
Electric field-induced amplification of graphene oxide's visible light photocatalytic activity†
Alsu G.
Nugmanova
 a,
Maxim R.
Sokolov
a,
Alexey E.
Alexandrov
a,
Maria A.
Kniazeva
b,
Ivan Yu.
Eremchev
b,
Andrey V.
Naumov
b,
Danil W.
Boukhvalov
cd,
Burkhard
König
e and
Maria A.
Kalinina
*a
a,
Maxim R.
Sokolov
a,
Alexey E.
Alexandrov
a,
Maria A.
Kniazeva
b,
Ivan Yu.
Eremchev
b,
Andrey V.
Naumov
b,
Danil W.
Boukhvalov
cd,
Burkhard
König
e and
Maria A.
Kalinina
*a
aA. N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, 31(4), Leninsky pr., Moscow 119071, Russia. E-mail: kalinina@phyche.ac.ru
bMoscow Pedagogical State University (MPGU), 29, M. Pirogovskaya Str., Moscow, 119992, Russia
cCollege of Science, Institute of Materials Physics and Chemistry, Nanjing Forestry University, No. 159 Longpan Road, Nanjing, 210037, China
dInstitute of Physics and Technology, Ural Federal University, Mira Street 19, 620002 Ekaterinburg, Russia
eInstitut für Organische Chemie, Universität Regensburg, D-93040 Regensburg, Germany
First published on 26th November 2024
Abstract
A static external electric field (EEF) is for the first time successfully applied to enhance the photocatalytic activity of graphene oxide (GO) photocatalysts functionalized by either zinc porphyrins or perylene diimide. The applied 4 kV EEF increases the reaction rate of the hybrid-assisted photodestruction of a model organic pollutant, 1,5-dihydroxynaphthalene (DHN), in water by 2.2–2.3 times in a contactless transparent static electric cell. A control material presenting zinc porphyrins on the non-polarizable MoS2 nanosheets does not change its activity in EEF. Ultrafast time-resolved photoluminescence spectroscopy, photoluminescent hydroxyl radical probing, and the GC-MS analysis of the supernatant solutions are used to confirm no effect of EEF on photochemical properties of the porphyrins as well as on the pathway of photodestruction of DHN. The DFT calculations show that the dielectric properties and polarizability of GO play a key role in the EEF-induced enhancement of photocatalysis due to the decrease in electron energy facilitating its transfer from GO into water or substrate. Our finding may provide a basis for an affordable alternative for conventional electrophotocatalysis schemes to advance this field towards more effective green-chemistry technologies and to encourage the rational design of new carbon-based photocatalysts, which can be applied for EEF-facilitated photocatalysis.
Introduction
Photocatalysis is the key to utilizing solar energy for useful chemical work such as CO2 transformation to fuels,1,2 hydrogen evolution,3 organic synthesis4,5 or oxidative degradation of organic pollutants6 in a recyclable and environmentally-friendly fashion.7–9 However, the effectiveness and scalability of heterogeneous photocatalysts remain challenging issues involving fundamental aspects of these materials,10 such as sensitivity to visible light,11 kinetics of photoinduced surface reactions,12 recombination rate and lifetimes of photoinduced charge-separated states.13 To widen the absorption range of solid photocatalysts, organic photosensitizers and/or surface heterojunctions are used.14–16 Introducing microporosity into conventional semiconductors or metal–organic networks (MOFs) can boost rates of photoinduced reactions.17,18 The most well-developed method providing enough energy to control recombination is electrophotocatalysis (EPC).19–21 However, the combination of high energy consumption, high current density in the reaction medium19 and the poisoning of metal electrodes is a major challenge for the integration of EPC technologies in industry.The use of external fields as alternative energy sources that do not directly affect the structure of photocatalysts can also be helpful in enhancing photocatalysis. Mechanical or ultrasound-induced activation22,23 as well as thermal assistance have been reported for increasing the activity of conventional ZnO and TiO2 photocatalysts.22,24 A magnetic field has also been used to suppress the recombination of photoinduced charges in such materials.24,25 Inner electric fields in heterogeneous photocatalysts can be initiated to improve their performance due to ferroelectric properties or heterojunctions.26
However, the application of a static external electric field (EEF) in photocatalysis remains largely unexamined. The EEF activation has only rarely been used either for pre-polarization of ferroelectrics to enhance their activity,27 or to promote photodegradation of vaporized pollutant in the gas phase.28
The EEF-induced enhancement of photocatalysis in liquids, especially in water, remains completely unexplored as of this writing. The most important limitation of this idea is a high dielectric constant of water decreasing the effect of EEF. However, a new class of hybrid materials composed of graphene oxide (GO)29 and reduced graphene oxide (rGO)30 sensitized by potent organic chromophores might compensate for this decrease, at least in part. The ability of GO to induce polarization of water molecules under applied EEF due to comparatively high dielectric constant (∼106,31) of the extended sp2-conjugated portions of carbon carcass is well-established.32–34 This ability may assist photoinduced charge transfer, stabilization of separated states as well as polarization of adsorbed substrates and products of the visible light photocatalytic reactions, thereby accelerating them.
Herein, we confirm this hypothesis by studying spectroscopically the kinetics of photocatalytic degradation of a canonical model substrate 1,5-dihydroxynaphthalene (DHN) with the well-established degradation pathways35 in the oxygenated and argon-rich aqueous solutions under applied EEF in the presence of several new solid photocatalysts (Fig. S1†).36–38 Two of them are the GO-based ones, sensitized by either SURMOF of Zn(II) meso-tetra(4-pyridyl)-porphine (ZnTPyP) (SURMOF/GO)36 (Fig. S1a†) or (N,N′-di(propanoic acid)-3,4,9,10-perylenetetracarboxylic diimide (PDI-PA) (PDI-PA/GO),37 both anchored to GO through similar coordination interactions with Zn(OAc)2 metal clusters (Fig. S1b†). The third material presents porphyrin-based SURMOF assembled from zinc(II) 5,10,15,20-tetrakis(4-carboxyphenyl)-porphine (ZnTCPP) and Zn(OAc)2 on the non-polarizable MoS2 nanosheets (SURMOF/MoS2)38 (Fig. S1c†). The crystal structure of ZnTCPP SURMOF on MoS2 is identical to that assembled from the same porphyrin linker on the surface of GO.37 We used two GO sensitizers with significantly different extinction (ZnTPyP36 with ε ≥ 2 × 105 and PDI-PA37 with ε ≥ 4 × 104) to investigate the role of chromophore chemistry in the EEF-accelerated photodegradation. Two different solids supporting similar porphyrin-based SURMOFs were used to evaluate the importance of polarizability of sensitized inorganic semiconductors in this process. The MoS2 material has low sensitivity to EEF due to negligibly small polarizability (∼4,39) compared to that of GO. The obtained experimental data suggest evidence of the accelerating effect of EEF on photocatalysis in water in the presence of visible-light-absorbing GO-based hybrids. The DFT calculations combined with ultrafast time-resolved fluorescent spectroscopy provide an understanding of this acceleration's origin.
Results and discussion
Spectroscopic studies of the EEF-accelerated photodegradation by the dye-sensitized GO
A custom-engineered photocatalytic electric cell for the kinetic studies of photodegradation of DHN with the dye-sensitized GO under applied EEF was assembled with a flat capacitor made of optically transparent ITO-based electrodes tightly adhering to a standard quartz cuvette with an optical path length of 10 mm and a distance gap between the electrodes 12.5 mm (Fig. 1a and b). The electrodes were connected to a DC power source with a nominal voltage of 10 kV. The voltage applied for kinetic photodegradation measurements using EEF was limited to 4 kV, which is the maximum value to avoid capacitor breakdown in our cell. The calculated EEF strength in the cuvette was approximately 240 kV m−1 under these conditions.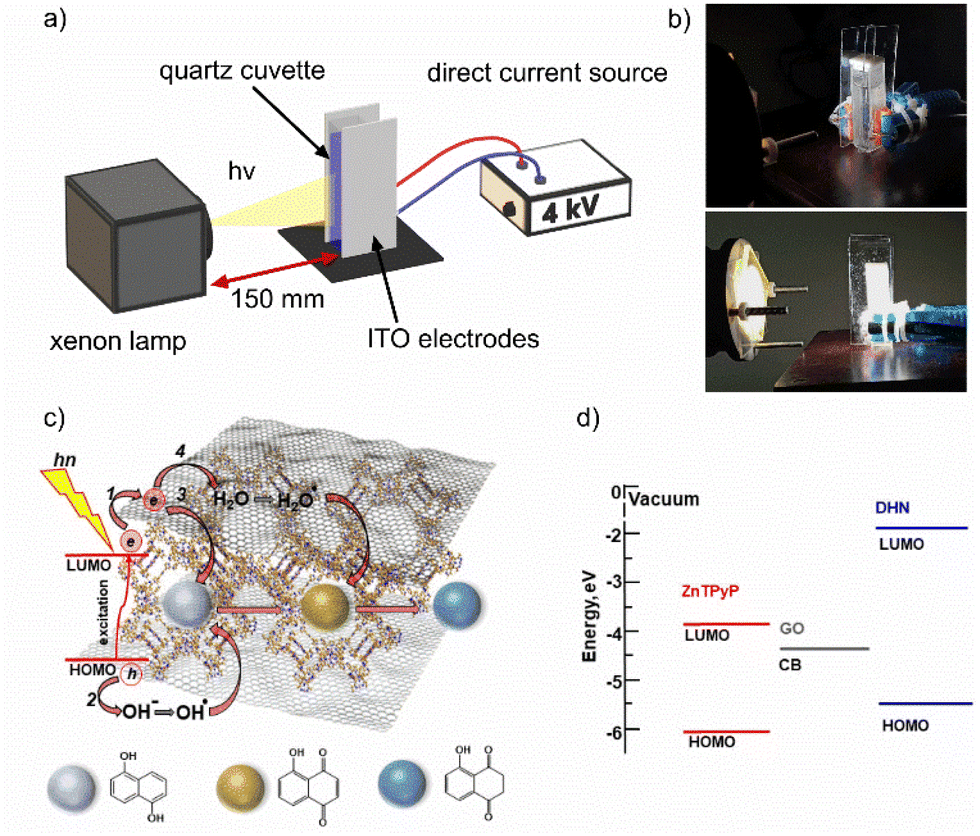 | ||
| Fig. 1 (a) Principal scheme and (b) digital photos of a contact-less photocatalytic EEF cell. (c) Schematically illustrated mechanism and (d) energy diagram of photoinduced sequential oxidation–reduction reactions of DHN in SURMOF/GO hybrid photocatalyst micropores. The conduction band edge (CB (−4.33 eV)) for GO and the HOMO energy level (−5.86 eV) for ZnTPyP are from ref. 40. For DNH, the HOMO energy level (−5.5 eV) and optical bandgap (3.6 eV) for calculation of the LUMO level are from ref. 41. | ||
A xenon lamp with AM1.5 G filter (Fig. S2†) irradiates the cuvette in a direction perpendicular to that of the electric field lines at a distance of 150 mm.
First of all, we studied the possible heating effect of EEF on water, the aqueous sol of pure GO and the SURMOF/GO aqueous suspension. The thermal effect of EEF in the dark was within the measurement error (Fig. S3†), whereas the increase of temperature in the cell under light irradiation did not exceed 1 ± 0.5 °C for 30 min (Fig. S3†). To completely eliminate the possible thermal contribution, further kinetic measurements were carried out in this time range.
The results of spectroscopic studies of the non-sensitized GO in the DHN solution (1 × 10−4 M) irradiated at λ > 420 nm under applied voltage suggest that EEF did not promote the adsorption of the organic substrate on GO and did not enhance its intrinsic visible-light photocatalytic activity towards DHN (Fig. S4 and S5†).
The ultrafast time-resolved photoluminescence spectroscopy setup equipped with the fluorescence microscope was used to provide evidence that EEF does not enhance absorption or photoexcitation of organic sensitizers at least under the conditions studied in this work (Fig. S6–S9†).42 A fluorescent test with terephthalic acid (TPA), which showed that EEF did not contribute to the photoinduced generation of hydroxyl-radicals (·ОН) by the SURMOF/GO hybrid material, complemented this assumption. (Fig. S10†).
The kinetic patterns of the EEF-assisted photodegradation of DHN in the presence of hybrid materials (Fig. 1c and d) were studied both in the ambient and argon atmosphere in the static cell without mixing to avoid any other possible EEF-associated effects, such as EEF-enhanced motion dynamics of polarizable GO particles in water. Solutions with added photocatalysts were kept in the dark until adsorption–desorption equilibrium was reached before the kinetic experiments. Probes with solutions were only removed from the cell every 10 min to minimize additional perturbation of the reaction mixture. Each measurement for each system was reproduced at least five times with and without EEF using fresh portions of materials (that is, five different samples of the same material were measured under identical conditions). The mean values of the relative decrease of the DHN absorption in the probes were used for plotting the kinetic dependencies.
A typical kinetic curve of the complete conversion of DHN using SURMOF/GO photocatalyst is shown in Fig. S11.† The initial portions of all kinetic curves are satisfactorily described by a pseudo-first order reaction equation and give linear dependencies on a semi-logarithmic scale for calculating corresponding rate constants (Fig. 2).
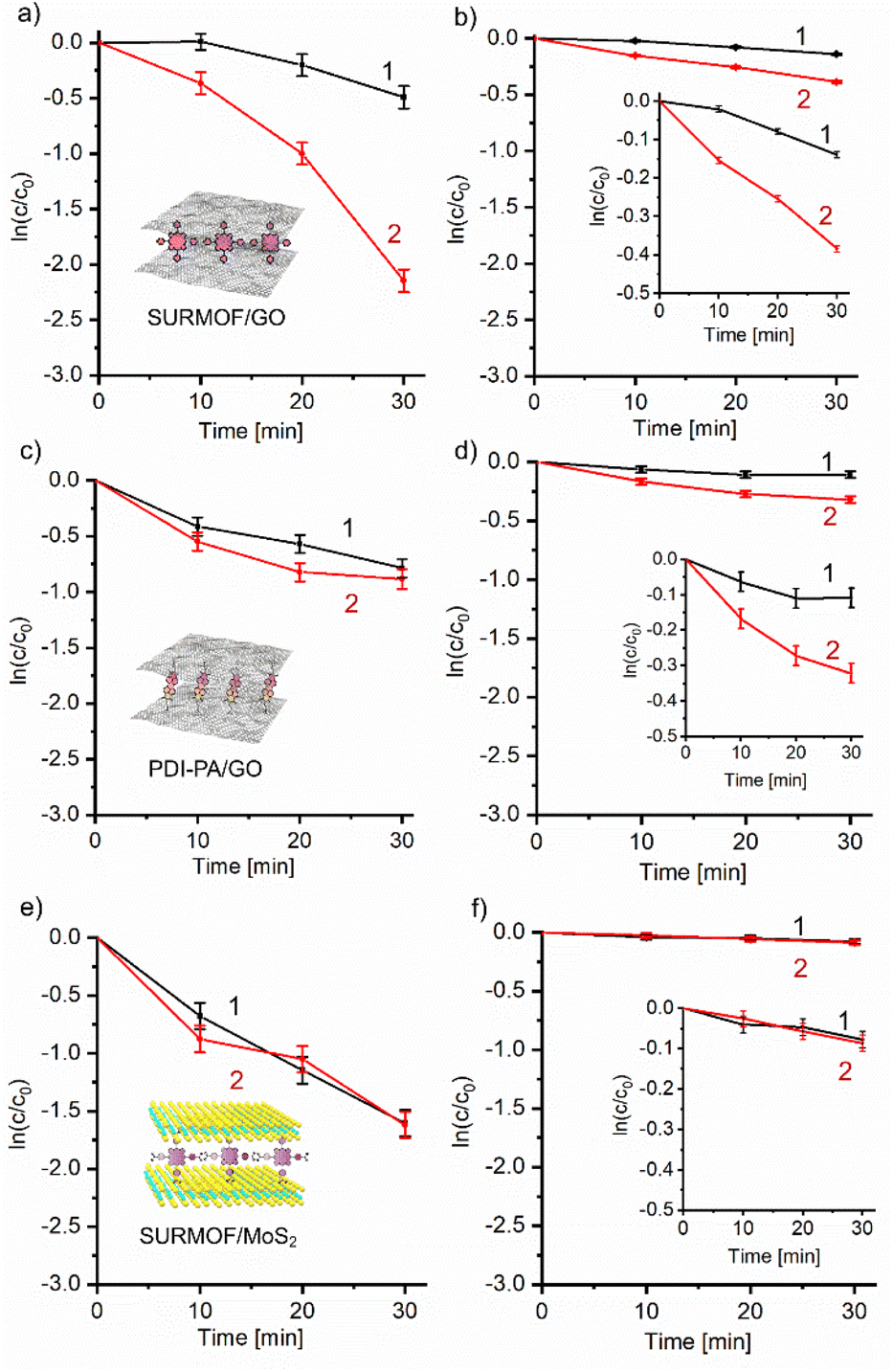 | ||
| Fig. 2 Kinetic dependencies of (a, c and e) aerobic and (b, d and f) anaerobic photodegradation of DHN (10−4 M) in the aqueous solutions with the SURMOF/GO (a and b), PDI-PA/GO (c and d) and SURMOF/MoS2 (e and f) hybrids in EEF-off (curves 1) and EEF-on (curves 2) modes. The kinetic dependencies were plotted using the values of the absorption decrease at λ = 298 nm. | ||
For the SURMOF/GO hybrid, the EEF-on photoinduced reaction proceeds 1.5 times faster than the photodegradation of DHN without EEF in the presence of oxygen (kEEF-ON = 2.9 × 10−2 min−1vs. kEEF-OFF = 2.0 × 10−2 min−1) (Fig. 2a). However, the rate of photodegradation can increase even more under anaerobic conditions. The EEF-assisted photoreduction proceeds 2.3 times faster than that without applied voltage (kEEF-ON = 1.6 × 10−2 min−1vs. kEEF-OFF = 6.9 × 10−3 min−1) (Fig. 2b). Different acceleration depending on the presence of oxygen implies that EEF affects reduction associated with the electron transfer from GO. This observation suggests that EEF-induced polarization of the carbon carcass plays a key role in enhancing photodegradation, especially under anaerobic conditions when the photoreduction dominates over the oxidative pathway through the generation of singlet oxygen and/or superoxide radicals.36 (The reaction rates measured herein are more than an order of magnitude smaller than those we reported earlier for the same GO hybrid measured without EEF in the convective cell under continuous mixing36 due to both diffusion limitations and a relatively small number of effectively irradiated photocatalytic particles on the bottom of the cuvette).
The rates of the same reactions measured with the PDI-PA/GO hybrid were lower than those of the SURMOF/GO photocatalysts (Fig. 2c and d) because PDI-PA has a relatively lower extinction and less effective charge separation between GO sensitizers compared to the ZnTPyP SURMOF. However, the PDI-PA/GO material as a photocatalyst also showed a confidently detectable sensitivity to the applied EEF. The rate of photooxidation of DHN increased under applied EEF by 1.4 times (kEEF-ON = 1.4 × 10−2 min−1vs. kEEF-OFF = 1,0 × 10−25 min−1) (Fig. 2c), whereas the EEF-assisted acceleration of photoreduction in the argon-rich solution of DHN was about 2.3 times (kEEF-ON = 1.26 × 10−2 min−1vs. kEEF-OFF = 5.4 × 10−3 min−1) (Fig. 2d), which is comparable to that observed for SURMOF/GO. This result suggests that the relative increase of the reaction rates is determined by the sensitivity of the oxidized carbon carcass to EEF rather than the sensitizer chemistry.
A direct relationship between the EEF-induced acceleration of photocatalysis and the polarizability of the sensitized inorganic matrix was further confirmed by using single-layer MoS2 sensitized by ZnTCPP SURMOF as photocatalyst (Fig. 2e and f).
We observed no EEF-induced acceleration of photodestruction of DHN assisted by the SURMOF/MoS2 material. The difference between rate constants determined in the EEF-on and EEF-off static regimes was only 1% in the presence of oxygen and 4% in argon-enriched DHN solution 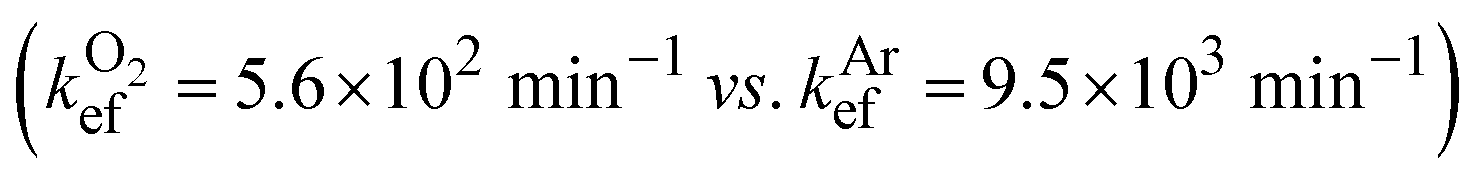 . These values are within the standard deviation for the series of samples studied.
. These values are within the standard deviation for the series of samples studied.
Thus, the dielectric properties of the inorganic component in sensitized hybrids determine the sensitivity of the photocatalyst to the external field in a contactless electric cell.
Analysis of the supernatant solutions by UV-vis spectroscopy (Fig. S12†) and GC-MS (Fig. S13 and S14†) after the EEF-assisted reactions confirmed that EEF does not alter the chemical pathways of photodestruction of DHN (Fig. 1c and d), which mainly depend on the chemistry of intermediates generated by the hybrid material as well as on the convection mode.
The mechanism of the EEF-induced acceleration of the photochemical process in the presence of the GO-based photocatalysts can be proposed as the result of first-principles calculations (see details in ESI†). The model of GO is depicted in Fig. 3a. The value of the bandgap after applying a semi-empirical correction to the calculated value (see Fig. 3b, and discussion in SV and SVI†) is 2.12 eV, which is in quantitative agreement with the measured value.36 The calculated work function is 0.116 eV, which is in reliable agreement with measured for different types of GO.43
 | ||
| Fig. 3 Optimized atomic structure of model supercell of GO (a), and corresponding densities of states for this system with and without electric field along the c axis. Changes in the charge densities after applying the electric field along (a), across sp2 channels (b), and along the c axis. The yellow and cyan “clouds” indicate an increase and decrease in electron density. The background cut-off (isosurface level) is 0.01 e− Å−3. The carbon atoms are shown in brown color, oxygen in red color, and hydrogen in pale pink color, hydrogen bonds shown by dashed lines. | ||
The next step of our modeling is to simulate the electric field with the strength of 200 kV m−1 applied along and across the GO sheet. Since our model of GO has sp2 channels (Fig. S15†), we also simulated the field applied along and across these channels. For both cases, only a tiny redistribution of the charge densities is observed when the electric field is applied along the flat of GO (see Fig. 3c and d). On the contrary, applying the field along the c-axis leads to a significant redistribution of electron density, as shown in Fig. 3e. From this picture, we can conclude that applying a vertical electric field corresponds with a decrease in electron density in carbon flat and an increase in electron density in oxygen-containing groups. Note that this harvesting of the electrons takes place in sp2 channels, too. This redistribution of the charges corresponds with the semiconductor-to-metal transition of GO (see Fig. 3b and S16). On the contrary, applying the same field across MoS2 monolayer corresponds with insignificant charge density redistribution (see Fig. S17†). This charge density also corresponds with the extension of the electron potential of graphene from 2.5 Å to 14.4 Å (see detailed discussion in SVI†). Thus, based on the results of our calculations, we can propose that the electric field applied along the c-axis of GO flat significantly facilitates the electron migration from GO.
However, this EEF sensitivity of GO does not lead to any noticeable structural changes in the GO-based photocatalyst, as evidenced by the XRD analysis of the SURMOF/GO before and after photodegradation of DHN in the EEF-on mode (Fig. S18†).
Conclusions
Our findings provide the first experimental evidence of the sensitivity of materials comprising graphene oxide and organic chromophores to an external electric field enhancing their performance as visible-light photocatalysts. The accelerating effect of static electric field on the model photodestruction reaction is related to the dielectric properties of graphene oxide experiencing field-induced polarization, which, in turn, promotes charge separation and facilitates electron transfer to the substrates.The photochemistry of chromophores, at least, those studied herein, seems not to play a role in the field-assisted acceleration of photocatalysis. The possibility of using different types of sensitizers to increase photocatalytic efficiency is important for extending the enhancement principle to a wide variety of graphene-based hybrids used for various processes. Field-assisted photocatalysis in static electric cells might be useful for advancing green chemical technologies exploiting photocatalysis such as hydrogen evolution, reduction of carbon dioxide and especially water purification. Rational design of novel carbon-based hybrids for use in nonpolar solvents with small dielectric constants can be applied for maximizing the field-induced effect in photocatalysis for organic synthesis.
Contact-less photocatalytic cells powered by external electric sources offer a more affordable environmentally friendly alternative to conventional electrophotocatalysis. Their performance can be optimized by targeted design adapting them to flow devices and to the structure of hybrid photocatalysts, which can achieve the best performance in removable cartridge systems with layered sensitized graphene oxide coatings oriented with respect to the field lines.
Data availability
The data supporting the findings of this study have been included as part of the ESI† and are also available from the authors upon reasonable request.Author contributions
Dr A. G. Nugmanova: methodology; investigation; visualization; writing; validation. M. R. Sokolov: methodology; investigation; visualization; writing; validation. Dr A. E. Alexandrov: methodology; visualization. M. A. Kniazeva: methodology; investigation; visualization; writing; validation. Dr I. Y. Eremchev: investigation; visualization; writing. Prof. A. V. Naumov: investigation; visualization; writing; validation. Prof. D. W. Boukhvalov: theoretical analysis; visualization; writing; validation. Prof. B. König: conceptualization; writing – review & editing writing. Prof. M. A. Kalinina: conceptualization, writing – review& editing; supervision; project administration; funding acquisition.Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
Analytical measurements were performed using the equipment of CKP FMI IPCE RAS. This work was financially supported by the RSF (Project No. 23-73-00095). M. A. K., I. Y. E., A. V. N. acknowledge support from MPGU (theme No. 124031100005-5) for the development of a technique for photoluminescence kinetics analysis of new materials. The authors would like to thank Reviewer 2 for helpful critical comments that improved the presentation of our findings.References
- H. L. Nguyen, Adv. Energy Mater., 2020, 10, 2002091 CrossRef CAS.
- K. Donabauer and B. König, Acc. Chem. Res., 2021, 54, 242–252 CrossRef CAS.
- L. Lin, T. Hisatomi, S. Chen, T. Takata and K. Domen, Trends Chem., 2020, 2, 813–824 CrossRef CAS.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- N. S. Shlapakov, A. D. Kobelev, J. V. Burykina, A. Y. Kostyukovich, B. König and V. P. Ananikov, Angew. Chem., Int. Ed., 2024, 63, e202314208 CrossRef CAS PubMed.
- W. S. Koe, J. W. Lee, W. C. Chong, Y. L. Pang and L. C. Sim, Environ. Sci. Pollut. Res., 2020, 27, 2522–2565 CrossRef CAS PubMed.
- S. S. Dhanabalan, S. R. Avaninathan, S. Rajendran and M. F. Carrasco, Green Photocatalysts for Energy and Environmental Process, Springer International Publishing, Cham, 2020, vol. 36 Search PubMed.
- B. König, Eur. J. Org. Chem., 2017, 2017, 1979–1981 CrossRef.
- E. Kabir, P. Kumar, S. Kumar, A. A. Adelodun and K.-H. Kim, Renew. Sustain. Energy Rev., 2018, 82, 894–900 CrossRef.
- S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 454 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909 RSC.
- B. Pattengale, S. Yang, J. Ludwig, Z. Huang, X. Zhang and J. Huang, J. Am. Chem. Soc., 2016, 138, 8072–8075 CrossRef CAS PubMed.
- Y. Chen, A. Li, Z.-H. Huang, L.-N. Wang and F. Kang, Nanomaterials, 2016, 6, 51 CrossRef PubMed.
- S. Silvestri, A. R. Fajardo and B. A. Iglesias, Environ. Chem. Lett., 2022, 20, 731–771 CrossRef CAS.
- Y.-P. Yuan, L.-W. Ruan, J. Barber, S. C. Joachim Loo and C. Xue, Energy Environ. Sci., 2014, 7, 3934–3951 RSC.
- W. Zhou, W. Li, J.-Q. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- J. Gao, Q. Huang, Y. Wu, Y.-Q. Lan and B. Chen, Adv. Energy Sustain. Res., 2021, 2, 2100033 CrossRef CAS.
- J. Li, J. Ren, S. Li, G. Li, M. M.-J. Li, R. Li, Y. S. Kang, X. Zou, Y. Luo, B. Liu and Y. Zhao, Green Energy Environ., 2024, 9, 859–876 CrossRef CAS.
- Y. Wang, D. He, H. Chen and D. Wang, J. Photochem. Photobiol. C Photochem. Rev., 2019, 40, 117–149 CrossRef.
- P. Alulema-Pullupaxi, P. J. Espinoza-Montero, C. Sigcha-Pallo, R. Vargas, L. Fernández, J. M. Peralta-Hernández and J. L. Paz, Chemosphere, 2021, 281, 130821 CrossRef CAS PubMed.
- C. Hu, S. Tu, N. Tian, T. Ma, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 16309–16328 CrossRef CAS PubMed.
- Y. Zhao, X. Huang, F. Gao, L. Zhang, Q. Tian, Z.-B. Fang and P. Liu, Nanoscale, 2019, 11, 9085–9090 RSC.
- T. Lv, J. Li, N. Arif, L. Qi, J. Lu, Z. Ye and Y.-J. Zeng, Matter, 2022, 5, 2685–2721 CrossRef CAS.
- Y. Areerob, Z. Meng, K. Ullah, K. Wijaya, Z. Otgonbayar and W.-C. Oh, Mater. Res. Bull., 2023, 168, 112483 CrossRef CAS.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- S. Park, C. W. Lee, M.-G. Kang, S. Kim, H. J. Kim, J. E. Kwon, S. Y. Park, C.-Y. Kang, K. S. Hong and K. T. Nam, Phys. Chem. Chem. Phys., 2014, 16, 10408–10413 RSC.
- Q. Fu, Y. Liu, J. Mo, Y. Lu, C. Cai, Z. Zhao, S. Wang and S. Nie, ACS Nano, 2021, 15, 10577–10586 CrossRef CAS PubMed.
- Z. Wang, J. Huang, J. Mao, Q. Guo, Z. Chen and Y. Lai, J. Mater. Chem. A, 2020, 8, 2934–2961 RSC.
- H. Li, Y. Zhang, G. Chang, S. Liu, J. Tian, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Chempluschem, 2012, 77, 545–550 CrossRef CAS.
- K. S. Kumar, S. Pittala, S. Sanyadanam and P. Paik, RSC Adv., 2015, 5, 14768–14779 RSC.
- S.-H. Hong, T.-Z. Shen and J.-K. Song, J. Phys. Chem. C, 2014, 118, 26304–26312 CrossRef CAS.
- M. Topsakal, H. H. Gürel and S. Ciraci, J. Phys. Chem. C, 2013, 117, 5943–5952 CrossRef CAS.
- Z. Qiao, C. Qin, Y. Gao, G. Zhang, R. Chen, L. Xiao and S. Jia, Sci. Rep., 2015, 5, 14441 CrossRef CAS PubMed.
- F. Bokaei, R. Rahimi and M. Rabbani, J. Nanoparticle Res., 2024, 26, 232 CrossRef CAS.
- A. G. Nugmanova, E. A. Safonova, A. E. Baranchikov, A. R. Tameev, A. V. Shkolin, A. A. Mitrofanov, A. A. Eliseev, I. N. Meshkov and M. A. Kalinina, Appl. Surf. Sci., 2022, 579, 152080 CrossRef CAS.
- M. Sokolov, A. Nugmanova, A. Shkolin, A. Zvyagina, I. Senchikhin and M. Kalinina, J. Compos. Sci., 2023, 7, 14 CrossRef CAS.
- M. R. Sokolov, K. A. Tumbinskiy, E. A. Varlamova, A. A. Averin, A. V. Shkolin and M. A. Kalinina, ACS Appl. Mater. Interfaces, 2023, 15, 49299–49311 CrossRef CAS PubMed.
- E. J. G. Santos and E. Kaxiras, ACS Nano, 2013, 7, 10741–10746 CrossRef CAS PubMed.
- A. I. Zvyagina, A. A. Shiryaev, A. E. Baranchikov, V. V. Chernyshev, Y. Yu. Enakieva, O. A. Raitman, A. A. Ezhov, I. N. Meshkov, D. A. Grishanov, O. S. Ivanova, Y. G. Gorbunova, V. V. Arslanov and M. A. Kalinina, New J. Chem., 2017, 41, 948–957 RSC.
- B. Lu, L. Zeng, J. Xu, Z. Le and H. Rao, Eur. Polym. J., 2009, 45, 2279–2287 CrossRef CAS.
- I. Y. Eremchev, M. Y. Eremchev and A. V Naumov, Phys. Usp., 2019, 62, 294–303 CrossRef CAS.
- A. Dey, P. Ghosh, J. Bowen, N. St. J. Braithwaite and S. Krishnamurthy, Phys. Chem. Chem. Phys., 2020, 22, 7685–7698 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06034b |
| This journal is © The Royal Society of Chemistry 2025 |