
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeactivation mechanisms of Nb2O5 catalyst for esterification of dimethyl adipate: crystal transformation and acid site coking†
Mengyue
Li
a,
Yang
Li
b,
Bin
He
a,
Lijun
Han
a,
Ruiyi
Yan
*a and
Jiayu
Xin
 *a
*a
aBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Innovation Academy for Green Manufacture, CAS, Beijing 100190, P. R. China. E-mail: ryyan@ipe.ac.cn; jyxin@ipe.ac.cn
bState Key Laboratory of Heavy Oil Processing, College of Chemical Engineering, China University of Petroleum, Beijing 102249, China
First published on 16th April 2025
Abstract
The deactivation mechanisms of Nb2O5 catalysts in fixed-bed reactors were systematically investigated, revealing that carbon deposition and structural phase transformations synergistically drive catalyst degradation. During adiponitrile synthesis, carbon deposits evolve progressively from monocyclic to polycyclic aromatic hydrocarbons, leading to blockage of critical acid sites and reduced catalytic activity. Simultaneously, the phase transformation of Nb2O5 to Nb12O29 generates oxygen vacancies, which form unsaturated metal coordination sites and alter the catalyst's acidity profile. Regeneration partially reverses this phase transformation, restoring Nb2O5 content while reducing Nb12O29, thereby improving stability Crucially, maintaining moderate acidity is essential for achieving high nitrile selectivity (>85%) at efficient conversion rates. The study proposes practical strategies to mitigate coke formation and stabilize active sites, including optimized ammonia flow rates and acidic site control. These findings provide a theoretical foundation for enhancing Nb2O5 catalyst stability, reducing energy consumption, and extending operational lifespan in industrial nitrile production processes.
Sustainability spotlightThis study systematically elucidates the deactivation mechanisms of Nb2O5 catalysts in fixed-bed reactors, offering a comprehensive understanding of structural and chemical transformations affecting catalyst performance. By identifying carbon deposition and structural phase transformations as the principal contributors to catalyst deactivation, the research provides actionable insights for improving catalytic stability in industrial adiponitrile production. These findings hold significant implications for reducing waste and energy inefficiencies in industrial chemical processes. |
1. Introduction
Adiponitrile (ADN) is a critical intermediate in the production of hexamethylene diamine, which is a key raw material for the manufacture of nylon-66.1,2 The primary industrial processes for large-scale ADN production rely on butadiene hydrocyanation3,4 and acrylonitrile electrolytic dimerization,5,6 both of which are widely adopted due to their high production efficiency and strong industrial scalability. However, despite the high efficiency of these processes, they face significant challenges in terms of sustainability and long-term feasibility, such as the use of toxic reagents, high energy consumption, and catalyst deactivation. For example, the butadiene hydrocyanation process, while having a relatively short reaction pathway (see Fig. 1), requires the use of highly toxic hydrogen cyanide (HCN), which introduces severe safety hazards and operational difficulties.7 Similarly, although the acrylonitrile electrolytic dimerization process benefits from a broad range of feedstocks and minimal environmental impact,6 it suffers from high energy consumption and complex reaction conditions, making it less suitable for large-scale industrial application. In contrast, adipic acid amination, while a promising alternative, typically operates at high temperatures (300–350 °C) with boron phosphate as a catalyst. This results in a longer process route, higher production costs, and significant NOx emissions, which contribute to air pollution and exacerbate the greenhouse effect.8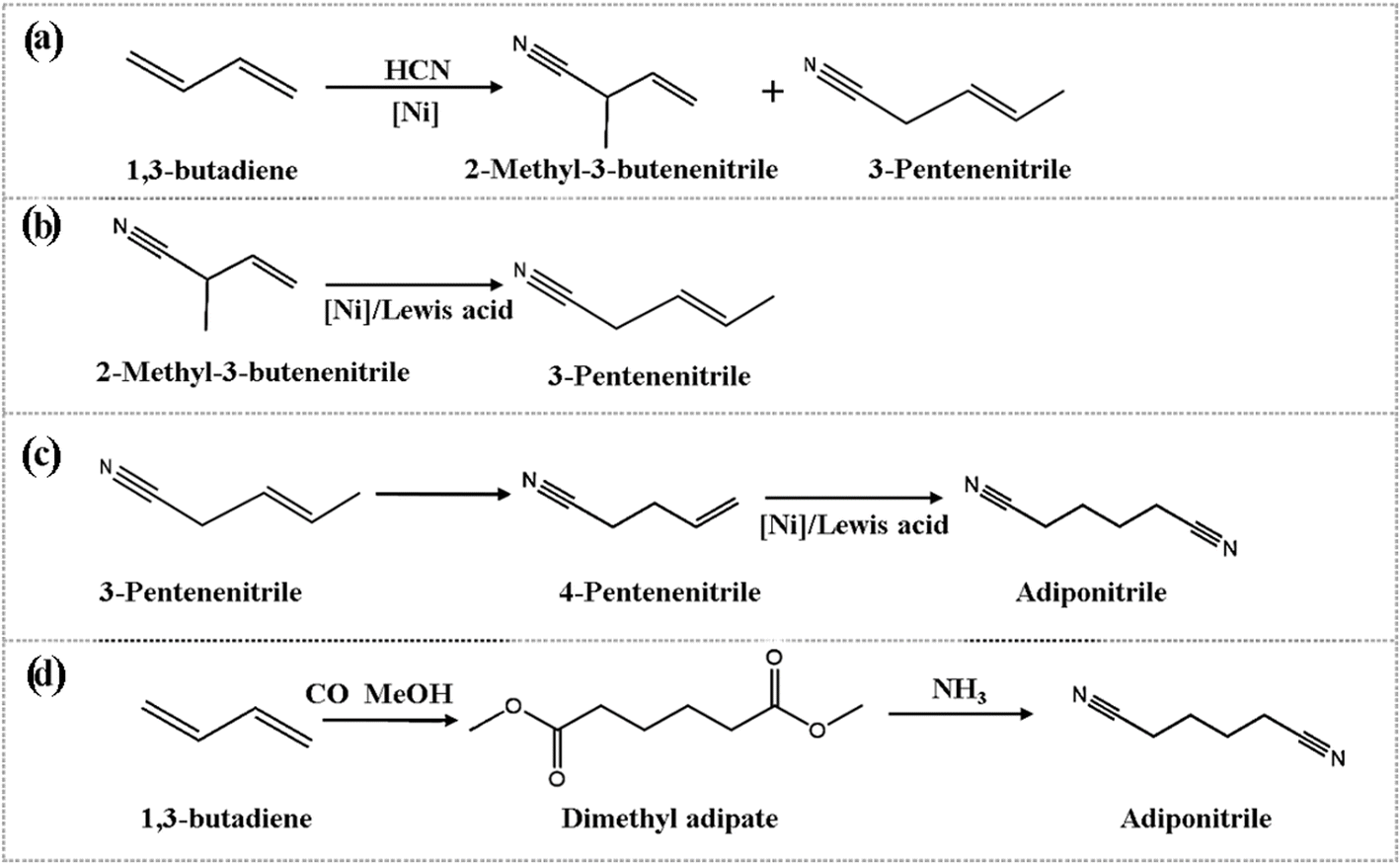 | ||
| Fig. 1 Cyanidation of butadiene for preparation of adiponitrile. | ||
In response to these challenges, recent research has focused on developing more environmentally friendly and efficient alternatives. The butadiene reacts with CO and methanol to obtain dimethyl adipate (DMA) followed with direct gas-phase amination of DMA to ADN represents a promising green chemistry-based approach (see in Fig. 1d).9–13 This method avoids the use of toxic reagents and has the potential for utilizing bio-based feedstocks, thereby reducing the environmental impact of the production process.14–17 The second direct gas-phase amination reaction is the crucial step for this route.11,13 Although this method has not yet been extensively studied in academic literature, preliminary studies have shown that the reaction of DMA with ammonia to form ADN is an endothermic process, with a positive reaction enthalpy change (ΔrH) of 42.3 kcal mol−1, indicating that the reaction requires significant energy input, presenting challenges for process optimization.18 Moreover, a key challenge in this reaction lies in the functionalized carbon chains of DMA and ADN, which contain highly reactive electron-withdrawing groups that may undergo intramolecular cyclization and intermolecular polycondensation, thus affecting reaction selectivity and efficiency.
In this context, acidic metal oxide catalysts, such as Nb2O5, V2O5, and Ta2O5, have demonstrated excellent catalytic activity for the gas-phase conversion of DMA to ADN. Studies have shown that Nb2O5 catalysts exhibit high DMA conversion (over 97.7%) and good nitrile selectivity (over 84.3%) in a fixed-bed reactor, making them ideal for this reaction.18 Compared to other metal oxide catalysts, Nb2O5 not only provides excellent conversion but also demonstrates significant advantages in terms of nitrile selectivity. For instance, although Ta2O5 shows excellent reactivity, its selectivity towards nitrile products is relatively low, at only 67.8%; V2O5 shows similar selectivity to Ta2O5 but with a lower DMA conversion.18 Therefore, Nb2O5 catalysts not only offer high conversion but also effectively avoid issues such as equipment corrosion and product separation caused by acidic reactants.19–21 Nb2O5 has shown significant catalytic activity in various reaction systems; however, its stability remains a challenge, particularly under high-temperature conditions. Studies have indicated that the deactivation of Nb2O5 catalysts is primarily attributed to sintering, poisoning by non-metal pollutants, and carbon deposition. During high-temperature sintering, Nb2O5 particles tend to agglomerate, leading to an increase in particle size, which significantly reduces the specific surface area and catalytic activity of the catalyst.22,23 Additionally, non-metal pollutants, such as sulfur, nitrogen, phosphorus, and chlorine, react with the catalyst surface to form non-target products. These products can adsorb on the active sites, thereby inhibiting the catalytic reaction rate.23,24 The impact of these non-target products on the catalyst varies, depending on the type of pollutant and the reaction conditions.25 In terms of carbon deposition, the carbon source present in the reactants may interact with Nb2O5 at elevated temperatures, leading to the formation of solid carbon deposits, which significantly degrade the catalytic performance.26,27
Further studies have revealed some unique features of Nb2O5 catalyst deactivation in specific reaction systems. For instance, in the ethanol dehydrogenation reaction, the addition of Nb2O5 to the ZrO2–Nb2O5 catalyst increased the density of acidic sites.28 However, an excessive density of acidic sites led to increased ethylene production, which exacerbated the accumulation of carbon sources, thereby impacting catalyst stability. Similarly, in the methane decomposition reaction, when Nb2O5 was used as a support for nickel and nickel–copper catalysts, the catalyst surface was found to be completely covered by carbon filaments (CFC), emphasizing the close relationship between carbon deposition at high temperatures and catalyst deactivation.29 These studies suggest that the deactivation of Nb2O5 catalysts is not only related to traditional mechanisms such as sintering and poisoning by non-metal pollutants but also to carbon deposition, which plays a decisive role in catalyst deactivation under specific reaction conditions.
Therefore, the aim of this study is to further explore the performance and deactivation mechanisms of Nb2O5 catalysts in gas-phase reactions, and to optimize their performance for large-scale industrial applications by tuning the catalyst's microstructure and adjusting reaction conditions. This research not only provides theoretical guidance for catalyst design and optimization but also offers valuable insights for the industrial application of DMA to ADN conversion as an environmentally friendly and efficient process.
2. Experimental
2.1 Chemicals and materials
DMA (≥99%) and niobium oxide (Nb2O5, 99.9%) were procured from Innochem. Ltd. Ethanol and acetonitrile were sourced from Fuchen Chemical Company (Tianjin, China), and NH3 was sourced from Beijing YongSheng Gas Technology Co. All reagents utilized in the experiments were of analytical grade.2.2 Catalyst preparation
Nb2O5 was synthesized via coprecipitation by first dissolving niobium oxalate in deionized water while stirring at room temperature. Subsequently, the solution's pH was adjusted to 9 using ammonia, resulting in the formation of a solid (Nb2O5·H2O), which was separated via filtration. Deionized water was used to wash the solid until the pH became neutral and then dried at 80 °C for 24 h. Finally, Nb2O5 was obtained through calcination in a tube furnace.The aforementioned precursors were positioned flat in a crucible and placed in a tube furnace. Air was supplied to the tube furnace, and the temperature was raised to 700 °C at a rate of 5 °C min−1. The desired sample was calcined at 700 °C for 3 h.
2.3 Equipment and characterization
A Smartlab diffractometer with Cu Kα radiation (40 kV, 150 mA) and a wavelength of 1.5406 Å (Rigaku, Japan) was used to obtain X-ray diffraction (XRD) patterns. The peak resolution error of the XRD data obtained from the Smartlab X-ray diffractometer is controlled within a range of ±0.2°. A micro-TOF II spectrometer with methanol as the mobile phase was used to record electrospray ionization high-resolution mass spectra. A JEM-2100F transmission electron microscope at an accelerating voltage of 200 kV (JEOL, Japan) was used to obtain transmission electron microscopy (TEM) images. SU-8020 scanning electron microscope at an accelerating voltage of 5 kV (Hitachi, Japan) was used to collect scanning electron microscopy (SEM) images. Al as the exciting source (ThermoFisher, USA) was used to perform X-ray photoelectron spectroscopy (XPS) measurements on an ESCALAB 250Xi X-ray photoelectron spectrometer.A BRUKER Tensor-27 FTIR spectrometer with a DTGS detector was used to perform pyridine adsorption Fourier transform infrared (Py-FTIR) spectroscopy. Each spectrum comprised 32 scans recorded at a resolution of 4 cm−1. Each sample was pressed into a self-supported wafer (20 mg), activated in an evacuated (10−3 Pa) chamber at 450 °C for 1 h, and then cooled to room temperature. A background spectrum was also collected. Subsequently, pyridine was introduced into the chamber for 30 min, followed by evacuation for 1 h. The sample was heated at 100 °C, 150 °C and 350 °C for 1 h to assess acid strength, with spectra recorded within the range of 1700–1400 cm−1 at room temperature.
2.4 Catalytic tests
A fixed-bed reactor with an internal diameter of 15 mm and a length of 595 mm was used to conduct the nitrile reaction between the monoester and NH3 (the process flow diagram is shown in Fig. S1†). The reaction airspeed was set at 2.4 h−1 (liquid hourly rate of air (LHSV)). 1.5 mL of catalyst (40 mesh) was loaded into the reactor before each experiment, which was then heated to the preheated temperature. A controlled flow rate of NH3 gas was subsequently introduced into the reactor and measured using a mass flow meter. DMA was introduced into the reactor using a pump after 1 h.The liquid product was collected by absorption in methanol at 10 °C after a stabilization period of 2 h and subsequently examined offline using an Agilent 4890 gas chromatograph. The column temperature was programmed as follows: initiating at 60 °C for 1 min, followed by a temperature gradient of 25 °C to 225 °C and a final hold of 13 min. The injector and flame ionization detector (FID) temperature were maintained at 250 °C. The following equations were used to calculate the conversion of dimethyl adipate (CDMA) and the selectivity of adiponitrile (SADN):
 | (1) |
 | (2) |
In addition to the calibration of the reactor and gas flow rates, we also performed periodic calibration of the mass flow meter used to measure the NH3 gas, ensuring a measurement error within ±1%. The gas chromatograph (Agilent 4890) was calibrated using standard samples, and its precision was confirmed to be within ±2% for the conversion and selectivity measurements.
3. Results and discussion
In this study, Nb2O5 was selected as a model catalyst to evaluate its long-term stability, catalyst deactivation, and regeneration mechanisms in a fixed-bed reactor, as shown in Fig. 2a. Non-precious metal oxides as catalysts demonstrate significant environmental benefits and high atomic efficiency in the synthesis of adiponitrile from dimethyladipate and ammonia, and they exhibit considerable industrial potential.13,30 During long-term operation tests, the selectivity of the main product and intermediate was not distinguished separately. This is because methyl 5-cyanovalerate (MCP), which is the intermediate, can easily be converted into ADN as demonstrated in our previous research. MCP serves as a key intermediate in the tandem reaction and can be recovered and reintroduced into the reaction system.18 Therefore, in the simulated industrial operation tests, MCP was treated as a recyclable component and was not included in the selectivity calculation. As a result, the selectivity data shown in Fig. 2a reflect the selectivity of the overall products, including the main product and the intermediate.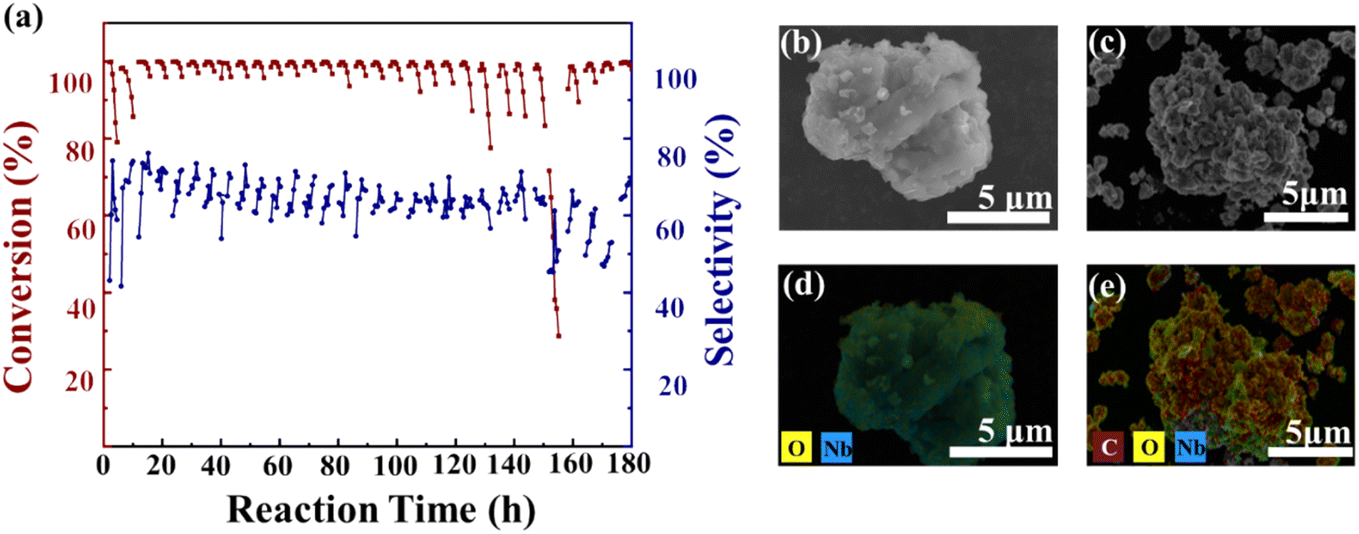 | ||
| Fig. 2 (a) Catalyst reaction test (T = 375 °C, DMA: 18.3 mmol h−1, NH3: 4.9 L h−1, NH3/DMA 12 and LHSV 2.4 h−1); SEM and EDS data of the catalyst before (b and d) and after the reaction (c and e). | ||
To ensure the validity of selectivity calculations and the reproducibility of the data, where the main products and by-products were clearly identified. The relevant GC-MS data are provided in the ESI Fig. S2.† The experimental results showed that NH3/DMA molar ratio of 12, the selectivity of ADN reached 87.6% (Fig. S3†). In this study, Nb2O5 was employed as a model catalyst to evaluate its multi-cycle stability in a fixed-bed reactor, as shown in Fig. 2a. During the first 7 h reaction with fresh Nb2O5 (F-Nb2O5), the conversion dropped from 99.8% to 79%, along with a sharp decline in adiponitrile selectivity. After the reaction, the catalyst (P-Nb2O5) appeared blackened carbon deposition, which is a well-known cause of catalyst deactivation.
SEM images (Fig. 2b and c) revealed significant structural collapse of the Nb2O5 (P-Nb2O5) catalyst surface, indicating that the catalyst framework may have been damaged due to prolonged operation. EDS mapping (Fig. 2d and e) confirmed a significant increase in surface carbon content, suggesting that carbon deposition plays a key role in catalyst deactivation. CHNS elemental analysis (Table S1†) further supported this observation, showing a substantial increase in carbon accumulation on the catalyst surface after the reaction, which may lead to the blockage of active sites, thereby reducing reaction selectivity and conversion. The catalyst lifetime assessment (Fig. 2a) demonstrates that, in addition to performance degradation due to carbon deposition, irreversible deactivation occurs following prolonged operation and repeated regeneration cycles. The crystalline structure of Nb-based catalysts plays a crucial role in determining their physicochemical properties and catalytic activity.31
To investigate the structural changes in the deactivated catalyst, X-ray diffraction (XRD) analysis was conducted, and the results are presented in Fig. 3a. The XRD patterns confirm that the primary structure of the catalyst is Nb2O5 before the reaction. After the reaction, new diffraction peaks appear at 18.6°, 26.3°, and 30.5°, corresponding to the formation of Nb12O29 (JCPDS 19-0863), indicating that the catalytic process induces crystalline modifications in Nb2O5.
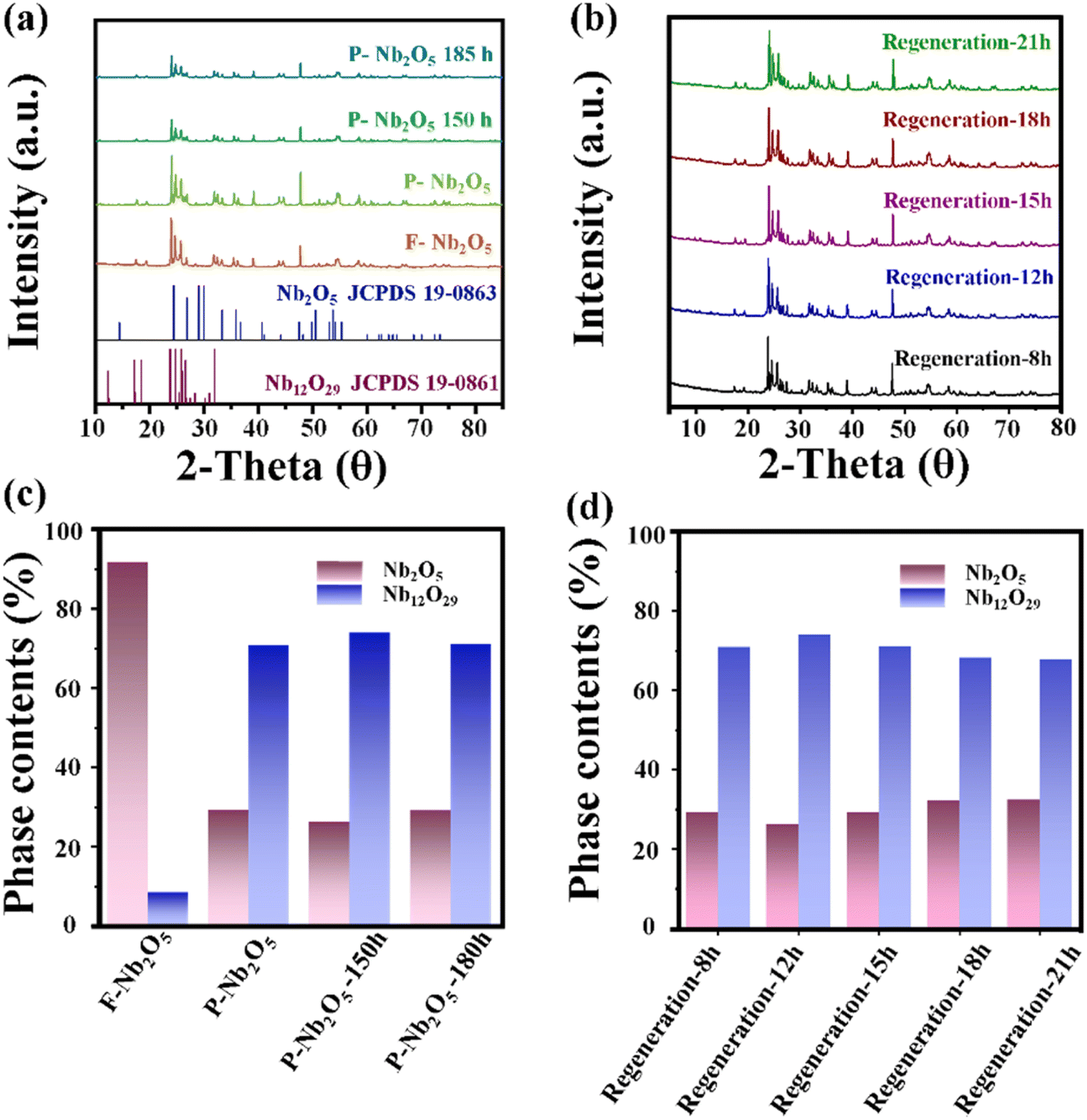 | ||
| Fig. 3 X-ray diffraction (XRD) patterns and phase content of F-Nb2O5, P-Nb2O5 and various catalysts to different reaction times (a and c); the XRD patterns and phase content of catalysts regenerated at 550 °C in an air atmosphere for varying durations (b and d). | ||
The phase composition of the catalysts, calculated using the Scherrer equation, is shown in Fig. 3c.25 After a single reaction (with a test duration of 7 h), the Nb12O29 phase content significantly increased. After extending the reaction time to 150 and 180 h, the catalyst's phase composition was predominantly Nb12O29. Regeneration experiments conducted after 150 h of reaction (Fig. 3b) show that, despite varying calcination times at 500 °C, the diffraction peak of Nb12O29 at 26.3° persists. Even with extended regeneration up to 21 h, the characteristic Nb12O29 peak remains unchanged. As shown in Fig. 3d, the calculated content of Nb2O5 and Nb12O29 across different catalysts indicates that after 21 h of regeneration, the Nb2O5 content does not increase with prolonged calcination, and the Nb12O29 content shows no significant variation. These observations suggest that the deactivation of Nb2O5 catalysts is not solely due to carbon deposition but also involves significant alterations in the catalyst's surface structure. To fully elucidate the deactivation mechanism, further analysis is required to quantify the individual and combined effects of carbon deposition and structural changes within the catalyst.
In this study, we analyzed carbon deposits from two locations: inside the fixed-bed reactor (Carbon-F) and on the catalyst surface (Carbon-C). Thermogravimetric Analysis (TGA) identified the coke species, as shown in Fig. S4.† Most carbon deposits, whether in the reactor or on the catalyst, consisted of hard, aromatic compounds, removable at 550 °C. We used Liquid Chromatography-Mass Spectrometry (LC-MS) to qualitatively analyze these coke species, confirming their types and pathways of accumulation.32–35 The identified carbon species ranged from aliphatic hydrocarbons and olefins to single-ring and fused-ring aromatic hydrocarbons, as well as amorphous and graphitic carbon.
Prior to LC-MS analysis, the carbon deposits on the catalyst were dissolved, with their solubility classified by their ability to dissolve in specific solvents.36,37 We employed the Guisnet method,38 where carbon deposits from the reactor (Carbon-F) and the post-reaction catalyst were treated with hydrofluoric acid to degrade the catalyst. Dichloromethane, an organic solvent, was used to extract soluble carbon, while insoluble carbon was separated by centrifugation. The LC-MS analysis of the dichloromethane extract differentiated the soluble carbon species, unaffected by inorganic acids.39 LC-MS chromatograms for Carbon-F and Carbon-C identified the main species, listed in Table 1. Carbon-F was primarily composed of malonic dihydrazide, with small amounts of pentanenitrile, likely due to ammonolysis and hydrolysis during the reaction.40 Myo-inositol was the major component of the soluble carbon deposits on the catalyst, possibly formed through cyclization during the reaction.
| Name | Formula | #CAS | |
|---|---|---|---|
| Carbon-C | |||
| Methanol | CH4O | 67-56-1 | |
| Hydrazine | H4N2 | 302-01-2 | |
| Oxalyl dihydrazide | C2H6N4O2 | 996-98-5 | |
| 1,2-Diformylhydrazine | C2H4N2O2 | 628-36-4 | |
| Malonic dihydrazide | C3H8N4O2 | 3815-86-9 | |
| N,N′′-Methyleneis-urea | C3H8N4O2 | 13547-17-6 | |
| Hexanedioyl azide | C6H8N6O2 | 25021-15-2 | |
| Pentanenitrile | C3H7NO | 33279-01-5 | |
| Myo-inositol | C6H12O6 | 87-89-8 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Carbon-F | |||
| 2,5-Dimethyl-3,4-hexanediol | C8H18O2 | 22067-11-0 | |
| 2,2-Dimethoxypropane | C5H12O2 | 77-76-9 | |
| 2,3-Diazabicyclo [2.2.1]hept-2-yl,3-(1,1-imethylethyl) – (9CI) | C9H17N2 | 40953-67-1 | |
| Propionamide | C3H7NO | 79-05-0 | |
| N-Methylacetamide | C3H7NO | 79-16-3 | |
Myo-inositol is identified as the primary component of Carbon-C, likely due to its formation during the reaction process. In catalytic reactions, especially those involving hydrocarbons, complex chemical transformations such as cyclization, polymerization, and condensation can occur.41,42 Myo-inositol, being a relatively stable cyclic compound, is more likely to persist under the reaction conditions and subsequently be detected in the soluble fraction of the carbon deposits.
During dichloromethane extraction, a black precipitate, termed “insoluble carbon”, was separated by centrifugation. This hydrocarbon, with a molecular weight exceeding that of pyrene,43,44 was identified as primarily amorphous carbon via X-ray diffraction (XRD) analysis (Fig. S5a†). Raman spectroscopy revealed G (1580–1600 cm−1) and D (1360–1400 cm−1) peaks (Fig. S5b†), indicating a disordered carbon structure with low graphitization, as shown by an ID/IG ratio of 1.88.40,45 Transmission electron microscopy (TEM) images (Fig. S5c and d†) further confirmed a block-like structure.
To monitor changes in carbon species during the nitridation reaction, the Nb2O5 catalyst was analyzed at different time intervals (3, 5, 10, and 25 h), with samples collected at distinct deactivation stages, labeled as Nb2O5 (3), Nb2O5 (5), Nb2O5 (10), and Nb2O5 (25). FT-IR spectroscopy was used to examine the vibrations of functional groups on the catalysts over time, as shown in Fig. 4a. Carbon deposition is typically observed between 1300 and 1800 cm−1, while the C–H vibration region appears at 2600–3200 cm−1. Peaks at 1460 and 1620 cm−1 correspond to fused-ring and single-ring aromatic hydrocarbons, respectively.40,46–48 The increasing intensities of these peaks indicate that aromatic hydrocarbons are the primary components of carbon deposition. Peaks between 1360 and 1460 cm−1 are associated with both aliphatic and aromatic hydrocarbons.49–51 The decreasing intensity of the aliphatic hydrocarbon peak at 1390 cm−1 and the increasing intensity of the aromatic hydrocarbon peak at 1460 cm−1 suggest that polycyclic aromatic hydrocarbons become the main constituents of carbon deposition over time. In the C–H vibration region (Fig. 4b), peaks at 2960, 2920, 2870, and 2830 cm−1 correspond to CH2 and CH3 groups,52,53 with their increasing intensities indicating a rise in aliphatic hydrocarbon content as the reaction progresses. LC-MS data (Table 1) further show that the soluble carbon species in the deposits shift from single-ring aromatic hydrocarbons to condensed-ring aromatic hydrocarbons as reaction time increases (Fig. 4c).
 | ||
| Fig. 4 (a and b) FT-IR spectroscopy and (c) soluble carbon species detectable with reaction time on Carbon-C. | ||
The impact of carbon build-up on the catalyst pore structure and its effect on catalytic performance can be thoroughly assessed through nitrogen adsorption and desorption analysis. As depicted in Fig. 5a, the nitrogen adsorption isotherms reveal a marked decrease in both microporous and total adsorption with increasing reaction time at low relative pressures (P/P0). This trend suggests that as the reaction progresses, surface micropores become increasingly obstructed by carbon deposits, thereby limiting the interaction between active sites and the reactants. To further explore the extent of carbon deposition, X-ray photoelectron spectroscopy (XPS) was employed to quantify the surface carbon content. Fig. 5b presents the XPS-derived C/Nb molar ratio for samples subjected to different reaction durations. The C/Nb ratio was calculated using eqn (3)
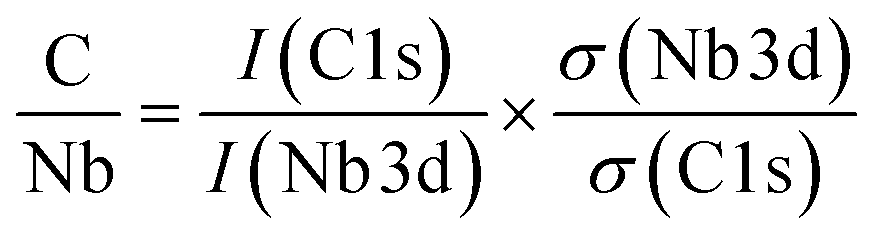 | (3) |
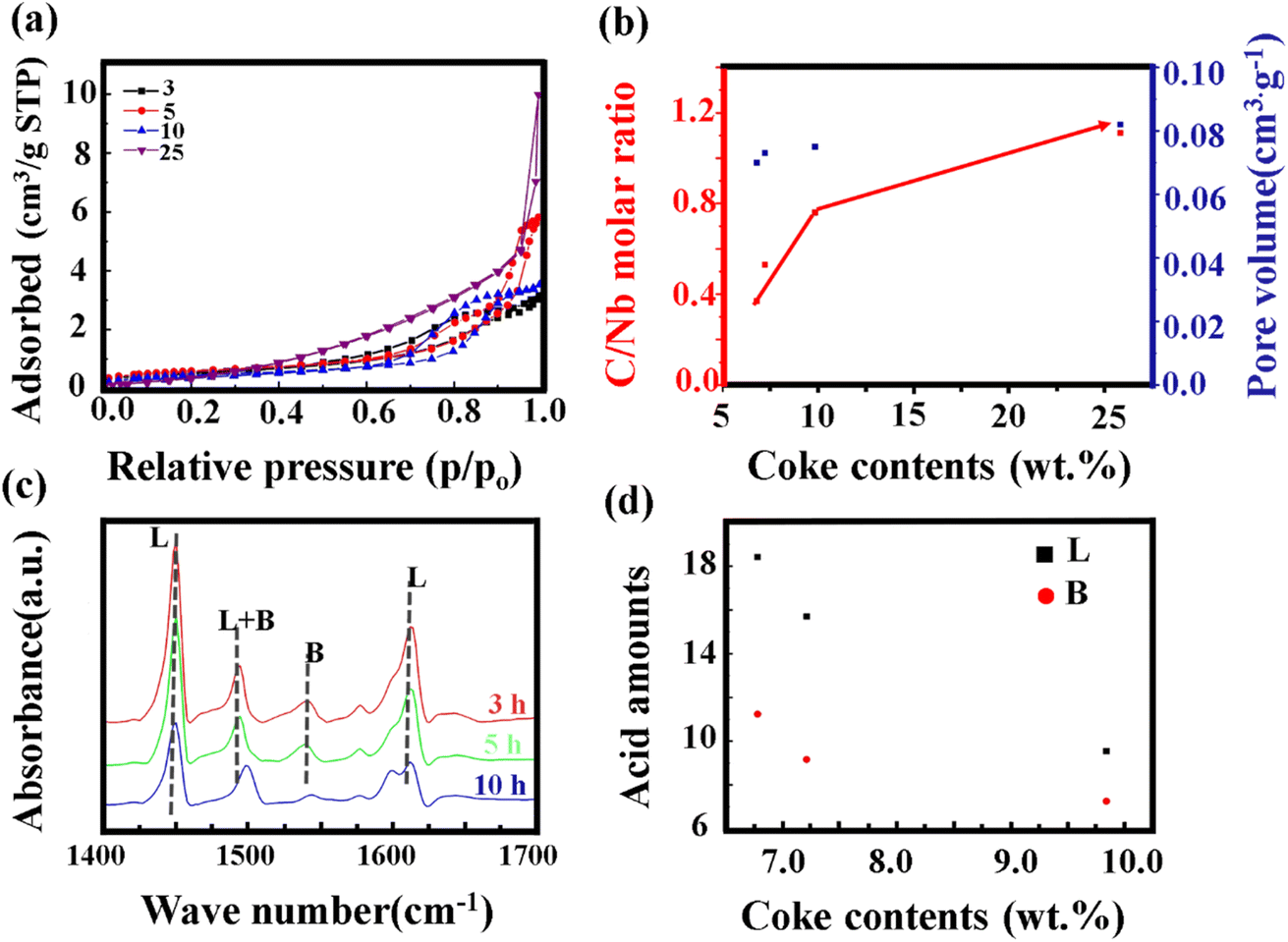 | ||
| Fig. 5 (a) Nitrogen adsorption and desorption isotherms of catalysts after various reaction times; (b) measured C/Nb ratios by XPS (red) and micropore volume (blue) of various samles as a function of coke weight content; (c) Py-IR spectra of catalysts after different reaction times; (d) peak area of the bands at 1450 and 1540 cm−1versus coke content similarities (b and c). | ||
Integration of the spectral data, as depicted in Fig. 5d, quantifies the impact of carbon accumulation on these acid sites. The integrated peak area for the L acid site shows a substantial decline with increasing carbon content, while the B acid exhibits a less pronounced reduction. This suggests that carbon deposition predominantly targets the L acid, impairing their functionality. Consequently, the reduction in available acid sites due to carbon deposition hinders the diffusion of reactants and products, thereby contributing to catalyst deactivation over time. This comprehensive analysis underscores the dual impact of carbon deposition: blocking of micropores and masking of crucial acid sites, both of which play pivotal roles in the observed catalyst deactivation. Further studies are required to explore mitigation strategies and optimize catalyst design for improved resistance to carbon-induced deactivation.
These findings indicate that carbon deposition plays a crucial role in blocking micropores and masking acid sites, leading to a gradual decline in catalytic performance. However, to fully elucidate the mechanisms of catalyst deactivation, it is essential not only to explore the impact of carbon accumulation but also to investigate the structural changes occurring within the Nb2O5 catalyst itself during the reaction.55,56 By examining the crystallographic phase transitions and surface structural modifications that occur under reaction conditions, we aim to gain a deeper understanding of the relationship between catalyst structure and its long-term stability.
The valence states of F-Nb2O5 and P-Nb2O5 were analyzed using XPS, as shown in Fig. 6a and b. For P-Nb2O5, the Nb 3d peaks were observed at 205.6 and 208.3 eV, corresponding to Nb 3d3/2 and Nb 3d5/2, respectively, which are characteristic of Nb5+ in Nb2O5.57,58 Additionally, companion peaks at 204.9 and 207.8 eV, associated with Nb4+, were also identified.59,60 These results suggest that during the reaction, a portion of Nb5+ was reduced to Nb4+, which aligns with the presence of Nb12O29 observed in the XRD analysis. Given these observations, it is crucial to further investigate how the reaction conditions, particularly the ammonia atmosphere and temperature, influence these structural changes in the catalyst. In the subsequent experiments, we will focus on examining the evolution of the catalyst structure under varying ammonia concentrations and thermal conditions, aiming to better understand the factors driving the reduction of Nb5+ to Nb4+.
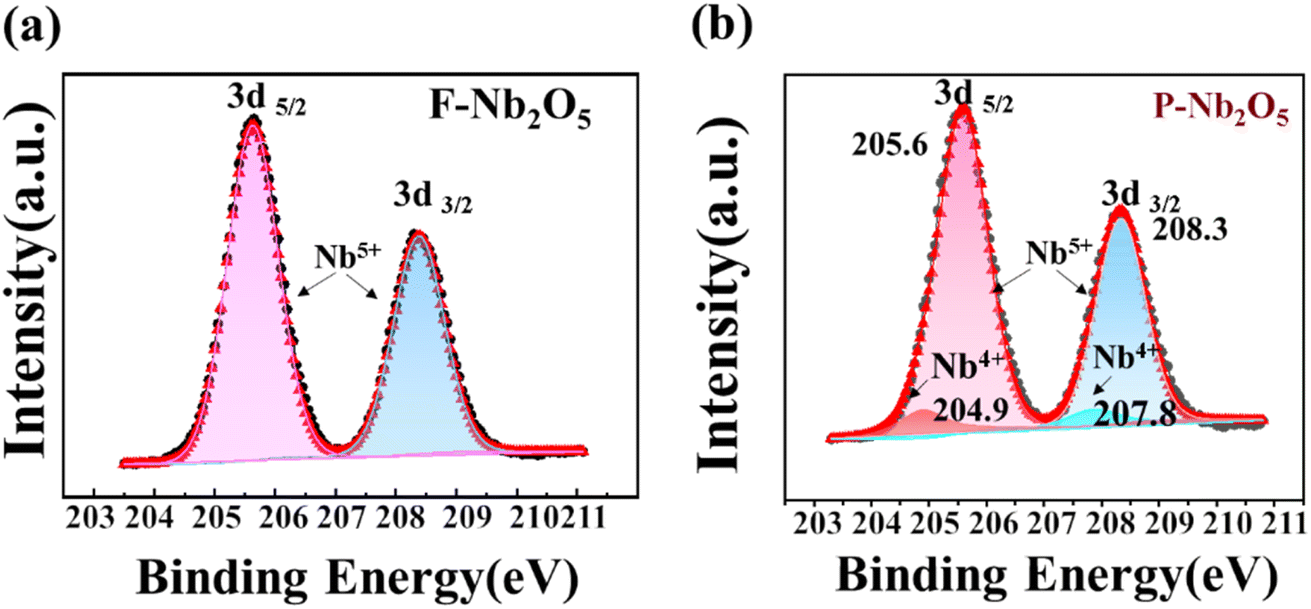 | ||
| Fig. 6 XPS patterns of Nb 3d (a and b) of the catalyst before and after the reaction. | ||
As illustrated in Fig. 7a and b, the XRD patterns reveal the impact of different reaction temperatures and durations on the crystalline structure of Nb2O5. The experiments were conducted using a Cu Kα radiation source (λ = 1.5406 Å) with a scanning range of 10°–80°, a step size of 0.02°, and a scanning rate of 2° min−1. As the reaction temperature increases or the duration extends, the diffraction peaks become sharper and more defined, indicating enhanced crystallinity. However, the peak positions remain consistent with Nb2O5 (JCPDS 19-0861), confirming its structural stability under the investigated conditions. This suggests that while higher temperatures or longer reaction times facilitate crystallization, they do not induce a phase transition within the tested parameter range.
 | ||
| Fig. 7 Effect of temperature (a and d), time (b and e), and NH3 (c and f) flow rate on the crystalline phases and oxidation states of niobium oxides: XRD and XPS. | ||
Further insights into the oxidation state of niobium under varying reaction conditions were obtained from XPS analysis (Fig. 7d and e). Across the temperature range of 200 to 500 °C and reaction times spanning 2 to 10 h, the Nb5+ peaks exhibit clear separation without significant shifts, indicating that niobium predominantly retains its Nb5+ oxidation state. This finding substantiates that temperature and reaction time alone do not contribute to the reduction of Nb5+.
In the NH3 reduction experiments, the XRD patterns under different NH3 flow rates (Fig. 7c) indicate that as the NH3 flow rate increases from 2.0 ml min−1 to 6.0 ml min−1, new diffraction peaks emerge at 18.6°, 26.3°, and 30.5°, corresponding to Nb12O29 (JCPDS 19-0863). This confirms a gradual phase transition from Nb2O5 to Nb12O29 with increasing NH3 flow rate. Furthermore, XPS spectra (Fig. 7f) reveal a shift in the Nb 3d binding energy toward lower values (204.9 eV and 207.8 eV), indicating the coexistence of Nb5+ and Nb4+ species. Notably, at higher NH3 flow rates, the Nb5+/Nb4+ ratio significantly decreases, suggesting that NH3 not only facilitates the reduction of Nb2O5 but may also lead to the formation of sub-stoichiometric niobium oxides such as NbO2. This alteration in oxidation state could subsequently influence the electronic structure and catalytic properties of the material.
All experiments were conducted under atmospheric pressure. According to the literature, pressure typically does not significantly impact phase composition in similar reaction systems under ambient conditions. Therefore, the effect of pressure on phase transitions was not explicitly analyzed in this study, as it was not expected to substantially influence the observed results. However, previous studies have reported that in the Nb–O system, pressure can modulate phase stability by altering oxygen vacancy concentration and Nb–O bond length. Specifically, under high-pressure conditions exceeding 1 GPa, Nb2O5 may undergo structural rearrangement and transition to NbO2 or NbO, primarily due to the reduced activation energy for Nb–O bond reconfiguration and oxygen vacancy formation.61 However, under atmospheric pressure (1 atm), the oxygen vacancy concentration remains relatively low, ensuring the structural stability of Nb2O5. Therefore, in the context of this study, we consider the impact of pressure on the phase transition of Nb2O5 to be negligible.
To investigate the effect of the Nb4+ structure on the acidity of the catalyst, pyridine adsorption infrared spectroscopy (FT-IR) was employed (Fig. 8). Pyridine, used as a probe molecule for acidic sites, allows for differentiation between Brønsted acid sites (B sites) and Lewis acid sites (L sites) based on its characteristic vibrational frequencies when adsorbed. FT-IR analysis was conducted on Nb2O5 samples treated with varying NH3 flow rates, with desorption temperatures of 100 °C, 150 °C, and 350 °C. The results demonstrate a clear increase in the intensity of peaks associated with both B and L acid sites as the NH3 flow rate increased. Notably, the characteristic L acid peaks at 1450 cm−1 and 1600 cm−1 became more pronounced, indicating that NH3 treatment not only facilitated the formation of B acid sites but also significantly enhanced the quantity of L acid sites.62,63 Additionally, it was observed that the sample treated at an NH3 flow rate of 2.5 ml min−1 exhibited a marked increase in the intensity of the L acid peaks. This detailed analysis highlights the critical role of NH3 flow rate in modulating the acid properties of Nb2O5, with particular emphasis on the enhancement of Lewis acidity, a key factor in catalytic performance.
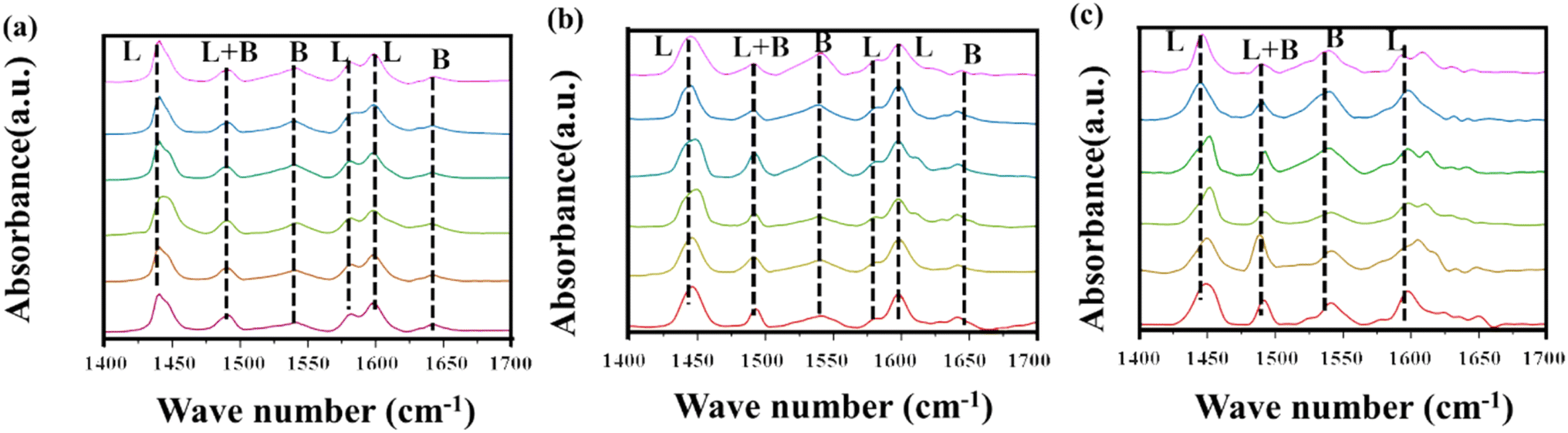 | ||
| Fig. 8 Infrared spectra of pyridine adsorption at different desorption temperatures after treatment with different ammonia flow rates: (a) 100 °C; (b) 150 °C; (c) 350 °C. | ||
Fig. 9a presents the O 1s XPS spectra reveal two key oxygen components in Nb2O5, lattice oxygen (O1) and oxygen vacancies (O2). The detailed O 1s data under different ammonia flow rates are presented in the Fig. S6.† The O1 component corresponds to highly ordered lattice oxygen atoms bound to niobium, which are stable and exhibit low reactivity.58 The O2 component is attributed to oxygen vacancies formed when Nb5+ is reduced to Nb4+ due to the loss of lattice oxygen. The presence of oxygen vacancies leads to the formation of coordinatively unsaturated metal centers, which are crucial for the catalytic properties of the material. As ammonia flow rate increases, the oxygen vacancy content initially rises, reaching a peak, and then decreases at higher ammonia flow rates due to the loss of surface-adsorbed oxygen. This volcano-shaped trend illustrates that excessive reduction beyond a certain point disrupts the balance of oxygen vacancies, thereby affecting the catalyst's ability to activate reactants.
 | ||
| Fig. 9 (a) The O 1s XPS spectra; (b) the different acid amount and the corresponding Ov ratio of the catalysts; and (c) the Raman shift. | ||
This relationship between oxygen vacancies and acidic sites is further confirmed by the measurements of Brønsted and Lewis acid sites in Fig. 9b. The optimal number of both B acid and L acid sites is achieved at an ammonia flow rate of 2 ml min−1, where the oxygen vacancy content is maximized. The formation of these vacancies not only reduces Nb5+ to Nb4+ but also facilitates the generation of a larger number of acidic sites. Brønsted acid sites (B acid) play a crucial role in activating DMA by providing proton transfer pathways, enhancing the reactivity of DMA with ammonia. On the other hand, Lewis acid sites (L acid) assist in the adsorption and activation of electron-donating molecules, promoting the overall reaction.
Raman spectroscopy further reveals the structural impact of ammonia treatment on Nb2O5 (Fig. 9c), particularly at a flow rate of 2 ml min−1, where the signal intensity corresponding to Nb–O–Nb bridging and edge sites is significantly enhanced.64,65 This observation indicates that the strengthening of Nb–O–Nb bridging bonds and the increase in edge sites under this condition promote the formation of oxygen vacancies.66 Changes in these bridging and edge sites imply that the reorganization of the Nb2O5 surface structure is beneficial for generating active sites, thereby enhancing the adsorption and activation of reactants and ultimately improving catalytic performance. Specifically, the enhancement of Nb–O–Nb bridging and edge sites is closely linked to the increased concentration of B acid sites, as these structural modifications provide more binding sites for protons, thus augmenting the strength and quantity of Brønsted acid sites.67 This suggests that the structural changes observed in Raman spectra are directly correlated with the formation of B acid sites.
In the reaction of dimethyl adipate with ammonia to form adiponitrile, an excess of B acid sites has a pronounced influence on the reaction mechanism.68,69 The B acid sites activate dimethyl adipate by providing proton transfer pathways, thereby facilitating its reactivity with ammonia. However, when B acid sites are in excess, the proton transfer reaction can become overly intense, leading to the rapid activation of intermediates and consequently accelerating adiponitrile formation. The acceleration of this reaction pathway is closely related to the formation of oxygen vacancies and the increased concentration of B acid sites. The presence of oxygen vacancies not only increases the number of active sites but also enhances the adsorption and conversion of reactants through the increased influence of surface Brønsted acid sites. Nevertheless, an excess of both B acid and L acid sites may result in side reactions, such as the over-acceleration of adiponitrile formation and rapid carbon deposition, which can cover the catalyst surface and hinder reactant adsorption, ultimately lowering the overall catalytic efficiency. Therefore, it is essential to control an appropriate ammonia-to-ester ratio during the reaction to avoid the side reactions and carbon deposition associated with excessive B and L acid sites.
4. Conclusion
This study systematically investigated the deactivation mechanisms of Nb2O5 catalysts in a fixed-bed reactor, revealing that carbon deposition and structural phase transformations are the primary contributors to catalyst deactivation. During the reaction, carbon deposits evolved from monocyclic to polycyclic aromatic hydrocarbons, leading to the blockage of critical acid sites and diminished catalytic activity. Simultaneously, Nb2O5 underwent a phase transformation to Nb12O29, and catalyst regeneration partially reversed this transformation, restoring Nb2O5 content and reducing Nb12O29. This phase change facilitated the formation of oxygen vacancies, which created unsaturated metal coordination sites, altering the catalyst's acidity profile. The findings emphasize the importance of maintaining moderate acidity to achieve efficient conversion and high nitrile selectivity. Additionally, strategies to mitigate coke formation and stabilize active catalyst sites are essential for improving the stability, reusability, and overall performance of Nb2O5 catalysts.Building on these findings, the study also highlights the practical implications for large-scale industrial applications. The stability and reusability of catalysts are crucial for optimizing industrial production, particularly in the manufacture of nitrile-based chemicals. This research provides a theoretical basis for improving catalyst stability, reducing energy consumption, and extending catalyst lifetime. By optimizing ammonia flow rates and controlling acidic sites, future research can enhance catalytic efficiency and minimize maintenance costs, thus contributing to more efficient and sustainable industrial processes.
Data availability
The data supporting this article have been included within the manuscript and its ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially funded by the National Key R&D Program of China (2022YFA1504902), National Natural Science Foundation of China (No. 22178358, 22078337, 22078336), the Young Scientists Fund of the National Natural Science Foundation of China (No. 22408376), the Postdoctoral Fellowship Program of CPSF (GZC20232700) and the “Special Research Assistant Project” of the Chinese Academy of Sciences.References
- K. Geng, H. Li, D. Zhang, B. Yu, H. Hu, J. Geng, Y. Wu and X. Hu, Producing hexamethylenediamine from caprolactam via 6-aminocapronitrile: a green production technology of the monomer of nylon-66, Green Chem., 2024, 8777–8784, 10.1039/d4gc02268h.
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Catalytic routes towards acrylic acid, adipic acid and ε-caprolactam starting from biorenewables, Green Chem., 2015, 17(3), 1341–1361, 10.1039/c4gc02076f.
- W. C. Drinkard and R. V. Lind sey Jr, Hydrocyanation of olefins using selected nickel phosphite catalysts, US Pat., 1970 Search PubMed.
- J. Yang, P. Wang, H. Neumann, R. Jackstell and M. Beller, Industrially applied and relevant transformations of 1,3-butadiene using homogeneous catalysts, Ind. Chem. Mater., 2023, 1(2), 155–174, 10.1039/d3im00009e.
- M. M. Baizer and J. L. Chruma, Electrolytic Reductive Coupling: XX. Mixed Reductive Couplings with Acrylonitrile Reductions at the Cathode Voltage Required for the More Difficultly Reduced Partner, J. Electrochem. Soc., 1971, 118, 3, DOI:10.1149/1.2408079.
- M. Seko, K. Mihara, S. Kumazaki, R. Komori and M. Yoshida, Adiponitrile production by the electrolytic hydrodimerization of acrylonitrile, US Pat., US3595764, 1971 Search PubMed.
- A. S. Nizami, X. Luo, J. Dou, A. Bai, L. Wang, M. Wei, M. J. K. Bashir and H. Ganjidoust, Production technology of adiponitrile, E3S Web Conf., 2023, 441, 01019, DOI:10.1051/e3sconf/202344101019.
- L. Du, Z.-T. Cao, X.-M. Wang, M.-Q. Wang, B.-C. Sun, G.-W. Chu and J.-F. Chen, Reaction kinetics and mechanism of adiponitrile preparation by adipic acid ammoniation, Chem. Eng. Sci., 2024, 290, 119851, DOI:10.1016/j.ces.2024.119851.
- H. Zhao, H. Wei, Y. Guo, L. Han, L. Wang, Y. Hu and J. Xin, Study on the stability of palladium complexes with Hemaraphos ligand and reaction kinetics in the bis-hydroalkoxycarbonylation of 1,3-butadiene, J. Catal., 2023, 428, 115160, DOI:10.1016/j.jcat.2023.115160.
- L. Wang, Z. Zhao, S. Wang, W. Zhou, X. Lu, R. Yan and J. Xin, Unveiling the hydroesterification of 1,3-butadiene with carbonyl cobalt ionic liquids: Facilitation of cationic protonic hydrogen for active species formation, J. Catal., 2024, 434, 115525, DOI:10.1016/j.jcat.2024.115525.
- T. Guo, F.-Y. Yan, Y.-F. Wang, X.-F. Xu, Q.-Z. Jia and B.-H. Xu, Boosted nitrilation of dimethyl adipate with NH3 to adiponitrile over bimetallic oxide: Synergetic effect between Nb and W, Chem. Eng. Sci., 2023, 281, 119121, DOI:10.1016/j.ces.2023.119121.
- Z. H. Zhang, L. J. Han, S. S. Ma, Y. R. Du, Z. K. Yu, J. Q. Lin and B. H. Xu, Scalable Palladium-Catalyzed Alkoxycarbonylation of Conjugated Dienes, J. Org. Chem., 2023, 88(2), 882–892, DOI:10.1021/acs.joc.2c02245.
- G.-R. Ding, Y.-F. Wang, G.-Y. Duan, T. Guo, X.-F. Xu, C.-S. Li and B.-H. Xu, Chemoselective nitrilation of dimethyl adipate with ammonia over carbon encapsulated WOx catalysts under continuous flow conditions, Catal. Sci. Technol., 2022, 12(12), 3982–3991, 10.1039/d2cy00733a.
- Y. Shirazi, H. Tafazolian, S. Viamajala, S. Varanasi, Z. Song and M. J. Heben, High-Yield Production of Fatty Nitriles by One-Step Vapor-Phase Thermocatalysis of Triglycerides, ACS Omega, 2017, 2(12), 9013–9020, DOI:10.1021/acsomega.7b01502.
- M. Eric, T. R. E. Karp, V. Sànchez i Nogué, V. Vorotnikov, M. J. Biddy, E. C. D. Tan, D. G. Brandner, R. M. Cywar, R. Liu, L. P. Manker, W. E. Michener, M. Gilhespy, Z. Skoufa, M. J. Watson, O. Stanley Fruchey, D. R. Vardon, R. T. Gill, A. D. Bratis and G. T. Beckham, Renewable acrylonitrile production, Science, 2017, 358(6368), 1307–1310 CrossRef PubMed.
- A. Mekki-Berrada, S. Bennici, J. P. Gillet, J. L. Couturier, J. L. Dubois and A. Auroux, Ammoniation-dehydration of fatty acids into nitriles: heterogeneous or homogeneous catalysis?, ChemSusChem, 2013, 6(8), 1478–1489, DOI:10.1002/cssc.201300210.
- A. Mekki-Berrada, S. Bennici, J. P. Gillet, J. L. Couturier, J. L. Dubois and A. Auroux, Fatty acid methyl esters into nitriles: Acid–base properties for enhanced catalysts, J. Catal., 2013, 306, 30–37, DOI:10.1016/j.jcat.2013.05.032.
- X.-F. Xu, Y.-F. Wang, T. Guo, L.-K. Luan, S.-S. Liu and B.-H. Xu, Synthesis of adiponitrile from dimethyl adipate and ammonia in the vapor-phase over niobium oxide, Catal. Sci. Technol., 2022, 12(12), 3947–3956, 10.1039/d2cy00734g.
- U. E. Amjad, A. Tajjamal, A. Ul-Hamid, A. Faisal, S. A. H. Zaidi, L. Sherin, A. Mir, M. Mustafa, N. Ahmad and M. Hussain, et al., Catalytic cracking of polystyrene pyrolysis oil: effect of Nb(2)O(5) and NiO/Nb(2)O(5) catalyst on the liquid product composition, Waste Manage., 2022, 141, 240–250, DOI:10.1016/j.wasman.2022.02.002.
- Y. Li, J. Guo, H. Liu, A. Liu and D. Li, In situ generated oxygen vacancy on Nb2O5 for boosted catalytic activities of M/Nb2O5 in photothermal CO2 reforming of CH4 J. CO2 Util., 2023, 67, 102330, DOI:10.1016/j.jcou.2022.102330.
- H. L. Kaiyi Su, Z. Gao, P. Fornasiero and F. Wang, Nb2O5-Based Photocatalysts, Adv. Sci., 2021, 2003156, DOI:10.1002/advs.202003156.
- C. H. Bartholomew, Mechanisms of catalyst deactivation, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- T. Z. Bo Yuan, Y. Han, X. Zhang, M. Wang and Li Chen, Deactivation Mechanism and Anti-Deactivation Measures of Metal Catalyst in the Dry Reforming of Methane: A Review, Atmosphere, 2023, 14(5), 770, DOI:10.3390/atmos14050770.
- F. Pio and L. Luca, Catalyst deactivation, Catal. Today, 1999, 52(2–3), 165–181 Search PubMed.
- M. Argyle and C. Bartholomew, Heterogeneous Catalyst Deactivation and Regeneration: A Review, Catalysts, 2015, 5(1), 145–269, DOI:10.3390/catal5010145.
- Z. Chen, Y. Ye, X. Feng, Y. Wang, X. Han, Y. Zhu, S. Wu, S. Wang, W. Yang and L. Wang, et al., High-density frustrated Lewis pairs based on Lamellar Nb(2)O(5) for photocatalytic non-oxidative methane coupling, Nat. Commun., 2023, 14(1), 2000, DOI:10.1038/s41467-023-37663-x.
- R. Pang, Z. Wang, J. Li and K. Chen, Polymorphs of Nb(2)O(5) Compound and Their Electrical Energy Storage Applications, Materials, 2023, 16(21), 6956, DOI:10.3390/ma16216956.
- V. G. F. Pereira, C. R. Moreira, C. P. Rodrigues and F. S. Toniolo, Influence of active sites and the reaction conditions on the ethanol upgrading over Nb2O5/ZrO2 based multifunctional catalysts, Braz. J. Chem. Eng., 2022, 40(4), 1039–1054, DOI:10.1007/s43153-022-00287-7.
- J. Li, G. Lu, K. Li and W. Wang, Active Nb2O5-supported nickel and nickel–copper catalysts for methane decomposition to hydrogen and filamentous carbon, J. Mol. Catal. A: Chem., 2004, 221(1–2), 105–112, DOI:10.1016/j.molcata.2004.06.015.
- G. R. Ding, Y. F. Wang, G. Y. Duan, Y. Q. Fan and B. H. Xu, Adjustment of W-O-Zr Boundaries Boosts Efficient Nitrilation of Dimethyl Adipate with Ammonia on WO(x)/ZrO(2) Catalysts, ACS Appl. Mater. Interfaces, 2023, 15(2), 3633–3643, DOI:10.1021/acsami.2c18908.
- L. Yan, Y. Liu, K. Zha, H. Li, L. Shi and D. Zhang, Deep insight into the structure–activity relationship of Nb modified SnO2–CeO2 catalysts for low-temperature selective catalytic reduction of NO by NH3, Catal. Sci. Technol., 2017, 7(2), 502–514, 10.1039/c6cy02242a.
- Y. Li, C. Zhang, Y. Liu, X. Hou, R. Zhang and X. Tang, Coke Deposition on Ni/HZSM-5 in Bio-oil Hydrodeoxygenation Processing, Energy Fuels, 2015, 29(3), 1722–1728, DOI:10.1021/ef5024669.
- M. J. Wulfers and F. C. Jentoft, Identification of carbonaceous deposits formed on H-mordenite during alkane isomerization, J. Catal., 2013, 307, 204–213, DOI:10.1016/j.jcat.2013.07.011.
- X.-Y. Ren, J.-P. Cao, S.-X. Zhao, X.-Y. Zhao, X.-B. Feng, T.-L. Liu, Y. Li, J. Zhang and X.-Y. Wei, Insights into coke location of catalyst deactivation during in-situ catalytic reforming of lignite pyrolysis volatiles over cobalt-modified zeolites, Appl. Catal., A, 2021, 613, 118018, DOI:10.1016/j.apcata.2021.118018.
- P. Magnoux, C. Canaff, F. Machado and M. Guisnet, Coking, Aging, and Regeneration of Zeolites, J. Catal., 1992, 134, 286–298 CrossRef CAS.
- L. Y. Jia, A. Farouha, L. Pinard, S. Hedan, J. D. Comparot, A. Dufour, K. Ben Tayeb, H. Vezin and C. Batiot-Dupeyrat, New routes for complete regeneration of coked zeolite, Appl. Catal., B, 2017, 219, 82–91, DOI:10.1016/j.apcatb.2017.07.040.
- M. Díaz, E. Epelde, J. Valecillos, S. Izaddoust, A. T. Aguayo and J. Bilbao, Coke deactivation and regeneration of HZSM-5 zeolite catalysts in the oligomerization of 1-butene, Appl. Catal., B, 2021, 291, 120076, DOI:10.1016/j.apcatb.2021.120076.
- L. Palumbo, F. Bonino, P. Beato, M. Bjørgen and A. Zecchina, Conversion of methanol to hydrocarbons: spectroscopic characterization of carbonaceous species formed over H-ZSM-5, J. Phys. Chem. C, 2008, 112(26), 9710–9716 CrossRef CAS.
- P. Rzepka, D. Sheptyakov, C. Wang, J. A. van Bokhoven and V. Paunović, How Micropore Topology Influences the Structure and Location of Coke in Zeolite Catalysts, ACS Catal., 2024, 14(8), 5593–5604, DOI:10.1021/acscatal.4c00025.
- F. F. Madeira, H. Vezin, N. S. Gnep, P. Magnoux, S. Maury and N. Cadran, Radical Species Detection and Their Nature Evolution with Catalyst Deactivation in the Ethanol-to-Hydrocarbon Reaction over HZSM-5 Zeolite, ACS Catal., 2011, 1(4), 417–424, DOI:10.1021/cs2000686.
- L. K. Shrestha, L. Adhikari, R. G. Shrestha, M. P. Adhikari, R. Adhikari, J. P. Hill, R. R. Pradhananga and K. Ariga, Nanoporous carbon materials with enhanced supercapacitance performance and non-aromatic chemical sensing with C(1)/C(2) alcohol discrimination, Sci. Technol. Adv. Mater., 2016, 17(1), 483–492, DOI:10.1080/14686996.2016.1219971.
- H. V. Lin, D. S. Chan, Y. H. Huang and W. C. Sung, Kinetics of Moisture Loss and Oil Absorption of Pork Rinds during Deep-Fat, Microwave-Assisted and Vacuum Frying, Foods, 2021, 10(12), 3025, DOI:10.3390/foods10123025.
- A. C. Ferrari, Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects, Solid State Commun., 2007, 143(1–2), 47–57, DOI:10.1016/j.ssc.2007.03.052.
- I Childres, L. A. Jauregui and Y. P. Chen, Raman spectra and electron-phonon coupling in disordered graphene with gate-tunable doping, J. Appl. Phys., 2014, 22, 243–250 Search PubMed.
- Z. Cheng, S. Huang, Y. Li, K. Cai, D. Yao, J. Lv, S. Wang and X. Ma, Carbonylation of dimethyl ether over MOR and Cu/H-MOR catalysts: Comparative investigation of deactivation behavior, Appl. Catal., A, 2019, 576, 1–10, DOI:10.1016/j.apcata.2019.02.032.
- X. Xian, C. Ran, C. Nai, P. Yang, S. Zhao and L. Dong, Characterization of the location of coke deposited on spent HZSM-5 zeolite by special temperature-programmed oxidation and isothermal oxidation methods, Appl. Catal., A, 2017, 547, 37–51, DOI:10.1016/j.apcata.2017.08.023.
- T. Vazhnova, S. P. Rigby and D. B. Lukyanov, Benzene alkylation with ethane in ethylbenzene over a PtH-MFI catalyst: Kinetic and IR investigation of the catalyst deactivation, J. Catal., 2013, 301, 125–133, DOI:10.1016/j.jcat.2013.02.002.
- C. Song, K. Liu, D. Zhang, S. Liu, X. Li, S. Xie and L. Xu, Effect of cofeeding n-butane with methanol on aromatization performance and coke formation over a Zn loaded ZSM-5/ZSM-11 zeolite, Appl. Catal., A, 2014, 470, 15–23, DOI:10.1016/j.apcata.2013.10.036.
- X.-G. Li, X. Huang, Y.-L. Zhang, H. Li, W.-D. Xiao and Z. Wei, Effect of n-butanol cofeeding on the deactivation of methanol to olefin conversion over high-silica HZSM-5: A mechanism and kinetic study, Chem. Eng. Sci., 2020, 226, 115859, DOI:10.1016/j.ces.2020.115859.
- T. Cordero-Lanzac, A. Ateka, P. Pérez-Uriarte, P. Castaño, A. T. Aguayo and J. Bilbao, Insight into the Deactivation and Regeneration of HZSM-5 Zeolite Catalysts in the Conversion of Dimethyl Ether to Olefins, Ind. Eng. Chem. Res., 2018, 57(41), 13689–13702, DOI:10.1021/acs.iecr.8b03308.
- X. Huang, X. Jiao, M. Lin, K. Wang, L. Jia, B. Hou and D. Li, Coke distribution determines the lifespan of a hollow Mo/HZSM-5 capsule catalyst in CH4 dehydroaromatization, Catal. Sci. Technol., 2018, 8(22), 5740–5749, 10.1039/c8cy01391h.
- H. Xu, Z. Wang, Z. Miao, Y. Zhu, A. Marianov, L. Wang, P. Castignolles, M. Gaborieau, J. Huang and Y. Jiang, Correlation between Acidity and Catalytic Performance of Mesoporous Zirconium Oxophosphate in Phenylglyoxal Conversion, ACS Sustain. Chem. Eng., 2019, 7(9), 8931–8942, DOI:10.1021/acssuschemeng.9b00989.
- H. T. Kreissl, K. Nakagawa, Y.-K. Peng, Y. Koito, J. Zheng and S. C. E. Tsang, Niobium oxides: Correlation of acidity with structure and catalytic performance in sucrose conversion to 5-hydroxymethylfurfural, J. Catal., 2016, 338, 329–339, DOI:10.1016/j.jcat.2016.03.007.
- M. Chakraborty and H.-J. Bart, Highly selective and efficient transport of toluene in bulk ionic liquid membranes containing Ag+ as carrier, Fuel Process. Technol., 2007, 88(1), 43–49, DOI:10.1016/j.fuproc.2006.08.004.
- V. V. Atuchin, I. E. Kalabin, V. G. Kesler and N. V. Pervukhina, Nb 3d and O 1s core levels and chemical bonding in niobates, J. Electron Spectrosc. Relat. Phenom., 2005, 142(2), 129–134, DOI:10.1016/j.elspec.2004.10.003.
- Z. Zhang, H. Xu and H. Li, Insights into the catalytic performance of Ni/Nb2O5 catalysts for vanillin hydrodeoxygenation in aqueous phase: the role of Nb2O5 crystal structures, Fuel, 2022, 324, 124400, DOI:10.1016/j.fuel.2022.124400.
- Z. Weibin, W. Weidong, W. Xueming, C. Xinlu, Y. Dawei, S. Changle, P. Liping, W. Yuying and B. Li, The investigation of NbO2 and Nb2O5 electronic structure by XPS, UPS and first principles methods, Surf. Interface Anal., 2013, 45(8), 1206–1210, DOI:10.1002/sia.5253.
- Y. Zou, Y. Hu, A. Uhrich, Z. Shen, B. Peng, Z. Ji, M. Muhler, G. Zhao, X. Wang and X. Xu, Steering accessible oxygen vacancies for alcohol oxidation over defective Nb2O5 under visible light illumination, Appl. Catal., B, 2021, 298, 120584, DOI:10.1016/j.apcatb.2021.120584.
- A. Barrera, J. A. Montoya, M. Viniegra, J. Navarrete, G. Espinosa, A. Vargas, P. del Angel and G. Pérez, Isomerization of n-hexane over mono- and bimetallic Pd–Pt catalysts supported on ZrO2–Al2O3–WOx prepared by sol–gel, Appl. Catal., A, 2005, 290(1–2), 97–109, DOI:10.1016/j.apcata.2005.05.011.
- L. Sun, J. Sun, S. Zhai, H. Yang, X. Chen, W. Q. Deng and H. Wu, Nb(2) CT(x) MXene Derived Polymorphic Nb(2)O(5), Small, 2023, 19(26), e2300914, DOI:10.1002/smll.202300914.
- Z. Guan, Q. Li, H. Zhang, P. Shen, L. Zheng, S. Chu, C. Park, X. Hong, R. Liu and P. Wang, et al., Pressure induced transformation and subsequent amorphization of monoclinic Nb(2)O(5) and its effect on optical properties, J. Phys.:Condens. Matter, 2019, 31(10), 105401, DOI:10.1088/1361-648X/aaf9bd.
- K. Stawicka, P. Decyk, A. Wojtaszek-Gurdak and M. Ziolek, Comparative study of acid-basic properties of MCF impregnated with niobium and cerium species, Catal. Today, 2019, 325, 2–10, DOI:10.1016/j.cattod.2018.05.025.
- C. A. Emeis, Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- S. M. A. H. Siddiki, M. N. Rashed, A. S. Touchy, M. A. R. Jamil, Y. Jing, T. Toyao, Z. Maeno and K.-i. Shimizu, Hydrolysis of amides to carboxylic acids catalyzed by Nb2O5, Catal. Sci. Technol., 2021, 11(5), 1949–1960, 10.1039/d0cy02230f.
- T. Ikeya and M. Senna, Change in the structure of ninbium pentoxide due to mechanical and thermal treatments, J. Non-Cryst. Solids, 1988, 105, 243–250 CrossRef CAS.
- G. D. Wilk, R. M. Wallace and J. M. Anthony, High-κ gate dielectrics: current status and materials properties considerations, J. Appl. Phys., 2001, 89, 5243–5275, DOI:10.1063/1.1361065.
- U. Balachandran and N. G. Eror, Raman spectrum of the high temperature form of Nb2O5, J. Mater. Sci. Lett., 1982, 374–376 CrossRef CAS.
- M. A. Ali, S. M. A. H. Siddiki, W. Onodera, K. Kon and K. i. Shimizu, Amidation of Carboxylic Acids with Amines by Nb2O5 as a Reusable Lewis Acid Catalyst, ChemCatChem, 2015, 7(21), 3555–3561, DOI:10.1002/cctc.201500672.
- M. A. Ali, S. K. Moromi, A. S. Touchy and K. i. Shimizu, Direct Synthesis of Cyclic Imides from Carboxylic Anhydrides and Amines by Nb2O5 as a Water-Tolerant Lewis Acid Catalyst, ChemCatChem, 2016, 8(5), 891–894, DOI:10.1002/cctc.201501172.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00731j |
| This journal is © The Royal Society of Chemistry 2025 |