
Dual-network bacterial cellulose-based separators with high wet strength and a dual ion transport mechanism for uniform lithium deposition†
Chen
Cheng
a,
Rendang
Yang
 a,
Yang
Wang
*ab,
Xiaohui
Guo
a and
Jie
Sheng
c
a,
Yang
Wang
*ab,
Xiaohui
Guo
a and
Jie
Sheng
c
aState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: misteryang@163.com
bSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China
cSchool of Environmental and Chemical Engineering, Foshan University, Foshan 528000, China
First published on 19th November 2024
Abstract
Inherently insulating and thermally stable nanocellulose membranes are prone to forming a three-dimensional porous structure, which is conducive to storing large amounts of electrolyte and providing ion migration pathways. However, the infiltration of polar electrolytes can compromise the mechanical strength of the nanocellulose membrane by disrupting hydrogen bonding, leading to a suboptimal interface and cycling stability of the batteries. This study effectively improves the mechanical strength and electrochemical performance of bacterial cellulose (BC) based separators by in situ constructing a dual-network structure polymerized from poly(ethylene glycol) diacrylate (PEGDA) and acrylic acid (AA). The distinctive dual-network structure not only significantly enhances the wet strength of the BC separator but also introduces a polymer-coordination transport mechanism, building upon the original lithium-ion pore transport system. The transport of lithium ions is regulated by the ether, ester and carboxyl groups in the polymer network, so that they are uniformly deposited on the surface of the lithium metal negative electrode, and finally form an SEI layer dominated by LiF, which greatly reduces the side reactions between the electrolyte and the electrode. The assembled lithium symmetric battery exhibits stable lithium deposition/stripping behavior, and the cycle stability and rate performance are far superior to those of commercial PP separators.
1. Introduction
In lithium-ion batteries (LIBs), separators are essential for ensuring both safety and efficiency by preventing direct contact between the anode and cathode while allowing ion transport through the electrolyte.1 Traditional commercial separators, predominantly composed of polyolefins like polypropylene (PP) and polyethylene (PE), exhibited several drawbacks. Despite their high mechanical strength and good chemical stability, these materials suffered from inadequate thermal stability. At elevated temperatures, polyolefin separators would shrink, leading to potential contact between electrodes, which might cause internal short circuits and pose significant safety hazards such as thermal runaway.2,3 Additionally, the low surface energy of polyolefin separators resulted in poor electrolyte wettability. This could lead to uneven current density distribution and the formation of irregular solid electrolyte interphases (SEI), which in turn promoted the rapid growth of lithium dendrites.4–6 In the typical single-layer pore structure of commercial separators, formed through stretching processes, the vertically penetrating pore structure could not prevent the continuous growth of lithium dendrites, increasing the risk of internal short circuits.These limitations had driven the search for alternative materials with better thermal stability and electrolyte wettability. Cellulose, the most abundant natural polymer, had emerged as a promising candidate for separator materials in LIBs.7,8 With inherent insulating properties and excellent thermal stability, cellulose-based separators could significantly enhance battery safety. Researchers had developed a diverse range of cellulose-based LIB separators, whose exceptional properties highlight the significant potential of cellulose as a substitute material for separators in LIBs.9–13
Compared to other cellulose sources, bacterial cellulose (BC) with its higher crystallinity exhibited superior strength and chemical stability.14,15 Its extremely high aspect ratio enables the formation of a three-dimensional porous network, while the abundant hydroxyl groups on its surface facilitate rapid electrolyte wetting.10,16 However, the infiltration of polar electrolytes could disrupt the hydrogen bonds between molecular chains, weakening the inter-fiber bonding and significantly reducing the mechanical strength of the separator, thereby increasing the risk of lithium dendrite penetration. Researchers had attempted to enhance the mechanical strength of cellulose membranes through graft modification,17–20 but this approach often compromised the degree of polymerization, diminishing the structural chemical stability. Another approach involved incorporating inorganic particles to utilize metal coordination bonds for crosslinking and hydrogen bond shielding, thereby reducing separator swelling and enhancing mechanical strength in solution.21,22 However, this method tended to decrease the wettability of the electrolyte. Thus, it was essential to find an appropriate strategy to improve the wet mechanical strength and electrochemical performance of the separator.
Inspired by dual-network hydrogels, a porous cellulose framework was employed as the first network to provide rigidity, while a second soft and resilient polymer network was formed through in situ crosslinking of polymer monomers within the framework.23–26 This approach maintained the porous structure of BC while enhancing its mechanical strength in electrolytes. Based on this concept, a dual-network separator was fabricated using UV curing. The three-dimensional porous BC framework served as the first network, with its high porosity capable of storing substantial amounts of electrolyte, facilitating rapid lithium-ion (Li+) migration through a pore transport mechanism. The second network was formed by the copolymerization of polyethylene glycol diacrylate (PEGDA) and acrylic acid (AA) monomers, enhancing the mechanical strength of the PEGDA/AA-bacterial cellulose (PABC) dual-network separator in the electrolyte. Simultaneously, as a derivative of polyethylene glycol, PEGDA exhibited excellent film-forming properties and compatibility at the electrode–electrolyte interface.27–29 Its abundant ether oxygen segments facilitated Li+ transport through a polymer-coordination transport mechanism, which complemented the pore transport mechanism of the BC network and helped to mitigate the issue of uneven lithium deposition caused by pore blockage. Leveraging this dual Li+ transport mechanism, the PABC separator effectively enhanced Li+ deposition/stripping behavior on the anode side, leading to the formation of an SEI layer predominantly composed of inorganic LiF. In various electrochemical tests, batteries using the PABC separator demonstrated superior electrochemical performance, establishing this unique dual-network separator as a promising candidate for advanced LIB separators.
2. Experimental
2.1 Materials
BC flakes were purchased from Hainan Yide Food Co., Ltd. Poly(ethylene glycol) diacrylate (PEGDA), acrylic acid (AA), the photoinitiator diphenyl-(2,4,6-trimethylbenzoyl) phosphine oxide (TPO), acetic acid, sodium hydroxide and ethanol were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Electrolyte (1 M LiPF6 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC:EMC = (1:1:1) wt%), cathode materials (the active substance was LiFePO4), lithium metal film and commercial PP separators (16 μm) were purchased from Canrd Laboratory Equipment Technology Co., Ltd.
:DMC:EMC = (1:1:1) wt%), cathode materials (the active substance was LiFePO4), lithium metal film and commercial PP separators (16 μm) were purchased from Canrd Laboratory Equipment Technology Co., Ltd.
2.2 Preparation of PABC separators
2 wt% of the total monomer mass of TPO was dissolved in 5 g of ethanol, different mass concentrations of monomers PEGDA and AA were added, the mass ratio was 1:1, and stirred well to make the mixture homogeneous. Subsequently, the BC separator was infiltrated in the monomer solution for 8 h, so that the monomer solution could be fully immersed into the separator structure. Then the separator was taken out, and the excess solution was wiped off with a non-woven cloth, and the monomer was fully polymerized by irradiation with a UV lamp for 10 min under nitrogen (1000 W and irradiation height 5 cm), and finally PABC separators were obtained. The wet PABC separators were transferred to ethanol by slow shaking for 48 h to remove the incompletely reacted monomers and the small molecule polymers, and finally dried with a plate heater at 70 °C and named PABC-2.5, PABC-5, and PABC-7.5 based on the concentration of monomers, respectively (2.5, 5 and 7.5 stand for monomer concentration).
2.3 Characterization
The molecular weights of PEGDA and AA copolymers were measured by gas permeation chromatography (GPC).
The 1H-NMR spectra of different samples after polymerization were measured using a Varian Inova 400 MHz Spectrometer (Agilent Technologies, USA). 1H-NMR shifts were referenced to deuterated dimethyl sulfoxide (d6-DMSO) using MestReNova software.
The crystal structure of the samples was analyzed using a D8 Advance X-ray diffractometer (Bruker, Germany) equipped with Cu Kα radiation (λ = 1.5406 Å), and the XRD measurements were carried out in the scattering range from 5° to 60° (2θ), with a scanning step of 1° min−1. The peak intensity method was used for the calculation of the crystallinity index (Xc) values:30
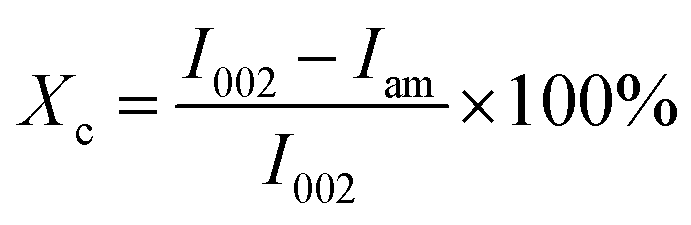 | (1) |
The PABC-5 separator and PP were heat-treated in an electric blast oven at 80 °C, 100 °C, 120 °C, 140 °C and 160 °C for 2 h, and then their Shrinkage was determined. Thermogravimetric (TG) analysis was performed on several PABC separators to evaluate their thermal stability using a NETZSCH TG209F3 instrument from Germany. The samples were heated at a constant rate of 10 °C min−1 in a nitrogen (N2) atmosphere over a temperature range of 40–600 °C.
The surface and back surface morphology of PABC was analyzed and studied by field emission scanning electron microscopy (FE-SEM). (Quanta FEG 250, USA).
The tensile strength of the samples under both dry and electrolyte-wetted conditions was studied using a tensile-compression tester with a tensile rate of 5 mm min−1.
The contact angle of the PABC separators with the electrolyte was measured at ambient temperature using an optical contact angle analyzer (OCA40 Microcontact Angle System, Germany) to assess the electrolyte wettability of the separators.
The porosity was determined using the n-butyl alcohol immersion method, as described by eqn (2):31
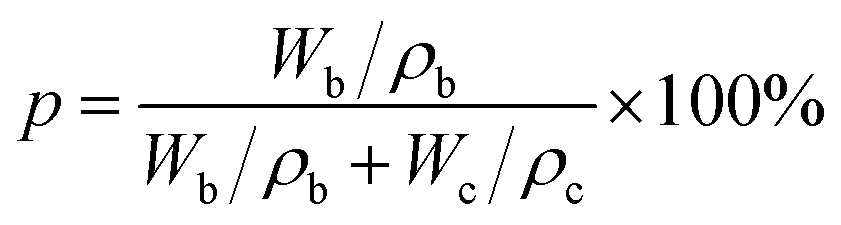 | (2) |
The electrolyte absorption rate was determined by immersing the separator in the electrolyte for 30 minutes. The rate was calculated by dividing the mass of the separator after absorbing the electrolyte by its mass before absorption.
The SEI layer on the metal side surface of the negative electrode was analyzed using an X-ray photoelectron spectrometer (XPS) to determine its components (Thermo Scientific K-Alpha, USA).
| σ = L/Rb × A | (3) |
Samples were assembled into Li/separators/SS for linear sweep voltammetry (LSV) testing of cells to determine the electrochemical stability with a potential range of 2.5 to 5.5 V and a scan rate of 10 mV s−1.
Tafel curves of Li/separator/Li cells were tested from −0.2 to 0.2 V with a scan rate of 10 mV·s−1, based on which the exchange current density was calculated employing the following equation (eqn (4)):
| η = a + blog(j) | (4) |
Activation energy was derived using the Arrhenius equation (eqn (5)):
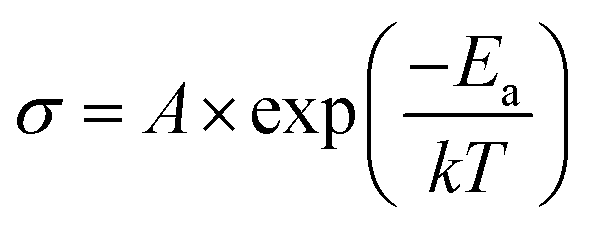 | (5) |
The Li+ transference number was calculated by combining the chronoamperometry (potentiostatic polarization method) and EIS methods, calculated according to the following formula:
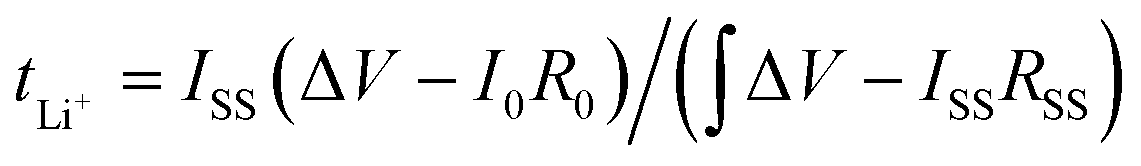 | (6) |
The stability of the Li electrode was tested with a current density of 0.5 mA cm−2, corresponding to a charge/discharge face capacity of 1.0 mA h·cm−2. The Li stripping and plating stability was then measured at current densities of 0.1, 0.2, 0.5, 1, 2 and 5 mA cm−2.
For the Aurbach CE test,33 lithium is first deposited on Cu at a current density of 0.5 mA cm−2 to a capacity of 5 mA h cm−2, then stripped to 1 V for an initial formation cycle. Next, 5 mA h cm−2 of lithium is deposited on Cu at 0.5 mA cm−2 as a lithium reservoir, followed by repeated stripping/deposition of 1 mA h cm−2 of lithium at a given 0.5 mA cm−2 for 10 cycles. Finally, the cell is charged to 1 V to strip all remaining lithium. The CE value can be calculated using the following formula:
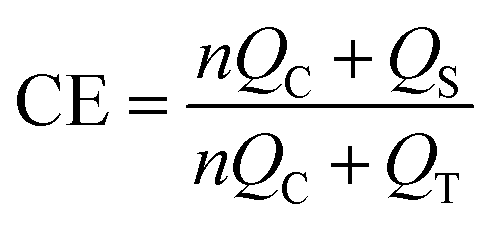 | (7) |
The samples were assembled into LiFePO4/separators/Li cells to analyze the cyclic voltammetry (CV) curves at a scan rate of 0.2 mV s−1 over the potential range of 2.3 to 4.5 V. The cyclic charge/discharge capability was tested using a NEWARE battery test system (CT-4008) with a charge/discharge current density of 0.5C/0.5C. In order to investigate the rate-discharge capability, button cell batteries were measured at different discharge current densities (0.2, 0.5, 1.0, 2.0 and 5.0C), while the charge current density was kept at 0.2C.
| E = ETotal − ELi − EMolecules | (8) |
3. Results and discussion
3.1 Physical and chemical properties of separators
PEGDA coordinated with Li+ through its ether oxygen segments, facilitating Li+ migration via segmental motion and polymer-coordination transport mechanism. However, the presence of PEGDA alone resulted in regular monomer alignment, which increased the proportion of crystalline regions and subsequently reduced ionic conductivity.36 The introduction of AA as a comonomer formed a random copolymer, effectively reducing the crystallinity of PEGDA. In the polymer network, the bifunctional PEGDA served as a monomer and crosslinker with AA to form a dual-network structure within the BC separator. As could be seen from Fig. S1,† the photoinitiator TPO decomposed into unstable radicals under UV irradiation, and these radicals reacted with the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in PEGDA and AA monomers, initiating a radical polymerization reaction, promoting monomer chain growth, and ultimately forming a crosslinked polymer network structure. Under the fixed monomer concentration of 5%, the crosslinking of the gel was compared by adding different amounts of TPO. From Fig. S2,† it was found that an insufficient amount of TPO resulted in incomplete monomer polymerization, failing to form a stable network structure, while excessive TPO produced an excess of radicals, causing quenching and counterproductive effects.37,38 Through a series of experimental studies, from Fig. S3† we found that when the TPO concentration was 2 wt% of the total monomer mass, the gel achieved a high degree of crosslinking and stable morphology.
C bonds in PEGDA and AA monomers, initiating a radical polymerization reaction, promoting monomer chain growth, and ultimately forming a crosslinked polymer network structure. Under the fixed monomer concentration of 5%, the crosslinking of the gel was compared by adding different amounts of TPO. From Fig. S2,† it was found that an insufficient amount of TPO resulted in incomplete monomer polymerization, failing to form a stable network structure, while excessive TPO produced an excess of radicals, causing quenching and counterproductive effects.37,38 Through a series of experimental studies, from Fig. S3† we found that when the TPO concentration was 2 wt% of the total monomer mass, the gel achieved a high degree of crosslinking and stable morphology.
GPC was used to determine the copolymer of PEGDA and AA. According to the molecular weight distribution shown in Fig. S4,† the copolymer had a broad molecular weight range, with the main concentration around 1000, which was higher than the molecular weights of the two monomers. As shown in Table S1,† the polydispersity Index of the copolymer reached 3, indicating a copolymer with a combination of long and short chains. The long chains enhanced mechanical strength by forming a stable three-dimensional network, whereas the short chains, with larger intermolecular gaps and greater mobility, facilitated electrolyte absorption and retention, offering more space and flexible channels for Li+ transport.
ATR spectroscopy was used to assess the polymerization state of the monomers within the separator. The data presented in Fig. 1(a) clearly demonstrate that both PEGDA and PABC exhibit characteristic absorption peaks at 1720 cm−1, attributed to the stretching vibration of the CO bond in the ester groups. The stretching vibration of the CO bond in the carboxyl group is observed at 1700 cm−1, indicating the successful incorporation of AA into PABC. Additionally, PEGDA and PABC shared a common characteristic peak at 1100 cm−1, corresponding to the stretching vibration of the C–O–C group present in the ether segment of PEGDA. Both AA and PABC displayed stretching vibration peaks at 1250 cm−1, associated with the C–O bond in the carboxyl groups. Moreover, the absorption peaks observed at 1630 cm−1 and 810 cm−1 in PABC are absent, indicating the disappearance of the CC bonds from PEGDA and AA. This observation confirmed that these monomers have been fully polymerized within the BC separator, with no residual small-molecule monomers remaining. Additionally, as shown in Fig. S5,† the hydroxyl peak in the range of 3000–3500 cm−1 for PABC was significantly broadened compared to that of BC, indicating the formation of hydrogen bonding interactions between BC and AA.39
 | ||
| Fig. 1 (a) ATR-FTIR spectrum and (b) X-ray diffraction pattern of separators; the tensile strength of separators (c) under electrolyte wetting conditions and (d) comparison of tensile strength; (e) TG curves of separators; (f) thermal stability at different temperatures of PP and PABC-5; (g) the SEM images and back surface contact angle of PABCs; (h) physical picture of PABC-5. | ||
Further structural analysis of the polymer was performed using 1H-NMR spectroscopy. Fig. S6† shows that AA alone tended to polymerize incompletely, whereas PEGDA exhibited minimal residual unsaturation after polymerization. Therefore, the few unsaturated bonds in the copolymer were predominantly from AA. The carboxyl peak in the 12–13 ppm region was almost absent in PABC, unlike in AA, due to hydrogen bonding interactions between the hydroxyl groups on cellulose and carboxyl groups in AA, which effectively shield the carboxyl hydrogen signal.40
The influence of the polymer network on the crystallinity of the cellulose framework was assessed using X-ray diffraction (XRD) analysis. In Fig. 1(b), the XRD patterns of BC and PABC displayed characteristic peaks at specific angles (14.4°, 16.8°, and 22.6°), indicative of BC-based materials.41 Since PEGDA and AA were amorphous polymers, the crystallinity in PABC was primarily attributed to the BC component. As depicted in Fig. S7,† the crystallinity values were calculated using an empirical formula, with BC at 72.2%, PABC-2.5 at 71.3%, PABC-5 at 70.4%, and PABC-7.5 at 68.7%, and the crystallinity of the PABC separator decreased with the increasing proportion of the polymer network. This indicated that the interaction between the polymer network and the cellulose network gradually intensifies, leading to the formation of more amorphous regions. Although this interaction may compromise the strength of the PABC separator, it also facilitated the coordination transport mechanism within the polymer network.42
To protect LIBs from external impacts and lithium dendrite penetration, the separator must possess sufficient mechanical strength. The dry cellulose-based separators exhibited excellent strength, but the hydrogen bonds between the cellulose molecular chains would break when immersed in polar electrolytes, leading to a reduction in intermolecular bonding forces.43,44 Therefore, enhancing the mechanical strength of cellulose-based separators in electrolyte environments was crucial. As shown in Fig. 1(c), under electrolyte-wetted conditions, the tensile strength of PABC-5 was 4.8 MPa, which was 1200% that of the BC separator. The significantly improved elongation demonstrated that the polymer, serving as the second reinforcing network, enhanced the toughness of PABC. As seen from Fig. 1(d), the strength of the separator under electrolyte-wetted conditions gradually increased with rising monomer concentration, while the tensile strength of PABC under dry conditions gradually decreased. The molecular structure depicted in Fig. S8† revealed that the polymer network was rich in oxygen-containing groups, which readily formed hydrogen bonds with the hydroxyl groups present in BC. As the cross-linking density of the polymer network increased, these interactions intensified, which not only limited the flexibility of the polymer network, but also disrupted the original network structure of BC due to the shrinkage effect, ultimately resulting in a reduction in mechanical strength. Additionally, the separators were assembled into a LiFePO4//Li cell and charged to 3.8 V. In Fig. S9,† the batteries were dropped from a height of 2 m onto a cement floor and the voltage measured to check for any short circuits. As indicated in Table S2,† PABC-5 maintained normal voltage after 70 drops, while the BC experienced an internal short circuit after 36 drops. This demonstrated PABC-5 had enhanced drop stability and safety performance compared to BC.
When a battery operated in high-power or high-temperature environments, the elevated temperature could cause deformation of polyolefin separators, increasing the risk of internal short circuits due to contact between the anode and cathode. Therefore, the separator must possess excellent thermal dimensional stability to ensure the thermal safety of the battery. As illustrated by the TGA curve in Fig. 1(e), the thermal behaviors of BC and PABC exhibited similarities, characterized by three distinct stages: (I) the thermal stability stage, (II) the intense pyrolysis stage, and (III) the slow pyrolysis stage. The primary differences between these materials emerged during stages II and III. Due to the faster pyrolysis rate of the polymer network compared to cellulose, the overall pyrolysis process for PABC accelerated with increasing monomer concentration. Although the initial decomposition temperature of PABC was slightly lower than that of BC, it remained high at approximately 250 °C, ensuring the separator's stability under high-temperature conditions. The reduction in initial decomposition temperature could be attributed to challenges in achieving uniform polymer network distribution, leading to the presence of low molecular weight oligomers in the separator that decomposed at lower temperatures. At around 450 °C, the residual mass stabilized, and this residual mass increased with higher monomer concentrations, further substantiating the formation of a dual network.
Additionally, we tested the morphological thermal stability of PP and PABC-5 at different temperatures. As shown in Fig. 1(f), PABC-5 maintained its original dimensions even at a high temperature of 160 °C, whereas PP began to deform at 100 °C and undergo horizontal shrinkage with increasing temperature until it became transparent and melted. Consequently, replacing polypropylene with PABC-5 as the separator could significantly reduce the safety risks of batteries operating at high temperatures.
In actual production, polyolefin separators were produced using stretching techniques, resulting in vertically penetrating pore structure. During battery operation, uneven lithium deposition occurs on the anode surface due to polarization effects, leading to the continuous growth of lithium dendrites, a problem that is more pronounced on lithium metal anodes.45 Irregular lithium dendrites can easily penetrate the monolayer pores of polyolefin separators, causing contact between the anode and cathode, ultimately resulting in internal short circuits. As shown in Fig. S10,† BC fibers could form a three-dimensional network structure through interlacing and stacking. The highly tortuous pore structure helped inhibit the growth and penetration of lithium dendrites while storing a large amount of electrolyte, aiding in the rapid conduction of Li+. Compared to BC, the front side of PABC exhibited a similar three-dimensional porous structure, which facilitates electrolyte infiltration. On the back side of PABC in Fig. 1(g), some pores were covered by the polymer, with the covered area increasing with higher monomer concentrations. This was due to the gravitational flow of the monomer solution toward one side of the bacterial cellulose separator during the polymer network formation process, leading to a relatively dense structure on the back side at higher monomer concentrations. As seen from Fig. S11,† contact angle tests indicated that the contact angle on the front side of PABC basically remained around 18°, and did not show significant variation with increasing monomer concentration. In contrast, the contact angle on the back side steadily increased due to the gradual densification of the polymer network, with PABC-7.5 exhibiting a contact angle of 47.94°, but still remained superior to that of the PP separator (92.28°).
Given its Janus-like characteristics, PABC could rapidly absorb the electrolyte from the front side, ensuring sufficient contact between the cathode material and the electrolyte. The polymer network on the back side not only provided the separator with excellent mechanical strength but also played a significant functional role in the formation of the SEI layer on the anode surface. This aspect was further discussed in subsequent electrochemical tests. In addition, as could be seen from Fig. 1(h), the prepared PABC still had good flexibility and could be folded into various shapes while maintaining structural stability, making it promising to be used in batteries with different structures.
The physical and chemical tests of the separator revealed that PABC significantly improved the mechanical strength under electrolyte-wetted conditions by incorporating a dual-network structure. Additionally, it exhibited excellent thermal stability and electrolyte wettability. Further tests were conducted to evaluate the impact of this dual-network structure on electrochemical performance.
3.2 Electrochemical characteristics of separators
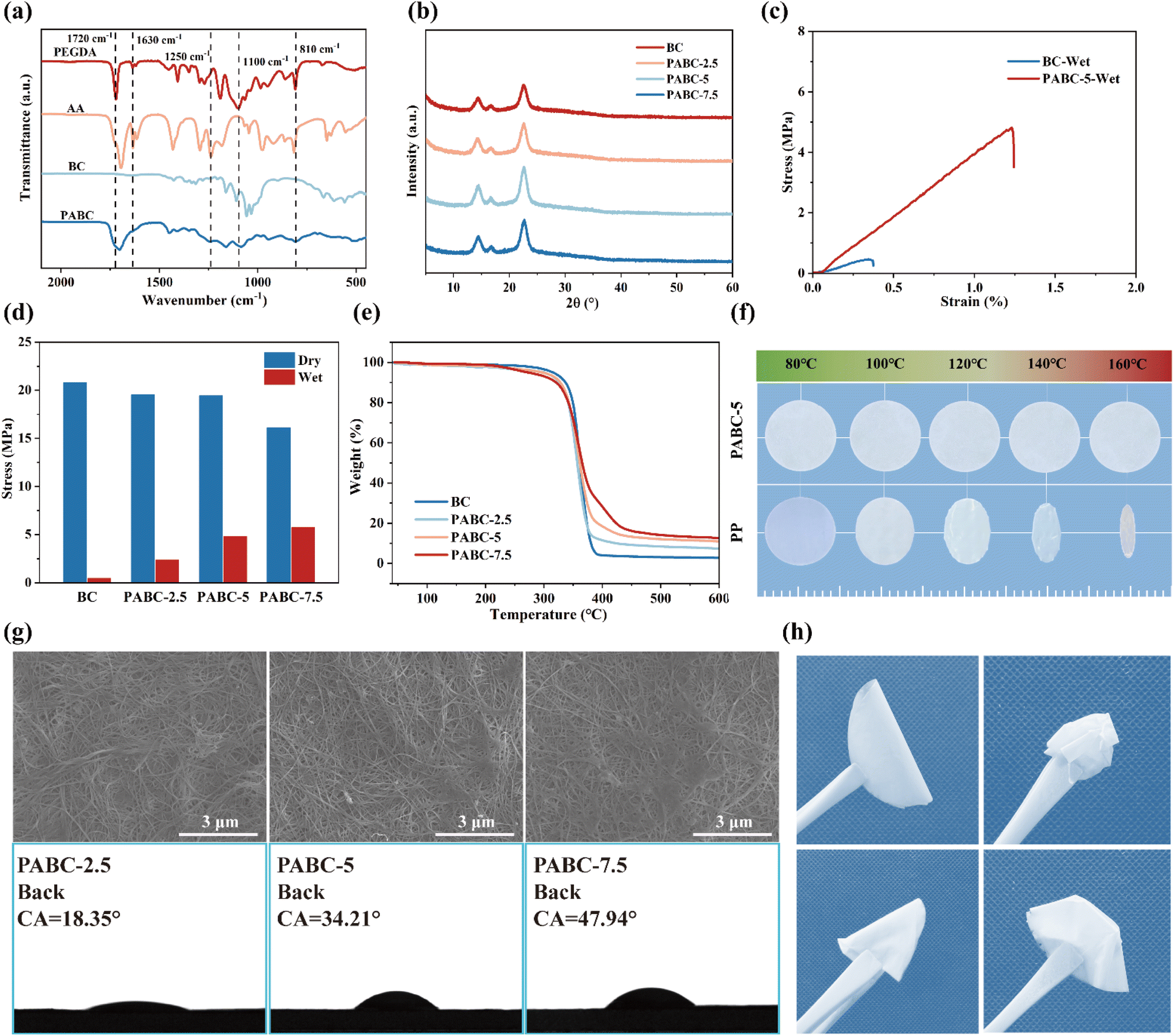
Subsequently, electrochemical impedance spectroscopy (EIS) tests were conducted on different separators. As shown in Fig. 2(a), the internal resistance of the separators gradually increased with rising monomer concentration. This was attributed to the clogging of separator pores by the excessive polymer network content. As shown in Fig. S12,† the porosity of the separator decreased with increasing monomer concentration, leading to insufficient electrolyte penetration and hindering the Li+ pore transport mechanism.46 This indicates that the introduction of the polymer network was a double-edged sword, making it crucial to select an appropriate polymer ratio to balance the ionic conductivity and mechanical strength performance of the dual network. The polymer network must enhance the strength of the BC framework in an electrolyte-wetted state without significantly disrupting the original pore transport mechanism. As illustrated in Fig. 2(b), increasing the polymer network ratio led to a decrease in the ionic conductivity of the separator while the wet strength gradually improved, approaching an optimal balance at PABC-5. Fig. S13† indicates that PABC-5 achieved a high liquid absorption rate of 214%, allowing the separator to retain a significant amount of electrolyte and exhibit excellent ionic conductivity of 0.57 mS cm−1. Simultaneously, it maintained sufficient mechanical strength in the electrolyte environment, reducing the risk of lithium dendrite penetration. Therefore, PABC-5 was selected for subsequent electrochemical performance comparisons alongside BC and commercial PP.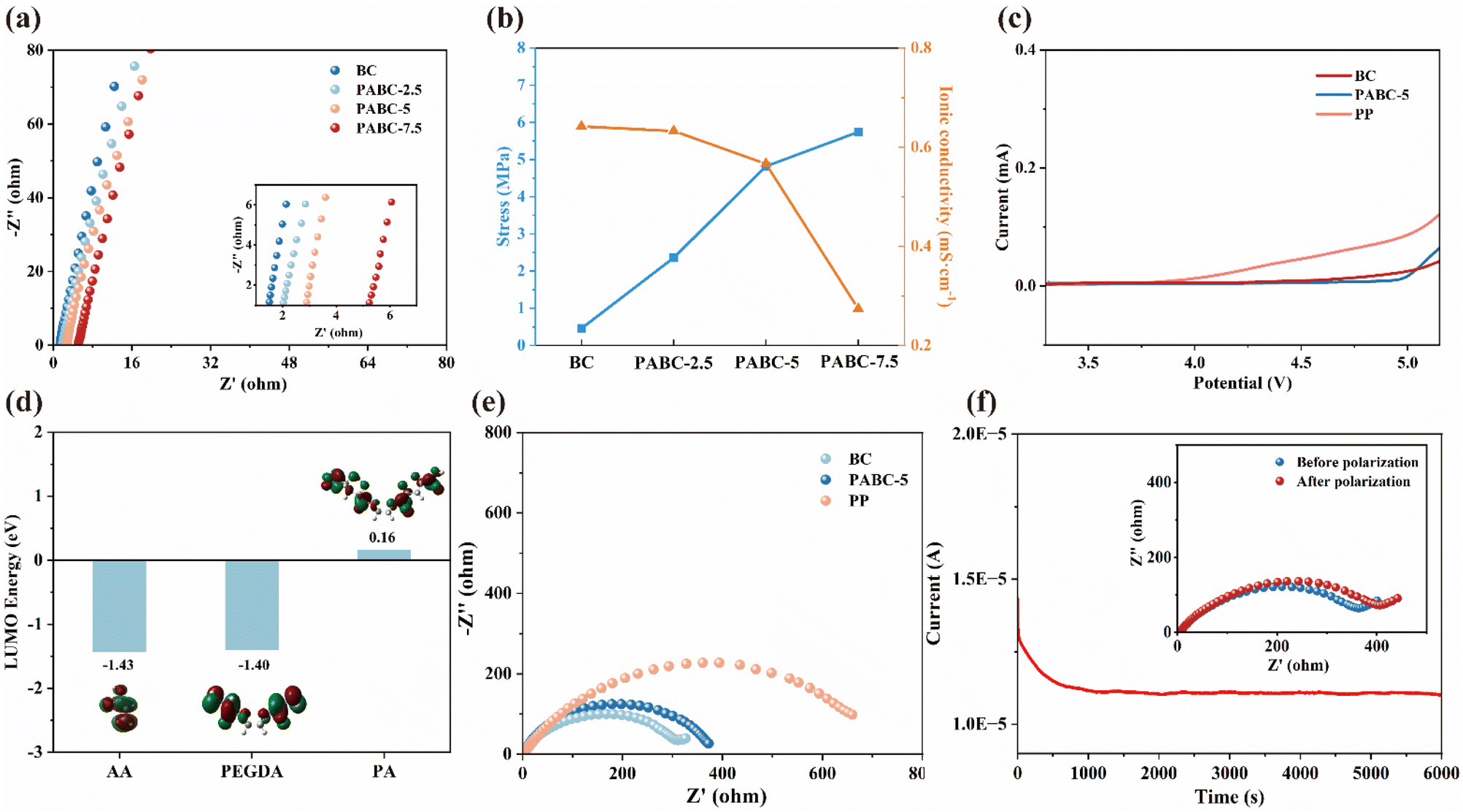 | ||
| Fig. 2 (a) Impedance plots of separators; (b) relationship between ionic conductivity and tensile strength of separators under electrolyte wetting conditions; (c) the LSV curves of separators; (d) LUMO energy level of different molecules; (e) Nyquist plots of the Li∥Li symmetric cells before cycling; (f) variation of current with time and EIS tests before and after the polarization of symmetric cells with PABC-5. | ||
Linear sweep voltammetry (LSV) was used to test the electrochemical stability of the separators. Fig. 2(c) shows that, compared to the PP separator, which began to show a voltage rise at 4 V, the BC separator exhibited a wider electrochemical window of 4.5 V. For PABC-5, the polymer network contained a significant number of –O– groups in PEGDA, which compared to –OH groups were more stable in an electric field,47 had stronger antioxidative properties, and were less likely to decompose at high voltages so PABC-5 finally showed a voltage window of up to 5 V. Additionally, since the polymer network on the anode side contacted metallic lithium, the separator required sufficient reduction stability to effectively minimize side reactions. Further calculations of the LUMO energy levels were conducted, as shown in Fig. 2(d). The results indicated that the polymerized PEGDA-AA (PA) exhibited a higher LUMO energy level, suggesting that the polymerization of PEGDA and AA enhanced their reduction stability. This improved stability was crucial for maintaining the separator's integrity and performance under operational conditions, ensuring that the PABC-5 separator could effectively reduce side reactions and maintain a stable electrochemical environment within the battery.48
The EIS comparison of different separators revealed that the impedance primarily arose from the electrolyte–electrode interface in Fig. 2(e). Although the introduction of the polymer network slightly reduced the electrolyte's wettability, leading to the impedance of PABC-5 increasing from 305 Ω of BC to 372 Ω, the Janus separator structure of PABC could still store a sufficient amount of electrolyte. This resulted in PABC exhibiting lower interfacial resistance compared to that of a commercial PP separator (659 Ω), demonstrating superior interfacial compatibility.
To investigate the impact of the polymer network on the ion migration process, the Li+ transference number was measured. As shown in Fig. 2(f) and S14,† although the ionic conductivity of PABC-5 (0.57 mS·cm−1) was lower than that of BC (0.64 mS·cm−1), PABC-5 exhibited the highest Li+ transference number (0.67) compared to BC (0.37) and PP (0.33). This demonstrated that the introduction of polymer networks could selectively enhance the transport of Li+via a polymer-coordination transport mechanism.
We combined simulation calculations and relevant experiments for analysis. Firstly, molecular models of the polymer chains and cellulose chains were established, and their electrostatic potentials (ESP) were calculated in Fig. 3(a). By examining the distribution of surface charges, we identified the binding sites for Li+ during ion transfer. The negative charges were primarily concentrated around the oxygen atoms, indicating that these sites have strong electron-donating effects and tend to coordinate with Li+ in the electrolyte.49,50 In Fig. 3(b), further calculations of the binding energies between various sites and Li+ revealed that the binding energies at the sites on PA chains were lower than those on BC molecules, and the relevant binding energy data are listed in Table S3.† This reduced binding energy means that Li+ experiences less constraint during transfer, resulting in a faster migration rate. Additionally, without the polymer network, Li+ could only migrate through the tortuous pores constructed by BC molecular chains. As could be seen from Fig. 3(c), due to the irregular arrangement of cellulose chains, not all pores are interconnected, which could easily lead to blockages in Li+ flow as it traversed the separator, similar to hitting a dead end. This could ultimately cause uneven deposition of Li+. However, with the polymer network present, it acted like a ladder over a wall, allowing blocked Li+ to bypass the unconnected pores through the coordination transport mechanism of the polymer network. This reduced the internal polarization of the separator, enabling more uniform deposition on the anode surface.
 | ||
| Fig. 3 (a) The ESP charge distribution of PEGDA-AA and BC; (b) binding energy of each binding site on PEGDA-AA and BC with Li+; (c) simple hole transport leading to uneven Li+ deposition (left) and dual transport mechanism promoting uniform Li+ deposition (right). | ||
3.3 Battery characteristics of LIBs
Further, Fig. 4(a and b) show the effect of separators on the Li+ deposition/stripping process. At 0.5 mA cm−2 and 1 mA h cm−2, the PP, BC and PABC-5 all exhibited similar polarization voltages initially. However, as Li+ continued to deposit and strip, the polarization voltage for PP rose due to its poor electrolyte wettability.51 Around 500 h cycling, BC started to show signs of internal short circuits, and the polarization voltage of PP increased significantly. In contrast, lithium symmetric cells assembled with PABC-5 maintained stable Li+ deposition/stripping for over 700 h. This stability was attributed to the formation of a stable SEI layer, resulting from the uniform deposition of Li+.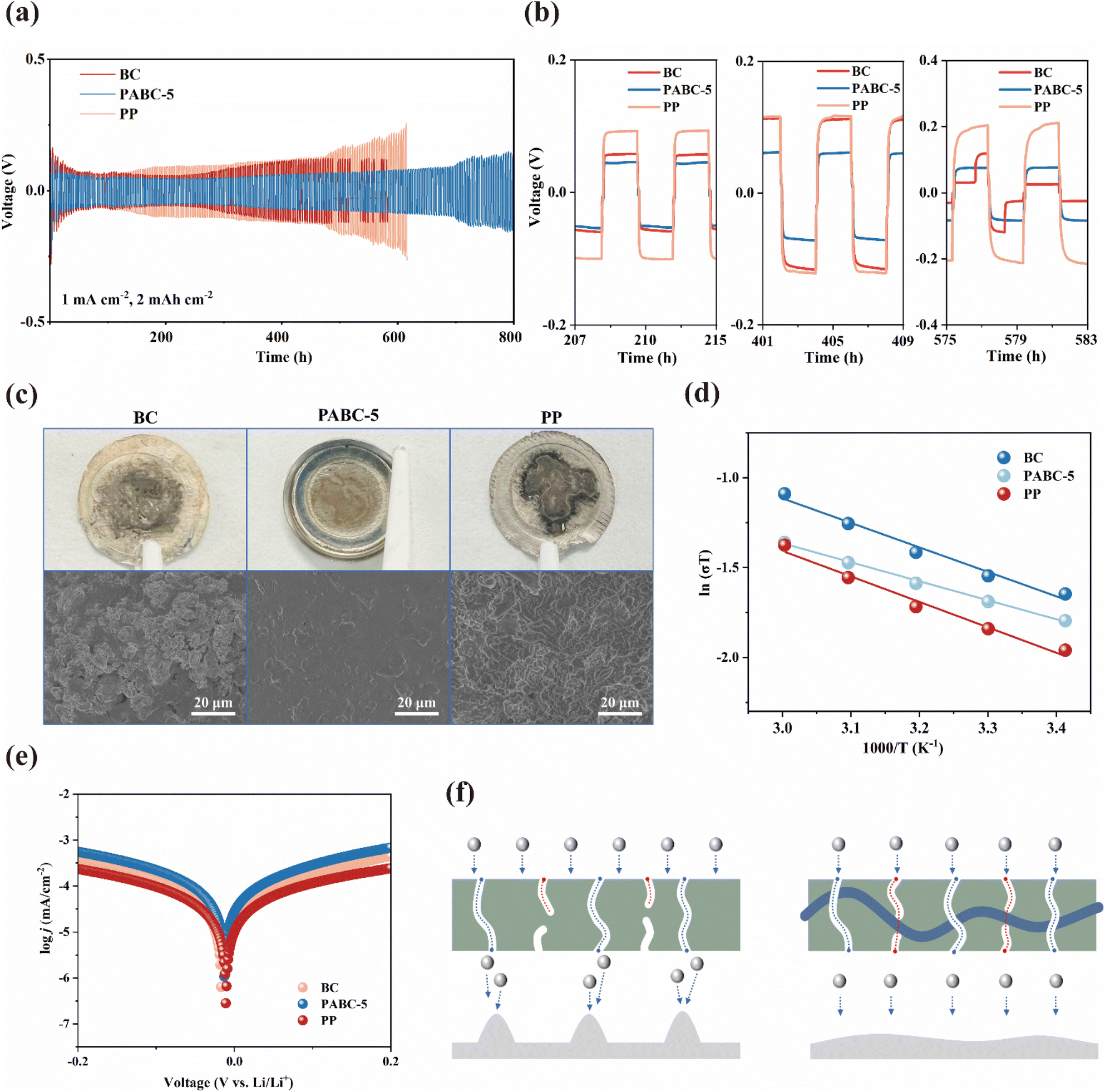 | ||
| Fig. 4 (a) Cycling performance of the Li∥Li cells at 1 mA cm−2 with a capacity of 2 mA h cm−2; (b) amplified voltage curves at different times; (c) optical images (upper) and SEM images (lower) of the lithium metal surface after cycling of lithium symmetric cells assembled with different separators. (d) The activation energies fitted by the Arrhenius equation with the temperature from 20 to 60 °C; (e) Tafel plot of separators; (f) dual transport mechanism promoting uniform Li+ deposition to reduce lithium dendrite growth. | ||
According to Fig. S15,† the modified Aurbach method was used to test the average CE of PP and PABC-5. Notably, PABC-5 achieved an average CE of 86.7%, which was significantly greater than that of PP (73.3%). The lower average CE of PP was due to its inability to form a stable interfacial layer during lithium deposition/stripping, resulting in continuous side reactions between the carbonate solvent and lithium metal, leading to more irreversible lithium loss.33 This suggested that PABC-5 played an important role in facilitating the formation of a stable interface during Li+ deposition/stripping, effectively reducing the occurrence of side reactions.
The rate performance of lithium stripping and deposition in Li∥Li symmetric cells with different separators was further investigated under current densities from 0.1 to 5 mA cm−2 and an areal capacity of 1 mA h cm−2. In Fig. S16,† the overpotential for lithium stripping and deposition increased with the rise in current density. Compared with BC and PP, PABC-5 consistently achieved the lowest overpotential across various current densities, maintaining stability even at a high current density of 5 mA cm−2. This demonstrated its potential for practical applications in LIBs, especially for rapid charging/discharging and high charge/discharge depth.52
The lithium deposition morphology on the anode surface was characterized using SEM. It could be observed from Fig. 4(c) that the anode surfaces with BC and PP separators exhibited moss-like lithium growth due to uneven deposition. This increased the risk of piercing and short circuits. Moreover, the loose and porous moss-like deposits enlarged the contact area with the electrolyte, increasing the likelihood of side reactions. In contrast, the anode surface with the PABC-5 separator primarily showed island-like lithium deposition.53,54 This indicated that under the influence of PABC-5, Li+ was deposited uniformly in large, even island-like structures, forming a dense surface. This dense surface reduced contact with the electrolyte, achieving stable deposition/stripping.
Further EIS tests at different temperatures, analyzed using the Arrhenius equation, calculated the activation energy required for lithium to traverse the SEI layer, shown in Fig. 4(d). Compared to the activation energy of BC (0.11 eV) and PP (0.12 eV), PABC-5 exhibited a significantly lower activation energy of 0.09 eV, indicating that the energy barrier for Li+ traversing the SEI layer formed by PABC-5 was the lowest, resulting in faster Li+ transport capability. This was primarily related to the structure and composition of the SEI. Additionally, Tafel curve fitting in Fig. 4(e) was used to study the Li+ transfer kinetics at the interface, with the exchange current density indicating the ability of lithium to gain or lose electrons at the interface. PABC-5 exhibited an exchange current density of 0.061 mA cm−2, higher than that of BC (0.035 mA cm−2) and PP (0.026 mA cm−2), demonstrating that the SEI formed with the PABC-5 separator possessed rapid Li+ transfer kinetics. These results indicated that there are two transport mechanisms in PABC as shown in Fig. 4(f), and through the synergistic effects of pore transport and polymer-coordination transport mechanisms, the PABC-5 separator exhibited superior Li+ migration kinetics compared to BC and PP. Consequently, PABC-5 could form a high-quality SEI on the lithium metal anode surface, facilitating stable Li+ deposition and stripping.
To elucidate the differences in SEI layers formed in lithium symmetric cells assembled with different separators, XPS analysis was performed to examine the composition of the SEI. In Fig. 5(a), the characteristic peaks in the C 1s spectra primarily originated from organic components such as carbonates in the SEI, with peaks at 284.8 eV, 286.5 eV, 288.6 eV and 289.0 corresponding to C–C, C–O, CO and ROCO2Li, respectively.55–57 It could be observed that the proportion of C-containing compounds in BC and PP was significantly higher than in PABC-5, indicating that solvent decomposition was suppressed in PABC. In the F 1s spectra shown in Fig. 5(b), peaks at 684.8 eV and 685.5 eV corresponded to LiF and LixPOyFz, respectively.58 It was evident that the content of the inorganic component LiF in the SEI of PABC significantly increased. The content of the inorganic component LiF in the SEI of PABC was notably higher, providing stable electronic insulation properties and significantly reducing side reactions between the lithium metal anode and the electrolyte, thus forming a stable interfacial layer.59,60 This explained why the lithium symmetric cells with PABC-5 could achieve stable Li+ deposition and stripping. The Li 1s spectra in Fig. 5(c) further confirmed this, with peaks at 54.9 eV and 56.2 eV corresponding to OC–OLi and LiF, respectively. Unlike the SEI layers in BC and PP, which were primarily composed of lithium carbonate, the SEI formed with PABC-5 was predominantly composed of LiF. This effectively inhibited the growth of lithium dendrites. It could also be seen from Fig. S17† that compared with BC and PP, the proportion of F and Li elements in the SEI formed by PABC-5 was significantly increased, indicating that the content of inorganic components with high mechanical strength and stability had increased.
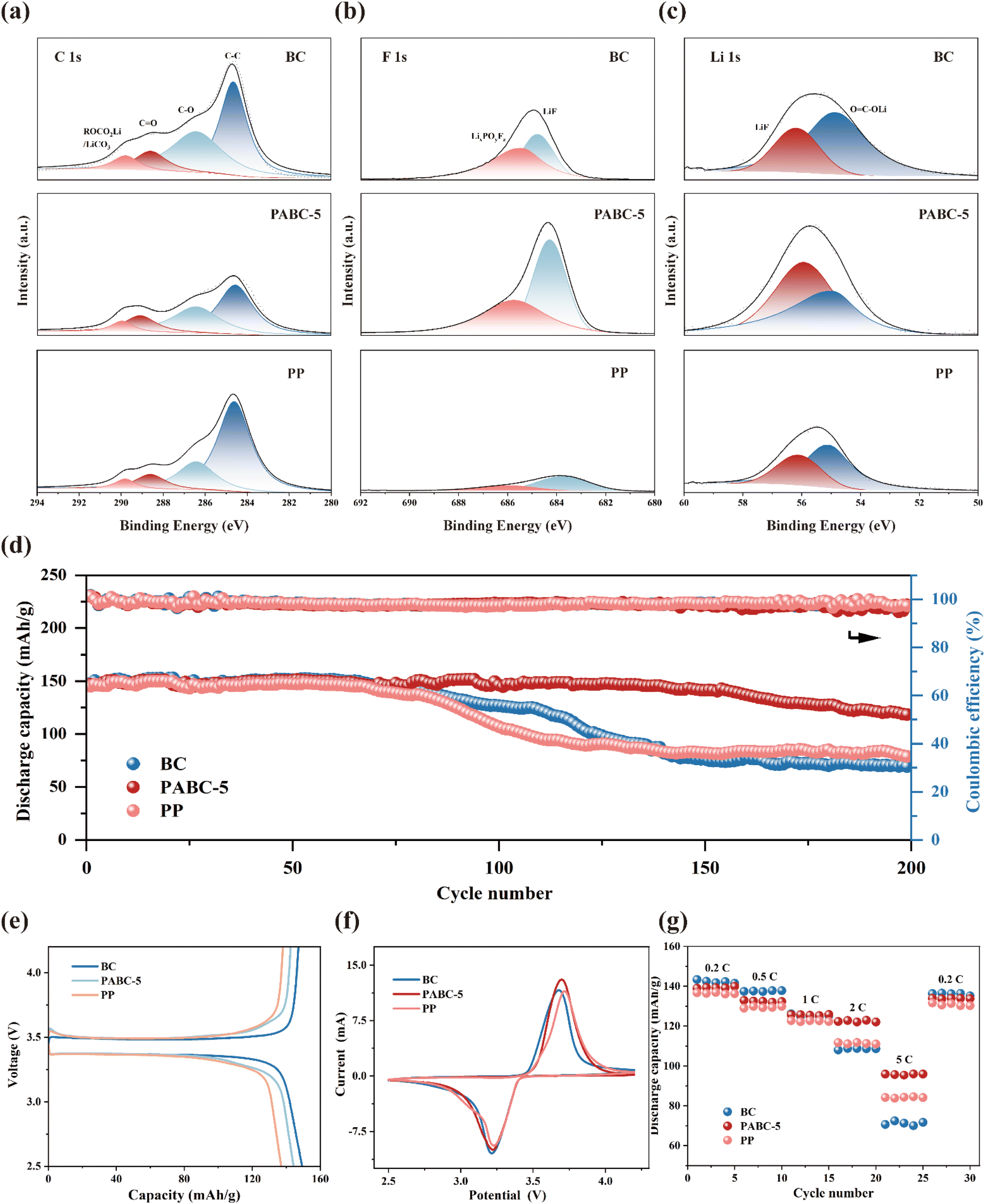 | ||
| Fig. 5 (a) XPS spectrum of C 1s; (b) XPS spectrum of F 1s; (c) XPS spectrum of Li 1s; (d) cycling performance of separators; (e) first charge and discharge capacity of separators; (f) CV curves of separators; (g) rate performance of separators. | ||
Furthermore, we analyzed the crystal orientation of deposited lithium after cycling by XRD. As shown in Fig. S18,† the peaks at 36° and 52° are the characteristic peaks of Li (110) and Li (200), respectively.61 Compared to the Li (200) plane, the Li (110) plane has a lower migration energy barrier for Li atoms, which facilitated rapid Li atom transport on the Li surface, resulting in a lower deposition overpotential, avoiding tip accumulation of Li atoms, and promoting planar deposition.62 The peak at 36° was considerably higher than the peak at 52°, demonstrating that PABC-5 regulated lithium deposition primarily along the Li (110) texture, which facilitates uniform, dense, and 2D planar Li deposition with better cycling stability.
Therefore, through the dual transport mechanism of the synergistic effect of mesopores and polymer-coordination transport in PABC-5, Li+ achieved uniform deposition and stripping on the lithium metal anode surface, forming a stable and high-quality SEI layer, thereby reducing side reactions between the solvent and the electrode.
To further explore the cycling stability and rate performance of the separators, LiFePO4/separators/Li cells were assembled and tested. As shown in Fig. 5(d), attributed to its stable SEI layer, the cell assembled with PABC-5 maintained a capacity retention rate of 79.9% after 200 cycles, significantly higher than those of PP (54.3%) and BC (47.17%). The initial charge/discharge curves in Fig. 5(e) revealed that BC, due to its excellent electrolyte wettability, exhibited the highest capacity of 149.3 mA h g−1. However, the uneven deposition/stripping of Li+ led to the formation of an unstable SEI layer, which was easily pierced by growing dendrites. This caused continuous side reactions between the electrolyte and lithium metal, resulting in substantial capacity loss. In contrast, PABC-5 promoted uniform Li+ deposition through its dual transport mechanism, forming an SEI layer predominantly composed of LiF, which effectively inhibited the continuous decomposition of the solvent, thereby demonstrating excellent cycling stability.
The cyclic voltammetry (CV) curves are shown in Fig. 5(f). The BC separator had a smaller redox peak separation, indicating stronger ion conductivity due to sufficient electrolyte wetting. However, compared with BC and PP, PABC-5 exhibited higher peak currents, suggesting a superior electrolyte/electrode interface.63 Moreover, as seen from the rate performance in Fig. 5(g), PABC-5 retained 68.3% of its capacity even at a high discharge rate of 5C, significantly higher than the capacity retention of PP (61.8%) and BC (51.1%). Additionally, when the rate returned to 0.2C, PABC-5 still maintained a capacity retention rate of 95.7%. These results confirmed that PABC, by regulating Li+ deposition behavior, promoted the formation of a stable SEI layer on the lithium metal anode surface, significantly enhancing various electrochemical performances.
4. Conclusions
The porous BC separator was infused with a polymer electrolyte monomer solution, forming an in situ dual-network structure through UV polymerization. This polymer network significantly enhanced the wet strength of the BC separator by 1200%, while also introducing a polymer-coordination transport mechanism alongside the original Li+ pore transport, thereby improving Li+ transport. PABC-5 exhibited excellent thermal stability and electrolyte wettability, with its dual Li+ transport mechanism regulating Li+ deposition behavior on the lithium metal anode surface via ether, ester and carboxyl groups. This facilitated efficient Li+ deposition and stripping kinetics. The assembled lithium symmetric battery demonstrated stable Li+ deposition and stripping for over 700 hours under conditions of 0.5 mA cm−2 and 1 mA h cm−2. The high-quality SEI layer, primarily composed of LiF formed by PABC-5, effectively minimized side reactions between the electrolyte and electrode, resulting in superior electrochemical performance. PABC-5 achieved a capacity retention rate of 79.9% after 200 charge–discharge cycles at 0.5C, and a retention rate of 68.3% at a 5C discharge rate, significantly outperforming commercial PP separators.Data availability
The data supporting this article have been included as part of the ESI† and all are available from the corresponding author (Yang Wang) upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare in this paper.Acknowledgements
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2022A1515111147, 2023A1515110431, 2024A1515011654) and National Natural Science Foundation of China (22308054).References
- H. Jia, C. Zeng, H. S. Lim, A. Simmons, Y. Zhang, M. H. Weber, M. H. Engelhard, P. Gao, C. Niu, Z. Xu, J. G. Zhang and W. Xu, Adv. Mater., 2023, e2311312, DOI:10.1002/adma.202311312.
- S. S. Zhang, J. Power Sources, 2007, 164, 351–364 CrossRef CAS.
- B. Yuan, K. Wen, D. Chen, Y. Liu, Y. Dong, C. Feng, Y. Han, J. Han, Y. Zhang, C. Xia, A. Sun and W. He, Adv. Funct. Mater., 2021, 31, 2101420 CrossRef CAS.
- X. Zhang, Q. Sun, C. Zhen, Y. Niu, Y. Han, G. Zeng, D. Chen, C. Feng, N. Chen, W. Lv and W. He, Energy Storage Mater., 2021, 37, 628–647 CrossRef.
- L. Peng, X. Kong, H. Li, X. Wang, C. Shi, T. Hu, Y. Liu, P. Zhang and J. Zhao, Adv. Funct. Mater., 2020, 31, 2008537 CrossRef.
- H. Du, Y. Wang, Y. Kang, Y. Zhao, Y. Tian, X. Wang, Y. Tan, Z. Liang, J. Wozny, T. Li, D. Ren, L. Wang, X. He, P. Xiao, E. Mao, N. Tavajohi, F. Kang and B. Li, Adv. Mater., 2024, 36, e2401482 CrossRef PubMed.
- D. Lv, J. Chai, P. Wang, L. Zhu, C. Liu, S. Nie, B. Li and G. Cui, Carbohydr. Polym., 2021, 251, 116975 CrossRef CAS PubMed.
- J. Sheng, S. Tong, Z. He and R. Yang, Cellulose, 2017, 24, 4103–4122 CrossRef CAS.
- H. Chen, Z. Wang, Y. Feng, S. Cai, H. Gao, Z. Wei and Y. Zhao, Chem. Eng. J., 2023, 471, 144593 CrossRef CAS.
- X. Du, Z. Zhang, W. Liu and Y. Deng, Nano Energy, 2017, 35, 299–320 CrossRef CAS.
- B. Huang, P. Lai, H. Hua, H. Ma, R. Li, X. Shen, P. Zhang, Y. Zhang and J. Zhao, J. Electroanal. Chem., 2022, 926, 116937 CrossRef CAS.
- C. Jin, J. Nai, O. Sheng, H. Yuan, W. Zhang, X. Tao and X. W. Lou, Energy Environ. Sci., 2021, 14, 1326–1379 RSC.
- C. H. Jo, N. Voronina, Y. K. Sun and S. T. Myung, Adv. Mater., 2021, 33, e2006019 CrossRef PubMed.
- C. Gilbert, T. C. Tang, W. Ott, B. A. Dorr, W. M. Shaw, G. L. Sun, T. K. Lu and T. Ellis, Nat. Mater., 2021, 20, 691–700 CrossRef CAS PubMed.
- J. Wang, J. Tavakoli and Y. Tang, Carbohydr. Polym., 2019, 219, 63–76 CrossRef CAS PubMed.
- C. Cheng, R. Yang, Y. Wang, D. Fu, J. Sheng and X. Guo, Carbohydr. Polym., 2023, 304, 120489 CrossRef CAS PubMed.
- T. W. Zhang, J. L. Chen, T. Tian, B. Shen, Y. D. Peng, Y. H. Song, B. Jiang, L. L. Lu, H. B. Yao and S. H. Yu, Adv. Funct. Mater., 2019, 29, 1902023 CrossRef.
- J. Hu, Y. Liu, M. Zhang, J. He and P. Ni, Electrochim. Acta, 2020, 334, 135585 CrossRef CAS.
- C. Huang, H. Ji, Y. Yang, B. Guo, L. Luo, Z. Meng, L. Fan and J. Xu, Carbohydr. Polym., 2020, 230, 115570 CrossRef CAS PubMed.
- X. Yang, S. K. Biswas, J. Han, S. Tanpichai, M. C. Li, C. Chen, S. Zhu, A. K. Das and H. Yano, Adv. Mater., 2021, 33, e2002264 CrossRef PubMed.
- H. C. Yu, X. P. Hao, C. W. Zhang, S. Y. Zheng, M. Du, S. Liang, Z. L. Wu and Q. Zheng, Small, 2021, 17, e2103836 CrossRef PubMed.
- S. Yang, Y. Zhang, Y. Zhang, J. Deng, N. Chen, S. Xie, Y. Ma and Z. Wang, Adv. Funct. Mater., 2023, 33, 2304280 CrossRef CAS.
- M. Rodin, J. Li and D. Kuckling, Chem. Soc. Rev., 2021, 50, 8147–8177 RSC.
- X. Huang, J. Li, J. Luo, Q. Gao, A. Mao and J. Li, Mater. Today Commun., 2021, 29, 102757 CrossRef CAS.
- Z. Wang, F. Yang, X. Liu, X. Han, X. Li, C. Huyan, D. Liu and F. Chen, Adv. Mater., 2024, e2313177, DOI:10.1002/adma.202313177.
- T. B. Schon, A. J. Tilley, C. R. Bridges, M. B. Miltenburg and D. S. Seferos, Adv. Funct. Mater., 2016, 26, 6896–6903 CrossRef CAS.
- J. Gou, W. Liu and A. Tang, Appl. Surf. Sci., 2021, 568, 150963 CrossRef CAS.
- X. Zheng, J. Wu, X. Wang and Z. Yang, Chem. Eng. J., 2022, 446, 137194 CrossRef CAS.
- X. Zeng, L. Dong, J. Fu, L. Chen, J. Zhou, P. Zong, G. Liu and L. Shi, Chem. Eng. J., 2022, 428, 131100 CrossRef CAS.
- F. Fu, L. Li, L. Liu, J. Cai, Y. Zhang, J. Zhou and L. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 2597–2606 CrossRef CAS PubMed.
- Y. Wang, X. Liu, J. Sheng, H. Zhu and R. Yang, ACS Sustain. Chem. Eng., 2021, 9, 14756–14765 CrossRef CAS.
- H. Lee, M. Yanilmaz, O. Toprakci, K. Fu and X. Zhang, Energy Environ. Sci., 2014, 7, 3857–3886 RSC.
- H. Zeng, K. Yu, J. Li, M. Yuan, J. Wang, Q. Wang, A. Lai, Y. Jiang, X. Yan, G. Zhang, H. Xu, J. Wang, W. Huang, C. Wang, Y. Deng and S. S. Chi, ACS Nano, 2024, 18, 1969–1981 CrossRef CAS PubMed.
- , M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Gao, G. Wu, W. Fang, Z. Qin, T. Zhang, J. Yan, Y. Zhong, N. Zhang and G. Chen, Angew. Chem., Int. Ed., 2024, 63, e202403668 CrossRef CAS PubMed.
- Y. Wang, X. Li, Y. Qin, D. Zhang, Z. Song and S. Ding, Nano Energy, 2021, 90, 106490 CrossRef CAS.
- H. Wang, J. Peng, Y. Yao and Z. Liang, Mater. Today Commun., 2024, 40, 109358 CrossRef CAS.
- H. Zheng, Y. Sun, C. Zhu, J. Guo, C. Zhao, Y. Liao and Q. Guan, Chem. Eng. J., 2013, 234, 318–326 CrossRef CAS.
- Z. Chen, X. Zhao, Y. Yu and J. Zhou, Macromol. Mater. Eng., 2024, 309, 2300454 CrossRef CAS.
- S. Zhang, B. Demir, X. Ren, S. D. Worley, R. M. Broughton and T. S. Huang, J. Appl. Polym. Sci., 2018, 136, 47426 CrossRef.
- F. Yu, H. Zhang, L. Zhao, Z. Sun, Y. Li, Y. Mo and Y. Chen, Carbohydr. Polym., 2020, 246, 116622 CrossRef CAS PubMed.
- J. L. Yang, X. X. Zhao, W. Zhang, K. Ren, X. X. Luo, J. M. Cao, S. H. Zheng, W. L. Li and X. L. Wu, Angew. Chem., Int. Ed., 2023, 62, e202300258 CrossRef CAS PubMed.
- L. Zhao, J. Fu, Z. Du, X. Jia, Y. Qu, F. Yu, J. Du and Y. Chen, J. Membr. Sci., 2020, 593, 117428 CrossRef CAS.
- M. F. Lagadec, R. Zahn and V. Wood, Nat. Energy, 2018, 4, 16–25 CrossRef.
- S. Gao, Z. Li, N. Liu, G. Liu, H. Yang and P. F. Cao, Adv. Funct. Mater., 2022, 32, 2202013 CrossRef CAS.
- W. Wang, Z. Li, H. Huang, W. Li and J. Wang, Chem. Eng. J., 2022, 445, 136568 CrossRef CAS.
- J. Zhang, S. Li, X. Wang, S. Mao, J. Guo, Z. Shen, J. Mao, Q. Wu, K. Shen, H. Cheng, Y. Tan and Y. Lu, Adv. Energy Mater., 2023, 14, 2302587 CrossRef.
- Z. Fang, Y. Luo, H. Liu, Z. Hong, H. Wu, F. Zhao, P. Liu, Q. Li, S. Fan, W. Duan and J. Wang, Adv. Sci., 2021, 8, e2100736 CrossRef PubMed.
- Y. Wu, Q. Hu, H. Liang, A. Wang, H. Xu, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2302587 Search PubMed.
- X. Li, M. Li, Y. Liu, Y. Jie, W. Li, Y. Chen, F. Huang, Y. Zhang, T. M. Sohail, S. Wang, X. Zhu, T. Cheng, M. D. Gu, S. Jiao and R. Cao, J. Am. Chem. Soc., 2024, 146, 17023–17031 CrossRef CAS PubMed.
- Z. Wen, W. Fang, F. Wang, H. Kang, S. Zhao, S. Guo and G. Chen, Angew Chem., Int. Ed., 2024, e202314876, DOI:10.1002/anie.202314876.
- Y. Li, S. Lin, D. Wang, T. Gao, J. Song, P. Zhou, Z. Xu, Z. Yang, N. Xiao and S. Guo, Adv. Mater., 2020, 32, e1906722 CrossRef PubMed.
- X. Wang, Z. Chen, K. Jiang, M. Chen and S. Passerini, Adv. Energy Mater., 2024, 14, 2302587 CrossRef.
- Y. Yin, H. Liu, F. Shen, J. Zuo, H. Guo, B. Xiao and X. Han, Electrochim. Acta, 2022, 421, 140520 CrossRef CAS.
- B. Zhou, I. Stoševski, A. Bonakdarpour and D. P. Wilkinson, ACS Appl. Energy Mater., 2023, 6, 6890–6895 CrossRef CAS.
- N. Schulz, R. Hausbrand, C. Wittich, L. Dimesso and W. Jaegermann, J. Electrochem. Soc., 2018, 165, A833–A846 CrossRef CAS.
- X. Q. Zhang, T. Li, B. Q. Li, R. Zhang, P. Shi, C. Yan, J. Q. Huang and Q. Zhang, Angew Chem. Int. Ed. Engl., 2020, 59, 3252–3257 CrossRef CAS PubMed.
- W. Cao, J. Lu, K. Zhou, G. Sun, J. Zheng, Z. Geng and H. Li, Nano Energy, 2022, 95, 106983 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- H. Chen, Z. Cao, J. Gu, Y. Cui, Y. Zhang, Z. Zhao, Z. Cheng, Q. Zhao, B. Li and S. Yang, Adv. Energy Mater., 2021, 11, 2003746 CrossRef CAS.
- Z. Sun, Y. Wang, S. Shen, X. Li, X. Hu, M. Hu, Y. Su, S. Ding and C. Xiao, Angew Chem. Int. Ed. Engl., 2023, 62, e202309622 CrossRef CAS PubMed.
- J. Tan, L. Ma, P. Yi, Y. Wang, Z. Li, Z. Fang, X. Li, S. He, X. Wang, M. Ye and J. Shen, Adv. Mater., 2024, 36, e2403570 CrossRef PubMed.
- L. Zuo, Q. Ma, P. Xiao, Q. Guo, W. Xie, D. Lu, X. Yun, C. Zheng and Y. Chen, Adv. Mater., 2024, 36, e2311529 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06151a |
| This journal is © The Royal Society of Chemistry 2025 |