
Atomic cation and anion co-vacancy defects boosted the oxide path mechanism of the oxygen evolution reaction on NiFeAl-layered double hydroxide†
Zhaoyan
Li
a,
Duo
Wang
b,
Hongguang
Kang
 b,
Zhongning
Shi
b,
Xianwei
Hu
b,
Hongbin
Sun
a and
Junli
Xu
*a
b,
Zhongning
Shi
b,
Xianwei
Hu
b,
Hongbin
Sun
a and
Junli
Xu
*a
aCollege of Science, Northeastern University, Shenyang, Liaoning 110819, China. E-mail: jlxu@mail.neu.edu.cn
bKey Laboratory for Ecological Metallurgy of Multimetallic Mineral (Ministry of Education), Northeastern University, Shenyang, Liaoning 110819, China
First published on 28th November 2024
Abstract
The oxide path mechanism (OPM) of the oxygen evolution reaction (OER) can overcome the scaling relation limit in the adsorbate evolution mechanism (AEM) and avoid forming oxygen vacancies in the lattice oxygen mechanism (LOM), which enables the catalyst to have both good OER activity and stability. However, there are currently few reports on the OPM for LDH catalysts. In this work, uniformly distributed atomic Al–O–Fe co-vacancies (VAl–O–Fe) on NiFeAl-LDH nanosheets are constructed by chemical etching and a successive electroreduction method. The obtained material only requires 223 mV to reach 500 mA cm−2 for the OER. The in situ electrochemical-Raman and chemical probe demonstrate the oxygen evolution reaction follows the OPM pathway. The theoretical calculation results show that the VAl–O–Fe can shorten the adjacent interatomic distance of bimetallic sites in Ni6Fe2Al-LDH, and thus triggers the O–O coupling on the bimetallic sites. Our study provides a novel strategy to trigger the OPM of the OER by introducing uniform co-vacancy defects on the catalysts.
Introduction
Water electrolysis in an alkaline electrolyte for hydrogen production has developed quickly in recent years because of its simplicity, abundant raw materials and zero carbon emission.1–4 However, the oxygen evolution reaction (OER), which involves a complex 4-electron transfer process, still limits its energy conversion efficiency.5–11 NiFe layered double hydroxides (NiFe-LDHs) are considered as the most promising OER electrocatalysts attributed to their unique layered structure, and flexible and adjustable chemical composition.12–15 Nevertheless, their electrocatalytic activities cannot meet the application demands yet due to their limited active sites, inherently poor conductivity and weak intrinsic activity.16–18Three distinctive O–O coupling mechanisms, including the adsorbate evolution mechanism (AEM), lattice oxygen mechanism (LOM), and oxide path mechanism (OPM), have been proposed for the OER. It has been widely acknowledged that the release of O2 by the LOM and OPM needs lower energy than the AEM since they can overcome the inherent scaling relationship of the binding energies between oxygen intermediates in the AEM: ΔGOOH = ΔGOH + 3.2 ± 0.2 eV.19–21 Nowadays, most reported works are focused on the boosting of LOM to lower the overpotential of the OER. Many strategies such as single atom loading,22 heteroatom doping23 and defect engineering18 have been proved to be effective to activate the lattice oxygen in LDHs and release O2 by the LOM. However, the participation of lattice oxygen will induce the formation of oxygen vacancies in the electrocatalysts, and the resulting unsaturated metal sites are easily dissolved in the electrolyte causing the stability problem of the electrocatalyst.24,25
Compared to LOM, OPM is more difficult to trigger since it requires dual-metal sites with appropriate interatomic distances. However, the OPM can void the generation of oxygen defects, which makes it possible to obtain high electroactivity with high stability performance. For instance, Lin et al.26 replaced some Mn atoms by Ru in MnO2 using the cation exchange method. The interatomic Ru–Ru distance in the prepared Ru/MnO2 material is shortened to 2.9 Å from the 3.1 Å of RuO2, which can boost the OPM as the *O radicals formed by the deprotonation of adsorbed *OH at adjacent Ru sites can directly couple to release O2. The prepared Ru/MnO2 material exhibits a low overpotential of 161 mV at 10 mA cm−2 and outstanding stability with little degradation after 200 h.
Defect engineering including cationic and anionic vacancy defects has been confirmed to adjust electronic interactions between surrounding atoms.27,28 Compared with the widely reported LDH catalysts with single anionic or cationic defects, the simultaneous presence of the two types of defects significantly enhances the electronic interaction between the metal and oxygen atoms around the defects, which may enhance the M–O bond and shorten the interatomic metal–metal (M–M) distance, which may promote the OPM for the OER. Herein, we construct well distributed atomic-scale Al–O–Fe co-vacancies on the NiFeAl-LDH (CR-Ni6Fe2Alx-LDH) via the integration of chemical etching and electrochemical reduction methods. In situ electrochemical-Raman spectroscopy and chemical probe prove the formation of the O–O bond at binuclear metal sites and release O2 through OPM. The theoretical calculation results indicate that the interatomic M–M distance is only 2.80 Å in CR-Ni6Fe2Al-LDH, which can promote O–O radical coupling. The CR-Ni6Fe2Al-LDH with optimized VAl–O–Fe content exhibits a remarkable OER catalytic activity and excellent stability in 1 mol L−1 KOH. Our work sheds new light on the surface defect engineering of catalysts.
Results and discussion
As depicted in Fig. 1a, Ni6Fe2Alx-LDH was synthesized by the hydrothermal method, and then it was etched to produce cation vacancies on Ni6Fe2Alx-LDH, which was named C-Ni6Fe2Alx-LDH. Subsequently, the C-Ni6Fe2Alx-LDH was reduced via an electrochemical method to further construct oxygen vacancies, and the obtained material was denoted as CR-Ni6Fe2Alx-LDH. Scanning electron microscopy (SEM) images indicate that the prepared Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH and CR-Ni6Fe2Alx-LDH all display 3D spherical structures, which are assembled by 2D nanosheets (Fig. S1a–f†). Fig. S1g–l† show that Ni, Fe, Al, and O elements uniformly distribute over the surface of CR-Ni6Fe2Al-LDH nanosheets. Using inductively coupled plasma-optical emission spectrometry (ICP-OES), the Ni, Fe, and Al mass percent of CR-Ni6Fe2Al-LDH is determined to be 43.71%, 7.62% and 1.06%, respectively (Fig. S2†). The atomic force microscopy (Fig. 1b and S3†) images reveal that the average nanosheet thicknesses are 3.6 nm, 8.1 nm and 7.1 nm for CR-Ni6Fe2Al-LDH, Ni6Fe2Al-LDH and C-Ni6Fe2Al-LDH, respectively. The thinner nanosheets in CR-Ni6Fe2Al-LDH are attributed to the strip of H2, which is generated in the electroreduction process.29 The transition electron microscopy (TEM) image of CR-Ni6Fe2Al-LDH confirms its nanosheet morphology (Fig. 1c). Moreover, the high-resolution TEM (HRTEM) diagram reveals that concave defects with an average diameter of about 2.86 nm disperse uniformly on the nanosheet (Fig. 1d and e). Fig. 1g displays the three-line profiles in Fig. 1f with disparate orientation, and it reveals that the concave defect is induced by atomic vacancy clusters as the slightly weaker atomic intensity is ascribed to the vacancy clusters.30Fig. 1h and k show there are abundant amorphous/crystalline interfaces (blue dashed lines) and dislocation defects in the nanosheets. The labeled lattice fringes (0.197 and 0.230 nm) match the (018) and (015) crystal facets of NiFe-LDH in Fig. 1i and j. Two electron spin resonance (ESR) signals at g = 2.16 and 2.09, which correspond to the cation and oxygen vacancies, respectively, are detected in C-Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH (Fig. 1l).31,32 The stronger ESR signal intensity of cation and oxygen vacancies in CR-Ni6Fe2Al-LDH indicates that more cation and oxygen vacancies are produced in the electroreduction process attributed to the high reduction ability of active H*.29,33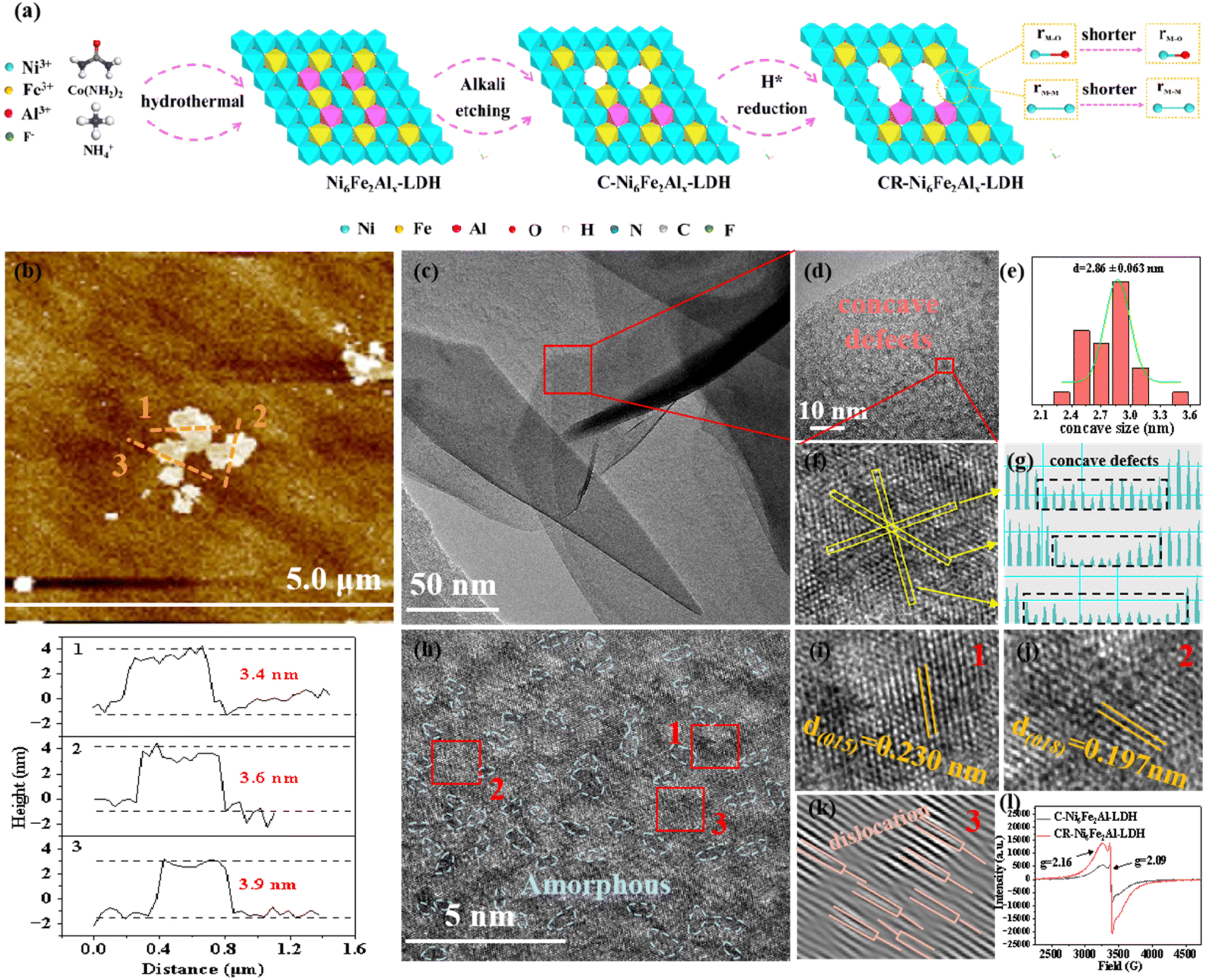 | ||
| Fig. 1 Synthesis scheme and morphological characterization of the prepared catalysts. (a) Synthesis schematic of CR-Ni6Fe2Alx-LDH. (b and c) AFM image (b) and TEM image (c) of CR-Ni6Fe2Al-LDH ultrathin nanosheets. (d and h) The HRTEM images of the selected area in (c). (e–k) VAl–O–Fe size distribution plot (e), VAl–O–Fe defects and the corresponding atomic intensity profile of the line section (f and g), lattice fringes (i and j), and dislocation (k) of CR-Ni6Fe2Al-LDH. (l) ESR spectra of C-Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH. | ||
The chemical composition and crystal structure of the catalyst were characterized by X-ray diffraction (XRD). As shown in Fig. 2a and S4b,† Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH, and CR-Ni6Fe2Alx-LDH all show two sets of diffractions, where the peaks at 11.4°, 22.9°, 34.4°, 38.9°, 45.9°, 59.9°, 61.2° and 44.5°, 50.7°, 51.8°, 76.4° are attributed to NiFe-LDH (JCPDS: PDF#40-0215) and nickel foam (Fig. S5†), respectively. Moreover, the CR-Ni6Fe2Al-LDH sample presents the highest intensity of diffraction peaks, which indicates it has the highest crystallinity or thickness.34–36 As the full-widths at half-maximum of the diffraction peaks at 22.9° for the samples are close to each other (Fig. S6†), we deduce that the increased intensity of diffraction peaks is caused by the increased detected thickness of the sample. The chemical states of Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH and CR-Ni6Fe2Alx-LDH were further measured by X-ray photoelectron spectroscopy (XPS). The survey spectra of all catalysts confirm the coexistence of Ni, Fe, Al and O elements (Fig. S4a†). In the Ni 2p XPS spectrum of Ni6Fe2Al-LDH (Fig. 2b), the peaks at 856.2 eV and 873.9 eV with the satellite peaks at 861.9 eV and 880.0 eV belong to Ni2+.37–39 For the Fe 2p XPS spectrum of Ni6Fe2Al-LDH (Fig. 2c), a pair of Fe3+ characteristic peaks can be fitted at 713.0 eV and 725.4 eV with the corresponding satellite peaks at 718.0 eV and 734.5 eV.40,41 Besides, a low-intensity peak is discovered on the low binding energy side (707.7 eV, named as Pre peak), which suggests the formation of Fe ions with a lower oxidation state due to the oxygen defect of the synthesized catalyst itself.42 For C-Ni6Fe2Al-LDH, the binding energy of Ni2+ has a positive shift (0.1 eV), while that of Fe3+ exhibits a negative shift (0.1 eV), indicating that the introduction of cation vacancies adjusts the electronic structure. After electrochemical reduction treatment, the Ni 2p peaks do not have an obvious shift, while the Fe 2p peaks move towards lower binding energy, which proves that the oxygen atoms near the Fe sites are more easily reduced forming oxygen vacancies in the electrochemical reduction process. Moreover, the Ni 2p and Fe 2p XPS spectra of CR-Ni6Fe2Alx-LDH (x = 0.5, 1 and 2) with different Al contents (Fig. S4c and d†) suggest that CR-Ni6Fe2Al-LDH has both the highest Ni2+ binding energy and the lowest Fe3+ binding energy, which is attributed to the highest content of VAl–O–Fe in the CR-Ni6Fe2Al-LDH sample. The O 1s XPS spectra of all catalysts (Fig. 2d and S4e†) are all fitted with three characteristic peaks matching M–O, M–OH and adsorbed water molecules at 530.8 eV, 531.6 eV and 532.3 eV,43,44 and the calculated peak area ratio of M–OH/M–O is presented in Table S1.† The maximum M–OH/M–O ratio in CR-Ni6Fe2Al-LDH further confirms it has the highest oxygen vacancy content.45,46
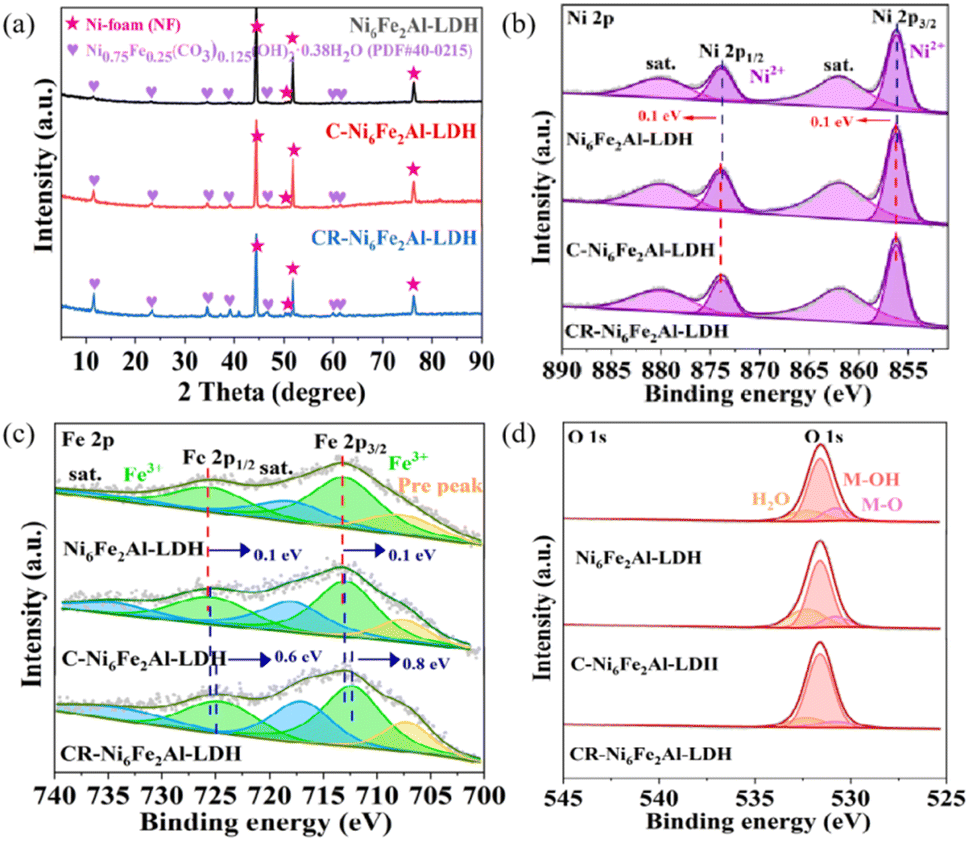 | ||
| Fig. 2 Composition and structural characterization. (a–d) XRD patterns (a), Ni 2p (b), Fe 2p (c) and O 1s spectra (d) of Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH. | ||
The electrocatalytic performances of the prepared samples were studied in 1 mol L−1 KOH. Fig. S8† shows the linear sweep voltammetry (LSV) curves measured with forward scanning. There is an obvious oxidation peak at ∼1.43 V, which is related to the oxidation of the catalyst.47 To obtain the overpotential at low reaction current density of the OER, LSV curves were measured with reverse scanning (Fig. 3a), and the obtained corresponding overpotentials at different current densities are shown in Fig. 3b. The CR-Ni6Fe2Al-LDH sample exhibits the highest electrocatalytic activity with overpotentials of 189, 209, 223 and 227 mV at 10, 100, 500 and 700 mA cm−2, respectively, which is also better than that of most reported LDHs as shown in Table S2.† Moreover, the CR-Ni6Fe2Al-LDH sample exhibits the lowest Tafel slope and highest TOF values, which indicates it has the fastest kinetics performance and highest intrinsic catalytic activity as shown in Fig. 3c and d. Besides, the derived Tafel slopes at low current density are relatively small, being even smaller than the reported low values as shown in Table S3.† The faster kinetics may be attributed to the rapid mass transport in the uniformly distributed network structure of vacancy clusters (VAl–O–Fe), which is available for the penetration of electrolyte to the active sites. Moreover, the value of the Tafel slope can be used to determine the rate-determining step (RDS). According to the formula of Tafel slope = 2.303RT/(0.5 + n)F, where n is the number of electrons transferred before the RDS, R is the universal gas constant, T is the absolute temperature and F is the Faraday constant;48 the calculated Tafel slope is 118, 40, 24, and 17 mV dec−1 when the first, second, third and fourth electron transfer is the RDS, respectively. The derived Tafel slopes on all the prepared samples are all below 20 mV dec−1, which indicates the fourth electron transfer reaction is the RDS.
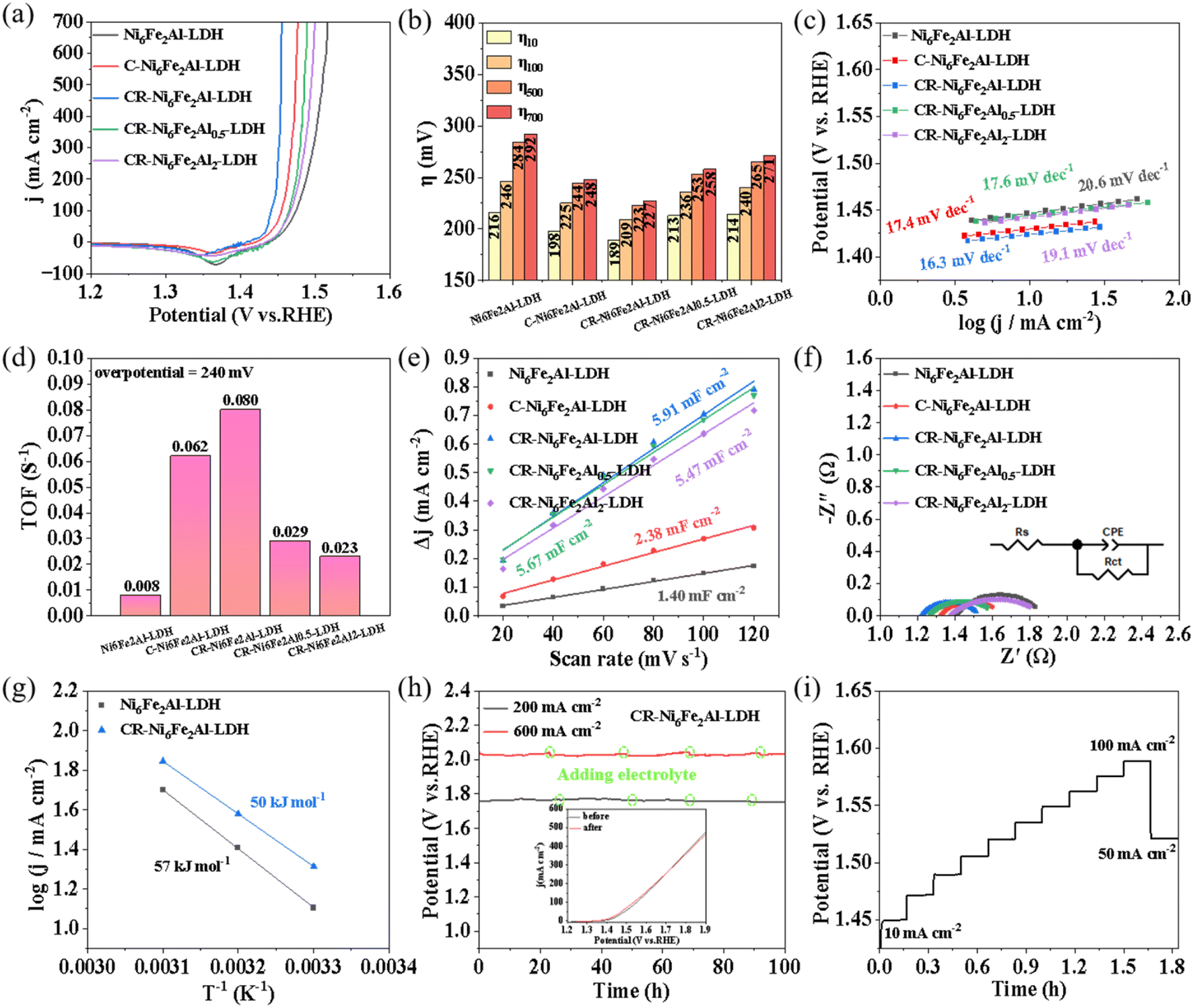 | ||
| Fig. 3 Electrocatalytic OER performance. (a–f) LSV curves (a), overpotentials at 10, 100, 500 and 700 mA cm−2 (b), Tafel plots (c), turnover frequency (TOF) at 1.47 V vs. RHE (d), Cdl values (e), EIS spectra (f) of Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH and CR-Ni6Fe2Alx-LDH (x = 0.5, 1 and 2), the inset in (f) is the equivalent circuit. (g) Arrhenius plots of the kinetic currents at 0.52 V vs. Hg/HgO for Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH. (h and i) Chronopotentiometric response at 200 mA cm−2, 600 mA cm−2 and at multicurrent densities for CR-Ni6Fe2Al-LDH, the inset in (h) shows the LSV curves before and after the stability test. | ||
The double-layer capacitance (Cdl), charge transfer resistance (Rct), carrier density (NA), and electrochemical activation energy (Ea) were measured to explore the sources of catalytic activity of CR-Ni6Fe2Al-LDH. The Cdl was calculated based on cyclic voltammetry (CV) curves at different sweep rates in the non-Faraday region (Fig. S9†). The CR-Ni6Fe2Al-LDH catalyst delivers a Cdl of 5.91 mF cm−2, which is around 4.2 times that of Ni6Fe2Al-LDH (1.40 mF cm−2), mirroring that it has more catalytic active centers (Fig. 3e). The smallest Rct (0.32 Ω) and Rs (1.22 Ω) of CR-Ni6Fe2Al-LDH indicates the most rapid charge transfer ability and highest electrical conductivity, respectively (Fig. 3f and Table S4†). The Mott–Schottky curves were tested to evaluate the conductivity of catalysts by calculating the carrier concentration according to eqn (3) in the Experimental section. As presented in Fig. S10,† the carrier concentration of CR-Ni6Fe2Al-LDH (1.30 × 1022) is significantly higher than that of Ni6Fe2Al-LDH (2.39 × 1021) and C-Ni6Fe2Al-LDH (2.55 × 1021), which further proves its higher conductivity. The activation energy (Ea) of the OER was obtained according to the Arrhenius equation (eqn (4)) in the Experimental section and their OER polarization curves at different temperatures (Fig. S11†). The CR-Ni6Fe2Al-LDH sample exhibits a lower Ea (50 kJ mol−1) than Ni6Fe2Al-LDH (57 kJ mol−1), revealing its lower catalytic activation energy barrier (Fig. 3g).
Stability is another important index to estimate the practical application potential of catalysts. Fig. 3h shows that the potential fluctuation in a 100 h test at 200 and 600 mA cm−2, and the inserted figure in Fig. 3h is the LSV curves before and after the durability test. The results indicate that the CR-Ni6Fe2Al-LDH exhibits good stability and has potential application in industry. The multistep chronopotentiometry measurement (Fig. 3i) shows that as the current density increases from 10 to 100 mA cm−2 at a rate of 10 mA cm−2, the electrode potential increases rapidly and reaches a stable state quickly. Moreover, as the current density decreases from 100 to 50 mA cm−2, the potential drops from 1.59 to 1.52 V vs. RHE, which is comparable to the initial potential, demonstrating its exceptional mass transportation ability and mechanical robustness during the OER process.
The optimal CR-Ni6Fe2Al-LDH catalyst was characterized after the stability test as shown in Fig. S12–S14.† The Ni 2p spectrum (Fig. S12b†) displays a new peak of Ni3+, and the binding energy of Fe3+ in the Fe 2p spectrum (Fig. S12c†) has a slight positive shift, manifesting that metal ions are partially oxidized during the continuous OER process. The slightly decreased ratio of M–OH/M–O (4.73) in the O 1s diagram (Fig. S12d†) and the two ESR signals corresponding to cation and oxygen vacancies (Fig. S13†) both demonstrate a well-maintained uniform VAl–O–Fe defect structure. The ICP-OES results (Fig. S14†) show that the content of Ni, Fe and Al in KOH solution after a 100 h durability test at 200 mA cm−2 is 0, 0.49 and 2.34 mg L−1, respectively, which indicates that the CR-Ni6Fe2Al-LDH has good durability. The SEM images (Fig. S12e†) and XRD pattern (Fig. S12f†) suggest that the morphology and phase structure have no obvious change after the stability test, which indicates its good structural stability.
As the overpotential of CR-Ni6Fe2Al-LDH is only 209 mV at 100 mA cm−2, the OER should mainly follow the LOM or OPM. The *O22− species was detected to recognize the OER mechanism since it is only produced by the LOM pathway. LSV was performed in 1 mol L−1 TMAOH since the tetramethylammonium cation (TMA+) can strongly bind to *O22− species and thus hinder OER kinetics.49 As shown in Fig. 4a–c, the OER activity of Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH is not inhibited by TMAOH, while it is significantly reduced for C-Ni6Fe2Al-LDH. Combined with the overpotential values, it can be deduced that the OER on Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH mainly obeys the AEM, LOM and OPM, respectively.
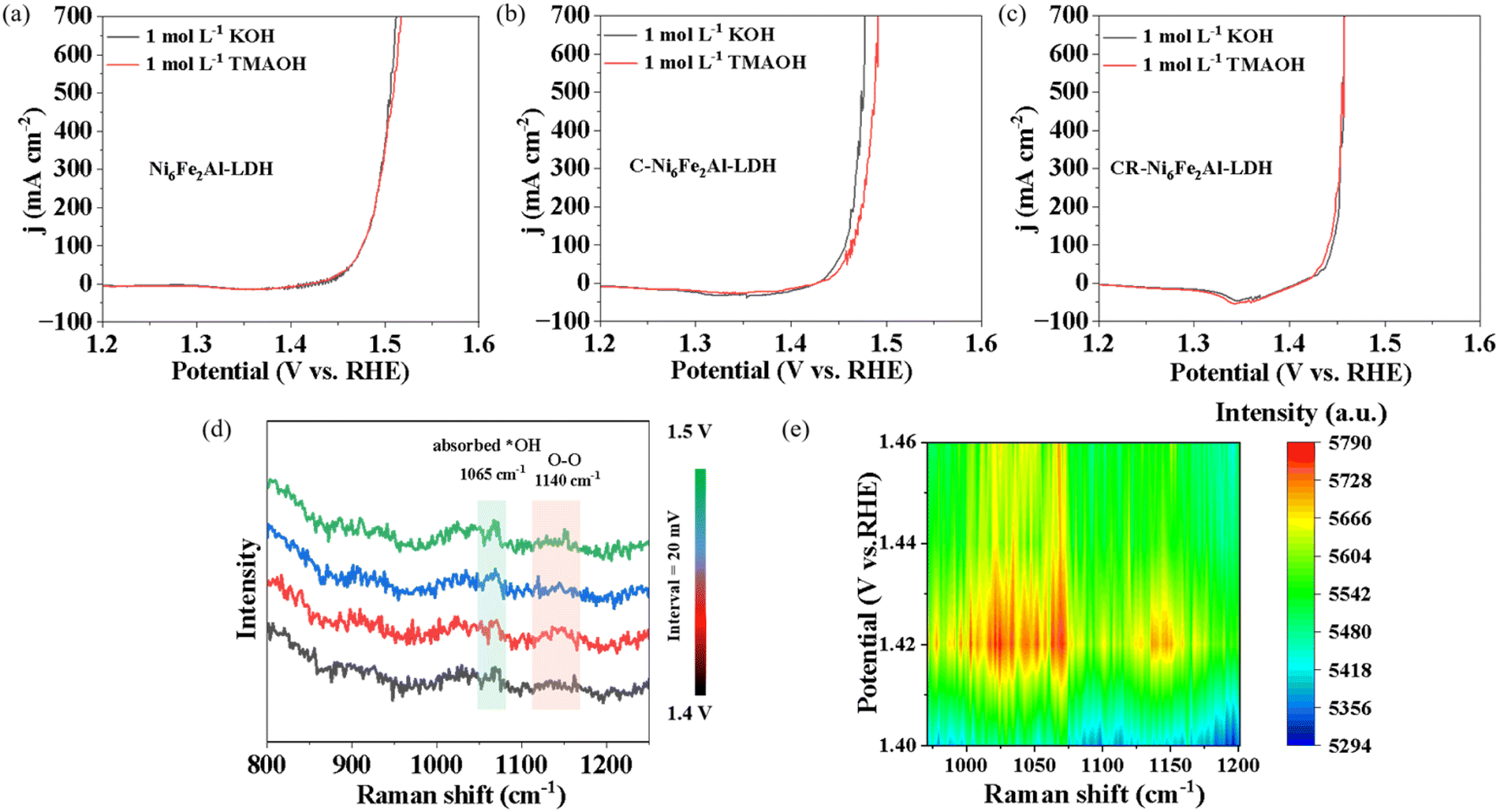 | ||
| Fig. 4 Mechanism analysis. (a–c) LSV curves in 1 mol L−1 KOH and 1 mol L−1 TMAOH of Ni6Fe2Al-LDH (a), C-Ni6Fe2Al-LDH (b) and CR-Ni6Fe2Al-LDH (c). (d and e) In situ electrochemical-Raman spectra and corresponding 2D Raman intensity map plot of CR-Ni6Fe2Al-LDH. | ||
In situ electrochemical-Raman tests were further performed on Ni6Fe2Al-LDH, C-Ni6Fe2Alx-LDH and CR-Ni6Fe2Alx-LDH to detect OER intermediates. As the theoretical potential of the OER is 1.23 V (vs. RHE), and the current density is about zero from 1.23 V to the open circuit potential (Fig. S15 and S8†), the Raman spectra were collected at 50 mV intervals over a potential range of 1.23–1.60 V as shown in Fig. S16 and S17.† Raman peaks associated with Ni2+–O(H) (454 cm−1), Ni2+–O (533 cm−1) and adsorbed *OH (1065 cm−1) are detected on both catalysts.50 Moreover, with increasing potential, the peaks of Ni2+–O(H) and Ni2+–O vibration continuously shift towards higher wave numbers of 465 cm−1 and 544 cm−1, which indicates that the catalysts are oxidized and transformed to γ-NiOOH during the OER.51 It is reported that the Raman signal of the three OER mechanisms, AEM, LOM and OPM, are located at 906 cm−1,52 1089 cm−1 (ref. 20) and 1140 cm−1,53 respectively. There are no Raman signal peaks of the AEM and LOM in all samples, which may be caused by the high thickness of the quartz cell wall. However, compared to other samples, a Raman signal at 1140 cm−1 is obviously observed on CR-Ni6Fe2Alx-LDH (Fig. 4d, e, S16 and S17†), which indicates the OER obeys the OPM. Moreover, the chemical probe results suggest that the OER on Ni6Fe2Al-LDH and C-Ni6Fe2Al-LDH follows the AEM and LOM, respectively (Fig. 4a and b). Raman spectra were further measured from 1.4 V to 1.5 V at intervals of 20 mV on CR-Ni6Fe2Al-LDH to amplify this phenomenon as shown in Fig. 4d. The corresponding 2D Raman intensity map (Fig. 4e) clearly shows that there is an obvious peak at 1140 cm−1, which verifies the existence of M–O–O–M species and reveals that the OER follows the OPM pathway.
Density functional theory calculations were used to further reveal the OER mechanism. The calculation results show that the Ni–O and Ni–Ni distances of CR-Ni6Fe2Al-LDH (1.84 Å, 2.80 Å) are lower than those of C-Ni6Fe2Al-LDH (2.05 Å, 3.10 Å) and pristine Ni6Fe2Al-LDH (2.07 Å, 3.12 Å) as exhibited in Fig. 5a–c. The density of states (DOS) (Fig. 5d) and band centers (Table S5†) were calculated to investigate the effect of defects on the electronic structure. Compared with that of Ni6Fe2Al-LDH, the Ni 3d band center (εNi 3d) of CR-Ni6Fe2Al-LDH moves towards the Fermi level, which enhances the adsorption between the metal center and the intermediate OH*. Moreover, a shift towards the Fermi level of the O 2p band center (εO 2p) is also observed, resulting in the significant increase of metal–oxygen orbital hybridization, which induces the shortened M–O bond and the interatomic M–M distance, thus triggering the OPM of the OER (Fig. 5e).
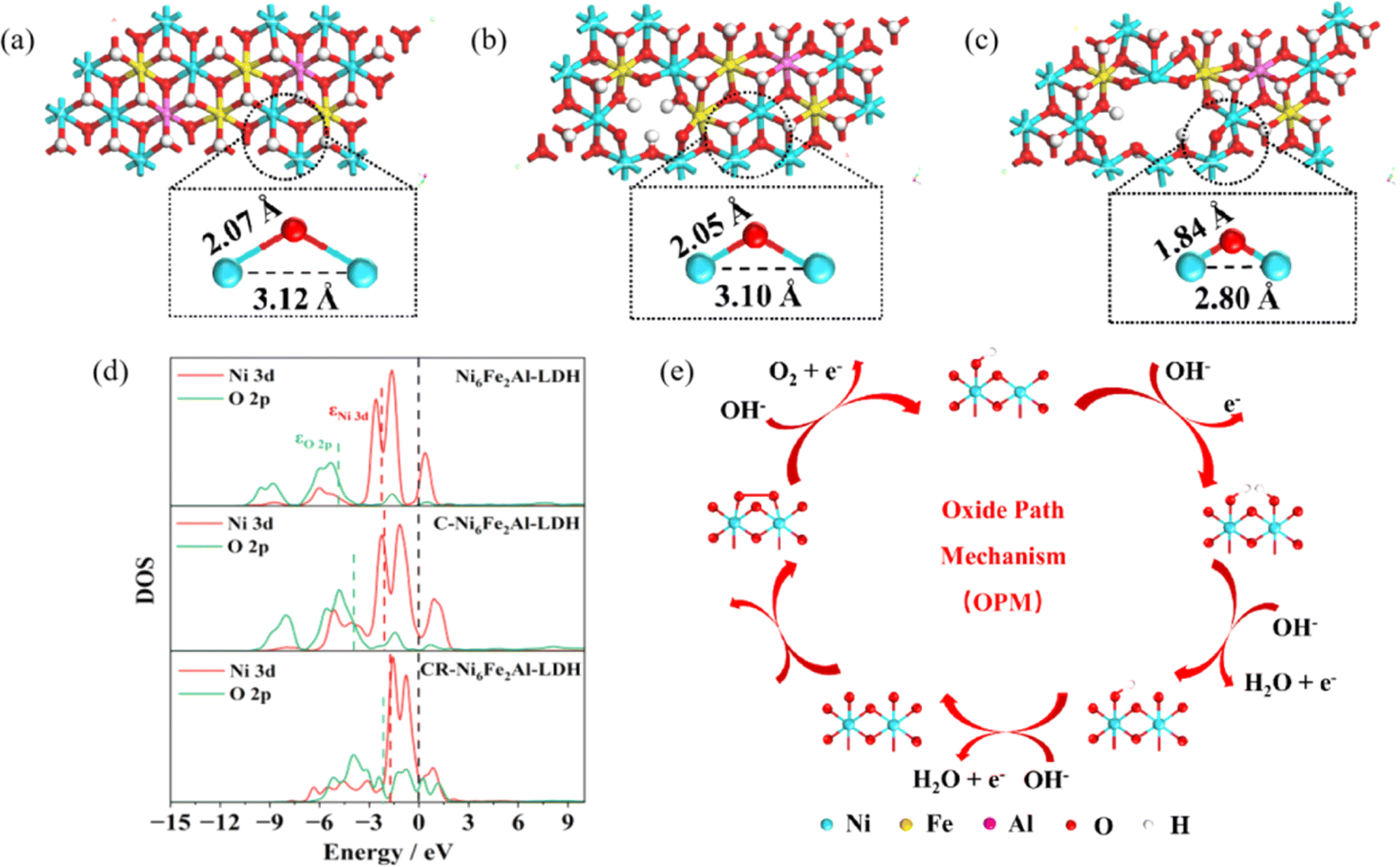 | ||
| Fig. 5 DFT calculations. (a–c) Calculated local ball-and-stick models of Ni6Fe2Al-LDH (a), C-Ni6Fe2Al-LDH (b) and CR-Ni6Fe2Al-LDH (c) demonstrating the metal–oxygen bond lengths and metal–metal distances. (d) DOS band centers of the Ni 3d and O 2p orbitals in Ni6Fe2Al-LDH, C-Ni6Fe2Al-LDH and CR-Ni6Fe2Al-LDH. (e) The oxide path mechanism (OPM) illustration. | ||
Conclusions
In conclusion, we have successfully prepared NiFeAl-LDH with homogeneous atomic-scale Al–O–Fe vacancy defects on the surface via successive hydrothermal–etching–electrochemical reduction processes. The in situ electrochemical-Raman and chemical probe together with theoretical calculation prove that the synergistic effect of anionic and cationic defects in Ni6Fe2Al-LDH adjusts the surface electronic structure of the material and optimizes the adsorption between the metal center and the oxygen-containing intermediate as well as obtaining the appropriate interatomic M–M distance to promote the direct O–O radical coupling at bimetallic sites. Such a unique reaction path makes the catalyst achieve a significant enhancement of OER activity with good stability. Our results reveal that constructing atomic scale cationic and anionic defects in LDHs is an effective strategy to regulate the interatomic M–M distance and trigger the OPM of the OER.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. L. prepared and tested the catalysts and analyzed the results. D. W. performed the DFT calculations. H. K. and X. H. collected the in situ electrochemical-Raman spectra. H. S. and Z. S. supervised the experiments and contributed to the data analysis. Z. L. and J. X. wrote, reviewed and edited the manuscript before submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (52074084).References
- J. Zhu, J. Qian, X. Peng, B. Xia and D. Gao, Nano-Micro Lett., 2023, 15, 30 CrossRef CAS PubMed.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- H. Cui, H.-X. Liao, Z.-L. Wang, J.-P. Xie, P.-F. Tan, D.-W. Chu and P. Jun, Rare Met., 2022, 41, 2606–2615 CrossRef CAS.
- X. Liang, J. Xiao, W. Weng and W. Xiao, Angew. Chem., Int. Ed., 2021, 60, 2120–2124 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, L. Liu, W. Qin, S. Liu, J. Tu, Y. Liu, Y. Qin, J. Liu, H. Wu, D. Zhang, A. Chu, B. Jia, X. Qu, M. Qin and J. Xue, J. Am. Chem. Soc., 2023, 145, 20261–20272 CrossRef CAS PubMed.
- K. Wan, J. Luo, W. Liu, T. Zhang, J. Arbiol, X. Zhang, P. Subramanian, Z. Fu and J. Fransaer, Chin. J. Catal., 2023, 54, 290–297 CrossRef CAS.
- W. Xu, G. M. Haarberg, F. Seland, S. Sunde, A. P. Ratvik, S. Holmin, J. Gustavsson, Å. Afvander, E. Zimmerman and T. Åkre, Corros. Sci., 2019, 150, 76–90 CrossRef CAS.
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, I. Furó, M. S. G. Ahlquist and L. Sun, Nat. Catal., 2022, 5, 414–429 CrossRef CAS.
- B. Guo, Y. Ding, H. Huo, X. Wen, X. Ren, P. Xu and S. Li, Nano-Micro Lett., 2023, 15, 57 CrossRef CAS PubMed.
- C. Chang, Y. Zhou, J. Wang, H. Zhang, P. Yan, H. B. Wu and X.-Y. Yu, Small, 2023, 19, 2206768 CrossRef PubMed.
- S. Du, Z. Ren, X. Wang, J. Wu, H. Meng and H. Fu, ACS Nano, 2022, 16, 7794–7803 CrossRef CAS PubMed.
- Y. Wei, Z. Han, T. Liu, X. Ding and Y. Gao, Chem. Commun., 2023, 59, 11572–11575 RSC.
- Q. Zhou, Y. Chen, G. Zhao, Y. Lin, Z. Yu, X. Xu, X. Wang, H. K. Liu, W. Sun and S. X. Dou, ACS Catal., 2018, 8, 5382–5390 CrossRef CAS.
- L. Xu, P. Yang, R. Ye, X. Wu and Y. Tao, J. Mater. Chem. A, 2024, 12, 9714–9722 RSC.
- Y. Du, D. Liu, T. Li, Y. Yan, Y. Liang, S. Yan and Z. Zou, Appl. Catal., B, 2022, 306, 121146 CrossRef CAS.
- M. Yan, Y. Zeng, Y. Mou, J. Hu, W. Tang, C. Jiang, Q. Li and Y. Zhao, J. Alloys Compd., 2023, 931, 167465 CrossRef CAS.
- Y.-H. Wang, L. Li, J. Shi, M.-Y. Xie, J. Nie, G.-F. Huang, B. Li, W. Hu, A. Pan and W.-Q. Huang, Adv. Sci., 2023, 10, 2303321 CrossRef CAS PubMed.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- F. Wang, P. Zou, Y. Zhang, W. Pan, Y. Li, L. Liang, C. Chen, H. Liu and S. Zheng, Nat. Commun., 2023, 14, 6019 CrossRef CAS.
- H. Wang and S. Lu, Energy Environ. Mater., 2023, 6, e12558 CrossRef CAS.
- Y. Wu, Y. Zhao, P. Zhai, C. Wang, J. Gao, L. Sun and J. Hou, Adv. Mater., 2022, 34, 2202523 CrossRef CAS PubMed.
- Q. Su, P. Wang, Q. Liu, R. Sheng, W. Cheng, J. Ding, Y. Lei and Y. Huang, Appl. Catal., B, 2024, 351, 123994 CrossRef CAS.
- H. Wu, Q. Huang, Y. Shi, J. Chang and S. Lu, Nano Res., 2023, 16, 9142–9157 CrossRef.
- C. Wang, P. Zhai, M. Xia, Y. Wu, B. Zhang, Z. Li, L. Ran, J. Gao, X. Zhang, Z. Fan, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2021, 60, 27126–27134 CrossRef CAS PubMed.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- D. Yan, C. Xia, W. Zhang, Q. Hu, C. He, B. Y. Xia and S. Wang, Adv. Energy Mater., 2022, 12, 2202317 CrossRef CAS.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- Z. Li, D. Wang, J. Xu, H. Sun and Z. Shi, Chem. Eng. J., 2024, 482, 148858 CrossRef CAS.
- Y. Liu, H. T. D. Bui, A. R. Jadhav, T. Yang, S. Saqlain, Y. Luo, J. Yu, A. Kumar, H. Wang, L. Wang, V. Q. Bui, M. G. Kim, Y. D. Kim and H. Lee, Adv. Funct. Mater., 2021, 31, 2010718 CrossRef CAS.
- Y. Zhai, X. Ren, Y. Sun, D. Li, B. Wang and S. Liu, Appl. Catal., B, 2023, 323, 122091 CrossRef CAS.
- L. Zheng, W. Ye, Y. Zhao, Z. Lv, X. Shi, Q. Wu, X. Fang and H. Zheng, Small, 2023, 19, 2205092 CrossRef CAS PubMed.
- X. Chen, M. Yu, Z. Yan, W. Guo, G. Fan, Y. Ni, J. Liu, W. Zhang, W. Xie, F. Cheng and J. Chen, CCS Chem., 2020, 3, 675–685 CrossRef.
- T. E. Abioye, H. Zuhailawati, S. Aizad and A. S. Anasyida, Trans. Nonferrous Met. Soc. China, 2019, 29, 667–679 CrossRef CAS.
- Q. Fan, L. Wang, Y. Fu and Z. Wang, J. Hazard. Mater., 2023, 442, 130060 CrossRef CAS PubMed.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- X. Yin, Y.-N. Hua and Z. Gao, Mater. Today Chem., 2023, 32, 101632 CrossRef CAS.
- W. Wang, Y. Liu, J. Li, J. Luo, L. Fu and S. Chen, J. Mater. Chem. A, 2018, 6, 14299–14306 RSC.
- S. A. Chala, M.-C. Tsai, W.-N. Su, K. B. Ibrahim, B. Thirumalraj, T.-S. Chan, J.-F. Lee, H. Dai and B.-J. Hwang, ACS Nano, 2020, 14, 1770–1782 CrossRef CAS PubMed.
- Y. Wang, M. Qiao, Y. Li and S. Wang, Small, 2018, 14, 1800136 CrossRef.
- D. Wang, L. Liu, Y. Liu, W. Luo, Y. Xie, T. Xiao, Y. Wang, Z. Song, H. Zhang and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 16479–16490 CrossRef CAS.
- H. Zhao, Y. Wang, Y. Wang, T. Cao and G. Zhao, Appl. Catal., B, 2012, 125, 120–127 CrossRef CAS.
- T. X. Nguyen, C.-C. Tsai, V. T. Nguyen, Y.-J. Huang, Y.-H. Su, S.-Y. Li, R.-K. Xie, Y.-J. Lin, J.-F. Lee and J.-M. Ting, Chem. Eng. J., 2023, 466, 143352 CrossRef CAS.
- Y. Gan, Y. Ye, X. Dai, X. Yin, Y. Cao, R. Cai and X. Zhang, J. Colloid Interface Sci., 2023, 629, 896–907 CrossRef CAS.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- L. Wu, M. Ning, X. Xing, Y. Wang, F. Zhang, G. Gao, S. Song, D. Wang, C. Yuan, L. Yu, J. Bao, S. Chen and Z. Ren, Adv. Mater., 2023, 35, 2306097 CrossRef CAS PubMed.
- J. Xie, X. Zhang, H. Zhang, J. Zhang, S. Li, R. Wang, B. Pan and Y. Xie, Adv. Mater., 2017, 29, 1604765 CrossRef PubMed.
- S. Sun, Y. Sun, Y. Zhou, J. Shen, D. Mandler, R. Neumann and Z. J. Xu, Chem. Mater., 2019, 31, 8106–8111 CrossRef CAS.
- Z.-F. Huang, S. Xi, J. Song, S. Dou, X. Li, Y. Du, C. Diao, Z. J. Xu and X. Wang, Nat. Commun., 2021, 12, 3992 CrossRef CAS PubMed.
- Z. Li, Y. Yao, S. Sun, J. Liang, S. Hong, H. Zhang, C. Yang, X. Zhang, Z. Cai, J. Li, Y. Ren, Y. Luo, D. Zheng, X. He, Q. Liu, Y. Wang, F. Gong, X. Sun and B. Tang, Angew. Chem., Int. Ed., 2024, 63, e202316522 CrossRef CAS PubMed.
- H. Liu, W. Shen, H. Jin, J. Xu, P. Xi, J. Dong, Y. Zheng and S.-Z. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202311674 CrossRef CAS PubMed.
- C.-F. Li, L.-J. Xie, J.-W. Zhao, L.-F. Gu, J.-Q. Wu and G.-R. Li, Appl. Catal., B, 2022, 306, 121097 CrossRef CAS.
- C. Hu, Y. Hu, C. Fan, L. Yang, Y. Zhang, H. Li and W. Xie, Angew. Chem., Int. Ed., 2021, 60, 19774–19778 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05839a |
| This journal is © The Royal Society of Chemistry 2025 |