
Potassium batteries for low temperature applications using high energy density organic cathodes†
Elena V.
Shchurik
a,
Alexander V.
Mumyatov
 a,
Ivan S.
Zhidkov
bc,
Tatiana A.
Savinykh
a,
Guzaliya R.
Baymuratova
a,
Alexander F.
Shestakov
ad,
Olga A.
Kraevaya
*a and
Pavel A.
Troshin
*ea
a,
Ivan S.
Zhidkov
bc,
Tatiana A.
Savinykh
a,
Guzaliya R.
Baymuratova
a,
Alexander F.
Shestakov
ad,
Olga A.
Kraevaya
*a and
Pavel A.
Troshin
*ea
aFederal Research Center for Problems of Chemical Physics and Medicinal Chemistry RAS, Academician Semenov Ave. 1, Chernogolovka, Moscow Region, 142432, Russia. E-mail: okraevaya@inbox.ru; troshin2003@inbox.ru
bInstitute of Physics and Technology, Ural Federal University, 620002 Yekaterinburg, Russia
cM. N. Mikheev Institute of Metal Physics of Ural Branch of Russian Academy of Sciences, 620108 Yekaterinburg, Russia
dFaculty of Fundamental Physics & Chemical Engineering, Lomonosov Moscow State University, GSP 1, 1-51 Leninskie Gory, 119991 Moscow, Russia
eZhengzhou Research Institute of HIT, Longyuan East 7th 26, Jinshui District, 450003 Zhengzhou, China
First published on 19th November 2024
Abstract
We introduce two highly promising polymeric cathode materials for potassium batteries prepared using simple and scalable single-step synthesis from triquinoyl and tetraaminophenazine as precursors. The obtained materials have been extensively characterized in the solid state using a set of complementary spectroscopic and physicochemical techniques and also computational methods to unravel the peculiarities of their molecular structures and also the nature of the formed structural defects. Importantly, both polymers were capable of complete utilization of all redox centers in their chemical structures (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups and pyrazine rings) with respect to electrochemical reduction and potassium ion storage, thus enabling theoretical specific capacities of 638 and 574 mA h g−1. Even though the achieved practical capacities of 400–475 mA h g−1 were considerably lower than the theoretical values, they enabled record-high energy densities of 800–950 W h kg−1 for organic cathodes in potassium cells. Furthermore, the fabricated batteries have demonstrated excellent operation at low temperatures between −20 and −55 °C with minimal capacity depression. These findings feature a considerable potential of organic potassium batteries for a broad range of applications, e.g., in the aerospace industry and other fields where stable battery performance at low temperature is crucially required.
O groups and pyrazine rings) with respect to electrochemical reduction and potassium ion storage, thus enabling theoretical specific capacities of 638 and 574 mA h g−1. Even though the achieved practical capacities of 400–475 mA h g−1 were considerably lower than the theoretical values, they enabled record-high energy densities of 800–950 W h kg−1 for organic cathodes in potassium cells. Furthermore, the fabricated batteries have demonstrated excellent operation at low temperatures between −20 and −55 °C with minimal capacity depression. These findings feature a considerable potential of organic potassium batteries for a broad range of applications, e.g., in the aerospace industry and other fields where stable battery performance at low temperature is crucially required.
Introduction
Organic small molecules and polymers possessing multiple accessible redox sites have drawn much attention as promising electrode materials for rechargeable batteries.1 Potassium-ion batteries (PIBs) represent one of the most promising emerging post-lithium battery technologies. Indeed, PIBs have advantages such as the high natural abundance of potassium, and low cost and relatively high energy density resulting from the low standard reduction potential of potassium (−2.93 V versus E0), which is close to that of lithium (−3.04 V versus E0).The development of PIBs is severely hindered by the lack of inorganic cathodes matching the requirements of both performance and operational safety. Typical intercalation-based compounds utilized in Li cells are not suitable for potassium, as they generally suffer from low cycle life and structural stress during cycling. Due to the soft nature of the redox-active organic compounds, they can easily accommodate a lot of potassium ions, providing high capacity and good rate capability. Additional benefits of organic electrode materials include mild synthesis conditions, abundant raw materials, tunability, low cost, potential for recyclability, and relatively low toxicity.2
Typical organic redox-active compounds utilized as cathodes in PIBs include ketones, imines and aromatic amines, which demonstrate satisfactory specific discharge capacities reaching 100–300 mA h g−1. Most of the materials also demonstrate rather low potentials vs. K+/K, so the resulting energy densities typically do not exceed 400–700 W h kg−1 (Fig. 1).
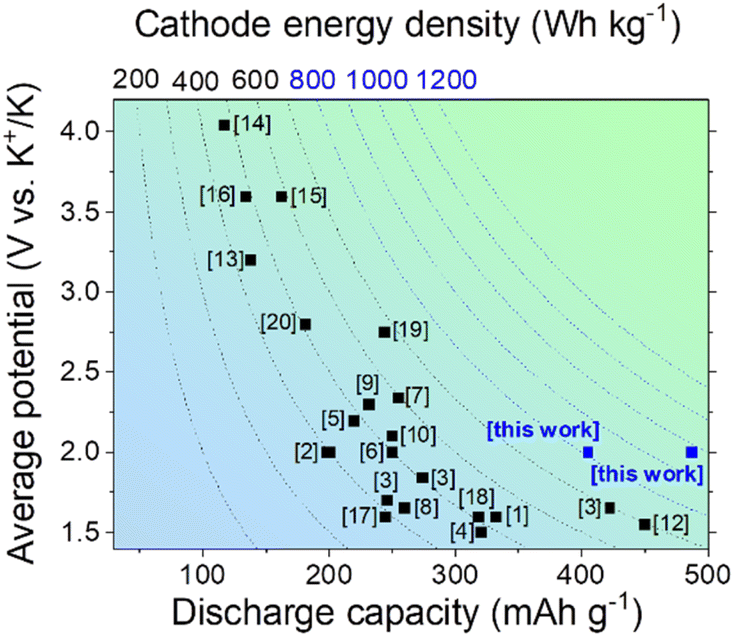 | ||
| Fig. 1 A comparison of specific capacities, average discharge potentials and energy densities of the most promising organic cathode materials for PIBs. | ||
Among the organic electrode materials enabling high energy densities, several small molecules and polymers should be mentioned, in particular, octahydroxytetraazapentacene (OHTAP) with a practical capacity of 332 mA h g−1 and an average discharge potential of 1.6 V,1 octahydroxytetraazapentacenedione (OHTAPQ, ∼200 mA h g−1, ∼2 V),2 polymers derived from triquinoyl and 3,3′-diaminobenzidine (422 mA h g−1, 1.65 V), triquinoyl and 1,2,4,5-benzenetetramine (274 mA h g−1, 1.84 V), triquinoyl and 1,4-diaminobenzene (246 mA h g−1, 1.7 V),3 perylenetetracarboxylic diimide (320 mA h g−1, 1.5 V),4 [N,N-bis(2-anthraquinone)]-perylene-3,4,9,10-tetracarboxydiimide (PTCDI-DAQ, 219 mA h g−1, ∼2.2 V),5 [N,N′-bis(2-anthraquinone)]-1,4,5,8-naphthalenetetracarboxydiimide (NTCDI-DAQ, 250 mA h g−1, ∼2.0 V),6 the benzoquinone-tetrathiafulvalene triad (Q-TTF-Q, 231 mA h g−1, 2.3 V),7 poly(pentacenetetraone sulfide) (PPTS, 260 mA h g−1, 1.65 V),8 poly(2,6-anthraquinonyl sulfide) (PAQS, 198 mA h g−1, ∼2 V),9 anthraquinone-based polymer with a polyaniline core (PANQ, 250 mA h g−1, 2.1 V),10 poly[anthraquinone-alt-dihydrophenazine] in the MXene matrix (PAD@MX (M = Ti, X = C), 255 mA h g−1, 2.34 V),11 fluorinated triazine-based covalent organic nanosheets (450 mA h g−1, 1.55 V),12 polyaniline (138 mA h g−1, 3.2 V),13 poly(N-vinylcarbazole) (117 mA h g−1, 4.05 V),14 poly(N-phenyl-5,10-dihydrophenazine) (p-DPPZ, 162 mA h g−1, 3.6 V),15 poly[6,6-(phenylazanediyl)bis(naphthol)] (poly(DNap-OH), 133.6 mA h g−1, ∼3.6 V),16 hexaazatriphenylene-based polymer (245 mA h g−1, ∼1.6 V),17 hexaazanonaphthalene (HATN)-based 2D MOF (Cu-HATNH, 317.5 mA h g−1, 1.6 V),18 CuTCNQ (244 mA h g−1, 2.75 V),19 and [5,15-bis(ethynyl)-10,20-diphenylporphinato]copper(II) (CuDEPP, 181 mA h g−1, ∼2.8 V).20 Chemical structures of the mentioned materials are provided in the ESI (Fig. S1),† while the overview of their electrochemical performance is given in Fig. 1.
The data presented in Fig. 1 suggest that in spite of considerable progress achieved in the field, the current generation of organic electrode materials for PIBs offers specific capacities usually below 400 mA h g−1 and energy densities of <700 W h kg−1. Herein, we present polymers P1–P2 (Fig. 2) prepared by a single-step condensation of triquinoyl and 2,3,7,8-tetraaminophenazine as highly promising organic electrode materials for PIBs delivering specific capacities of 400–490 mA h g−1 and record-high energy densities approaching 947 W h kg−1. Furthermore, potassium batteries assembled using P1 and P2 as electrode materials demonstrated efficient operation at low temperatures (down to −55 °C), which provides many opportunities for their practical implementation in Arctic area and aerospace missions.
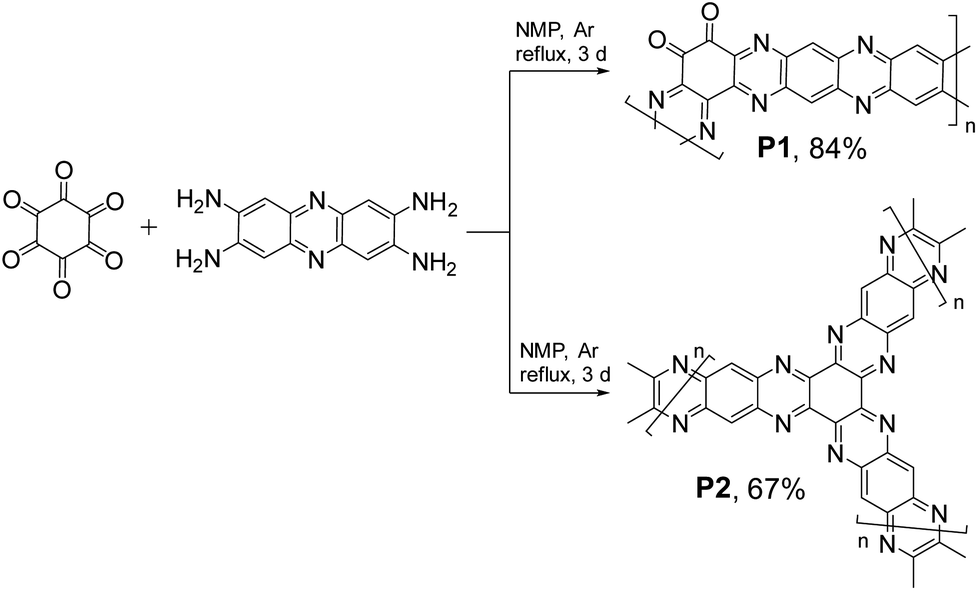 | ||
| Fig. 2 Synthesis of the polymers P1 and P2. | ||
Results and discussion
Both triquinoyl and tetraaminophenazine represent promising redox-active molecules that could be utilized as building blocks for designing advanced organic electrode materials with excellent electrochemical performances. This strategy has been explored recently when covalent organic framework P2 was synthesized and evaluated as a cathode material for Li-metal batteries.21 In this work, we reproduced the synthesis of P2 and also obtained the unknown to date polymer P1 by controlling the stoichiometric ratios between triquinoyl and tetraaminophenazine monomers introduced in the polycondensation reactions (Fig. 2).Both P1 and P2 precipitated from the reaction mixtures during the synthesis and were isolated in high yields. To remove low molecular weight impurities, they were purified by Soxhlet extractions with N-methylpyrrolidone (NMP) and diethylcarbonate and then were dried in a vacuum. Since P1–P2 were insoluble in any common solvents, their characterization could be performed only in the solid state using such techniques as Fourier transform infrared spectroscopy (FTIR), solid-state nuclear magnetic resonance (SS NMR) and chemical analysis (Fig. 3).
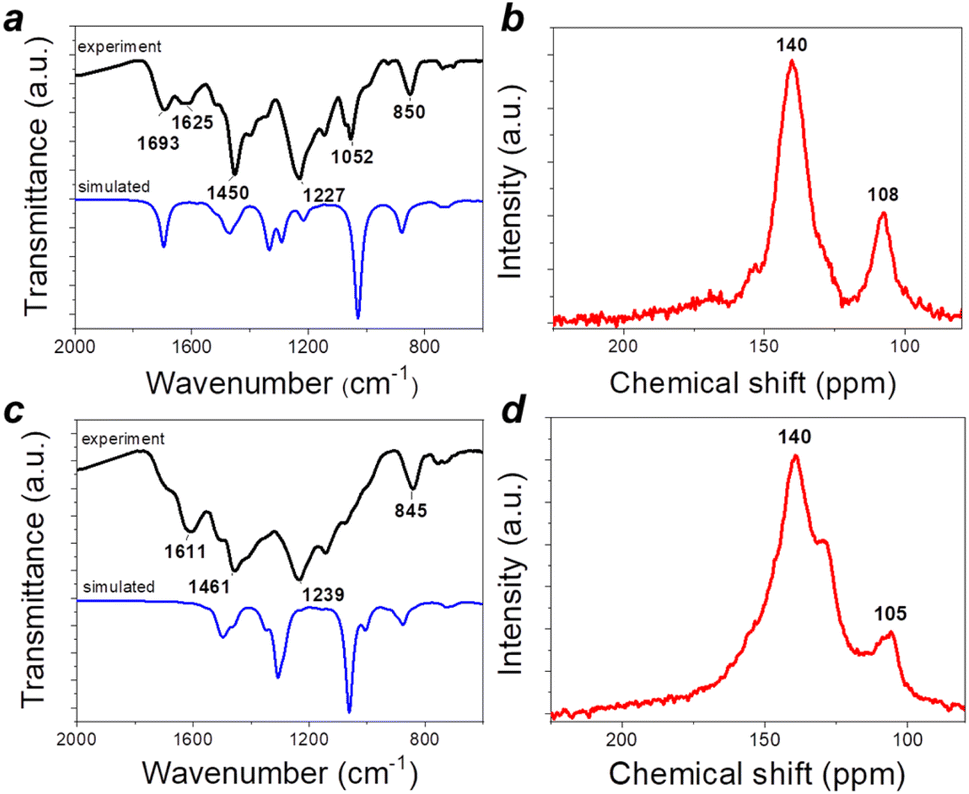 | ||
| Fig. 3 Characterization of polymers P1 (a and b) and P2 (c and d): experimental and calculated ATR-FTIR spectra (a and c) and 13C SS NMR spectra (b and d). | ||
Polymer P1 showed an intense vibration band at 1693 cm−1, which corresponds to the quinone-type carbonyl groups CO (Fig. 3a). This band was significantly suppressed in the case of P2, but the remaining weak shoulder still pointed to the presence of some non-functionalized CO groups (Fig. 3c). This result is expectable since sequential functionalization of carbonyl groups of the triquinoyl moiety creates steric hindrance effects, which make the remaining CO groups less accessible for further reaction. Furthermore, the condensation of triquinoyl with tetraaminophenazine may involve carbonyl groups at 1,2- and 4,5- positions, thus leading to the formation of a linear type condensation product with a p-benzoquinone moiety, which is expected to show low reactivity with respect to further functionalization of CO groups (Fig. S2, ESI†). The theoretically predicted (based on DFT calculations) FTIR spectrum of P1 strongly resembled the experimental spectrum with some notable differences suggesting the simultaneous realization of several reaction pathways leading to the formation of several types of molecular frameworks (e.g., with para-quinone units instead of ortho-quinone and probably some others such as Schiff type adducts). The computed FTIR spectrum of P2 just partially matched the experimental spectrum, thus pointing to a more sophisticated real molecular structure of P2 as compared to that expected and shown in Fig. 2. We have also performed a computational analysis of the FTIR spectra of P1 and P2 structures with various types of structure defects (Fig. S3, ESI†). The simulated spectra for a model structure with incomplete functionalization of carbonyl groups revealed the presence of symmetrical and antisymmetrical scissor-mode oscillations of NH2 groups at 1607 and 1574 cm−1 in the experimental spectrum of P1. Since the model structures with ortho- and para-quinone units show very similar spectra, both types of defects can be present in the real structure of P1. Similar conclusions can also be made for electrode material P2 after comparison of the experimental FTIR spectrum with the simulated spectra of the optimized model structures (Fig. S4 and S5, ESI†).
The SS NMR data are consistent with the analysis of the FTIR spectra. Particularly, the 13C SS NMR spectrum of P1 revealed two distinct broad peaks at ca. 155 and 170 ppm, which can be attributed to two types of carbonyl groups (presumably incorporated within ortho- and p-quinone blocks) (Fig. 3b). The two most intense peaks correspond to the carbon atoms of phenazine units (CN type, 140 ppm, 12 atoms per repeating unit) and benzenoid units (CC type, 108 ppm, 4 atoms per repeating unit). Thus, the SS NMR spectrum of P1 is fully consistent with the chemical structure of this material. The 13C SS NMR spectrum of P2 shows a distinct shoulder at ca. 155 ppm, which may correspond to the residual unfunctionalized CO groups within p-quinone units (Fig. 3d). The major peak in the 13C SS NMR spectrum of P2 is split into two bands, which may be explained by the appearance of non-pyrazine CN groups, e.g., due to the formation of Schiff type adducts. Thus, both FTIR and SS NMR spectra suggest that the formulae of polymers P1 and P2 shown in Fig. 2 give just a very basic idea about their chemical compositions and molecular structures. More realistic structures of these materials deduced based on the analysis of the FTIR and SS NMR spectral data are given in Fig. S6 (ESI).† The chemical analysis data obtained for P1 and P2 (Table S1†) are consistent with the structure representations given in Fig. S3† and also indicate that both materials incorporate some solvation water molecules, thus representing hydrates. Such behaviour is not surprising considering that the parent triquinoyl is stable only in the form of hydrates with several water molecules, while the polymers P1 and P2 both have multiple sites for the formation of stable hydrogen bonds. Therefore, the hydrates of P1 and P2 appear to be quite stable and release solvated water molecules only upon heating above 250–300 °C, as revealed by thermal gravimetry data presented in Fig. S7 (ESI).† The thermal decomposition of the polymer frameworks of P1–P2 occurs only at temperatures of >600 °C, thus pointing at the ultrahigh stability of these materials. Interestingly, P1 decomposes entirely to volatile products by 1000 °C, whereas P2 retains ca. 15–20% of its initial weight due to partial graphitization of the material. Obviously, different decomposition behaviours of P1 and P2 are associated with the different contents of non-functionalized CO groups promoting the formation of volatile species.
Polymers P1 and P2 were obtained as fine powders with broad particle size distributions as revealed by laser diffraction analysis. Fig. 4a and b show that P2 is characterized by smaller particle sizes as compared to P1, but both materials demonstrate the presence of particles approaching 10–20 micrometres in size, which are too big for efficient material operation in batteries. SEM provided more optimistic results by revealing that both materials have a mostly submicrometric particle size with random inclusion of bigger particles approaching a few micrometres in size. Bigger average particle sizes revealed by laser diffraction may be due to the aggregation effects and also a higher sensitivity of the optical technique to bigger particles.
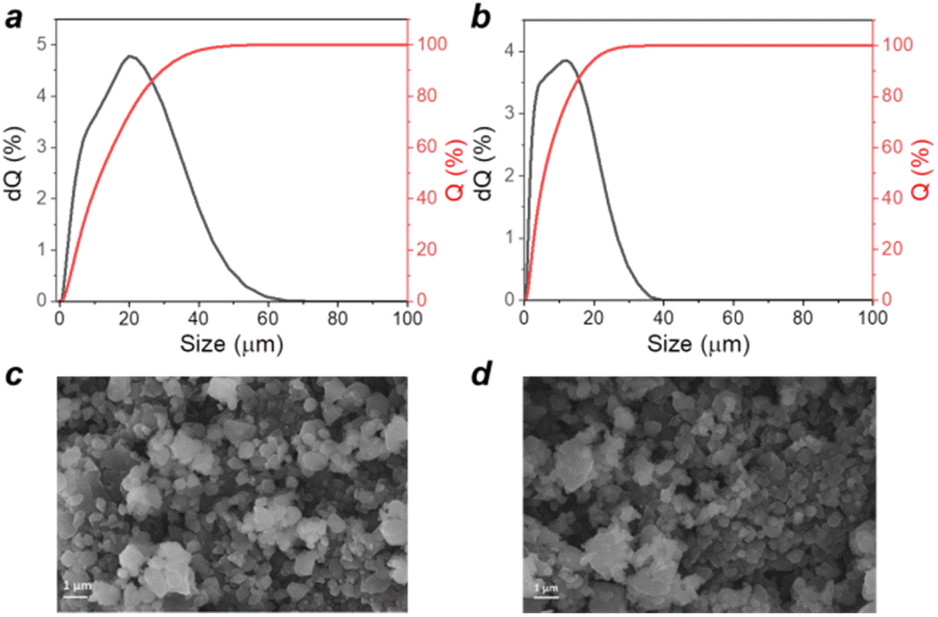 | ||
| Fig. 4 Particle size distribution obtained using laser diffraction analysis for P1 (a) and P2 (b) polymer powders; SEM images of P1 (c) and P2 (d) powders. | ||
The surface properties of the materials were characterized by the nitrogen porosimetry technique. The estimated Brunauer–Emmett–Teller (BET) surface areas for P1 and P2 were quite high and approached 75.6 and 25.5 m2 g−1, respectively. Both materials revealed an average pore size of 3.42 nm (Table S2†).
The electrochemical properties of P1–P2 were evaluated in the coin-type CR2032 cells using metallic potassium anodes, a glass fibre separator and 1 M KPF6 in 1,2-dimethoxyethane (DME) as the electrolyte. The cyclic voltammograms (CVs) of the cells with P1–P2 revealed broad oxidation and reduction waves in the potential range of 1.75–3.75 V (Fig. S8, ESI†). Such behavior is very typical for conjugated polymers with multiple redox centers since the electrochemical conversion of some of the functional groups strongly affects the oxidation or reduction potentials of the neighboring ones.3,5,6 Thus, multiple peaks with continuously shifted reduction or oxidation potentials overlap and form broad waves. We also analysed the evolution of the CV profiles at different voltage sweep rates and estimated b coefficients from the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ipvs. logν dependences, where Ip is the peak current (A) and ν is scan rate in mV s−1. The obtained b values were 0.83 and 0.82 for the cells with P1 and P2, thus indicating a significant contribution of the pseudocapacitive behavior to the cell current. This could originate from high surface area of organic electrode materials as discussed above and also their soft nature allowing for facile diffusion of ions into the bulk of the electrode, which is in sharp contrast with inorganic cathodes where ions have to intercalate into the crystal lattice.
Ipvs. logν dependences, where Ip is the peak current (A) and ν is scan rate in mV s−1. The obtained b values were 0.83 and 0.82 for the cells with P1 and P2, thus indicating a significant contribution of the pseudocapacitive behavior to the cell current. This could originate from high surface area of organic electrode materials as discussed above and also their soft nature allowing for facile diffusion of ions into the bulk of the electrode, which is in sharp contrast with inorganic cathodes where ions have to intercalate into the crystal lattice.
The performance of the potassium batteries using P1 and P2 polymers as cathode components has been evaluated by charge–discharge cycling in the galvanostatic regime at the current densities ranging from 50 to 1000 mA g−1. Both types of cells demonstrated a clearly pronounced activation behavior manifested in a gradual capacity increase over the first several tens of cycles (Fig. 5 and S9, ESI†). This kind of activation correlates with the morphological characteristics of P1–P2 due to the presence of rather big particles, which require multiple charge–discharge cycles to form ion conduction channels and involve the active material inside these big domains in redox transitions.
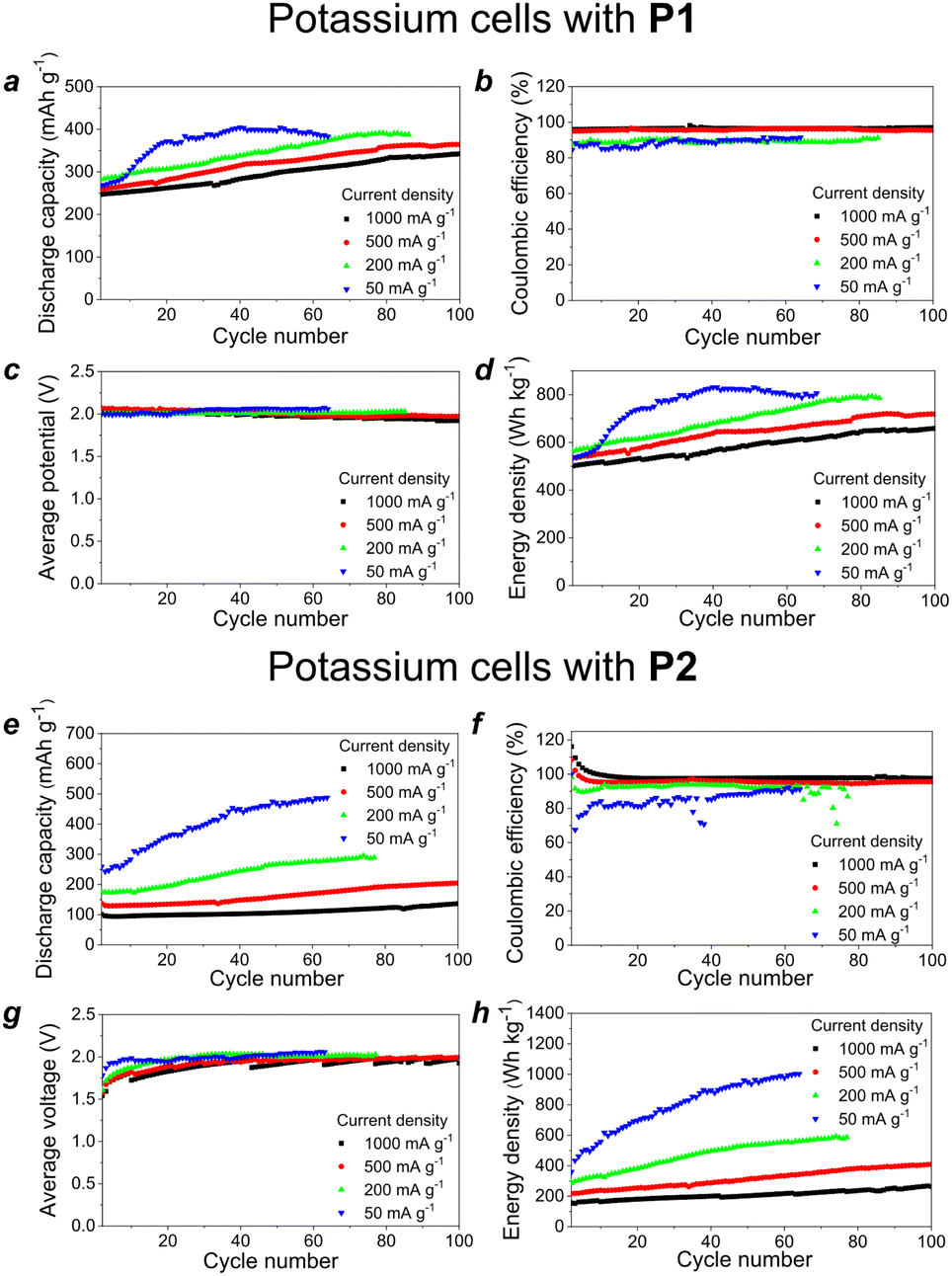 | ||
| Fig. 5 Discharge capacity (a and e), coulombic efficiency (b and f), average discharge potential (c and g) and energy density (d and h) of the cells with a potassium anode and P1- (a–d) or P2-based (e–h) cathode cycled at different current densities from 50 to 1000 mA g−1. | ||
The batteries with P1 as the cathode material delivered specific capacities of 320–400 mA h g−1, average discharge potentials of around 2.0 V and resultant impressive energy densities ranging from ∼650 to ∼800 W h kg−1 depending on the current density (Fig. 5d). The cells assembled with P2 showed an even higher specific capacity of almost 500 mA h g−1 at a low current density of 50 mA g−1, which leads to a record-high energy density of ca. 950 W h kg−1 (Fig. 5h). The operation of the cells at higher current density was hindered due to the slow material activation upon charge–discharge cycling.
The rate capability of the batteries using P1 and P2 as cathodes was evaluated at the current densities ranging from 0.02 A to 2 A g−1 (Fig. 6a). One could notice that both types of cells could still deliver high specific capacities of 200–250 mA h g−1 even at the highest current density of 2 A g−1, which illustrates the ability of the batteries to support the ultrafast charge/discharge regime with minimal capacity depression (less than twice upon a 100-fold increase in the current density). Furthermore, the cells showed continuously increasing specific capacity upon cycling at 10 A g−1 (slow activation behavior), which approached a plateau at 125–150 mA h g−1 after 800–1000 charge–discharge cycles (Fig. 6b).
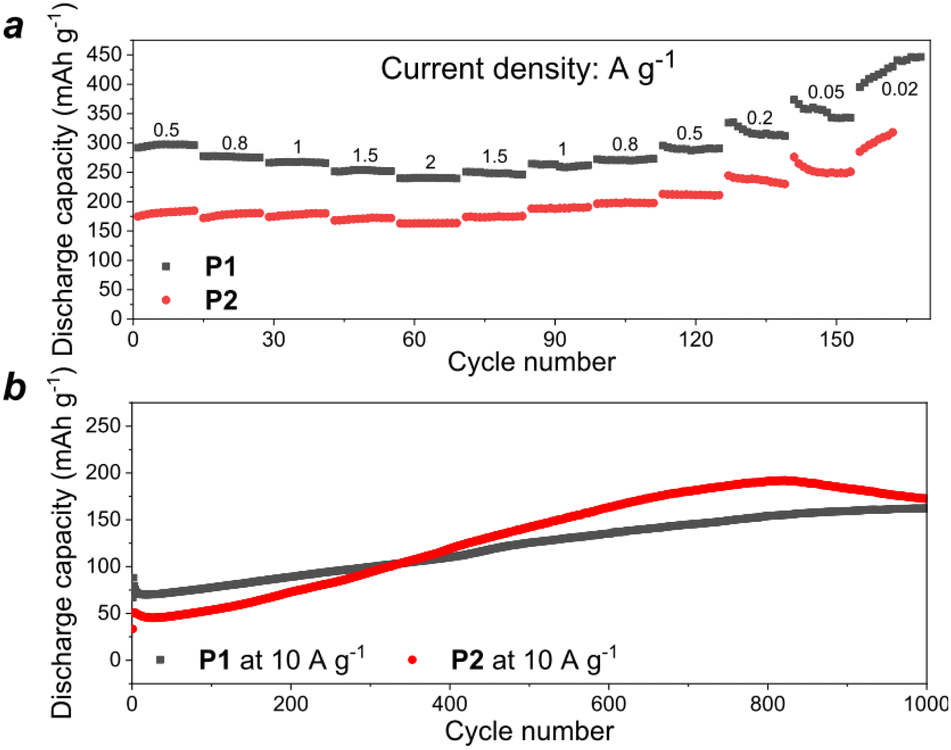 | ||
| Fig. 6 Rate capability of P1 and P2-based cells (cells were precycled for 30 cycles at 200 mA h g−1) (a) and long term cycling at high current density (b). | ||
During the cycling at 10 A g−1, the initial capacity of the P2-based cells was lower than that of P1, which is in good agreement with the theoretical calculations. However, after ∼350 cycles, the performance of the P2-based cells exceeded that of P1. There are several possible factors that could explain the observed distinct behavior of these materials, including the formation of different SEIs on the surface of the electrodes,22,23 and different conductivity,24 wettability,25 or morphology26 of the materials. Most likely, the smaller average particle size of P2 makes the active sites of this material more accessible to redox reactions thus leading to faster electrode activation as compared to the behavior of P1. Such a phenomenon is commonly observed for a wide range of organic electrode materials.27,28 Additionally, P2 has a honeycomb-like structure with large pores, which may contribute to the effective ion transport. Notably, after ∼800 cycles, the capacity of P2 starts to degrade, probably due to the dissolution of the reduced active material.
Since material P1 demonstrated more stable behavior in K half-cells, the battery operational stability has been evaluated at a high current density of 10 A g−1 for 2400 cycles (Fig. S10, ESI†). The capacity first increased due to the activation behavior, approached the maximal value of 163 mA h g−1 and then remained quite stable for ∼1500 cycles with a minor decay down to 134 mA h g−1.
The mechanistic aspects of the redox transitions and potassium storage in polymers P1–P2 were studied experimentally and using density functional theory (DFT) calculations. Within the experimental approach, we first performed chemical ex situ metalation of P1 and P2 using different equivalents of the alkali metal by careful and thorough grinding of the powders in a mortar inside an argon-filled glove box under anoxic conditions (O2, H2O < 0.1 ppm). The FTIR spectra of the metalated and pristine polymers were also measured inside the glove box in the attenuated total reflectance (ATR) mode using a KRS5 prism. The obtained FTIR spectra (Fig. 7a) clearly show that P1 first undergoes complete metalation of carbonyl groups (vibrations above 1600 cm−1 partially disappear) and metalation of about half the pyrazine units as defined by the P1 to metal stoichiometry ratio (6 M per repeating unit). Using larger amounts of alkali metal (8 M per repeating unit) results in a complete reduction of P1 with utilization of all redox groups (CO and pyrazine rings). Similarly, polymer P2 undergoes first partial and them complete chemical metalation with 6 and 9 equivalents of alkali metal per repeating unit (Fig. 7b).
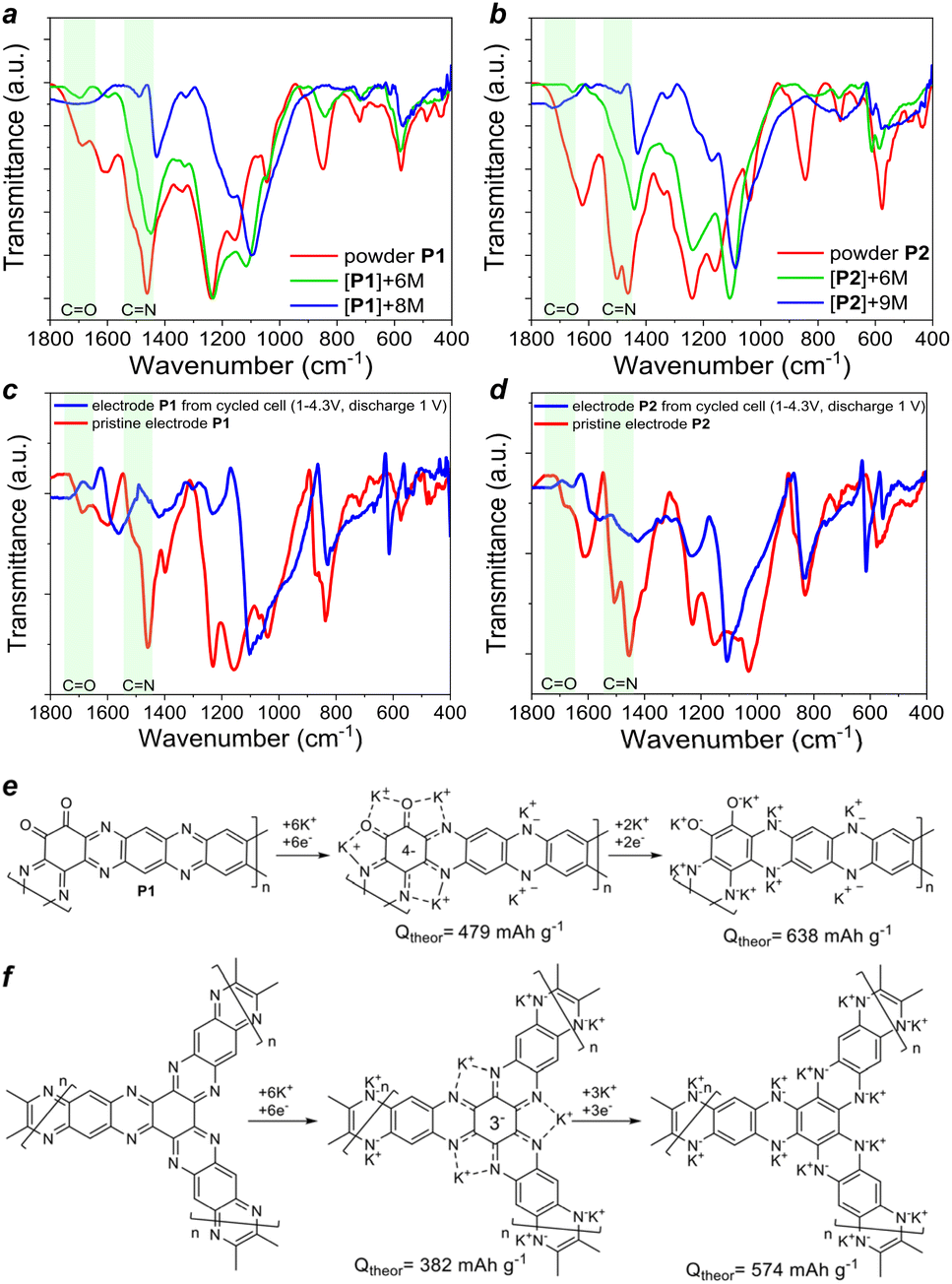 | ||
| Fig. 7 ATR-FTIR spectra of P1 (a) and P2 (b) powders before and after treatment with a K/Na liquid alloy; ATR-FTIR spectra of P1- (c) and P2-based (d) cathodes before and after cycling and proposed charge–discharge mechanisms for P1 (e) and P2 (f). | ||
To elucidate the degree of the metalation of P1 and P2 as electrode materials in operating batteries, we disassembled the cells discharged to 1.0 V vs. K+/K and compared their FTIR spectra to those of the pristine electrodes (Fig. 7c and d). We note that the spectra of the electrodes are slightly different from the spectra of pristine P1–P2 powders due to the presence of the polyvinylidene fluoride (PVDF) binder and potential formation of passivation layers (solid electrolyte interphase, SEI) involving electrolyte species upon battery cycling. Still, the evolution of the FTIR spectra of the electrodes clearly shows complete reductive metalation of P1 and P2 upon cell discharge to 1.0 V vs. K+/K. This behavior suggests that theoretical capacities of P1 and P2 correspond to 638 and 574 mA h g−1.
The practical specific capacities achieved are still notably lower than the theoretical values, which we believe is mostly related to non-optimal morphology and the presence of large-size particles since both materials clearly demonstrate activation behavior. This type of behavior is manifested as a slow increase in the capacity value upon cycling due to the gradual decrease in the effective particle size and the appearance of ion diffusion pathways enhancing the accessibility of redox sites. Since the increase in the current density leads to discharge capacity fading for both materials, slow ion diffusion could also be one of the factors limiting the practical capacity. The electrochemical performance of the electrode is not solely determined by the active material but is also influenced by other factors such as electrode thickness, porosity, composition, preparation method and cycling conditions, so a thorough optimization of these parameters may lead to a further increase in the capacity. Therefore, there is room for improvement of the electrochemical performance of the potassium batteries based on P1 and P2.
We also compared X-ray photoelectron spectra (XPS) of the electrodes extracted from non-cycled cells and the cells after 5 cycles finally discharged to 1.0 V. The survey spectra (Fig. S11, ESI†) first revealed the presence of a considerable amount of potassium in all samples, which suggests that polymers P1–P2 are at least partially reduced and metalated. The metalation of cathodes is most likely caused by the elevated pressure during the cell opening that induces metal diffusion from the anode through the separator. Second, we did not observe any notable differences in the spectra of the non-cycled and cycled cells, which could be consistent with the formation of any surface passivation layers. This finding is surprising since usually a SEI is formed atop both the cathode and anode during the first few charge–discharge cycles.29 The high-resolution C 1s spectra (Fig. S12, ESI†) revealed a significant contribution of the carbonyl group (CO) signal for both polymers, which becomes notably smaller for the discharged samples. In particular, there is a high-energy shoulder at ca. 289 eV characteristic of O–CO groups in the spectra of the electrodes from non-cycled cells, which is almost vanished in the spectra of the discharged samples. These results proved the presence of some amount of unfunctionalized carbonyl groups in the structure of P2, which is consistent with the FTIR and SS NMR data discussed above. Furthermore, both polymers seem to have some amount of carboxylic groups (COOH) in the structure that could be formed due to partial oxidation of the 1,2-diketone moiety while handling P1–P2 or, more likely, the triquinoyl precursor. The O 1s spectra (Fig. S13, ESI†) were consistent with the interpretation of C 1s data. The high-energy shift of O 1s bands for discharged samples is caused by the reduction of CO carbonyl groups to C–OK, which concurs with the redox mechanism shown in Fig. 7. The N 1s spectra (Fig. S14, ESI†) revealed the increased contribution of the amine-type single C–N bonds and decrease in the contribution of the pyridine-type double CN bonds for discharged electrodes (particularly P2), which is consistent with the reduction of pyrazine rings upon metalation (–CN– transforms into C–N(K)–). Thus, the XPS data fully support the redox pathways of polymers P1–P2 presented in Fig. 7.
DFT calculations with the ab initio PBE functional were performed to gain an additional insight into the mechanism of the redox transitions of polymers P1–P2. Reductive metalation of P1 has been calculated for the H-terminated dimer (Fig. 8a). The first two K expectedly reduce the carbonyl groups and the formed K+ ions additionally coordinate with the N atoms of the adjacent pyrazine fragment (site A). The most favorable geometry involves the intercalation of potassium between the planes of two P1 molecules, which enables the coordination of each K+ ion with two O and two N atoms. Subsequent metalation occurs at the N atoms of site A, where each K+ can be coordinated with four N atoms of two stacked P1 molecules. Finally, the metalation also occurs at site B. The reductive intercalation increases the distance between the stacked P1 molecules by 0.35 Å as compared to the non-metalated form of P1. The metalation energies were also calculated for the stepwise introduction of potassium atoms and the ΔEnK value decreased from 3.39 eV for the first potassium atom (n = 1) to 1.36 eV for the 6th (n = 6) (Fig. S15, ESI†).
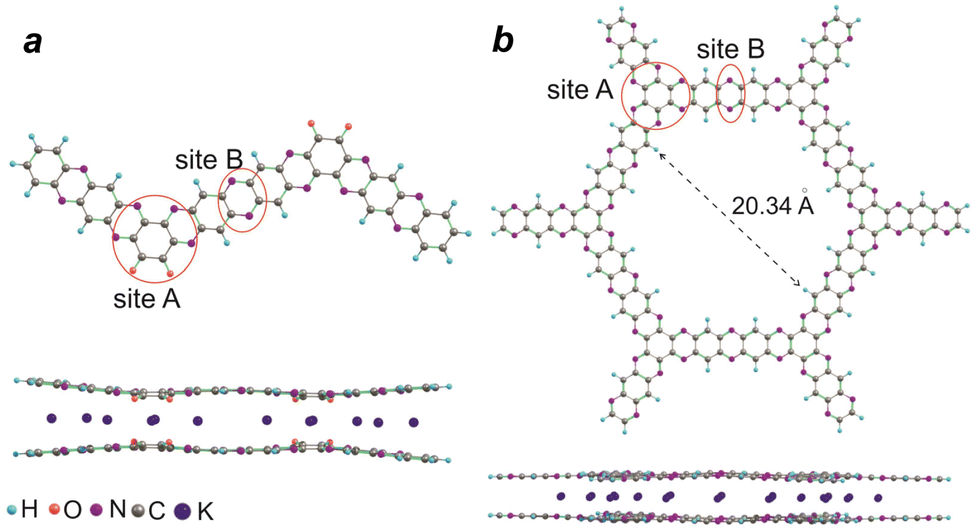 | ||
| Fig. 8 DFT optimized molecular structures of oligomer frameworks representing P1 (a) and P2 (b) showing different sites for coordination of potassium ions and the corresponding metalation products modelled by bimolecular stacks (bottom). | ||
DFT calculations were also used to obtain the optimized 2D molecular honeycomb-like structure of P2. The size of the “pore” was estimated to be 17.94 Å (distance shown in Fig. 8b minus two times the van der Waals radii of H), which is two times smaller than the experimental pore size determined by nitrogen porosimetry. Most probably, the real structure of the pores is also affected by the defects and irregularities of the P2 structure as discussed above.
The reductive metalation of P2 has been calculated for the stack of two H-terminated hexamer molecules (Fig. 8b). The distance between the molecular planes in the stack was estimated to be 4.0–4.1 Å. Like in the case of P1, the insertion of potassium occurs between the molecules within the stack. The first three potassium ions per unit coordinate to site A (Fig. 8b), where each metal ion binds to four N atoms of pyrazine fragments of two stacked molecules. Further incorporation of K+ occurs at the B-sites. The interplanar distance for the heavily potassiated P2 model stack is estimated to be 4.67 Å, which is ∼0.6 Å bigger than that for the pristine non-metalated material. The energy of the gradual reductive potassiation of P2 model ΔEnK decreased from 2.68 eV for the first 3 atoms (n = 3) to 1.39 for 5 atoms (n = 5) per repeating unit (Fig. S15, ESI†).
The decrease in energy gain upon metalation of B sites versus A sites in P1–P2 should strongly affect the metalation reaction rate. Indeed, at high current rates of 500 mA h g−1 (Fig. 6a) specific discharge capacities of 296 mA h g−1 for P1 and 184 mA h g−1 for P2 were obtained, which correspond to 3.7 and 3.1 K+ ions per unit, respectively. At low current rates, the highest specific capacities (487 and 405 mA h g−1) corresponding to 6.1 and 6.3 K+ ions per unit were reached accordingly for P1 and P2, thus confirming the metalation of B sites. To summarize, the results of the theoretical calculations supported the redox pathways of polymers P1–P2 shown in Fig. 7.
The promising electrochemical performances of polymers P1–P2 as electrode materials for potassium batteries inspired us to explore their behavior at low temperatures.
At low temperatures, the charge transfer kinetics at the electrode/electrolyte interface and the diffusion rates of ions in the electrode material are decreased, thus resulting in a poor performance of the battery.30
Organic redox-active materials have soft amorphous structures, which enable facile redox reactions and easy ion diffusion with minimal activation barriers in contrast to inorganic materials mostly operating via intercalation/deintercalation of ions in the cathode crystal lattice and exhibiting low lithium diffusion coefficients and poor reaction kinetics.31
As demonstrated recently, ion desolvation is one of the rate-limiting steps in charge transfer and ion insertion into the electrode material at low temperatures.32,33 Thus, switching to electrode reactions that are not completely controlled by the desolvation process represents a promising approach for low-temperature batteries. Most organic electrodes, including P1 and P2, demonstrate pseudocapacitive behavior (Fig. S8†), meaning that charge storage sites are mainly located on the surface or in the interstitial space, where materials could easily accommodate solvated ions.31 Thus, the combination of rapid reaction kinetics, fast ion transport and the ability to accommodate solvated ions leads to superior operation of organic batteries at low temperatures, as has already been demonstrated in several studies.34–36
Herein, we investigated the operation of potassium batteries using P1 or P2 as cathode materials at low temperatures ranging from −20 to −55 °C (Fig. 9 and S16, ESI†).
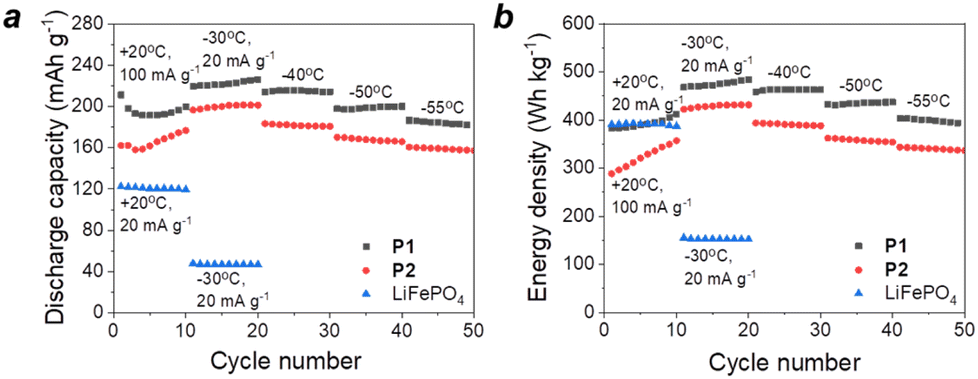 | ||
| Fig. 9 Specific discharge capacities (a) and energy densities (b) of the cathodes based on P1–P2 in potassium cells with 1 M KPF6 DME electrolyte and LiFePO4 in Li cells at low temperatures. | ||
The reference lithium batteries using LiFePO4 (LFP) cathodes showed strong (almost 3-fold) capacity depression when the temperature was reduced from +20 °C to −30 °C. In contrast, potassium batteries with organic electrodes showed minimal capacity roll-off at low temperature demonstrating impressive values of >220 mA h g−1 at −30 °C and >180 mA h g−1 at −55 °C (Fig. 9). Furthermore, both organic cathodes showed energy densities of >400 W h kg−1 at a temperature of −30 °C, which are more than two times higher in comparison with that of inorganic LFP. It is also worth noting that lithium cells with LFP cathodes failed to operate at temperatures below −30 °C, whereas organic potassium batteries showed advanced performances even at −55 °C. The obtained results highlight the remarkable potential of organic batteries, in particular the potassium-ion cells, to find important applications in aerospace technologies or in Arctic or Antarctic areas where stable battery operation at low temperatures is crucially required. A more in-depth analysis of charge transfer and ion diffusion processes in low-temperature batteries based on P1 and P2 should be performed in the future following the approaches developed recently.37
Experimental
Materials and equipment
All utilized materials (chemicals and solvents) were purchased from Acros Organics or Sigma-Aldrich. Cyclohexanehexone octahydrate (triquinoyl)38 and 2,3,6,7-tetraaminophenazine hydrochloride were prepared following previously reported procedures.39A Bruker Avance instrument (400 MHz for 1H and 101 MHz for 13C) (Bruker, Billerica, MA, USA) using a 3.2 mm MAS probe was used to record SS NMR spectra at room temperature. A PerkinElmer Spectrum 100 (ZnSe prism) (PerkinElmer, Waltham, MA, USA) and Spectrum Two (KRS5 prism) were used to obtain the FTIR spectra in ATR mode. A simultaneous thermal analyzer STA 8000 (PerkinElmer, Waltham, MA, USA) was used for TGA analysis (nitrogen atmosphere). The scanning electron microscopy (SEM) images were obtained using a ZEISS LEO Supra 25 scanning autoemission electron microscope (Carl Zeiss AG, Oberkochen, Germany). The surface properties of the materials were analysed using a Quadrasorb SI instrument (USA).
Synthesis of P1 and P2
Polymer P1 (1,2,3,4,5,6-cyclohexanehexone:phenazine-2,3,7,8-tetraamine copolymer (1:1)) was synthesized using the following procedure: cyclohexanehexone octahydrate (312 mg, 1 mmol), 2,3,6,7-tetraaminophenazine hydrochloride (296 mg, 1 mmol), N-methyl-2-pyrrolidone (NMP) (30 mL) and a catalytic amount of sulfuric acid were placed in a two-necked round-bottom flask filled with argon. The mixture was heated at reflux for 3 days under an argon atmosphere. Then the reaction mixture was cooled to room temperature and the precipitated polymer was isolated by filtration, vigorously washed with NMP and water and dried. The obtained crude material was further purified by Soxhlet extraction with NMP, methanol and then with diethyl carbonate. Afterwards, the material was dried at 60 °C in a vacuum for 12 h to produce P1 with 84% yield.
P2 (1,2,3,4,5,6-cyclohexanehexone:phenazine-2,3,7,8-tetraamine copolymer (1:1.5)) was synthesized following the same procedure but using different molar ratios of cyclohexanehexone octahydrate (312 mg, 1 mmol) and 2,3,6,7-tetraaminophenazine hydrochloride (444 mg, 1.5 mmol). The yield of the purified polymer was 67%.
Fabrication of potassium and lithium half-cells
The cathode composition consisted of the active material P1 or P2 (50 wt%), conductive carbon black (40 wt%, Timical Super C65), and polyvinylidene difluoride (PVDF) binder (10 wt%, Gelon). PVDF binder (10 mg) was stirred with 0.7 mL of NMP for 1 hour. The well-ground active material P1 or P2 (50 mg) and carbon black (40 mg) were added to the PVDF solution. The resulting mixture was vigorously stirred at room temperature for 24 h to form a slurry, which was tape cast on a piece of carbon-coated Al foil (Gelon Lib). The obtained coatings were dried in air at 150 °C for 10 h, in a vacuum oven at 110–120 °C for 5–7 h, and then calendered at room temperature. The mass loading of P1–P2 was in the range of 1.0–1.2 mg cm−2. The counter electrodes were made from metallic potassium pressed onto a stainless steel disc. Three layers of glass fiber filter (Whatman GF/A glass microfiber filters, GE Healthcare, Chicago, IL, USA) were used as a separator. 1 M KPF6 in DME was used as the electrolyte (50 μL per cell). The CR2032 coin-type cells K//P1 and K//P2 were assembled inside an MBraun argon-filled glove box (H2O, O2 < 0.1 ppm). The Li cells were fabricated similarly, except the mass of the LFP was 75 mg, PVDF – 5 mg, and carbon black – 20 mg. The counter electrode was a lithium disc, 1 M LiTFSI DOL/DME (2:1 v/v) was used as the electrolyte (20 μL per cell). The mass loading of LFP was 1.7 mg cm−2.
Electrochemical characterization
Cyclic voltammograms were recorded with an Elins P40 potentiostat (Elins, Russia) at scan rates of 1–100 mV s−1. The galvanostatic measurements were carried out on a Neware BTS3000 workstation (Neware, China) at the current rates ranging from 20 mA g−1 to 10 A g−1 in the potential range of 1.0–4.3 V (vs. K+/K). The galvanostatic cycling was started from the discharge for all potassium cells. LFP-based cells were cycled in the potential range of 2.6–3.8 V (vs. Li+/Li).Quantum chemical calculations
Structures of oligomers were optimized using the PBE exchange–correlation functional40 and the SBK41 pseudopotential with an extended C,N,O basis: [5s, 5p, 2d/3s, 3p, 2d], H [5s, 1p/3s, 1p], K [4s, 1p/2s, 1p] for valence electrons. The Hirshfeld method42 was used to calculate atomic charges. All molecular structures were optimized using the PRIRODA software package43 at the PBE/SBK level, followed by the calculation of vibration frequencies to confirm their stability. Structures of P1–P2 model oligomers and their metalated forms with the lowest energies were chosen (Fig. S17 and S18, ESI†). The Cartesian coordinates of the metalated stacks of the model oligomers are also given in the ESI.† The Chemcraft software was used to visualize the molecular structures and IR spectra. All calculations were carried out at the Joint Supercomputer Center of the Russian Academy of Sciences.Conclusions
We have designed two promising organic redox-active polymers and explored for the first time their performance as cathode materials in potassium batteries. Both polymers showed impressive specific capacities of 400–490 mA h g−1 and energy densities approaching 800–950 W h kg−1, thus setting new records for organic (and to the best of our knowledge also inorganic) cathodes for potassium-ion batteries. A thorough experimental and computational study evidenced the utilization of all redox centers of polymers P1–P2 in the electrochemical transformations. Still, the practical capacities we reached amount only to 65–83% of the corresponding theoretical values, thus leaving substantial room for further improvement. Among the feasible strategies, we believe that the optimization of the morphology of polymers P1–P2, e.g., achieving smaller particle sizes with a narrowed distribution, represents one of the most promising directions. Furthermore, FTIR, SS NMR and XPS data revealed that the real structures of polymers P1–P2 are more complicated than the simplified drawings shown in Fig. 2 above and incorporate a number of defects, including even carboxylic groups. The role of these defects and their impact on the electrochemical performance of the materials are the issues to be further investigated.In the context of promising practical applications, we have demonstrated the advanced performance of the potassium batteries with polymers P1–P2 used as cathodes at low temperatures ranging from −20 to −55 °C. The impressive specific capacities and energy densities achieved under cryogenic conditions evidence that organic potassium batteries can play an important role in the development of aerospace technologies and exploration of Arctic and Antarctic areas, where a stable energy supply at low temperature is vitally important.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
E. V. S. – data curation, formal analysis, investigation, visualization, validation; A. V. M. – methodology, investigation; I. S. Z. – methodology, investigation, visualization; T. A. S. – investigation, visualization, methodology, software; G. R. B. – investigation, visualization; A. F. S. – supervision, methodology, software; O. A. K. – writing – original draft, writing – review & editing, funding acquisition, visualization; P. A. T. – writing – original draft, writing – review & editing, project administration, resources, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation: studies on P1 within the project FFSG-2024-0008 (124013000743-3) and research on P2 within the project FFSG-2022-0001 (122111700046-3), “Laboratory of perspective electrode materials for chemical power sources”. The XPS measurements were supported by the Ministry of Science and Higher Education of the Russian Federation: Theme “Electron” No. AAAA-A18-118020190098-5 and Project FEUZ-2023-0013.Notes and references
- A. Slesarenko, I. K. Yakuschenko, V. Ramezankhani, V. Sivasankaran, O. Romanyuk, A. V. Mumyatov, I. Zhidkov, S. Tsarev, E. Z. Kurmaev, A. F. Shestakov, O. V. Yarmolenko, K. J. Stevenson and P. A. Troshin, J. Power Sources, 2019, 435, 226724 CrossRef CAS.
- V. Ramezankhani, I. K. Yakuschenko, A. V. Mumyatov, S. G. Vasil'ev, I. S. Zhidkov, E. Z. Kurmaev, A. F. Shestakov and P. A. Troshin, J. Power Sources, 2022, 517, 230711 CrossRef CAS.
- V. Ramezankhani, I. K. Yakuschenko, S. Vasilyev, T. A. Savinykh, A. V. Mumyatov, I. S. Zhidkov, E. V. Shchurik, E. Z. Kurmaev, A. F. Shestakov and P. A. Troshin, J. Mater. Chem. A, 2022, 10, 3044 RSC.
- Y. Bai, W. Fu, W. Chen, Z. Chen, X. Pan, X. Lv, J. Wu and X. Pan, J. Mater. Chem. A, 2019, 7, 24454 RSC.
- Y. Hu, W. Tang, Q. Yu, X. Wang, W. Liu, J. Hu and C. Fan, Adv. Funct. Mater., 2020, 30, 2000675 CrossRef CAS.
- M. Guo, W. Tang, C. Tang, X. He, J. Hu and C. Fan, ChemSusChem, 2023, 16, e202300343 CrossRef CAS PubMed.
- M. Kato, T. Masese, M. Yao, N. Takeichi and T. Kiyobayashi, New J. Chem., 2019, 43, 1626 RSC.
- M. Tang, Y. Wu, Y. Chen, C. Jiang, S. Zhu, S. Zhuo and C. Wang, J. Mater. Chem. A, 2019, 7, 486 RSC.
- Y. Hu, Y. Gao, L. Fan, Y. Zhang, B. Wang, Z. Qin, J. Zhou and B. Lu, Adv. Energy Mater., 2020, 10, 2002780 CrossRef CAS.
- E. V. Shchurik, O. A. Kraevaya, S. G. Vasil'ev, I. S. Zhidkov, E. Z. Kurmaev, A. F. Shestakov and P. A. Troshin, Molecules, 2023, 28, 5351 CrossRef CAS PubMed.
- X. He, B. Wei, W. Tang, M. Guo, J. Hu, Z. Lin and C. Fan, Adv. Funct. Mater., 2024, 34, 2311740 CrossRef CAS.
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252 CrossRef CAS PubMed.
- H. Gao, L. Xue, S. Xin and J. B. Goodenough, Angew. Chem., Int. Ed., 2018, 57, 5449 CrossRef CAS PubMed.
- C. Li, J. Xue, A. Huang, J. Ma, F. Qing, A. Zhou, Z. Wang, Y. Wang and J. Li, Electrochim. Acta, 2019, 297, 850 CrossRef CAS.
- F. A. Obrezkov, V. Ramezankhani, I. Zhidkov, V. F. Traven, E. Z. Kurmaev, K. J. Stevenson and P. A. Troshin, J. Phys. Chem. Lett., 2019, 10, 5440 CrossRef CAS PubMed.
- T. Kim, T. Lee, Y. R. Yoon, W. S. Heo, S. Chae, J. W. Kim, B.-K. Kim, S. Y. Kim, J. Lee and J. H. Lee, Small, 2024, 2400333 CrossRef CAS PubMed.
- R. R. Kapaev, I. S. Zhidkov, E. Z. Kurmaev, K. J. Stevenson and P. A. Troshin, J. Mater. Chem. A, 2019, 7, 22596 RSC.
- J. Wang, H. Jia, Z. Liu, J. Yu, L. Cheng, H.-G. Wang, F. Cui and G. Zhu, Adv. Mater., 2024, 36, 2305605 CrossRef CAS PubMed.
- J. Ma, E. Zhou, C. Fan, B. Wu, C. Li, Z.-H. Lu and J. Li, Chem. Commun., 2018, 54, 5578 RSC.
- S. Lv, J. Yuan, Z. Chen, P. Gao, H. Shu, X. Yang, E. Liu, S. Tan, M. Ruben, Z. Zhao-Karger and M. Fichtner, ChemSusChem, 2020, 13, 2286 CrossRef CAS PubMed.
- J. Wang, J. C. Z. En, S. N. Riduan and Y. Zhang, Chem.–Eur. J., 2020, 26, 2581 CrossRef CAS PubMed.
- M. Zhu, H. Wang, H. Wang, C. Li, D. Chen, K. Wang, Z. Bai, S. Chen, Y. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2024, 136, e202316904 CrossRef.
- H. Wang, M. Zhu, H. Wang, C. Li, Z. Ren, Y. Zhang, S. Chen, H. Li, D. Chen, Z. Bai, Y. Zhang and Y. Tang, Energy Storage Mater., 2024, 67, 103238 CrossRef.
- J. Moon, H. Yun, J. Ukai, C. Chokradjaroen, S. Thiangtham, T. Hashimoto, K. Kim, Y. Sawada and N. Saito, Carbon, 2023, 215, 118479 CrossRef CAS.
- L. Zhao, Y. Li, M. Yu, Y. Peng and F. Ran, Adv. Sci., 2023, 10, e2300283 CrossRef PubMed.
- S. Oswald, M. Bock and H. A. Gasteiger, J. Electrochem. Soc., 2023, 170, 090505 CrossRef CAS.
- J. Li, H. Yao, H. Guan, Z. Sun, X. Ling and S. Guan, ACS Sustainable Chem. Eng., 2024, 12, 4328 CrossRef CAS.
- H. Lyu, X.-G. Sun and S. Dai, Adv. Energy Sustainability Res., 2021, 2, 2000044 CrossRef CAS.
- L. Caracciolo, L. Madec and H. Martinez, ACS Appl. Energy Mater., 2021, 4(10), 11693–11699 CrossRef CAS.
- M. Chen, Y. Zhang, G. Xing, S.-L. Chou and Y. Tang, Energy Environ. Sci., 2021, 14, 3323 RSC.
- Y. Hong, Z. Ma, K. Li, J. Li, S. Tang, Z. Xu, D. Yu, D. Chen, L. Qin, J. Xie and Q. He, J. Mater. Chem. A, 2023, 11, 7898 RSC.
- K. Xu and A. V. Cresce, J. Mater. Res., 2012, 27, 2327 CrossRef CAS.
- Q. Y. Li, D. P. Lu, J. M. Zheng, S. H. Jiao, L. L. Luo, C. M. Wang, K. Xu, J. G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 42761 CrossRef CAS PubMed.
- Y. Hong, Z. Ma, K. Li, J. Li, S. Tang, Z. Xu, D. Yu, D. Chen, L. Qin, J. Xie and Q. He, J. Mater. Chem. A, 2023, 11, 7898 RSC.
- X. Dong, Z. Guo, Z. Guo, Y. Wang and Y. Xia, Joule, 2018, 2, 902 CrossRef CAS.
- J. Chen, Y. Peng, Y. Yin, Z. Fang, Y. Cao, Y. Wang, X. Dong and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 23858 CrossRef CAS PubMed.
- F. Wang, F. Li, H. Gong, Y. Zhang, X. Liu, Z. Jiang, L. Chen, J. Huang, Y. Zhang, Y. Jiang, B. Chen and Y. Tang, J. Alloys Compd., 2024, 981, 173668 CrossRef CAS.
- K. Kanakarajan and A. W. Czarnik, J. Org. Chem., 1986, 26, 5241 CrossRef.
- T. Ashirov, T. Puangsamlee, A. Robles, P. W. Fritz, K. Piech, O. Š. Miljanić and A. Coskun, Helv. Chim. Acta, 2023, 106, e202300072 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhov, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- W. J. Stevens, H. Basch and M. J. Krauss, Chem. Phys., 1984, 81, 6026 Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- D. N. Laikov, Chem. Phys. Lett., 1997, 281, 151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization, quantum-chemical calculations and electrochemical performance. See DOI: https://doi.org/10.1039/d4ta05744a |
| This journal is © The Royal Society of Chemistry 2025 |