
Solventless, rapid-polymerizable liquid resins from solid carboxylic acids through low-viscosity acid/base complexes†
Grant M.
Musgrave‡
 a,
Eden Y.
Yau‡
a,
Sijia
Huang
b,
Caleb J.
Reese
a and
Chen
Wang
*a
a,
Eden Y.
Yau‡
a,
Sijia
Huang
b,
Caleb J.
Reese
a and
Chen
Wang
*a
aDepartment of Materials Science and Engineering, University of Utah, Salt Lake City, Utah 84112, USA. E-mail: chen.wang@utah.edu
bLawrence Livermore National Laboratory, Livermore, California 94550, USA
First published on 21st November 2024
Abstract
Bio-based carboxylic acids are some of the most available renewable chemicals, but since they are solids with high melting temperatures, they cannot be directly used as liquid resins. To this end, we report the formation of supramolecular complexes between an amino methacrylate and various solid carboxylic acids. The ionically bonded methacrylates exhibit low viscosities and rapid reaction kinetics for free-radical mediated polymerization, showing quantitative methacrylate conversions within one minute of irradiation at 5 mW cm−2 405 nm light. We demonstrate the implementation of these acid-base complexes as a neat resin system that comprises orthogonal polymerization reactions (free-radical methacrylate polymerization and epoxy-acid polymerization reactions), which yields high-strength network polymer materials.
 Chen Wang | Chen Wang is an Assistant Professor of Materials Science and Engineering at the University of Utah. He received BS in Polymer Materials and Engineering from Beijing University of Chemical Technology. He earned PhD in Chemical Engineering from University of Colorado Boulder and his thesis was focused on polymer colloids and dynamic covalent polymer chemistry. Prior to his academic career, he received training in industry (Formlabs Inc.) and national labs (National Renewable Energy Laboratory). His current research focuses on “orthogonal reactions in neat polymerization” for applications such as 3D printing and composite materials processing. |
Introduction
Carboxylic acids are important feedstock chemicals for the synthesis of a variety of polymer materials, including polyesters, polyamides (i.e., nylon), polyols, and photopolymers.1,2 Today, carboxylic acids are mostly produced by oxidating hydrocarbons—adding oxygen atoms into the petrochemicals.1 While petrochemicals are mostly hydrocarbons that contain few oxygen atoms, oxygen-rich molecules are ubiquitously found in biological systems.3 Producing bio-based carboxylic acids is often advantageous as it reduces the embodied supply chain energy and greenhouse gas emissions.4 Several multifunctional bio-acids have been commercialized, including succinic acid, itaconic acid, and adipic acid (Scheme 1A).3 Recent studies also show carboxylic acids as products from chemical recycling and upcycling of plastics, including polyolefins and polyesters.7,8 However, it is often extremely challenging for such emerging green chemical technologies to compete with the existing petrochemical supply chain. Therefore, we must develop new high-value, high-volume applications from these renewable chemicals to help decarbonize the chemical industry.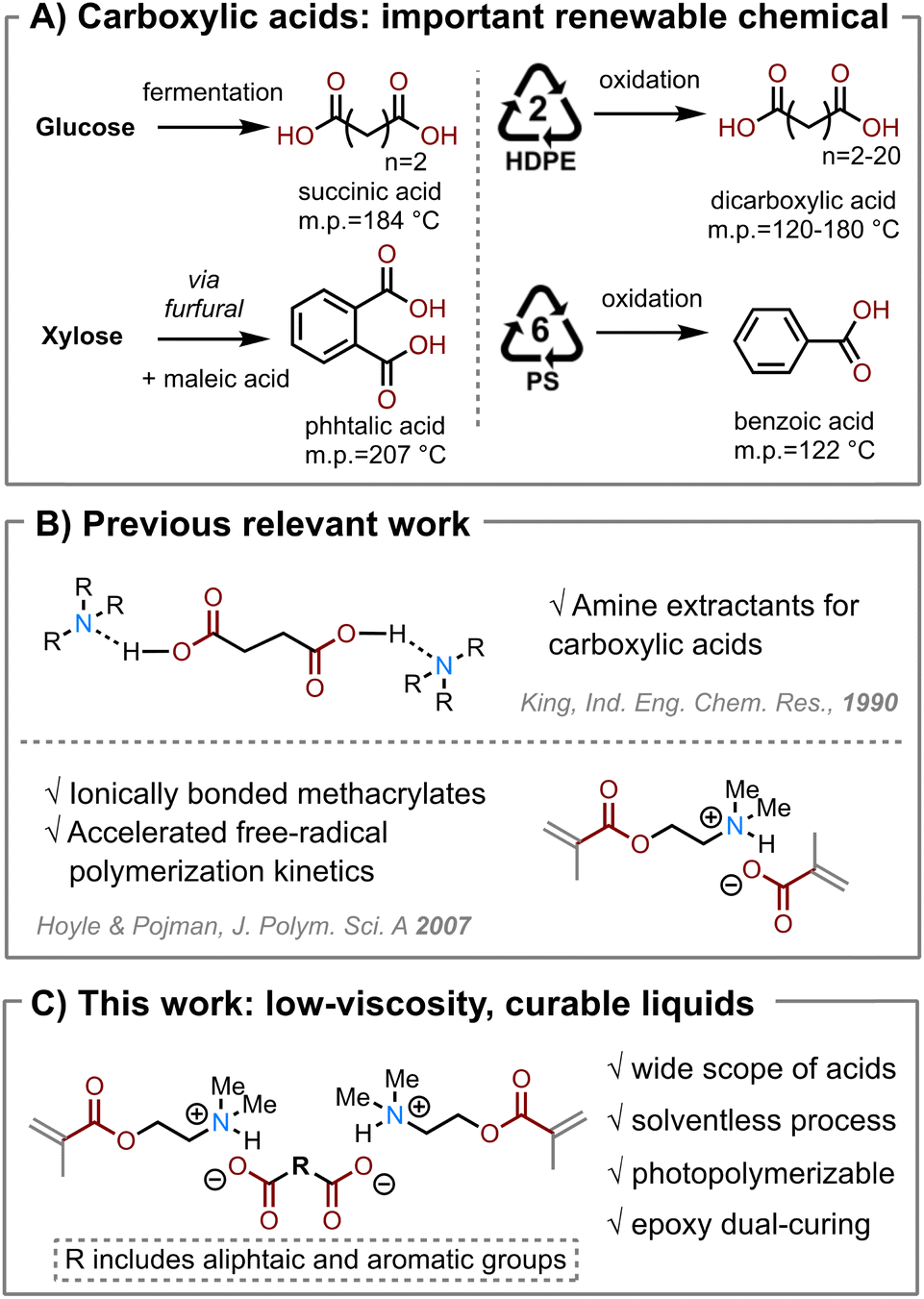 | ||
| Scheme 1 (A) Carboxylic acids are important renewable chemicals, including ones derived from biological feedstocks such as glucose and hemicellulose (left) and ones from plastic upcycling (right). (B) Previous work that inspires our hypothesis. King et al.5 studied the formation of ionic liquid between aliphatic acids with amines. Hoyle and Pojman et al.7 found the fast reaction kinetics of the ionic liquid formed by 2-(dimethylamino)ethyl methacrylate (DMAEMA) and methacrylic acid. (C) Hypothesis of forming ionic liquids of low viscosity by solventless processing of DMAEMA and solid dicarboxylic acids. | ||
Network polymers, i.e. crosslinked polymers, make up many high-volume polymers for high-value applications such as fiber-reinforced composites.9 Epoxy resins alone are consumed at >4 million metric tons (MMT) annually.10 In practice, epoxy monomers are used by performing step-growth polymerizations with a co-monomer, a process often described as “curing”. Suitable comonomers include thiols,11,12 amines,13 and anhydrides14—often described as a “hardener”. Carboxylic acids can readily polymerize with epoxy but are rarely used as a hardener. These epoxy-hardener polymerization reactions are thermally controlled, which limits a practitioner's capability to initiate curing promptly. For example, in the manufacturing of glass fiber composites used for wind turbine blades, after transferring the epoxy resin into the molds, work must be halted for hours for the curing to complete.15 Therefore, it is beneficial to introduce a curing mechanism that enables prompt controllability.
Photopolymerization is a technique where light acts as a contactless stimulus, creating free radicals that rapidly convert low-viscosity liquid monomers into network polymers.16 The monomers used for photopolymerization are typically multifunctional acrylate (or methacrylate) monomers that contain two or more of the vinyl groups. The ability to form covalent crosslinking is instrumental to the rapid reaction kinetics, which is the key enabler for several photopolymerization applications such as coatings, adhesives, and dental restorative materials (“fillings”).17 Photopolymerization reactions can be spatially and temporally controlled through the generation of a catalyst from the incident light with remarkable precision. Such spatiotemporal control is highlighted in the application of 3D printing by vat photopolymerization processes that create 3D shapes with high resolution.18
We sought to develop epoxy hardeners from carboxylic acids with desirable capabilities of photo-mediated curing. Given the recently discovered benefits of epoxy-anhydride polymers,19 we envisioned that implementing carboxylic acids would advance the field by broadening the available chemical structures in these network polymers. One of the prohibitive challenges of using carboxylic acids for epoxy curing is their high melting temperatures (Tm). As shown in Scheme 1A, virtually all multifunctional carboxylic acids are solids at ambient conditions, with Tms in the range of 120–300 °C. Due to their high polarity, high hydrophilicity, and high crystallinity, these acids also have poor solubility in many organic solvents and most organic monomers used for synthesizing hydrophobic, mechanically robust materials.
We hypothesize that supramolecular-bonded, stable liquids at ambient conditions can be formed between a basic (meth)acrylate and a solid carboxylic acid. This hypothesis is inspired by using tertiary amines as extractants to recover carboxylic acid from aqueous solutions (Scheme 1B).20 King et al. have comprehensively studied the solubility and extraction of a variety of carboxylic acids in various organic solvents.5 We identify an amino methacrylate monomer, namely 2-(dimethylamino)ethyl methacrylate (DMAEMA), a suitable platform base compound to investigate the acid–base interactions with various carboxylic acids (Scheme 1C). Hoyle and Pojman et al. reported that neat DMAEMA showed slow polymerization reaction, but showed fast reaction kinetics after it was neutralized with methacrylic acid (Scheme 1B).6 The effect of neutralization of DMAEMA by various carboxylic acids on its reaction kinetics has not been investigated. We further designed a photo/heat “dual-cure” method for DMAEMA/acid/epoxy systems: first, the methacrylate is photopolymerized by free-radical reactions; second, the acid is reacted with epoxy after being freed from the acid-base complexes. We chose two bio-derived epoxy monomers derived from glycerol and sorbitol to investigate the relationship between the monomer chemical structures and the material properties.
Results and discussions
Viscosity measurement of stoichiometric DMAEMA-acid complexes
As a first attempt, we investigated a series of combinations by mixing DMAEMA with various carboxylic acids with a stoichiometric ratio between the amine and the acid groups. To explore a wide range of acids, in this study, we investigated aromatic acids, linear aliphatic diacids, cyclic aliphatic diacids, and trifunctional aliphatic acids, shown in Scheme 2. We hypothesized that since the tertiary amine on DMAEMA has a pKa of 8.44 (conjugated), complexes should be formed between DMAEMA and the carboxylic acids, which typically have pKa values of 3–5. Such acid–base complexes could be formed by hydrogen bonding or the formation of ionic bonds. The DMAEMA-acid mixtures remained separated until they were heated to 120 °C, and in some cases to even higher temperature to melt the acid. As a result, the DMAEMA-acid complexes formed a homogeneous clear liquid. When the mixtures were cooled to ambient conditions, we found that many of them remained as bench-stable liquids, though some of the acids recrystallized in the mixture. The acids that recrystallized when mixed with DMAEMA included terephthalic acid (tPA), oxalic acid, malonic acid, and citric acid. To our surprise, no stable liquid could be formed with oxalic acid (n = 0), nor malonic acid (n = 1), despite their high acidity and their moderate melting temperatures (190 °C and 136 °C, respectively). We were delighted to observe that several dicarboxylic acids of interest formed stable liquids, including glutaric acid (GA), adipic acid (AA, shown in Fig. 1A), suberic acid (SuA), and phthalic acid (PA, shown in Fig. 1B). Interestingly, succinic acid (SA) formed a stable liquid but recrystallized after a few days of storage under ambient conditions. All observations are summarized in Table 1.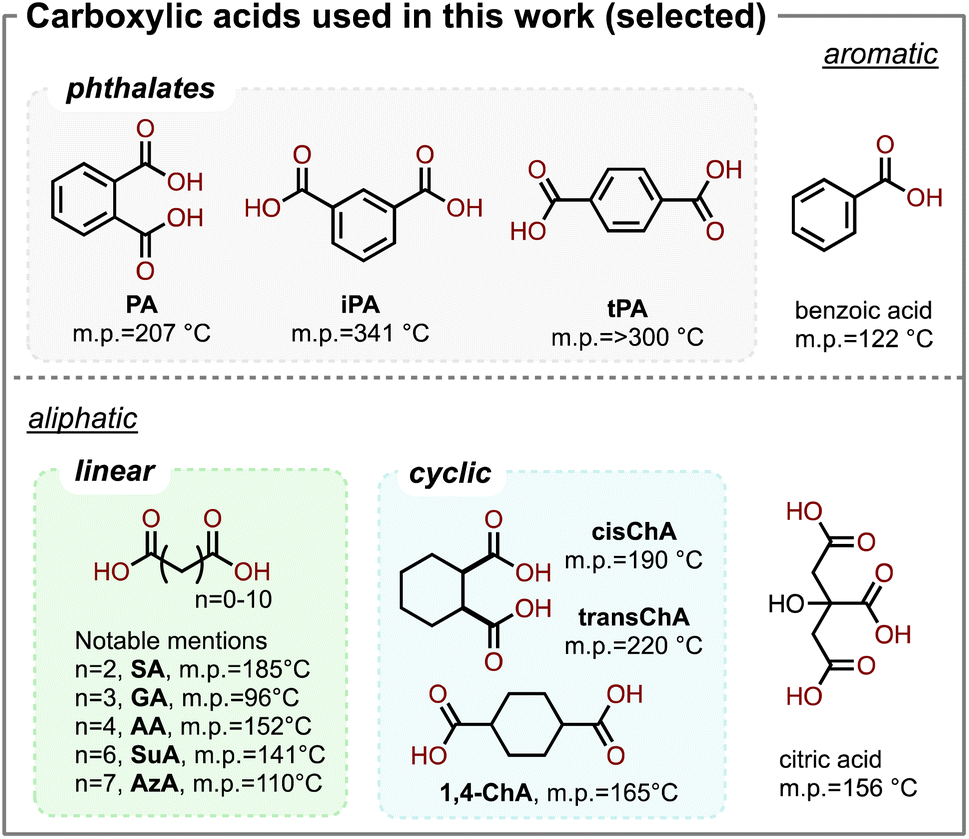 | ||
| Scheme 2 Chemical structures and melting temperatures (m.p.) of some of the carboxylic acids used in this work. Aromatic acids include phthalates with three isomers: phthalic acid (PA), isophthalic acid (iPA), and terephthalic acid (tPA); and benzoic acid. Aliphatic acids include linear dicarboxylic acids such as succinic acid (SA), glutaric acid (GA), adipic acid (AA), suberic acid (SuA), and azelaic acid (AzA). Cycloaliphatic acids include cis-cyclohexane-1,2-dicarboxylic acid (cChA), trans-cyclohexane-1,2-dicarboxylic acid (tChA), and cyclohexane-1,4-dicarboxylic acid (1,4ChA). Trifunctional acids include citric acid. | ||
 | ||
| Fig. 1 Stoichiometric acid/base mixtures formed bench-stable liquids after brief heating. (A) Pictures of DMAEMA and adipic acid mixture. (B) Pictures of DMAEMA and phthalic mixture. (C) Viscosities of complexes using aromatic, linear aliphatic, and cyclic aliphatic diacids. Measurement was taken at 22 °C at 60 s−1 shear rate on a rheometer equipped with parallel plates. Numerical data is listed in Table S1.† | ||
| Acid | pKa | Acid wt% | Soluble |
|---|---|---|---|
| a Predicted. | |||
| PA | 2.89, 5.51 | 34.6 | Yes |
| iPA | 3.46, 4.46 | 34.6 | Yes |
| tPA | 3.54, 4.34 | 34.6 | No |
| Benzoic acid | 4.20 | 43.7 | Yes |
| Oxalic acid | 1.27, 4.27 | 22.3 | No |
| Malonic acid | 2.85, 5.05 | 24.9 | No |
| SA | 4.21, 5.41 | 27.3 | Yes, unstable |
| GA | 4.34, 5.41 | 29.6 | Yes |
| AA | 4.41, 5.41 | 31.7 | Yes |
| Pimelic acid | 4.50, 5.43 | 33.8 | Yes |
| SuA | 4.53, 5.50 | 35.7 | Yes |
| Azelaic acid | 4.55, 5.50 | 37.5 | Yes |
| Sebacic acid | 4.72, 5.45 | 39.1 | Yes |
| cChA | 3.89, 6.79 | 35.4 | Yes |
| tChA | 4.18, 5.93 | 35.4 | Yes |
| 1,4ChA | 4.38a | 35.4 | Yes |
| Citric acid | 3.15, 4.78, 6.40 | 28.9 | No |
| Levulinic acid | 4.59 | 42.5 | Yes |
Many of the bench-stable DMAEMA-diacid complexes were found to be liquids with moderate viscosities. The viscosities were measured on a TA DHR-20 rheometer equipped with 25 mm parallel plates at a steady shear rate of 60 s−1 and an ambient temperature of 22 °C. For aromatic acids, the stoichiometric DMAEMA-PA complex exhibited a viscosity of 214 cP. The complex was found to be bench-stable. We found its viscosity showed <10% change when it was stored at 22 °C for a week. Also, as an “accelerated storage test”, we found its viscosity also within 10% change after being heated at 60 °C for 24 hours. Interestingly, the DMAEMA-iPA complex was viscous (4350 ± 110 cP). Despite iPA being an isomer of PA, iPA has a high melting temperature at 341 °C and a slightly higher pKa, which may result in incomplete bonding of the carboxylic acid group with the tertiary amine. The unbonded carboxylic acid can form intermolecular hydrogen bonding, which results in increased viscosity. For linear aliphatic diacids, when n = 3–8 (Fig. 1C), liquids were formed with low viscosities ranging from 77 to 144 cP; these low viscosities are desirable for our intended use of these acid-base complexes as “hardeners” for epoxy. For cyclic aliphatic diacids, we tested three commercially available dicarboxylic acids which are based on cyclohexane. The 1,2-dicarboxylic acids exhibited low viscosities, at 220 ± 2 cP and 399 ± 6 cP for cis and trans isomers, respectively. The 1,4-dicarboxylic acid resulted in a highly viscous liquid (58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 ± 1000 cP). Given the similarities of the three cyclic diacids in their chemical structures, melting temperature, and pKa, it is likely that the viscosity is affected by other factors. In the following study, we decided to focus on investigating AA and PA, given their ability to form low-viscous liquids with DMAEMA and their profound impact on the (bio)chemical industry.21,22
400 ± 1000 cP). Given the similarities of the three cyclic diacids in their chemical structures, melting temperature, and pKa, it is likely that the viscosity is affected by other factors. In the following study, we decided to focus on investigating AA and PA, given their ability to form low-viscous liquids with DMAEMA and their profound impact on the (bio)chemical industry.21,22
Verification of ionic bonding in DMAEMA-acid complexes by 1H NMR
The acid/base complexes, including DMAEMA-AA and DMAEMA-PA, were examined by 1H NMR conducted with various solvents. First, all samples were taken 24 hours after they were prepared in deuterated water (D2O), shown in Fig. 2. DMAEMA exhibited spontaneous hydrolysis since the ester is prone to hydrolysis in basic conditions. Such hydrolysis was absent in the DMAEMA-AA complex. The peaks associated with the aliphatic backbone of AA shifted upfield from 1.63 and 2.41 ppm to 1.54 and 2.18 ppm in the DMAEMA-AA complex, respectively, likely due to the deprotonation of the carboxylic acid. The peaks associated with the tertiary amine on DMAEMA shifted downfield from 4.30, 2.78, and 2.32 ppm to 4.52, 3.56, and 2.96 ppm, respectively. Similarly, for the DMAEMA-PA complex, the aromatic peaks on PA shifted upfield from 7.83 and 7.71 ppm to 7.50 and 7.44 ppm, respectively. We believe that these two complexes are bonded ionically since other non-covalent mechanisms, such as hydrogen bonding, would have disappeared in water.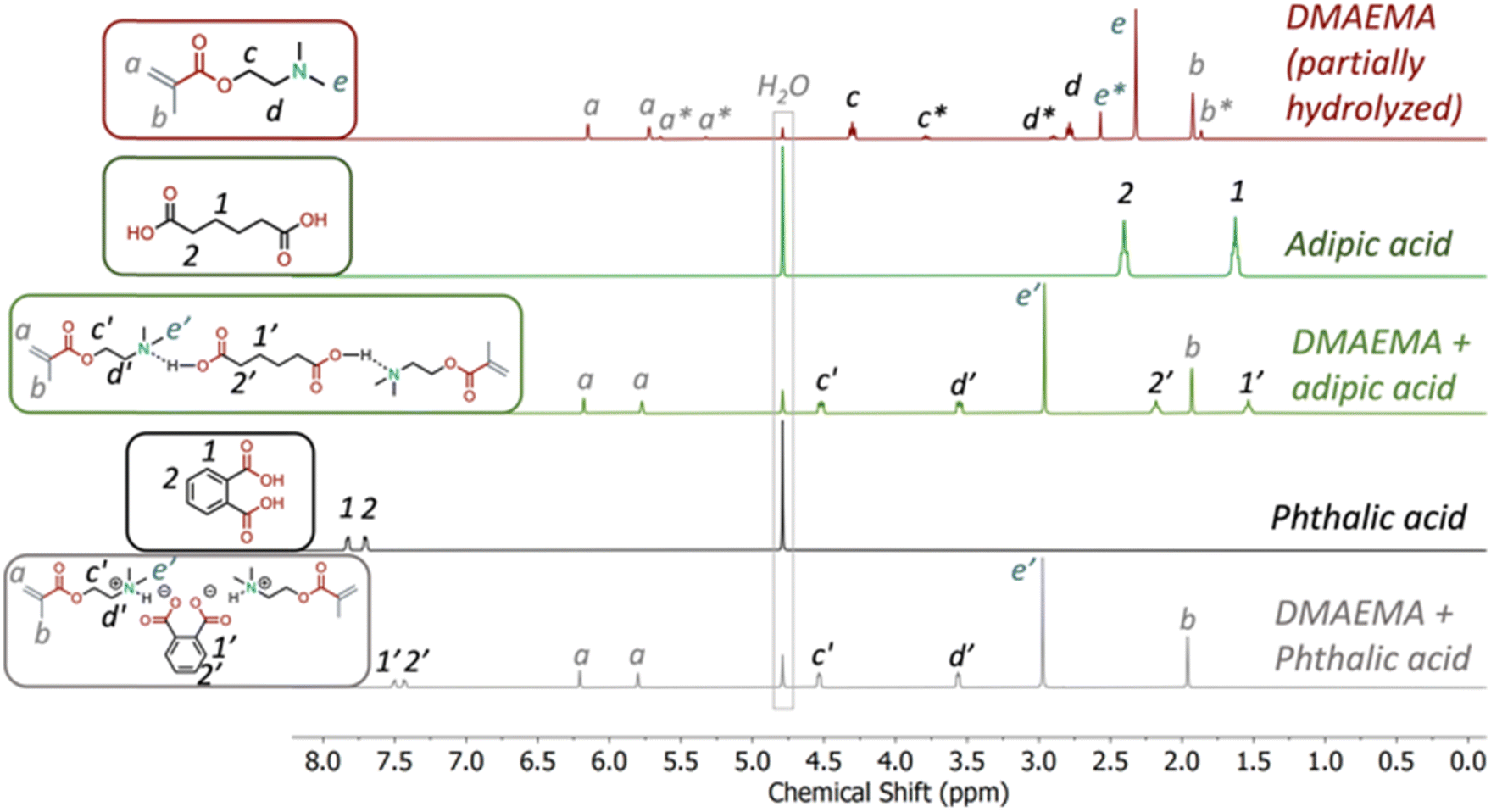 | ||
| Fig. 2 1H NMR spectrum of 2-(dimethylamino)ethyl methacrylate (DMAEMA), adipic acid, DMAEMA-adipic acid complex (stoichiometric), phthalic acid, DMAEMA-phthalic acid (stoichiometric) complex in D2O. NMRs taken 24 h after sample preparation. DMAEMA showed partial hydrolysis. Both acid/base complexes remained bonded in D2O. | ||
We conducted 1H NMR measurements in non-polar and polar organic solvents. In deuterated chloroform (CDCl3), the peaks associated with the tertiary amine on DMAEMA shifted downfield (Fig. S1 and S2†). Both AA and PA do not dissolve in CDCl3. Similarly, in deuterated dimethyl sulfoxide (DMSO-D6), the DMAEMA-PA complex was bonded (Fig. S3†). However, interestingly, the DMAEMA-AA complex dissociated, likely because AA became less acidic in DMSO (Fig. S4†). As an aprotic polar solvent, DMSO cannot donate hydrogen bonds to stabilize carboxylic anions. We reason that in neat conditions, these acid-base complexes are ionically bonded.
Thermal properties of off-stoichiometric DMAEMA-acid complexes
We further examined thermal properties of the complexes. Differential scanning calorimetry (DSC) revealed that the complexes are glass formers, showing “glass transitions” at −80 to −60 °C. As shown in Fig. 3A, the temperature of “glass transitions” differed consistently with the structure of the acids. For aliphatic acids, when the diacids are shorter, the concentration of ionic bonds increases in the stoichiometric DMAEMA-acid complexes. As a result, the “glass transition” increased from −80 °C for DMAEMA-AA to −79 °C and −76 °C for DMAEMA-GA and DMAEMA-SA complexes, respectively. The DMAEMA-SA showed a crystallization temperature of 9 °C, consistent with our observation that it recrystallizes at ambient conditions. DMAEMA-PA showed a much higher “glass transition” at −60 °C, likely due to stronger ionic bonding. The formation of glass was also evident in the cooling cycles (Fig. S5†). We believe the formation of glass suggests these acid-base complexes are supramolecular macromolecules, which has been evidenced in many examples of ionic liquids.23–25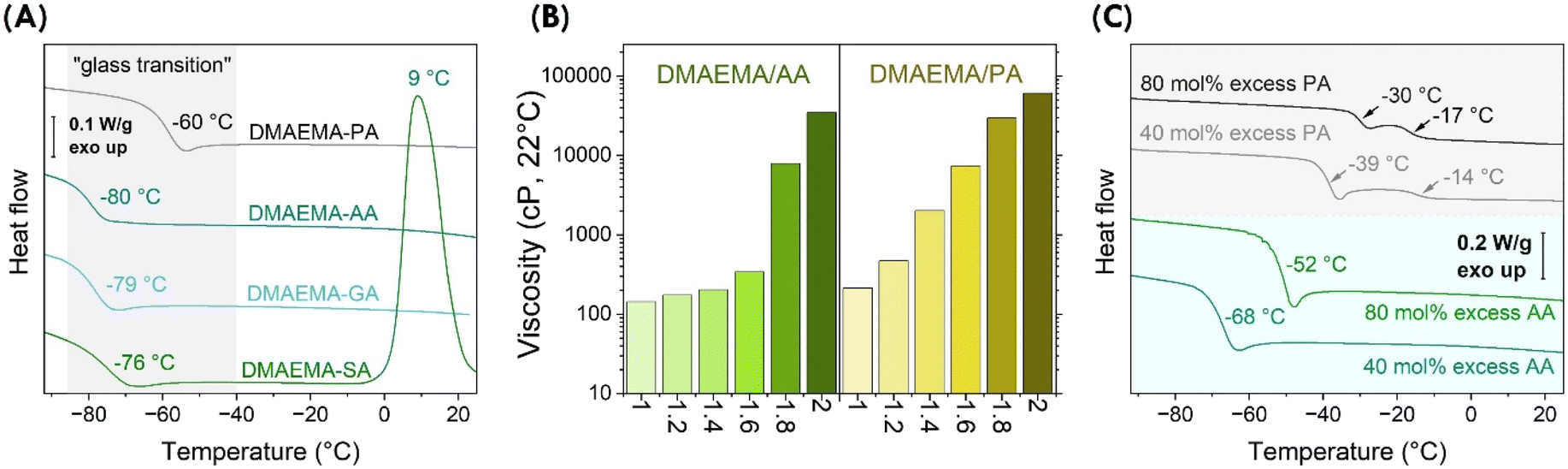 | ||
| Fig. 3 Physical properties of DMAEMA-AA and DMAEMA-PA complexes. (A) These acid/base complexes form glass with transition temperatures varying from −80 to −60 °C. (B) Acid-excess, off-stoichiometric complexes remain as bench stable liquids. Viscosity increases dramatically with additional acids at various molar ratios showing on the x axis. Numerical data is listed in Table S2.† (C) Acid excess complexes exhibit higher “glass transition” temperatures measured by DSC (10 °C min−1). | ||
Off-stoichiometric DMAEMA-acid complexes were examined to determine the effect of excess acids since it is desirable to “solubilize” more solid bio-based acids. A series of DMAEMA-AA and DMAEMA-PA samples were prepared with varying acid/base ratios. All samples with excess acids remained as bench-stable liquids. Interestingly, the viscosities increased consistently with increased ratios of acids. As shown in Fig. 3B, viscosities of DMAEMA-AA complexes increased from 143 ± 1, 175 ± 5, 204 ± 5, to 341 ± 4 cP for acid/base ratios of 1.0, 1.2, 1.4, and 1.6, respectively. When the acid/base ratios increased to 1.8, viscosity abruptly increased to 7900 ± 140 cP; at a ratio of 2.0, the viscosity increased further to 34600 ± 2500 cP. In the case of aromatic acids, off-stoichiometric DMAEMA-PA complexes showed a consistent logarithmic change: for acid/base ratios from 1.2 to 2.0 at an interval of 0.2, viscosities increased from 472 ± 5, 2026 ± 45, 7380 ± 180, 29600 ± 210, to 60800 ± 700 cP. The presence of additional acids may contribute to intermolecular hydrogen bonding, which results in increased viscosity. The additional hydrogen bonding also affected the “glass transitions”. Shown in Fig. 3C, DMAEMA-AA complexes showed transition temperatures at −68 °C and −52 °C with 40 mol% and 80 mol% excess AA, respectively; DMAEMA-PA complexes showed transition temperatures at −39 °C and −30 °C with 40 mol% and 80 mol% excess PA, respectively. It is worth noting that there were additional Tg's for DMAEMA-PA systems, likely because PA has two distinct pKa values (2.89 and 5.51). In a word, the physical properties of DMAEMA-acid complexes can be tuned by acid/base stoichiometric ratios, which enables a wider landscape of attainable chemical structures and properties of their polymers. In the following, we investigate their formation of polymer materials.
The ionic bonding accelerated methacrylate free-radical photopolymerization kinetics
Fourier transform infrared spectroscopy (FTIR) was used to investigate the reaction kinetics of free-radical polymerization of methacrylate. Photopolymerization of methacrylate monomers has widespread use in applications like coatings, adhesives, and additive manufacturing, given their straightforward processing, excellent spatiotemporal control, and remarkably fast reaction kinetics. Covalently bonded multifunctional methacrylate monomers are largely used because the formation of crosslinking accelerates reaction kinetics.26,27 Monofunctional methacrylate, however, is typically slow-reacting and thus unsuitable for rapid photopolymerization. We examined the photopolymerization of neat DMAEMA that contains 1 wt% ethyl (2,4,6-trimethylbenzoyl) phenylphosphinate (TPOL) as a photoinitiator, under 5 mW cm−2 405 nm light from a LED source. As expected, polymerization of neat DMAEMA was slow, reaching <20% conversion within three minutes of continuous irradiation, monitored by the alkene peak at 1637 cm−1. Under the same conditions, the DMAEMA-AA complex exhibited a much faster reaction rate, reaching 90% conversion within one minute of irradiation (Fig. 4A). We reason that the ionic bonding creates non-covalent crosslinking, enabling a gelation effect that is similar to covalent crosslinking. The FTIR spectra revealed that as the DMAEMA-AA polymerized, the carbonyl peak shifted from 1721 cm−1 to 1726 cm−1, indicating a slightly shorter C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, likely due to reduced intermolecular interactions in the polymer (Fig. 4B).
O bond, likely due to reduced intermolecular interactions in the polymer (Fig. 4B).
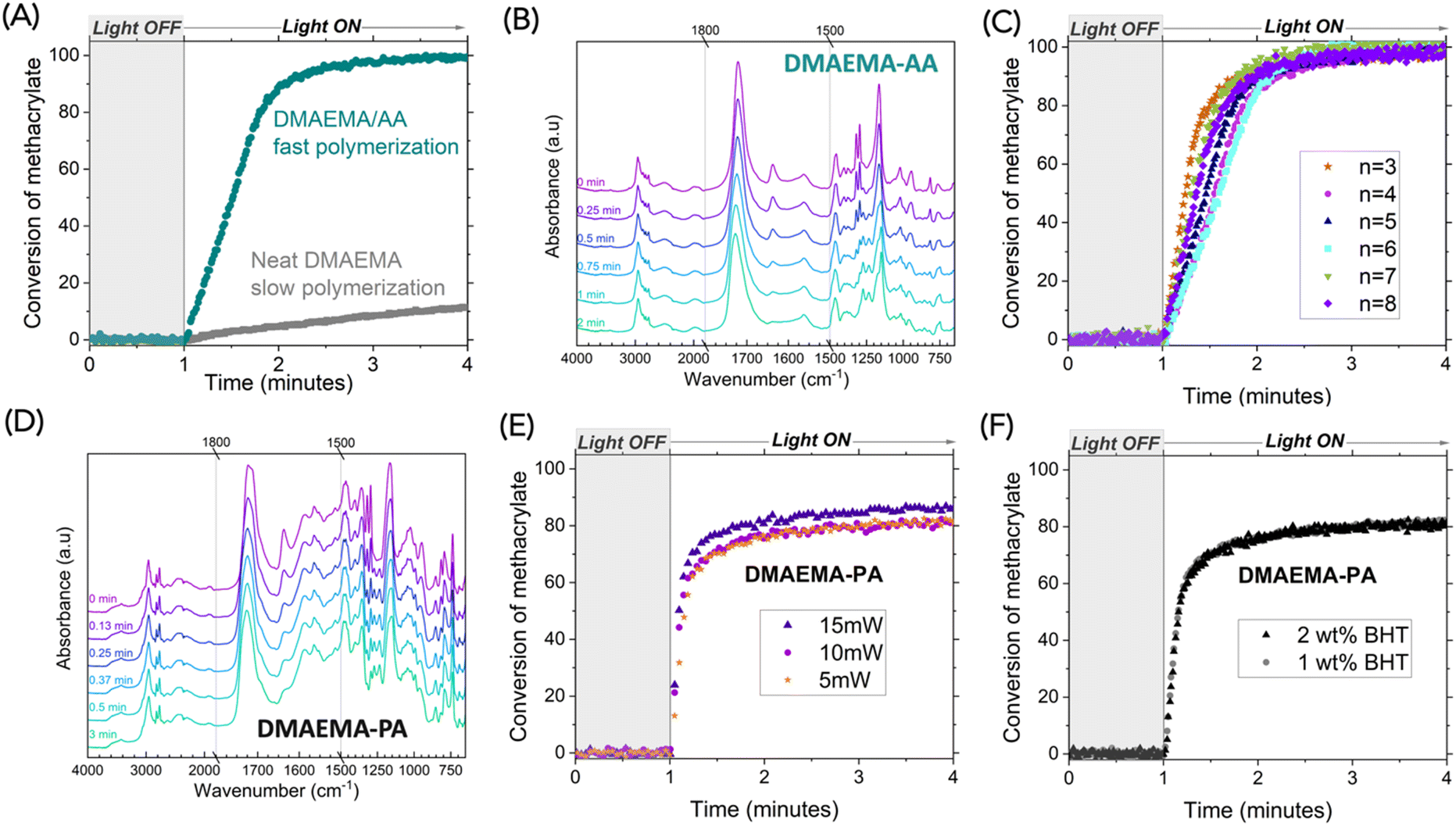 | ||
| Fig. 4 Polymerization reaction kinetics measured by real-time FTIR. (A) DMAEMA-AA complex exhibited much accelerated reaction than neat DMAEMA. (B) Stacked FTIR spectra of DMAEMA-AA complex at various durations of irradiation. (C) Polymerization reaction kinetics of complexes with various aliphatic diacids (structures shown in Scheme 2). (D) Stacked FTIR spectra of DMAEMA-PA complex at various durations of irradiation. (E) Polymerization reaction kinetics of DMAEMA-PA at various light intensities. (F) Polymerization reaction kinetics of DMAEMA-PA with additional inhibitor (BHT). Curing conditions: 5 mW cm−2 405 nm light from a LED source unless otherwise specified; light turned on after 1 min. | ||
Fast polymerizations were observed for all DMAEMA-acid complexes. Fig. 4C shows polymerization kinetics for the complexes with aliphatic acids with varying carbon counts (structures shown in Scheme 2). The polymerization reaction kinetics were comparable, confirming the non-covalent crosslinking that accelerated methacrylate polymerization. We further investigated the aromatic acid system. As shown in Fig. 4D, the alkene peak at 1635 cm−1 was reduced to nearly full disappearance after half a minute of irradiation. We further investigated the effect of light intensity. As the light intensity increased from 5, 10, to 15 mW cm−2, slightly faster reactions were observed (Fig. 4E). Interestingly, higher final conversions were also observed with higher light intensity. We suspect that vitrification may have occurred when the propagating reactions were prohibited as the free radicals lost their mobility. A faster reaction can result in a higher internal temperature within the thin film sample (∼20 μm) since the polymerization reaction is exothermic. The fast polymerization coincided with the undesired spontaneous polymerization that we sometimes experienced. When we added an additional free radical inhibitor, butylated hydroxytoluene (BHT), we were able to stably store these resins. We confirmed that the addition of BHT did not hinder the polymerization (Fig. 4F).
In addition, inspired by the glass-forming properties of these complexes, we sought to investigate the effect of intermolecular interactions among the DMAEMA-acids in photopolymerization kinetics, since the complexes with monoacids may also benefit from the gelation effect. We picked levulinic acid and benzoic acid to represent monofunctional aliphatic and aromatic acids, respectively. The polymerization of stoichiometric complexes showed much more accelerated kinetics than that of the neat DMAEMA (Fig. S6†). It is worth noting that the DMAEMA-levulinic acid showed a slower reaction (37% conversion with 1 min irradiation) than DMAEMA-AA, suggesting that difunctional ion pairs can build more crosslinking than the monofunctional ion pairs. The same trend was also observed for DMAEMA-benzoic acid, which showed a slower reaction (50% conversion with 0.6 min irradiation) than DMAEMA-PA (50% conversion with 0.2 min irradiation). We expect the contribution of ionic bonding strength (quantified by pKa) and supramolecular crosslinking (quantified by carbonyl shift on FTIR) can be deconvoluted, which is worth further study.
Characterization of poly(DMAEMA-acid) complexes
We characterized the polymer material's thermomechanical properties by DSC, tensile test, and thermogravimetric analysis (TGA). The photopolymerized poly(DMAEMA-AA) was rubbery, while the poly(DMAEMA-PA) was stiff (Fig. 5A). The glass transition temperatures (Tg) of DMAEMA-AA and DMAEMA-PA were measured to be 4 °C and 19 °C, respectively, by DSC (Fig. 5B). The increased Tg of the aromatic acid system is in good agreement with covalently crosslinked network polymers, where the incorporation of stiff aromatic linkages increases backbone rigidity. The mechanical properties were measured by tensile test following ASTM D638 standard. Poly(DMAEMA-AA) was indeed rubbery (tensile strength of 3.7 MPa) but also exhibited an impressive elongation before break (>250%), which is comparable to covalent network polymers with low crosslinking densities (Fig. 5C). | ||
| Fig. 5 Physical properties of poly(DMAEMA-AA) and poly(DMAEMA-PA) materials. (A) Pictures of the polymers showing rubbery and stiff materials. (B) Glass transition temperatures measured by DSC at 10 °C min−1. (C) Example tensile stress–strain curves showing compliant and brittle materials. (D) TGA measurement (10 °C min−1) showing that the polymers have increased thermal stability. | ||
The poly(DMAEMA-PA) was stiff and exhibited a tensile modulus of 0.6 GPa, but was brittle (elongation-at-break of 1.5%). Such brittleness is typically seen in highly crosslinked covalent networks. We were surprised by the brittleness in poly(DMAEMA-PA) as it lacked covalent crosslinking. The effect of ionic crosslinking on mechanical properties is worth further investigation.
TGA measurement under nitrogen also confirmed the formation of polymethacrylates. As shown in Fig. 5D, the neat DMAEMA was volatile, and nearly complete evaporation was observed at 175 °C. Neat AA became volatile shortly above its melting temperature, and its nearly complete evaporation was observed at 305 °C. It is interesting that the formation of supramolecular polymer in the ionic complex was also evident in their thermal stability. The stoichiometric DMAEMA-AA complex gradually lost mass but 24 wt% mass remained at 305 °C. The remaining mass gradually decomposed at higher temperatures, eventually reaching near disappearance at 580 °C. The poly(DMAEMA-AA) showed enhanced thermal stability, with 5 wt% decomposition observed at 220 °C and 50 wt% decomposition observed at 260 °C. These decomposition temperatures are generally below covalently crosslinked networks and are comparable to linear polymers.28
Dual-cure reactions and thermomechanical properties of polymers made from DMAEMA-acid complex and epoxy monomers
We sought to implement these polymerizable acid/base complexes as hardeners for epoxy resins. The widely controllable viscosities are particularly attractive for epoxy curing applications since epoxy material processing techniques, including resin transfer molding, filament winding, prepreg, and pultrusion, require vastly different viscosities.29 Meanwhile, we are interested in epoxy resins that are derived from renewable resources, so that the target materials can obtain high renewable content, given that many carboxylic acids are bio-derivable. Two epoxy monomers are chosen as demonstrations in this work including a glycerol-based epoxy and a sorbitol-based epoxy. Given the inherent reversibility of ionic bonds, we hypothesize that the deprotonated carboxylate will react with the free epoxy, which will push the equilibrium towards dissociation of the ionic bonds (Scheme 3). The first stage – free radical polymerization of methacrylate – can be triggered by either photopolymerization or heat; the second stage – epoxy-carboxylic acid polymerization – will be mediated by heat. For the polymerization process, we chose “flood curing” with 405 nm light for 10 min as the first stage and 130 °C for 3 h as the second stage. The detailed formulation of the polymer samples is listed in Table S3.†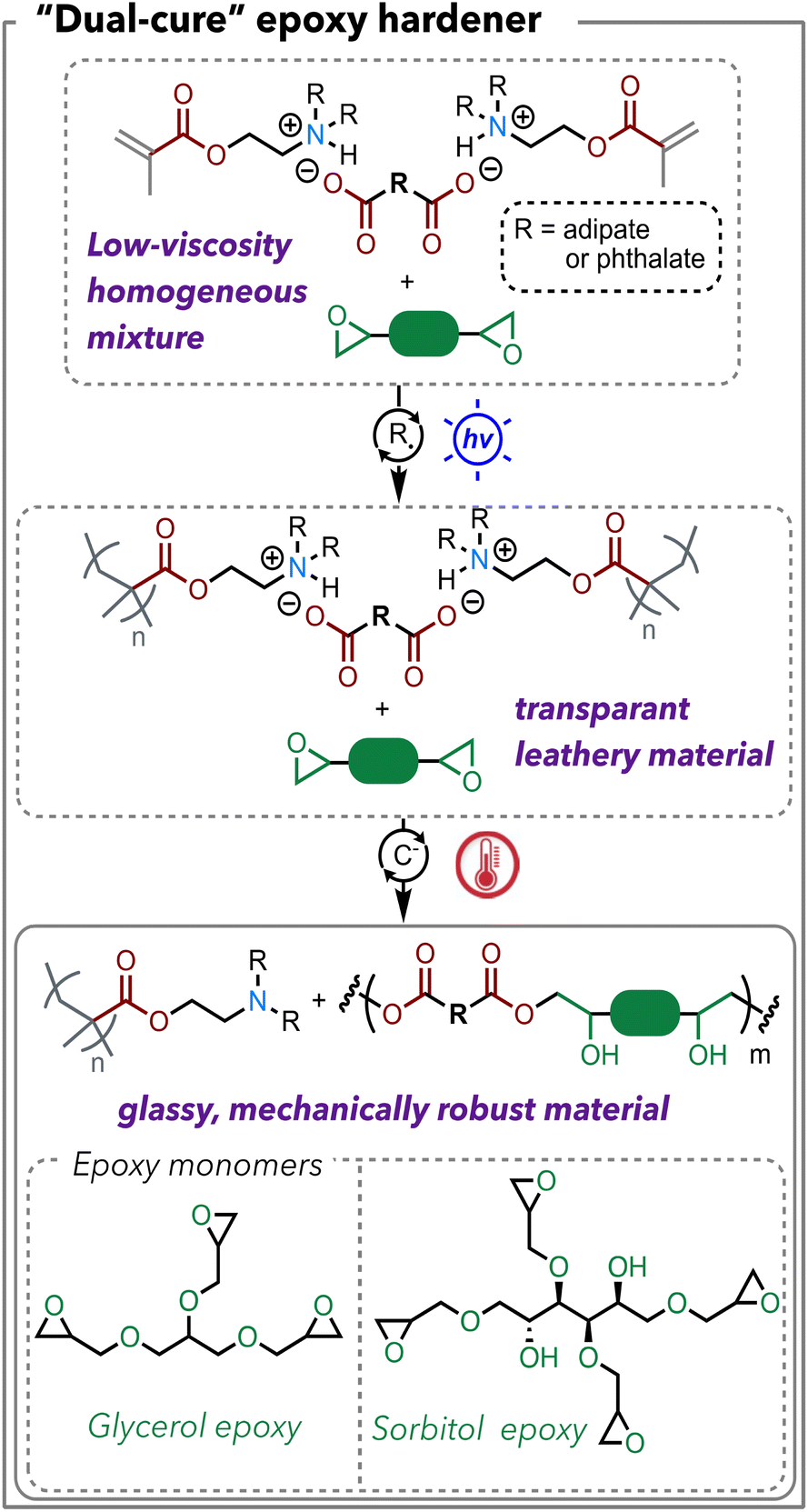 | ||
| Scheme 3 Reaction scheme of photo/heat “dual-cure” of DMAEMA-acid and epoxy resin mixtures. The first stage is photopolymerization of the methacrylate group; the second stage is epoxy-acid polymerization catalyzed by the tertiary amines. | ||
We examined the two stages of curing by FTIR. Similarly to the DMAEMA-AA complex, the alkene peak in DMAEMA-AA-glycerol epoxy disappeared after photocuring (Fig. 6A). After the heat cure, a significant increase of O–H stretch was observed around 3400 cm−1 because of the epoxy-acid reaction that generates hydroxy groups. Unfortunately, in this case the epoxy groups were difficult to monitor by FTIR. For the DMAEMA-PA-glycerol epoxy system, we observed the same phenomenon of increased O–H stretch after the heat cure. We also observed a shift of carbonyl peaks from 1724 to 1717 cm−1, likely due to the conversion of deprotonated phthalic acid to phthalate esters (Fig. 6B).30 In epoxy-anhydride or epoxy-acid curing, the shift of the carbonyl peak is often used to monitor the polymerization reaction conversion.12 In this case, the carbonyl stretches from the methacrylate, and the carboxylic acids merge into one peak, so it is difficult to determine the polymerization conversion by FTIR. Instead, we sought to evaluate the thermomechanical properties of the cured polymers.
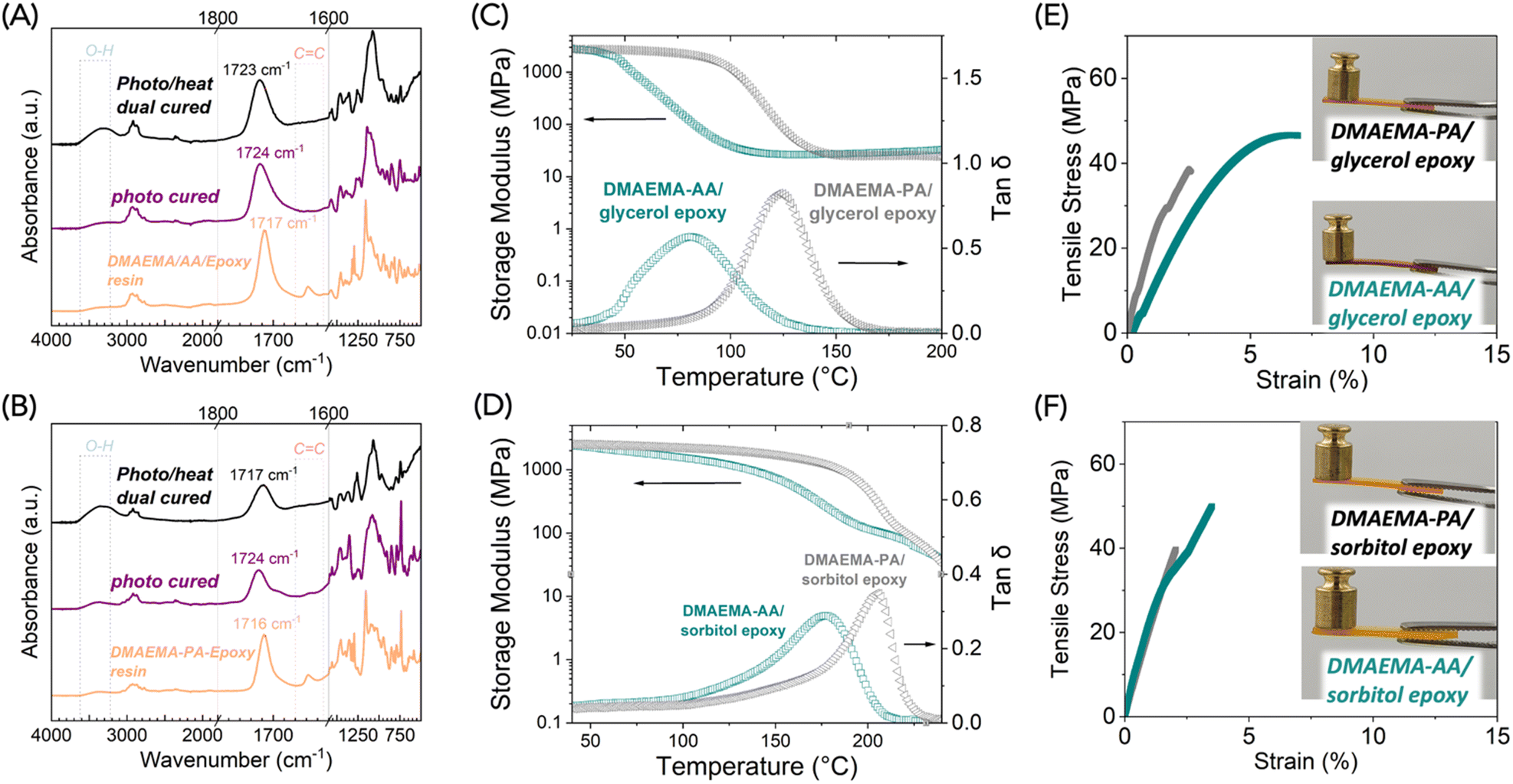 | ||
| Fig. 6 Materials characterization of dual-cured DMAEMA-acids-epoxy polymers. (A and B) Stacked FTIR spectra epoxy at uncured, photo cured, and dual-cured stages of DMAEMA-AA-glycerol epoxy resin (A) and DMAEMA-PA-glycerol epoxy resin (B). (C and D) Dynamic mechanical analysis of dual-cured polymers made with glycerol epoxy (C) and sorbitol epoxy (D). (E and F) Tensile properties of dual-cured polymers made with glycerol epoxy (E) and sorbitol epoxy (F). | ||
Dynamic mechanical analysis (DMA) showed that the glycerol epoxy was cured into highly crosslinked polymers (Fig. 6C and D). The DMAEMA-AA-glycerol epoxy polymer exhibited a Tg of 81 °C and a rubbery modulus of 27 MPa (determined at Tg + 40 °C). The stiff aromatic groups in PA causes the DMAEMA-PA-glycerol epoxy polymer to have a higher Tg of 124 °C while showing a similar rubbery modulus of 25 MPa. The comparable rubbery moduli suggested that the two network polymers had similar crosslinking densities. The stiff aromatic linkages in PA resulted in an increase of >40 °C in Tg. The Tg values are comparable to epoxy-anhydride polymers that contain flexible aliphatic linkages.
Crosslinking density significantly affected the material properties. When we switched from the glycerol epoxy (a mixture of di- and tri-functional) to the sorbitol epoxy (tetra-functional), we saw increased Tg values. The DMAEMA-AA-sorbitol epoxy polymer exhibited a Tg of 177 °C and a rubbery modulus of 84 MPa (Fig. 6D). The increased rubbery moduli indicated an increased crosslinking density. Similarly, DMAEMA-PA-sorbitol epoxy polymer had an impressively high Tg of 206 °C. Although preliminary, this data suggests a robust structure–property relationship of using free-radical polymerizable acid/base complexes as “hardeners” for epoxy resins. In both cases, we noticed an absence of the rubbery plateau. The storage modulus continued to decrease at T > Tg. A plausible explanation of this phenomenon is that at T > 200 °C, the ester bonds become covalently reversible, thus making these networks behave as the Covalent Adaptable Networks, also often described as “vitrimers”. A stronger nucleophilic catalyst such as triazabicyclodecene is often used for vitrimers with transition temperatures of 150–200 °C.31,32 Herein, the tertiary amine on DMAEMA serves as a weak nucleophile catalyst. We observed classic stress relaxation behavior of DMAEMA-PA-sorbitol epoxy polymer (Fig. S7†). The heat-induced plasticity of these network polymers is worth further study.
The dual-cured polymers were found to be mechanically strong polymers by tensile test. All four of the polymers showed typical tensile stress–strain behavior of glassy network polymers: high modulus and small elongation-at-break (numerical data in Table S4†). The DMAEMA-AA-glycerol epoxy polymer exhibited yielding and an averaged elongation-at-break of 7.6 ± 2.3%, an ultimate tensile strength of 45.8 ± 7.2 MPa, and a Young's modulus of 1050 ± 230 MPa. The other polymers exhibited higher Young's moduli at >2000 MPa, but were rather brittle, with elongation-at-break <3%. It is worth noting that aromatic epoxy monomers such as bisphenol A-based glycidyl ethers are ubiquitously used in commercial products, which generally offer superior mechanical properties than aliphatic epoxy monomers. Mechanical properties of materials cured from bio-based aromatic epoxy monomers such as lignin derivatives are worth further study.
Conclusions
We for the first time demonstrate that solid carboxylic acids are incorporated in neat polymerizable liquid resins by the formation of ionic bonded complexes. The polymerization of the supramolecular bonded methacrylate monomers exhibits unusually fast reaction kinetics—comparable to the covalently bonded multifunctional methacrylate monomers. The resin viscosity is highly tunable by off-stoichiometric acid/base complexes, where excess acids significantly increase viscosities. We demonstrate that these acid/base complexes are efficient “hardeners” for epoxy resins, as demonstrated by curing two types of bio-based epoxy monomers through dual-cure reactions. We believe that this study enables a new platform of directly utilizing solid carboxylic acids in valuable materials for a wide range of thermoset applications such as fiber reinforced composite materials. Further study of a material structure-processing-property relationship (such as acrylate versus methacrylate), optimization of maximizing the content of the bio-based acids, the effect of various amine groups (such as ethyl and propyl substituted tertiary amines that are similar to DMAEMA), enhancement of material properties, implementation to mixed acids, and demonstration of carbon fiber reinforced composites are currently ongoing in our laboratory and will be reported elsewhere.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: C. W.; investigation: E. Y. Y., G. M. M, S. H. and C. W.; writing – G. M. M, C. J. R. and C. W.; funding acquisition: C. W.; supervision: C. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the start-up fund provided by the Price College of Engineering, as well as the College of Science Seed program, at the University of Utah. E. Y. Y. thanks the Undergraduate Research Opportunity Program at the University of Utah. C. J. R. and C. W. thank a gift from Meta Inc. donated to the University of Utah. We thank Michael Thompson and Tyler Kirk for assisting with resin processing and characterization. We thank Dom Porcincula for assisting with DSC measurement and Brandon Wells for assisting with TGA measurement. Portions of this work were performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344 with support from the Laboratory Directed Research and Development (LDRD) program under projects 22-ERD-047. Portions of this work were performed under the support from U.S. Department of Interior Office of Surface Mining Reclamation and Enforcement Applied Science Program (S24AC00035).Notes and references
- J. Kubitschke, H. Lange and H. Strutz, in Ullmann's Encyclopedia of Industrial Chemistry, 2014, pp. 1–18 Search PubMed.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. M. Alonso, de R. Mariscal and M. L. Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- E. de Jong, A. Higson, P. Walsh and M. Wellisch, Bio-based chemicals value added products from biorefineries, IEA Bioenergy, Task42 Biorefinery, vol. 34, pp. 1–33 Search PubMed.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y.-X. Chen, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS.
- J. A. Tamada and C. J. King, Ind. Eng. Chem. Res., 1990, 29, 1327–1333 CrossRef CAS.
- Z. Jiménez, C. Bounds, C. E. Hoyle, A. B. Lowe, H. Zhou and J. A. Pojman, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3009–3021 CrossRef.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- D. D. Chung and D. Chung, Carbon Fiber Composites, Elsevier, 2012 Search PubMed.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- F. Gamardella, V. Sabatini, X. Ramis and À. Serra, Polymer, 2019, 174, 200–209 CrossRef CAS.
- D. Guzmán, X. Ramis, X. Fernández-Francos and A. Serra, Eur. Polym. J., 2014, 59, 377–386 CrossRef.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- S. Ma, X. Liu, Y. Jiang, Z. Tang, C. Zhang and J. Zhu, Green Chem., 2013, 15, 245–254 RSC.
- R. E. Murray, D. Penumadu, D. Cousins, R. Beach, D. Snowberg, D. Berry, Y. Suzuki and A. Stebner, Appl. Compos. Mater., 2019, 26, 945–961 CrossRef.
- M. Kaur and A. K. Srivastava, J. Macromol. Sci., Part C, 2002, 42, 481–512 CrossRef.
- J. He, L. Lassila, S. Garoushi and P. Vallittu, Biomater. Invest. Dent., 2023, 10, 2191621 Search PubMed.
- J. Zhang and P. Xiao, Polym. Chem., 2018, 9, 1530–1540 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- S. Pandey, D. Pal and S. Kumar, Chem. Data Collect., 2022, 41, 100908 CrossRef CAS.
- N. A. Rorrer, S. F. Notonier, B. C. Knott, B. A. Black, A. Singh, S. R. Nicholson, C. P. Kinchin, G. P. Schmidt, A. C. Carpenter and K. J. Ramirez, Cell Rep. Phys. Sci., 2022, 3, 100840 CrossRef CAS.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167–175 RSC.
- M. A. Harris, T. Kinsey, D. V. Wagle, G. A. Baker and J. Sangoro, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020878118 CrossRef CAS PubMed.
- H. L. Ngo, K. LeCompte, L. Hargens and A. B. McEwen, Thermochim. Acta, 2000, 357, 97–102 CrossRef.
- W. Xu, E. I. Cooper and C. A. Angell, J. Phys. Chem. B, 2003, 107, 6170–6178 CrossRef CAS.
- K. S. Anseth, C. N. Bowman and N. A. Peppas, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 139–147 CrossRef CAS.
- K. S. Anseth, C. M. Wang and C. N. Bowman, Polymer, 1994, 35, 3243–3250 CrossRef CAS.
- E. Kandare, H. Deng, D. Wang and J. M. Hossenlopp, Polym. Adv. Technol., 2006, 17, 312–319 CrossRef CAS.
- H. Andersson, T. Lundström, B. Gebart and R. Långström, Polym. Compos., 2002, 23, 895–901 CrossRef CAS.
- G. M. Musgrave, et al., Polyester networks from structurally similar monomers: recyclable-by-design and upcyclable to photopolymers, Polym. Chem., 2023, 14, 2964–2970 RSC.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05417b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |