
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid one-pot microwave-assisted synthesis and defect engineering of UiO-66 for enhanced CO2 capture†
Dong A.
Kang
a,
Amro M. O.
Mohamed
 c,
Christian
Murphy
a,
Andres
Ramos
a,
Ioannis G.
Economou
c,
Jinsoo
Kim
*d and
Hae-Kwon
Jeong
*abd
c,
Christian
Murphy
a,
Andres
Ramos
a,
Ioannis G.
Economou
c,
Jinsoo
Kim
*d and
Hae-Kwon
Jeong
*abd
aArtie McFerrin Department of Chemical Engineering, Texas A&M University, 3122 TAMU, College Station, TX 77843-3122, USA. E-mail: hjeong7@tamu.edu
bDepartment of Materials Science and Engineering, Texas A&M University, 3122 TAMU, College Station, TX 77843-3122, USA
cChemical Engineering Program, Texas A&M University at Qatar, PO Box 23874, Doha, Qatar
dDepartment of Chemical Engineering (Integrated Engineering), Kyung Hee University, 1732 Deogyeong-daero, Yongin, Gyeonggi-do 17104, Republic of Korea. E-mail: jkim21@khu.ac.kr
First published on 29th November 2024
Abstract
UiO-66 and its derivative consisting of zirconium oxide clusters and terephthalate-based linkers stand out as some of the most extensively studied metal–organic frameworks (MOFs) for various applications owing to their exceptional stability as compared with other MOFs. However, practical applications often require the rapid synthesis of highly crystalline UiO-66 and its derivatives and the facile engineering of their defects. Herein, we present the rapid formation of UiO-66 at ambient pressure under microwave irradiation. More importantly, we control the defectivity of UiO-66 simply by modulating microwave power. Lower microwave power results in more defective UiO-66, exhibiting higher textural properties than theoretical values, attributable to the concurrent increase in the linker and cluster defects in the framework. The most defective UiO-66 in this work exhibits unexpectedly high CO2/N2 adsorption selectivity (ca. 41), far surpassing that of all other previously reported UiO-66 (<ca. 25). Both the experimental and computational results confirm that the unusually high CO2/N2 selectivity of the most defective UiO-66 is likely due to the relatively high concentration of energetically favorable adsorption sites generated under microwave irradiation. Computational studies at the molecular level confirm that the unexpectedly high CO2 heat of adsorption is due to surface heterogeneity, specifically the local distribution of defective sites with varying terminations, rather than the overall concentrations of each terminal group in the UiO-66 crystal.
1 Introduction
UiO-66, consisting of zirconium oxide clusters and terephthalate linkers, exhibits exceptional structural stability under thermal, acidic, aqueous and high-pressure conditions, attributed to the robustness of the zirconium-oxide clusters in its crystalline framework and the high coordination number of zirconium centers.1 This inherent stability renders it an ideal candidate for many applications including acidic gas adsorption,2 catalysis,3 sensors4 and water purification.5 Furthermore, the terephthalate linker of UiO-66 can be readily substituted with its functionalized counterparts such as 2-aminoterephthalates (i.e., UiO-66-NH2), thereby enabling precise modification of pore sizes and surface functionalities.6Conventional solvothermal synthesis of UiO-66 requires reaction temperatures above 80 °C typically for more than 24 hours.6,7 For example, Lillerud et al.1 first synthesized UiO-66 solvothermally at 120 °C for 24 hours. Later, the same group8 examined the effects of synthesis temperatures ranging from 100 °C to 220 °C for a fixed synthesis time of 20 hours on the UiO-66 structure. They found that lower temperatures created more defects in the framework, consequently increasing the surface area to ca. 1475 m2 g−1 from ca. 1110 m2 g−1. It was hypothesized that higher synthesis temperatures helped “iron out” these defects by shifting the solution equilibrium in favor of linker-Zr bonds. Farha et al.9 reported a facile solvothermal synthesis of UiO-66 at 80 °C overnight by adding hydrochloric acid (HCl) to the synthesis solution. They observed that UiO-66 was formed within 2 hours with HCl, while no UiO-66 was formed without HCl in the same period. UiO-66 synthesized with higher HCl concentrations exhibited a surface area of up to ca. 1580 m2 g−1, compared to ca. 730 m2 g−1 of UiO-66 synthesized with the least amount of HCl. This increase in surface area was attributed to the creation of more defects due to the loss of linkers in the framework. It is reported that strong acid inhibits deprotonation of the linkers, thereby increasing defects in the UiO-66 structure.6
To reduce required temperature for the solvothermal synthesis of UiO-66, Farha et al.10 proposed a two-step synthesis approach where the time-consuming second step (18 hours) could occur at a lower temperature than the conventional solvothermal method. First, they started with zirconium propoxide (Zr(OC3H7)4) as a zirconium source and then converted it into Zr6O4(OH4)(OAc)12 (OAc = CH3COO−, acetate ligand) as secondary building units (SBUs) by carrying out a solvothermal reaction at 130 °C for 2 h. In the second step, UiO-66 linkers were added to the solution of the pre-formed SBUs to replace the labile acetate ligands coordinated to the zirconium oxide cluster, thereby forming UiO-66 crystals even at 25 °C for 18 hours. They reported that the textural properties of the obtained UiO-66 were similar to textural properties of those previously reported from the conventional solvothermal synthesis method. Furthermore, they engineered defects in UiO-66 by varying temperature in the second step from 25 °C to 130 °C, where more defective MOFs were obtained at the lower temperature.
As a rapid and energy-efficient alternative, microwave-assisted synthesis methods have been investigated for the synthesis of various MOFs including UiO-66 and its derivatives.11–17 Microwaves (MWs) can provide rapid volumetric heating throughout the synthesis solution, leading to shorter reaction time and improved energy efficiency.11,18 However, most reported MW-based synthesis approaches required the reaction temperature to be fixed at certain elevated temperature throughout reactions, mimicking conventional solvothermal synthesis, while their detailed conditions such as MW power, radiation time, and temperature may vary (see Table S1†).13,15–17,19,20 For example, Li et al.17 first synthesized UiO-66 using MW heating with acidic modulators. The synthesis solutions were irradiated at 100 °C for 2 hours, resulting in the highest surface area of ca. 1661 m2 g−1 when benzoic acid was used. Efforts have also been made to develop continuous flow reactor synthesis of MOFs to increase production throughput compared to the conventional batch method in an autoclave.21 Kim et al.13 proposed a MW-assisted continuous tubular reactor for the synthesis of UiO-66, achieving this with a MW residence time of 10 minutes and a reaction temperature of 120 °C. These advancements highlight the potential of MW-assisted synthesis methods in the rapid production of MOFs, offering both efficiency and scalability. It is noted that all MW-assisted synthesis of UiO-66 reported so far used ZrCl4 as the metal precursor as summarized in Table S1,† indicating that MW heating was effective in overcoming the relatively high activation energy for forming SBUs.
Over the last decade or so, UiO-66 has garnered significant interest for CO2 capture due to its remarkable moisture stability along with its relatively high surface area.22 Moisture stability is crucial for practical CO2 capture where water vapor is commonly present in gas streams such as post-combustion flue gases.23 Walton et al.24 reported that solvothermally synthesized UiO-66 showed a CO2 uptake capacity of ca. 0.55 mmol g−1 at 0.15 bar CO2 and a CO2/N2 selectivity of ca. 22.8 at 25 °C. D'Alessandro et al.15 investigated defective UiO-66 synthesized by MW heating in the presence of acid modulators for CO2 capture applications. The defective UiO-66 showed lower CO2 uptake compared to its pristine counterpart synthesized without acids. They suggested that although defective UiO-66 samples possessed a higher surface area, the larger pore size created by defects hindered the confinement of CO2 molecules within the framework, resulting in reduced CO2 uptake in defective samples. However, there are no studies on systematic generation of defects and their structures and their effects on the carbon capture properties of the resulting defective UiO-66.
Here, we report a rapid one-pot MW-assisted approach, where UiO-66 could be rapidly formed under much shorter MW irradiation (i.e., 90 seconds) than typical cases (i.e., several minutes to hours). Since zirconium propoxide is used as the metal precursor, unlike previous reports, our MW synthesis does not require preheating of the precursor solution, thereby considerably simplifying the synthesis process. More importantly, the defectivity of UiO-66 was systematically varied by controlling MW power. Finally, the CO2/N2 adsorptive separation properties of MW-synthesized UiO-66 with different defectivities were measured and compared with those of their counterparts synthesized under conventional heating. Moreover, we employed computational techniques to investigate the structure of UiO-66 with different types of defects, focusing on properties such as geometric pore volume and surface area. Molecular simulations were conducted to examine how CO2 binding energy varies with the defect number and termination type. The variation in CO2 heat of adsorption is an important metric to explain the presence of defective structures with high CO2/N2 selectivity.
2 Experimental section
2.1 Materials
Zirconium(IV) propoxide solution (Zr(OC3H7)4, 70 wt% in 1-propanol), terephthalic acid (TA, 98%), acetic acid (>99.7%), dimethylformamide (DMF, >99%), and methanol (MeOH, >99.8%) were purchased from Sigma-Aldrich. All chemicals were used as received without further purification.2.2 Microwave-assisted synthesis of UiO-66
Precursor solutions were prepared by dissolving 0.2 g of TA and 0.6 g of the Zr(OC3H7)4 solution in a mixture of 16 ml of acetic acid and 28 ml of DMF. The prepared solution was then transferred to a MW-inert glass tube, followed by MW radiation at a fixed MW power for 90 s in a MW synthesizer (CEM, Discover-SP w/ActiVent®). The MW power was varied from 50 W to 200 W. The resulting samples are denoted as xW where x is the MW power. After the reaction, the glass tube was allowed to cool naturally to room temperature (RT). The synthesized powders were subsequently collected through centrifugation at 8000 rpm for 15 min and subjected to a single wash with DMF (20 ml) followed by washing twice with MeOH (20 ml each). Finally, the collected powders were dried at 150 °C for 1 day in a preheated oven for characterization. The detailed conditions are summarized in Table S2.† For comparison, synthesis of UiO-66 was performed in a heated oil bath with magnetic stirring under the same conditions. Syntheses were conducted at room temperature, 50 °C, and 100 °C in closed round bottom flasks for 3 h to obtain sufficient powders for further characterization. The resulting samples are denoted as TH-y where y is the synthesis temperature.2.3 Characterization
Powder X-ray diffraction (PXRD) patterns were acquired using a Miniflex II X-ray diffractometer from Rigaku, employing CuKα radiation (λ = 1.5406 Å). Scanning electron microscope (SEM) images were captured using a JEOL JSM-7500F instrument, operating at an acceleration voltage of 5 keV and utilizing a lower secondary electron image (LEI) detector. Thermogravimetric analysis (TGA) was performed with a Q5000 apparatus (TA Instruments) in a temperature range of 25–800 °C with a ramp rate of 5 °C min−1 under an air flow rate of 50 cm3 min−1. Nitrogen isotherms at 77.3 K were recorded using an Autosorb iQ-C-MP instrument from Anton Paar Quanta Tec. Before obtaining isotherms, powder samples were degassed at 150 °C for 24 h. BET surface area was estimated at p/p0 of 0.01–0.1 and micropore volume was estimated by the t-plot method at p/p0 of 0.2–0.5. Pore size distribution was estimated by the non-local density functional theory (NLDFT) method. Adsorption isotherms for CO2 and N2 were obtained using an ASAP 2020 Plus instrument (Micromeritics) at temperatures of 30 °C and 40 °C, over an absolute pressure range of 0.0002 to 1 bar. Before conducting the gas adsorption experiments, the samples were degassed under vacuum at 150 °C. The isosteric heat of adsorption (ΔHads) for CO2 was determined by applying eqn (1) and (2) and fitting the adsorption isotherms to a virial-type equation (eqn (3)).25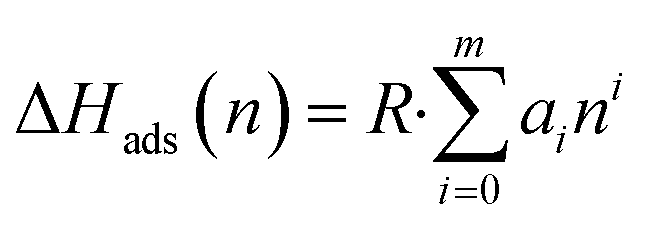 | (1) |
| ΔH0ads = R·a0 | (2) |
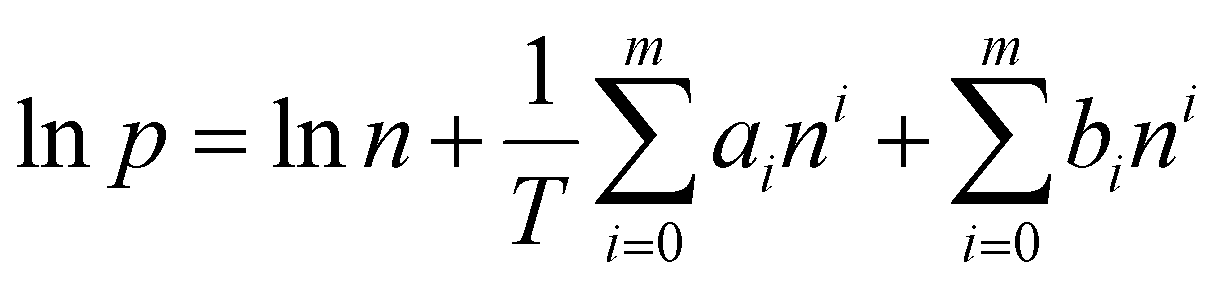 | (3) |
2.4 Molecular simulation
Static binding energies for CO2 at 0 K were determined using the dispersion-corrected semi-empirical DFT-D3 density functional theory method. All computations were carried out with the CP2K/QUICKSTEP simulation package,26 employing a plane-wave energy cut-off of 500 eV and a Gamma-point mesh for Brillouin zone sampling. The projector-augmented-wave (PAW) method described the interactions between core and valence electrons, while the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) functional27 was used for the exchange–correlation functional. Core electrons were represented by Goedecker–Teter–Hutter (GTH) PBE pseudopotentials.28 The optimized Gaussian basis sets (MOLOPT)29 used TZV2P-MOLOPT contracted Gaussian basis sets.The initial position of CO2 in the periodic cell of defective and defect-free UiO-66 tetrahedral cages was determined via classical simulated annealing, where the temperature was progressively lowered to enable the gas molecule to achieve an optimal configuration through rotations, translations, and repositioning with predetermined probabilities. This heating and cooling process was repeated in several cycles, with forty heating cycles performed, reaching a maximum temperature of 1000 K and a final temperature of 100 K.
Static binding energies (ΔE) at 0 K were calculated using the formula:
| ΔE = Ecluster+CO2 − Ecluster − ECO2 | (4) |
To study the effect of defects on structural properties of UiO-66, Zeo++ was used to estimate the surface area, the total pore volume and other structural properties including the largest cavity parameter (LCD) and pore limiting diameter (LPD).30 Surface area and atomistic pore size distribution were estimated using a N2 probe of 1.86 Å.
3 Results and discussion
3.1 One-pot ambient pressure MW-assisted synthesis of UiO-66
Fig. S1 and Table S1† compare our MW-assisted approach with previously reported MW methods. A couple of differences can be observed. First, our approach utilizes Zr(OC3H7)4 as a metal source with acetic acid as a modulator, whereas conventional MW-assisted methods use ZrCl4. Zr(OC3H7)4 is chosen because it enables formation of UiO-66 even at temperature as low as 20 °C due to the formation of more labile intermediate zirconium oxide clusters capped with acetate ligands while hydroxylated intermediate zirconium oxide clusters are formed with ZrCl4.6,10,31,32 This difference allows for much milder synthesis conditions such as 50 W for 90 s without the need for relatively high reaction temperatures above 100 °C, typically required in previous MW-assisted syntheses (see Table S1†).13,15–17,19 Second, our MW synthesis is carried out under ambient pressure whereas most of the previous MW syntheses were under autogenous pressures. Ambient pressure synthesis combined with the above-mentioned lower MW power and shorter synthesis time provides our MW synthesis with a significant advantage in cost-effective large-scale production of MOFs. In summary, our MW synthesis is carried out at autogenous temperature under ambient pressure while conventional MW syntheses are conducted at fixed temperature under autogenous pressure (i.e., solvothermal conditions).Fig. S2† shows photographs captured during the UiO-66 synthesis process under MW irradiation. As depicted in Fig. S2a–d,† the higher the MW power the faster the reaction, leading to the formation of a more turbid dispersion. For instance, MW radiation at 200 W enabled formation of UiO-66 crystals in 90 seconds (see Fig. S2a†). It is noted that the final temperature of the solution, when irradiated at 200 W, reached approximately 126 °C. No significant volume change was observed before and after MW irradiation (see Fig. S3†), suggesting that though the reaction pressure was maintained at ambient pressure, there was no concentration change. Without MW, however, it took over 90 minutes for the precursor solution to transition into a translucent dispersion of UiO-66 particles at RT (Fig. S4†). The yield of UiO-66 is presented in Fig. S5,† which increases as MW power increases. This can be attributed to the faster reaction due to more rapid heating of solutions as MW power increased.
3.2 Crystallinity and morphology of UiO-66
Fig. 1 presents the XRD patterns of the obtained powders at different MW powers. While the diffraction patterns closely resemble the simulated pattern of UiO-66, variations in peak intensities are observed with changes in MW power. It is known that the defects of UiO-66 (i.e., crystallinity) influence diffraction peak intensities with higher defects corresponding to lower intensities.33 As can be seen in the figure, the peak intensity gradually increases as MW power increases from 50 W to 200 W, particularly noticeable at a 2θ of approx. 7.4°, corresponding to the (111) plane. This increase in the (111) peak suggests a decrease in defects as MW increases. The quantitative analysis of the defects of the obtained powders is presented in Section 3.3. Furthermore, an additional broad peak in the 2θ range of 3–5° emerges and becomes more prominent as MW power increases (see the inset of Fig. 1). As with other MOFs, UiO-66 exhibits two types of defects: linker defects (missing linkers) and cluster defects (missing metal clusters). Unlike linker defects, cluster defects in UiO-66 result in reo nanoregions, inducing an additional broad peak at 2θ of 2–7°.6,34,35 This observation indicates that as MW power increases, cluster defects increase while defectivity (both linker and cluster defects) decreases.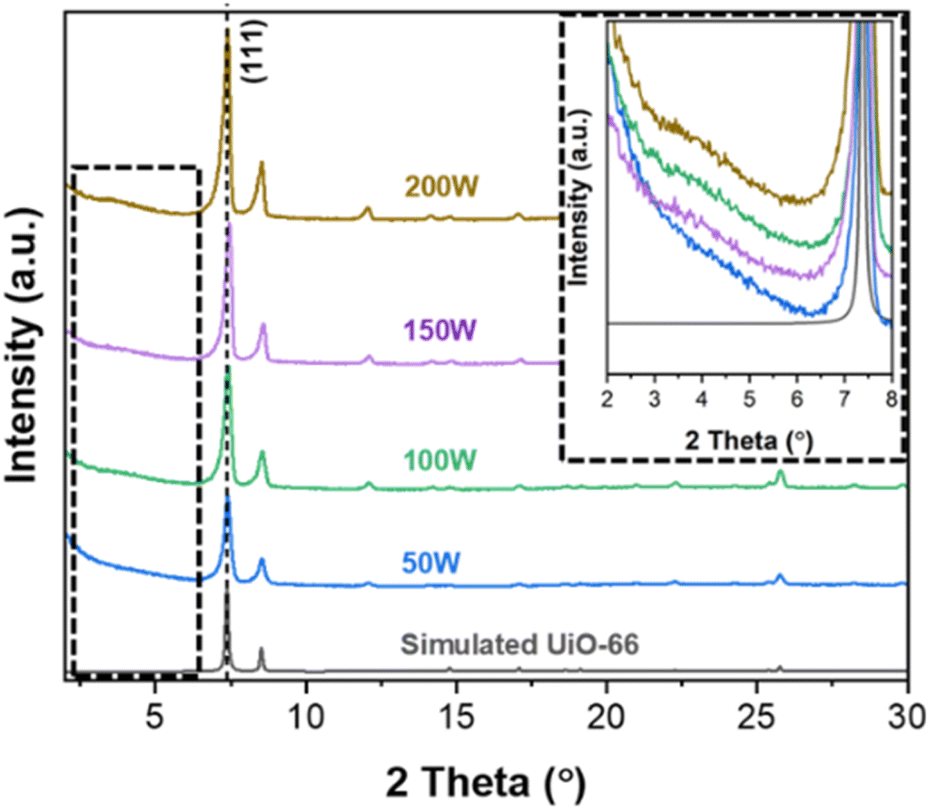 | ||
| Fig. 1 PXRD patterns of UiO-66 synthesized under MW irradiation with varying MW power. | ||
Fig. 2 presents the SEM images of the obtained UiO-66 powders at varying MW power. As seen in the figure, the particle size increases from ca. 330 nm to ca. 1 μm as MW power increases (see Fig. S7†). As explained in the previous section, higher MW power means a faster temperature increase and higher final temperature, leading to faster crystal growth. The samples prepared at 150 and 200 W display a typical well-defined octahedral morphology while the morphology of the 100 W sample is slightly less defined. In contrast, the sample at 50 W shows a poorly defined octahedral morphology with macroscopic defects (see the inset image in Fig. 2a). The macroscopic defects of the 50 W sample (Fig. 2a) are likely due to its highest defectivity (i.e., poor crystallinity).
 | ||
| Fig. 2 SEM images of UiO-66 prepared under MW irradiation with varying MW power: (a) 50 W, (b) 100 W, (c) 150 W, and (d) 200 W. | ||
3.3 The defectivity of UiO-66
The defectivity of MOFs has significant effects on the properties of MOFs. As such, we attempted to control the defectivity of UiO-66 by simply modulating MW power rather than by varying acidic/basic modulators as reported in other previous studies. According to Farha et al.,10 when Zr(OC3H7)4 is used as the metal source, labile intermediate zirconium oxide clusters (Zr6O4(OH)4(OAc)12) are formed. At elevated synthesis temperatures, the capping ligands (i.e., acetates) of these clusters become more labile, facilitating increased linker replacement and resulting in less defective UiO-66. Fig. 3a presents the TGA thermograms of UiO-66 samples synthesized with varying MW power. Fig. S8† outlines a comprehensive procedure to estimate linker deficiencies per Zr6 unit (i.e., defectivity) from the TGA results.6,8,35Fig. 3b shows the estimated defectivity (denoted as d) of UiO-66 as a function of MW power. The estimated defectivity aligns well with the XRD analysis, indicating a decrease in the defectivity as MW power increases (i.e., as temperature increases), consistent with the observation by Farha et al.10 As presented in Fig. 3a and S8b,† the weight loss up to ca. 390 °C, which is attributed to the dehydroxylation and the decomposition of acetate ligands, decreases with increasing MW power. It is noted that the loss of acetate groups (ca. 325–390 °C) is markedly reduced as MW power increases (Fig. S8b†), indicating that the samples prepared at higher MW power have fewer capping acetate ligands present. For example, approximated weight ratios of decomposed acetate/hydroxyl are 3.5 in the 50 W sample and 0.94 in the 200 W sample respectively, which were estimated from integrating DTG peaks in Fig S8b.† In other words, capping acetate ligands of the intermediate clusters are more readily replaced by linkers at higher MW power (i.e., more rapid heating and higher temperature), consequently forming less defective UiO-66.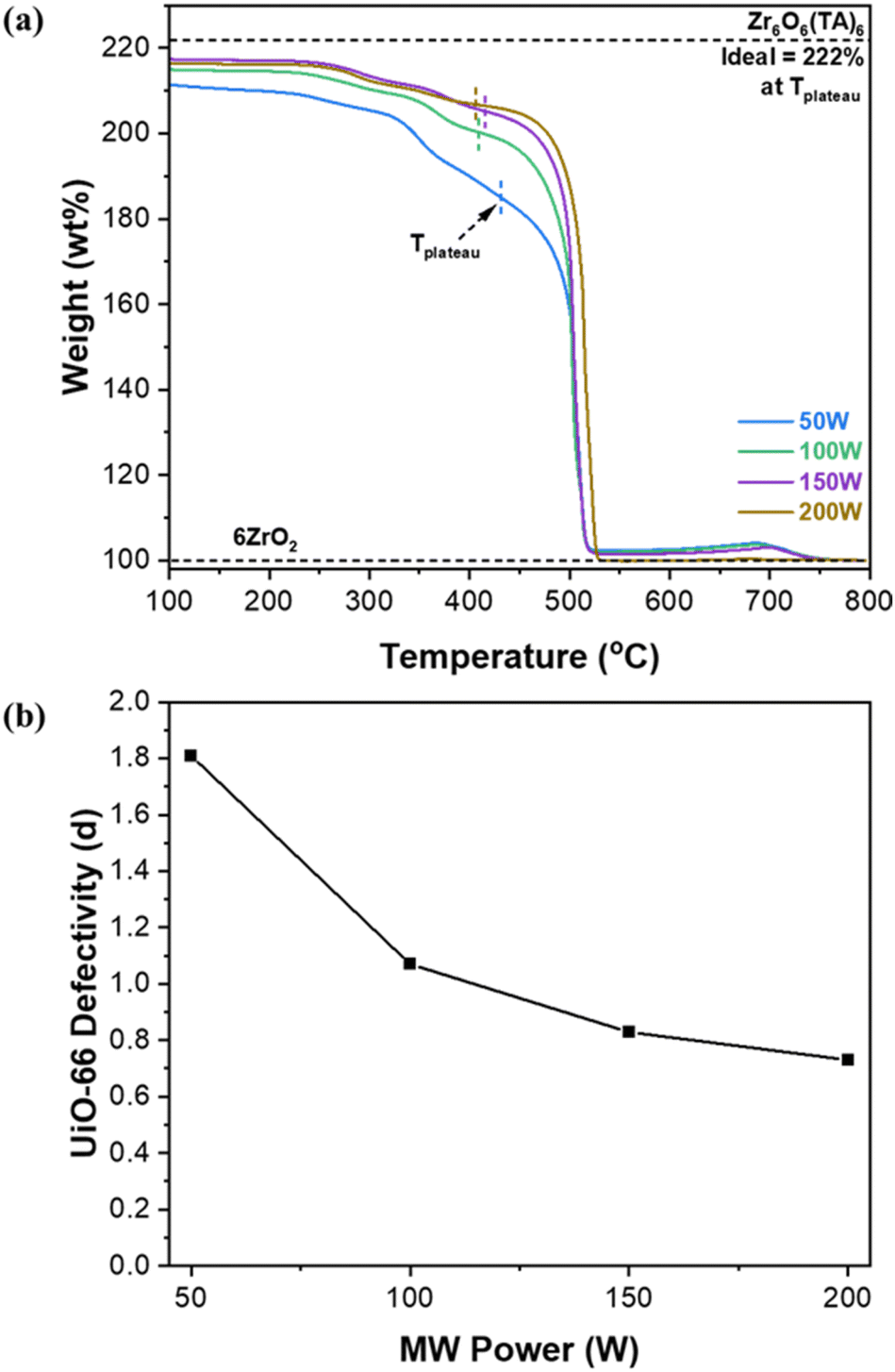 | ||
| Fig. 3 (a) TGA thermograms of UiO-66 synthesized under MW irradiation with varying MW power and (b) defectivity (d) of MW-assisted synthesized UiO-66 as a function of MW-radiation power. | ||
3.4 Textural properties of UiO-66
Fig. S9† and 4a present N2 isotherms at 77 K and estimated BET (Brunauer–Emmett–Teller) surface area and micropore volume of UiO-66 samples as a function of MW power. Table S3† summarizes the textural properties of the samples along with their defectivity. The theoretical surface area and pore volume of defect-free UiO-66 are known to be ca. 954 m2 g−1 and ca. 0.426 cm3 g−1, respectively.36 As shown in Fig. 4a and Table S3,† as MW power increases (i.e., as defectivity decreases), both BET surface area and micropore volume decrease from ca. 1731 m2 g−1 to ca. 1148 m2 g−1 and from ca. 0.638 cm3 g−1 to ca. 0.444 cm3 g−1, respectively. Notably, the obtained surface areas are comparable to or higher than the theoretical values and the previously reported results by MW-assisted synthesis methods (see Table S1†). As discussed above, UiO-66 samples synthesized at lower MW powers are more defective and contain more acetate terminal groups than hydroxyl terminal groups. It is surmised that higher defectivity in the UiO-66 framework would generate more micropores (with pore widths less than 2 nm), resulting in increased microporosity (i.e., higher BET surface area and higher micropore volume), as shown in Table S3.†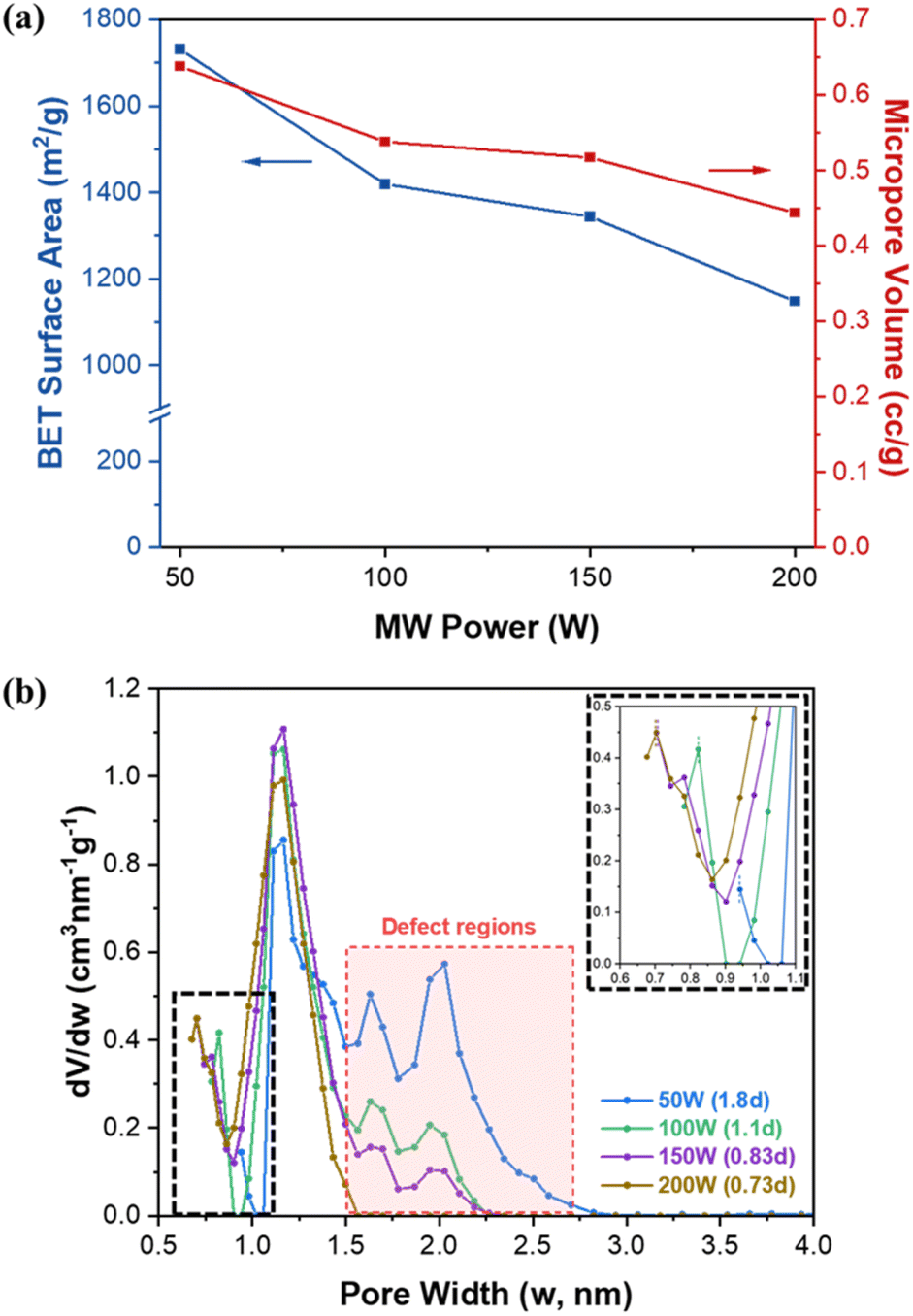 | ||
| Fig. 4 (a) BET surface area and micropore volume of UiO-66 synthesized under MW irradiation as a function of MW power and (b) pore size distribution of UiO-66 with varying MW power. | ||
Fig. 4b shows the pore size distribution of UiO-66 synthesized at different MW powers. UiO-66 possesses two different kinds of cages with diameters of ca.7.5 Å (tetrahedral cage) and ca. 12 Å (octahedral cage).6 The least defective sample (200 W) exhibits a pore size distribution similar to theoretical values with two peaks located at ca. 7.0 Å and 12 Å. As defectivity increases with decreasing MW power, the peak corresponding to the tetrahedral cage shifts towards larger pore widths from ca. 7.0 Å to ca. 9.4 Å with a corresponding decrease in pore volume, while the peak of the octahedral cage remains at ca.12 Å. From 150 W to 50 W, two additional peaks emerge at ca. 16 Å and 20 Å, indicating the presence of larger pores. Moreover, the pore volume of these larger pores increases as MW power decreases. It has been reported that the disordered nanoregions created by cluster defects in UiO-66 form larger cavities with a width of ca. 17 Å, while linker defects create smaller cavities with a width of ca. 8.9 Å.37 Based on the results discussed above, it seems that the density of both linker defects and cluster defects (i.e., defectivity) of UiO-66 increases as MW power decreases.
3.5 CO2 and N2 isotherms of UiO-66
Fig. 5a presents the single gas adsorption isotherms of CO2 and N2 at 30 °C. Fig. 5b shows the corresponding Ideal Adsorbed Solution Theory (IAST) CO2 uptake at 0.15 bar and CO2/N2 adsorption selectivity as a function of MW power. The IAST calculation using GraphIAST38 is based on the CO2 and N2 partial pressures of 0.15 bar and 0.85 bar, respectively. As shown in Fig. 5b, the CO2 uptake capacity decreases from ca. 0.39 mmol g−1 to ca. 0.23 mmol g−1 (ca. 41% decrease) as MW power decreases (i.e., as defectivity increases). This trend is somewhat counter-intuitive because the N2 porosimetry shows increasing surface areas and pore volumes with increasing defectivity (Fig. 4a and Table S3†). This reduction in the CO2 capacity could, however, be attributed to a diminished gas confinement effect resulting from enlarged pores associated with increased defectivity. Davis et al.39 emphasized the importance of the confinement effect for CO2 adsorption at low pressures, and D'Alessandro et al.15 observed a similar trend, with CO2 uptake decreasing at 1 bar but increasing at 35 bar as UiO-66 became more defective and its surface area increased. In other words, narrower pores are more effective up to a certain limit where the adsorbate size exceeds the pore size.40 As defectivity (d) increases, the enlarged pores up to ca. 9.4 Å resulting from linker defects, along with the larger pores of ca. 16 Å and 20 Å due to cluster defects, increase, thereby reducing the confinement effect. This leads to decreased CO2 uptake despite the increase in surface area and pore volume.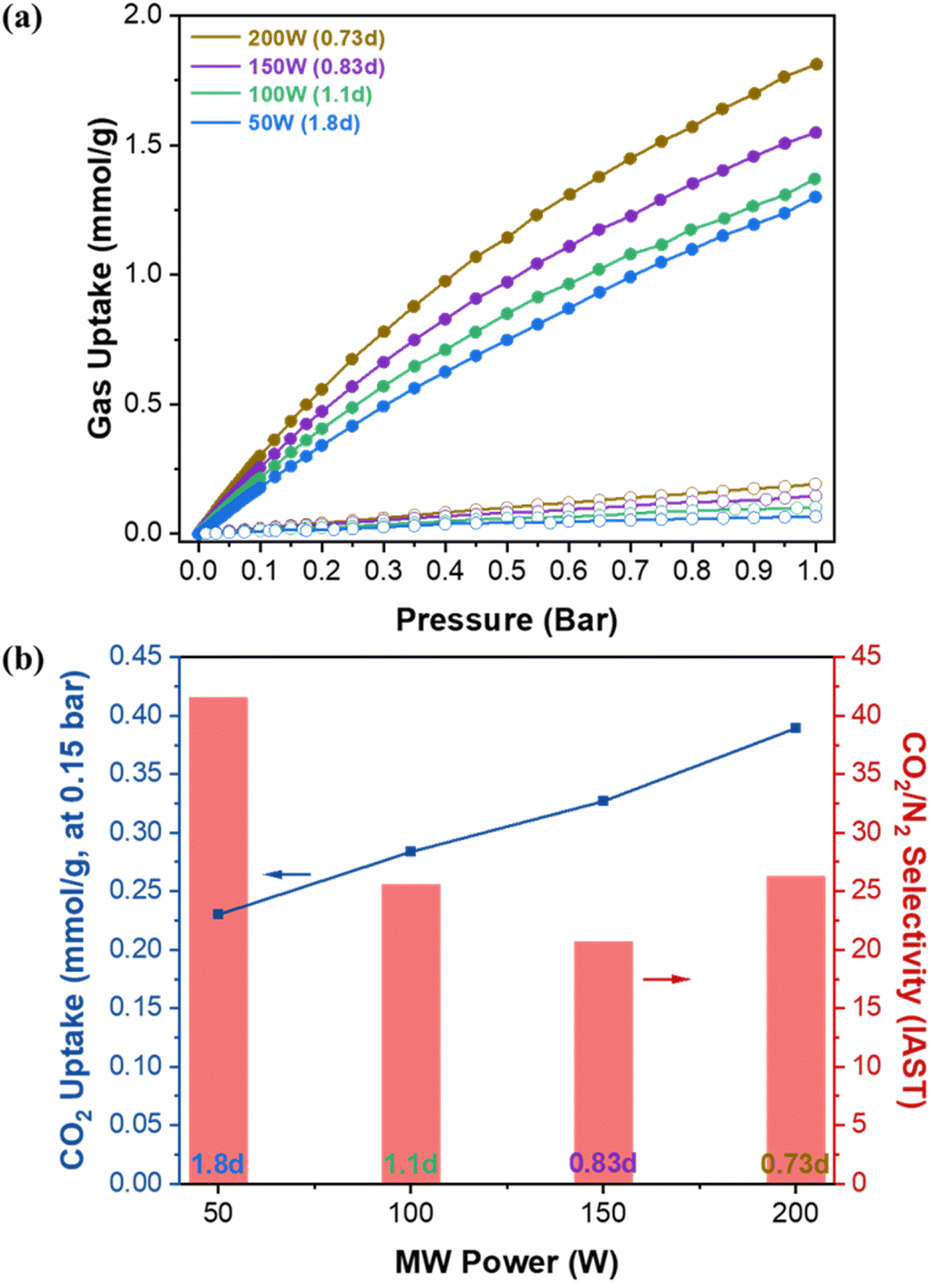 | ||
| Fig. 5 (a) CO2 and N2 pure gas isotherms at 30 °C of UiO-66 synthesized under MW irradiation with varying MW power and (b) IAST CO2 uptake (line) at 0.15 bar and CO2/N2 selectivity (bar) as a function of MW power. | ||
Fig. 5b shows the CO2/N2 selectivity as a function of MW power exhibiting a different trend from the CO2 uptake capacity. From 200 W (0.73d) to 150 W (0.83d), the selectivity decreases from ca. 26 to ca. 21. When further decreased to 50 W (1.8d), the selectivity is enhanced significantly from ca. 21 to ca. 41 (approx. 95% increase). Fig. 6 presents the isosteric heat of adsorption of CO2 calculated based on the isotherms obtained at 30 °C and 40 °C using virial analysis (eqn (1)–(3)).25 It is noted that all isotherms fit well to the virial equation (eqn (3)) with R2 values exceeding 0.999 (Fig. S11 and S12†). Both 200 W (0.73d) and 150 W (0.83d) samples exhibit a similar heat of adsorption and a typical trend where the CO2 heat of adsorption decreases slightly as the CO2 uptake increases. When examined at lower CO2 coverage (<ca. 0.1 mmol g−1), the 150 W sample shows slightly higher heat of adsorption than the 200 W sample while the trend becomes reversed at higher CO2 coverage (>ca. 0.1 mmol g−1). This is likely due to the reduced CO2 confinement effect as pore sizes increase, resulting in decreased CO2/N2 selectivity from ca. 26 (200 W) to ca. 21 (150 W). The 100 W sample shows lower heat of adsorption across the entire CO2 coverage than the samples prepared at 150 W and 200 W, likely due to further enlarged pores. The increase in the selectivity from ca. 21 (150 W) to ca. 25 (100 W) may be attributed to a greater reduction in N2 uptake than CO2 resulting from a more pronounced reduction in the gas confinement effect. However, the 50 W (1.8d) sample displays unexpectedly high CO2 heat of adsorption at low CO2 coverage (<ca. 0.2 mmol g−1) and much pronounced surface heterogeneity where the heat of adsorption decreases from ca. 41 kJ mol−1 to ca. 24 kJ mol−1 as the CO2 coverage increases. Consequently, the most defective sample (50 W) exhibits the highest CO2/N2 selectivity (ca. 41) at 0.15 bar CO2 and 0.85 bar N2 (Fig. 5b). As CO2 loading increases, however, CO2 heat of adsorption ultimately decreases comparable to less defective samples (100 W, 1.1d) due to reduced CO2 confinement effects from larger pore sizes.
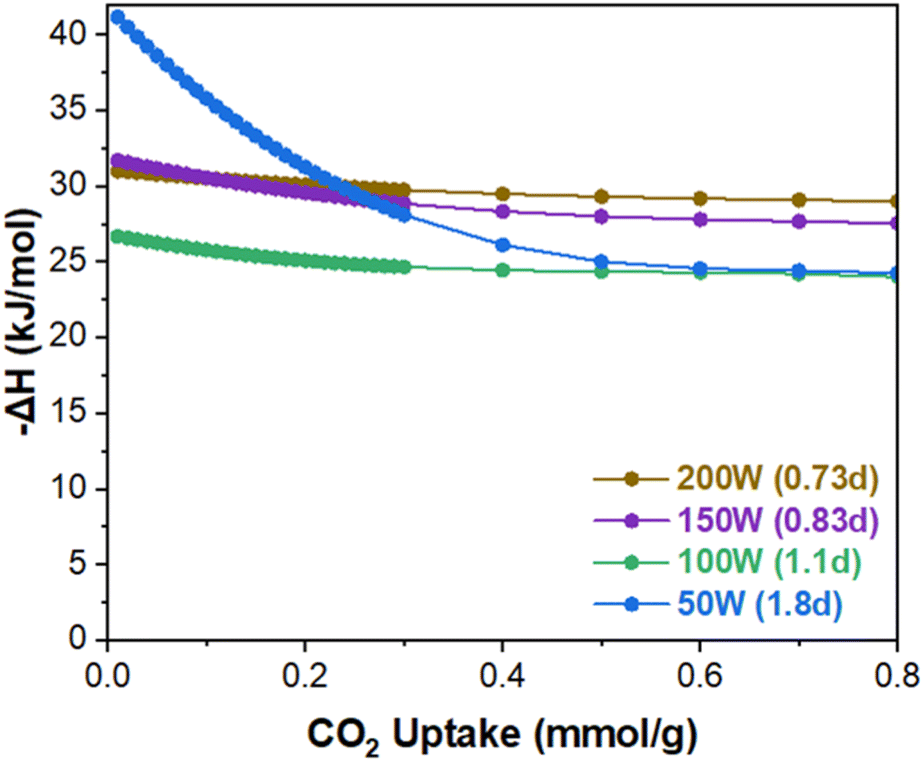 | ||
| Fig. 6 CO2 isosteric heat of adsorption of the MW-assisted synthesized UiO-66 with varying MW power. | ||
To examine the nature of the unexpectedly high CO2 heat of adsorption and the surface heterogeneity of the 50 W sample, UiO-66 samples were synthesized using a thermal (TH) method at varying temperatures. As presented in Fig. S10a,† the defectivity of the sample increases and the CO2/N2 selectivity decreases as temperature increases. Fig. S10d† presents the CO2 isosteric heat of adsorption of the thermally prepared UiO-66 samples, showing the expected trend of the heat of adsorption. Fig. 7 compares the CO2 heat of adsorption at zero CO2 coverage and CO2/N2 selectivity of samples with the highest defectivity prepared by MW (50 W-1.8d) and thermal (RT-1.5d) methods. As observed, the MW sample (50 W) exhibits a CO2 uptake capacity of ca. 0.23 mmol g−1 and a CO2/N2 selectivity of ca. 41 while the TH sample (RT) shows a CO2 uptake capacity of ca. 0.28 mmol g−1 and a CO2/N2 selectivity of ca. 18.4. It is worth mentioning that the 50 W sample displays the highest IAST CO2/N2 selectivity, surpassing all UiO-66 reported so far (Table S4†).14,24,31,41 This surprisingly high CO2/N2 selectivity of the 50 W sample strongly suggests that though similar in the defectivity, the MW sample contains different defects and their distribution as compared to the TH sample.
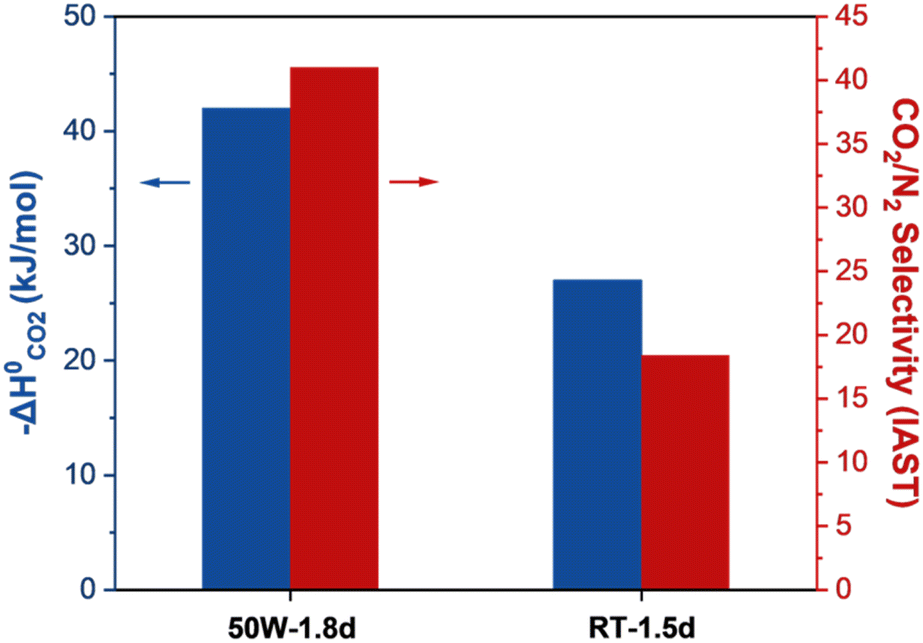 | ||
| Fig. 7 Comparison of the MW (50 W-1.8d) and TH (RT-1.5d) samples with the highest defectivity respectively: CO2 heat of adsorption at zero CO2 coverage (blue bar) and CO2/N2 IAST selectivity (red bar). | ||
3.6 Molecular simulation results for defects in UiO-66
To understand the distribution of different defects and their effects on CO2/N2 adsorption in the MW sample, computational studies were conducted to identify adsorption sites and binding energies. Consistent with previous studies,36,37,42 the hydroxyl group inside the tetrahedral cages of UiO-66 was identified as the strongest site for CO2 adsorption. Consequently, a cluster model was created from a Zr metal–oxide structure with 12 linkers, cleaved from the UiO-66 cell. Both defective and perfect cluster models were simulated using a 35 × 35 × 35 Å cell size. The impact of missing linkers on CO2 interaction energy was modelled by removing one BDC linker at a time and replacing it with acetate or hydroxyl groups to balance the charge and study the effect of different capping. Fig. 8 illustrates the procedure for removing a single organic linker at a time, while terminating the defect with either hydroxyl (Fig. 8b) or acetate groups (Fig. 8c). The results show that the binding energy increases with the presence of defective sites terminated with –OH groups, while acetate terminations result in lower binding energies compared to the pristine tetrahedral cage of UiO-66. The distribution of defects and type of defect influence the availability of high-binding CO2 sites, leading to improved CO2/N2 selectivity. In each unit cell, the distribution of defects can lead to concentrated defects (e.g., three neighbouring missing organic linkers), resulting in higher heat of adsorption.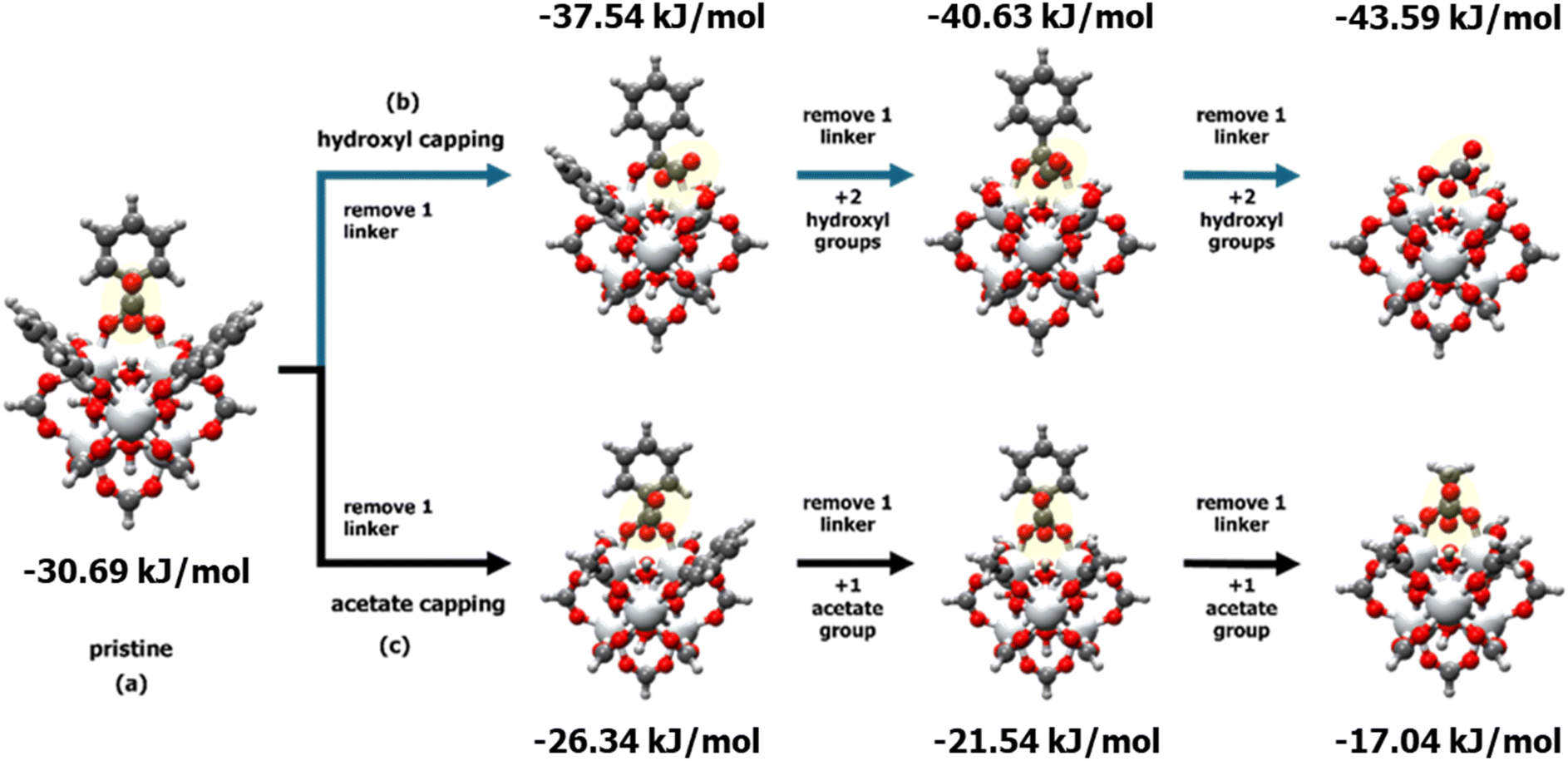 | ||
| Fig. 8 Comparison of the heat of adsorption for (a) pristine and defective UiO-66 clusters terminated with (b) hydroxyl groups and (c) acetate groups, at varying numbers of adjacent ligand defects. | ||
Therefore, the highest CO2 heat of adsorption observed in the 50 W sample at low loading can be attributed to surface heterogeneity, specifically the local distribution of defective sites with different terminations. The 50 W sample also showed the greatest variability in the heat of adsorption as a function of CO2 loading, further highlighting the impact of heterogeneous local defect concentration. Combination of experimental and simulation studies suggests that the 50 W sample likely possesses a more uneven distribution of –OH and –acetate terminations compared to other samples, with a higher local concentration of –OH terminations. This leads to the highest CO2 heat of adsorption at low coverage, despite the 50 W sample having the lowest overall concentration of CO2-philic hydroxyl groups among the UiO-66 samples. As these high-energy sites (i.e., hydroxyl-terminated sites) become occupied by CO2, the heat of adsorption converges with that of the 100 W sample, leaving acetate-terminated and defect-free sites.
We have also investigated the surface area of UiO-66 as a function of the number of linker deficiencies per Zr6 unit (i.e., defectivity) and the type of termination. As shown in Fig. 9, the computational results align well with the experimental data for both surface area (Fig. 9a) and geometric pore volume (Fig. 9b), especially when defects are terminated with hydroxyl groups. It is worth noting that when organic ligands are completely removed, creating Lewis acid sites, the surface area is higher than the values reported in the experiments. The computational results also highlight the increase in PLD values as the number of defects increases (Table S5†). The increase in PLD values complements the observed increase in the geometric pore volume.
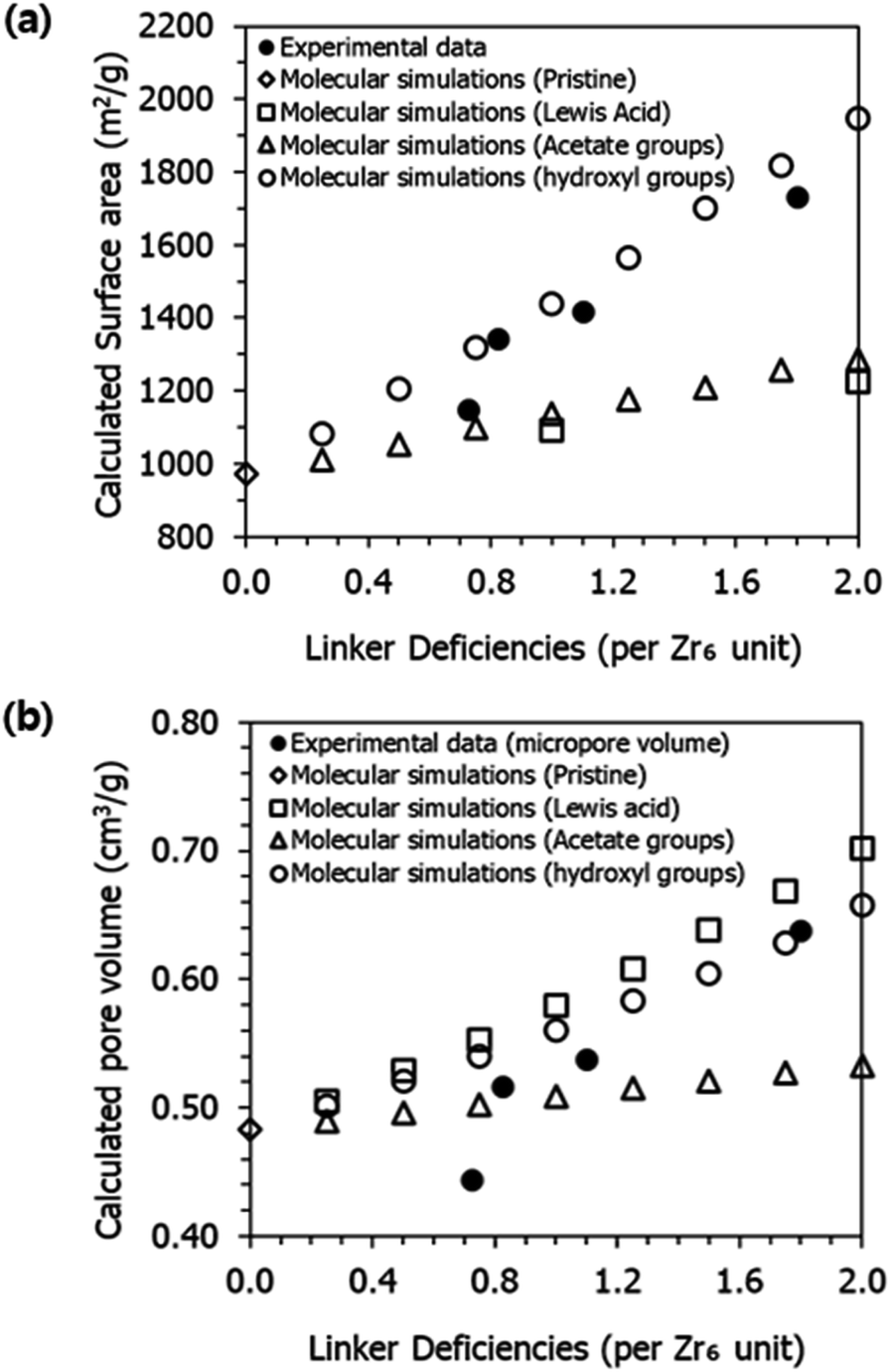 | ||
| Fig. 9 (a) Gravimetric surface area (m2 g−1) and (b) pore volume (cm3 g−1) for pristine and defective UiO-66 structures with various terminations: experimental data and molecular simulations. The positions of the terminated groups were optimized using geometry optimization with the UFF force field. | ||
4 Conclusions
In this study, we have introduced a novel one-pot MW-assisted synthesis of UiO-66. It was found that highly crystalline UiO-66 could be rapidly formed as fast as ca. 90 s of MW irradiation, yielding textural properties comparable to textural properties of those previously prepared using MW methods that require much longer time and higher MW power. These improvements were enabled by the use of zirconium propoxide as the metal precursor, which created more labile reaction intermediates for UiO-66 formation, along with MW irradiation that provided rapid, uniform heating throughout the solution. More importantly, the defectivity of UiO-66 can be controlled by simply varying MW power. As MW power decreases, the defectivity of UiO-66 increases and its textural properties increase. Surprisingly, the most defective UiO-66 prepared at 50 W of MW power showed unexpectedly high CO2 heat of adsorption at zero coverage and unusual surface heterogeneity, displaying the highest CO2/N2 IAST selectivity (ca. 41) outperforming all other UiO-66 reported previously. Molecular simulations confirm that the unexpectedly high CO2 heat of adsorption of the 50 W sample might be due to surface heterogeneity, specifically the local distribution of defective sites with different terminations. These results suggest that our MW-assisted synthesis method enables creation of high-energy defect sites for CO2 adsorption, not possible by conventional thermal methods.While the rapid synthesis protocol presented here is expected to enhance UiO-66 production by improving energy efficiency and reducing synthesis time, further studies are needed to assess the suitability of this method with the organic zirconium salt for large-scale production of UiO-66 and its derivatives. Additionally, the variations in gas adsorption properties by adjusting UiO-66 defectivity, as clarified in this work, would provide guidelines for future applications of UiO-66 produced by the proposed synthesis method. These insights extend not only to CO2 separation using UiO-66 as the adsorbent and membrane but also to CO2 conversion as a heterogeneous catalyst.
Data availability
Data supporting this study are included within the article and/or ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
H.-K. J. acknowledges the financial support from the Korea Evaluation Institute of Industrial Technology (KEIT) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea) (Project: 20018346). This work was supported in part by the Brain Pool Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (RS-2024-00400935). The authors would like to thank Dr Abdoulaye Djire, Dr Manish Shetty, Dr Denis Johnson, Mr Ray Yoo and Ms. Jenna Vito at Texas A&M University for their assistance in obtaining N2 porosimetries of MOF samples.Notes and references
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- K. A. Adegoke, K. G. Akpomie, E. S. Okeke, C. Olisah, A. Malloum, N. W. Maxakato, J. O. Ighalo, J. Conradie, C. R. Ohoro and J. F. Amaku, Sep. Purif. Technol., 2024, 331, 125456 CrossRef CAS.
- A. Dhakshinamoorthy, A. Santiago-Portillo, A. M. Asiri and H. Garcia, ChemCatChem, 2019, 11, 899–923 CrossRef CAS.
- T. Guo, Q. Wei, H. Gao, J. Chen, Z. Liu, J. Li and G. Wang, Mater. Today Chem., 2023, 34, 101693 CrossRef CAS.
- F. Ahmadijokani, H. Molavi, M. Rezakazemi, S. Tajahmadi, A. Bahi, F. Ko, T. M. Aminabhavi, J.-R. Li and M. Arjmand, Prog. Mater. Sci., 2022, 125, 100904 CrossRef CAS.
- J. Winarta, B. Shan, S. M. Mcintyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2019, 20, 1347–1362 CrossRef.
- C. S. Cox, E. Slavich, L. K. Macreadie, L. K. McKemmish and M. Lessio, Chem. Mater., 2023, 35, 3057–3072 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef CAS.
- R. U. Rajesh, T. Mathew, H. Kumar, A. Singhal and L. Thomas, Inorg. Chem. Commun., 2024, 112223 CrossRef.
- S. Xiao, Y. Guan, H. Shang, H. Li, Z. Tian, S. Liu, W. Chen and J. Yang, J. CO2 Util., 2022, 55, 101806 CrossRef CAS.
- T. K. Vo, V. N. Le, K. S. Yoo, M. Song, D. Kim and J. Kim, Cryst. Growth Des., 2019, 19, 4949–4956 CrossRef CAS.
- A. Huang, L. Wan and J. Caro, Mater. Res. Bull., 2018, 98, 308–313 CrossRef CAS.
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D'Alessandro and R. Babarao, Dalton Trans., 2016, 45, 4496–4500 RSC.
- M. Taddei, P. V. Dau, S. M. Cohen, M. Ranocchiari, J. A. van Bokhoven, F. Costantino, S. Sabatini and R. Vivani, Dalton Trans., 2015, 44, 14019–14026 RSC.
- Y. Li, Y. Liu, W. Gao, L. Zhang, W. Liu, J. Lu, Z. Wang and Y.-J. Deng, CrystEngComm, 2014, 16, 7037–7042 RSC.
- A. de la Hoz, A. Díaz-Ortiz and P. Prieto, Alternative Energy Sources for Green Chemistry, Royal Society of Chemistry, 2016 Search PubMed.
- M. Yahia, L. A. Lozano, J. M. Zamaro, C. Téllez and J. Coronas, Sep. Purif. Technol., 2024, 330, 125558 CrossRef CAS.
- L. H. T. Nguyen, T. T. T. Nguyen, Y. T. Dang, P. H. Tran and T. Le Hoang Doan, Asian J. Org. Chem., 2019, 8, 2276–2281 CrossRef CAS.
- Z. Hu and D. Zhao, Dalton Trans., 2015, 44, 19018–19040 RSC.
- M. Usman, A. Helal, M. M. Abdelnaby, A. M. Alloush, M. Zeama and Z. H. Yamani, Chem. Rec., 2021, 1771–1791 CrossRef CAS.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- C. Hartwigsen, S. Gœdecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- J. Yan, T. Ji, Y. Sun, S. Meng, C. Wang and Y. Liu, J. Membr. Sci., 2022, 661, 120959 CrossRef CAS.
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Férey, Chem. Commun., 2010, 46, 767–769 RSC.
- X. Shi, X. Zhang, F. Bi, Z. Zheng, L. Sheng, J. Xu, Z. Wang and Y. Yang, J. Mol. Liq., 2020, 316, 113812 CrossRef CAS.
- P. Chammingkwan, G. Y. Shangkum, P. Mohan, A. Thakur, T. Wada and T. Taniike, RSC Adv., 2020, 10, 28180–28185 RSC.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F.-X. Coudert, Dalton Trans., 2016, 45, 4352–4359 RSC.
- E. Dautzenberg, S. van Hurne, M. M. Smulders and L. C. de Smet, Comput. Phys. Commun., 2022, 280, 108494 CrossRef CAS.
- D. Fu, Y. Park and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2211544119 CrossRef CAS.
- E. J. García, J. Pérez-Pellitero, C. Jallut and G. D. Pirngruber, Phys. Chem. Chem. Phys., 2013, 15, 5648–5657 RSC.
- N. C. Pham, T. K. Vo, Q. B. Nguyen, T. K. Nguyen, T. H. C. Nguyen, N. N. Dao, J. Kim and V. C. Nguyen, Inorg. Chem. Commun., 2023, 158, 111476 CrossRef CAS.
- H. Chevreau, W. Liang, G. J. Kearley, S. G. Duyker, D. M. D'Alessandro and V. K. Peterson, J. Phys. Chem. C, 2015, 119, 6980–6987 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06814a |
| This journal is © The Royal Society of Chemistry 2025 |