
High-voltage symmetric supercapacitors developed by engineering DyFeO3 electrodes and aqueous electrolytes†
Mohasin
Tarek
 ,
Ferdous
Yasmeen
and
M. A.
Basith
*
,
Ferdous
Yasmeen
and
M. A.
Basith
*
Department of Physics, Nanotechnology Research Laboratory, Bangladesh University of Engineering and Technology, Dhaka-1000, Bangladesh. E-mail: mabasith@phy.buet.ac.bd
First published on 22nd November 2024
Abstract
Aqueous supercapacitors (SCs) are often constrained by low operational voltage and energy density due to the low decomposition voltage of water. In this work, we address these limitations by fabricating symmetric SCs using nanoporous dysprosium orthoferrite (DyFeO3) electrodes in dilute, neutral aqueous electrolytes. The nanoporous architecture of the DyFeO3 electrode material, with an average pore size of 3.41 nm, was confirmed using Brunauer–Emmett–Teller analysis and comprehensively characterized through XRD, FESEM, TEM, XPS, Raman spectroscopy, EPR, and zeta potential measurements. The fabricated SC, operating in a 0.5 M Na2SO4 aqueous electrolyte, exhibited a high working voltage of 2.5 V, delivering an energy density of 41.81 W h kg−1 at a power density of 1250 W kg−1, with 90% capacitance retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Furthermore, the addition of 20% acetonitrile (AN) to the 0.5 M Na2SO4 electrolyte extended the potential window to 3.1 V, increasing the energy density to 84.43 W h kg−1 at a power density of 1550 W kg−1. The fabricated symmetric SC demonstrated excellent long-term stability, retaining approximately 99% capacitance and Coulombic efficiency after a 600 hours float voltage test. These findings, for the first time, reveal the potential of nanoporous DyFeO3 as electrode material in a 0.5 M Na2SO4(aq.)/20%AN electrolyte for advancing symmetric SCs, featuring an unprecedented ultra-wide electrochemical stability window along with significantly enhanced energy and power densities.
000 cycles. Furthermore, the addition of 20% acetonitrile (AN) to the 0.5 M Na2SO4 electrolyte extended the potential window to 3.1 V, increasing the energy density to 84.43 W h kg−1 at a power density of 1550 W kg−1. The fabricated symmetric SC demonstrated excellent long-term stability, retaining approximately 99% capacitance and Coulombic efficiency after a 600 hours float voltage test. These findings, for the first time, reveal the potential of nanoporous DyFeO3 as electrode material in a 0.5 M Na2SO4(aq.)/20%AN electrolyte for advancing symmetric SCs, featuring an unprecedented ultra-wide electrochemical stability window along with significantly enhanced energy and power densities.
1 Introduction
Electrochemical supercapacitors (SCs) are emerging as promising alternatives for sustainable and reliable energy storage devices.1 Their advantages, including high power density, long cycle life, fast charge–discharge rates, wide operating temperature range, and safety, make them particularly compelling when compared to traditional batteries.2–4 However, the limited energy densities, typically around 1–10 W h kg−1, observed in commercially available SCs, significantly constrain their broader application.5,6 Consequently, there is a pressing need for extensive research into various parameters, particularly electrode materials and electrolytes, to enhance energy densities, as well as related factors like cost, safety, and lifetime, to unlock the full potential of SCs.1 In this context, maximizing energy density (E) can be achieved by effectively expanding the voltage window (V), and leveraging the square proportionality between V and E for a specific capacitance. Notably, the electrochemical stability window (ESW) of aqueous SCs is restricted by the narrow stability range of water, approximately 1.23 V.7,8 To achieve a significant expansion of ESW in SCs, careful selection of electrode materials and judicious choice of electrolytes are key factors.6,9To enhance the electrochemical energy storage capabilities of electrode materials, various strategies are employed, including doping,10 nanocomposite formation,11 molecular cross-linking,12 intricate structural design,13 and chemical modifications.14 While these strategies can improve electrochemical performance, they often introduce complexity, reduce reliability, and increase costs. Specifically, in designing asymmetric SCs, a common approach to enhance voltage windows involves using different, complex electrode materials for the cathode and anode.15 This approach, along with intentional imbalances in mass and charge between the electrodes, increases both the complexity and cost of fabrication and functionality.15 Therefore, adopting a unified, perovskite-like electrode material for both the cathode and anode—one capable of demonstrating a hybrid charge storage mechanism involving both electrochemical double-layer capacitance and pseudocapacitance—may offer significant advantages.16–18
Notably, perovskite oxides with the formula ABO3 (where A is a lanthanide or alkaline earth element and B is a transition metal) have garnered significant attention in electrode research due to their structural stability, compositional flexibility, and inherent oxygen vacancies.16,17 Among these materials, dysprosium orthoferrite (DyFeO3), which crystallizes in an orthorhombic perovskite structure,19 has been extensively studied for its potential in magnetic data storage, spintronics devices, and multiferroic systems.20–22 However, DyFeO3's unique electronic structure—characterized by the diverse oxidation states of its constituent elements, magnetic and dielectric properties, thermal stability, ion mobility, and porous morphology—positions it as a strong candidate for applications beyond spintronics, particularly in electrochemical energy storage systems.16,23 The variable oxidation states of Dy and Fe facilitate redox reactions, improving charge transfer in electrochemical systems.16 Additionally, the unpaired electrons in both Dy and Fe contribute to DyFeO3's magnetic properties. Specifically, Dy3+ ions, with their large magnetic moment due to the 4f-electron configuration, interact with the magnetic moments of Fe3+ ions, influencing the overall electronic structure and local environment. This magnetic interaction may enhance pseudocapacitive behavior, potentially increasing the material's overall capacitance.
Furthermore, DyFeO3's high dielectric constant improves its charge storage capacity, making it an attractive material for SCs. The elevated dielectric constant also enhances the electrolyte/electrode interface, facilitating better ion diffusion and improving energy storage efficiency. DyFeO3's thermal stability is another key advantage, ensuring structural integrity and performance in high-temperature environments, which is crucial for the long-term cycling stability of SCs. Additionally, the incorporation of Dy into the FeO3 matrix can create pathways for enhanced ion diffusion, which is essential for rapid charge–discharge cycles.
Notably, the inherent oxygen vacancies in DyFeO3 promote more efficient redox reactions at the electrode–electrolyte interface, enhancing surface reactivity and ion diffusion, which ultimately impacts the specific capacitance of the electrode material.16,24–26 The porous structure of DyFeO3, achieved through controlled fabrication, provides numerous electroactive sites, significantly boosting the charge storage capacity.16 While dysprosium (Dy), a rare earth metal, is more costly than commonly used materials like carbon or manganese due to its limited availability and complex extraction processes, it is still less expensive than elements such as platinum (Pt) or ruthenium (Ru). In contrast, iron is abundant and inexpensive, contributing minimally to the overall cost of DyFeO3. Despite the moderate cost of Dy, its exceptional properties—such as high energy density, thermal stability, and long cycle life—make DyFeO3 a highly suitable choice for high-performance SCs, where these advantages surpass cost considerations.
Apart from the electrode material, selecting a suitable electrolyte is crucial, as it significantly impacts the stability and overall effectiveness of SCs.9,27 Currently, non-aqueous electrolytes are widely used in advanced energy storage systems for portable electronics and automotive applications due to their wide electrochemical ESW and high energy density. However, challenges such as flammability, high economic costs, and environmental concerns limit their widespread adoption.27,28 In contrast, aqueous electrolytes offer a promising alternative, providing cost-effectiveness, high ionic conductivity, intrinsic safety, and environmental compatibility.7–9,11,29 Given these advantages, a 0.5 M Na2SO4 aqueous solution is used as the electrolyte in this investigation, chosen for its neutral pH, noncorrosive nature, small hydrated ion size, excellent conductivity, and cost-efficiency.27,30,31
Various approaches have been explored to expand the ESW of aqueous electrolytes, including adjusting the pH, increasing electrolyte concentration, modifying the potential of zero voltage of electrodes, introducing redox-active additives, balancing electrode masses, and passivating electrode surfaces.7,11,32 However, these strategies, whether applied individually or in combination, have achieved only modest success in expanding the ESW. For example, using a super-concentrated electrolyte with 21 m lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) produced a ‘water-in-salt’ (WIS) system, extending the ESW to 2.4 V.33 Another study utilized a 17 m sodium perchlorate aqueous electrolyte, achieving an ESW of 2.3 V with a Coulombic efficiency of 66% after 10000 cycles.7 Another study employed a 17 m sodium perchlorate aqueous electrolyte, achieving an ESW of 2.3 V with a Coulombic efficiency of 66% after 10000 cycles.8,34 Conversely, the addition of organic solvents such as acetonitrile (AN), ethanol, and ethylene glycol (EG) to a WIS electrolyte has shown promise in enhancing the ESW. Notably, incorporating organic solvents to increase the ESW—primarily by lowering salt concentration and decreasing the activity of free water molecules—proves to be a simple and cost-effective technique.8
Therefore, in addition to using a pure aqueous 0.5 M Na2SO4 electrolyte, we developed an aqueous-dominant, dilute salt-containing solid electrolyte interface (SEI) system. This involved incorporating 10% and 20% volumetric AN with water as the solvent for the 0.5 M Na2SO4 electrolyte. Subsequently, we fabricated coin cell SCs using DyFeO3 nanoparticles as the electrode material for both the cathode and anode, with electrolyte solutions of 0.5 M Na2SO4(aq.) and 0.5 M Na2SO4(aq.)/20%AN. Notably, the diverse oxidation states, porous structure, and inherent oxygen vacancies of the DyFeO3 electrode material, combined with the 0.5 M Na2SO4 aqueous electrolyte solution, collectively mitigate water splitting. This breakthrough extends the ESW to 2.5 V, marking, to the best of our knowledge, the first instance of such an achievement for a hybrid aqueous symmetric supercapacitor (ASSC). Furthermore, the electrolyte solution with a 20% AN additive, i.e., 0.5 M Na2SO4(aq.)/20%AN, remarkably extended the ESW to 3.1 V. As a result, the as-fabricated aqueous electrolyte-dominant hybrid symmetric SC demonstrates an impressive energy density of 84.43 W h kg−1 at a power density of 1550 W kg−1, along with excellent rate capability and cycle performance. These findings represent a significant improvement over the performance of previously reported aqueous electrolyte-dominated symmetric supercapacitors.
2 Experimental section
2.1 Synthesis of DyFeO3 nanoparticles
Nanoparticles of the DyFeO3 perovskite were synthesized using a sol–gel technique, as illustrated in Fig. S1 (ESI†).35 Initially, stoichiometric amounts of Dy(NO3)3·5H2O (Sigma-Aldrich, 99.80%) and Fe(NO3)3·9H2O (Sigma-Aldrich, 98.00%) were individually dissolved in 100 ml of deionized water and stirred for 15–20 minutes using a magnetic stirrer. Subsequently, the solutions were combined, and citric acid (C6H8O7) was introduced into the mixture. The citric acid served as a chelating agent, forming stable complexes with the metal ions. The solution's pH was adjusted to 7 by incorporating NH4OH. Subsequently, the chelating agent, ethylene glycol, was introduced to generate a polymeric metal cationic network, serving as the precursor for the gel. After four hours, the temperature was raised to 200 °C, leading to the complete combustion of the gel and the formation of the desired powder. The combustion process entails the decomposition of the organic components within the gel, liberating gases and leaving behind metal oxides. The resulting material was finely grounded using an agate mortar. To achieve the desired crystallization and porous structure, the material underwent calcination at 750 °C for 6 hours, with a heating rate of 5 °C per minute in a nitrogen environment. The controlled heating rate of 5 °C per minute minimizes thermal stress, facilitating controlled decomposition. Nitrogen gas flow, particularly during the initial stages of the calcination process, ensures an oxygen-free environment. This controlled atmosphere is crucial for preventing unwanted oxidation reactions and influencing the evolution of gases during decomposition. The reproducible formation of porosity in DyFeO3 was meticulously achieved through the judicious selection of solvent (water), systematic adjustment of gel precursor concentration (ethylene glycol), precise temperature control, and optimization of the calcination temperature in an N2 environment.2.2 Structural and morphological characterization
The crystal structure of the synthesized materials was determined using a Rigaku SmartLab X-ray diffractometer with copper K-Alpha radiation. Further insights were gained through a thorough analysis of the powder X-ray diffraction (XRD) patterns, employing the Rietveld refinement method with the Fullprof software program. The surface morphology was analyzed using both Field Emission Scanning Electron Microscopy (FESEM) imaging (GeminiSEM 360, Zeiss, Germany) and Transmission Electron Microscopy (TEM) imaging (Talos F200X, Thermo Fisher Scientific, USA). Elemental insights were obtained through energy-dispersive X-ray (EDX) spectra (Bruker, USA), meticulously revealing the elemental composition of the synthesized samples, and the nature of the surface charge was analyzed by zeta potential analyzer (zeta potential analyzer Horiba SZ-100V2). The presence of unpaired electrons in our synthesized sample was carried out by high-field, high-frequency electron paramagnetic resonance (EPR) measurements. Further, Fourier transform infrared (FTIR) spectroscopy (Shimadzu, IRSpirit, Japan), Raman spectroscopy (macroRam spectrometer, Horiba Scientific, Japan), Nuclear Magnetic Resonance (NMR) spectroscopy (Advance 800 MHz NMR spectrometer, Bruker, USA) analyses were perform using a Horiba Scientific, macroRam spectrometer to evaluate the molecular structure of various electrolyte solutions.2.3 Electrochemical characterizations
Cyclic voltammetry (CV), linear sweep voltammetry (LSV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) analyses were systematically performed on the DyFeO3 electrode material across diverse electrolyte solutions. These assessments were conducted using both three-electrode and symmetric two-electrode configurations, including our fabricated coin cell supercapacitor. The experiments were carried out utilizing an electrochemical workstation (Metrohm Autolab PGSTAT302N). The detailed procedure for preparing the working electrode slurry is outlined in the ESI,† and Fig. S2† schematically illustrates both the preparation process of the working electrode slurry and the electrochemical setup of the three-electrode system.2.4 Assembly of coin cell supercapacitors in symmetric configuration
ESI Fig. S3† depicts the configuration of the CR2032 coin cell and the corresponding fabrication steps. In the context of the coin cell supercapacitor, DyFeO3 nanoparticles served as the electrode material for both the anode and cathode. The electrolyte comprised of 0.5 M Na2SO4(aq.) and 0.5 M Na2SO4(aq.)/20%AN solutions, Whatman filter paper functioned as the separator, and disc-shaped graphite sheets served as current collectors for both the anode and cathode. In the coin cell assembly, a slurry of the active material was uniformly applied to two individual graphite sheets (diameter 1.65 cm) at a consistent loading of 10 mg cm−2. The treated graphite sheets, designated as the anode and cathode, were then dried in an oven at 80 °C for 12 hours. A circular Whatman filter paper, saturated for 24 hours with 0.5 M Na2SO4(aq.) and 0.5 M Na2SO4(aq.)/20%AN electrolyte solutions, was positioned between the modified graphite sheets to finalize the coin cell arrangement. Subsequently, the coin cell supercapacitor was sealed under a pressure of 800 kg using an automatic coin cell assembling machine (TMAX-160S).2.5 Electrochemical measurement
The specific capacitance value of our fabricated ASSC-1 and ASSC-2 is calculated using the following equation (detailed derivation is provided in the ESI†).36–38 | (1) |
The specific energy density (Esp, measured in W h kg−1), and specific power density (Psp, measured in W kg−1) for our fabricated ASSC-1 and ASSC-2 were computed based on the GCD data using eqn (2) and (3), respectively.36,39,40
 | (2) |
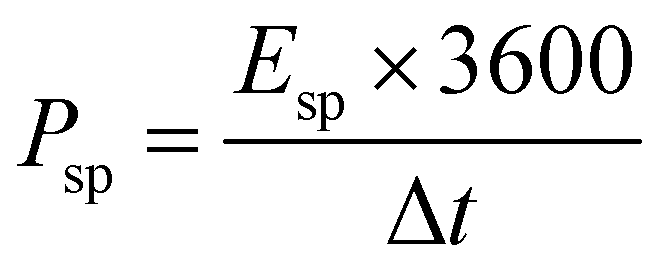 | (3) |
 | (4) |
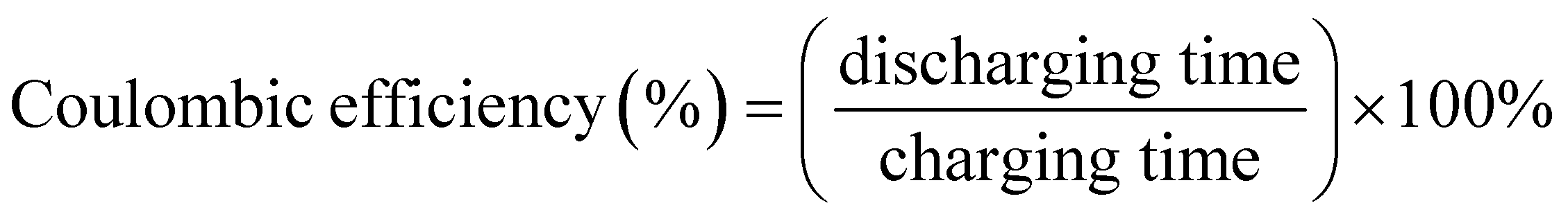 | (5) |
3 Results and discussion
The as-synthesized DyFeO3 crystallizes in an orthorhombic structure with the space group Pnma,4,41 as confirmed by the Rietveld refinement of the powder XRD pattern presented in Fig. 1(a). The calculated crystallinity (93%) confirms the formation of highly crystalline DyFeO3 particles. The successful formation of DyFeO3 nanoparticles with a porous structure is evident in the FESEM and TEM images, as depicted in Fig. 1(b) and (c), respectively. Fig. 1(d) shows a high-resolution transmission electron microscopy (HRTEM) image of porous DyFeO3 nanoparticles, revealing d-spacing values corresponding to various crystal planes. The measured lattice spacings are 0.280 nm and 0.385 nm, corresponding to the (020) and (110) planes, respectively, aligning with the results from XRD analysis. In Fig. 1(e), the pore sizes estimated from the electron microscopy images range from approximately 20 to 40 nm, indicating a high surface-area-to-volume ratio, which is crucial for achieving high specific capacitance in electrode materials.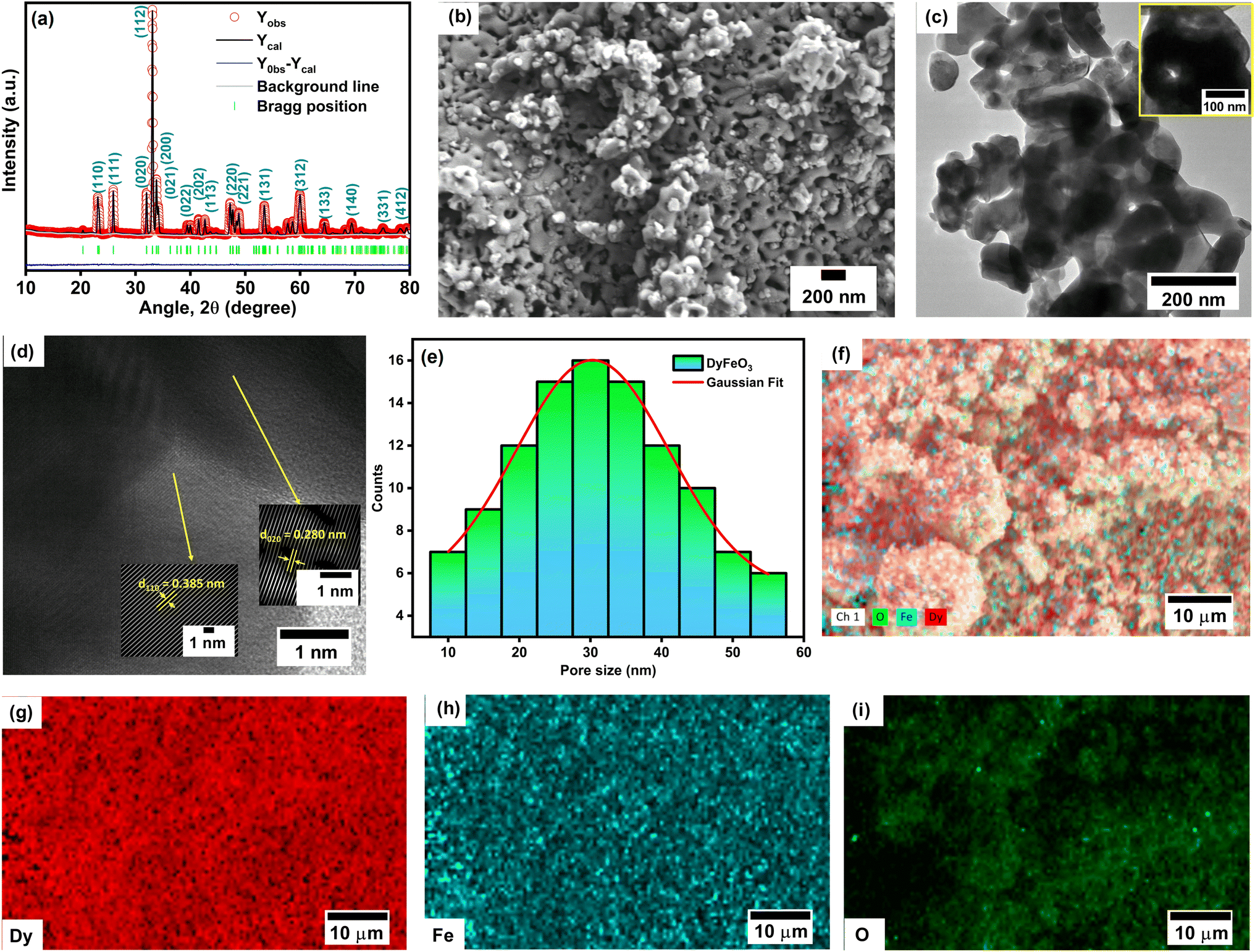 | ||
| Fig. 1 Structural and morphological characterizations of DyFeO3 nanoparticles. (a) Rietveld-refined powder XRD pattern showing the crystalline nature of DyFeO3 in the orthorhombic phase with the Pnma space group. (b) FESEM image revealing the porous structure of DyFeO3 nanoparticles. (c) TEM image confirming the porous structure observed in FESEM imaging, with an inset highlighting a pore within a nanoparticle. (d) HRTEM image displaying d-spacing values corresponding to various crystal planes of DyFeO3 nanoparticles. (e) Pore size distribution histogram indicating that the DyFeO3 sample contains nanoscale pores with an average size of 30 nm. (f)–(i) Elemental mapping images revealing the distribution of Dy, Fe, and O atoms in DyFeO3 nanoparticles. | ||
To accurately estimate the pore size of the as-synthesized DyFeO3, we also performed BET analysis using nitrogen gas adsorption measurements. The BET analysis revealed a pore size of 3.41 nm, which is significantly smaller than the size observed in FESEM images.42 This discrepancy stems from the different methods used: BET analysis measures smaller internal pores by assessing monolayer gas adsorption, while FESEM captures larger surface-accessible pores or voids between particles. Therefore, the smaller pore size from BET reflects internal porosity, whereas the larger pores observed in FESEM likely correspond to surface voids or interparticle gaps. Subsequently, EDX analysis, as illustrated in Fig. S4 (ESI†), was conducted to confirm the presence of each element in the synthesized material. The experimentally determined mass and atomic percentages of the constituent elements were found to closely match their theoretical values, as detailed in Table S1.† Following the EDX analysis, the elemental mapping of DyFeO3 in Fig. 1(f)–(i) reveals a uniform distribution of Dy, Fe, and O throughout the material, confirming its homogeneity.
Fig. 2(a) displays the Raman spectrum of porous DyFeO3 nanoparticles, highlighting characteristic peaks corresponding to Fe–O and Dy–O bonds, along with the F2g band associated with oxygen ion vibrations.43–46 These features offer valuable insights into the material's structural and electronic properties. Notably, a distinct band at 560 cm−1 signals the presence of oxygen vacancies46 within the DyFeO3 lattice. These vacancies, resulting from missing oxygen atoms, cause localized disruptions in the lattice, altering the vibrational modes detected by Raman spectroscopy. Beyond structural effects, these oxygen vacancies also enhance ion adsorption, contributing to the material's overall capacitance in SCs. The structural information is further supported by the Electron Paramagnetic Resonance (EPR) spectrum in Fig. 2(b), which confirms the presence of oxygen vacancies with a signal at a g value of 2.019 (inset of Fig. 2(b)).47,48 This signal is characteristic of unpaired electron spins associated with oxygen vacancies, where trapped electrons create localized magnetic moments detectable by EPR. Collectively, this evidence strengthens the conclusions drawn from the Raman spectrum.
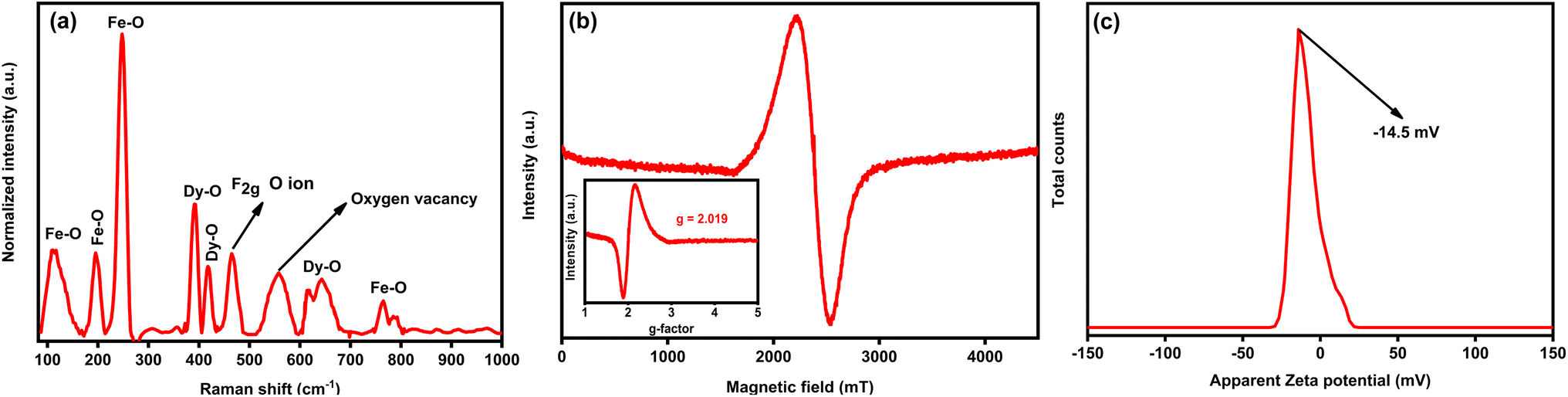 | ||
| Fig. 2 Raman, EPR, and zeta potential analyses of DyFeO3 nanoparticles. (a) Raman spectra of DyFeO3 nanoparticles displaying peaks corresponding to Fe–O and Dy–O bonds, with a band at 560 cm−1 indicative of oxygen vacancies. (b) EPR spectra confirm the presence of oxygen vacancies with a g-factor of 2.019 before cycling. (c) Zeta potential analysis reveals a surface charge of −14.5 mV, which facilitates Na+ ion adsorption and enhances the formation of an EDL, thereby improving the material's electrochemical performance. | ||
The zeta potential analysis of DyFeO3 nanoparticles, presented in Fig. 2(c), shows a value of approximately −14.5 mV. While this value is below the ±30 mV threshold typically associated with high colloidal stability, it still contributes to moderate stabilization under controlled conditions, such as specified electrolyte concentrations and external influences. The observed negative surface charge is instrumental in enhancing the electrochemical performance of this material when used as an electrode in supercapacitor applications. It facilitates the formation of a dense EDL at the electrode–electrolyte interface by attracting and adsorbing Na+ ions onto the electrode surface, which increases the EDL capacitance. This attraction of Na+ ions is vital for facilitating redox reactions at the electrode surface during charge–discharge cycles, as the enhanced ion adsorption increases the number of active sites, thereby improving the pseudocapacitive behavior of the material.
XPS was employed to analyze the chemical state of synthesized DyFeO3 nanoparticles, as illustrated in Fig. 3. The survey spectrum of the DyFeO3 nanoparticles depicted in Fig. 3(a) confirmed the presence of Dy, Fe, and O, consistent with the results from EDX analysis. High-resolution XPS spectra of DyFeO3 nanoparticles, presented in Fig. 3(b)–(f), revealed the coexistence of Dy4+, Dy3+, Fe2+, Fe3+, and O2− ions.49–52 These oxidation states might enhance the charge storage capacity of the nanoparticles through a pseudocapacitive mechanism involving faradaic redox reactions. The specific binding energies of all the XPS peaks for DyFeO3 nanoparticles are documented in Table S2 in the ESI.† Note that, in Fig. 3(f), the high-resolution XPS spectra of O-1s were deconvoluted, yielding three distinct peaks at binding energies of 532.26 eV, 530.53 eV and 529.47 eV. These peaks correspond to the adsorption of OH on the surface, oxygen vacancy and metal–oxygen bonds,52,53 with molar ratios of 4.22%, 59.35% and 36.43%, respectively. The oxygen vacancies likely result from the charge imbalance caused by the mixed oxidation states of rare earth ions (Dy3+ and Dy4+) and transition metal ions (Fe2+ and Fe3+) within the crystal lattice. The presence of oxygen vacancies in DyFeO3 is likely to enhance charge carrier generation and improve charge transfer, contributing to the material's potential for increased capacitance.
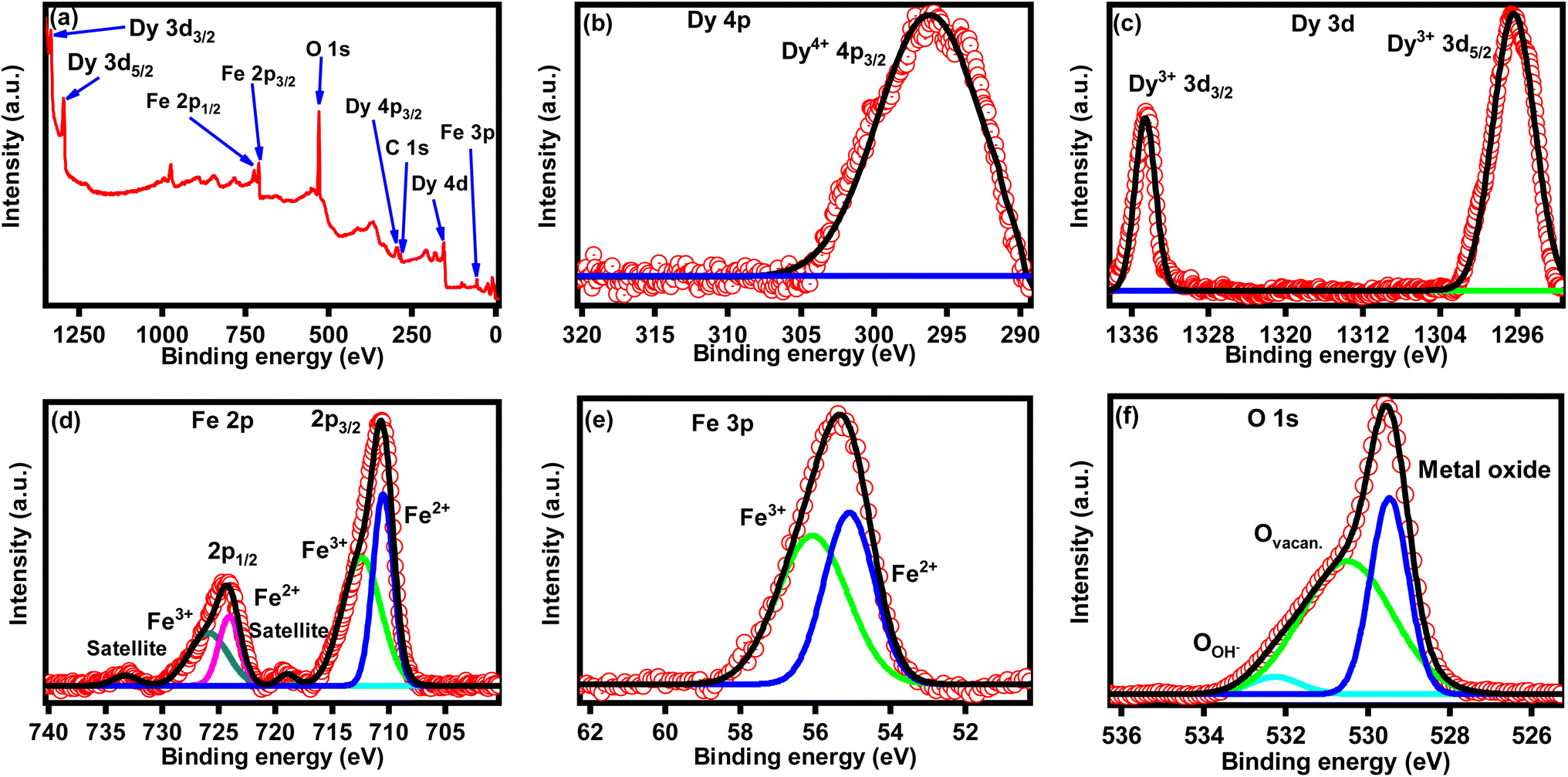 | ||
| Fig. 3 Chemical states analyses of DyFeO3 nanoparticles via XPS. (a) Full survey spectrum revealing distinct peaks corresponding to the oxidation states of Dy4+, Dy3+, Fe2+, Fe3+, and O2−. These oxidation states may enhance the electrochemical charge storage capability of DyFeO3 as an electrode material. High-resolution XPS spectra of (b) Dy 4p, (c) Dy 3d, (d) Fe 2p, (e) Fe 3p, and (f) O 1s provide detailed information on the specific binding energies of each element. The high-resolution XPS spectrum of the O 1s region shows a satellite peak at 530.53 eV, indicating the presence of oxygen vacancies in DyFeO3 nanoparticles. These vacancies can potentially improve the electrochemical performance of DyFeO3 as an electrode material, as discussed in the text. | ||
Notably, in synthesizing DyFeO3 nanoparticles using the sol–gel method, we used EG as a chelating agent, forming a polymeric metal cationic network for the gel precursor. EG's thermal decomposition released gases, including water vapor, carbon dioxide, and volatile organic compounds,54,55 creating internal pressure in the gel matrix. This pressure sought paths of least resistance, leading to void or pore formation. Simultaneously, thermal stress from decomposition initiated microcracks or voids where internal pressure exceeded the material's structural strength.54 Moreover, the produced gas bubbles serve as nucleation sites, initiating pore formation and coalescing throughout the synthesis process to create more substantial voids.56 The calcination at 750 °C additionally aids in removing volatile components, including organic residues and solvent remnants, crucially contributing to the porous structure in DyFeO3. The controlled atmosphere with N2 gas during calcination at high temperatures extracts oxygen atoms from the lattice, generating oxygen vacancies.
3.1 Electrochemical performance evaluation in three-electrode system
We aim to develop ASSCs using electrode materials with hybrid charge storage capability and electrolytes with a wide ESW, predominantly composed of water. In pursuit of our objectives, we first selected an aqueous solution of 0.5 M Na2SO4 as the electrolyte with DyFeO3 electrode material. The selection of Na2SO4 was based on considerations such as hydrated ion size, ionic conductivity, cost-effectiveness, and non-corrosive properties, as outlined in Table S3 (ESI†). To further enhance the ESW, particularly for commercial applications, small proportions (10% and 20% relative to water) of organic additives such as AN and EG were introduced into the 0.5 M Na2SO4(aq.) electrolyte solution. Subsequently, we conducted cyclic voltammetry (CV), linear sweep voltammetry (LSV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) analyses on the DyFeO3 electrode material utilizing a three-electrode system. To investigate the potential window of porous DyFeO3 electrode material, CV experiments were conducted at various potential windows (−1.4 to 1.5 V) using a fixed scan rate of 50 mV s−1 in a three-electrode system with 0.5 M Na2SO4(aq.) electrolyte solution, which illustrated in Fig. S5(a).† Among the various potential windows, −1.4 V to 1.1 V was selected based on the observed symmetric and quasi-rectangular shape of the CV curve within this range. Similarly, the chosen potential window of DyFeO3 electrode material in 0.5 M Na2SO4(aq.)/10%AN, 0.5 M Na2SO4(aq.)/10%EG, 0.5 M Na2SO4(aq.)/20%AN and 0.5 M Na2SO4(aq.)/20%EG electrolyte solutions were (−1.6 to 1.3) V, (−1.5 to 1.2) V, (−1.8 to 1.3) V, and (−1.6 to 1.3) V, respectively. All subsequent electrochemical measurements, including CV and GCD, were conducted within their respective selected potential windows. The integrated CV area of DyFeO3 electrode material in 0.5 M Na2SO4(aq.)/20%AN electrolyte solution, as depicted in Fig. 4(a), exhibited a significantly larger value compared to other electrolyte solutions, indicating the heightened charge storage capacity of DyFeO3 nanoparticles in that particular electrolyte solution. To gain deeper insights, CV curves of DyFeO3 nanoparticles were presented at scan rates of 5, 10, 30, 50, 70, and 100 mV s−1, as shown in Fig. S6(a)–(e).† In 0.5 M Na2SO4(aq.) electrolyte solution, the rectangular-shaped CV curves signify a typical capacitive dominant behavior.57 In contrast, quasi-rectangular-shaped CV curves observed in other electrolyte solutions indicate a characteristic pseudocapacitive dominant behavior. To evaluate the ESW of the DyFeO3 electrode material across various electrolyte solutions, an LSV test was conducted with a scan rate of 0.5 mV s−1, as depicted in Fig. S5(b).† The LSV curves reveal an impressive and broad ESW of 2.5 V (−1.4 to 1.1 V) in the 0.5 M Na2SO4(aq.) electrolyte and 3.1 V (−1.8 to 1.3 V) in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution. The broad ESW of the DyFeO3 electrode material is attributed to its distinctive features: (i) diverse oxidation states enabling redox reactions, (ii) oxygen vacancies that act as adsorption sites for Na+ and SO42− ions within the electrolyte, influencing the interaction between the electrode and electrolyte ions and providing alternative pathways for charge transfer that avoid water-splitting reactions, and (iii) a porous structure that facilitates efficient ion diffusion, allowing Na+ and SO42− ions to access the interior of the porous structure.58,59 These features, combined with the Na2SO4 electrolyte characterized by high conductivity and dielectric constant, low viscosity, and effective solvation structure,16 collectively contribute to achieving this ultra-wide ESW. To enhance the ESW of the WIS (0.5 M Na2SO4(aq.)) electrolyte solution, AN emerges as the most favorable organic additive. This consideration is based on its favorable electrochemical performance and desirable physical and chemical properties such as a high dielectric constant, low viscosity, high conductivity, and cost-effectiveness, as documented in Table S4 (ESI).† Note that the distinct charge storage mechanism of the DyFeO3 electrode material was also identified by analyzing the value of Tafel slope “b” using CV curve data as shown in Fig. 4(b).36 As depicted in Fig. 4(b), the Tafel slope “b” of the DyFeO3 electrode material in a 0.5 M Na2SO4(aq.) electrolyte solution has a value of 0.94, indicating the prevalence of the EDL charge storage mechanism. In contrast, the Tafel slope “b” in other electrolyte solutions reflects values corresponding to pseudo-capacitive dominant charge storage processes.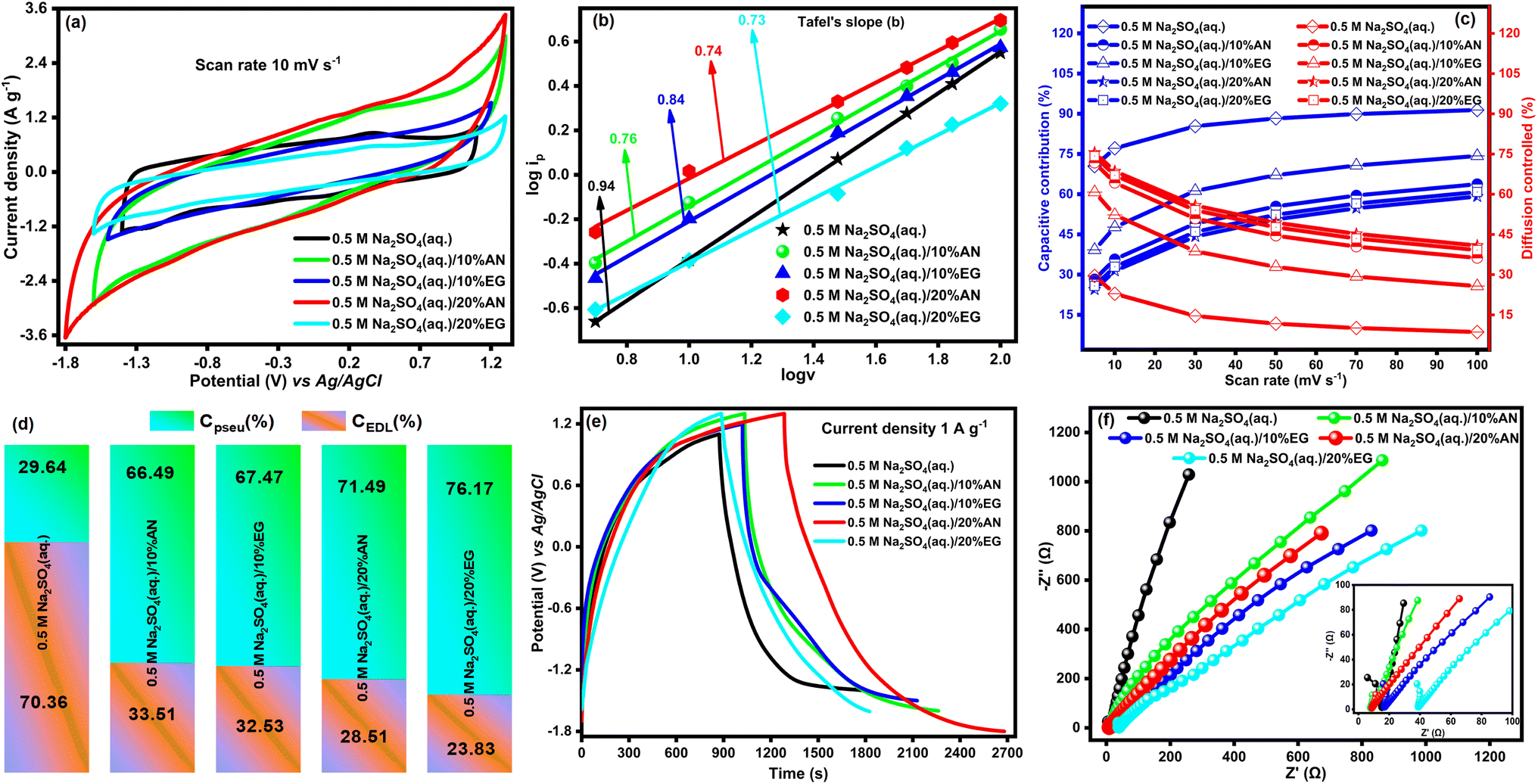 | ||
| Fig. 4 Electrochemical evaluation of DyFeO3 in aqueous electrolyte using a three-electrode system. (a) CV curves at 10 mV s−1 show a large integrated area and an ultra-high potential window (3.1 V) for DyFeO3 in 0.5 M Na2SO4(aq.)/20%AN electrolyte, outperforming other solutions. (b) Tafel slope analysis indicates that DyFeO3 primarily exhibits an EDL charge storage mechanism in 0.5 M Na2SO4(aq.), while pseudo-capacitance dominates in hybrid electrolytes. (c) Dunn's method shows balanced contributions from capacitive and diffusion-controlled currents in 0.5 M Na2SO4(aq.)/20%AN, highlighting its reliability for energy storage. (d) Trasatti's method reveals that EDL capacitance is dominant in 0.5 M Na2SO4(aq.), while pseudo-capacitance prevails in hybrid electrolytes. (e) GCD curves at 1 A g−1 demonstrate significantly longer charge–discharge times for DyFeO3 in 0.5 M Na2SO4(aq.)/20%AN. (f) Nyquist plots show very low impedance, with minimal solution resistance (Rs) and charge transfer resistance (Rct), with an inset providing an enlarged view of the high-frequency region. | ||
Using Dunn's method, we determined the capacitive resultant and diffusion-controlled current (details methodology is shown in ESI† and related graphs are shown in Fig. S7†) of DyFeO3 electrode material in various electrolyte solutions and the obtained results are shown in Fig. 4(c).36 The results depicted in Fig. 4(c) indicate that the DyFeO3 electrode material showed a high capacitive contribution and a low diffusion contribution in a 0.5 M Na2SO4(aq.) electrolyte solution. Conversely, in other electrolyte solutions, the DyFeO3 electrode exhibited a combination of capacitive and diffusion currents. This combination is desirable for an energy storage device, ensuring efficient energy consumption, rapid energy delivery, stability, and a long operational lifespan.36
Using Trassatt's method,36 we calculated the percentages of EDL and pseudo capacitance (the graphs related to the calculation are shown in Fig. S8 and S9†) and the obtained results are displayed in Fig. 4(d). In Fig. 4(d), the substantial EDL capacitance observed in the DyFeO3 electrode material in 0.5 M Na2SO4(aq.) electrolyte solution can be attributed to its porous structure, high dielectric constant, low viscosity, and the high conductivity of Na+, SO42−, and water. These characteristics facilitate rapid adsorption and desorption of electrolyte ions at the electrode surface, promoting efficient charge separation and thereby enhancing the EDL capacitance.36 Conversely, in hybrid electrolyte solutions, DyFeO3 exhibits higher pseudocapacitance compared to the 0.5 M Na2SO4(aq.) electrolyte solution. The additives (AN and EG) in hybrid electrolytes create a less polar, less conductive environment with different dielectric properties and viscosity compared to water. These distinct properties facilitate ion intercalation (Na+ and SO42−) and enable redox reactions between electrolyte ions and the electrode material, thereby contributing to higher pseudo-capacitance than EDL capacitance.
The charged-discharged time of DyFeO3 electrode material in 0.5 M Na2SO4(aq.)/20%AN electrolyte solution, as depicted in Fig. 4(e), exhibited a significantly larger value compared to other electrolyte solutions, indicating the superior charge storage capability compared to other electrolyte solutions. To gain deeper insights, GCD curves of DyFeO3 nanoparticles were presented at current densities of 1, 2, 3, 4, 5, 7 and 10 A g−1, as shown in Fig. S10.† Additionally, specific capacitance (Csp) values were calculated from the GCD curves at different current densities, and these values are presented in Fig. S11(a).† It is observed that the Csp values of the DyFeO3 electrode material in 0.5 M Na2SO4(aq.), 0.5 M Na2SO4(aq.)/10%AN, 0.5 M Na2SO4(aq.)/10%EG, 0.5 M Na2SO4(aq.)/20%AN and 0.5 M Na2SO4(aq.)/20%EG electrolyte solutions at 1 A g−1 current density are 370.9, 423.6, 410, 450, and 324.5 F g−1, respectively. Remarkably, in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution, the DyFeO3 electrode material exhibits substantial specific capacitance (Csp) values over a wide range of current densities. Additionally, the capacitance retention of the DyFeO3 electrode material in various electrolyte solutions was evaluated by conducting 10000 charge–discharge cycles at a current density of 10 A g−1 (as depicted in Fig. S11(b)†). It is noteworthy that the DyFeO3 electrode material shows exceptionally high capacitance retention across all tested electrolyte solutions.
To examine the impedance characteristics of the DyFeO3 electrode material in various electrolyte solutions EIS technique was employed and the results are displayed in Fig. 4(f) by Nyquist plot.36 The solution resistance (Rs) values of the DyFeO3 electrode material in 0.5 M Na2SO4(aq.), 0.5 M Na2SO4(aq.)/10%AN, 0.5 M Na2SO4(aq.)/10%EG, 0.5 M Na2SO4(aq.)/20%AN and 0.5 M Na2SO4(aq.)/20%EG electrolyte solutions are determined to 5.8, 8.99, 16.08, 11.34, and 38.08 Ω, respectively. In 0.5 M Na2SO4(aq.) electrolyte solution, the low impedance value is attributed to the high conductivity of electrolyte ions and water, low viscosity, and high dielectric constant of water, facilitating efficient and rapid charge transfer at the electrode–electrolyte interface.36 Conversely, in hybrid electrolyte solutions, increased impedance levels are attributed to the presence of organic additives, which reduce both conductivity and ionic mobility. Notably, the DyFeO3 electrode material demonstrates relatively lower impedance in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution compared to other hybrid electrolytes, indicating that a 20% concentration of AN is optimal for 0.5 M Na2SO4(aq.) electrolyte solutions. Moreover, the steep slope observed in the Warburg region (low-frequency range of the Nyquist plot) of the DyFeO3 electrode material in 0.5 M Na2SO4(aq.) electrolyte solution suggests high ion mobility and diffusion, indicative of favorable high-rate capacitive behavior. In contrast, the slow slope observed in hybrid electrolyte solutions reflects a transition from capacitive behavior to diffusion-limited behavior influenced by ion transport within the electrolyte.36
To reveal the cause of ESW expansion with increasing concentration of AN, the solvation structure of various concentration AN incorporated electrolyte solutions were characterized using Raman, FTIR, and NMR spectroscopy as shown in Fig. 5. In Fig. 5(a), the comparison of O–H stretching vibrations in different electrolyte solutions reveals notable changes: the strong hydrogen-bonded Raman peak at 3200 cm−1 decreases in intensity with the addition of AN, signifying the disruption of the water–water hydrogen bond network, while the peak at 3400 cm−1, indicating partially hydrogen-bonded water molecules, shifts to higher wavenumbers, suggesting an increase in the partially hydrogen-bonded network.7,8 The vibrational peak at around 2263 cm−1 of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group60 of AN molecule (exhibited in Fig. 5(b)) broadens and shifts to higher wavenumbers due to increased hydrogen bonding with water molecules, further disrupting the strong hydrogen-bond structure and increasing the partially hydrogen-bonded network, ultimately decreasing the activity of free water molecules and increasing the ESW of the electrolyte solution. Additionally, the ion solvation of SO42− anions with Na+ cations in hybrid electrolyte solutions inhibit water molecule decomposition, as evidenced by a blue-shift in the S–O stretching vibrational peak from 981 cm−1 to 988 cm−1 with increasing AN concentration (depicted in Fig. 5(c)). This shift indicates a transition from solvent-separated ion pairs (SSIP) to contact ion pairs (CIP) and anion–cation aggregates (AGG), enhancing the electrolyte structure and reducing the activity of free water molecules. Ultimately, this transition contributes to an increase in the ESW of the hybrid electrolyte solution.7,8 FTIR analysis reveals shifts in O–H bending and H-bending stretching vibrational modes with increasing AN concentration: in Fig. 5(d), a shift from 1640.45 cm−1 to 1649.55 cm−1 indicates stronger O–H bonds.8 Meanwhile, in Fig. S12,† a shift from 3337.6 cm−1 to 3356.1 cm−1 suggests a disruption of strong water–water hydrogen bonds and the growth of partially hydrogen-bonded structures,8 indicating reduced water molecule activity. 1H NMR spectroscopy reveals shifts in the peaks of water molecules and AN molecules with increasing AN concentration: in Fig. 5(e), a shift from 4.77 ppm to 4.42 ppm suggests higher electron density around hydrogen atoms, weakening the water–water hydrogen bond network and strengthening O–H bonds,8,61 while in Fig. 5(f), a shift from 1.95 ppm to 1.84 ppm indicates disruption of water–water hydrogen bonds and enhancement of AN-water hydrogen bonds.62 Overall, the results obtained from Raman, FTIR, and NMR spectroscopy are consistent with each other, revealing that AN addition improves ESW by reducing the activity of free water molecules in the electrolyte solution. Moreover, AN reduces the viscosity of electrolyte solutions, facilitating the formation of a strong SEI layer in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution.11 Based on findings from Raman, FTIR, and NMR spectroscopy, schematic representations in Fig. 5(g) illustrate solvent-separated ion pairs (SSIP), contact ion pairs (CIP), and anion–cation aggregates (AGG) coordination structures of Na+ and SO42− ions. In Fig. 5(h), solvation and interfacial interactions between Na+ and SO42− ions with the DyFeO3 electrode material are depicted, aiding in the comprehension of the hybrid electrolyte's solvation structure and its interaction with the electrode. Overall, AN's role in enlarging the voltage window is multifaceted, involving the modulation of ion solvation, stabilization of the electrolyte, suppression of water electrolysis, and the potential formation of a protective SEI layer. These effects collectively enable the SC to operate at higher voltages with improved performance, which is critical for enhancing the energy density and overall efficiency of the device.
N group60 of AN molecule (exhibited in Fig. 5(b)) broadens and shifts to higher wavenumbers due to increased hydrogen bonding with water molecules, further disrupting the strong hydrogen-bond structure and increasing the partially hydrogen-bonded network, ultimately decreasing the activity of free water molecules and increasing the ESW of the electrolyte solution. Additionally, the ion solvation of SO42− anions with Na+ cations in hybrid electrolyte solutions inhibit water molecule decomposition, as evidenced by a blue-shift in the S–O stretching vibrational peak from 981 cm−1 to 988 cm−1 with increasing AN concentration (depicted in Fig. 5(c)). This shift indicates a transition from solvent-separated ion pairs (SSIP) to contact ion pairs (CIP) and anion–cation aggregates (AGG), enhancing the electrolyte structure and reducing the activity of free water molecules. Ultimately, this transition contributes to an increase in the ESW of the hybrid electrolyte solution.7,8 FTIR analysis reveals shifts in O–H bending and H-bending stretching vibrational modes with increasing AN concentration: in Fig. 5(d), a shift from 1640.45 cm−1 to 1649.55 cm−1 indicates stronger O–H bonds.8 Meanwhile, in Fig. S12,† a shift from 3337.6 cm−1 to 3356.1 cm−1 suggests a disruption of strong water–water hydrogen bonds and the growth of partially hydrogen-bonded structures,8 indicating reduced water molecule activity. 1H NMR spectroscopy reveals shifts in the peaks of water molecules and AN molecules with increasing AN concentration: in Fig. 5(e), a shift from 4.77 ppm to 4.42 ppm suggests higher electron density around hydrogen atoms, weakening the water–water hydrogen bond network and strengthening O–H bonds,8,61 while in Fig. 5(f), a shift from 1.95 ppm to 1.84 ppm indicates disruption of water–water hydrogen bonds and enhancement of AN-water hydrogen bonds.62 Overall, the results obtained from Raman, FTIR, and NMR spectroscopy are consistent with each other, revealing that AN addition improves ESW by reducing the activity of free water molecules in the electrolyte solution. Moreover, AN reduces the viscosity of electrolyte solutions, facilitating the formation of a strong SEI layer in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution.11 Based on findings from Raman, FTIR, and NMR spectroscopy, schematic representations in Fig. 5(g) illustrate solvent-separated ion pairs (SSIP), contact ion pairs (CIP), and anion–cation aggregates (AGG) coordination structures of Na+ and SO42− ions. In Fig. 5(h), solvation and interfacial interactions between Na+ and SO42− ions with the DyFeO3 electrode material are depicted, aiding in the comprehension of the hybrid electrolyte's solvation structure and its interaction with the electrode. Overall, AN's role in enlarging the voltage window is multifaceted, involving the modulation of ion solvation, stabilization of the electrolyte, suppression of water electrolysis, and the potential formation of a protective SEI layer. These effects collectively enable the SC to operate at higher voltages with improved performance, which is critical for enhancing the energy density and overall efficiency of the device.
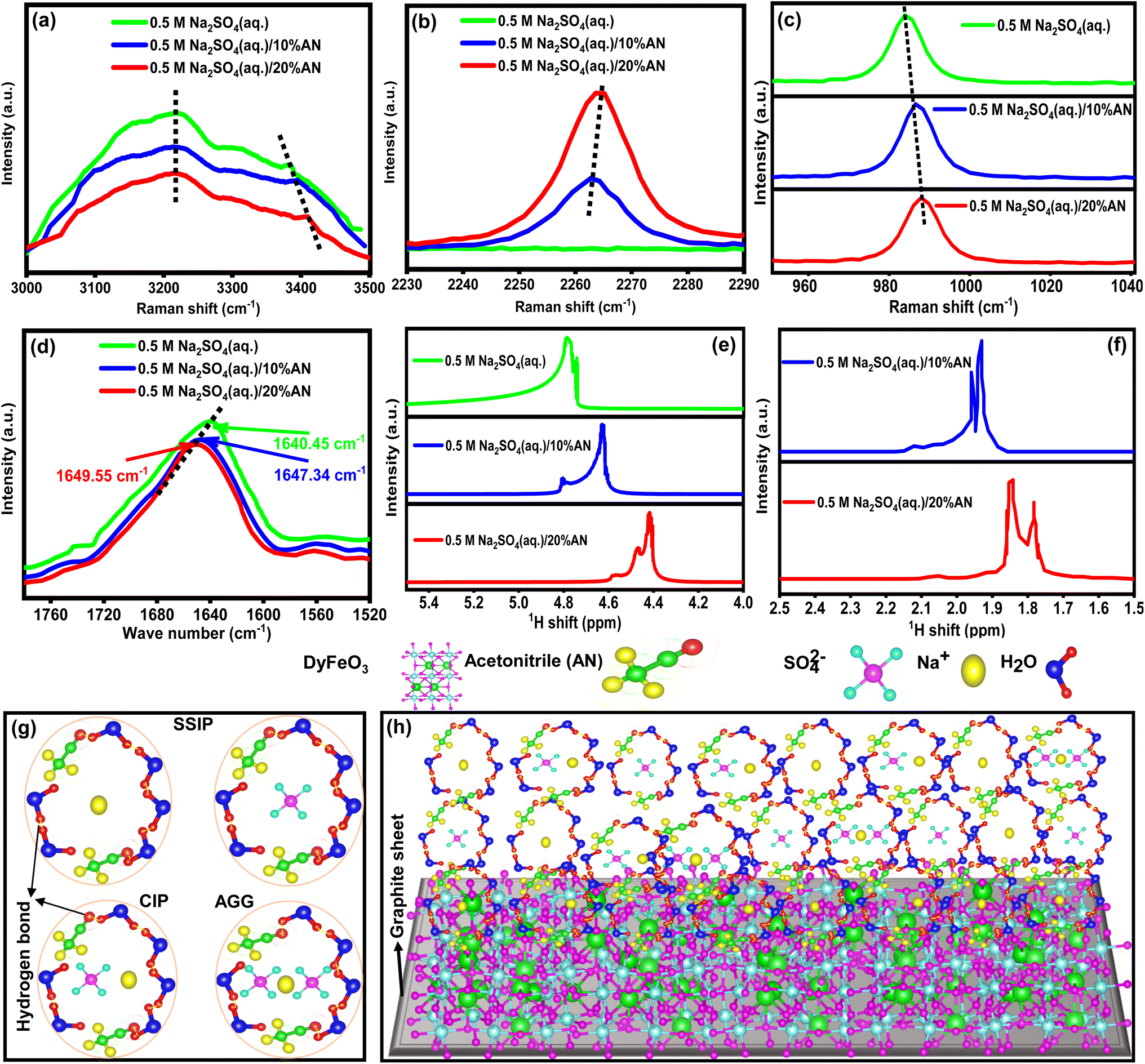 | ||
| Fig. 5 Raman, FTIR, and NMR spectroscopy analyses of various electrolyte solutions. (a) The O–H stretching vibrations consistently peak at 3200 cm−1, representing strongly hydrogen-bonded water, with intensity decreasing as AN concentration increases. The partially hydrogen-bonded peak at 3400 cm−1 shifts to higher wavenumbers, indicating disrupted water–water hydrogen bonds and more partially hydrogen-bonded structures. (b) The –CN group of AN shows a broadened vibrational peak at 2260 cm−1, shifting to higher wavenumbers due to strong hydrogen bonding between AN's N atoms and water's H atoms as AN concentration rises. (c) The S–O stretching peak of the SO42− anion shifts from 981 cm−1 to 988 cm−1 with increasing AN concentration, signaling the transition from SSIP to CIP and AGG of Na+ and SO42− ions. (d) The peak at 1640.45 cm−1, corresponding to the O–H vibrational mode of water, blue-shifts with increasing AN, indicating stronger O–H bonds. (e) The downshift in NMR peaks of water suggests a weakening of water–water hydrogen bonds and stronger O–H bonds with rising AN concentration. (f) AN's NMR peak also downshifts, reflecting disrupted water–water hydrogen bonds and enhanced AN–water hydrogen bonds. (g and h) The study explores SSIP, CIP, and AGG solvation structures and interfacial interactions between electrolyte ions and electrodes. | ||
3.2 Electrochemical performance of as-fabricated aqueous symmetric supercapacitors (ASSCs)
In light of the favorable electrochemical performance in three-electrode system, we fabricated the ASSCs using a coin cell arrangement to extend our investigation into the electrochemical properties of the DyFeO3 electrode material in both 0.5 M Na2SO4(aq.) and 0.5 M Na2SO4(aq.)/20%AN electrolyte solutions. To enhance clarity, for the remainder of this manuscript, the aqueous symmetric SC featuring DyFeO3 electrode material and 0.5 M Na2SO4(aq.) electrolyte solution is denoted as ASSC-1, while the SC utilizing 0.5 M Na2SO4(aq.)/20%AN electrolyte solution as ASSC-2. Initially, we conducted CV measurements of ASSC-1 at 2.5 V and ASSC-2 at 3.1 V across different scan rates to assess cyclic reversibility, as illustrated in Fig. S13(a) and (b) (ESI†). A substantial increase in the integrated CV area of ASSC-2, compared to ASSC-1, was observed, as shown in Fig. 6(b). This enhancement is attributed to the incorporation of 20% AN in 0.5 M Na2SO4(aq.) electrolyte solution. To assess the ESW of the ASSC-1 and ASSC-2, an LSV test was conducted at a constant scan rate of 0.5 mV s−1, as illustrated in Fig. 6(c). The LSV curves reveal an impressive ultra-wide ESW of 2.5 V in additive-free ASSC-1 and 3.1 V in ASSC-2. Remarkably, minimal water splitting is observed within these potential windows. The preceding section extensively elaborated on the factors contributing to the ultra-wide ESW observed in ASSC-1 within a 100% aqueous electrolyte solution and ASSC-2 within an aqueous electrolyte solution containing a low percentage of AN (organic additive). Moreover, to investigate gas evolution, we conducted CV and GCD studies with and without additives at identical cutoff potentials, as presented in ESI Fig. S15.† In the CV curve of ASSC-1 at 3.1 V (Fig. S15(b)†), a pronounced increase in current density signifies substantial gas evolution attributed to the decomposition of water molecules, driven by the electrolysis of free water at higher potentials. This observation is corroborated by the GCD curve of ASSC-1 at 3.1 V (Fig. S15(d)†), which exhibits a non-linear potential drop, indicative of gas formation and increased internal pressure within the cell. Conversely, ASSC-2, containing acetonitrile additives, demonstrates stable CV (Fig. S15(a) and (b)†) and GCD (Fig. S15(c) and (d)†) behavior at both 2.5 V and 3.1 V, with no abrupt increase in current density or irregular potential drops. This enhanced stability can be attributed to the ability of acetonitrile to diminish the activity of free water molecules, thereby inhibiting water decomposition even at elevated potentials.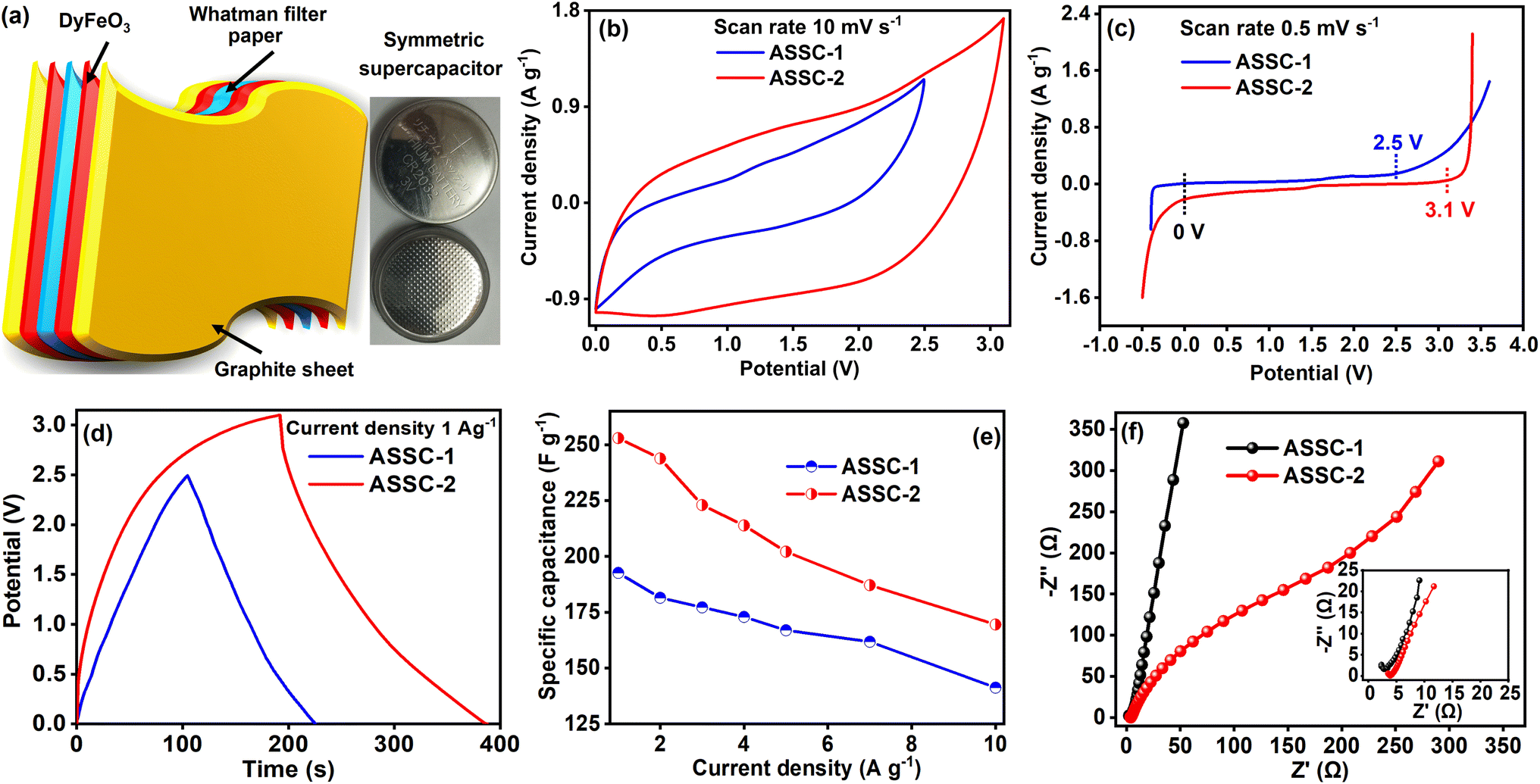 | ||
| Fig. 6 Electrochemical performance of fabricated ASSCs (ASSC-1 and ASSC-2) using DyFeO3 nanoparticles as electrode material in 0.5 M Na2SO4(aq.) and 0.5 M Na2SO4(aq.)/20%AN electrolytes, with graphite sheet as current collector and Whatman filter paper as separator. (a) Schematic and real illustration of the ASSCs. (b) CV curves at a fixed scan rate of 10 mV s−1 show the large integrated CV area with ultra-high (3.1 V) potential window of ASSC-2 compared to ASSC-1. (c) LSV curves demonstrate that the water oxidation potential of the ASSC-1 and ASSC-2 exceeds the theoretical water oxidation potential (1.23 V). (d) GCD curves at a fixed current density of 1 A g−1. (e) High specific capacitance (Csp) of up to 253 F g−1 exhibited by ASSCs at various current densities. (f) Nyquist plots show the very low impedance value i.e. low value of Rs and Rct, inset shows the enlarged view of the high-frequency region of Nyquist plots. | ||
In Fig. 6(d), the comparative GCD curves of ASSC-1 and ASSC-2 exhibit that the charging–discharging duration of the ASSC-2 surpasses that of the ASSC-1. To a comprehensive understanding of the charging–discharging time under varied current densities (1, 2, 3, 4, 5, 7, and 10 A g−1), the GCD curves of the ASSC-1 and ASSC-2 are presented in Fig. S14(a) and (b) (ESI†). Notably, GCD curves of ASSC-1 exhibit a triangular shape, indicative of EDL capacitive-dominant behavior.57 On the other hand, the GCD curves of ASSC-2 indicate pseudo-capacitive-dominant behavior. Moreover, the Csp values were determined at various current densities based on the GCD curves, and the findings are depicted in Fig. 6(e).36,39 The ASSC-1 demonstrates a Csp of 192.6 F g−1 at low current density (1 A g−1) and 141.3 F g−1 at high current density (10 A g−1), whereas for ASSC-2, the corresponding Csp values are 253 F g−1 and 169.5 F g−1, respectively. In Fig. 6(f), the Nyquist plots reveal that the series resistance (Rs) of the ASSC-1 and ASSC-2 is 2.33 and 3.51 Ω, respectively and the charge transfer resistance (Rct) is 0.7 and 0.4 Ω, respectively. The reduced impedance parameters observed can be ascribed to the presence of highly conductive small-sized ions (Na+ and SO42−), along with the low viscosity and high dielectric constant of the solvent medium, which facilitates the easy penetration of electrolyte ions into the porous DyFeO3 nanoparticles.
The performance of energy storage devices is significantly influenced by their energy and power density, as these metrics play a crucial role in determining operational efficiency. To evaluate these key parameters, we calculated the energy and power density of the ASSCs using GCD data, the results are depicted in Fig. 7(a) through Ragone plot.36 Specifically, at a power density of 1250 W kg−1, ASSC-1 shows an energy density of 41.81 W h kg−1, and ASSC-2 exhibits an energy density of 84.43 W h kg−1 at a power density of 1550 W kg−1. Remarkably, even at higher power densities, ASSC-1 and ASSC-2 maintain substantial energy density. For instance, at a power density of 12500 W kg−1, ASSC-1 achieves an energy density of 30.65 W h kg−1, and similarly, ASSC-2 exhibits an energy density of 56.57 W h kg−1 at 15500 W kg−1. These findings highlight the excellent energy storage and delivery capabilities of our fabricated ASSCs.
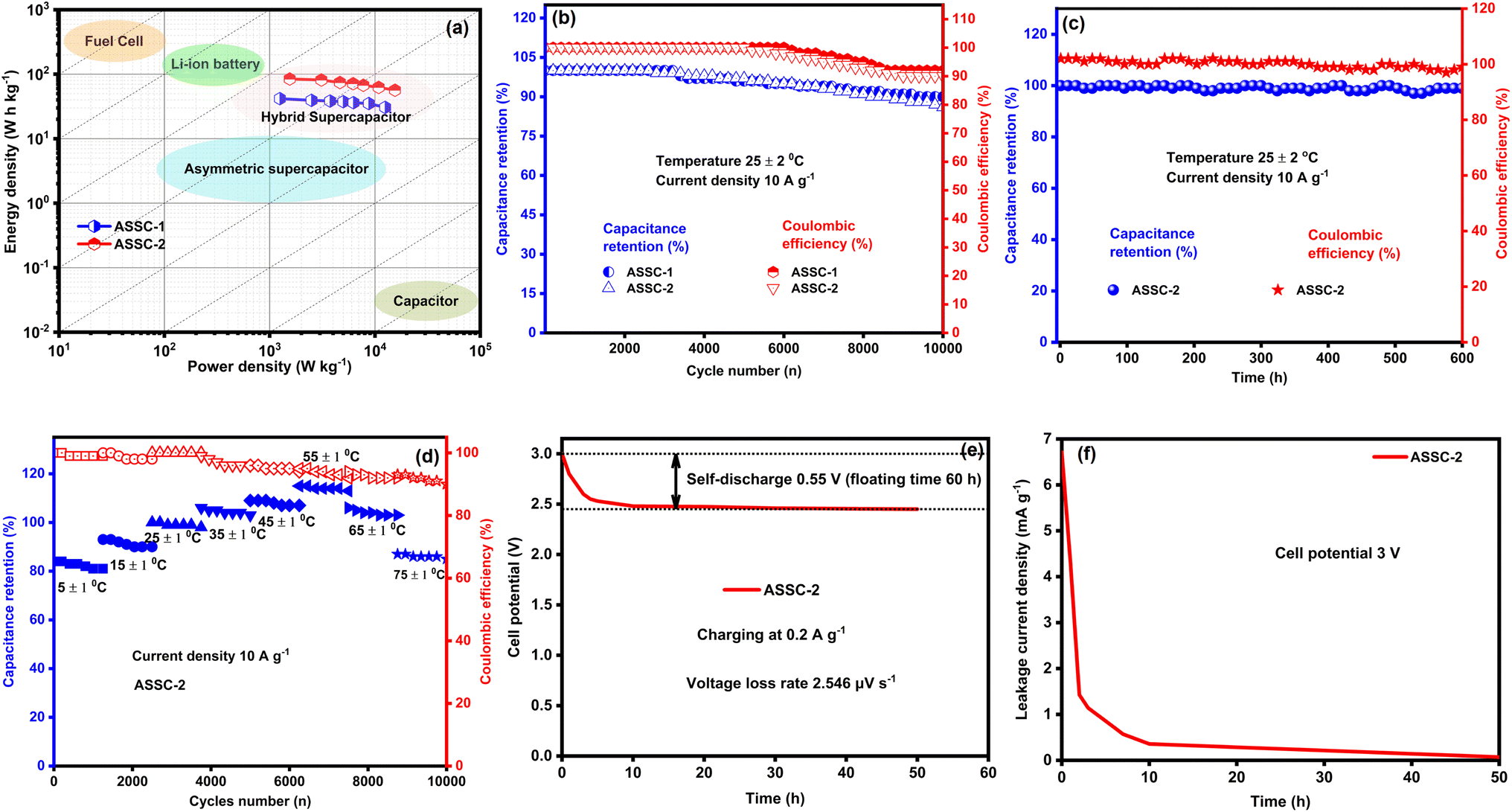 | ||
| Fig. 7 Energy storage and durability performance of ASSC-1 and ASSC-2. (a) Ragone plots showing high energy density comparable to Li-ion batteries and high power density similar to EDL capacitors. (b) Room-temperature performance of the fabricated aqueous symmetric SCs, exhibiting impressive capacitance retention of up to 90% and Coulombic efficiency of up to 92% after 10000 GCD cycles. (c) Float voltage test reveals enhanced capacitance retention and Coulombic efficiency of ASSC-2. (d) Excellent capacitance retention and Coulombic efficiency are observed across a wide temperature range (5–75 °C). (e) Self-discharge profile of ASSC-2 charged to 3 V at 0.2 A g−1, showing a 0.55 V drop over 60 hours with a voltage loss rate of 2.546 μV s−1. (f) Leakage current of ASSC-2 at 3 V in potentiostatic mode, stabilizing at 6.71 mA g−1 after 50 hours. | ||
Our fabricated ASSCs exhibit notable rate capability and durability at room temperature, as shown in Fig. 7(b), retaining 90% and 86% of the initial capacitance after 10000 GCD cycles for ASSC-1 and ASSC-2, respectively. This performance is particularly significant for applications with frequent cycling, such as electric vehicles and renewable energy systems. Additionally, ASSC-1 and ASSC-2 exhibit Coulombic efficiencies of 92% and 90% after 10000 GCD cycles, respectively, indicating efficient charge–discharge processes and minimal energy losses. The observed high Coulombic efficiency is crucial for the overall performance of the energy storage devices, ensuring effective retrieval of a substantial proportion of stored energy during discharge. The stability of the ASSC-2 was rigorously examined through the float voltage test, offering a more reliable assessment compared to repeated GCD cycling.50 In this experiment, the assembled coin cell SC underwent a voltage application of 3.1 V, and a series of 15 charge and discharge cycles were carried out at a constant current density of 10 A g−1 with intervals of 12 hours, as illustrated in Fig. 7(c). The ASSC-2 exhibited remarkable stability over an extended testing period of 600 hours, maintaining approximately 99 ± 5% of its initial capacitance and Coulombic efficiency. These results demonstrate the robustness and efficiency of the ASSC-2 within a wide potential window of 3.1 V, which is essential for long-term stability under challenging operational conditions. To investigate the practical applicability of our fabricated ASSC-2 across diverse temperature environments, the capacitance retention and Coulombic efficiency at high and low temperatures (5 to 75 °C) were calculated as shown in Fig. 7(d). All calculations were conducted relative to results obtained at 25 °C. The values of capacitance retention and Coulombic efficiency decreased at lower temperatures, with the lowest values observed at 5 °C, being 81% and 99%, respectively, attributed to sluggish kinetics and diffusion processes.26,63 Conversely, values increased up to 55 °C due to enhanced kinetics and efficient diffusion, reaching a maximum at this temperature of 113% and 92%, respectively. However, above 55 °C, both capacitance retention and Coulombic efficiency decreased, reaching their lowest points at 75 °C—85% and 90%, respectively. This suggests that at higher temperatures, the advantages gained from improved kinetics are offset by heightened internal resistance. Despite these fluctuations, the ASSC-2 demonstrated superior durability and Coulombic efficiency across the wide temperature range, highlighting its promising practical applicability.
The data presented in Fig. 7(e) and (f) highlight the remarkable performance of the ASSC-2 device, particularly in minimizing self-discharge and leakage current—two critical factors that impact the practical application of supercapacitors. The self-discharge test in Fig. 7(e), conducted by charging the cell to 3.0 V at a current density of 0.2 A g−1, reveals a voltage drop of only 0.55 V over 60 hours, resulting in a low voltage loss rate of 2.546 μV s−1. This performance indicates excellent suppression of electrochemical side reactions, likely due to a stable electrode–electrolyte interface and minimal faradaic losses, which are common in aqueous systems. Similarly, the leakage current in Fig. 7(f), measured in potentiostatic mode at 3 V for 50 hours, stabilized at a minimal value of 6.71 mA g−1, reflecting a low rate of energy loss through internal pathways. Compared to existing aqueous supercapacitors, as detailed in ESI Table S5,† ASSC-2 demonstrates a significant reduction in energy loss mechanisms, positioning it as a superior high-voltage supercapacitor with both high efficiency and long-term energy retention capabilities.
The data in Table 1, including specific capacitance, ESW, capacitance retention, and energy density, demonstrate the superior performance of our fabricated ASSCs compared to other assembled SCs. The majority of SCs detailed in Table 1 exhibit an asymmetric configuration, utilizing costly and highly concentrated electrolyte solutions. Asymmetric configurations utilizing identical active components as symmetrical arrangements provide an expanded voltage range compared to their symmetrical counterparts, thereby enhancing power and energy density.16 For instance, the asymmetric system consisting of Na0.5MnO//Fe3O4@C and Na0.25MnO2//ERPC, as outlined in Table 1, demonstrated elevated energy density with voltage windows of 2.6 V and 2.7 V, respectively, when immersed in a 1 M Na2SO4(aq.) electrolyte solution. A comprehensive analysis of Table 1 substantiates the assertion that, considering various factors such as the merits of symmetric configuration, utilization of environmentally friendly electrolyte solutions, increased capacitance, durability, as well as enhanced energy and power density, our DyFeO3//DyFeO3 symmetric SC surpasses both previously documented symmetric and asymmetric SCs. For instance, when tested in a costly 17 m NaClO4/H2O electrolyte solution, the AC//AC symmetric SC retains 66.1% capacitance after 10000 GCD cycles within a 2.3 V potential window.7 In contrast, ASSC-1, operated in a cost-effective and highly conductive 0.5 M Na2SO4(aq.) electrolyte solution with an ESW of 2.5 V, exhibits an impressive 90% capacitance retention after 10000 GCD cycles. Comparatively, a porous carbon symmetric SC, operating in a high-density 20 m LiTFSI WIS electrolyte solution with a wide 2.4 V ESW, exhibits a specific capacitance of 63 F g−1 at a current density of 0.5 A g−1, accompanied by an energy density of 44 W h kg−1 at 564 W kg−1 power density.64 In contrast, our fabricated ASSC-2, featuring an extended ESW of 3.1 V, demonstrates a specific capacitance of 253 F g−1 and an energy density of 84.43 W h kg−1 at a power density of 1550 W kg−1, significantly surpassing the performance of numerous similarly assembled SCs. In addition to its exceptional electrochemical properties, our fabricated electrolyte system (refer to Table S6 in the ESI†) demonstrates a significantly lower production cost compared to many other electrolyte systems used previously. Although the cost of DyFeO3 nanoparticles as an electrode material is slightly higher than that of commercially available carbon derivatives, the integrated production cost of our fabricated ASSCs might be more economical, considering their distinctive energy storage performances and the use of a low-cost, environmentally friendly electrolyte system. It should also be noted that in comparison to the performance of various perovskite oxides, including single, double, and Ruddlesden-Popper perovskite oxides as discussed in a recent review article,16 our investigation highlights DyFeO3 as an exceptional electrode material for symmetric SCs, even when operating in an aqueous electrolyte.
| Electrode material | Electrochemical cell configuration | Electrolyte | Potential window | Specific capacitance (F g−1) at current density (A g−1) | Energy density (W h kg−1) at power density (W kg−1) | Capacitance retention (%) after cycles | Ref. |
|---|---|---|---|---|---|---|---|
| La2CoMnO6//AC | Asymmetric | 1 M Na2SO4(aq.) | 2 V | 120 at 0.5 | 65.8 at 1000 | 89.2 after 3000 | 65 |
| NiCo2S4//G/CS | Asymmetric | 6 M KOH(aq.) | 1.6 V | 119 at 5 mV s−1 | 42.3 at 476 | 78.6 after 10000 |
66 |
| NiCo2S4//graphene flim | Asymmetric | 1 M KOH(aq.) | 1.8 V | 332 at 4 | 60 at 1800 | 63.2 after 50000 |
67 |
| NiMo2O4//AC | Asymmetric | 2 M KOH(aq.) | 1.7 V | 151.7 at 1 | 60.9 at 850 | 85.7 after 10000 |
68 |
| Ni(OH)2//graphene | Asymmetric | 6 M KOH(aq.) | 1.6 V | 218 at 1 mV s−1 | 7708 at 174.7 | 94.3 after 3000 | 69 |
| MnO2//AC | Asymmetric | PVA/1 M Na2SO4(aq.) | 1.7 V | 100.2 at 0.36 | 40.2 at 340 | — | 70 |
| GrMnO2//GrMoO3 | Asymmetric | 1 M Na2SO4(aq.) | 2 V | 307 at 0.2 | 42.6 at 276 | — | 71 |
| Na0.5MnO//Fe3O4@C | Asymmetric | 1 M Na2SO4(aq.) | 2.6 V | 88 at 0.5 | 81 at 647 | 93 after 10000 |
72 |
| Na0.25MnO2//ERPC | Asymmetric | 1 M Na2SO4(aq.) | 2.7 V | 82.9 at 1 | 61.1 at 982.5 | 93.7 after 10000 |
73 |
| MnO2/GO//HPC | Asymmetric | 1 M Na2SO4(aq.) | 2 V | 84 at 0.1 | 46.7 at 100 | 93 after 4000 | 74 |
| Graphene//MGC | Asymmetric | 1 M Na2SO4(aq.) | 2 V | 31 at 0.5 | 30.4 at 100 | 79 after 1000 | 75 |
| Porous carbons | Symmetric | 20 M LiTFSI(aq.) | 2.4 V | 63 at 0.5 | 44 at 564 | 84 after 5000 | 64 |
| Porous carbon monoliths | Symmetric | 5 M LiTFS(aq.) | 2.4 V | 143 at 2 | 24 at 480 | 81 after 10000 |
33 |
| δ-MnO2//AC | Asymmetric | 5 M NaNO3(aq.) | 2.4 V | 48 at 0.5 | 38.4 at 599.7 | 88.6 after 5000 | 76 |
| AC//AC | Symmetric | 17 M NaClO4(aq.) | 2.3 V | 33 at 1 | — | 66.1 after 10000 |
7 |
| DyFeO3//DyFeO3 | Symmetric | 0.5 M Na2SO4(aq.) | 2.5 V | 192.6 at 1 | 41.81 at 1250 | 90 after 10000 |
This work |
| DyFeO3//DyFeO3 | Symmetric | 0.5 M Na2SO4(aq.)//20%AN | 3.1 V | 253 at 1 | 84.43 at 1550 | 86 after 10000 |
This work |
To evaluate the stability of DyFeO3 as an electrode material after 10000 GCD cycles, we conducted, Raman, EPR, zeta potential, and electrochemical analyses (CV, GCD, and EIS) on ASSC-2, as shown in Fig. 8. The XRD analysis of the electrode material, both before and after the cycling process (Fig. 8(a) and (b)), shows no peak shifts, indicating that the material retained its structural integrity without any detectable phase transformation (see Table S7† for details). This stability highlights DyFeO3 as a reliable choice for electrode materials in durable energy storage devices, particularly symmetric SCs. However, changes in peak intensity at specific positions suggest a reduction in the material's crystallinity. In Fig. 8(c), the Raman spectra show that after 10000 GCD cycles, the peak at 468 cm−1, associated with metal–oxygen bonds, weakens, while the peak at 560 cm−1, linked to oxygen vacancies, becomes more pronounced. This suggests that repeated cycling leads to the formation of additional oxygen vacancies within the DyFeO3 lattice, likely due to the stress and strain experienced during charge–discharge cycles. This observation is reinforced by the EPR spectra in Fig. 8(d), where the post-cycling signal shows a significant increase in intensity, indicating a higher concentration of unpaired electron spins related to these oxygen vacancies. These vacancies act as critical adsorption sites for Na+ and SO42− ions from the electrolyte, providing more active sites for reversible faradaic reactions, thereby enhancing charge storage.
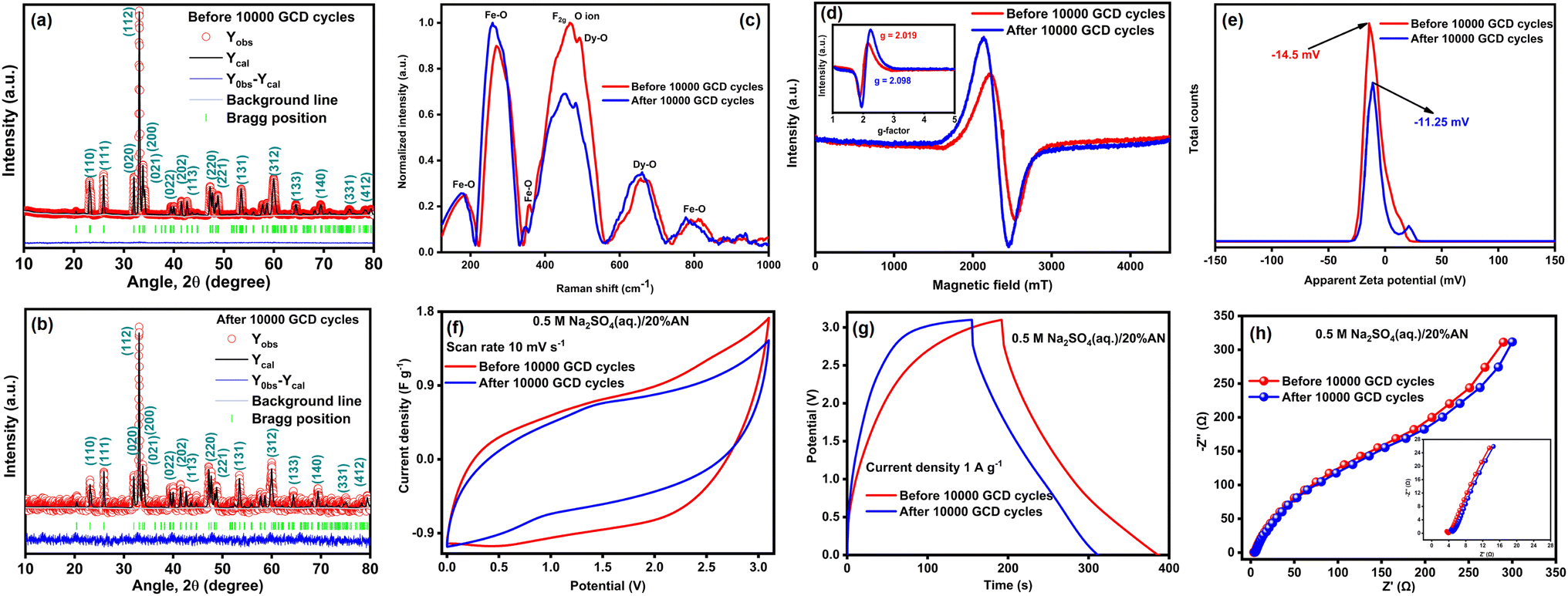 | ||
| Fig. 8 Structural, morphological, and electrochemical analyses of DyFeO3 electrode material after 10000 GCD cycles in 0.5 M Na2SO4(aq.)/20%AN electrolyte solution. (a) Rietveld-refined XRD spectra showing structural stability of DyFeO3 before and (b) after 10000 GCD cycles. (c) Raman spectra illustrate a decrease in the 468 cm−1 peak (metal–oxygen bonds) and an increase in the 560 cm−1 peak (oxygen vacancies) before and after 10000 GCD cycles. (d) EPR spectra reveal a marked increase in signal intensity, suggesting a higher concentration of oxygen vacancies; inset shows a shift in g-factor from 2.019 to 2.098, reflecting alterations in the local electronic environment due to Fe3+/Fe2+ redox activity. (e) Zeta potential curve shifting towards less negative values after 10000 GCD cycles. (f) CV curves at 10 mV s−1, (g) GCD curves at 1 A g−1 current density, and (h) Nyquist plots spanning a frequency range from 100 kHz to 0.1 Hz with a 1 mV alternating potential, demonstrating the superior electrochemical performance of DyFeO3 in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution. | ||
As a result, the pseudocapacitive behavior of the electrode material is greatly improved. Additionally, the EPR spectra of the DyFeO3 electrode material after 10000 GCD cycles show a shift in the g-factor from 2.019 to 2.098, as highlighted in the inset of Fig. 8(d). This shift reflects changes in the local electronic environment, primarily driven by the intercalation and deintercalation of electrolyte ions during cycling. Specifically, redox reactions between Fe3+ and Fe2+ ions within the DyFeO3 lattice, which typically exhibit a g-factor between 2.1 and 2.3,77 enhance spin–orbit coupling, leading to the increased g-value observed after cycling. These combined effects—oxygen vacancies and Fe redox activity—are key factors that enhance both the capacitance and stability of the material over extended cycling. Fig. 8(e) shows that after 10000 cycles, the zeta potential curve shifts towards less negative values. This shift is likely due to an increased presence of oxygen vacancies and surface modifications, such as the partial neutralization of surface charges caused by the repeated intercalation and deintercalation of Na+ ions during cycling. However, the zeta potential remains significantly negative, indicating that the DyFeO3 nanoparticles retain a substantial negative surface charge. This persistent negative charge is essential for facilitating the continued adsorption of Na+ ions, which is crucial for the formation and stability of the EDL. The maintained negative surface charge also prevents agglomeration, ensuring electrostatic stability and preserving the high surface area required for efficient ion adsorption. Fig. 8(f) and (g) show a slight reduction in the integrated CV area and charge/discharge time after 10000 cycles. However, the consistent shape within the ultra-wide potential window demonstrates stable electrochemical behavior, which is critical for practical applications. The Nyquist plot in Fig. 8(h) indicates only a minor increase in charge transfer resistance (Rct) and series resistance (Rs), confirming good conductivity even after prolonged cycling.
Fig. 9 shows high-resolution XPS spectra of DyFeO3 nanoparticles before and after 10000 GCD cycles, highlighting significant changes in their oxidation states and surface chemistry. In the Dy 3d spectrum (Fig. 9(a)), the Dy3+ 3d5/2 peak exhibits only a slight increase, indicating that Dy sites remain largely stable and unaffected by the cycling process. However, in Fig. 9(b) and (c), there is a notable increase in Fe2+ concentration after cycling, suggesting that iron plays an active role in redox reactions during charge–discharge cycles, which is essential for pseudocapacitive charge storage. The most significant changes are observed in the O 1s spectrum (Fig. 9(d)), where the oxygen vacancy peak increases substantially to 78.56% after 10000 cycles, while the metal oxide peak decreases to 17.76%. This rise in oxygen vacancies aligns with the results from the Raman (Fig. 8(c)) and EPR (Fig. 8(d)) analyses, indicating that electrochemical cycling generates additional oxygen vacancies, thereby enhancing the material's charge storage capacity. Furthermore, the OH group peak decreases from 4.22% to 3.68% after cycling, suggesting a reduction in free water molecule activity near the electrode surface. This decrease in water activity likely contributes to an increased water-splitting potential, enabling the ESW to extend up to 3.1 V in the 0.5 M Na2SO4(aq.)/20%AN electrolyte solution. The post-cycling analysis of DyFeO3 demonstrates that although an increase in oxygen vacancies contributes to enhanced charge storage capacity by promoting pseudocapacitive behavior, an excessive concentration of vacancies adversely affects overall electrochemical performance. This is evidenced by increased impedance and a reduction in crystallinity, as well as a decline in the integrated CV area after cycling. Despite these effects, the DyFeO3 electrode maintained its structural integrity, chemical stability, and charge storage functionality. These findings demonstrate the necessity of carefully balancing porosity and oxygen vacancies to optimize both the performance and durability of the electrode over extended cycling.
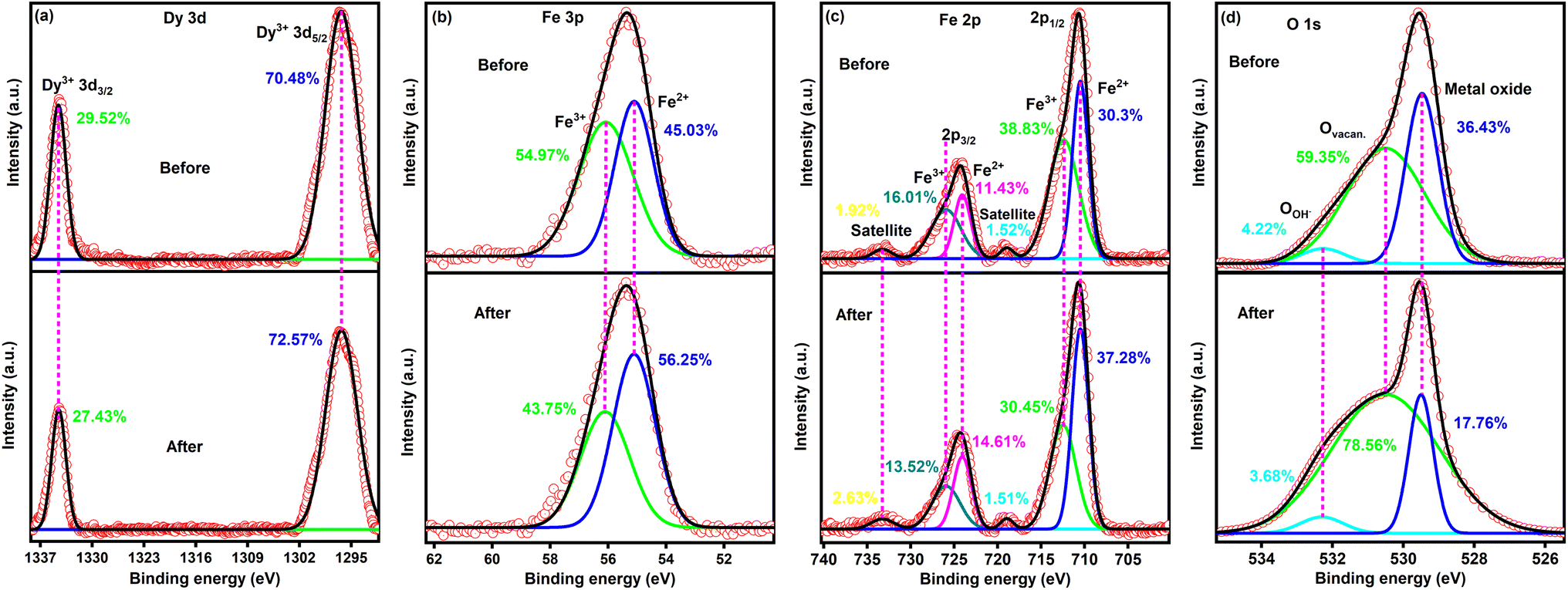 | ||
| Fig. 9 XPS spectra of DyFeO3 nanoparticles before and after 10000 GCD cycles in 0.5 M Na2SO4(aq.)/20%AN electrolyte. (a) The Dy 3d spectrum remains largely unchanged, indicating stable Dy sites. (b) and (c) The Fe 3p and Fe 2p spectra show an increase in Fe2+ concentration after cycling, highlighting iron's active role in redox reactions. (d) The O 1s spectrum reveals a significant increase in oxygen vacancies (from 59.35% to 78.56%) and a decrease in the metal oxide peak (from 36.43% to 17.76%), suggesting enhanced charge storage capacity. The reduction in OH groups indicates decreased free water activity, likely contributing to the extended ESW up to 3.1 V. | ||
3.3 Charge storage mechanism of ASSC-2
The charge storage mechanism of ASSC-2, illustrated schematically in Fig. 10, involves a synergistic combination of EDL capacitance and pseudocapacitance. The system uses porous DyFeO3 nanoparticles as the electrode material, a 0.5 M Na2SO4(aq.)/20%AN mixture as the electrolyte, Whatman filter paper as the separator, and graphite sheets as current collectors. During charging, processes such as ion intercalation, surface adsorption, and redox reactions take place, and these processes are reversed during discharging.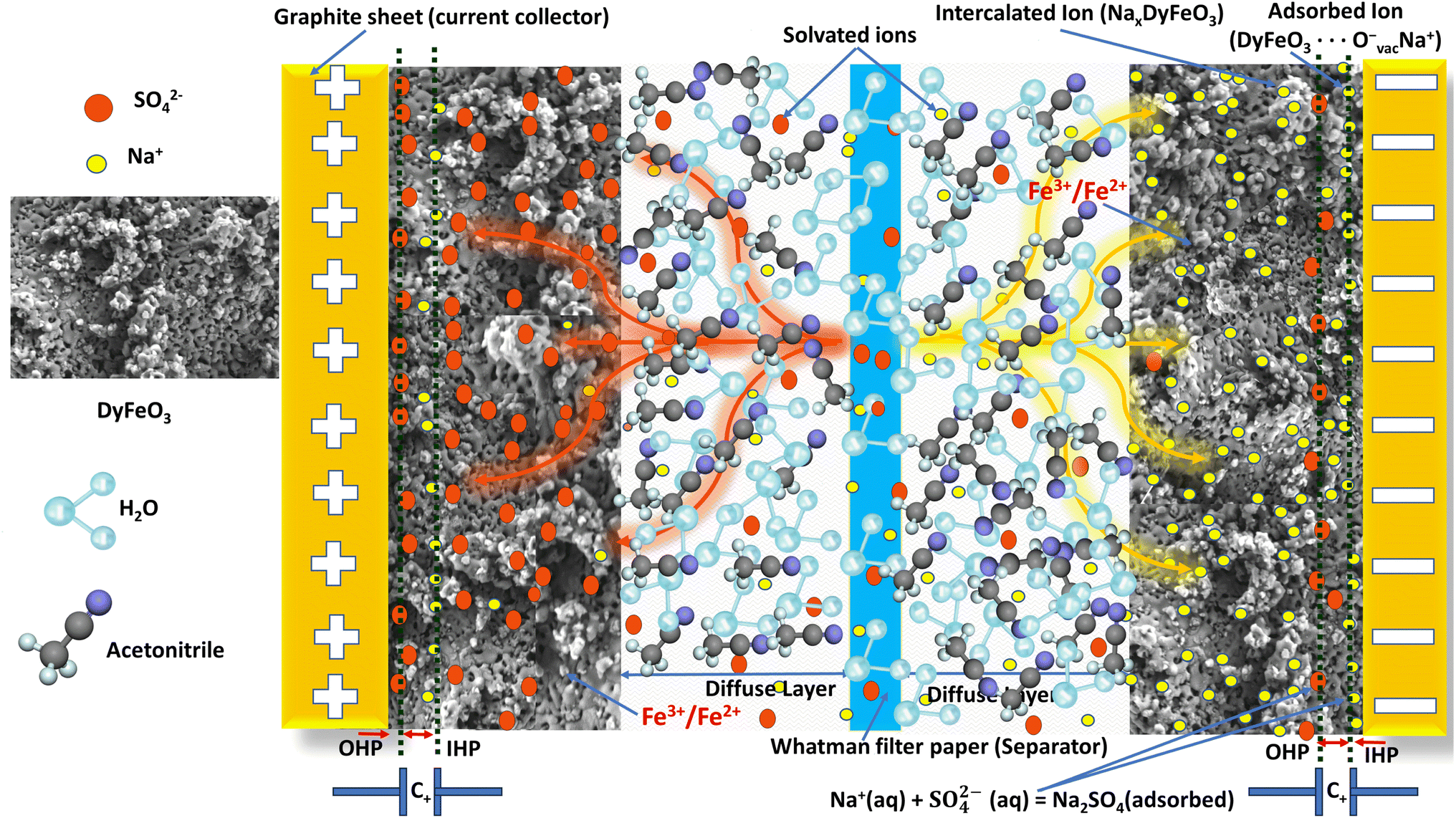 | ||
| Fig. 10 Schematic illustration of charge storage mechanism of the ASSC-2. Upon the application of voltage, the electrolyte ionizes, producing Na+ and SO42− ions. These ions migrate toward the electrodes, forming an inner Helmholtz plane (IHP) adjacent to the electrode surface and an outer Helmholtz plane (OHP) extending into the bulk electrolyte. The interaction between the IHP and OHP defines the EDL architecture. This setup enables efficient charge separation, creating a high-capacitance region near the electrode surface. DyFeO3 nanoparticles, known for their porous structures, play a crucial role by intercalating electrolyte ions. This intercalation enhances the system's pseudocapacitive behavior and charge storage capacity. The electrolyte's solvation structure, along with DyFeO3's porous structure and oxygen vacancies, collectively contribute to increasing the ESW to 3.1 V. Thus, the intricate charge storage mechanism and the energy storage potential of the system are demonstrated. | ||
Na+ ions from the electrolyte are attracted to the electrode and intercalate into the DyFeO3 structure, specifically targeting sites associated with oxygen vacancies. These oxygen vacancies, confirmed by Raman, EPR, and XPS analyses, create the necessary sites for accommodating Na+ ions. This intercalation stabilizes the electrode material through electrostatic interactions. The intercalation process is represented by:
| DyFeO3 + xNa+ + xe− → NaxDyFeO3 |
Simultaneously, Na+ ions are adsorbed onto the surface of DyFeO3 nanoparticles, contributing to the formation of the IHP. Oxygen vacancies are crucial in this process, as they act as negatively charged sites that attract and retain Na+ ions, further stabilizing the double layer. This can be expressed as:
| DyFeO3 + Ovac−⋯Na+ → DyFeO3⋯Ovac−Na+ |
Beyond the IHP, the OHP forms as solvated Na+ and SO42− ions loosely adhere to the electrode surface. The porous structure of DyFeO3 facilitates the approach of these ions, allowing them to interact with the surface mainly through electrostatic forces. This interaction contributes to the non-faradaic capacitance of the system:
| Na+(aq.) + SO42−(aq.) ⇌ Na2SO4(adsorbed) |
Simultaneously, pseudocapacitance is generated through faradaic redox reactions at the electrode surface. The applied potential reduces Fe3+ to Fe2+ within the DyFeO3 lattice, a process that is enhanced by the oxygen vacancies, which serve as active sites for electron transfer. This reaction can be described as:
| Fe3+ + e− → Fe2+ |
The reduction of Fe3+ to Fe2+ plays a crucial role in the pseudocapacitance, which is a key component of the overall charge storage mechanism. The reversible nature of this redox process is demonstrated by the shifts in the Fe 2p and Fe 3p peaks in XPS spectra before and after 10000 GCD cycles, underscoring the durability and stability of the electrode material under repeated cycling.
| NaxDyFeO3 → DyFeO3 + xNa+ + xe− |
Similarly, the Na+ ions that were adsorbed onto the electrode surface during charging are desorbed, leading to the discharge of the EDL:
| DyFeO3⋯Ovac−Na+ → DyFeO3 + Ovac− + Na+ |
The ions within the OHP, which had adhered to the electrode surface, are also released back into the bulk electrolyte, contributing to the reduction in potential across the SC, completing the discharge cycle:
| Na2SO4(adsorbed) ⇌ Na+(aq.) + SO42−(aq.) |
Furthermore, during discharging, the Fe2+ ions in DyFeO3 are oxidized back to Fe3+, a reaction that releases electrons contributing to the discharge current:
| Fe2+ → Fe3+ + e− |
This reversible redox process is critical for maintaining the pseudocapacitance over numerous cycles.
The inclusion of AN in the electrolyte significantly extends the ESW of the SC up to 3.1 V. AN's low dielectric constant reduces the solvation energy of ions like Na+ and SO42−, which promotes ion pairing. This stabilizes the electrolyte and reduces the likelihood of water splitting at higher potentials:
| Na+(aq.) + AN ⇌ Na+(solvated by AN) |
Additionally, AN molecules adsorb onto the electrode surface, forming a hydrophobic layer that effectively blocks water molecules from reaching the electrode. This barrier prevents unwanted reduction and oxidation reactions, enabling the SC to operate at higher voltages without compromising stability:
| DyFeO3⋯AN(adsorbed) + H2O → DyFeO3⋯AN(blocking water) |
This mechanism reduces water activity near the electrode, enabling the device to operate stably under elevated potentials and thereby improving the performance and reliability of ASSC-2. As shown in Fig. S15,† our fabricated ASSC-2 successfully illuminated an LED, demonstrating the practical utility of DyFeO3 as an electrode material for high-voltage ASSCs.
4 Conclusions
In aqueous-based symmetric systems, the operational voltage is typically limited to less than 1.23 V due to the thermodynamic breakdown potential of water. However, we present a significant breakthrough: the first demonstration of a 2.5 V aqueous symmetric SC. This innovative design employs single-structured porous DyFeO3 as both the cathode and anode in a pure aqueous 0.5 M Na2SO4 electrolyte. Building on this advancement, our study further demonstrates that introducing just 20% acetonitrile (AN) into the 0.5 M Na2SO4 aqueous electrolyte significantly extends the ESW of the fabricated aqueous symmetric supercapacitor from 2.5 V to 3.1 V, surpassing the limits of typical aqueous systems. The remarkable performance of this aqueous symmetric supercapacitor is attributed to the synergistic interaction of several factors, including oxygen vacancies, the porous nanostructure, varied oxidation states of DyFeO3's constituent elements, and the solvation structure of the electrolyte. Specifically, the use of AN with the 0.5 M Na2SO4 aqueous electrolyte creates an ordered solvation structure and a robust electrode/electrolyte interface, reducing the free activity of water molecules and thus extending the ESW of the electrolyte to 3.1 V. The as-fabricated aqueous symmetric supercapacitor exhibited exceptional metrics, achieving a high capacitance of 253 F g−1 at a current density of 1 A g−1 and an energy density of 84.43 W h kg−1 at a power density of 1550 W kg−1 in the 0.5 M Na2SO4/20%AN electrolyte. Impressively, it retained 86% of its capacitance and 90% Coulombic efficiency after 10000 cycles. The stability and durability of DyFeO3 as an electrode material were further validated through post-cycle characterization using Raman spectroscopy, EPR, zeta potential measurements, electrochemical analysis, and XPS, following 10000 GCD cycles. Our findings are poised to inspire further research into high-performance energy storage devices using porous, single-structured perovskite-based electrode materials in aqueous or aqueous-dominant electrolytes.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors contributed equally to this work. This manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely acknowledge the University Grants Commission of Bangladesh for financial support, which has been instrumental in the successful execution of this research. We also acknowledge Bangladesh University of Engineering and Technology for providing essential resources crucial for conducting our experiments.Notes and references
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, eaan8285 CrossRef CAS.
- J. T. Frith, M. J. Lacey and U. Ulissi, Nat. Commun., 2023, 14, 420 CrossRef CAS.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- Y. Du, Z. X. Cheng, X. L. Wang and S. X. Dou, J. Appl. Phys., 2010, 107, 09D908 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- X. Bu, L. Su, Q. Dou, S. Lei and X. Yan, J. Mater. Chem. A, 2019, 7, 7541–7547 RSC.
- S. Huang, Z. Li, P. Li, X. Du, M. Ma, Z. Liang, Y. Su and L. Xiong, J. Mater. Chem. A, 2023, 11, 15532–15539 RSC.
- B. Pal, S. Yang, S. Ramesh, V. Thangadurai and R. Jose, Nanoscale Adv., 2019, 1, 3807–3835 RSC.
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS.
- J. Xu, X. Ji, J. Zhang, C. Yang, P. Wang, S. Liu, K. Ludwig, F. Chen, P. Kofinas and C. Wang, Nat. Energy, 2022, 7, 186–193 CrossRef CAS.
- D. Jung, L. M. Saleh, Z. J. Berkson, M. F. El-Kady, J. Y. Hwang, N. Mohamed, A. I. Wixtrom, E. Titarenko, Y. Shao and K. McCarthy, et al. , Nat. Mater., 2018, 17, 341–348 CrossRef CAS PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- J. C. Russell, V. A. Posey, J. Gray, R. May, D. A. Reed, H. Zhang, L. E. Marbella, M. L. Steigerwald, Y. Yang and X. Roy, et al. , Nat. Mater., 2021, 20, 1136–1141 CrossRef CAS.
- J. Huang, K. Yuan and Y. Chen, Adv. Funct. Mater., 2022, 32, 2108107 CrossRef CAS.
- Y. Cao, J. Liang, X. Li, L. Yue, Q. Liu, S. Lu, A. M. Asiri, J. Hu, Y. Luo and X. Sun, Chem. Commun., 2021, 57, 2343–2355 RSC.
- J. T. Mefford, W. G. Hardin, S. Dai, K. P. Johnston and K. J. Stevenson, Nat. Mater., 2014, 13, 726–732 CrossRef CAS PubMed.
- X. Zhang, C. Jiang, J. Liang and W. Wu, J. Mater. Chem. A, 2021, 9, 8099–8128 RSC.
- D. Afanasiev, J. Hortensius, B. Ivanov, A. Sasani, E. Bousquet, Y. Blanter, R. Mikhaylovskiy, A. Kimel and A. Caviglia, Nat. Mater., 2021, 20, 607–611 CrossRef CAS PubMed.
- S. Cao, L. Chen, W. Zhao, K. Xu, G. Wang, Y. Yang, B. Kang, H. Zhao, P. Chen and A. Stroppa, et al. , Sci. Rep., 2016, 6, 37529 CrossRef CAS.
- B. Biswas, V. F. Michel, Ø. S. Fjellvåg, G. Bimashofer, M. Döbeli, M. Jambor, L. Keller, E. Müller, V. Ukleev and E. V. Pomjakushina, et al. , Phys. Rev. Mater., 2022, 6, 074401 CrossRef CAS.
- Z. Zeng, Y. Xu, Z. Zhang, Z. Gao, M. Luo, Z. Yin, C. Zhang, J. Xu, B. Huang and F. Luo, et al. , Chem. Soc. Rev., 2020, 49, 1109–1143 RSC.
- V. Manju, R. Rohith, A. T. Prasannakumar, B. Bhavija and S. J. Varma, New J. Chem., 2022, 46, 19874–19887 RSC.
- S. K. Hussain and J. H. Bang, Phys. Chem. Chem. Phys., 2023, 25, 11892–11907 RSC.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- P. M. Shafi, N. Joseph, A. Thirumurugan and A. C. Bose, Chem. Eng. J., 2018, 338, 147–156 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- J. Park, Y.-E. Yoo, L. Mai and W. Kim, ACS Sustain. Chem. Eng., 2019, 7, 7728–7735 CrossRef CAS.
- J. Liang and W. Wu, Appl. Phys. Rev., 2024, 11, 011301 CAS.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 RSC.
- Z. Chang, Y. Yang, M. Li, X. Wang and Y. Wu, J. Mater. Chem. A, 2014, 2, 10739–10755 RSC.
- S.-E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- G. Hasegawa, K. Kanamori, T. Kiyomura, H. Kurata, T. Abe and K. Nakanishi, Chem. Mater., 2016, 28, 3944–3950 CrossRef CAS.
- Q. Dou, S. Lei, D.-W. Wang, Q. Zhang, D. Xiao, H. Guo, A. Wang, H. Yang, Y. Li and S. Shi, et al. , Energy Environ. Sci., 2018, 11, 3212–3219 RSC.
- A. A. Ansari, N. Ahmad, M. Alam, S. F. Adil, S. M. Ramay, A. Albadri, A. Ahmad, A. M. Al-Enizi, B. F. Alrayes and M. E. Assal, et al. , Sci. Rep., 2019, 9, 7747 CrossRef.
- M. Tarek and M. A. Basith, J. Mater. Chem. C, 2023, 11, 16605–16622 RSC.
- F. Yasmeen, M. Tarek and M. A. Basith, ACS Appl. Mater. Interfaces, 2024, 16, 47535–47550 CrossRef CAS.
- M. Islam, M. Tarek, M. A. Adib and M. A. Basith, J. Phys. D: Appl. Phys., 2024, 57, 215302 CrossRef CAS.
- S. A. Melchior, K. Raju, I. S. Ike, R. M. Erasmus, G. Kabongo, I. Sigalas, S. E. Iyuke and K. I. Ozoemena, J. Electrochem. Soc., 2018, 165, A501–A511 CrossRef CAS.
- S. Umezawa, T. Douura, K. Yoshikawa, D. Tanaka, V. Stolojan, S. R. P. Silva, M. Yoneda, K. Gotoh and Y. Hayashi, Energy Environ. Mater., 2023, 6, e12320 CrossRef CAS.
- A. Jaiswal, R. Das, S. Adyanthaya and P. Poddar, J. Mater. Chem. C, 2011, 115, 2954–2960 CAS.
- M. Tarek, F. Yasmeen and M. Basith, J. Mater. Chem. A, 2024, 12, 25475–25490 RSC.
- P. Kumar, H. No-Lee and R. Kumar, J. Mater. Sci.: Mater. Electron., 2014, 25, 4553–4561 CrossRef CAS.
- N. D. Sharma, J. Singh, S. Dogra, D. Varandani, H. K. Poswal, S. Sharma and A. Bandyopadhyay, J. Raman Spectrosc., 2011, 42, 438–444 CrossRef CAS.
- A. T. Nguyen, A. K. Do, D. K. Nguyen, T. N. Tran, C. H. N. Hoang, D. T. Tran, V. T. N. Anh, T. G. Pham, T. H. Bui and X. V. Bui, J. Mater. Sci.: Mater. Electron., 2024, 35, 1404 CrossRef CAS.
- L. Li, F. Chen, J.-Q. Lu and M.-F. Luo, J. Phys. Chem. A, 2011, 115, 7972–7977 CrossRef CAS PubMed.
- D. Chen, F. Niu, L. Qin, S. Wang, N. Zhang and Y. Huang, Sol. Energy Mater. Sol. Cells, 2017, 171, 24–32 CrossRef CAS.
- F.-t. Li, Q. Wang, J. Ran, Y.-j. Hao, X.-j. Wang, D. Zhao and S. Z. Qiao, Nanoscale, 2015, 7, 1116–1126 RSC.
- X. Liu, J. You, R. Wang, Z. Ni, F. Han, L. Jin, Z. Ye, Z. Fang and R. Guo, Sci. Rep., 2017, 7, 13085 CrossRef PubMed.
- K. A. Owusu, L. Qu, J. Li, Z. Wang, K. Zhao, C. Yang, K. M. Hercule, C. Lin, C. Shi and Q. Wei, et al. , Nat. Commun., 2017, 8, 14264 CrossRef.
- D.-Q. Yang and E. Sacher, J. Mater. Chem. C, 2009, 113, 6418–6425 CAS.
- R. Liu, Z. Wang, S. Peng, J. Bi, J. Wu and Z.-G. Ye, J. Am. Ceram. Soc., 2020, 103, 1097–1104 CrossRef CAS.
- J. Liu, C. Wu, I. D. Gates, B. Jia, Z. Huang and T. Ma, Energy Environ. Mater., 2023, 6, e12520 CrossRef CAS.
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- S. A. Al-Muhtaseb and J. A. Ritter, Adv. Mater., 2003, 15, 101–114 CrossRef CAS.
- A. Feinle, M. S. Elsaesser and N. Huesing, Chem. Soc. Rev., 2016, 45, 3377–3399 RSC.
- C. Lei, N. Amini, F. Markoulidis, P. Wilson, S. Tennison and C. Lekakou, J. Mater. Chem. A, 2013, 1, 6037–6042 RSC.
- A. Vu, Y. Qian and A. Stein, Adv. Energy Mater., 2012, 2, 1056–1085 CrossRef CAS.
- Z. Chen, D. L. Danilov, R.-A. Eichel and P. H. Notten, Adv. Energy Mater., 2022, 12, 2201506 CrossRef CAS.
- M. D. Sonntag, Raman Spectroscopy in The Undergraduate Curriculum, American Chemical Society, Washington, DC, 2018 Search PubMed.
- N. Matubayasi, C. Wakai and M. Nakahara, Phys. Rev. Lett., 1997, 78, 2573 CrossRef CAS.
- S. Budavari, The Merck Index, an encyclopedia of chemical drug, and biologicals, Merck technical report, 1989 Search PubMed.
- C. Masarapu, H. F. Zeng, K. H. Hung and B. Wei, ACS Nano, 2009, 3, 2199–2206 CrossRef CAS.
- S.-W. Xu, M.-C. Zhang, G.-Q. Zhang, J.-H. Liu, X.-Z. Liu, X. Zhang, D.-D. Zhao, C.-L. Xu and Y.-Q. Zhao, J. Power Sources, 2019, 441, 227220 CrossRef CAS.
- Z. Meng, J. Xu, P. Yu, X. Hu, Y. Wu, Q. Zhang, Y. Li, L. Qiao, Y. Zeng and H. Tian, J. Chem. Eng., 2020, 400, 125966 CrossRef CAS.
- L. Shen, L. Yu, H. B. Wu, X.-Y. Yu, X. Zhang and X. W. Lou, Nat. Commun., 2015, 6, 6694 CrossRef CAS PubMed.
- W. Chen, C. Xia and H. N. Alshareef, ACS Nano, 2014, 8, 9531–9541 CrossRef CAS PubMed.
- S. Peng, L. Li, H. B. Wu, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2015, 5, 1401172 CrossRef.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- N. Liu, Y. Su, Z. Wang, Z. Wang, J. Xia, Y. Chen, Z. Zhao, Q. Li and F. Geng, ACS Nano, 2017, 11, 7879–7888 CrossRef CAS PubMed.
- J. Chang, M. Jin, F. Yao, T. H. Kim, V. T. Le, H. Yue, F. Gunes, B. Li, A. Ghosh and S. Xie, et al. , Adv. Funct. Mater., 2013, 23, 5074–5083 CrossRef CAS.
- N. Jabeen, A. Hussain, Q. Xia, S. Sun, J. Zhu and H. Xia, Adv. Mater., 2017, 29, 1700804 CrossRef.
- T. Xiong, T. L. Tan, L. Lu, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2018, 8, 1702630 CrossRef.
- Y. Zhao, W. Ran, J. He, Y. Huang, Z. Liu, W. Liu, Y. Tang, L. Zhang, D. Gao and F. Gao, Small, 2015, 11, 1310–1319 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, D.-W. Wang, F. Li, B. Liu and H.-M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed.
- J. Wang, L. Tian, W. Xie, X. Wang, X. Long, K. Sun, A. Emin, D. Liu, Y. Fu and Q. Chen, et al. , ACS Appl. Energy Mater., 2020, 3, 2614–2622 CrossRef CAS.
- C. P. Poole, Electron spin resonance: a comprehensive treatise on experimental techniques, Wiley, New York, 2nd edn, 1983 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06769j |
| This journal is © The Royal Society of Chemistry 2025 |