost-4-ene, occurs in the [Rh2(μ-OMe)2(COD)2]/P(O-o-tBuC6H4)3 catalytic system at 100 °C and 20 bar of syngas pressure in toluene (Scheme 53).219 The [Rh(acac)(CO)2]/tris[2′-benzyloxy-1,1′-binaphthyl-2-yl]phosphite catalytic system (Rh(I)/L2-OBn) also acts as an efficient catalytic system in the hydroformylation of 17β-acetoxyandrost-4-ene with similar reaction conditions (Scheme 53).220 The studies on Δ4-steroids, namely, cholest-4-ene and 3β-acetoxycholest-4-ene, and that on Δ5-steroids, namely, 3β-acetoxycholest-5-ene and 3β-acetoxypregn-5-en-20-one, revealed that Δ4-steroids give 4-formyl derivatives with regiospecificity, while Δ5-steroids do not undergo hydroformylation.221 Androstene and pregnene derivatives undergo hydroformylation–amidocarbonylation in rhodium or rhodium–cobalt binary catalytic systems to grant amido steroidal derivatives (Scheme 54).222
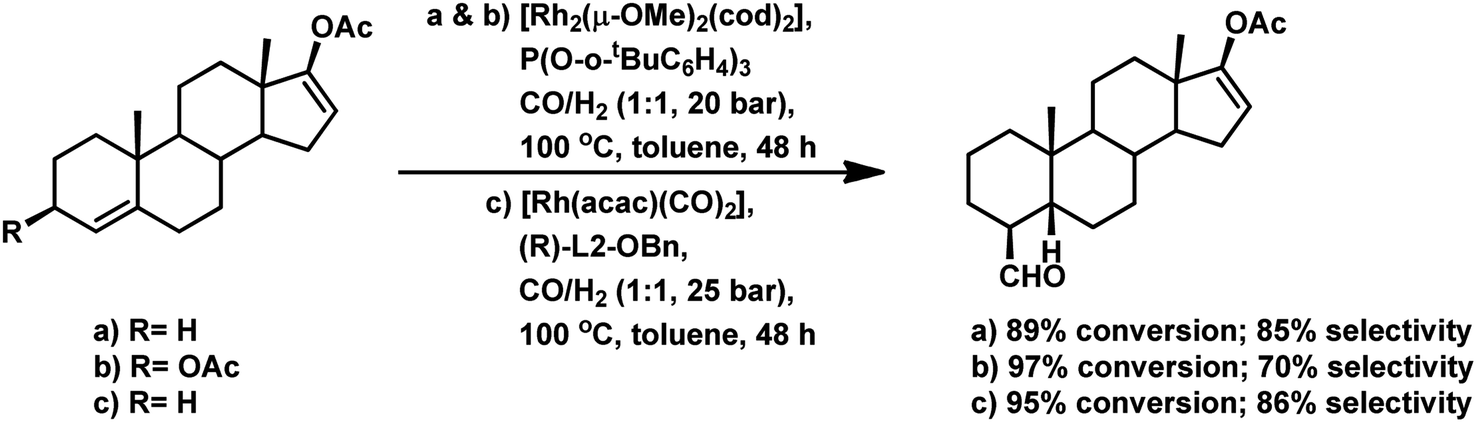 | ||
| Scheme 53 Hydroformylation of 17β-acetoxyandrost-4-ene 1 and 3β,17β-diacetoxyandrost-4-ene. | ||
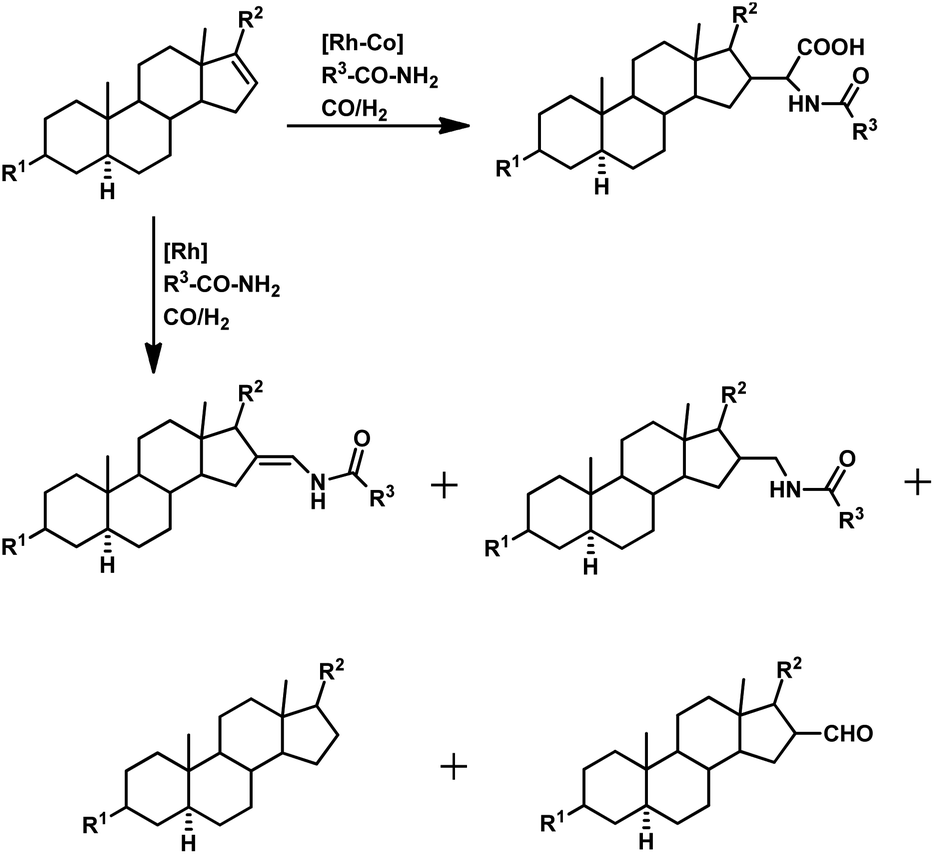 | ||
| Scheme 54 Hydroformylation–amidocarbonylation of a steroid. | ||
3 Hydroaminomethylation of natural olefins
3.1 Terpenes
An early report on the hydroformylation/reductive amination of (R)-(+)-limonene provided by Kranemann et al. showed the formation of tertiary amines in high yield of 93% with morpholine and 81% with diethylamine (Table 27).24 These amines act as growth regulators for tobacco plants.223|
|
||||
|---|---|---|---|---|
| Amine counterpart | Reaction conditions | Product | Conversion (%) | Yield (%) |
| Morpholine | [Rh(COD)Cl]2, CO/H2 (1:1, 80 atm), acetonitrile, 80 °C, 20 h |

|
— | 93 |
| Diethylamine | [Rh(COD)Cl]2, CO/H2 (1:1, 80 atm), acetonitrile, 80 °C, 20 h |
|
— | 81 |
| Di-n-butylamine | [Rh(COD)(μ-OMe)]2, PPh3, CO/H2 (1:1, 60 atm), toluene, 100 °C, 22 h |
|
100 | 81 |
| Morpholine | [Rh(COD)(μ-OMe)]2, PPh3, CO/H2 (1:1, 60 atm), toluene, 100 °C, 22 h |
|
100 | 88 |
| n-butylamine | [Rh(COD)(μ-OMe)]2, PPh3, CO/H2 (1:1, 60 atm), toluene, 100 °C, 4 h |
|
97 | 77 |
| 4-Methylpiperidine | [Rh(COD)(OMe)]2, (2,4-di-t BuPhO)3P, CO/H2 (1:1, 60 atm), solvent, 100 °C |
|
(a) 100 | (a) 98 |
| (a) Solvent: toluene, 6 h | (b) 100 | (b) 98 | ||
| (b) Solvent: ethanol, 48 h | ||||
| 1,2,3,4-Tetrahydroisoquinoline | [Rh(COD)(OMe)]2, (2,4-di-tBuPhO)3P, CO/H2 (1:1, 60 atm), solvent, 100 °C |
|
(a) 100 | (a) 98 |
| (a) Solvent: Toluene, 6 h | (b) 100 | (b) 95 | ||
| (b) Solvent: ethanol, 24 h | ||||
| Ammonia | [Rh(COD)Cl]2/TPPTS/CTAB, CO/H2 (1:1, 60 bar), water/toluene, 130 °C, 6 h |
|
— | 25 |
| Aniline | Ru3CO12, [BMMI]Cl, H3PO4, CO2/H2 (1:1, 60 bar), 120 °C, 36 h |
|
88 | 93 |
| Piperidine | Ru3CO12, (2-(dicyclohexylphosphanyl)-1-(2-methoxyphenyl)-1H-imidazole), Na2CO3, CO (40 bar), THF/water, 140 °C, 20 h |
|
— | 58 |
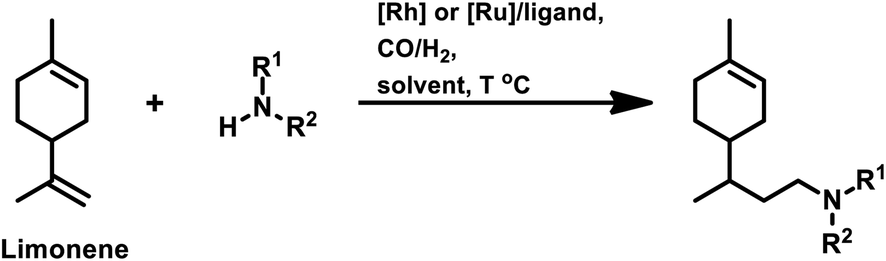
Years later, Graebin et al. demonstrated the one-pot hydroformylation/reductive amination of limonene with the use of different amine substrates, namely, isopropylamine, n-propylamine, benzylamine, piperidine, piperazine, morpholine, aniline and ethylenediamine, which gave the corresponding amines in moderate to good yields (Scheme 55).224
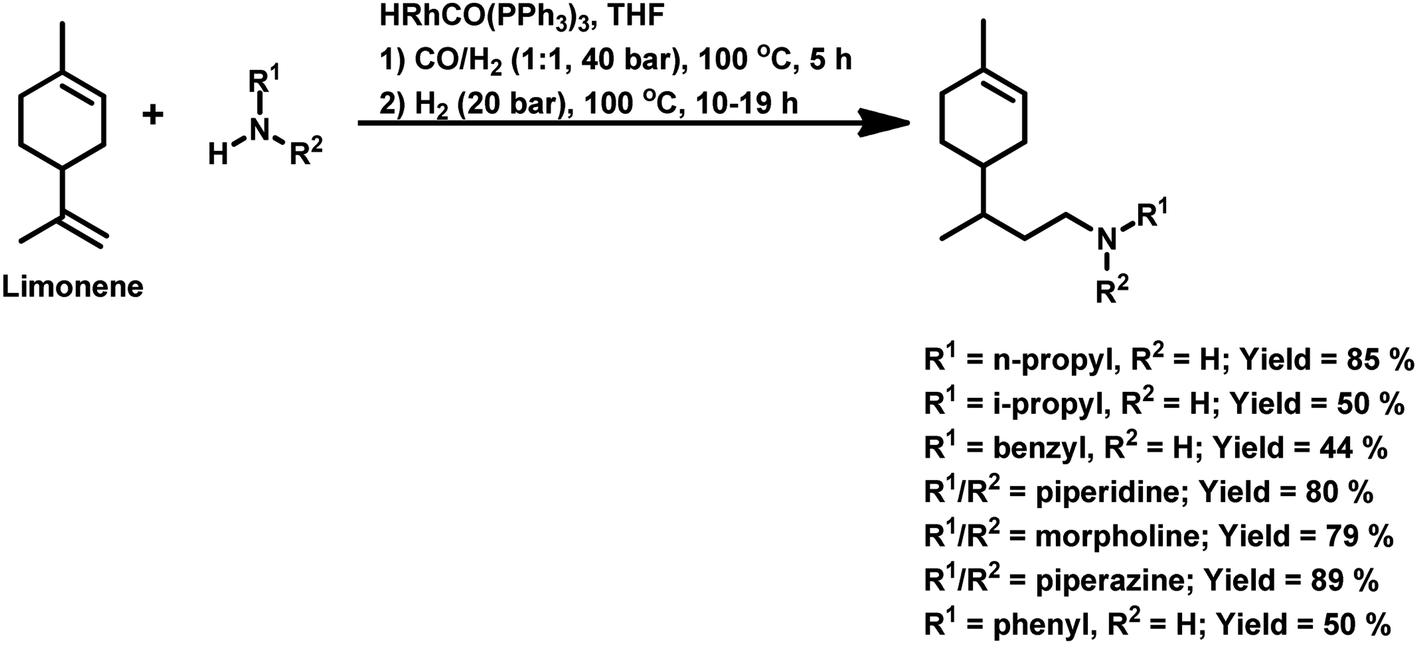 | ||
| Scheme 55 Two-step protocol for the tandem hydroaminomethylation of limonene. | ||
Melo et al. reported the hydroaminomethylation of the sterically encumbered natural olefins camphene, limonene, and β-pinene with their amine counterparts such as di-n-butylamine, morpholine and n-butylamine, obtaining moderate to good yields (75–94%) of the corresponding amines (Tables 27–29).25 The absence of ancillary ligands led to the double-bond isomerisation in these substrates, while, the presence of PPh3 retarded the isomerisation but resulted in the accumulation of aldehydes and enamines as the enamine/imine hydrogenation was slowed down, making the rate-determining reductive elimination step troublesome. Replacing PPh3 with PBn3 (tribenzylphosphine) in the hydroaminomethylation of β-pinene gave better results given that the bulkiness of the latter prevented the formation of inactive bis(ligand) rhodium species. The formation of the intermediates and the products showed a delicate balance based on the type and concentration of the ligands and the reaction time. These substrates were subjected to similar reaction conditions using morpholine, di-n-butylamine, 4-methylpiperidine or 1,2,3,4-tetrahydroisoquinoline as the condensation counterparts in green solvents such as anisole, p-cymene and ethanol to find an alternative to the conventional toxic solvent toluene.225 The novel amines were obtained in good to excellent yields. Anisole and p-cymene were observed to give results comparable with that of toluene, while ethanol rendered even better yields (in case of β-pinene). A bimetallic rhodium/cobalt catalytic system in glycerol endowed tertiary amines with good selectivity with (R)-limonene, β-pinene and camphene being the substrates.226 In these systems, the rhodium-based catalyst assists the hydroformylation step, while the cobalt-based catalyst favours the reductive amination step.
|
|
||||
|---|---|---|---|---|
| Amine counterpart | Reaction conditions | Product | Conversion (%) | Yield (%) |
| Di-n-butylamine | [Rh(COD)(μ-OMe)]2, PPh3, CO/H2 (1:1, 60 atm), toluene, 100 °C, 22 h |
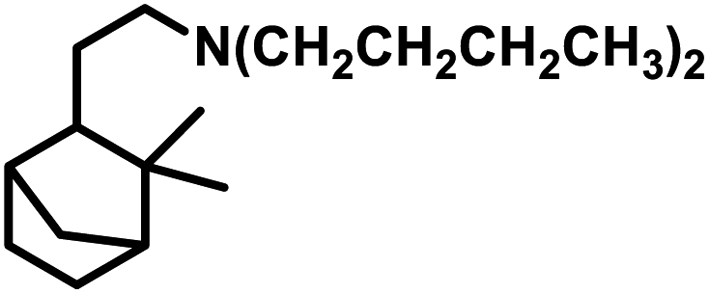
|
92 | 90 |
| Morpholine | [Rh(COD)(μ-OMe)]2, PPh3, CO/H2 (1:1, 60 atm), toluene, 100 °C, 22 h |
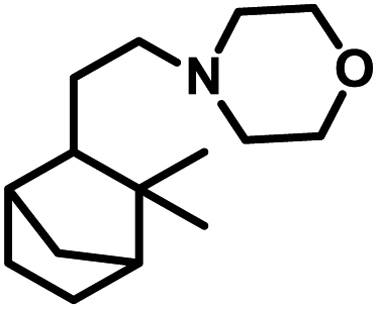
|
87 | 100 |
| n-Butylamine | [Rh(COD)(μ-OMe)]2, CO/H2 (1:1, 60 atm), toluene, 100 °C, 20 h |
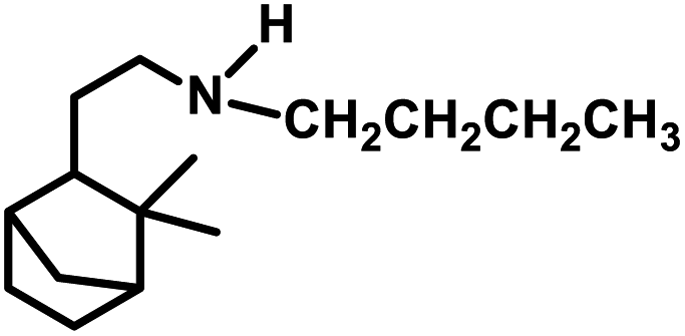
|
100 | 94 |
| 4-Methylpiperidine | [Rh(COD)(OMe)]2, CO/H2 (1:1, 60 atm), solvent, 100 °C |
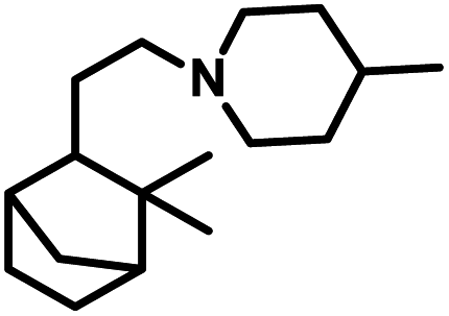
|
(a) 100 | (a) 98 |
| (a) Solvent: toluene, 24 h | (b) 100 | (b) 99 | ||
| (b) Solvent: ethanol, 48 h | ||||
| 1,2,3,4-Tetrahydroisoquinoline | [Rh(COD)(OMe)]2, CO/H2 (1:1, 60 atm), solvent, 100 °C |
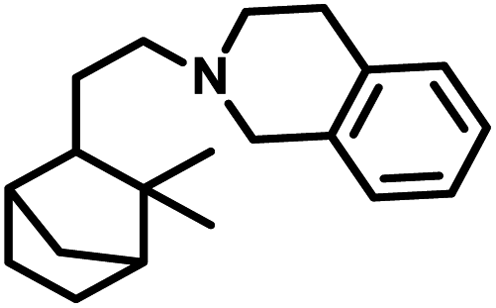
|
(a) 100 | (a) 98 |
| (a) Solvent: toluene, 24 h | (b) 87 | (b) 99 | ||
| (b) Solvent: ethanol, 48 h | ||||
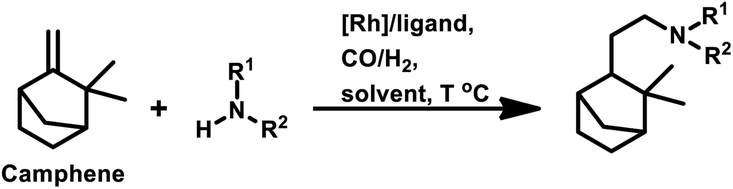
|
|
||||
|---|---|---|---|---|
| Amine counterpart | Reaction conditions | Product | Conversion (%) | Yield (%) |
| Di-n-butylamine | [Rh(COD)(μ-OMe)]2, PBn3, CO/H2 (1:1, 60 atm), toluene, 100 °C, 4 h |
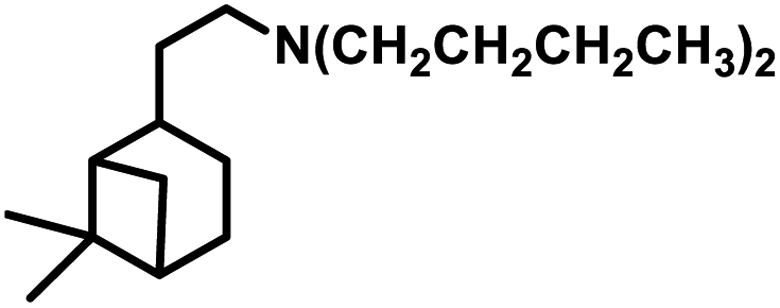
|
83 | 77 |
| 4-Methylpiperidine | [Rh(COD)(OMe)]2, L, CO/H2 (1:1, 60 atm), solvent, 100 °C |
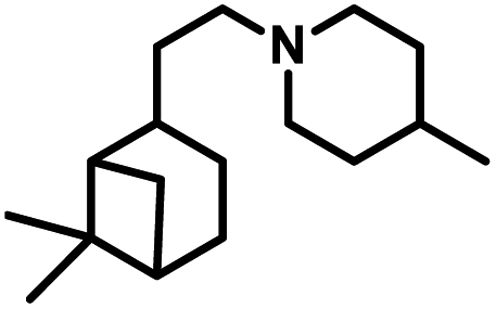
|
(a) 56 | (a) 84 |
| (a) Solvent: toluene, 24 h, PPh3 | (b) 97 | (b) 81 | ||
| (b) Solvent: ethanol, 24 h, (2,4-di-tBuPhO)3P | ||||
| 1,2,3,4-Tetrahydroisoquinoline | [Rh(COD)(OMe)]2, L, CO/H2 (1:1, 60 atm), solvent, 100 °C |
|
(a) 64 | (a) 67 |
| (a) Solvent: toluene, 48 h, PPh3 | (b) 99 | (b) 76 | ||
| (b) Solvent: ethanol, 24 h, (2,4-di-tBuPhO)3P | ||||
Limonene also undergoes hydroaminomethylation with ammonia as its counterpart amine in an aqueous biphasic system using the [Rh(COD)Cl]2/TPPTS catalyst complex together with the surfactant CTAC as a phase transfer promoter.227 The maximum amount of primary amine obtained is only 25% given that the reaction system is complicated with side-products such as isoterpinolene (from substrate isomerisation) and secondary and tertiary amines (obtained from further reaction of primary amines) (Table 27).
An efficient protocol for the hydroaminomethylation of limonene was designed by Ali et al. to procure the corresponding secondary amine in excellent yields by making use of the primary amine aniline (Table 27).228 Herein, an Ru3(CO)12/imidazolium salt ionic liquid was the catalytic system, while CO2 acted as a green source of CO, which makes the use of syngas unnecessary. The steps involved are sequential reversed water-gas shift, followed by hydroformylation, which is followed by the reduction of imines/enamines to give the final amine product.
Another protocol proceeds through a domino reversed water-gas shift/hydroaminomethylation reaction for limonene with the amine piperidine to give moderate yield of the resultant tertiary amine product (Table 27).229 This sequence is ruthenium catalysed and makes use of (2-(dicyclohexylphosphanyl)-1-(2-methoxyphenyl)-1H-imidazole) as the ligand. Under identical conditions, β-myrcene gets hydroaminomethylated to give 90% yield of the corresponding amine (Scheme 56).
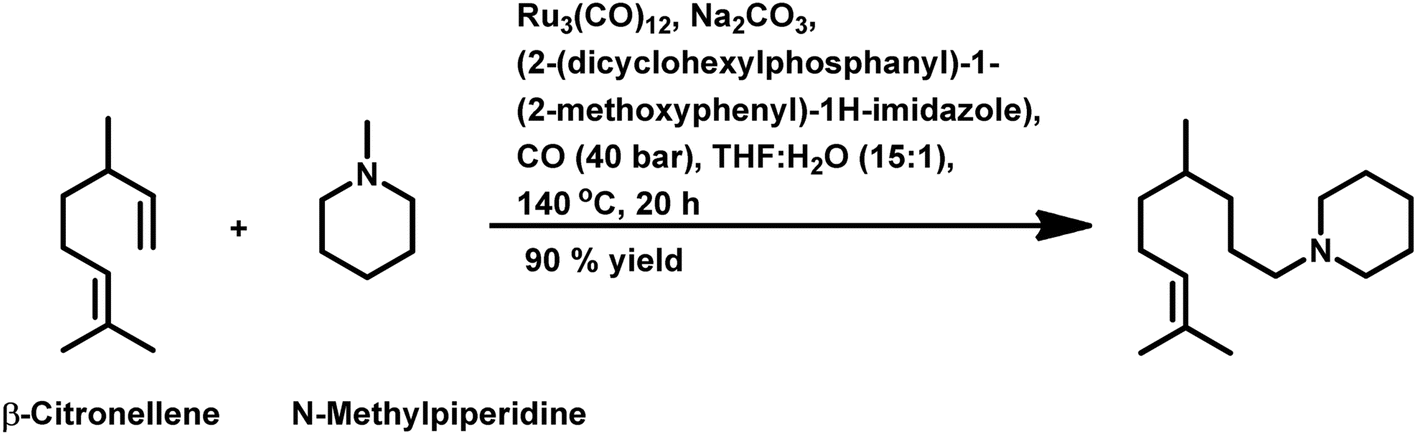 | ||
| Scheme 56 Hydroaminomethylation of β-citronellene. | ||
An Ru-catalysed system assisted by imidazolium salt ionic liquid gave a di-amine upon the hydroaminomethylation of carvone with aniline.228 This di-amine is formed by the hydroformylation/reductive amination of the alkene group and simultaneously the reductive amination of the carbonyl group already present in the substrate (Scheme 57). The source of CO is CO2, which serves as a greener alternative to syngas and the first step in this pathway is the reverse water-gas shift.
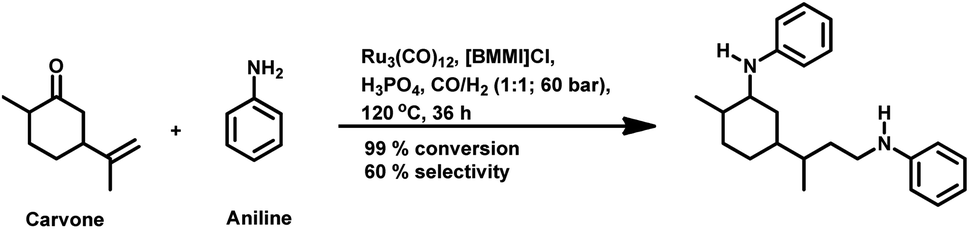 | ||
| Scheme 57 Hydroaminomethylation of carvone. | ||
β-Myrcene and β-farnesene were hydroformylated in the [Rh(COD)Cl]2/dppe system with diethylamine as the substrate amine in toluene solutions to obtain the corresponding amine in 70% yield (Scheme 58).230 The quaternary ammonium compounds derived from these amines were used to synthesise cationic surfactants via a green approach.
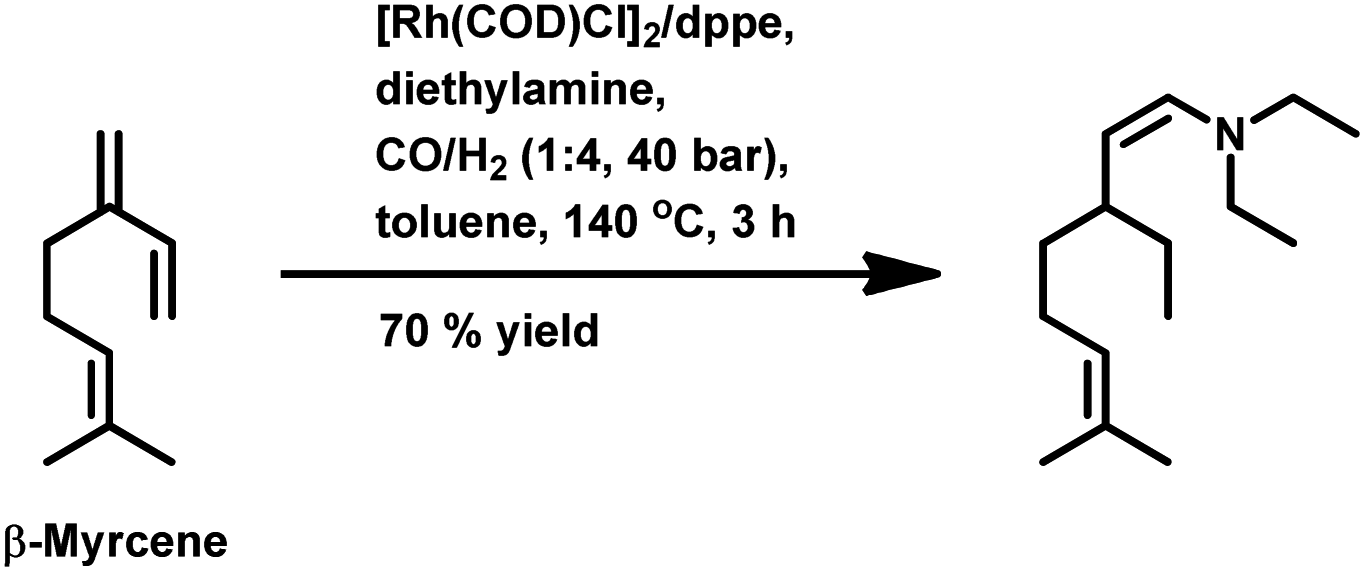 | ||
| Scheme 58 Hydroaminomethylation of β-myrcene. | ||
3.2 Ally/propenyl benzene derivatives
The first report on the hydroaminomethylation of eugenol by Oliveira et al. revealed an efficient process to obtain complete conversion, 95% yield of amines and 98% selectivity for the linear amine upon reaction with di-n-butylamine as the condensation counterpart, with the aid of NAPHOS as an ancillary and triflic acid as a promoter (Scheme 59).26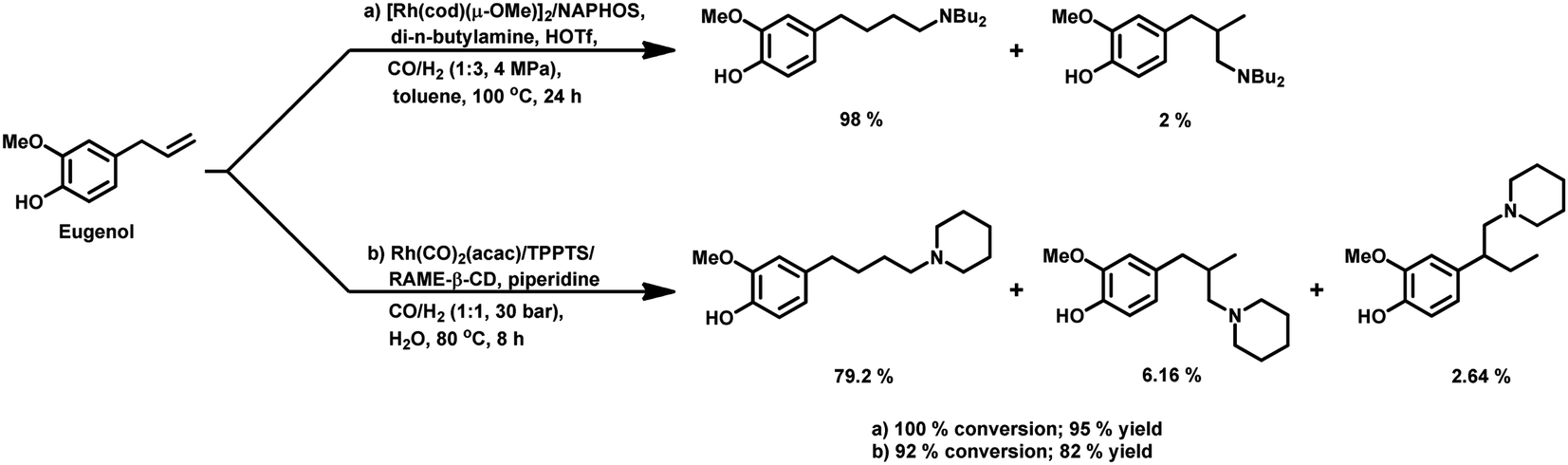 | ||
| Scheme 59 Hydroaminomethylation of eugenol. | ||
Later, Bhanage and co-workers demonstrated that this process is also feasible with a recyclable and greener system consisting of Rh/TPPTS/RAME-β-CD with piperidine as the substrate amine.231 Different cyclodextrins such as α-CD, β-CD, γ-CD, RAME-β-CD and hp-β-CD were screened, and eventually RAME-β-CD proved to be the most suitable mass transfer promoter. With eugenol as the substrate, this approach provided 92% conversion and 79.2% selectivity toward the linear amine (Scheme 59). With the same system and under almost the same conditions, estragole exhibited 96% conversion and 74% selectivity for the linear amine (Scheme 60b), while anethole resulted in 91% conversion and 81% of total amines (Scheme 61). This catalyst was recyclable for up to five cycles without any remarkable change in its activity or selectivity.
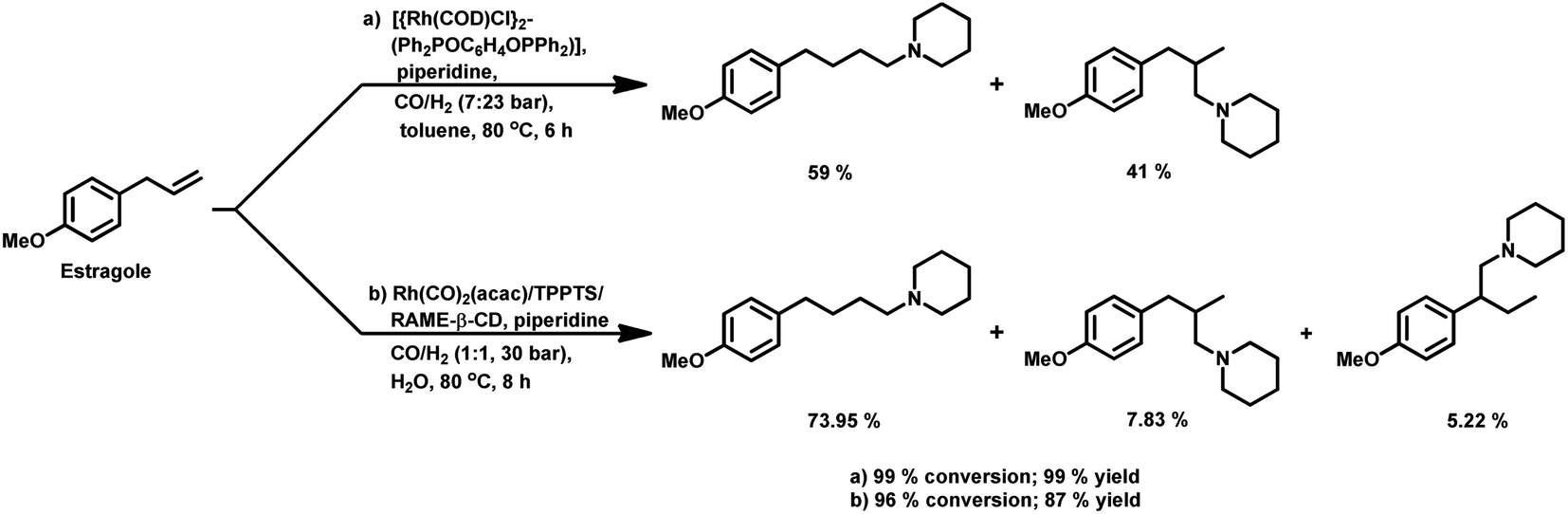 | ||
| Scheme 60 Hydroaminomethylation of estragole. | ||
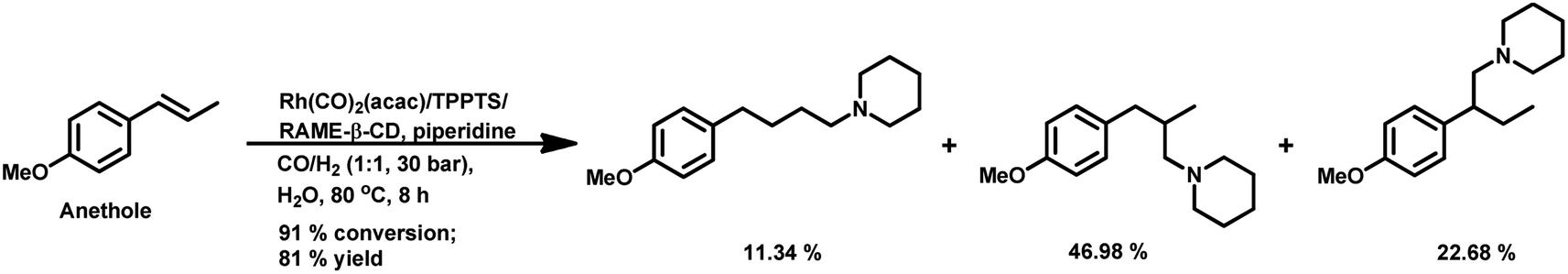 | ||
| Scheme 61 Hydroaminomethylation of anethole. | ||
One of the earliest mentions on the hydroaminomethylation of estragole was given by Khan et al. in 2013, wherein an Rh–diphosphinite complex, [{Rh(COD)Cl}2(Ph2POC6H4OPPh2)], in THF assisted this process.232 The condensation counterpart was piperidine and both 99% conversion and yield of amines were obtained (Scheme 60a). Another report brought revealed the efficiency of phospholes such as 1,2,3-triphenylphosphole (TPP), 1-phenydibenzophosphole (DBP) and 1,2,3,4,5-pentaphenylphosphole (PPP) in improving the chemo- and regio-selectivity in the hydroaminomethylation of estragole.233 With di-n-butylamine as the condensation counterpart in the hydroformylation/reductive amination of estragole, phospholes enhance the overall performance of the system. These systems have higher selectivity in the hydroformylation step compared to non-promoted or phosphite-promoted systems, while being more efficient in the reductive amination step than PPh3 (Table 30). Estragole gets hydroformylated in environmentally friendly solvents such as anisole, p-cymene and ethanol as an alternative to the toxic solvent toluene.225 These solvents show comparable results to that in toluene in terms of conversion, yield of amines and selectivity toward the resultant linear amine with morpholine, di-n-butylamine and 4-methyl piperidine being the amine counterparts (Table 31). The sequential hydroaminomethylation/hydrogenolysis of estragole with benzylamine in Rh/Pd catalytic system resulted in primary and secondary amines.234 The rhodium catalyst resulted in hydroaminomethylation, while the Pd/C catalyst played a role in hydrogenolysis (Scheme 62).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Ligand | Conversion (%) | Product distribution (%) | Regioselectivity (%) | ||||
| Substrate hydrogenation | Aldehydes | Amines | α | β | γ | ||
| None | 100 | 10 | 0 | 79 | 2 | 31 | 67 |
| PPh3 | 100 | 3 | 47 | 49 | 3 | 41 | 56 |
| TBPP | 100 | 0 | 0 | 87 | 10 | 32 | 58 |
| P(OPh)3 | 100 | 6 | 0 | 85 | 5 | 34 | 61 |
| TPP | 100 | 1 | 23 | 70 | 1 | 28 | 71 |
| PPP | 100 | 1 | 24 | 67 | 1 | 25 | 74 |
| DBP | 100 | 7 | 13 | 80 | 2 | 41 | 57 |
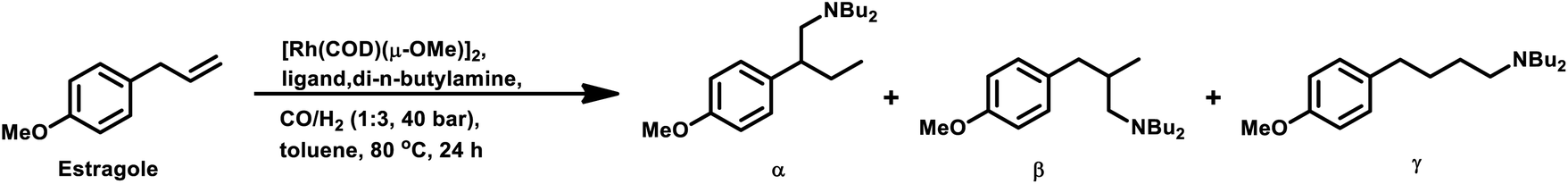
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Amine counterpart | Solvent | Time (h) | Conversion (%) | Total amines (%) | Product distribution (%) | ||
| α | β | γ | |||||
| Di-n-butylamine | Toluene | 24 | 100 | 75 | — | 39 | 60 |
| p-Cymene | 24 | 100 | 87 | — | 39 | 61 | |
| Anisole | 24 | 100 | 63 | 6 | 26 | 67 | |
| Ethanol | 24 | 100 | 83 | — | 42 | 58 | |
| Morpholine | Toluene | 6 | 100 | 95 | 16 | 38 | 46 |
| p-Cymene | 6 | 100 | 96 | 23 | 37 | 40 | |
| Anisole | 6 | 100 | 97 | 12 | 36 | 52 | |
| Ethanol | 6 | 100 | 99 | 3 | 40 | 57 | |
| 4-Methylpiperidine | Toluene | 24 | 100 | 97 | — | 47 | 53 |
| p-Cymene | 24 | 100 | 98 | 2 | 48 | 50 | |
| Anisole | 24 | 100 | 98 | — | 47 | 53 | |
| Ethanol | 24 | 100 | 97 | 3 | 46 | 51 | |
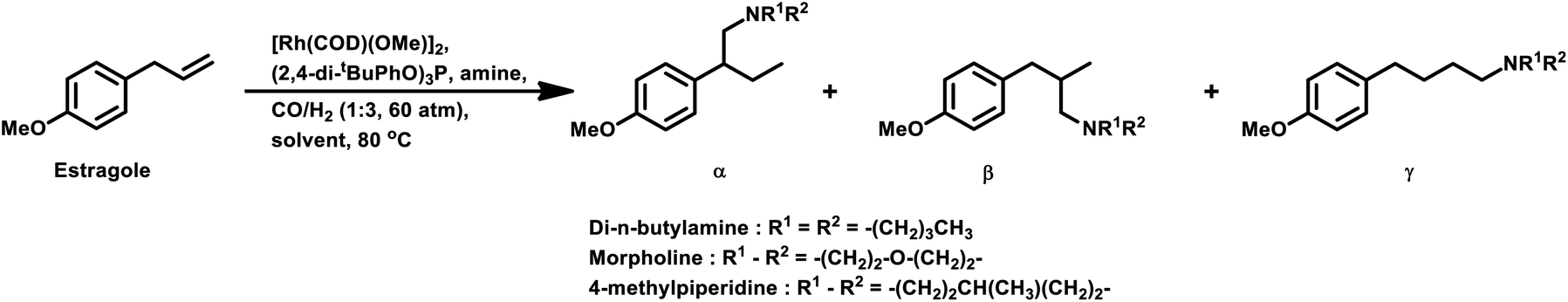
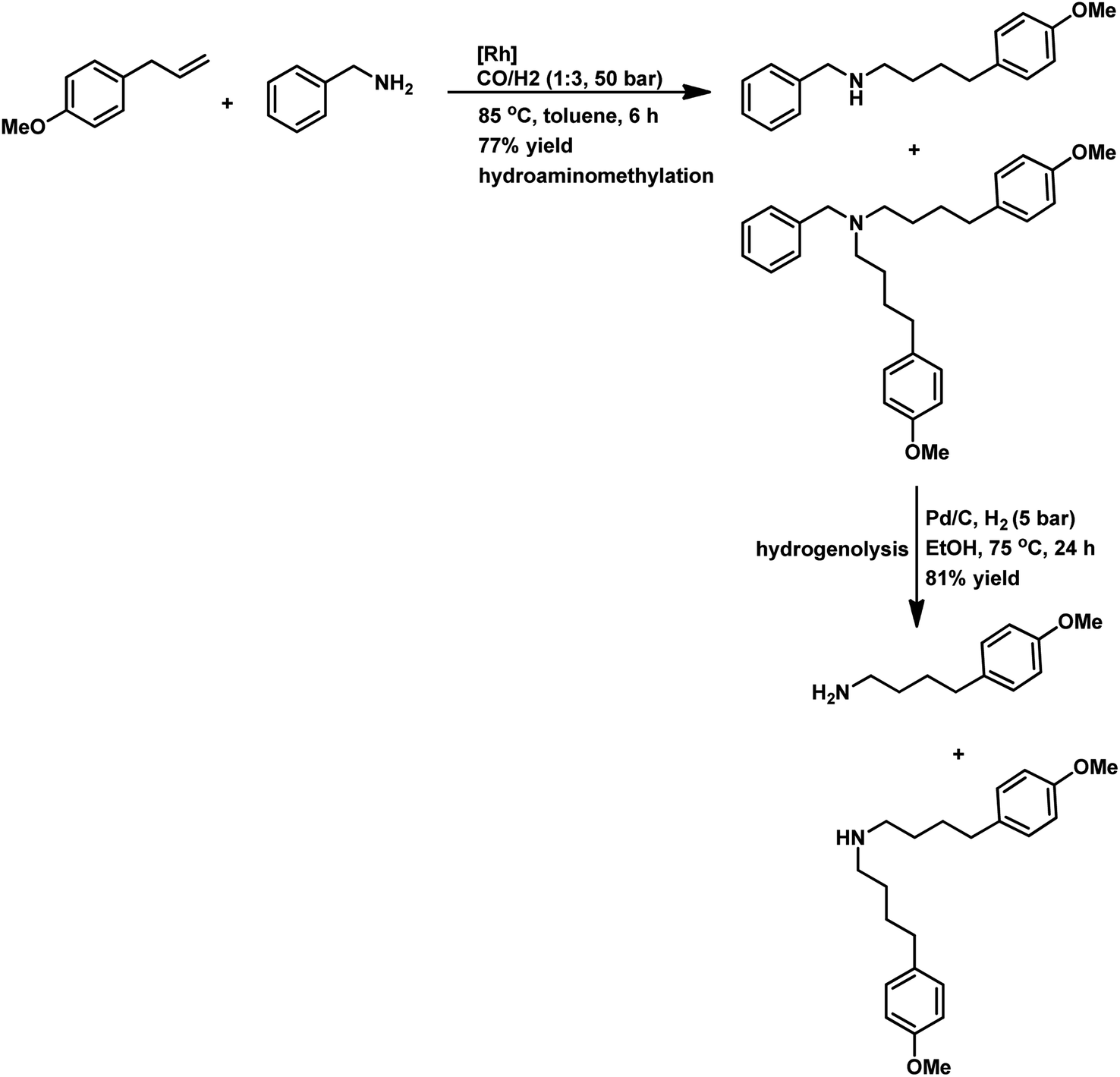 | ||
| Scheme 62 Sequential hydroaminomethylation/hydrogenolysis of estragole. | ||
3.3 Oleo compounds
HAM provides a very feasible route toward the synthesis of amino oleocompounds80,235 and these compounds have found applications as surface-active agents and in polymer chemistry.29The hydroaminomethylation of ethyl oleate takes place with primary or secondary amines when [Rh(COD)Cl]2 is used as the metal precursor under harsh conditions to give up to 99% yield of product amines (Scheme 63).236 Amines such as morpholine, benzylamine, hexylamine, diisopropylamine, valinol and aspartic diethyl acid can be employed. One unit of secondary amine reacts with one unit of ethyl oleate. On the contrary, one unit of primary amine reacts with two units of ethyl oleate and forms a dimer of fatty acid ester bridged by an amine group (Scheme 64). However, when primary amine is present in excess, the formation of this dimer is restricted and the reaction of one unit of ethyl oleate with only one unit of primary amine dominates. The same reaction of oleyl alcohol with morpholine gives a low yield of 47% for amines. The use of bis(ditertbutylphosphinomethyl)benzene (1,2-DTBPMB) as a ligand in the hydroaminomethylation of oleyl alcohol proved to be efficient under the optimised reaction conditions, giving a conversion of 98% and yield of 89% for the product amine.237 With the use of thermomorphic multicomponent solvents (TMS), recovering and recycling of the precious catalyst becomes possible. The one-pot [Rh(COD)Cl]2-catalysed hydroaminomethylation/esterification of methyl oleate with proline led to the formation of carboxylic diesters in a recyclable TMS system (Scheme 65).238 The introduction of phosphanes in this system promoted hydroformylation but retarded esterification. However, when 1 equivalent (with respect to proline) of methane sulphonic acid (MSA) was used in place of phosphane, 77% conversion and 77% selectivity of product amine were obtained. The hydroaminomethylation of methyl oleate with 3-(methylamino)-propionitrile (Scheme 66) resulted in the formation of the corresponding amino ester in a heptane/acetonitrile biphasic system; although the issues with catalyst separation and recyclability of the system are a concern.239 The reaction of soybean fatty acid methyl ester with n-butyl amine and the catalyst precursor HRhCO(PPh3)3 at 100 °C and 40 bar (CO/H2 = 2/1) in the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate yielded the intermediate enamine but failed to yield the HAM products.240 The hydroaminomethylation/hydrohydroxymethylation (HHM) cascade reaction (Scheme 67) works efficiently, wherein the triglyceride-grafted tertiary amine acts as a ligand in the presence of a rhodium catalyst and reduces the formyl groups in the remaining aldehydes to give the corresponding alcohols.241 The synthesis of aminohydroxylated triglycerides was reported to occur through this cascade process under aqueous biphasic conditions.242
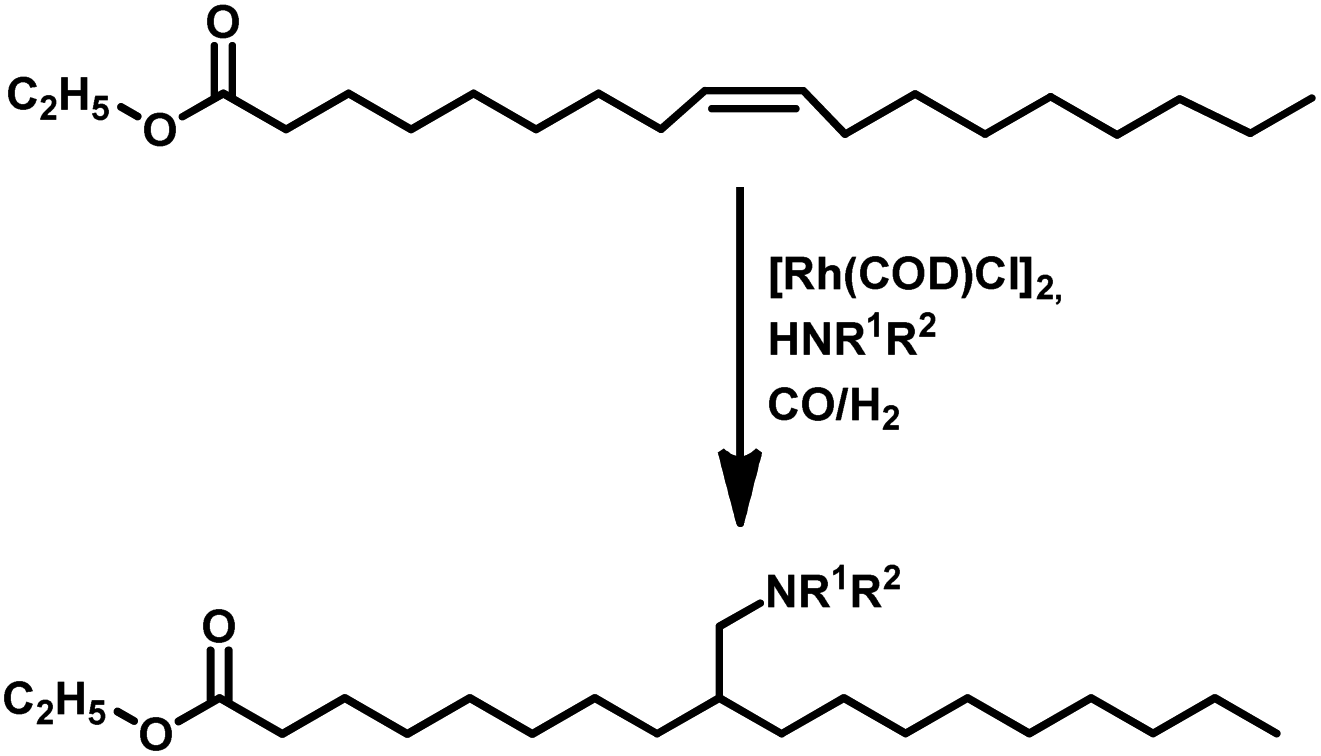 | ||
| Scheme 63 Hydroaminomethylation of ethyl oleate. | ||
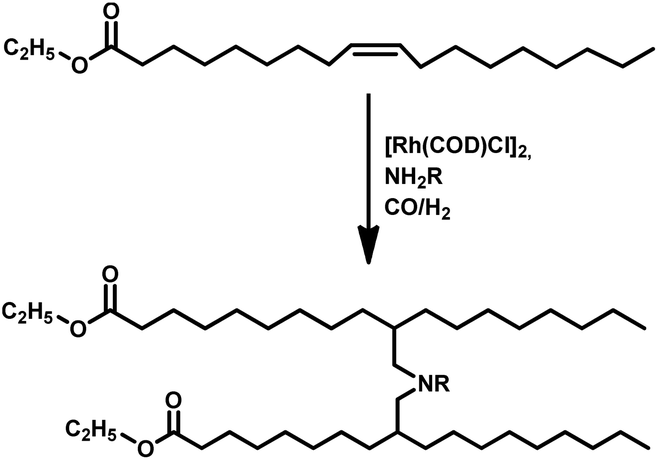 | ||
| Scheme 64 Hydroaminomethylation of ethyl oleate with a primary amine. | ||
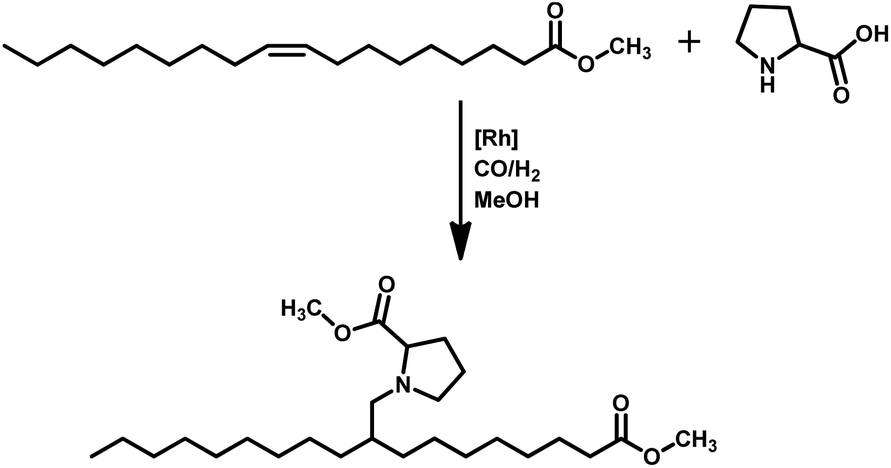 | ||
| Scheme 65 Hydroaminomethylation/esterification of methyl oleate using proline. | ||
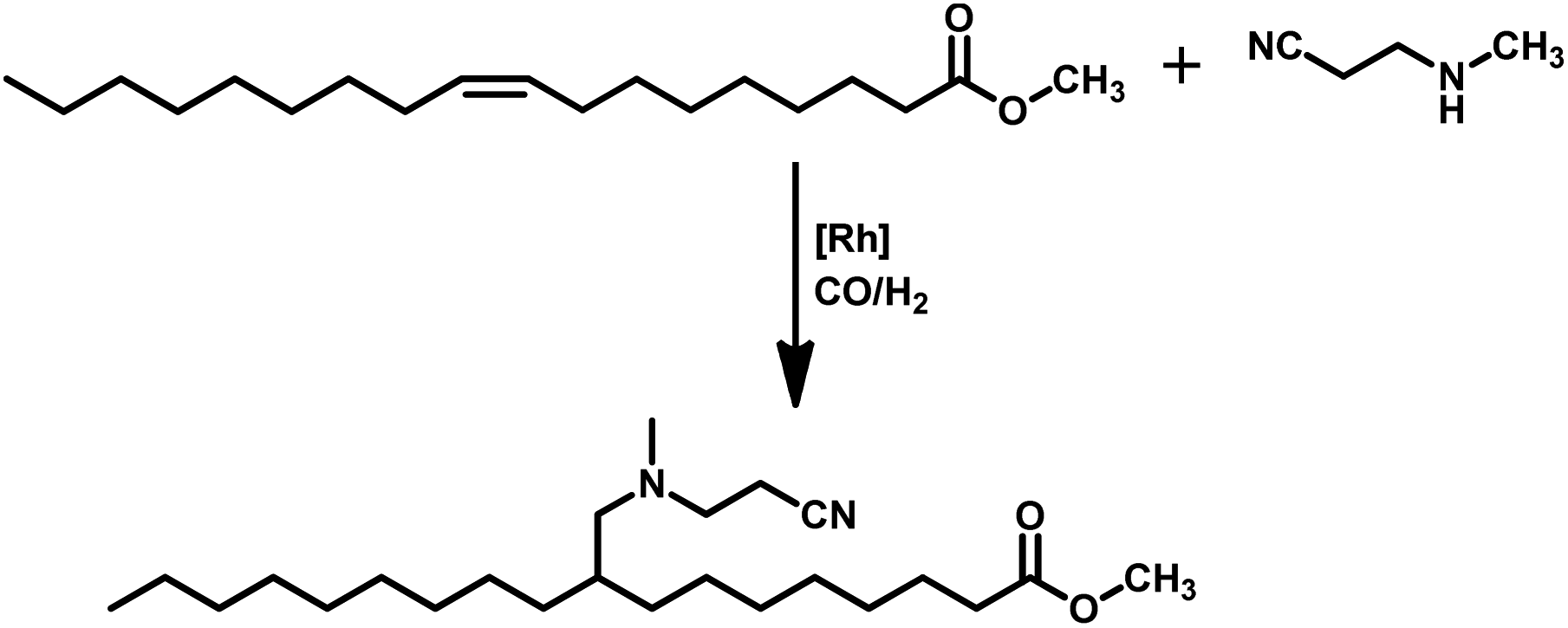 | ||
| Scheme 66 Hydroaminomethylation of methyl oleate with 3-(methylamino)-propionitrile. | ||
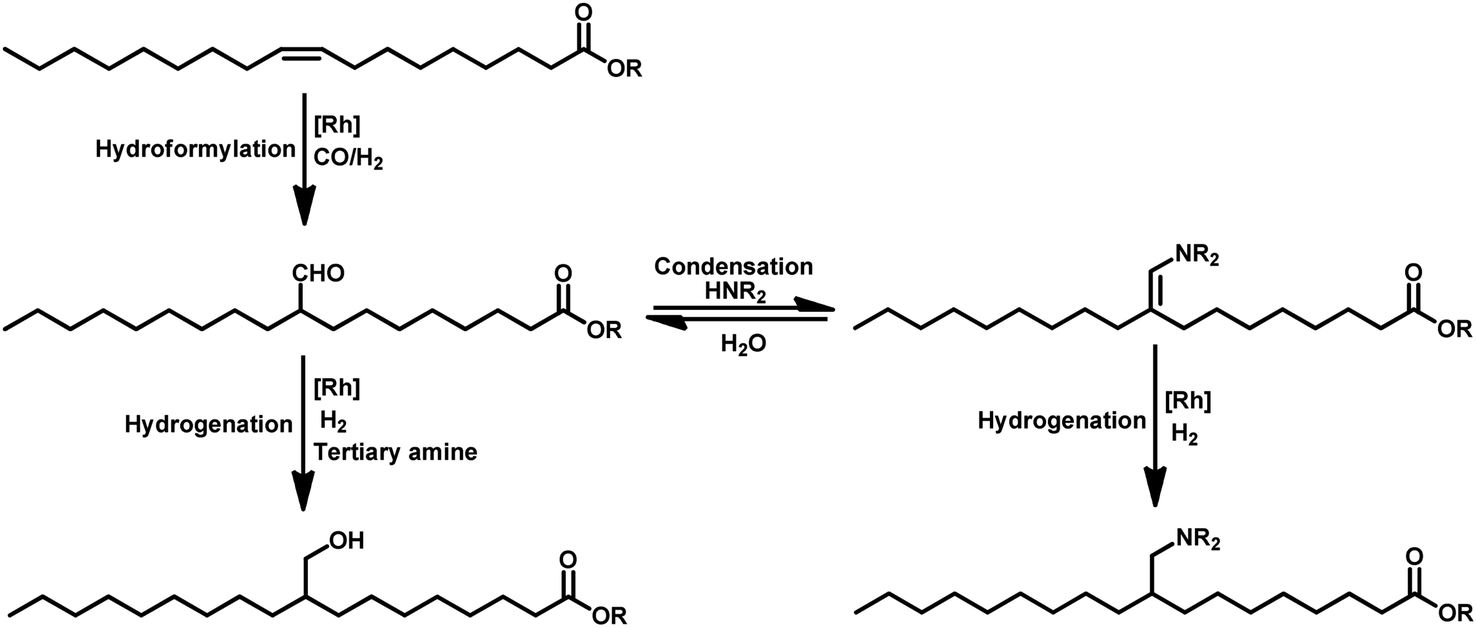 | ||
| Scheme 67 Hydroaminomethylation/hydrohydroxymethylation of triglycerides. | ||
3.4 Steroids
The hydroaminomethylation of Δ16-steroids with aminoalcohols in a rhodium-catalysed one-pot catalytic system provided hydroxy-aminomethyl derivatives of steroids with high chemo- and regio-selectivity (Scheme 68).243 | ||
| Scheme 68 Hydroaminomethylation of a steroid with aminoalcohol. | ||
4 Conclusion
This review discussed the use of sustainable, bio-renewable feedstocks as starting materials in hydroformylation and associated one-pot processes to obtain aldehydes and their derivatives, which act as intermediates in the production of other useful chemicals utilised in the food, flavours, fragrances, cosmetics and other industries. Four categories of natural substrates were summarised for this purpose, namely, terpenes, allyl/propenyl benzene derivatives, oleo compounds and steroids. The use of these starting materials is a smart move toward sustainability through SDG 12 “Responsible Consumption and Production” and SDG 9 “Industry, Innovation and Infrastructure” in an effort to prevent resource depletion in the near future.Hydroformylation reaction follows the green principles of atom economy and waste prevention. With the emergence of new catalyst precursors and ligands, smart catalysis comes into picture and provides highly efficient chemo- and regio-selective systems, which work under mild conditions of temperatures and pressures, leading to energy efficiency and safer chemistry for accident prevention. Furthermore, some of the systems make use of eco-friendly solvents, thus fulfilling the green principle of safer solvents.
Numerous systems have been developed with the vision of catalyst recovery and recyclability, and hence reducing waste. Tandem reactions such as hydroformylation-acetalisation, hydroformylation-cyclisation, hydroaminomethylation and others are based on the principles of waste prevention and reduction of derivatives with added advantages of labour minimisation and time reduction.
Exploration of new substrates and tandem reactions involving hydroformylation, development of new ligands to provide high enantiomeric purity to the products and recyclable systems with the benefits of homogenous systems have a huge future scope. Also, more cost-effective ligands that provide high regioselectivity to the system with respect to either the linear or branched aldehyde need to be designed. Rhodium complexes are the most commercially utilised catalysts for this process. However, considering the cost of this precious metal, it is necessary to search for cheaper metal catalysts that work better or at least as efficiently as rhodium catalysts. Designing catalytic systems that utilize CO surrogates or CO2 gas in place of CO gas would help reduce the risk involved with the handling of toxic syngas.
In conclusion, bio-renewable olefins act as sustainable sources in hydroformylation- and hydroformylation-derived tandem processes through SDG 12 and SDG 9 to obtain valuable chemicals by observing the principles of green chemistry.
Data availability
The data supporting this article are provided within this manuscript.Conflicts of interest
Authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The author (Rupali S. Prajapati) is grateful for the Research Fellowship from the University Grants Commission (UGC, India). We acknowledge financial support from Department of Science and Technology sponsored Indo-Russian Project No. DST/INT/RUS/RSF/P54/2021.References
- (a) A. Behr and L. Johnen, Alternative Feedstocks for Synthesis, Ch-J. Li Handbook of Green Chemistry, Green Synthesis, Wiley-VCH, Weinheim, 1st edn, 2012, vol. 7, pp. 69–92 Search PubMed; (b) H. Wu, H. Li and Z. Fang, Green Chem., 2021, 23, 6675–6697 RSC; (c) J. Li, P. Sudarsanam and H. Li, Trends Chem., 2023, 5, 649–652 CrossRef CAS.
- (a) K.-D. Wiese and D. Obst, Hydroformylation, in Catalytic Carbonylation Reactions, ed. M. Beller, Springer, Berlin, Heidelberg, 2006, vol. 18, pp. 1–33 Search PubMed; (b) K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2008, pp. 127–144 Search PubMed; (c) C. W. Kohlpainter, M. Schulte, J. Falbe, P. Lappe and J. Weber, Ullmann's Encyclopedia of Industrial Chemistry, Wiley–VCH, Weinheim, 7th edn, 2005 Search PubMed; (d) E. F. Lutz, J. Chem. Educ., 1986, 63, 202–203 CrossRef CAS.
- (a) O. Roelen, DE Pat., DE 849548, 1952 Search PubMed; (b) O. Roelen, US Pat., US 2327066, 1943 Search PubMed; (c) B. Cornils, W. A. Herrmann and M. Rasch, Angew Chem., Int. Ed., 1994, 33, 2144–2163 CrossRef.
- B. Breit and W. Seiche, Synthesis, 2001, 1–36 CrossRef CAS.
- (a) B. Breit, Acc. Chem. Res., 2003, 36, 264–275 CrossRef CAS PubMed; (b) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (c) B. M. Bhanage, S. S. Divekar, R. M. Deshpande and R. V. Chaudhari, J. Mol. Catal. A: Chem., 1997, 115, 247–257 CrossRef CAS; (d) R. V. Chaudhari, B. M. Bhanage, R. M. Deshpande and H. Delmas, Nature, 1995, 373, 501–503 CrossRef CAS.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS; (b) B. M. Trost, Angew Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC; (b) Y. Kim and C. J. Li, Green Synth. Catal., 2020, 1, 1–11 CrossRef.
- F. E. Paulik, Catal. Rev., 1972, 6, 49–84 CrossRef CAS.
- (a) J. A. Osborn, G. Wilkinson and J. F. Young, Chem. Commun., 1965, 17 RSC; (b) D. Evans, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 33, 3133–3142 RSC.
- A. Behr and P. Neubert, Applied Homogeneous Catalysis, Wiley, Weinheim, 2012 Search PubMed.
- (a) M. L. Clarke, Curr. Org. Chem., 2005, 9, 701–718 CrossRef CAS; (b) Y. Ning, T. Ohwada and F.-E. Chen, Green Synth. Catal., 2021, 2, 247–266 CrossRef.
- (a) P. T. Anastas and I. T. Horváth, Chem. Rev., 2007, 107, 2167–2168 CrossRef; (b) P. T. Anastas, Handbook of Green Chemistry, Wiley-VCH, Weinheim, 2009 Search PubMed; (c) L. Tietze and U. Beifuss, Angew. Chem., 1993, 105, 137–170 CrossRef CAS; (d) P. T. Anastas and J. C. Warner, 12 Principles of Green Chemistry – American Chemical Society, Oxford University Press, 1998 Search PubMed.
- (a) L. F. Tietze and U. Beifuss, Angew Chem., Int. Ed., 1993, 32, 131–163 CrossRef; (b) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS; (c) L. F. Tietze, G. Brasche, K. M. Gericke and L. Friedjan, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef.
- (a) E. N. Frankel, J. Am. Oil Chem. Soc., 1976, 53, 138–141 CrossRef CAS; (b) S. R. Khan and B. M. Bhanage, Tetrahedron Lett., 2013, 54, 5998–6001 CrossRef CAS.
- K. Soulantica, S. Sirol, S. Koïnis, G. Pneumatikakis and P. Kalck, J. Organomet. Chem., 1995, 498, 10–13 CrossRef.
- C. G. Vieira, J. G. da Silva, C. A. A. Penna, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2010, 380, 125–132 CrossRef CAS.
- R. Roggenbuck and P. Eilbracht, Tetrahedron Lett., 1999, 40, 7455–7456 CrossRef CAS.
- S. Sirol and P. Kalck, New J. Chem., 1997, 21, 1129–1137 CAS.
- P. N. Rylander and H. Greenfield, Catalysis of Organic Reactions, ed. A. J. Chalk, Dekker, New York, 1988, pp. 43–63 Search PubMed.
- A. O. de Dias, R. Augusti, E. N. dos Santos and E. V. Gusevskaya, Tetrahedron Lett., 1997, 38, 41–44 CrossRef.
- C. G. Vieira, M. C. de Freitas, E. N. dos Santos and E. V. Gusevskaya, ChemCatChem, 2012, 4, 795–801 CrossRef CAS.
- W. H. Clement and M. Orchin, Ind. Eng. Chem. Process Res. Dev., 1965, 4, 283–286 CAS.
- (a) A. Behr and J. P. Gomes, Eur. J. Lipid Sci. Technol., 2010, 112, 31–50 CrossRef CAS; (b) A. Börner, M. Beller and B. Wünsch, Sci. Synth., 2009, 40a, 111–B Search PubMed; (c) S. R. Khan, M. V. Khedkar, Z. S. Qureshi, D. B. Bagal and B. M. Bhanage, Tetrahedron Lett., 2011, 15, 141–145 CAS.
- C. L. Kranemann and P. Eilbracht, Synthesis, 1998, 71–77 CrossRef CAS.
- D. S. Melo, S. S. Pereira-Júnior and E. N. dos Santos, Appl. Catal., A, 2012, 411–412, 70–76 CrossRef CAS.
- K. C. B. Oliveira, A. G. Santos and E. N. dos Santos, Appl. Catal., A, 2012, 445–446, 204–208 CrossRef CAS.
- (a) Z. Sahli, B. Sundararaju, M. Achard and C. Bruneau, Org. Lett., 2011, 13, 3964–3967 CrossRef CAS; (b) A. Behr and A. Westfechtel, Chem. Ing. Tech., 2007, 79, 621–636 CrossRef CAS.
- A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC.
- T. Vanbésien, J. Le Nôtre, E. Monflier and F. Hapiot, Eur. J. Lipid Sci. Technol., 2018, 120, 1700190 CrossRef.
- (a) M. Taddei and A. Mann, Hydroformylation for Organic Synthesis, Springer, Berlin, Heidelberg, 2013 CrossRef; (b) A. C. Brezny and C. R. Landis, Acc. Chem. Res., 2018, 51, 2344–2354 CrossRef CAS PubMed; (c) A. Cunillera, C. Godard and A. Ruiz, Asymmetric hydroformylation using rhodium, in Rhodium Catalysis: Topics in Organometallic Chemistry, ed. C. Claver, Springer, Cham, 2017, vol. 61, pp. 99–143 Search PubMed; (d) B. P. Bondzic, J. Mol. Catal. A: Chem., 2015, 408, 310–334 CrossRef CAS.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS.
- (a) T. Kégl, RSC Adv., 2015, 5, 4304–4327 RSC; (b) A. Börner and R. Franke, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2016 CrossRef; (c) S. S. Nurttila, P. R. Linnebank, T. Krachko and J. N. H. Reek, ACS Catal., 2018, 8, 3469–3488 CrossRef CAS PubMed; (d) R. Lazzaroni, R. Settambolo, G. Alagona and C. Ghio, Coord. Chem. Rev., 2010, 254, 696–706 CrossRef CAS; (e) E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 382–411 CrossRef CAS.
- A. Behr and A. J. Vorholt, Top. Organomet. Chem., 2012, 39, 103–128 CrossRef CAS.
- (a) D. Evans, G. Yagupsky and G. Wilkinson, J. Chem. Soc. A, 1968, 2660–2665 RSC; (b) G. Yagupsky, C. K. Brown and G. Wilkinson, J. Chem. Soc. A, 1970, 1392–1401 RSC; (c) C. K. Brown and G. Wilkinson, J. Chem. Soc. A, 1970, 2753–2764 RSC.
- R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1961, 83, 4023–4027 CrossRef.
- (a) C. Dwyer, H. Assumption, J. Coetzee, C. Crause, L. Damoense and M. Kirk, Coord. Chem. Rev., 2004, 248, 653–669 CrossRef CAS; (b) L. Damoense, M. Datt, M. Green and C. Steenkamp, Coord. Chem. Rev., 2004, 248, 2393–2407 CrossRef CAS.
- P. W. N. M. van Leeuwen, Introduction to hydroformylation, Rhodium Catalyzed Hydroformylation: Catalysis by Metal Complexes, ed. C. Claver, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000, vol. 22, pp. 1–13 Search PubMed.
- G. Dümbgen and D. Neubauer, Chem. Ing. Tech., 1969, 41, 974–980 CrossRef.
- A. M. Trzeciak and J. J. Ziółkowski, Coord. Chem. Rev., 1999, 190–192, 883–900 CrossRef CAS.
- (a) D. W. P. Tay, J. D. Nobbs, C. Romain, A. J. P. White, S. Aitipamula, M. Van Meurs and G. J. P. Britovsek, ACS Catal., 2020, 10, 663–671 CrossRef CAS; (b) Y. Zhang, S. Torker, M. Sigrist, N. Bregović and P. Dydio, J. Am. Chem. Soc., 2020, 142, 18251–18265 CrossRef CAS PubMed.
- (a) G. Franciò, R. Scopelliti, C. G. Arena, G. Bruno, D. Drommi and F. Faraone, Organometallics, 1998, 17, 338–347 CrossRef; (b) S. Rojas, J. L. García Fierro, R. Fandos, A. Rodríguez and P. Terreros, J. Chem. Soc., Dalton Trans., 2001, 2316–2324 RSC; (c) M. L. Clarke, Angew. Chem., Int. Ed., 2016, 55, 13377 CrossRef CAS.
- (a) D. Evans, J. A. Osborn, F. H. Jardine and G. Wilkinson, Nature, 1965, 208, 1203–1204 CrossRef CAS; (b) H. F. Schulz and F. Bellstedt, Ind. Eng. Chem. Prod. Res. Dev., 1973, 12, 176–183 CrossRef CAS; (c) I. Fleischer, L. Wu, I. Profir, R. Jackstell, R. Franke and M. Beller, Chem.–Eur. J., 2013, 19, 10589–10594 CrossRef CAS PubMed; (d) A. Kämper, P. Kucmierczyk, T. Seidensticker, A. J. Vorholt, R. Franke and A. Behr, Catal. Sci. Technol., 2016, 6, 8072–8079 RSC.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS PubMed.
- (a) L. J. Kehoe and R. A. Schell, J. Org. Chem., 1970, 35, 2846–2848 CrossRef CAS; (b) E. Billig and R. L. Pruett, Hydroformylation of Olefins, in Inorganic Reactions and Methods, 2007, vol. 16, pp. 353–354 Search PubMed.
- (a) J. Palágyi and L. Markó, J. Organomet. Chem., 1982, 236, 343–347 CrossRef; (b) Z. He, N. Lugan, D. Neibecker, R. Mathieu and J.-J. Bonnet, J. Organomet. Chem., 1992, 426, 247–259 CrossRef CAS.
- R. Kumar and S. H. Chikkali, J. Organomet. Chem., 2022, 1960, 122231 CrossRef.
- T. Tewari, R. Kumar, A. C. Chandanshive and S. H. Chikkali, Chem. Rec., 2021, 21, 1182–1198 CrossRef CAS.
- S. H. Chikkali, I. J. Van Der Vlugt and J. N. H. Reek, Coord. Chem. Rev., 2014, 262, 1–15 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- (a) C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. A. Gavney and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 5535–5543 CrossRef CAS; (b) M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- (a) P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton trans., 1999, 1519–1529 RSC; (b) P. W. N. M. van Leeuwen, P. C. J. Kamer, L. A. van der Veen and J. N. H. Reek, Chin. J. Chem., 2001, 19, 1–8 CrossRef CAS.
- M. Diéguez, O. Pàmies and C. Claver, Tetrahedron: Asymmetry, 2004, 15, 2113–2122 CrossRef.
- (a) J. Huang, E. Bunel, A. Allgeier, J. Tedrow, T. Storz, J. Preston, T. Correll, D. Manley, T. Soukup, R. Jensen, R. Syed, G. Moniz, R. Larsen, M. Martinelli and P. J. Reider, Tetrahedron Lett., 2005, 46, 7831–7834 CrossRef CAS; (b) T. P. Clark, C. R. Landis, S. L. Freed, J. Klosin and K. A. Abboud, J. Am. Chem. Soc., 2005, 127, 5040–5042 CrossRef CAS PubMed; (c) Y. Yan and X. Zhang, J. Am. Chem. Soc., 2006, 128, 7198–7202 CrossRef CAS.
- S. Gladiali, J. Carles Bayón and C. Claver, Tetrahedron: Asymmetry, 1995, 6, 1453–1474 CrossRef CAS.
- (a) J. Klosin and C. R. Landis, Acc. Chem. Res., 2007, 40, 1251–1259 CrossRef CAS; (b) A. Gual, C. Godard, S. Castillón and C. Claver, Tetrahedron: Asymmetry, 2010, 21, 1135–1146 CrossRef CAS; (c) Y. Deng, H. Wang, Y. Sun and X. Wang, ACS Catal., 2015, 5, 6828–6837 CrossRef CAS.
- (a) X.-S. Chen, C.-J. Hou and X.-P. Hou, Synth. Commun., 2016, 46, 917–941 CrossRef CAS; (b) S. Chakrabortty, A. A. Almasalma and J. G. de Vries, Catal. Sci. Technol., 2021, 11, 5388–5411 RSC.
- D. N. Gorbunov, M. V. Nenasheva, M. V. Terenina, Y. S. Kardasheva, E. R. Naranov, A. L. Bugaev, A. V. Soldatov, A. L. Maximov, S. Tilloy, E. Monflier and E. A. Karakhanov, Appl. Catal., A, 2022, 647, 118891 CrossRef CAS.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis, Kluwer, Dordrecht, The Netherlands, 2004, pp. 1–28 Search PubMed.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- P. Purwanto and H. Delmas, Catal. Today, 1995, 24, 135–140 CrossRef CAS.
- (a) M. J. H. Russell, Platinum Met. Rev., 1998, 32, 179–186 CrossRef; (b) M. J. H. Russell and B. A. Murrer, GB Pat., GB 2085874, 1982 Search PubMed.
- (a) H. Chen, Y. Z. Li, J. R. Chen, P. M. Cheng, Y. E. He and H. J. Li, J. Mol. Catal. A: Chem., 1999, 149, 1–6 CrossRef CAS; (b) A. Riisager and B. E. Hanson, J. Mol. Catal. A: Chem., 2002, 189, 195–202 CrossRef CAS.
- B. Cornils and W. A. Hermanns, Aqueous Phase Organometallic Catalysis, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Curr. Org. Chem., 2010, 14, 1296–1307 CrossRef CAS; (b) F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, C. R. Chim., 2011, 14, 149–166 CrossRef CAS; (c) M. Ferreira, H. Bricout, T. F. H. Roth, T. Seidensticker, S. Tilloy and E. Monflier, Catal. Today, 2024, 442, 114951 CrossRef CAS.
- (a) J. R. Anderson, E. M. Campi and W. R. Jackson, Catal. Lett., 1991, 9, 55–58 CrossRef CAS; (b) E. Monflier, G. Fremy, Y. Castanet and A. Mortreux, Angew Chem., Int. Ed., 1995, 34, 2269–2271 CrossRef CAS.
- (a) F. Hapiot, L. Leclercq, N. Azaroual, S. Fourmentin, S. Tilloy and E. Monflier, Curr. Org. Synth., 2008, 5, 162–172 CrossRef CAS; (b) L. Leclercq, H. Bricout, S. Tilloy and E. Monflier, J. Colloid Interface Sci., 2007, 307, 481–487 CrossRef CAS PubMed.
- N. Kania, B. Leger, S. Fourmentin, E. Monflier and A. Ponchel, Chem.–Eur. J., 2010, 16, 6138–6141 CrossRef CAS PubMed.
- B. Fell and G. Papadogianakis, J. Mol. Catal., 1991, 66, 143–154 CrossRef CAS.
- E. A. Karakhanov, Y. S. Kardasheva, A. L. Maksimov, V. V. Predeina, E. A. Runova and A. M. Utukin, J. Mol. Catal. A: Chem., 1996, 107, 235–240 CrossRef CAS.
- (a) H. Zhu, Y. Ding, H. Yin, L. Yan, J. Xiong, Y. Lu, H. Luo and L. Lin, Appl. Catal., A, 2003, 245, 111–117 CrossRef CAS; (b) A. G. Panda, M. D. Bhor, S. R. Jagtap and B. M. Bhanage, Appl. Catal., A, 2008, 347, 142–147 CrossRef CAS.
- (a) L. Obrecht, P. C. J. Kamer and W. Laan, Catal. Sci. Technol., 2013, 3, 541–551 RSC; (b) W. Alsalahi and A. M. Trzeciak, Coord. Chem. Rev., 2021, 430, 213732 CrossRef CAS.
- C. C. Brasse, U. Englert, A. Salzer, H. Waffenschmidt and P. Wasserscheid, Organometallics, 2000, 19, 3818–3823 CrossRef CAS.
- K. W. Kottsieper, O. Stelzer and P. Wasserscheid, J. Mol. Catal. A: Chem., 2001, 175, 285–288 CrossRef CAS.
- (a) P. Wasserscheid, H. Waffenschmidt, P. Machnizti, K. W. Kottsieper and O. Stelzer, Chem. Commun., 2001, 451–452 RSC; (b) W. Keim, D. Vogt, H. Waffenschmidt and P. Wasserscheid, J. Catal., 1999, 186, 481–484 CrossRef CAS.
- (a) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef; (b) M. Haumann and A. Riisager, Chem. Rev., 2008, 108, 1474–1497 CrossRef CAS.
- (a) P. J. Walsh, H. Li and C. A. de Parrodi, Chem. Rev., 2007, 107, 2503–2545 CrossRef CAS; (b) A. C. J. Koeken, M. C. A. van Vliet, L. J. P. van den Broeke, B. J. Deelman and J. T. F. Keurentjes, Adv. Synth. Catal., 2008, 350, 179–188 CrossRef CAS; (c) M. L. Clarke and G. J. Roff, Green Chem., 2007, 29, 792–796 RSC.
- (a) T. W. Smith, US Pat., US 4252678, 1981 Search PubMed; (b) L. Tuchbreiter and S. Mecking, Macromol. Chem. Phys., 2007, 208, 1688–1693 CrossRef CAS; (c) M. Guerrero, N. T. Than Chau, S. Noël, A. Denicourt-Nowicki, F. Hapiot, A. Roucoux, E. Monflier and K. Philippot, Curr. Org. Chem., 2013, 17, 364–399 CrossRef CAS; (d) S. A. Jagtap, M. A. Bhosale, T. Sasaki and B. M. Bhanage, Polyhedron, 2016, 120, 162–168 CrossRef CAS; (e) S. S. Bhagade, S. R. Shivkumar and B. M. Bhanage, Catal. Today, 2018, 309, 147–152 CrossRef CAS.
- (a) Â. C. B. Neves, M. J. F. Calvete, T. M. V. D. P. E. Melho and M. M. Pereira, Eur. J. Org. chem., 2012, 6309–6320 CrossRef; (b) C. Li, W. Wang, L. Yan and Y. Ding, Front. Chem. Sci. Eng., 2018, 12, 113–123 CrossRef CAS.
- (a) A. F. M. Iqbal, Helv. Chim. Acta, 1971, 54, 1440–1445 CrossRef CAS; (b) B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2732–2735 CrossRef; (c) J. R. Briggs, J. Klosin and G. T. Whiteker, Org. Lett., 2005, 7, 4795–4798 CrossRef CAS.
- (a) D. Crozet, M. Urrutigoïty and P. Kalck, ChemCatChem, 2011, 3, 1102–1118 CrossRef CAS; (b) C. Chen, X.-Q. Dong and X. Zhang, Org. Chem. Front., 2016, 3, 1359–1370 RSC; (c) P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS.
- (a) P. P. Brahmkshatriya and P. S. Brahmshatriya, Terpenes: Chemistry, Biological Role, and Therapeutic Applications, in, Natural Products, ed. J. M. Mérillon and K. Ramawat, Springer, Berlin, Heidelberg, 2013, pp. 2665–2691 Search PubMed; (b) J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408–414 CrossRef CAS.
- (a) B. Steenackers, L. de Cooman and D. de Vos, Food Chem., 2015, 172, 742–756 CrossRef CAS PubMed; (b) S. Koyama and T. Heinbockel, Int. J. Mol. Sci., 2020, 21, 1558 CrossRef CAS PubMed.
- A. J. Vieira, F. P. Beserra, M. C. Souza, B. M. Totti and A. L. Rozza, Chem.-Biol. Interact., 2018, 283, 97–106 CrossRef CAS PubMed.
- L. Kollár, J. Bakos, B. Heil, P. Sándor and G. Szalontai, J. Organomet. Chem., 1990, 385, 147–152 CrossRef.
- E. V. Gusevskaya, E. N. dos Santos, R. Augusti, A. D. O. Dias and C. M. Foca, J. Mol. Catal. A: Chem., 2000, 152, 15–24 CrossRef CAS.
- E. V. Gusevskaya, E. N. dos Santos, R. Augusti, A. O. Dias, P. A. Robles-Dutenhefner, C. M. Foca and H. J. V. Barros, Stud. Surf. Sci. Catal., 2000, 130, 563–568 CrossRef.
- L. Kollár and G. Bóadi, Chirality, 1995, 7, 121–127 CrossRef.
- J. Hagen, R. Lehmann and K. Bansemir, DE Pat., DE 2914090, 1980 Search PubMed.
- J. Hagen and K. Bruns, DE Pat., DE 2849742, 1980 Search PubMed.
- I. Ciprés, P. Kalck, D.-C. Park and F. Serein-Spirau, J. Mol. Catal., 1991, 66, 399–407 CrossRef.
- H. J. V. Barros, C. C. Guimarães, E. N. dos Santos and E. V. Gusevskaya, Catal. Commun., 2007, 8, 747–750 CrossRef CAS.
- C. Botteghi, M. Lenarda, R. Ganzerla and G. Moretti, J. Mol. Catal., 1987, 40, 129–182 CrossRef CAS.
- J. C. Chalchat, R. Ph. Gary, E. Lecomte and A. Michet, Flavour Fragrance J., 1991, 6, 179–182 CrossRef CAS.
- H. J. V. Barros, J. G. da Silva, C. C. Guimarães, E. N. dos Santos and E. V. Gusevskaya, Organometallics, 2008, 27, 4523–4531 CrossRef CAS.
- P. W. N. M. van Leeuwan and C. F. Roobeek, J. Organomet. Chem., 1983, 258, 343–350 CrossRef.
- H. J. V. Barros, B. E. Hanson, E. V. Gusevskaya and E. N. dos Santos, Appl. Catal., A, 2004, 278, 57–63 CrossRef CAS.
- P. Pongrácz, B. Bartal, L. Kollár and L. T. Mika, J. Oragnomet. Chem., 2017, 847, 140–145 CrossRef.
- M. P. dee Oliveira, F. G. Delolo, J. A. A. Villarreal, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2021, 616, 118082 CrossRef.
- M. Winnacker, Angew. Chem., Int. Ed., 2018, 57, 14362–14371 CrossRef CAS PubMed.
- H. Mimoun, Chimia, 1996, 50, 620–625 CrossRef CAS.
- E. N. dos Santos, C. U. Pittman Jr and H. Toghiani, J. Mol. Catal., 1993, 83, 51–65 CrossRef CAS.
- H. J. V. Barros, M. L. Ospina, E. Arguello, W. R. Rocha, E. V. Gusevskaya and E. N. dos Santos, J. Organomet. Chem., 2003, 671, 150–157 CrossRef CAS.
- V. D. Silva, E. N. dos Santos, E. V. Gusevskaya and W. R. Rocha, J. Mol. Struct.: THEOCHEM, 2007, 816, 109–117 CrossRef CAS.
- P. Pino, Proc. Symp. on Rhodium in Homogeneous Catalysis, Veszprem, 1978, vol. 98F Search PubMed.
- A. Ceriotti, L. Garlaschelli, G. Longoni, M. C. Malatesta, D. Strumolo, A. Fumagalli and S. Martinengo, J. Mol. Catal., 1984, 24, 309–321 CrossRef CAS.
- F. Azzaroni, P. Biscarini, S. Bordoni, G. Longoni and E. Venturini, J. Organomet. Chem., 1996, 508, 59–67 CrossRef CAS.
- M. C. de Freitas, C. G. Vieira, E. N. dos Santos and E. V. Gusevskaya, ChemCatChem, 2013, 5, 1884–1890 CrossRef CAS.
- A. R. Almeida, R. D. Dias, C. J. P. Monteiro, A. R. Abreu, P. M. P. Gois, J. C. Bayon and M. M. Pereira, Adv. Synth. Catal., 2014, 356, 1223–1228 CrossRef CAS.
- H. Siegel and W. Himmele, Angew Chem., Int. Ed., 1980, 19, 178–183 CrossRef.
- J. G. da Silva, H. J. V. Barros, A. Balanta, A. Bolaños, M. . L. Novoa, M. Reyes, R. Contreras, J. C. Bayón, E. V. Gusevskaya and E. N. dos Santos, Appl. Catal., A, 2007, 326, 219–226 CrossRef CAS.
- J. A. Pino, J. Mesa, Y. Muñoz, M. P. Martí and R. Marbot, J. Agric. Food Chem., 2005, 53, 2213–2223 CrossRef CAS PubMed.
- J. C. LoCicero and R. T. Johnson, J. Am. Chem. Soc., 1952, 74, 2094–2097 CrossRef CAS.
- C. M. Foca, E. N. dos Santos and E. V. Gusevskaya, J. Mol. Catal. A: Chem., 2002, 185, 17–23 CrossRef CAS.
- N. S. Pagar and R. M. Deshpande, Asian J. Chem., 2019, 31, 213–219 CrossRef CAS.
- N. S. Pagar, K. B. Rajurkar and R. M. Deshpande, Int. J. Chem. Kinet., 2020, 52, 485–495 CrossRef CAS.
- M. Eggersdorfer, Terpenes, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2000 Search PubMed.
- K. Schmid and D. Hoff, DE Pat., DE 10205835, 2003 Search PubMed.
- J. G. da Silva, C. G. Vieira, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2009, 365, 231–236 CrossRef CAS.
- F. Fujioka, R. M. Boden and W. L. Schreiber, US Pat., US 4491537, 1985 Search PubMed.
- Q. An, J.-N. Ren, X. Li, G. Fan, S.-S. Qu, Y. Song, Y. Li and S.-Y. Pan, Food Funct., 2021, 12, 10370–10389 RSC.
- (a) K. A. D. Swift, Top. Catal., 2004, 27, 143–155 CrossRef CAS; (b) V. A. Semikolenov, I. I. Ilyna and I. L. Simakova, Appl. Catal., A, 2001, 211, 91–107 CrossRef CAS.
- W. M. Lawrence, B. D. Mookherjee and B. J. Willis, Flavors and Fragrances: A World Perspective, Washington, DC, USA, 1986 Search PubMed.
- M. Benaissa, U. J. Jáuregui-Haza, I. Nikov, A. M. Wilhelm and H. Delmas, Catal. Today, 2003, 79–80, 419–425 CrossRef CAS.
- J. G. da Silva, H. J. V. Barros, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2006, 309, 169–176 CrossRef CAS.
- C. G. Vieira, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2013, 466, 208–215 CrossRef CAS.
- C. G. Vieira, M. C. de Freitas, K. C. B. de Oliveira, A. C. Faria, E. N. dos Santos and E. V. Gusevskaya, Catal. Sci. Technol., 2015, 5, 960–966 RSC.
- D. H. Pybus and C. S. Sell, The Chemistry of Fragances, RSC Paperbacks, Cambridge, 1999 Search PubMed.
- M. I. Dawson, P. D. Hobbs, K. Kuhlmann, V. A. Fung, C. T. Helmes and W. R. Chao, J. Med. Chem., 1980, 23, 1013–1022 CrossRef CAS PubMed.
- C. M. Foca, H. J. V. Barros, E. N. dos Santos, E. V. Gusevskaya and J. C. Bayón, New J. Chem., 2003, 27, 533–539 RSC.
- M. T. Reetz, S. R. Waldvogel and R. Goddard, Heterocycles, 2000, 52, 935–938 CrossRef CAS.
- S. P. Bhatia, D. McGinty, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2008, 46, S237–S240 CrossRef CAS PubMed.
- F. G. Delolo, K. C. B. Oliveira, E. N. dos Santos and E. V. Gusevskaya, Mol. Catal., 2019, 462, 1–9 CrossRef CAS.
- F. G. Delolo, G. M. Vieira, J. A. Avendaño-Villarreal, A. D. O. Dias, E. N. dos Santos and E. V. Gusevskaya, J. Catal., 2023, 421, 20–29 CrossRef CAS.
- J. Leffingwell and D. Leffingwell, Chiral chemistry in flavours and fragrances, Speciality Chemicals Magazine, 2011, pp. 30–33 Search PubMed.
- G. Klein, D. Arlt and R. Braden, DE Pat., DE 3135948, 1983 Search PubMed.
- B. Schäfer, Naturstoffe der Chemischen Industrie, Elsevier Spektrum Akademischer Verlag, Heidelberg, 2007 Search PubMed.
- R. S. Prajapati and B. M. Bhanage, Mol. Catal., 2024, 569, 114607 CrossRef CAS.
- S. S. Bhagade and B. M. Bhanage, Catal. Commun., 2018, 112, 21–25 CrossRef CAS.
- M. Eggersdorfer, Terpenes, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- T. J. McGenity, A. T. Crombie and J. C. Murrell, ISME J., 2018, 12, 931–941 CrossRef CAS PubMed.
- G. M. Whited, F. J. Feher, D. A. Benko, M. A. Cervin, G. K. Chotani, J. C. McAuliffe, R. J. LaDuca, E. A. Ben-Shoshan and K. J. Sanford, Ind. Biotechnol., 2010, 6, 152–163 CrossRef CAS.
- D. Bartschat and A. Mosandl, Eur. Food Res. Technol., 1998, 206, 165–168 CAS.
- H. Adkins and J. L. R. Williams, J. Org. Chem., 1952, 17, 980–987 CrossRef CAS.
- M. F. Mirbach, Transition Met. Chem., 1984, 9, 465–468 CrossRef CAS.
- C. Botteghi, M. Branca and A. Saba, J. Organomet. Chem., 1980, 184, C17–C19 CrossRef CAS.
- S. Bertozzi, N. Campigli, G. Vitulli, R. Lazzaroni and P. Salvadori, J. Organomet. Chem., 1995, 487, 41–45 CrossRef CAS.
- H. J. V. Barros, C. C. Guimarães, E. N. dos Santos and E. V. Gusevskaya, Organometallics, 2007, 26, 2211–2218 CrossRef CAS.
- A. Behr, S. Reyer and N. Tenhumberg, Dalton Trans., 2011, 40, 11742–11747 RSC.
- K. I. Galkin, S. E. Lubimov, I. A Godovikov, F. M. Dolgushin, A. F. Smol’yakov, E. A. Sergeeva, V. A. Davankov and I. T. Chizhevsky, Organometallics, 2012, 31, 6080–6084 CrossRef CAS.
- J. Chappell and R. M. Coates, Comprehensive Natural Products II: Chemistry and Biology, 2010, vol. 1, pp. 609-641 Search PubMed.
- (a) A. F. Barrero, J. Q. del Moral and A. Lara, Tetrahedron, 2000, 56, 3717–3723 CrossRef CAS; (b) G. Stork and R. Breslow, J. Am. Chem. Soc., 1953, 75, 3291 CrossRef CAS; (c) P. Walter, Adv. Cycloaddit., 1841, 39, 247–251 Search PubMed.
- F. E. Huelin and K. E. Murray, Nature, 1966, 210, 1260–1261 CrossRef CAS PubMed.
- J. J. Scheibel and R. E. Shumate, WO Pat., WO 2010/033976, 2010 Search PubMed.
- (a) T. E. Furia, Handbook of Flavor Ingredients, CRC Press, 2019, vol. I Search PubMed; (b) E. Guenther, The Essential Oils, D. Van Nostrade Company, Inc.Toronto , New York and London, 1950, vol. IV Search PubMed.
- T. C. Chen, C. O. da Fonseca and A. H. Schönthal, Am. J. Cancer Res., 2015, 5, 1580–1593 Search PubMed.
- R. M. Boden, W. L. Schreiber, F. Fujioka, P. Chant and L. Dekker, EP Pat., EP 0155973, 1984 Search PubMed.
- (a) I. G. Collado, J. R. Hanson and A. J. Macías-Sáncheza, Nat. Prod. Rep., 1998, 15, 187–204 RSC; (b) M. R. R. Tappin, J. F. G. Pereira, L. A. Lima, A. C. Siani, J. L. Mazzei and M. F. S. Ramos, Quim. Nova, 2004, 27, 236–240 CrossRef CAS.
- (a) A. V. Tkachev, Chem. Nat. Compd., 1987, 23, 393–412 CrossRef; (b) D. H. Pybus and C. S. Sell, The Chemistry of Fragrances, RSC Publishing, Dorset, UK, 2nd edn, 2006, vol. 2, pp. 52–88 Search PubMed.
- K. C. B. Oliveira, A. de Camargo Faria, A. C. Monteiro, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2016, 523, 139–145 CrossRef CAS.
- A. de Camargo Faria, K. C. B. Oliveira, A. C. Monteiro, E. N. dos Santos and E. V. Gusevskaya, Catal. Today, 2020, 344, 24–31 CrossRef CAS.
- (a) N. P. Lopes, M. J. Kato, E. H. D. A. Andrade, J. G. S. Maia, M. Yoshida, A. R. Planchart and A. M. Katzin, J. Ethanopharmacol., 1999, 67, 313–319 CrossRef CAS; (b) J. Koudou, A. A. Abena, P. Ngaissona and J. M. Bessière, Fitoterapia, 2005, 76, 700–703 CrossRef CAS PubMed; (c) D. C. Arruda, F. L. D'Alexandri, A. M. Katzin and S. R. B. Uliana, Antimicrob. Agents Chemother., 2005, 49, 1679–1687 CrossRef CAS; (d) F. C. Klopell, M. Lemos, J. P. B. Sousa, E. Comunello, E. L. Maistro, J. K. Bastos and S. F. de Andrade, J. Biosci., 2007, 62, 537–542 CAS.
- (a) M. Grande, I. S. Bellido, P. Torres and F. Piera, J. Nat. Prod., 1992, 55, 1074–1079 CrossRef CAS; (b) A. Baruah and S. C. Nath, J. Essent. Oil Res., 2006, 18, 200–202 CrossRef CAS.
- M. C. de Freitas, K. C. B. de Oliveira, A. de Camargo Faria, E. N. dos Santos and E. V. Gusevskaya, Catal. Sci. Technol., 2014, 4, 1954–1959 RSC.
- M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. van Otterlo, Chem. Rev., 2015, 115, 5462–5569 CrossRef CAS PubMed.
- (a) M. N. I. Bhuiyan, J. Begum, N. C. Nandi and F. Akter, Afr. J. Plant Sci., 2010, 4, 451–454 CAS; (b) G. R. Mallavarapu, S. Ramesh, R. S. Chandrasekhara, B. R. Rajeswara Rao, P. N. Kaul and A. K. Bhattacharya, Flavour Fragrance J., 1995, 10, 239–242 CrossRef CAS.
- J. Barnes, L. A. Anderson and J. S. Phillipson, Herbal Medicines, Pharmaceutical Press, London, 2007 Search PubMed.
- P. Kalck, D. C. Park, F. Serein and A. Thorez, J. Mol. Catal., 1986, 36, 349–357 CrossRef CAS.
- A. C. da Silva, K. C. B. de Oliveira, E. V. Gusevskaya and E. N. dos Santos, J. Mol. Catal. A: Chem., 2002, 179, 133–141 CrossRef CAS.
- S. A. Jagtap and B. M. Bhanage, Appl. Organomet. Chem., 2018, 32, e4478 CrossRef.
- M. Alhaffar, R. Suleiman and B. E. Ali, Catal. Commun., 2010, 11, 778–782 CrossRef CAS.
- P. J. Baricelli, M. Rodriguez, L. G. Melean, M. Modroño-Alonso, M. Barusiak, M. Rosales, B. Gonzalez, K. C. B. Oliveira, E. V. Gusevskaya and E. N. dos Santos, Appl. Catal., A, 2015, 490, 163–169 CrossRef CAS.
- L. G. Melean, M. Rodriguez, M. Romero, M. L. Alvarado, M. Rosales and P. J. Baricelli, Appl. Catal., A, 2011, 394, 117–123 CrossRef CAS.
- S. A. Jagtap, E. Monflier, A. Ponchel and B. M. Bhanage, Mol. Catal., 2017, 436, 157–163 CrossRef CAS.
- R. S. Prajapati, A. R. Kapdi, R. Sahu and B. M. Bhanage, Catal. Today, 2024, 438, 114804 CrossRef CAS.
- P. J. Baricelli, M. Rodríguez, L. G. Melean, M. Barusiak, I. Crespo, J. C. Pereira and M. Rosales, Mol. Catal., 2020, 497, 111189 CrossRef CAS.
- F. G. Delolo, G. M. Vieira, J. A. A. Villarreal, E. N. dos Santos and E. V. Gusevskaya, Catal. Today, 2021, 381, 272–279 CrossRef CAS.
- A. D. O. Dias, F. G. Delolo, J. A. Avendaño-Villarreal, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2023, 665, 119369 CrossRef CAS.
- F. M. S. Rodrigues, V. Masliy, F. M. C. Silva, A. P. Felgueiras, R. M. B. Carrilho and M. M. Pereira, Catal. Today, 2023, 418, 114055 CrossRef CAS.
- M. Concha-puelles, A. Cortínez, N. Lezana, M. Vilches-Herrera and S. Lühr, Catal. Sci. Technol., 2022, 12, 6883–6890 RSC.
- R. L. Smith, T. B. Adams, J. Doull, V. J. Feron, J. I. Goodman, L. J. Marnett, P. S. Portoghese, W. J. Waddell, B. M. Wagner, A. E. Rogers, J. Caldwell and I. G. Sipes, Food Chem. Toxicol., 2002, 40, 851–870 CrossRef CAS.
- M. R. Axet, S. Castillon and C. Claver, Inorg. Chim. Acta, 2006, 359, 2973–2979 CrossRef CAS.
- L. Kollár, E. Farkas and J. Bâtiu, J. Mol. Catal. A: Chem., 1997, 115, 283–288 CrossRef.
- S. R. Khan and B. M. Bhanage, Appl. Organomet. Chem., 2013, 27, 313–317 CrossRef CAS.
- M. J. Hickey, J. Org. Chem., 1948, 13, 443–446 CrossRef CAS.
- (a) Z. S. Petrović, I. Cvetković, D. P. Hong, X. Wan, W. Zhang, T. W. Abraham and J. Malsam, Eur. J. Lipid Sci. Technol., 2010, 112, 97–102 CrossRef; (b) D. Babb, A. Larre, A. Schrock, D. Bhattacharjee and M. Sonnenschein, Polym. Prepr., 2007, 48, 855–856 CAS; (c) W.-J. Peng, D. Babb, A. Sanders, C. Derstine, J. Jimenez, Z. Lysenko, K. Olson, J. Phillips, B. Roesch and A. Schrock, WO Pat., WO 2008/073729, 2008 Search PubMed; (d) J. Argyropoulos, P. Popa, G. Spilman, D. Bhattacharjee and W. Koonce, J. Coat. Technol. Res., 2009, 6, 501–508 CrossRef CAS; (e) T. Vanbesien, F. Hapiot and E. Monflier, Lipid Technol., 2013, 25, 175–178 CrossRef CAS; (f) J. Hong, D. Radojčić, X.-Q. Yang, X. Wan and Z. S. Petrovic, J. Appl. Polym. Sci., 2020, 137, 48509 CrossRef CAS.
- W. F. Gresham, R. E. Brooks and W. M. Bruner, US Pat., US 2437600, 1948 Search PubMed.
- (a) R. Lai, M. Naudet and E. Ucciani, Rev. Fr. Corps Gras, 1996, 13, 737–745 Search PubMed; (b) R. Lai, M. Naudet and E. Ucciani, Rev. Fr. Corps Gras, 1968, 15, 15–21 CAS.
- E. N. Frankel, S. Meltin, W. K. Rohwedder and I. Wender, J. Am. Oil Chem. Soc., 1969, 46, 133–138 CrossRef CAS.
- E. Frankel, J. Am. Oil. Chem. Soc., 1971, 48, 248–253 CrossRef CAS.
- J. P. Friedrich, Ind. Eng. Chem. Prod. Res. Dev., 1978, 17, 205–207 CrossRef CAS.
- E. N. Frankel, W. E. Neff, F. L. Thomas, T. H. Khoe and E. H. Pryde, J. Am. Oil Chem. Soc., 1975, 52, 498–504 CrossRef CAS.
- E. N. Frankel and E. H. Pryde, J. Am. Oil Chem. Soc., 1977, 54(11), A873–A881 CrossRef.
- E. H. Pryde, J. Am. Oil Chem. Soc., 1984, 61, 419–425 CrossRef CAS.
- K. F. Muilwijk, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Oil Chem. Soc., 1997, 74, 223–228 CrossRef CAS.
- P. Kandanarachchi, A. Guo and Z. Petrovic, J. Mol. Catal. A: Chem., 2002, 184, 65–71 CrossRef CAS.
- P. Kandanarachchi, A. Guo, D. Demydov and Z. Petrovic, J. Am. Oil Chem. Soc., 2002, 79, 1221–1225 CrossRef CAS.
- A. N. F. Mendes, J. R. Gregório and R. G. da Rosa, J. Braz. Chem. Soc., 2005, 16, 1124–1129 CrossRef CAS.
- A. Behr, D. Obst and A. Westfechtel, Eur. J. Lipid Sci. Technol., 2005, 107, 213–219 CrossRef CAS.
- J. P. Friedrich, G. R. List and V. E. Sohns, J. Am. Oil Chem. Soc., 1973, 50, 455–458 CrossRef CAS.
- B. Fell, D. Leckel and C. Schobben, Fat Sci. Technol., 1995, 97, 219–228 CAS.
- (a) R. E. Hefner and M. L. Tulchinsky, WO Pat., WO 2009/091670, 2009 Search PubMed; (b) M. L. Tulchinsky and R. R. Peterson, WO Pat., WO 2009/091669, 2009 Search PubMed; (c) M. L. Tulchinsky and A. G. Abatjoglou, WO Pat., WO 2009/091671, 2009 Search PubMed.
- B. Fell, C. Schobben and G. Papadogianakis, J. Mol. Catal. A: Chem., 1995, 101, 179–186 CrossRef CAS.
- Z. Xia, U. Kloeckner and B. Fell, Fett/Lipid, 1996, 98, 313–321 CrossRef CAS.
- J. P. Arhancet, M. E. Davis, J. S. Merola and B. E. Hanson, Nature, 1989, 339, 454–455 CrossRef CAS.
- Z. S. Petrović, I. Cvetković, J. Milić, D. Hong and I. Javni, J. Appl. Polym. Sci., 2012, 125, 2920–2928 CrossRef.
- J. Boulanger, A. Ponchel, H. Bricout, F. Hapiot and E. Monflier, Eur. J. Lipid Sci. Technol., 2012, 114, 1439–1446 CrossRef CAS.
- J. Bianga, N. Herrmann, L. Schurm, T. Gaide, J. M. Dreimann, D. Vogt and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2020, 122, 1900317 CrossRef CAS.
- J. Ternel, J.-L. Couturier, J.-L. Dubois and J.-F. Carpentier, ChemCatChem, 2015, 7, 513–520 CrossRef CAS.
- L. le Goanvic, J.-L. Couturier, J.-L. Dubois and J.-F. Carpentier, J. Mol. Catal. A: Chem., 2016, 417, 116–121 CrossRef CAS.
- T. Gaide, J. Bianga, K. Schlipköter, A. Behr and A. J. Vorholt, ACS Catal., 2017, 7, 4163–4171 CrossRef CAS.
- R. C. Dutra, T. V. S. Martins, D. D. G. Rocha, M. R. Meneghetti, S. M. P. Meneghetti, M. G. Sulman, V. G. Matveeva and P. A. Z. Suarez, Catalysts, 2023, 13, 630 CrossRef CAS.
- T. F. H. Roth, M. Häusler, D. Vogt and T. Seidensticker, Catal. Today, 2024, 439, 114803 CrossRef CAS.
- (a) R. O. Greep, Recent Progress in Hormone Research, Elsevier Science, 3rd edn, 1977, vol. 33, pp. 345–391 Search PubMed; (b) D. Lednicer, Steroid Chemistry at Glance, Wiley, 2011 Search PubMed; (c) T. Rhen and J. A. Cidlowski, N. Engl. J. Med., 2005, 353(16), 1711–1723 CrossRef CAS.
- (a) A. L. Nussbaum, T. L. Popper, E. P. Oliveto, S. Friedman and I. Wender, J. Am. Chem. Soc., 1959, 81, 1228–1231 CrossRef CAS; (b) P. F. Beal, M. A. Rebenstorf and J. E. Pike, J. Am. Chem. Soc., 1959, 81, 1231–1234 CrossRef CAS.
- R. Skoda-Földes, L. Kollár, B. Heil, G. Gálik, Z. Tuba and A. Arcadi, Tetrahedron: Asymmetry, 1991, 2, 633–634 CrossRef.
- S. Törös, L. Kollár, B. Heil and Z. Tuba, XIIIth International Conference on Organometallic Chemistry, Torino, 1988, p. 359 Search PubMed.
- S. Törös, I. Gémes-Pécsi, B. Heil, S. Mahó and Z. Tuba, Chem. Commun., 1992, 858–859 RSC.
- L. Kollár, R. Skoda-Földes, S. Mahó and Z. Tuba, J. Organomet. Chem., 1993, 453, 159–162 CrossRef.
- Z. Freixa, M. M. Pereira, J. C. Bayón, A. M. S. Silva, J. A. R. Salvador, A. Matos Beja, J. A. Paixão and M. Ramos, Tetrahedron: Asymmetry, 2001, 12, 1083–1087 CrossRef CAS.
- G. N. Costa, R. M. B. Carrilho, L. D. Dias, J. C. Viana, G. L. B. Aquino, M. Pineiro and M. M. Pereira, J. Mol. Catal. A: Chem., 2016, 416, 73–80 CrossRef CAS.
- A. F. Peixoto, M. M. Pereira, A. M. S. Silva, C. M. Foca, J. C. Bayón, M. J. S. M. Moreno, A. Matos Beja, J. A. Paixão and M. Ramos Silva, J. Mol. Catal. A: Chem., 2007, 275, 121–129 CrossRef CAS.
- E. Nagy, C. Benedek, B. Heil and S. Törös, Appl. Organomet. Chem., 2002, 16, 628–634 CrossRef CAS.
- J. G. Strong, US Pat., US 3890384, 1975 Search PubMed.
- C. S. Graebin, V. L. Eifler-Lima and R. G. da Rosa, Catal. Commun., 2008, 9, 1066–1070 CrossRef CAS.
- A. de Oliveira Dias, M. G. P. Gutiérrrez, J. A. A. Villarreal, R. L. L. Carmo, K. C. B. Oliveira, A. G. Santos, E. N. dos Santos and E. V. Gusevskaya, Appl. Catal., A, 2019, 574, 97–104 CrossRef CAS.
- A. Pérez Alonso, D. Pham Minh, D. Pla and M. Gómez, ChemCatChem, 2023, 15, e202300501 CrossRef.
- A. Behr and A. Wintzer, Chem. Eng. Technol., 2015, 38, 2299–2304 CrossRef CAS.
- M. Ali, A. Gual, G. Ebeling and J. Dupont, ChemSusChem, 2016, 9, 2129–2134 CrossRef CAS.
- J. Liu, C. Kubis, R. Franke, R. Jackstell and M. Beller, ACS Catal., 2016, 6, 907–912 CrossRef CAS.
- T. A. Faßbach, T. Gaide, M. Terhorst, A. Behr and A. J. Vorholt, ChemCatChem, 2017, 9, 1359–1362 CrossRef.
- S. A. Jagtap, S. P. Gowalkar, E. Monflier, A. Ponchel and B. M. Bhanage, Mol. Catal., 2018, 452, 108–116 CrossRef CAS.
- S. R. Khan and B. M. Bhanage, Appl. Organomet. Chem., 2013, 27, 711–715 CrossRef CAS.
- K. C. B. Oliveira, S. N. Carvalho, M. F. Duarte, E. V. Gusevskaya, E. N. dos Santos, J. El Karroumi, M. Gouygou and M. Urrutigoïty, Appl. Catal., A, 2015, 497, 10–16 CrossRef CAS.
- J. October and S. F. Mapolie, Catal. Lett., 2021, 70, 153018 CAS.
- E. Petricci and E. Cini, Top. Curr. Chem., 2013, 342, 117–149 CrossRef CAS.
- A. Behr, M. Fiene, C. Buss and P. Eilbracht, Eur. J. Lipid Sci. Technol., 2000, 102, 467–471 CrossRef CAS.
- A. J. Vorholt, P. Neubert and A. Behr, Chem. Ing. Tech., 2013, 85, 1540–1547 CrossRef CAS.
- A. Behr, T. Seidensticker and A. J. Vorholt, Eur. J. Lipid Sci. Technol., 2014, 116, 477–485 CrossRef CAS.
- A. J. Vorholt, S. Immohr, K. A. Ostrowski, S. Fuchs and A. Behr, Eur. J. Lipid Sci. Technol., 2017, 119, 1600211 CrossRef.
- H. F. Ramalho, K. M. C. Ferreira, P. M. A. Machado, T. B. Silva, E. T. Rangel, M. J. Prauchner and P. A. Z. Suarez, J. Braz. Chem. Soc., 2016, 27, 321–333 CAS.
- (a) T. Vanbésien, E. Monflier and F. Hapiot, Green Chem., 2017, 19, 1940–1948 RSC; (b) F. Hapiot, E. Monflier and T. Vanbésien, WO Pat., WO 2016/128390, 2016 Search PubMed.
- K. Cousin, T. Vanbésien, E. Monflier and F. Hapiot, Catal. Commun., 2019, 125, 37–42 CrossRef CAS.
- E. Nagy, B. Heil and S. Torös, J. Organomet. Chem., 1999, 586, 101–105 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |